3.1. General Experimental Procedures
All commercial materials (Aldrich, Fluka, St. Louis, MO, USA) were used without further purification. All solvents were of reagent grade or HPLC grade. All reactions were carried out under a nitrogen atmosphere unless otherwise noted. All reactions were monitored by thin layer chromatography (TLC) on precoated silica gel 60 F254; spots were visualized with UV light or by treatment with ninhydrin solution in ethanol. Products were purified by flash chromatography (FC) on silica gel 60 (230–400 mesh). 1H-NMR spectra and 13C-NMR spectra were recorded on 300 and 400 MHz spectrometers (AVANCE, Bruker, Billerica, MA, USA). Chemical shifts are reported in parts per million relative to the residual solvent. Multiplicities in 1H-NMR are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, br s = broad singlet. 13C-NMR spectra have been recorded using the APT pulse sequence. The number of carbons reported in the 13C data are derived from Heteronuclear Multiple Bond Correlation (HMBC) experiments. High-resolution MS spectra were recorded with a Micromass Q-ToF micro TM mass spectrometer (Waters, Milford, MA, USA), equipped with an ESI source. Chiral HPLC analysis was performed on a PU-2080 system (JASCO Europe, Cremella (LC), Italy) (UV Detector and binary HPLC pump) at 254 nm. Chiralcel AD columns were purchased from Daicel Chemical Industries® (Osaka, Japan). Optical rotator power was measured with a Jasco P-1030 polarimeter, equipped with a cell of 1 dm path length and 1 mL capacity. The light used has a wavelength of 589 nm (sodium D line).
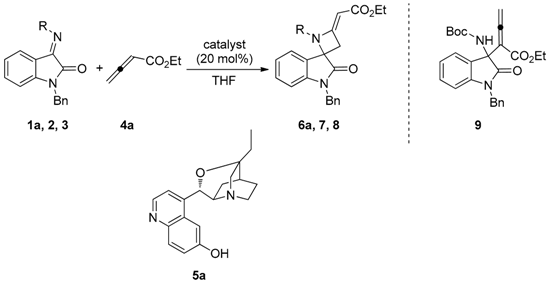
3.1.1. General Procedure for the Synthesis of N-tert-Butylsulfonyl Ketimines 1a–j
To a solution of N-substituted isatin (1.17 mmol, 1.0 eq) in anhydrous CH2Cl2 (2.9 mL, 0.4 M), Ti(OiPr)4 (2.34 mmol, 2.0 eq) and 2-methyl-2-propanesulfinamide (1.4 mmol, 1.2 eq) were added. The solution was refluxed until complete disappearance of the starting materials (monitored by TLC). The reaction was quenched by adding saturated aq. NaHCO3 (15 mL) and diluted with CH2Cl2 (15 mL). The biphasic solution was filtered through a pad of Celite and the organic phase washed with water (2 × 15 mL), dried over Na2SO4 and the solvent was evaporated under reduced pressure. The crude was purified by flash chromatography (FC), using hexane/EtOAc/CH2Cl2 (gradient from 9:1:10 to 5/5/10) as eluent.
New compounds:
(E)-N-(1-Propyl-2-oxoindolin-3-ylidene)-2-methylpropane-2-sulfinamide. Prepared starting from N-1-propylisatin. Orange foam, yield: 82%. 1H-NMR (300 MHz, CDCl3) δ 8.37 (m, br, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.02 (t, J = 7.8 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 3.66 (t, J = 7.4 Hz, 2H), 1.70 (sext, J = 7.5 Hz, 2H), 1.38 (s, 9H), 0.96 (t, J = 7.4 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 161.0, 148.9, 144.2, 136.1, 124.9, 123.9 (2C), 109.9, 54.1, 42.5, 23.8 (3C), 21.3, 12.0; HRMS (ESI): [M + Na]+, Calcd. for C15H20N2NaO2S+ 315.1138, found 315.1142.
(E)-N-(1-Isopropyl-2-oxoindolin-3-ylidene)-2-methylpropane-2-sulfinamide. Prepared starting from N-isopropylisatin. Orange foam, yield: 88%. 1H-NMR (300 MHz, CDCl3) δ 8.40 (m, br, 1H), 7.41 (t, J = 7.8 Hz, 1H), 7.03 (t, J = 7.8 Hz, 1H), 6.93 (d, J = 7.7 Hz, 1H), 4.50 (hept, J = 6.8 Hz, 1H), 1.50 (d, J = 6.8 Hz, 6H), 1.39 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 162.3, 148.3, 143.9, 136.0, 123.8, 123.6 (2C), 110.9, 55.9, 45.4, 23.7 (3C), 22.8 (2C); HRMS (ESI): [M + Na]+, Calcd. for C15H20N2NaO2S+ 315.1138, found 315.1147.
(E)-2-Methyl-N-(2-oxo-1-phenylindolin-3-ylidene)propane-2-sulfinamide. Prepared starting from N-phenylisatin. Orange foam, yield: 76%. 1H-NMR (300 MHz, CDCl3) δ 8.46 (m, br, 1H), 7.54 (t, J = 7.6 Hz, 2H), 7.49–7.33 (m, 4H), 7.10 (t, J = 7.8 Hz, 1H), 6.80 (d, J = 7.8 Hz, 1H), 1.43 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 158.2, 152.3, 149.4, 136.1, 133.9, 130.5 (3C), 129.3 (2C), 127.0, 124.9, 124.7, 111.1, 55.9, 23.9 (3C); HRMS (ESI): [M + Na]+, Calcd. for C18H18N2NaO2S+ 349.0981, found 349.0989.
To a solution of the desired sulfinamide in CH2Cl2 (6 mL), m-CPBA (1.5 eq) was slowly added at room temperature and the mixture was stirred until completion of the reaction (monitored by TLC). The reaction was quenched by adding saturated aq. NaHCO3 (15 mL) and diluted with CH2Cl2 (15 mL). The organic phase was washed with sat. NaHCO3 (2 × 40 mL), dried (Na2SO4), and concentrated. The corresponding sulfonamide was used without further purification.
New compounds:
(E)-2-Methyl-N-(2-oxo-1-tritylindolin-3-ylidene)propane-2-sulfonamide (1c). Red foamy solid, yield: 97%. 1H-NMR (300 MHz, CDCl3) δ 8.42 (d, J = 7.7 Hz, 1H), 7.49–7.37 (m, 6H), 7.37–7.19 (m, 9H), 7.11 (t, J = 7.8 Hz, 1H), 6.97 (d, J = 7.8 Hz, 1H), 6.35 (d, J = 7.8 Hz, 1H), 1.44 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 162.1, 151.5, 144.5, 141.7 (3C), 136.9, 130.1 (6C), 128.6 (6C), 128.0 (3C), 123.9, 123.8, 117.9, 114.9, 75.9, 60.7, 24.5 (3C); HRMS (ESI): [M + Na]+, Calcd. for C31H28N2NaO3S+ 531.1716, found 531.1709.
(E)-2-Methyl-N-(2-oxo-1-propylindolin-3-ylidene)propane-2-sulfonamide (1d). Red foamy solid, yield: 95%. 1H-NMR (300 MHz, CDCl3, 7:3 mixture of two rotamers) δ 8.40 (m, br, 0.3H), 8.06 (d, J = 7.8 Hz, 0.7H), 7.50 (t, J = 7.8 Hz, 0.3H), 7.45 (t, J = 7.8 Hz, 0.7H), 7.11 (t, J = 7.8 Hz, 0.7H), 7.07 (t, J = 7.8 Hz, 0.3H), 6.93 (d, J = 7.8 Hz, 0.7H), 6.82 (t, J = 7.8 Hz, 0.3H), 3.74–3.62 (m, 2H), 1.74 (sext, br, J = 6.8 Hz, 2H), 1.62 (s, 6.3H), 1.60 (s, 2.7H), 0.99 (t, br, J = 6.8 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ 162.6, 150.4, 147.4, 139.0 and 138.5 (1C), 124.0 (2C), 116.2, 110.4 and 110.3 (1C), 60.6, 43.1 and 42.7 (1C), 24.5 (3C), 21.2 and 20.8 (1C), 12.0; HRMS (ESI): [M + Na]+, Calcd. for C15H20N2NaO3S+ 331.1087, found 331.1096.
(E)-2-(1-Isopropyl-2-oxoindolin-3-ylidene)-2-methylpropane-2-sulfonamide (1e). Red foamy solid, yield: 99%. 1H-NMR (400 MHz, CDCl3, 17:3 mixture of two rotamers) δ 8.19 (dd, J = 7.8 and 1.7 Hz, 0.15H), 8.12 (dd, J = 7.8 and 1.7 Hz, 0.85H), 7.76 (ddd, J = 8.2, 7.5 and 1.7 Hz, 0.15H), 7.73 (ddd, J = 8.2, 7.5 and 1.7 Hz, 0.85H), 7.34–7.21 (m, 2H), 4.77 (hept, J = 6.8 Hz, 1H), 1.62 (d, J = 6.9 Hz, 6H), 1.62 (s, 1.35H) 1.57 (s, 7.65H); 13C-NMR (101 MHz, CDCl3) δ 162.6, 158.0, 149.9, 138.9 and 138.5 (1C), 124.9 and 124.7 (1C), 124.0 and 123.7 (1C), 113.3, 112.0 and 111.3 (1C), 60.0, 45.7 and 45.5 (1C), 24.7 and 24.5 (3C), 19.9 (2C); HRMS (ESI): [M + Na]+, Calcd. for C15H20N2NaO3S+ 331.1087, found 331.1081.
(E)-2-Methyl-N-(2-oxo-1-phenylindolin-3-ylidene)propane-2-sulfonamide (1f). Red foamy solid, yield: 98%. 1H-NMR (400 MHz, CDCl3, 7:3 mixture of two rotamers) δ 8.21 (dd, J = 7.9 and 1.4 Hz, 0.7H), 8.00 (d, br, J = 7.9 Hz, 0.3H), 7.69–7.53 (m, 4H), 7.44 (t, J = 7.8 Hz, 0.3H), 7.41–7.26 (m, 2.7H), 6.58 (d, J = 7.8 Hz, 1H), 1.47 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 162.2, 150.9, 143.1, 138.5 and 137.5 (1C), 133.7, 130.6 (3C), 129.6 and 129.5 (2C), 127.1, 125.0 and 124.7 (1C), 115.9, 111.4, 60.8, 24.9–24.5 (3C); HRMS (ESI): [M + Na]+, Calcd. for C18H18N2NaO3S+ 365.0930, found 365.0939.
(E)-N-(1-Benzyl-5-methoxy-2-oxoindolin-3-ylidene)-2-methylpropane-2-sulfonamide (1g). Red foamy solid, yield: 98%. 1H-NMR (400 MHz, CDCl3) δ 7.42–7.26 (m, 6H), 6.99 (dd, J = 8.2 and 2.6 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 4.89 (s, 2H), 3.79 (s, 3H), 1.64 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 162.7, 156.7, 145.2, 144.1, 135.3, 129.7 (2C), 128.8, 128.0 (2C), 126.7, 125.6, 116.7, 111.9, 60.7, 56.7, 44.9, 24.6 (3C); HRMS (ESI): [M + Na]+, Calcd. for C20H22N2NaO4S+ 409.1192, found 409.1186.
(E)-N-(1-Benzyl-5-chloro-2-oxoindolin-3-ylidene)-2-methylpropane-2-sulfonamide (1h). Red foamy solid, yield: 92%. 1H-NMR (400 MHz, CDCl3, 4:1 mixture of two rotamers) δ 7.42–7.26 (m, 7H), 6.76 (d, J = 8.2 Hz, 0.2H), 6.68 (d, J = 8.2 Hz, 0.8H), 4.93 (s, 2H), 1.67 (s, 1.8H), 1.65 (s, 7.2H); 13C-NMR (101 MHz, CDCl3) δ 161.2, 148.2, 145.6, 138.3 and 137.9 (1C), 134.7, 130.7 and 129.8 (2C), 129.9, 129.0 (2C), 128.0 (2C), 116.9, 112.2, 63.9, 45.5 and 45.0 (1C), 24.6 and 24.5 (3C); HRMS (ESI): [M + Na]+, Calcd. for C19H19ClN2NaO3S+ 413.0697, found 413.0674.
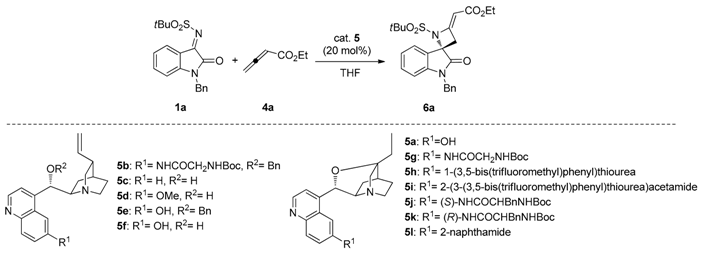
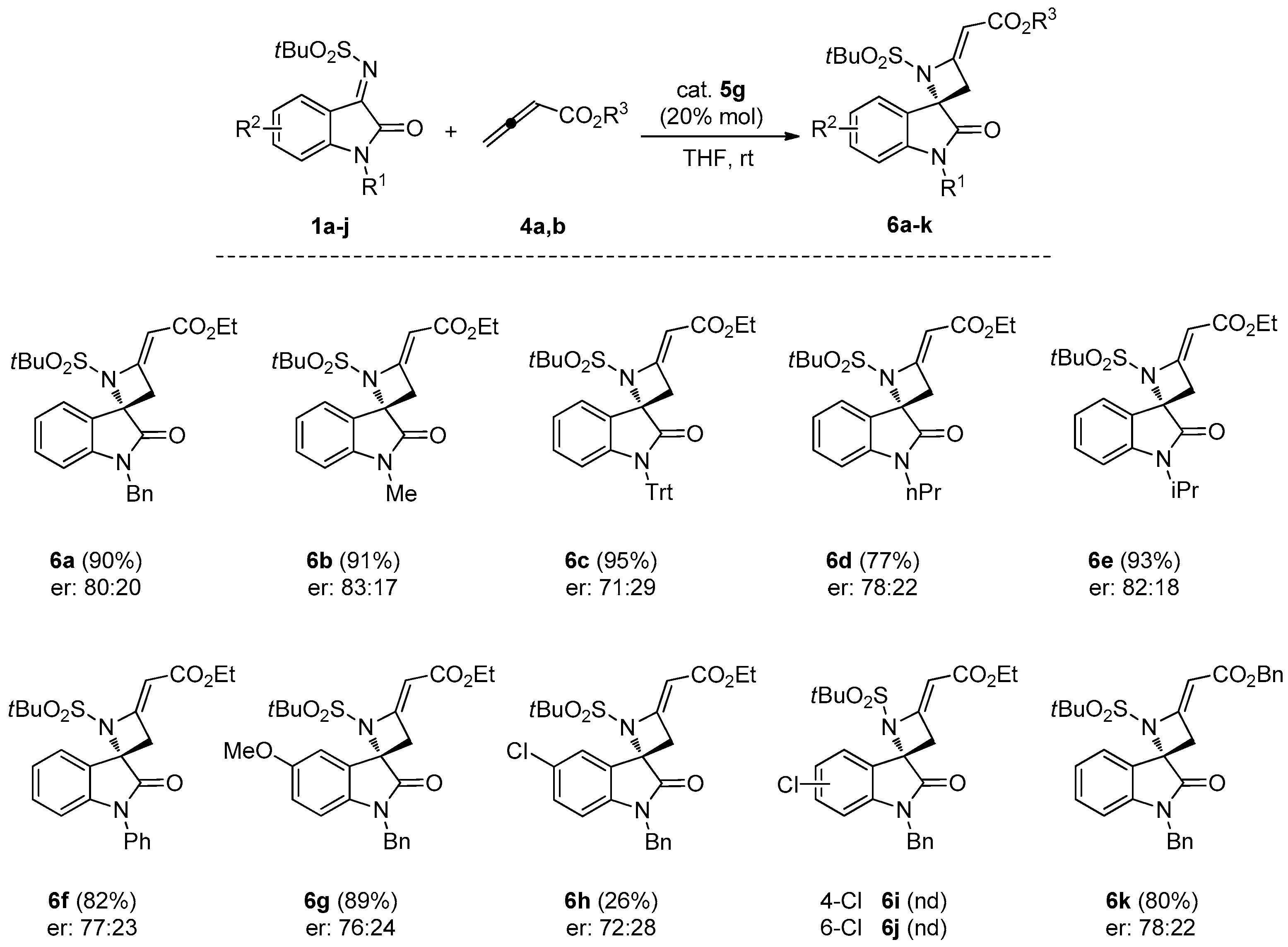
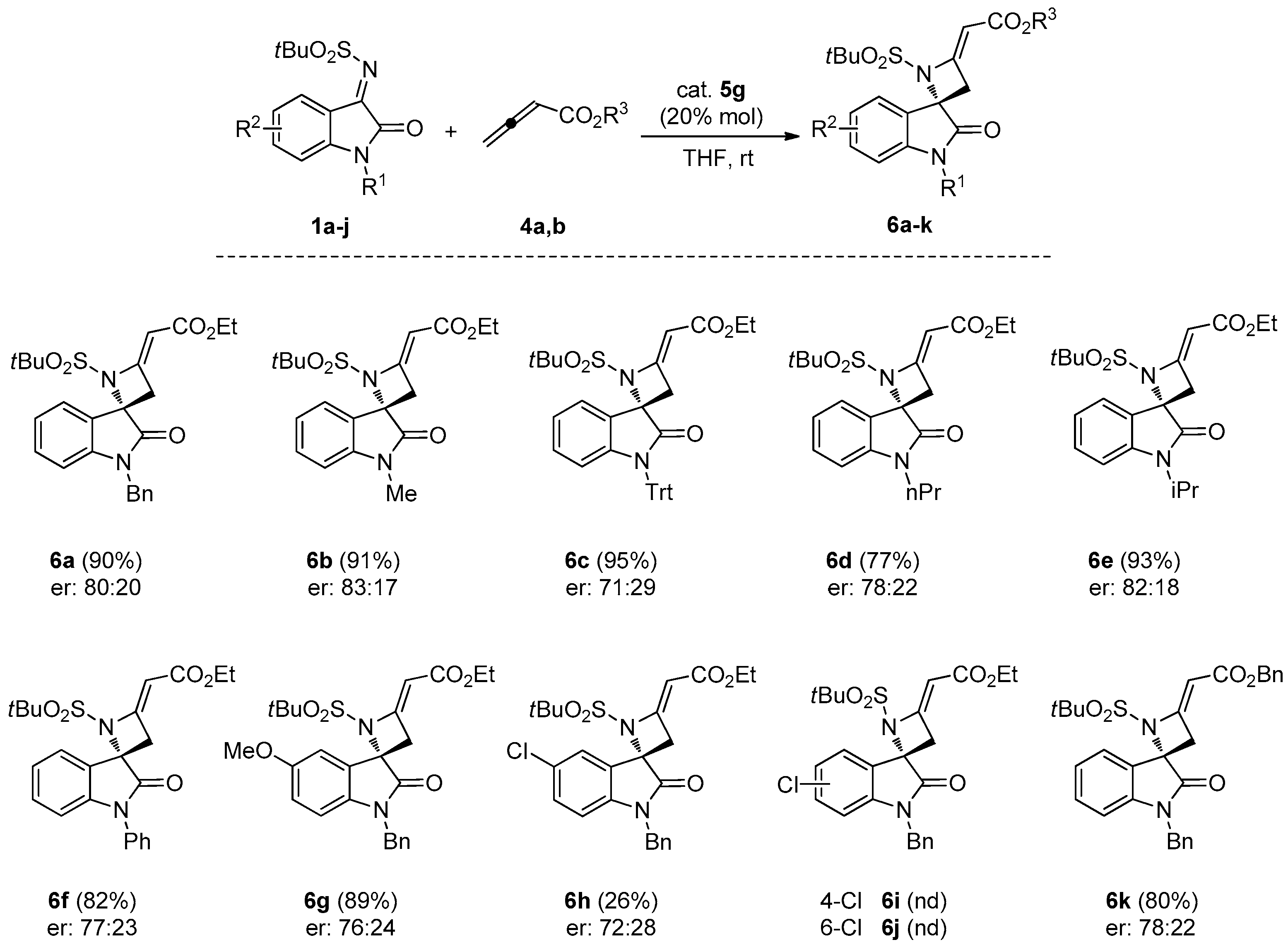
3.1.2. General Procedure for the Synthesis of Spiroazetidines 6a–h, 6k
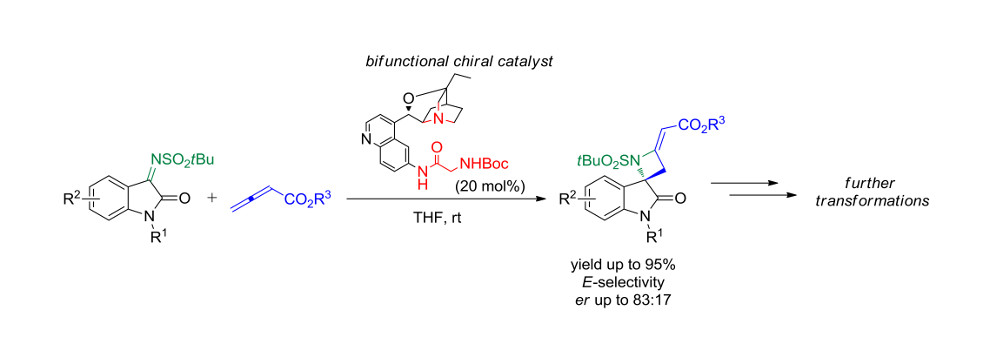
To a solution of the appropriate N-tert-butylsulfonyl ketimine 1a–j (0.10 mmol) and catalyst 5g (0.02 mmol) in THF (1.5 mL), allenoate 4a (or 4b, for spiroazetidine 6k) was added (0.20 mmol). The mixture was stirred at room temperature and the conversion was monitored by TLC. The solvent was evaporated under reduced pressure and the crude was purified by FC, using CH2Cl2/EtOAc 9/1 as eluent.
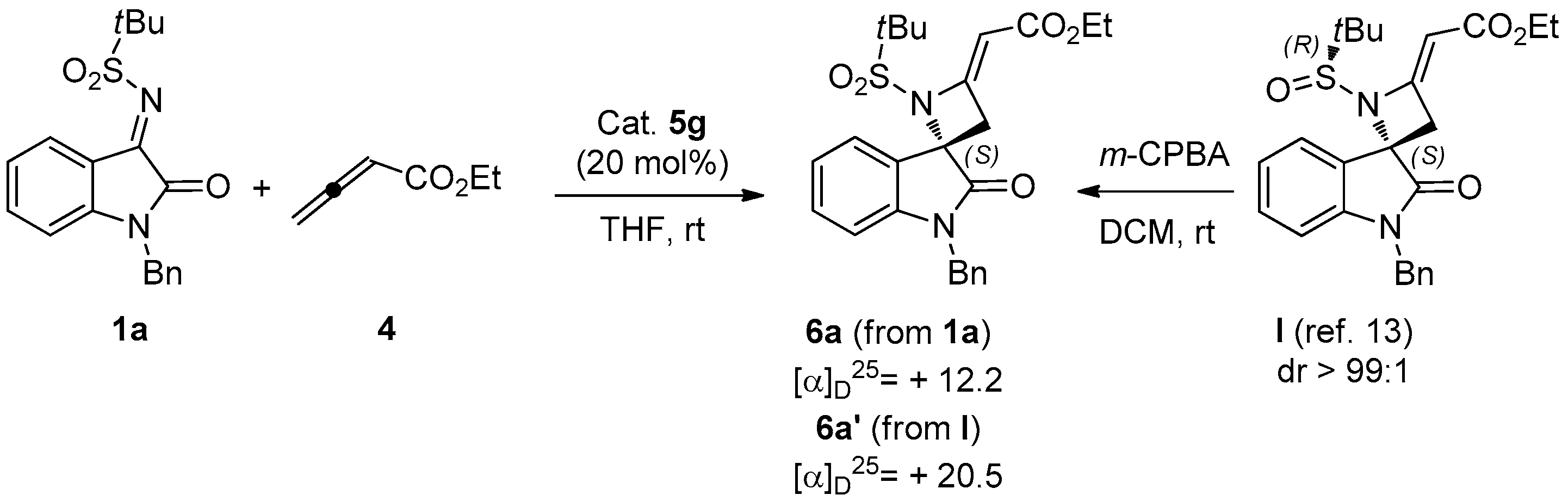
(S,E)-Ethyl-2-(1′-benzyl-1-(tert-butylsulfonyl)-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6a). Pale orange foam; yield: 90%. 1H-NMR (300 MHz, CDCl3) δ 7.50 (d, br, J = 7.8 Hz, 1H), 7.19–7.38 (m, 6H), 7.08 (t, br, J = 7.7 Hz, 1H), 6.69 (d, br, J = 7.8 Hz, 1H), 5.68 (s, br, 1H), 5.04 and 4.83 (AB system, J = 15.6 Hz, 2H), 4.16 (q, J = 7.1 Hz, 2H), 3.71 (dd, J = 15.6 and 1.9 Hz, 1H), 3.44 (dd, J = 15.6 and 1.9 Hz, 1H), 1.33 (s, 9H), 1.27 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 173.3, 167.3, 159.2, 143.6, 134.6, 129.0 (3C), 128.2, 127.5 (2C), 126.4, 126.2, 124.6, 113.2, 94.3, 70.6, 59.7, 58.9, 44.4, 41.8, 22.7 (3C), 14.5; = +12.2 (c 0.90, CHCl3); HRMS (ESI): [M + Na]+, Calcd. for C25H28N2NaO5S+ 491.1611, found 491.1604; enantiomeric ratio: 80:20, determined by HPLC (C-AD, Hexane/iPrOH 7:3, flow: 0.7 mL/min, λ = 254 nm): tR = 16.80 min (major) tR = 23.38 min (minor).
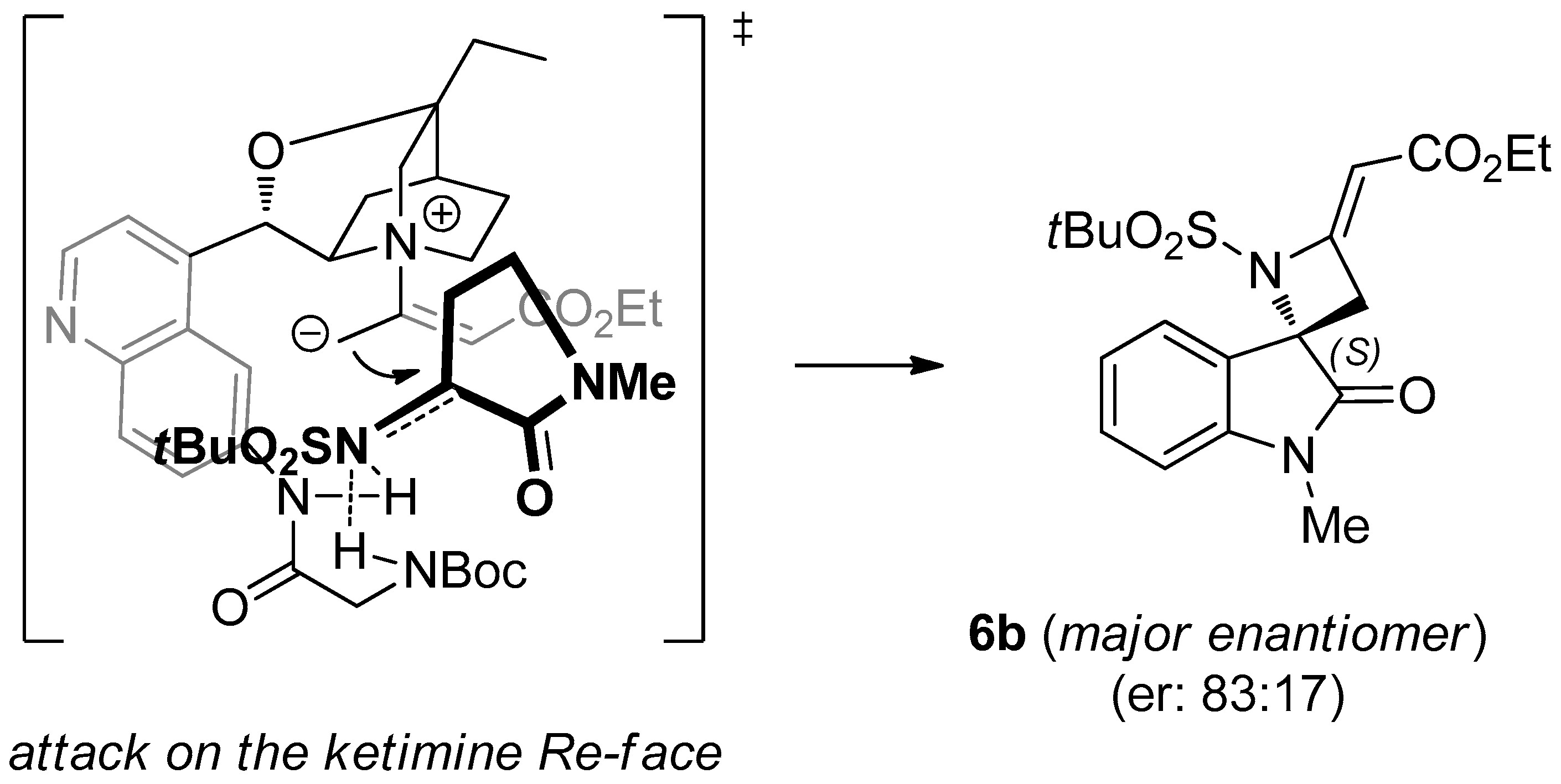
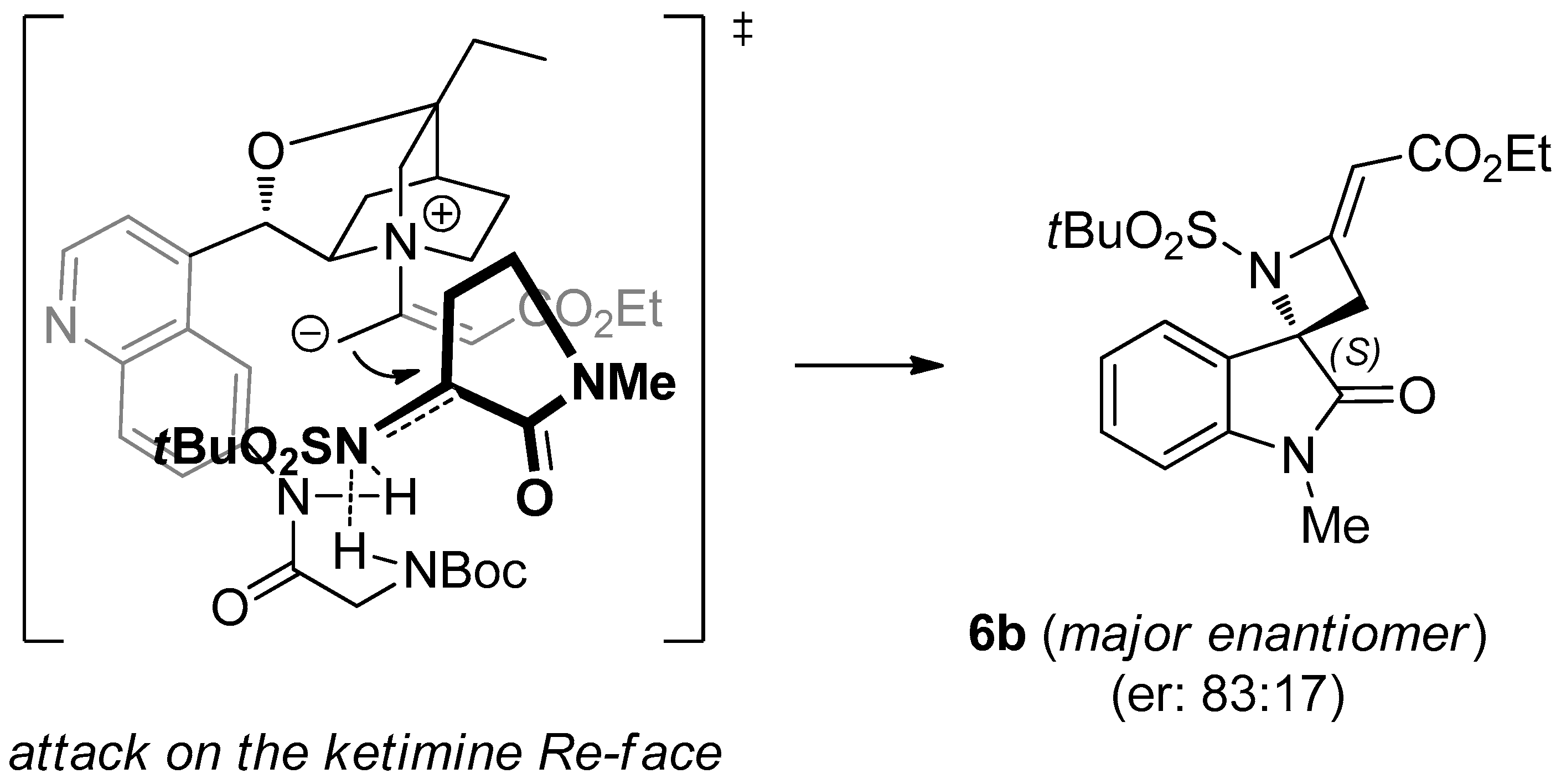
(S,E)-Ethyl-2-(1-(tert-butylsulfonyl)-1′-methyl-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6b). Pale orange foam; yield: 91%. 1H-NMR (400 MHz, CDCl3) δ 7.50 (d, J = 7.5 Hz, 1H), 7.38 (d, J = 7.5 Hz, 1H), 7.12 (t, J = 7.5 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 5.66 (t, J = 1.9 Hz, 1H), 4.16 (q, J = 7.1 Hz, 2H), 3.66 (dd, J = 16.0, 1.9 Hz, 1H), 3.41 (dd, J = 16.0, 1.9 Hz, 1H), 3.24 (s, 3H), 1.32 (s, 9H), 1.23–1.30 (m, 3H); 13C-NMR (101 MHz, CDCl3) δ 173.3, 167.7, 159.4, 144.7, 131.8, 125.9, 125.0, 123.7, 109.5, 95.9, 71.7, 62.2, 60.5, 39.8, 27.3, 24.5 (3C), 15.0; = +19.3 (c 0.80, CHCl3); HRMS (ESI): [M + Na]+, Calcd. for C19H24N2NaO5S+: 415.1298, found 415.1303; enantiomeric ratio: 83:17, determined by HPLC (C-AD, Hexane/iPrOH 7:3, flow: 0.7 mL/min, λ = 254 nm): tR = 24.96 min (major) tR = 29.58 min (minor).
(S,E)-Ethyl-2-(1-(tert-butylsulfonyl)-2′-oxo-1′-tritylspiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6c). Pale orange foam; yield: 95%. 1H-NMR (300 MHz, CDCl3) δ 7.50–7.39 (m, 7H), 7.30–7.16 (m, 9H), 7.03–6.89 (m, 2H), 6.28 (d, J = 7.4 Hz, 1H), 5.62 (t, J = 1.9 Hz, 1H), 4.19–4.05 (m, 2H), 3.58 (dd, J = 15.7, 1.9 Hz, 1H), 3.31 (dd, J = 15.7, 1.9 Hz, 1H), 1.34 (s, 9H), 1.24 (t, J = 6.8 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ 172.9, 167.2, 9.0, 144.0, 141.7 (3C), 129.5 (7C), 127.7 (6C), 127.1 (3C), 125.6, 123.9, 122.6, 116.6, 94.9, 74.9, 71.6, 61.7, 59.8, 40.1, 24.1 (3C), 14.4; = −0.67 (c 1.5, CHCl3); HRMS (ESI): [M + Na]+, Calcd. for C37H36N2NaO5S+: 643.2237, found 643.2240; enantiomeric ratio: 71:29, determined by HPLC (C-AD, Hexane/iPrOH 7:3, flow: 0.7 mL/min, λ = 254 nm): tR = 8.00 min (major) tR = 11.84 min (minor).
(S,E)-Ethyl-2-(1-(tert-butylsulfonyl)-2′-oxo-1′-propylspiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6d). Pale orange foam; yield: 77%. 1H-NMR (300 MHz, CDCl3) δ 7.49 (d, J = 7.4 Hz, 1H), 7.35 (td, J = 7.4 and 1.1 Hz, 1H), 7.10 (t, J = 7.4 Hz, 1H), 6.86 (d, J = 7.4 Hz, 1H), 5.65 (t, J = 1.9 Hz, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.75 (dt, J = 14.6 and 6.8 Hz, 1H), 3.63 (dd, J = 15.6 and 1.9 Hz, 1H), 3.58 (dt, J = 14.6 and 6.8 Hz, 1H), 3.39 (dd, J = 15.6 and 1.9 Hz, 1H), 1.72 (sext, J = 6.8 Hz, 2H), 1.32 (s, 9H), 1.27–1.22 (m, 3H) 0.97 (t, J = 6.8 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 173.1, 167.8, 159.6, 142.9, 131.6, 126.2, 125.1, 123.5, 109.8, 95.7, 71.8, 60.5, 42.7, 40.0, 30.4, 24.6 (3C), 21.2, 15.1, 12.0; = +5.5 (c 1.02, CHCl3); HRMS (ESI) calculated for C21H28N2NaO5S+ [M + Na]+ 443.1611, found 443.1617; enantiomeric ratio: 78:22, determined by HPLC (C-AD, Hexane/iPrOH 7:3, flow: 0.7 mL/min, λ = 254 nm): tR = 7.73 min (major) tR = 9.85 min (minor).
(S,E)-Ethyl-2-(1-(tert-butylsulfonyl)-1′-isopropyl-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6e). Pale orange foam; yield: 93%. 1H-NMR (300 MHz, CDCl3) δ 7.49 (d, J = 7.7 Hz, 1H), 7.34 (t, J = 7.7 Hz, 1H), 7.08 (t, J = 7.7 Hz, 1H), 6.99 (d, J = 7.7 Hz, 1H), 5.64 (t, J = 2.0 Hz, 1H), 4.56 (hept, J = 6.8 Hz, 1H), 4.15 (q, J = 7.6 Hz, 2H), 3.64 (dd, J = 15.9 and 2.0 Hz, 1H), 3.40 (dd, J = 15.9, 2.0 Hz, 1H), 1.50 (d, J = 6.8 Hz, 3H), 1.48 (d, J = 6.8 Hz, 3H), 1.30 (s, 9H), 1.26 (t, J = 7.6 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 172.4, 167.2, 159.1, 143.2, 130.9, 125.6, 124.8, 122.5, 110.4, 95.0, 71.1, 61.5, 59.9, 44.7, 39.1, 23.9 (3C), 19.2 (2C), 14.5; = +14.7 (c 0.99, CHCl3); HRMS (ESI) calculated for C21H28N2NaO5S+ [M + Na]+ 443.1611, found 443.1616; enantiomeric ratio 82:18, determined by HPLC (C-AD, Hexane/iPrOH 7:3, flow: 0.7 mL/min, λ = 254 nm): tR = 7.32 min (major) tR = 9.33 min (minor).
(S,E)-Ethyl-2-(1-(tert-butylsulfonyl)-2′-oxo-1′-phenylspiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6f). Pale orange foam; yield: 82%. 1H-NMR (300 MHz, CDCl3) δ 7.64–7.39 (m, 6H), 7.30 (t, J = 7.8 Hz, 1H), 7.15 (t, J = 7.8 Hz, 1H), 6.83 (d, J = 7.8 Hz, 1H), 5.68 (t, J = 2.0 Hz, 1H), 4.17 (q, J = 7.2 Hz, 2H), 3.77 (dd, J = 16.0, 2.0 Hz, 1H), 3.52 (dd, J = 16.0, 2.0 Hz, 1H), 1.33 (s, 9H), 1.27 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 172.3, 167.2, 158.9, 144.6, 134.0, 131.2, 129.8 (2C), 128.5, 126.6 (2C), 125.8, 124.9, 123.5, 110.2, 95.4, 71.3, 61.6, 60.0, 39.2, 24.0 (3C), 14.5; = +10.3 (c 1.04, CHCl3); HRMS (ESI) calculated for C24H26N2NaO5S+ [M + Na]+ 477.1455, found 447.1460; enantiomeric ratio: 77:23, determined by HPLC (C-AD, Hexane/iPrOH 4:1, flow: 0.7 mL/min, λ = 254 nm): tR = 9.63 min (major) tR = 14.79 min (minor).
(S,E)-Ethyl-2-(1′-benzyl-1-(tert-butylsulfonyl)-5′-methoxy-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)-acetate (6g). Pale orange foam; yield: 89%. 1H-NMR (400 MHz, CDCl3) δ 7.38–7.27 (m, 5H), 7.12 (d, J = 2.7 Hz, 1H), 6.78 (dd, J = 8.4 and 2.7 Hz, 1H), 6.60 (d, J = 8.4 Hz, 1H), 5.71 (t, J = 1.9 Hz, 1H), 5.05 and 4.81 (AB system, J = 15.8 Hz, 2H), 4.19 (q, J = 7.2 Hz, 2H), 3.78 (s, 3H), 3.75 (dd, J = 15.9 and 1.9 Hz, 1H), 3.45 (dd, J = 15.9 and 1.9 Hz, 1H), 1.38 (s, 9H), 1.30 (t, J = 7.2 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 172.5, 167.1, 158.8, 156.4, 136.4, 135.2, 128.8 (2C), 127.7, 127.3 (2C), 127.0, 115.4, 111.4, 110.5, 95.2, 71.5, 61.8, 59.9, 55.9, 44.3, 39.7, 24.0 (3C), 14.4; = +8.9 (c 0.95, CHCl3); HRMS (ESI) calculated for C26H30N2NaO5S+ [M + Na]+ 521.1717, found 521.1720; enantiomeric ratio: 76:24, determined by HPLC (C-AD, Hexane/iPrOH 4:1, flow: 0.7 mL/min, λ = 254 nm): tR = 15.00 min (major) tR = 20.63 min (minor).
(S,E)-Ethyl-2-(1′-benzyl-1-(tert-butylsulfonyl)-5′-chloro-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)-acetate (6h). Pale orange foam; yield: 26%. 1H-NMR (400 MHz, CDCl3) δ 7.52 (d, J = 2.1 Hz, 1H), 7.38–7.26 (m, 5H), 7.23 (dd, J = 8.2 and 2.1 Hz, 1H), 6.63 (d, J = 8.2 Hz, 1H), 5.71 (t, J = 2.1 Hz, 1H), 5.07 and 4.82 (AB system, J = 16.0 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 3.74 (dd, J = 15.9 and 2.1 Hz, 1H), 3.47 (dd, J = 15.9 and 2.1 Hz, 1H), 1.39 (s, 9H), 1.31 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 172.4, 166.9, 158.2, 141.6, 136.7, 134.6, 130.8, 129.0 (2C), 128.7, 127.9, 127.2 (2C), 124.7, 111.0, 95.6, 70.7, 61.8, 60.0, 44.4, 39.5, 23.9 (3C), 14.4; = +9.8 (c 1.05, CHCl3); HRMS (ESI) calculated for C25H27ClN2NaO5S+ [M + Na]+ 525.1221, found 525.1229; enantiomeric ratio: 72:28, determined by HPLC (C-AD, Hexane/iPrOH 4:1, flow: 0.7 mL/min, λ = 254 nm): tR = 10.31 min (major) tR = 16.59 (minor).
(S,E)-Benzyl-2-(1′-benzyl-1-(tert-butylsulfonyl)-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)acetate (6k). Pale yellow foam; yield: 80%. 1H-NMR (400 MHz, CDCl3) δ 7.53 (d, br, J = 7.8 Hz, 1H), 7.44–7.23 (m, 11H), 7.11 (t, br, J = 7.7 Hz, 1H), 6.72 (d, br, J = 7.8 Hz, 1H), 5.78 (s, br, 1H), 5.19 (s, br, 2H), 5.08 and 4.84 (AB system, J = 15.8 Hz, 2H), 3.74 (d, br, J = 16.0 Hz, 1H), 3.47 (d, br, J = 16.0 Hz, 1H), 1.36 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 173.4, 167.5, 160.0, 143.9, 136.9, 135.7, 131.7, 129.5 (3C), 129.3, 129.0, 128.8, 128.4, 128.0 (3C), 126.0, 125.1, 123.8, 110.7, 95.5, 71.8, 66.6, 62.3, 44.9, 40.3, 24.6 (3C); = +15.0 (c 0.8, CHCl3); HRMS (ESI): [M + Na]+, Calcd. for C30H30N2NaO5S+ 553.1768, found 553.1772; enantiomeric ratio: 78:22, determined by HPLC (C-AD, Hexane/iPrOH 7:3, flow: 0.7 mL/min, λ = 254 nm): tR = 16.80 min (major) tR = 23.38 min (minor).
3.1.3. Reactions of 6a
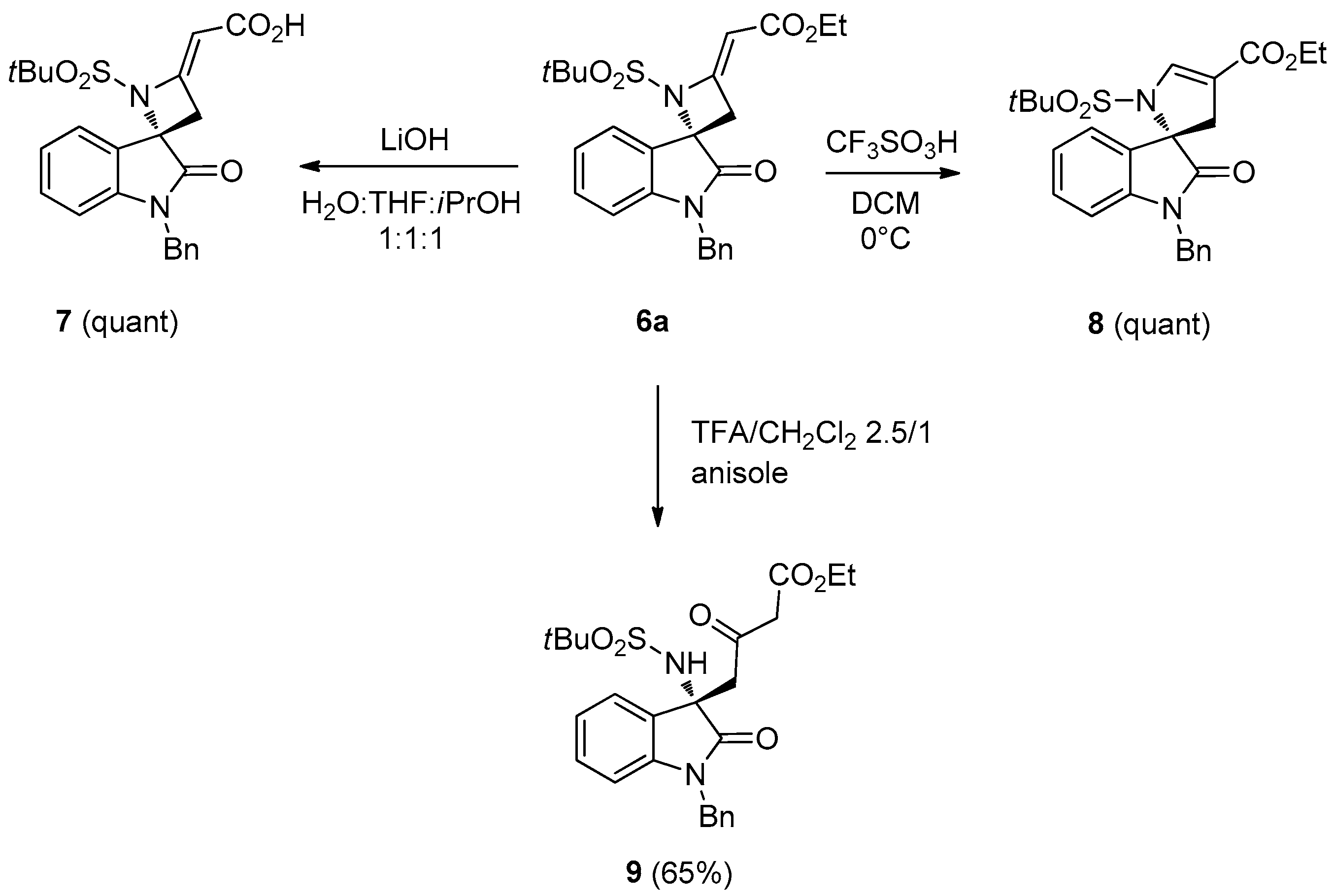
(S,E)-2-(1′-Benzyl-1-(tert-butylsulfonyl)-2′-oxospiro[azetidine-2,3′-indolin]-4-ylidene)acetic acid (7). To a solution of 6a (0.21 mmol) in water/THF/iPrOH (1:1:1, 1.5 mL), LiOH (0.63 mmol) was added and the mixture was stirred at room temperature. After the completion of the reaction (monitored by TLC), the reaction mixture was quenched with a 1 M aqueous solution of HCl and extracted with CH2Cl2. The organic layer was collected, dried over anhydrous sodium sulfate, and concentrated under reduced pressure affording the product as a white foam (quantitative yield). 1H-NMR (400 MHz, CDCl3) δ 7.54 (d, J = 7.4 Hz, 1H), 7.41–7.23 (m, 6H), 7.12 (t, J = 7.4 Hz, 1H), 6.73 (d, J = 7.4 Hz, 1H), 5.71 (t, J = 1.9 Hz, 1H), 5.07 and 4.87 (AB system, J = 15.8 Hz, 2H), 3.76 (dd, J = 16.3 and 2.0 Hz, 1H), 3.49 (dd, J = 16.3 and 2.0 Hz, 1H), 2.05 (m, br, 1H), 1.37 (s, 9H); 13C-NMR (101 MHz, CDCl3) δ 172.6, 172.5, 161.3, 143.2, 135.0, 131.1, 128.9 (2C), 127.8, 127.3 (2C), 125.1, 124.4, 123.2, 110.1, 94.4, 71.2, 61.8, 44.3, 39.7, 23.9 (3C); = +5.7 (c 0.79, CHCl3); HRMS (ESI) calculated for C23H24N2NaO5S+ [M + Na]+ 463.1298, found 463.1289.
(S)-Ethyl-1-benzyl-1′-(tert-butylsulfonyl)-2-oxo-1′,3′-dihydrospiro[indoline-3,2′-pyrrole]-4′-carboxylate (8). To a stirred solution of 6a (0.21 mmol) in CH2Cl2 (9 mL) at 0 °C, CF3SO3H (0.68 mmol) was added. After stirring at 0 °C for 2 h, the mixture was neutralized with 0.1 M NaOH and extracted with CH2Cl2 (4 × 15 mL). The combined organic phases were dried (MgSO4) and concentrated in vacuo, affording the product as a white foam (quantitative yield). 1H-NMR (400 MHz, CDCl3) δ 7.56 (d, br, J = 7.8 Hz, 1H), 7.40–7.25 (m, 6H), 7.05 (t, br, J = 7.7 Hz, 1H), 6.79 (d, br, J = 7.8 Hz, 1H), 6.15 (s, br, 1H), 4.97 and 4.91 (AB system, J = 15.5 Hz, 2H), 4.23 (q, J = 7.1 Hz, 2H), 3.49 (d, br, J = 16.4 Hz, 1H), 2.63 (d, br, J = 16.4 Hz, 1H), 1.46 (s, 9H), 1.38 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 171.1, 170.5, 169.7, 142.3, 134.7, 131.6, 129.1 (2C), 128.1, 127.3 (2C), 126.4, 125.1, 124.0, 81.2, 79.7, 66.6, 58.5, 44.2, 38.4, 24.0 (3C), 14.2; = +6.3 (c 0.53, CHCl3); HRMS (ESI): [M + Na]+, Calcd. for C25H28N2NaO5S+ 491.1611, found 491.1617.
(S)-Ethyl-4-(1-benzyl-3-(tert-butylsulfonamido)-2-oxoindolin-3-yl)-3-oxobutanoate (9). To a solution of 6a (0.20 mmol) in trifluoroacetic acid (5 mL) and CH2Cl2 (2 mL), anisole (4 mmol) was added, and the mixture was stirred for 1 h at 60 °C. To this saturated Na2CO3 was added and organic materials were extracted with EtOAc. Dried and concentrated extract was subjected to FC (Hexane/EtOAc 1/1) to give the product as a pale yellow foam (yield: 65%). 1H-NMR (400 MHz, CDCl3) δ 7.57 (d, br, J = 7.8 Hz, 1H), 7.43–7.24 (m, 5H), 7.21 (t, br, J = 7.7 Hz, 1H), 7.04 (t, J = 7.7 Hz, 1H), 6.72 (d, br, J = 7.8 Hz, 1H), 5.75 (s, br, 1H), 5.00 and 4.95 (AB system, J = 16.0 Hz, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.60 (d, J = 17.4 Hz, 1H), 3.45 and 3.40 (AB system, J = 16.0 Hz, 2H), 3.07 (d, J = 17.4 Hz, 1H), 1.34 (s, 9H), 1.22 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3) δ 201.0, 174.7, 166.1, 142.4, 135.6, 129.7, 128.8 (2C), 128.7, 127.7, 127.3 (2C), 125.3, 122.9, 109.7, 61.7, 60.6, 60.3, 50.1, 49.2, 44.4, 24.1 (3C), 14.0; = −57.4 (c 0.50, CHCl3); HRMS (ESI): [M + Na]+, Calcd. for C25H30N2NaO6S+ 509.1717, found 509.1711.

 and
and 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}