DNA G-Wire Formation Using an Artificial Peptide is Controlled by Protease Activity

 ,
,  , ,
, , 

Abstract
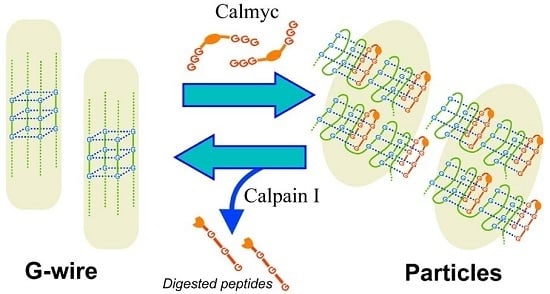
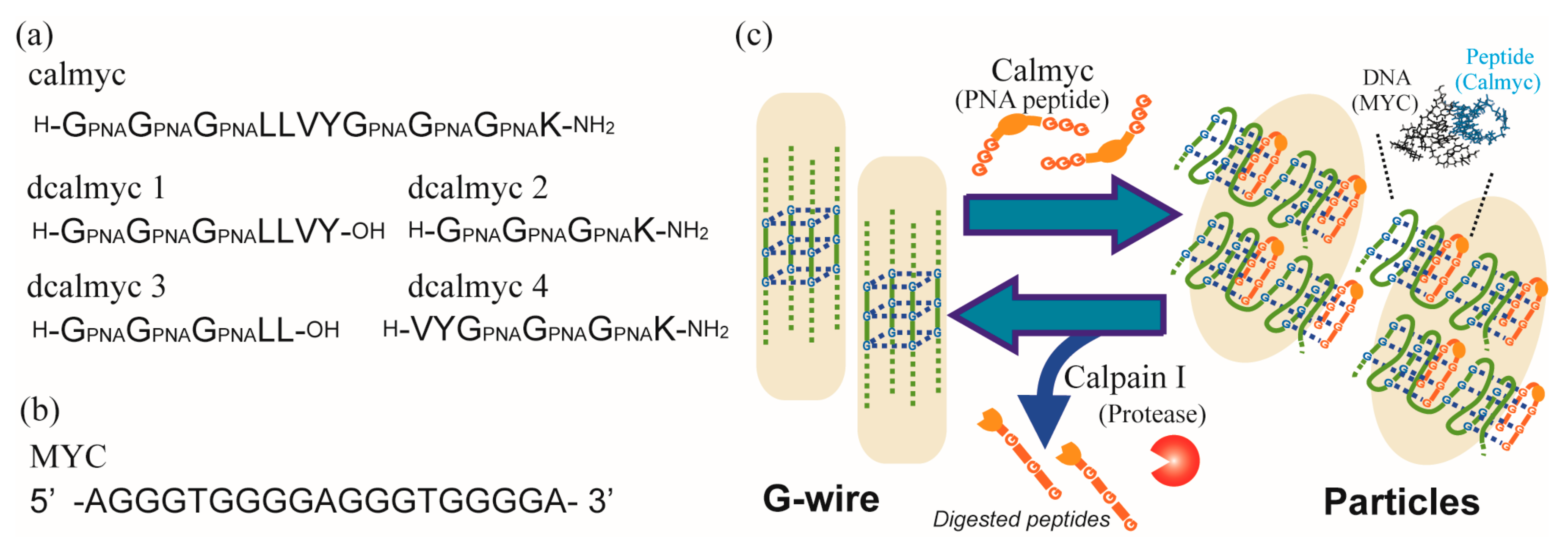
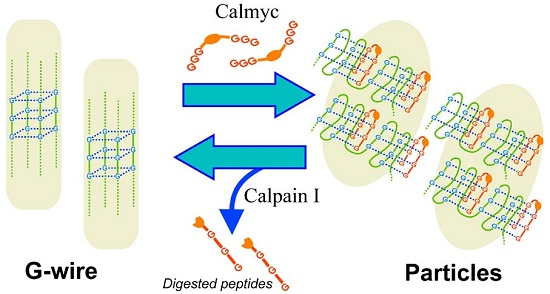{kind=link}
{kind=link}
{kind=link}
{kind=link}
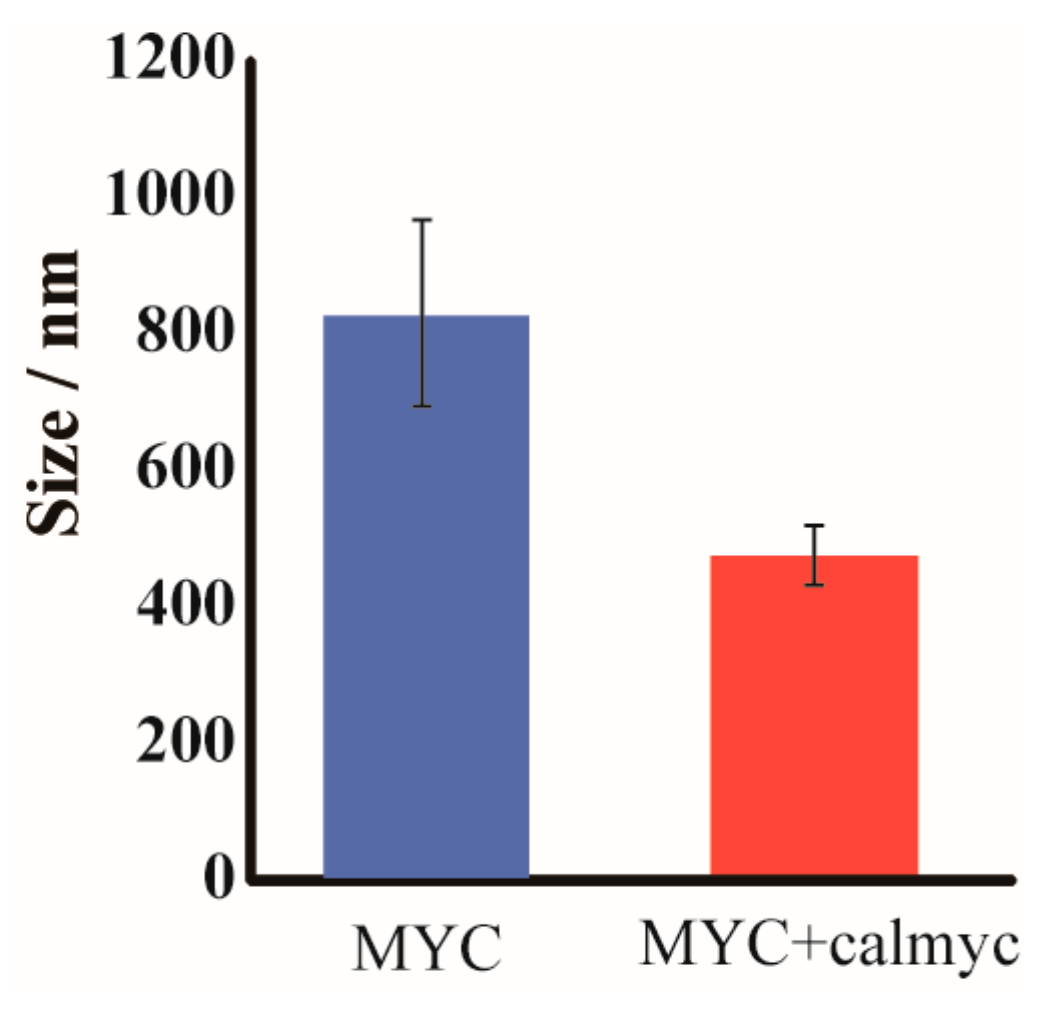{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
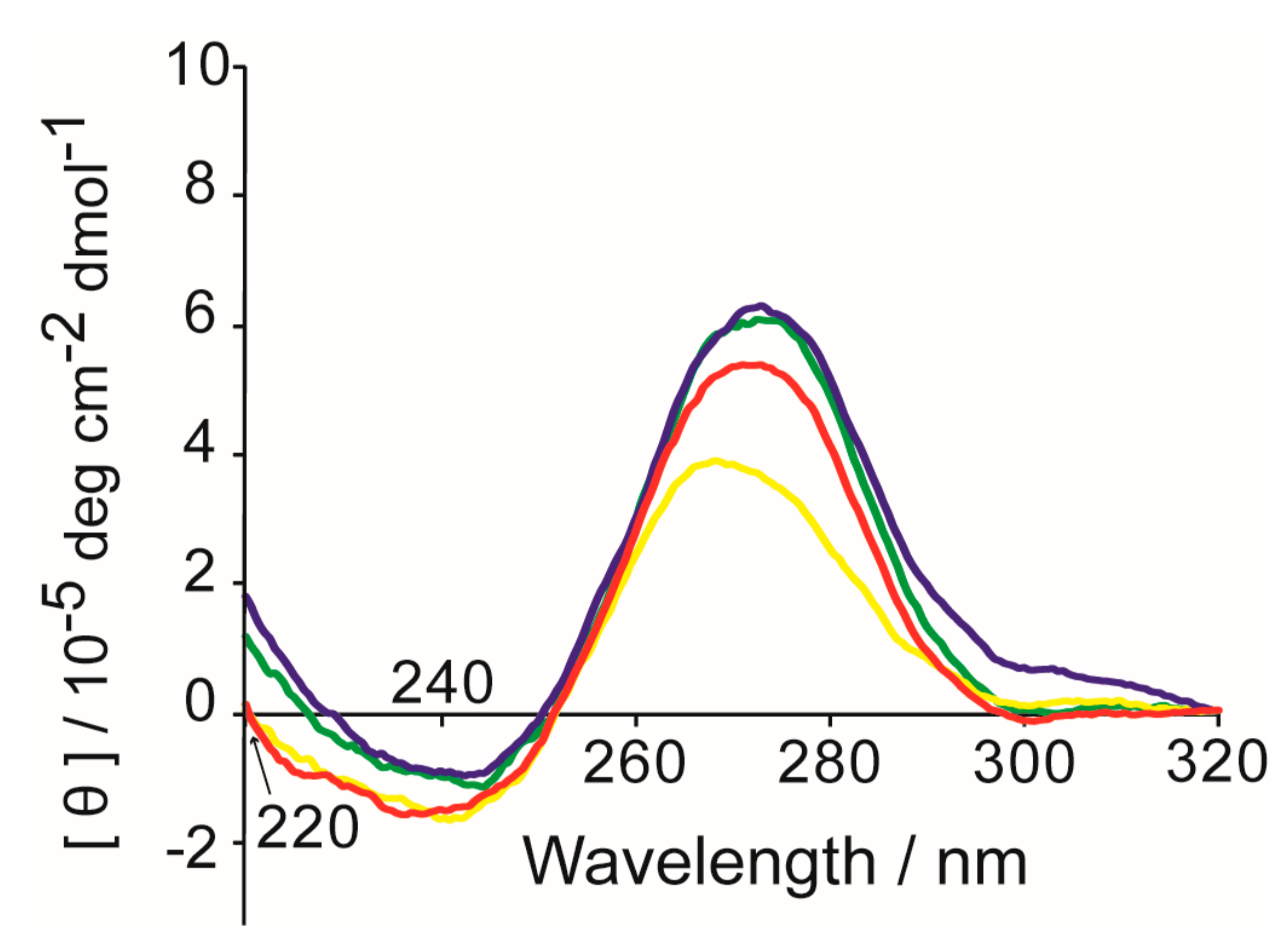
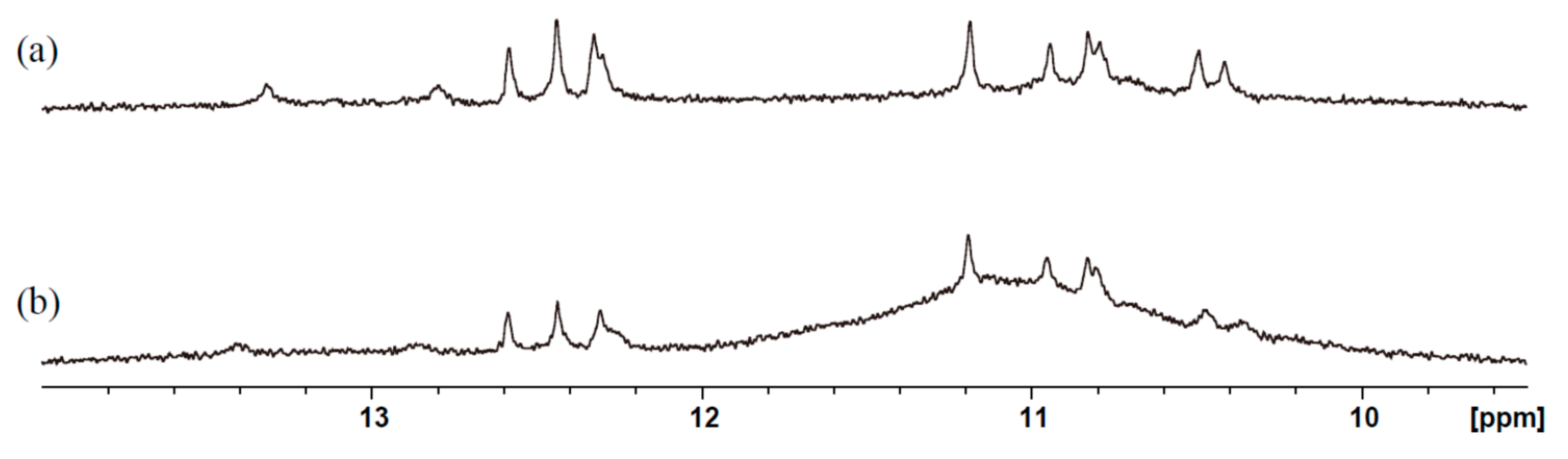
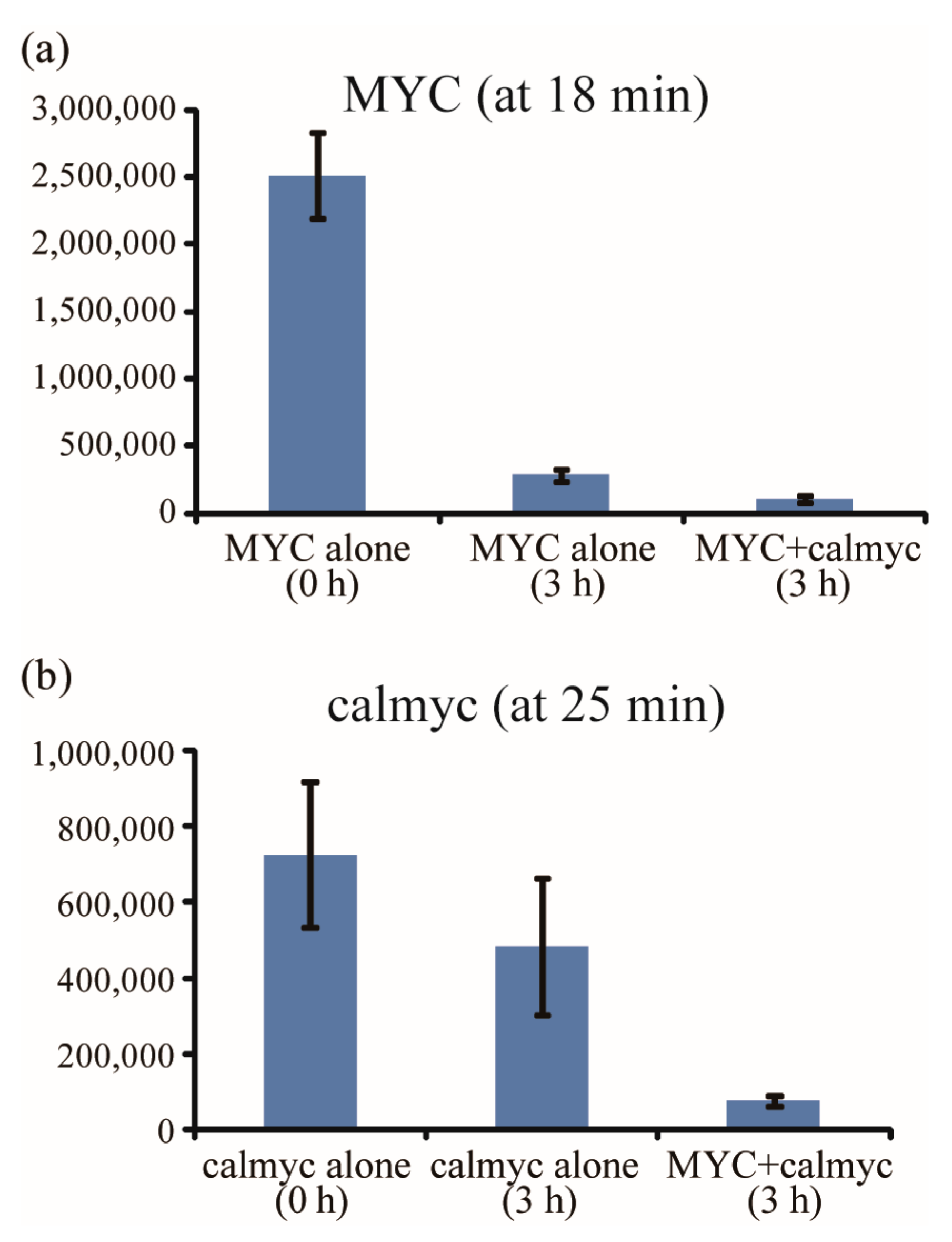
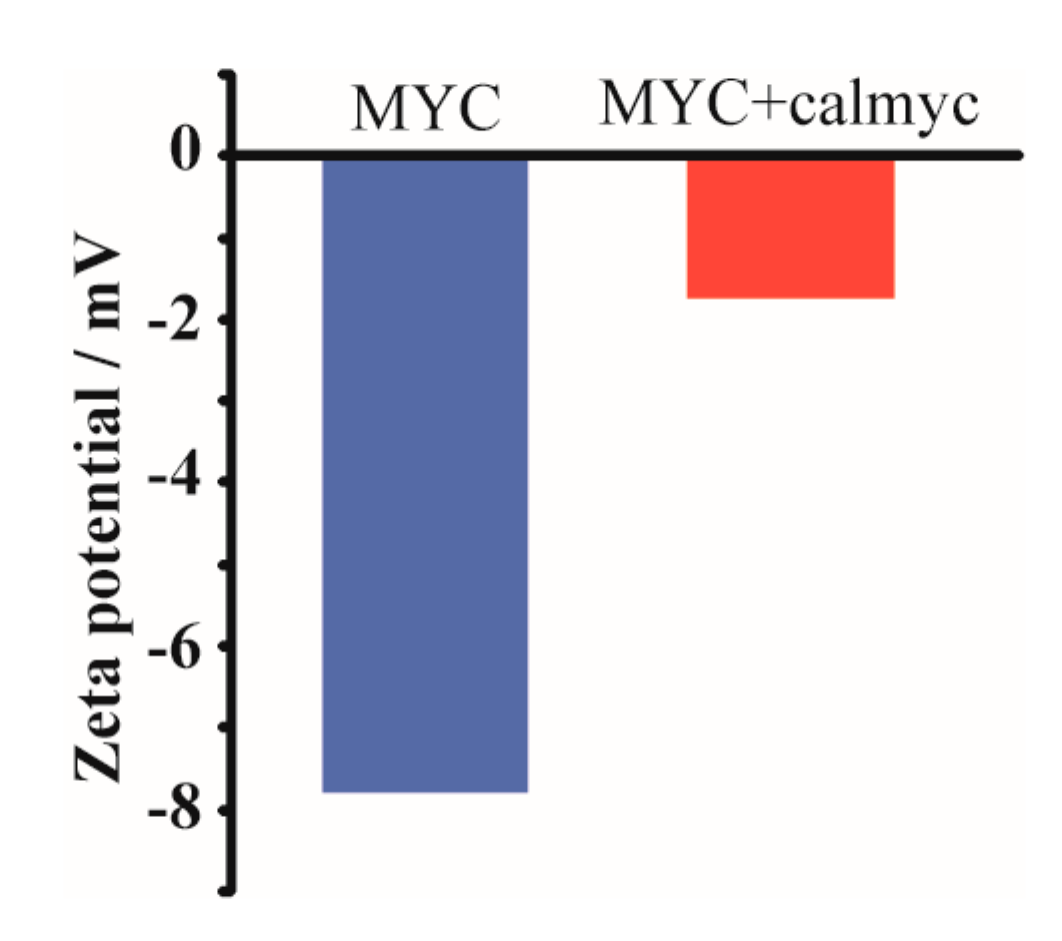
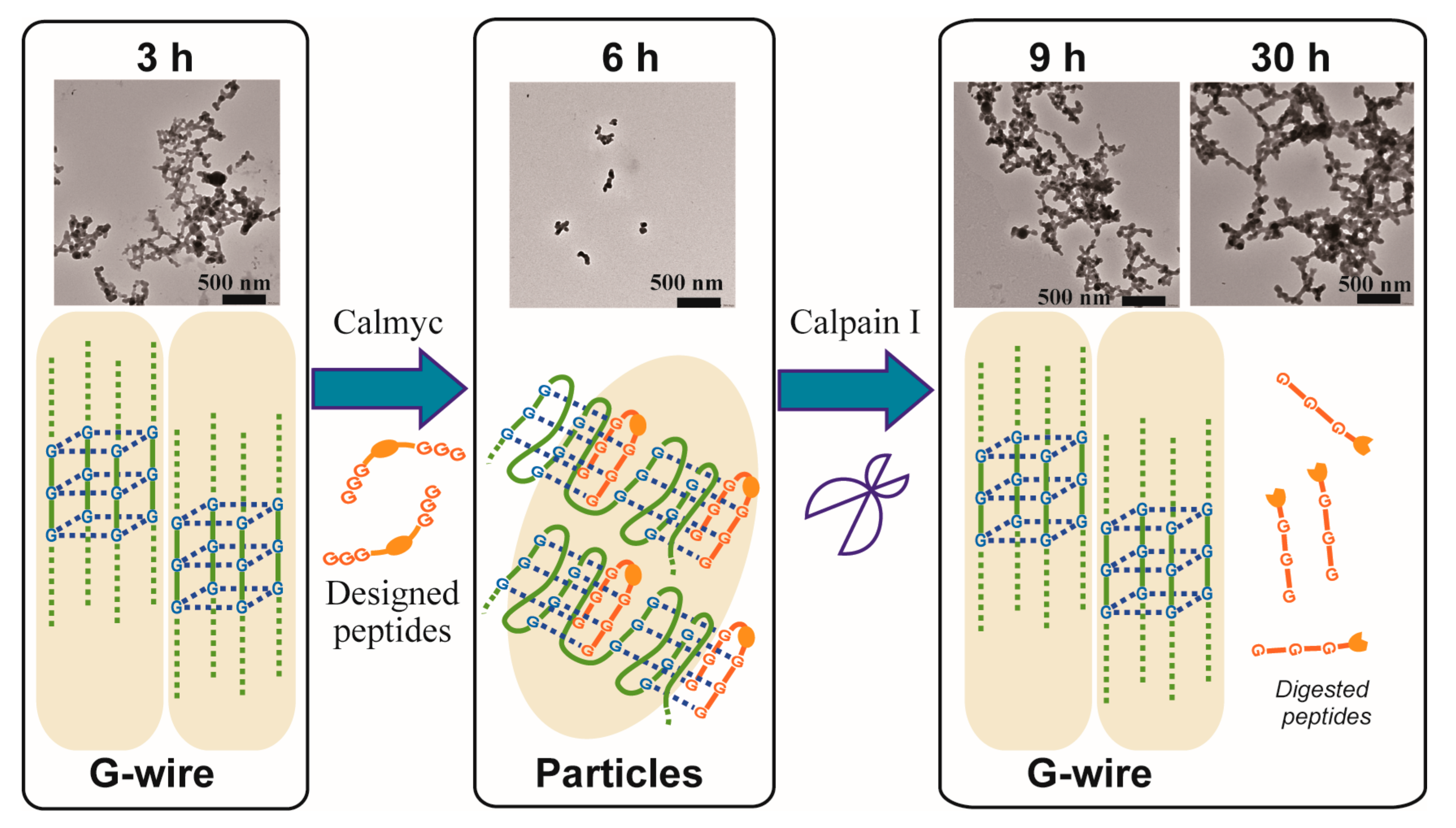
2. Results and Discussion
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Artificial Peptides
3.3. Atomic Force Microscope Measurements
3.4. Transmission Electron Microscope Measurements
3.5. Dynamic Light Scattering (DLS) Measurements and Zeta Potential Measurements
3.6. Circular Dichroism (CD) Spectroscopy
3.7. NMR Spectroscopy
3.8. Size-Exclusion Chromatography (SEC) Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seeman, N.C. DNA nanotechnology: Novel DNA constructions. Annu. Rev. Biophys. Biomol. Struct. 1994, 27, 225–248. [Google Scholar] [CrossRef] [PubMed]
- Storhoff, J.J.; Mirkin, C.A. Programmed Materials Synthesis with DNA. Chem. Rev. 1999, 99, 1849–1862. [Google Scholar] [CrossRef] [PubMed]
- Mao, C.; Sun, W.; Shen, Z.; Seeman, N.C. A nanomechanical device based on the B-Z transition of DNA. Nature 1999, 397, 144–146. [Google Scholar] [PubMed]
- Yurke, B.; Turberfield, A.J., Jr.; Mills, A.P.; Simmel, F.C.; Neumann, J.L. A DNA-fuelled molecular machine made of DNA. Nature 2000, 406, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Stoddart, J.F. The master of chemical topology. Chem. Soc. Rev. 2009, 38, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.M.; Forgan, R.S.; Friedman, D.C.; Gassensmith, J.J.; Smaldone, R.A.; Stoddart, J.F.; Sauvage, J.P. Donor–acceptor molecular figures-of-eight. Chem. Commun. 2011, 47, 11870–11872. [Google Scholar] [CrossRef] [PubMed]
- Carroll, G.G.; Pollard, M.M.; Delden, R.; Feringa, B.L. Controlled rotary motion of light-driven molecular motors assembled on a gold film. Chem. Sci. 2010, 1, 97–101. [Google Scholar] [CrossRef]
- Osada, E.; Suzuki, Y.; Hidaka, K.; Ohno, H.; Sugiyama, H.; Endo, M.; Saito, H. Engineering RNA—Protein Complexes with Different Shapes for Imaging and Therapeutic Applications. ACS Nano 2014, 8, 8130–8140. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. DNA in a material world. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Gothelf, K.V.; LaBean, T.H. DNA-programmed assembly of nanostructures. Org. Biomol. Chem. 2005, 3, 4023–4037. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Seeman, N.C. Synthesis from DNA of a Molecule with the Connectivity of a Cube. Nature 1991, 350, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Seeman, N.C. Construction of a DNA-Truncated Octahedron. J. Am. Chem. Soc. 1994, 116, 1661–1669. [Google Scholar] [CrossRef]
- Winfree, E.; Liu, F.; Wenzler, L.A.; Seeman, N.C. Design and self-assembly of two-dimensional DNA crystals. Nature 1998, 393, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Shih, W.M.; Quispe, J.D.; Joyce, G.F. A 1.7-kilobase single-stranded DNA that folds into a nanoscale octahedron. Nature 2004, 427, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Sugiyama, H. Recent progress in DNA origami technology. Curr. Protoc. Nucleic Acid Chem. 2011, 12, 1025–1029. [Google Scholar]
- Endo, M.; Sugita, T.; Rajendran, A.; Katsuda, Y.; Emura, T.; Hidaka, K.; Sugiyama, H. Two-dimensional DNA origami assemblies using a four-way connector. Chem. Commun. 2011, 47, 3213–3215. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Endo, M.; Sugiyama, H. Lipid-bilayer-assisted two-dimensional self-assembly of DNA origami nanostructures. Nat. Commun. 2015, 6, 8052. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, A.; Komiyama, M. DNA origami: Fold, stick, and beyond. Nanoscale 2010, 2, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Kuzuya, A.; Sakai, Y.; Yamazaki, T.; Xu, Y.; Komiyama, M. Nanomechanical DNA origami ‘single-molecule beacons’ directly imaged by atomic force microscopy. Nat. Commun. 2011, 2, 449. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.C.; Vesenka, J.; Henderson, E. A new DNA nanostructure, the G-wire, imaged by scanning probe microscopy. Nucleic Acids Res. 1995, 23, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, D.; Nakao, A.; Sugimoto, N. Structural transition from antiparallel to parallel G-quadruplex of d(G4T4G4) induced by Ca2+. Nucleic Acids Res. 2003, 31, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, D.; Karimata, H.; Wang, Z.M.; Koumoto, K.; Sugimoto, N. Artificial G-Wire Switch with 2,2′-Bipyridine Units Responsive to Divalent Metal Ions. J. Am. Chem. Soc. 2007, 129, 5919–5925. [Google Scholar] [CrossRef] [PubMed]
- Marsh, T.C.; Henderson, E. G-wires: Self-assembly of a telomeric oligonucleotide, d(GGGGTTGGGG), into large superstructures. Biochemistry 1994, 33, 10718–10724. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Changenet-Barret, P.; Improta, R.; Vaya, I.; Gustavsson, T.; Kotlyar, A.B.; Zikich, D.; Sket, P.; Plavec, J.; Markovitsi, D. Cation Effect on the Electronic Excited States of Guanine Nanostructures Studied by Time-Resolved Fluorescence Spectroscopy. J. Phys. Chem. 2012, 116, 14682–14689. [Google Scholar] [CrossRef]
- Livshits, G.I.; Stern, A.; Rotem, D.; Borovok, N.; Eidelshtein, G.; Migliore, A.; Penzo, E.; Wind, S.J.; Felice, R.D.; Skourtis, S.S.; et al. Long-range charge transport in single G-quadruplex DNA molecules. Nat. Nanotechnol. 2014, 9, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.P.; Weisbrod, S.H.; Tang, Z.; Marx, A.; Scheer, E.; Erbe, A. Direct measurement of electrical transport through G-quadruplex DNA with mechanic-ally controllable break junction electrodes. Angew. Chem. Int. Ed. 2010, 49, 3313–3316. [Google Scholar] [CrossRef] [PubMed]
- Macaya, R.F.; Schultze, P.; Smith, F.W.; Roe, J.A.; Feigon, J. Thrombin-bin-ding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. USA 1993, 90, 3745–3749. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.T. G-quartets 40 years later: From 5′-GMP to molecular biology and supramolecular chemistry. Angew. Chem. Int. Ed. 2004, 43, 668–698. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine rich motifs in DNA and its implications for meiosis. Nature 1998, 334, 364–366. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.M. Structure and Function of Telomeres. Nature 1991, 350, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, D.; Matsumura, S.; Nakano, S.; Sugimoto, N. Duplex dissociation of telomere DNAs induced by molecular crowding. J. Am. Chem. Soc. 2004, 126, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Keniry, M.A. Quadruplex structures in nucleic acids. Biopolymers 2001, 56, 123–146. [Google Scholar] [CrossRef]
- Takahashi, S.; Sugimoto, N. Effect of pressure on the stability of G-quadruplex DNA: Thermodynamics under crowding conditions. Angew. Chem. Int. Ed. 2013, 52, 13774–13778. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef] [PubMed]
- Endoh, T.; Kawasaki, Y.; Sugimoto, N. Suppression of gene expression by G-quadruplexes in open reading frames depends on G-quadruplex stability. Angew. Chem. Int. Ed. 2013, 52, 5522–5526. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.; Hardin, C.C.; Walk, S.W.; Tinoco, I.; Blackburn, E.H. Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine guanine base pairs. Cell 1987, 51, 899–908. [Google Scholar] [CrossRef]
- Rankin, S.; Reszka, A.P.; Huppert, J.; Zloh, M.; Parkinson, G.N.; Todd, A.K.; Ladame, S.; Balasubramanian, S.; Neidle, S. Putative DNA Quadruplex Format-ion within the Human c-kit Oncogene. J. Am. Chem. Soc. 2005, 127, 10584–10589. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.Q.; Miyoshi, D.; Sugimoto, N. Characterization of structure and stab-ility of long telomeric DNA G-quadruplexes. J. Am. Chem. Soc. 2006, 128, 15461–15468. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Matsui, N.; Usui, K. Use of a designed Peptide library to screen for binders to a particular DNA g-quadruplex sequence. J. Nucleic Acids 2011, 2011, 572873. [Google Scholar] [CrossRef] [PubMed]
- Okada, A.; Usui, K. Chemical Biology of Nucleic Acids: Fundamentals and Clinical Applications; Erdmann, V.A., Markiewicz, W., Barciszewski, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; p. 459. [Google Scholar]
- Calzolari, A.; Felice, R.D.; Molinari, E.; Garbesi, A. G-quartet biomolecular nanowires. Appl. Phys. Lett. 2002, 80, 3331–3333. [Google Scholar] [CrossRef]
- Ren, W.; Zhang, Y.; Chen, H.G.; Gao, Z.F.; Li, N.B.; Luo, H.Q. Ultrasensitive Label-Free Resonance Rayleigh Scattering Aptasensor for Hg (2+) Using Hg (2+)-Triggered Exonuclease III-Assisted Target Recycling and Growth of G-Wires for Signal Amplification. Anal. Chem. 2016, 88, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Hessari, N.M.; Spindler, L.; Troha, T.; Lam, W.C.; Drevenšek-Olenik, I.; da Silva, M.W. Programmed self-assembly of a quadruplex DNA nanowire. Chem. Eur. J. 2014, 20, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted poly-amide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Tomizaki, K.Y.; Usui, K.; Mihara, H. A PNA-DNA hybridization chip approach for the detection of beta-secretase activity. Bioorg. Med. Chem. Let. 2006, 16, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Lusvarghi, S.; Murphy, C.T.; Roy, S.; Tanious, F.A.; Sacui, I.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. Loop and backbone modifications of peptide nucleic acid improve g-quadruplex binding selectivity. J. Am. Chem. Soc. 2009, 131, 18415–18424. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Sengupta, P.; Krishnan, Y.; Ladame, S. Combining G-quadruplex targeting motifs on a single peptide nucleic acid scaffold: A hybrid (3+1) PNA-DNA bimolecular quadruplex. Chem. Eur. J. 2008, 14, 8682–8689. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tanious, F.A.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. High-affinity homologous peptide nucleic acid probes for targeting a quadruplex-forming sequence from a MYC promoter element. Biochemistry 2007, 46, 10433–10443. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Yang, J.; Komiyama, M.; Xu, Y. G-rich sequence-specific recognition and scission of human genome by PNA/DNA hybrid G-quadruplex formation. Angew. Chem. Int. Ed. 2012, 51, 7198–7202. [Google Scholar] [CrossRef] [PubMed]
- Datta, B.; Schmitt, C.; Armitage, B.A. Formation of a PNA2-DNA2 hybrid quadruplex. J. Am. Chem. Soc. 2003, 125, 4111–4118. [Google Scholar] [CrossRef] [PubMed]
- Usui, K.; Okada, A.; Kobayashi, K.; Sugimoto, N. Control of guanine-rich DNA secondary structures depending on the protease activity using a designed PNA peptide. Org. Biomol. Chem. 2015, 13, 2022–2025. [Google Scholar] [CrossRef] [PubMed]
- Ou, T.M.; Lu, Y.J.; Zhang, C.; Huang, Z.S.; Wang, X.D.; Tan, J.H.; Chen, Y.; Ma, D.L.; Wong, K.Y.; Tang, J.C.; et al. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J. Med. Chem. 2007, 50, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, D.; Nakao, A.; Toda, T.; Sugimoto, N. Effect of divalent cations on antiparallel G-quartet structure of d(G4T4G4). FEBS Lett. 2001, 496, 128–133. [Google Scholar] [CrossRef]
- Balagurumoorthy, P.; Brahmachari, S.K.; Mohanty, D.; Bansal, M.; Sasisekh, H. Hairpin and parallel quartet structures for telomeric sequences. Nucleic Acids Res. 1992, 20, 4061–4067. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Gaffney, B.L.; Wang, C.; Jones, R.A.; Breslauer, K.J. Thermodynamics and structure of a DNA tetraplex: A spectroscopic and calorimetric study of the tetramolecular complexes of d(TG3T) and d(TG3T2G3T). Proc. Natl. Acad. Sci. USA 1992, 89, 8832–8836. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Matsugami, A.; Nishikawa, F.; Nishikawa, S.; Katahira, M. Uniqu equadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 2009, 37, 6249–6258. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Nishikawa, F.; Kamatari, Y.O.; Fujiwara, H.; Saimura, M.; Nagata, T.; Kodaki, T.; Nishikawa, S.; Kuwata, K.; Katahira, M. Anti-prion activity of an RNA aptamer and its structural basis. Nucleic Acids Res. 2013, 41, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Podbevšek, P.; Plavec, J. KRAS promoter oligonucleotide with decoy activity dimerizes into a unique topology consisting of two G-quadruplex units. Nucleic Acids Res. 2016, 44, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.L.; Ishizuka, T.; Sakamoto, T.; Fujimoto, K.; Uechi, T.; Kenmochi, N.; Xu, Y. Characterization of human telomere RNA G-quadruplex structures in vitro and in living cells using 19F NMR spectroscopy. Nucleic Acids Res. 2017, 45, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.C.; White, P.D. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Slocik, J.M.; Kuang, Z.; Knecht, M.R.; Naik, R.R. Optical modulation of azobenzene-modified peptide for gold surface binding. ChemPhysChem 2016, 17, 3252–3259. [Google Scholar] [CrossRef] [PubMed]
- Aemissegger, A.; Hilvert, D. Synthesis and application of an azobenzene amino acid as a light-switchable turn element in polypeptides. Nat. Protoc. 2007, 2, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Bhatia, D.; Saminathan, A.; Chakraborty, S.; Kar, S.; Krishnan, Y. Controlled release of encapsulated cargo from a DNA icosahedron using a chemical trigger. Angew. Chem. Int. Ed. 2013, 52, 6854–6857. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Gao, J. Iminoboronate-based peptide cyclization that responds to pH, oxidation, and small molecule modulators. J. Am. Chem. Soc. 2016, 138, 2098–2101. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds studied in this research are available from the authors. |









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usui, K.; Okada, A.; Sakashita, S.; Shimooka, M.; Tsuruoka, T.; Nakano, S.-i.; Miyoshi, D.; Mashima, T.; Katahira, M.; Hamada, Y. DNA G-Wire Formation Using an Artificial Peptide is Controlled by Protease Activity. Molecules 2017, 22, 1991. https://doi.org/10.3390/molecules22111991
Usui K, Okada A, Sakashita S, Shimooka M, Tsuruoka T, Nakano S-i, Miyoshi D, Mashima T, Katahira M, Hamada Y. DNA G-Wire Formation Using an Artificial Peptide is Controlled by Protease Activity. Molecules. 2017; 22(11):1991. https://doi.org/10.3390/molecules22111991
Chicago/Turabian StyleUsui, Kenji, Arisa Okada, Shungo Sakashita, Masayuki Shimooka, Takaaki Tsuruoka, Shu-ichi Nakano, Daisuke Miyoshi, Tsukasa Mashima, Masato Katahira, and Yoshio Hamada. 2017. "DNA G-Wire Formation Using an Artificial Peptide is Controlled by Protease Activity" Molecules 22, no. 11: 1991. https://doi.org/10.3390/molecules22111991
APA StyleUsui, K., Okada, A., Sakashita, S., Shimooka, M., Tsuruoka, T., Nakano, S.-i., Miyoshi, D., Mashima, T., Katahira, M., & Hamada, Y. (2017). DNA G-Wire Formation Using an Artificial Peptide is Controlled by Protease Activity. Molecules, 22(11), 1991. https://doi.org/10.3390/molecules22111991