Novel α, β-Unsaturated Sophoridinic Derivatives: Design, Synthesis, Molecular Docking and Anti-Cancer Activities

 , and
, and
Abstract
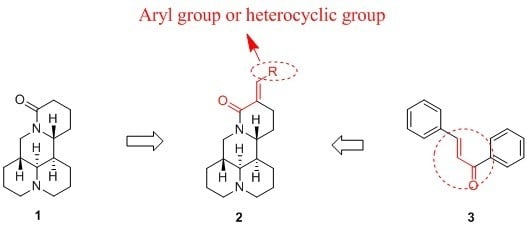
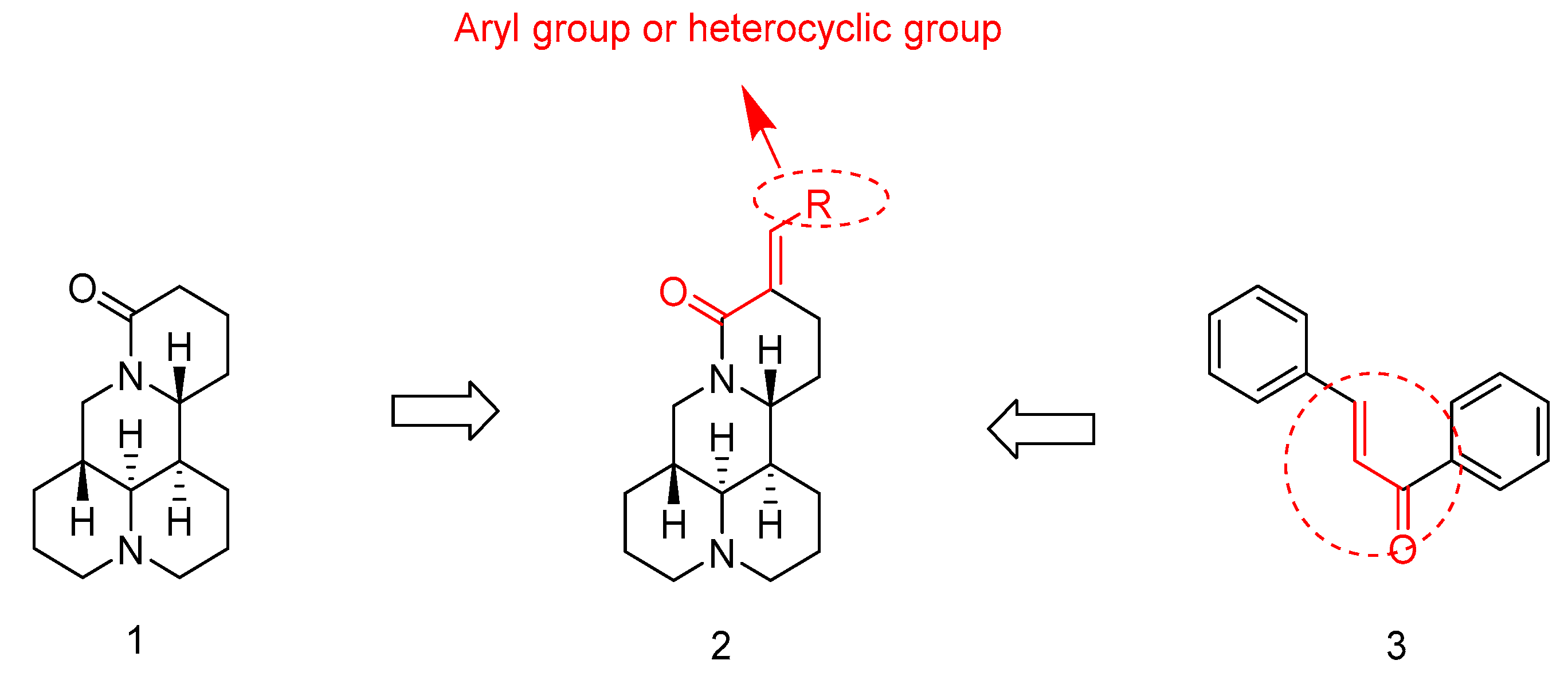
:
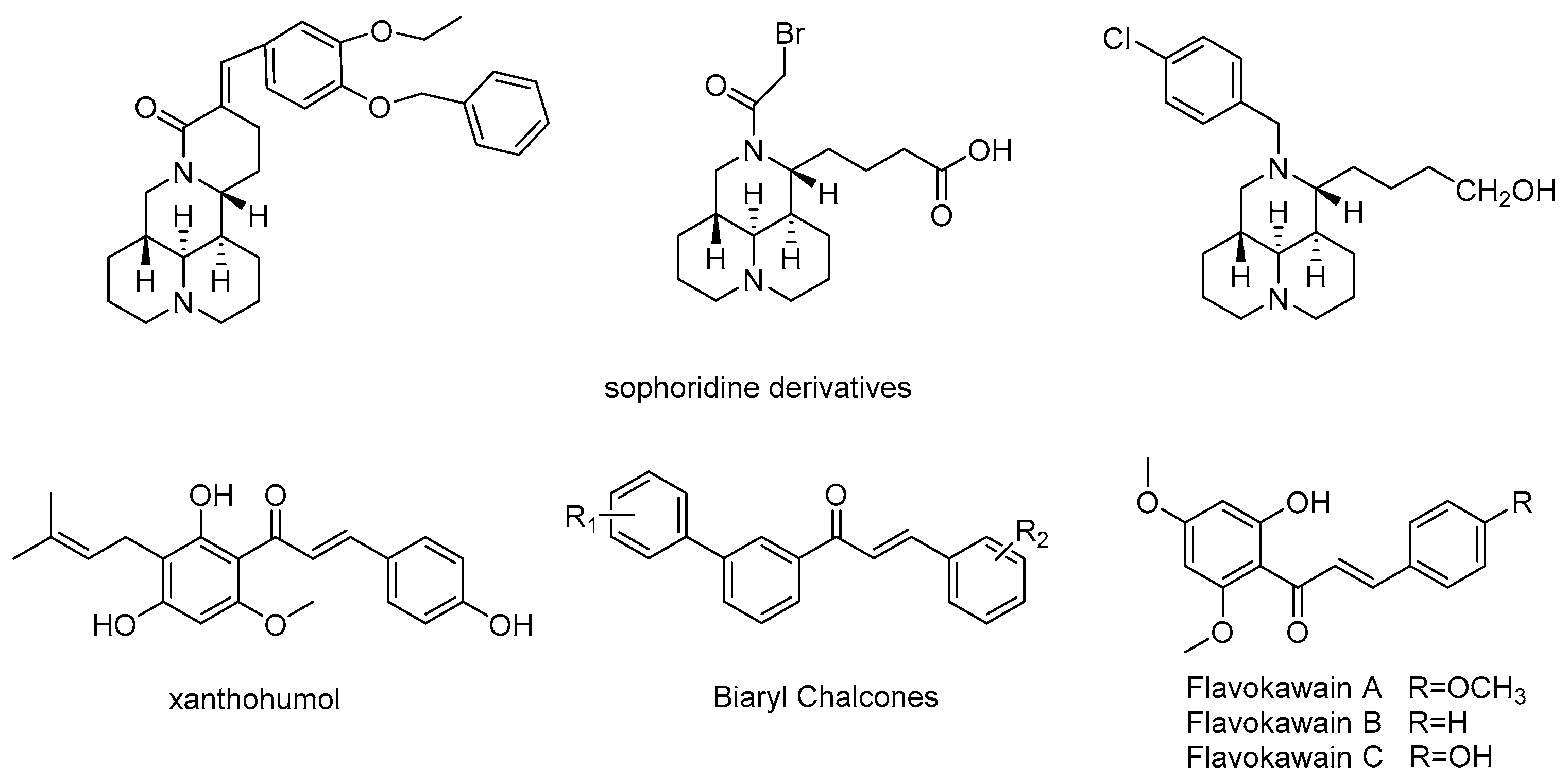
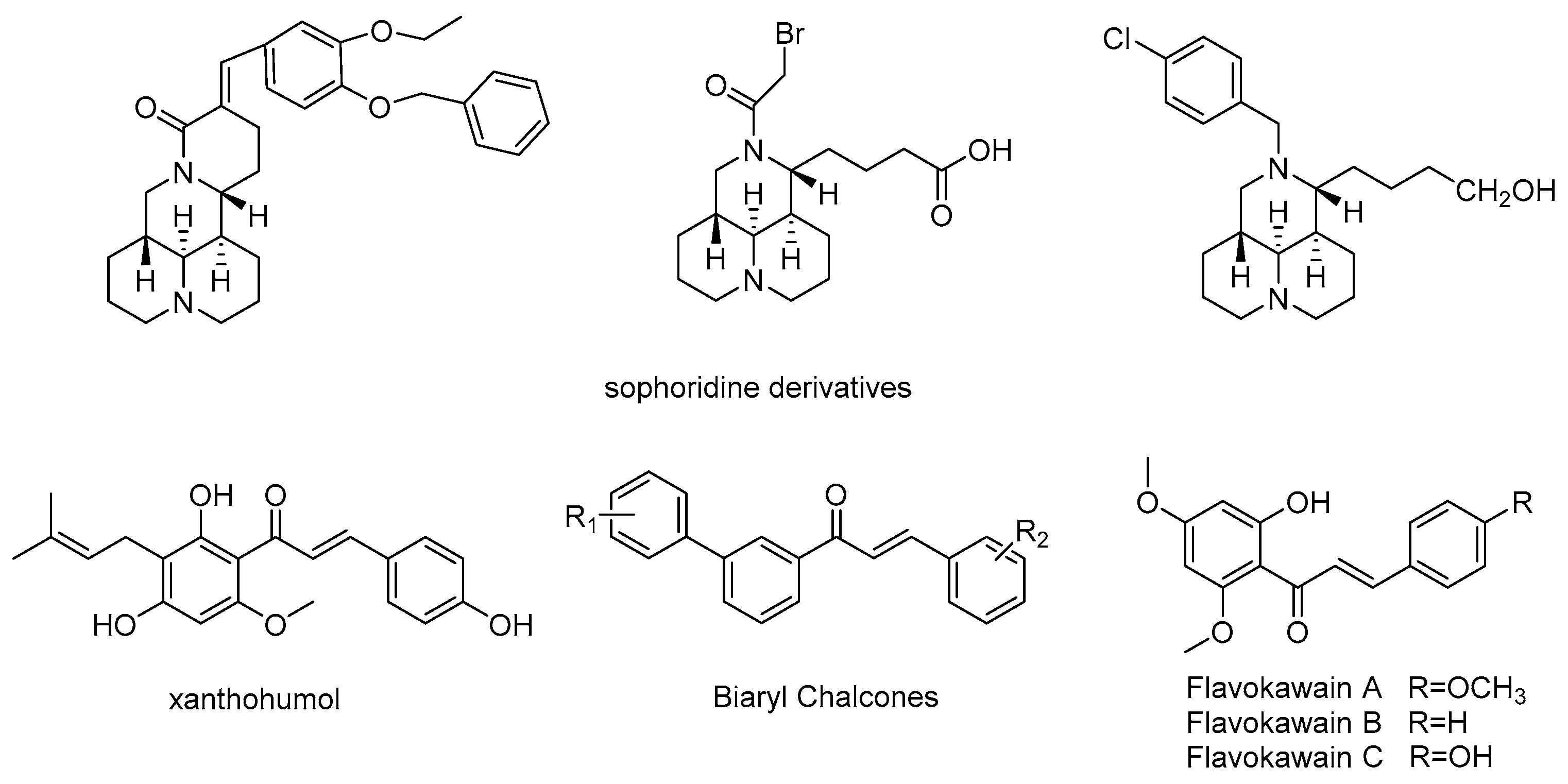
1. Introduction
2. Results
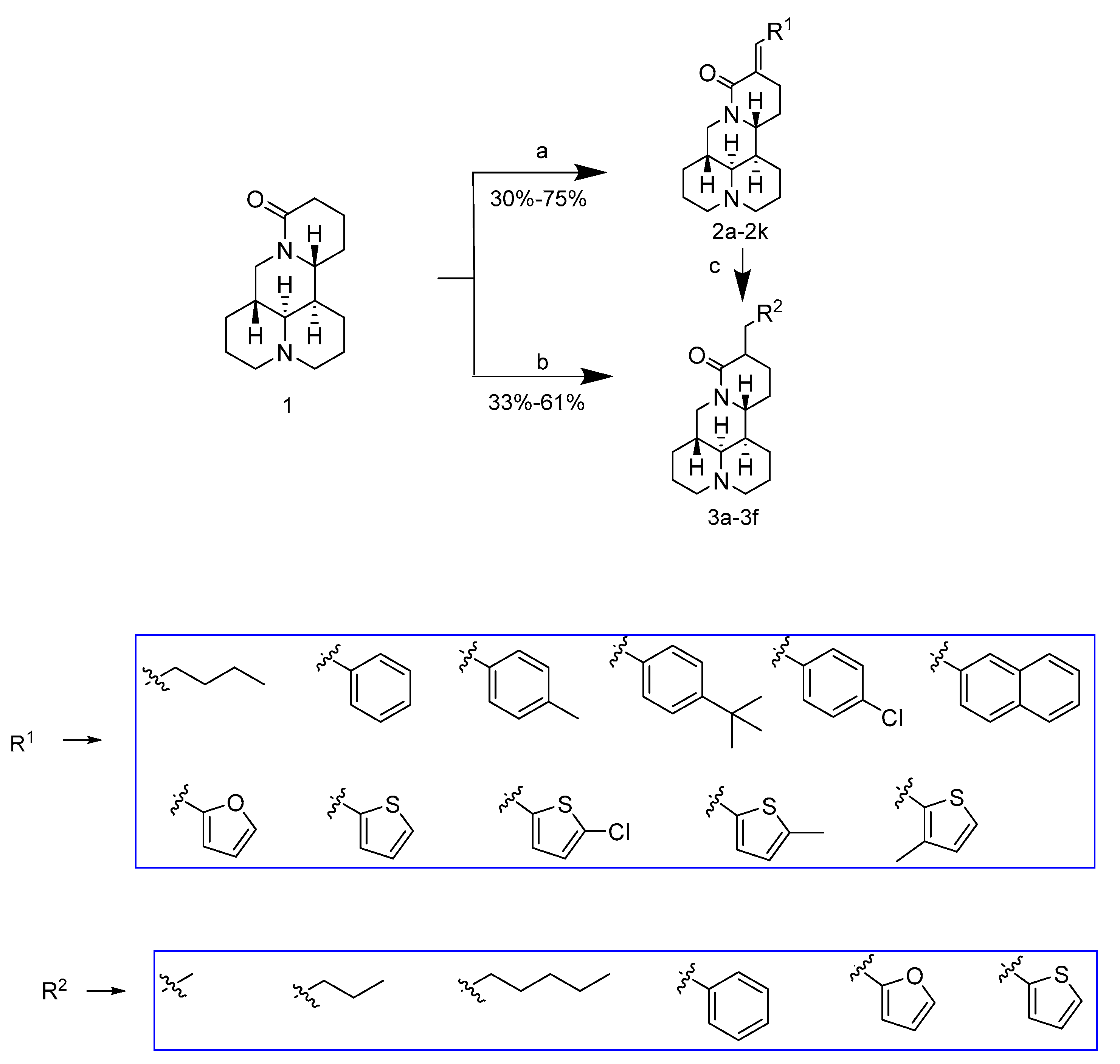
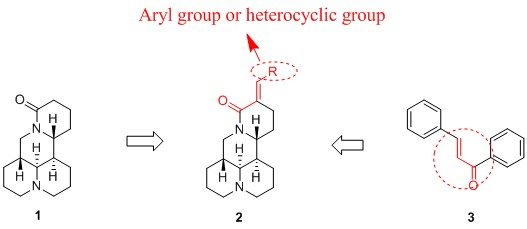
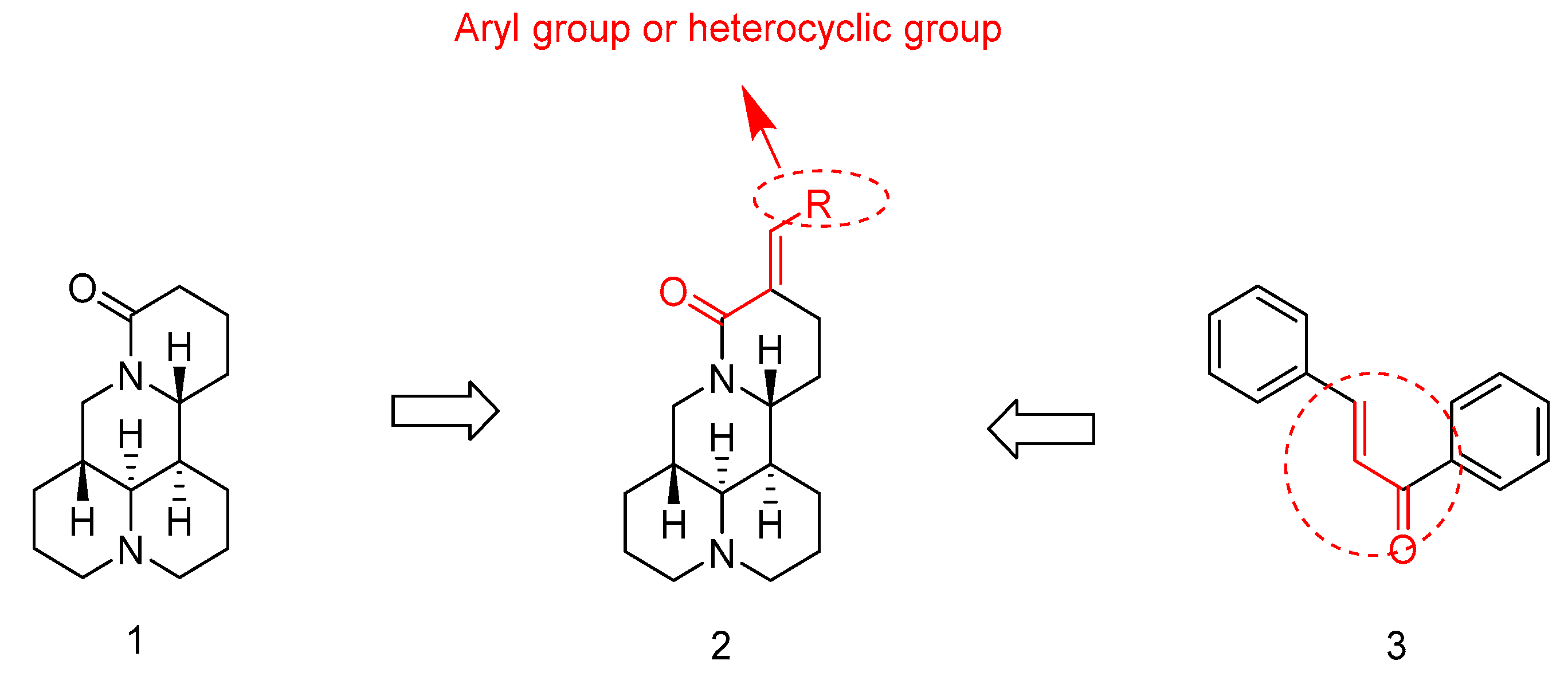
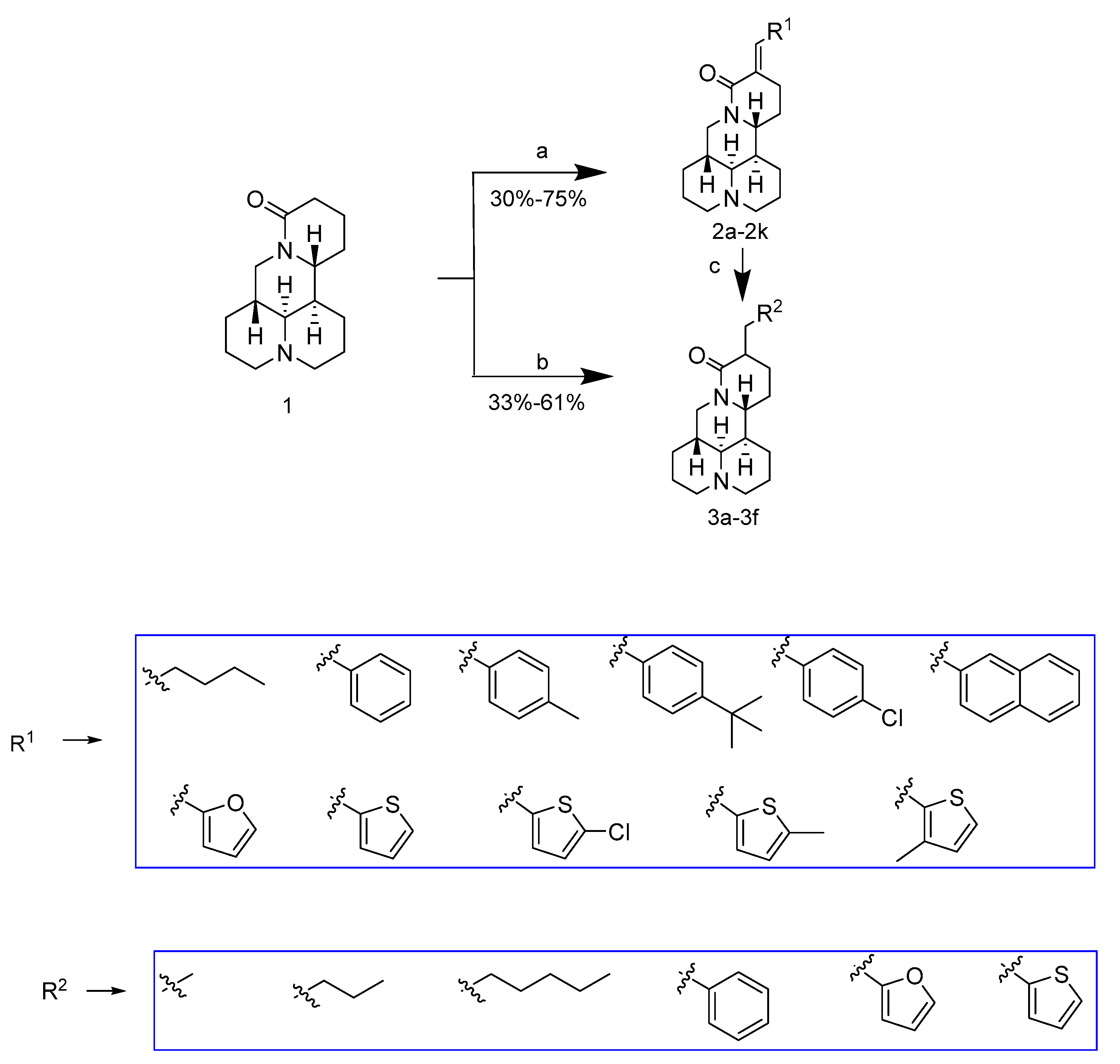
2.1. Chemistry
2.2. SAR Analysis for the Antiproliferative Activity
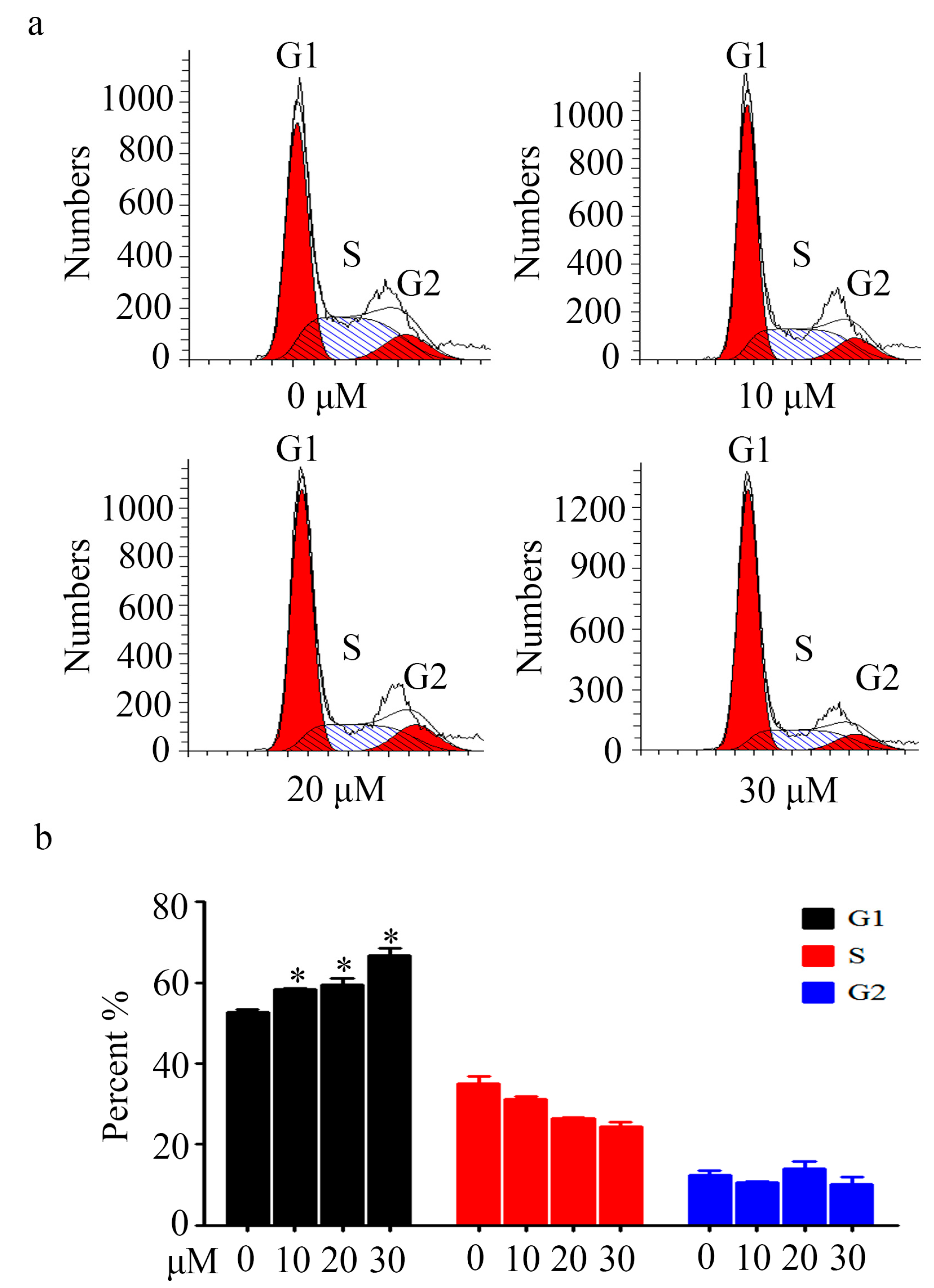
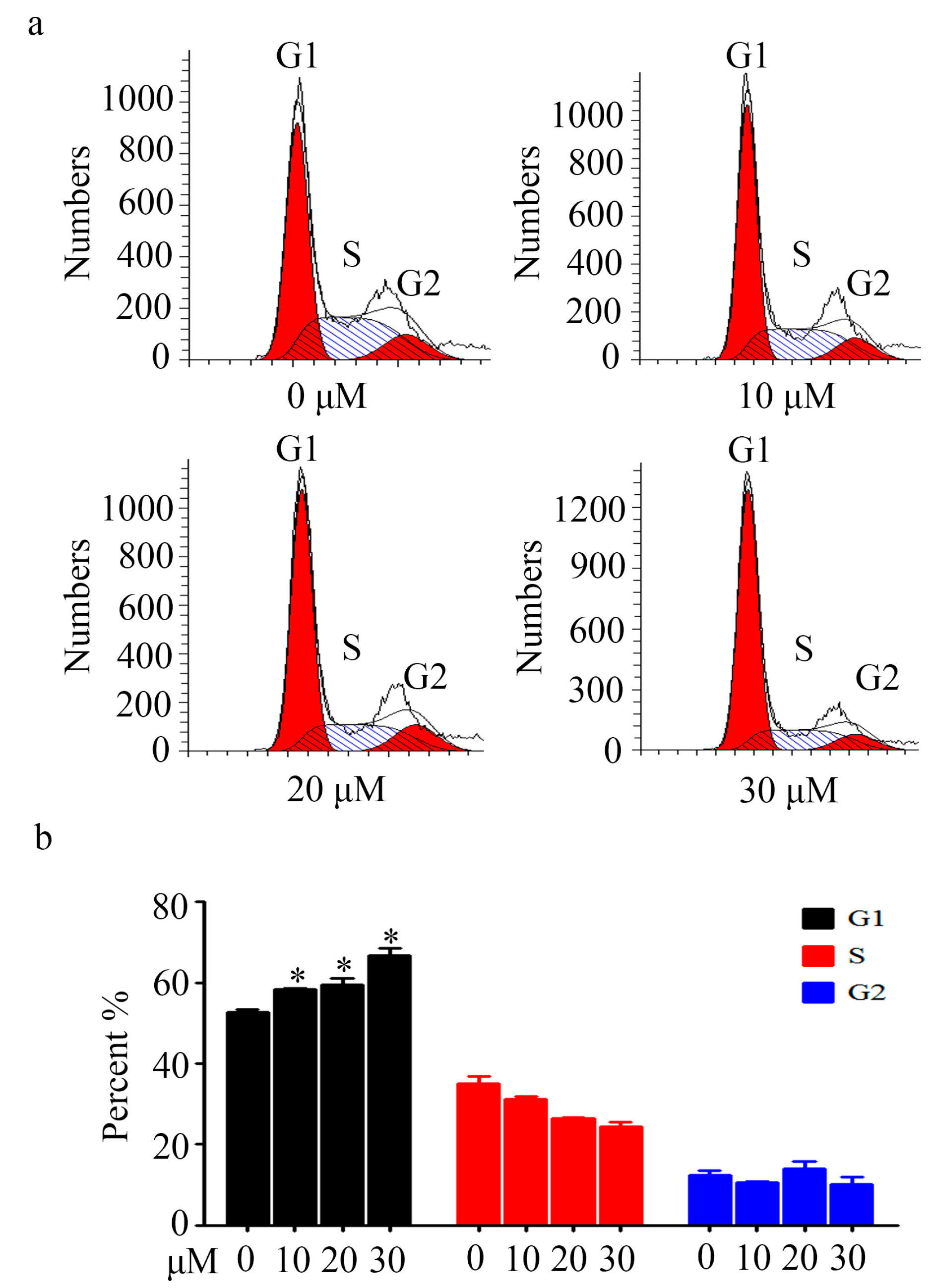
2.3. Cell Cycle Analysis
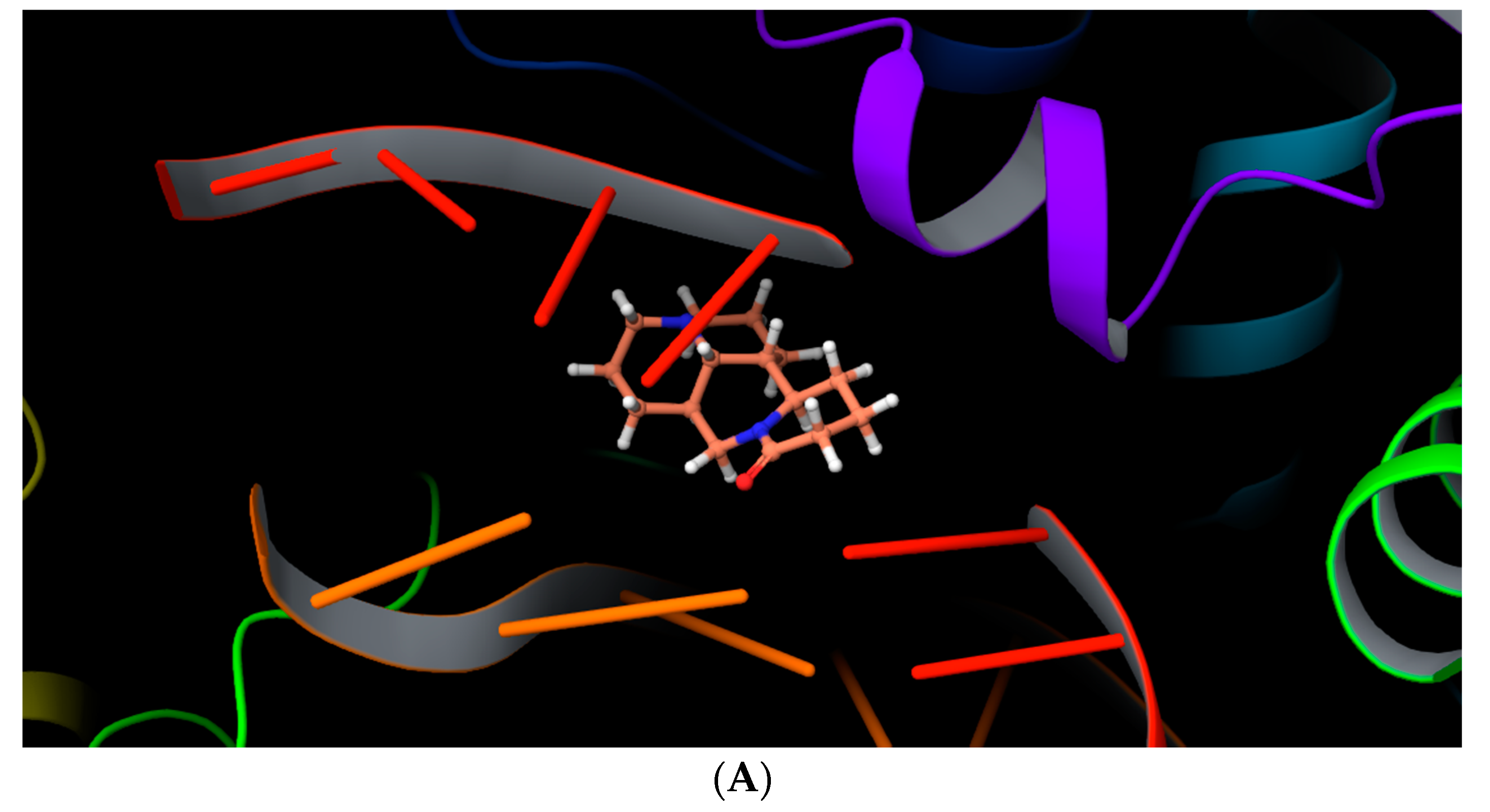
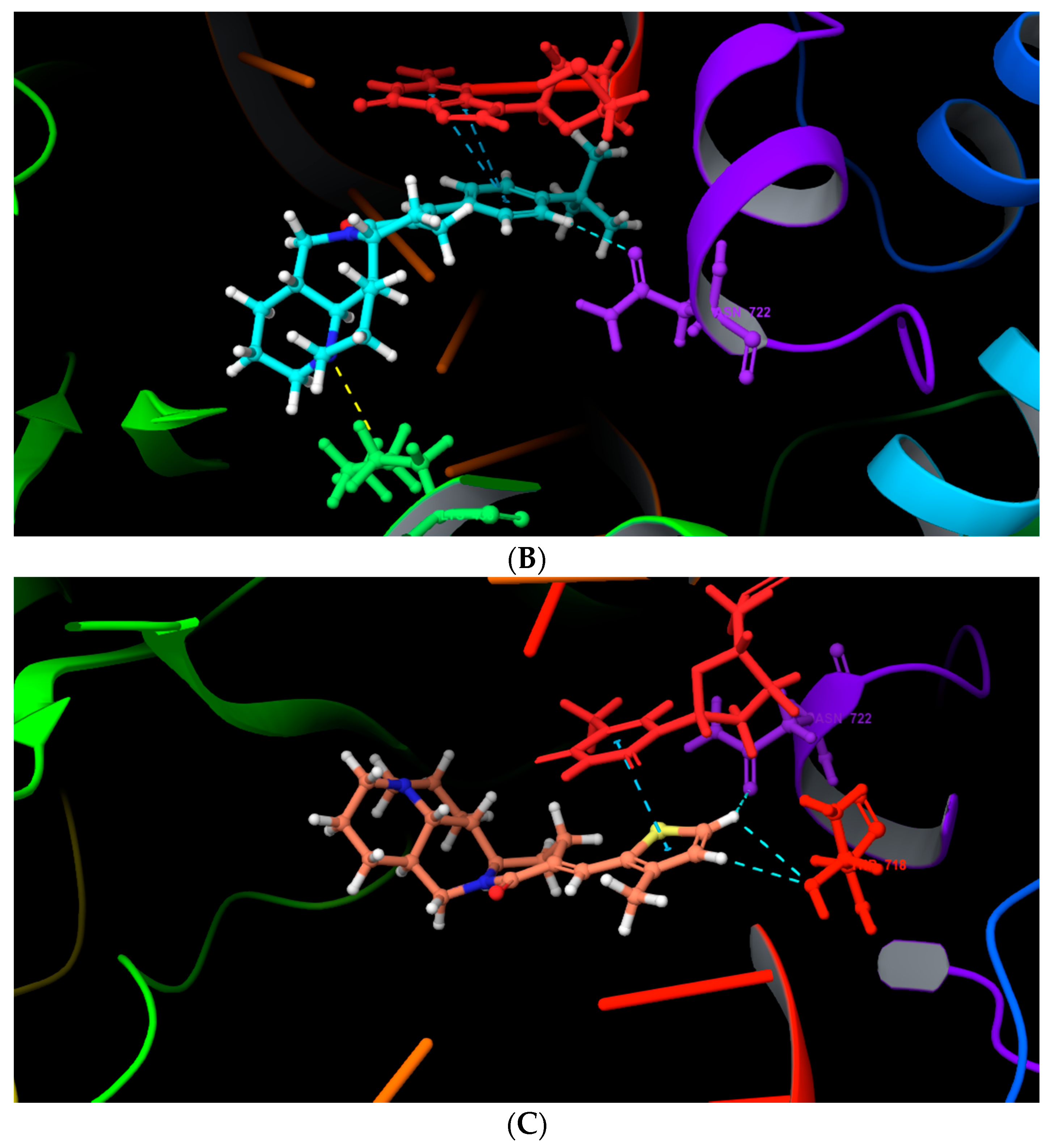
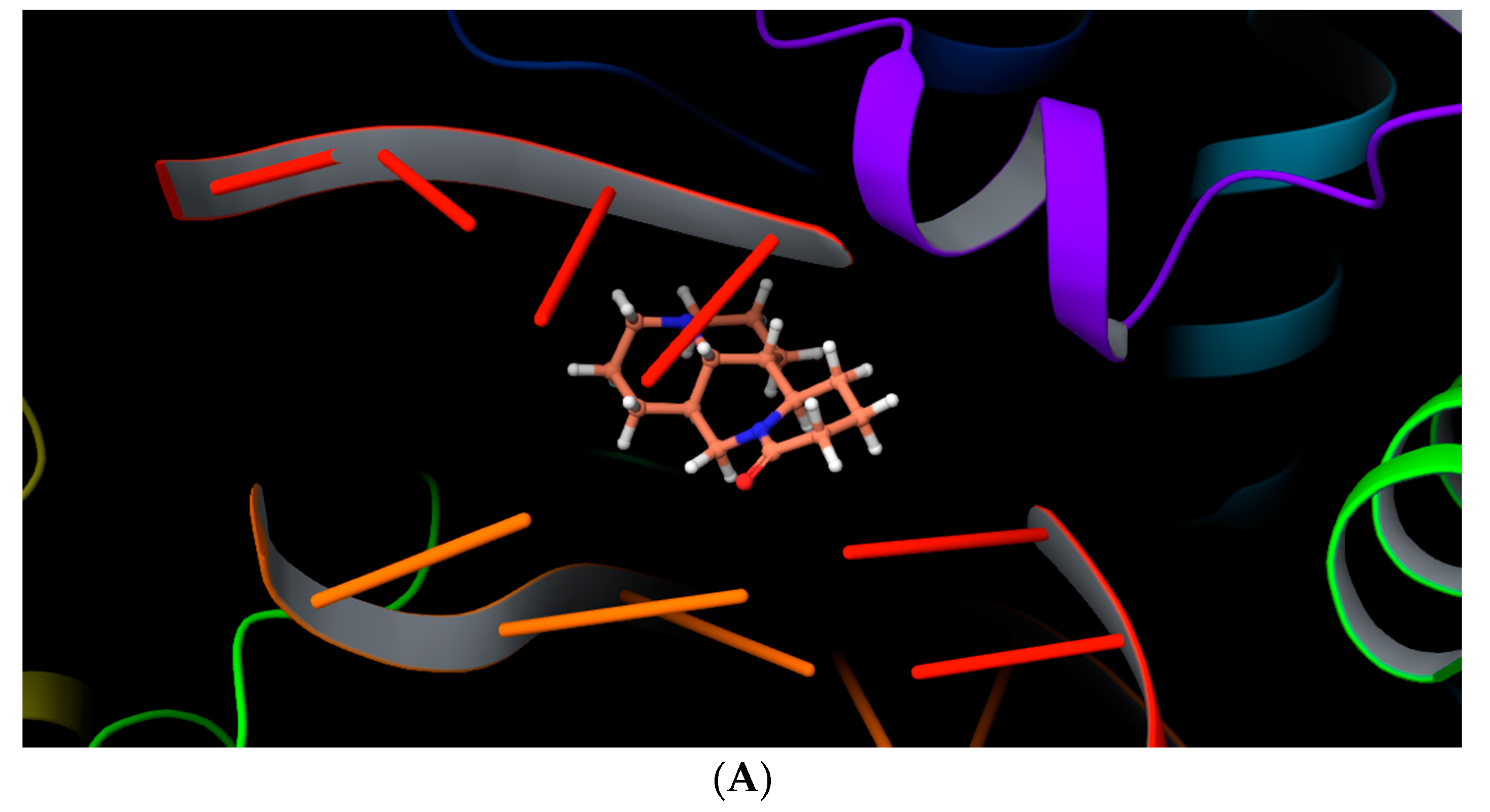
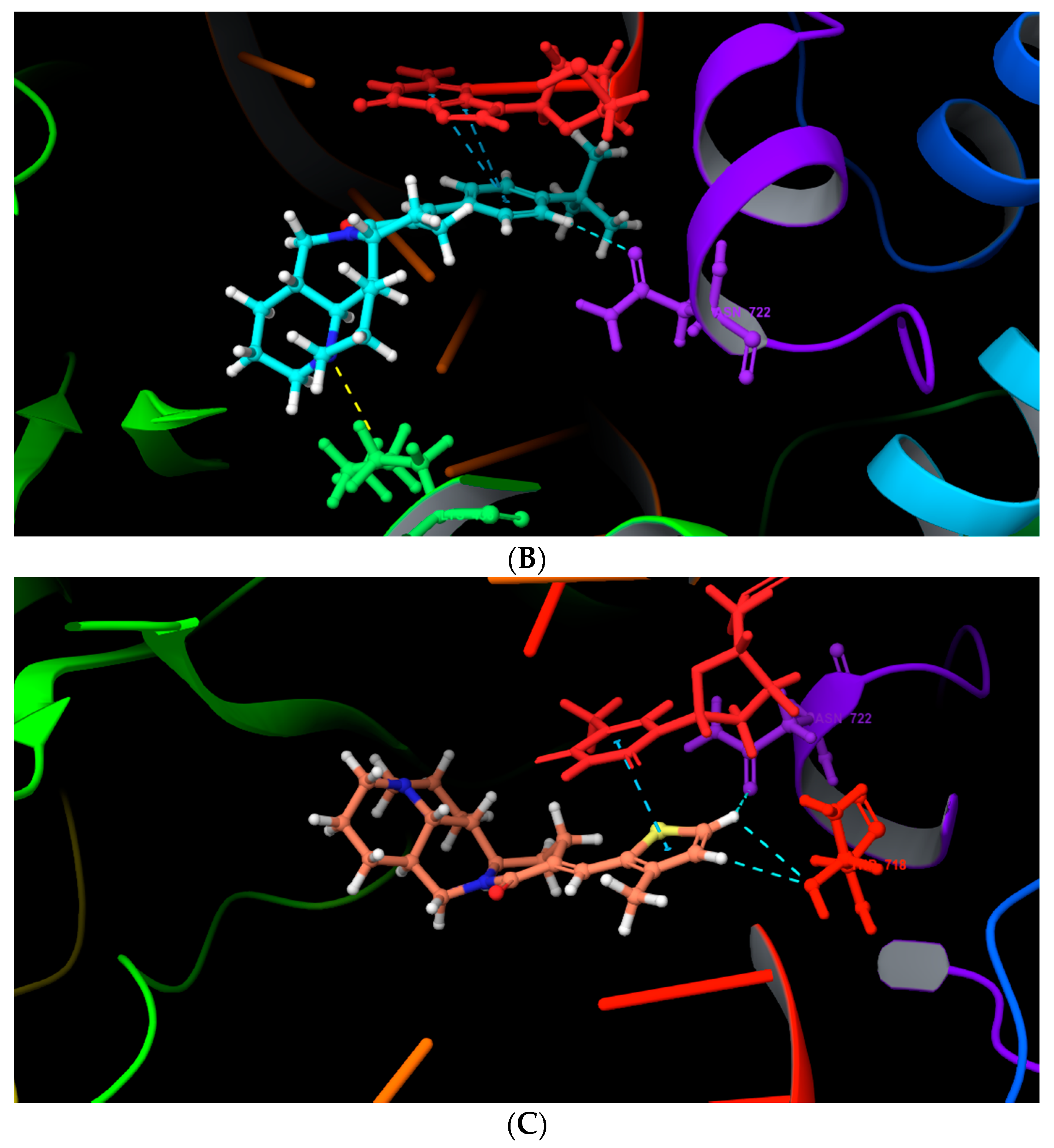
2.4. Molecular Docking
3. Materials and Methods
3.1. Instrumentation
3.2. Materials
3.3. Experimental Procedures and Characterization
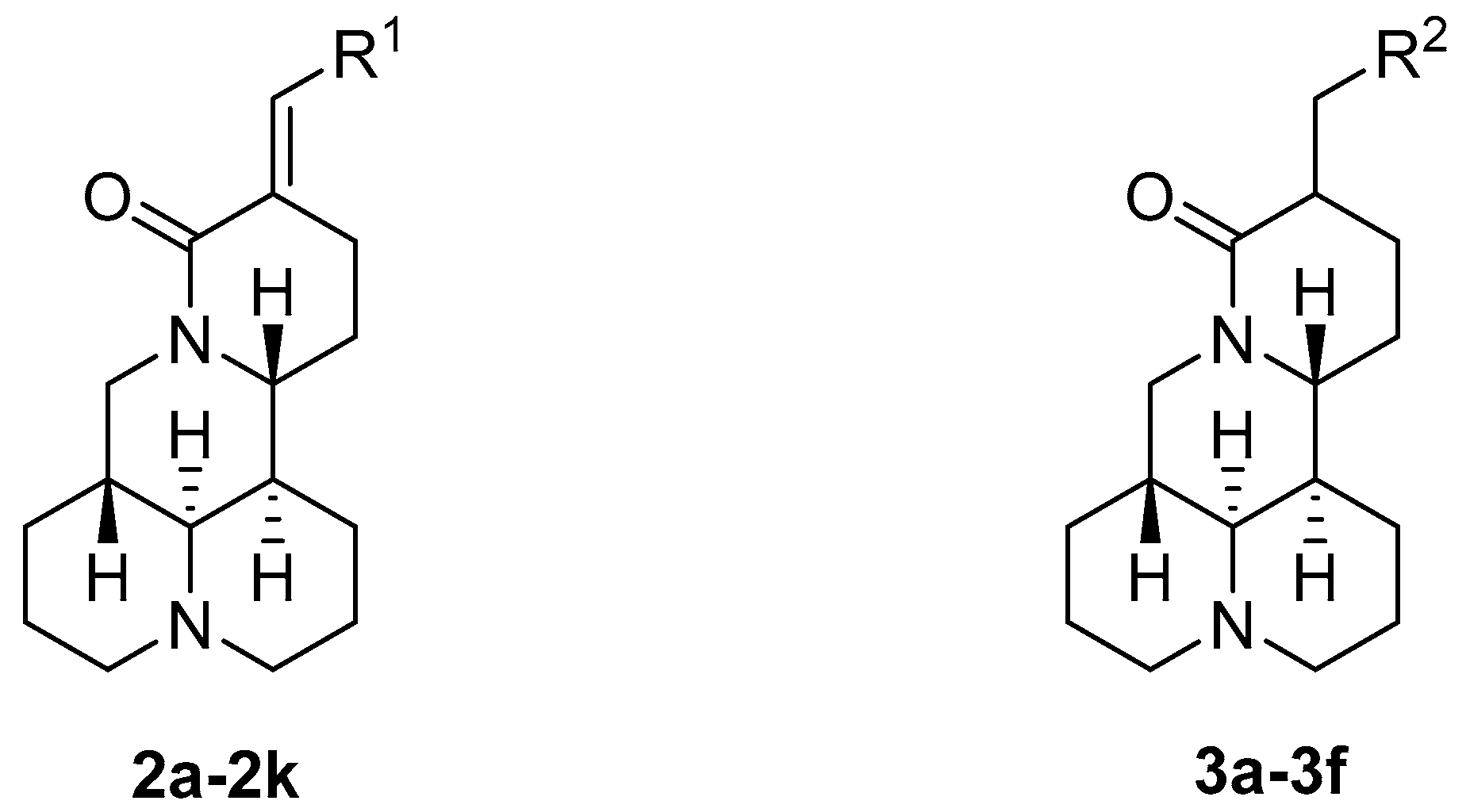
3.3.1. Synthesis of Compounds 2a–2k
3.3.2. Synthesis of Compounds 3a–3f
3.4. Anti-Proliferation Assay
3.5. Cell Cycle Analysis
3.6. Molecular Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Xu, Z.; Zhang, F.; Bai, C.; Yao, C.; Zhong, H.; Zou, C.; Chen, X. Sophoridine induces apoptosis and S phase arrest via ROS-dependent JNK and ERK activation in human pancreatic cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 124. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Cao, H.; Sun, L.; Dong, S.; Bian, Y.; Han, J.; Zhang, L.; Ren, S.; Hu, Y.; Liu, C.; et al. Antitumor Activities of Kushen: Literature Review. Evid. Based Complement. Alternat. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Bai, X.; Wang, C. Traditional Chinese Medicine: A Treasured Natural Resource of Anticancer Drug Research and Development. Am. J. Chin. Med. 2014, 42, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y. Influence of erbanxiao solution on inhibiting angiogenesis in stasis toxin stagnation of non-small cell lung cancer. J. Tradit. Chin. Med. 2013, 33, 303–306. [Google Scholar] [CrossRef]
- Zhuang, H.; Ren, J.; Zhou, Y.; Chen, X.; Wu, Y. Matrine injection combined with intrapleural cisplatin in treatment of 24 patients with hematologic malignancies complicated by pleural effusion. Zhongguo Zhong Yao Za Zhi 2012, 21, 1013–1015. [Google Scholar]
- Zhao, Z.; Fan, H.; Higgins, T.; Qi, J.; Haines, D.; Trivett, A.; Oppenheim, J.J.; Wei, H.; Li, J.; Lin, H.; et al. Fufang Kushen injection inhibits sarcoma growth and tumor-induced hyperalgesia via TRPV1 signaling pathways. Cancer Lett. 2014, 355, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, C.; Dong, R.; He, S.; Liu, T.; Zhao, T.; Wang, Z.; Shen, X.; Zhang, B.; Gao, X.; et al. Safety Evaluation of Chinese Medicine Injections with a Cell Imaging-Based Multiparametric Assay Revealed a Critical Involvement of Mitochondrial Function in Hepatotoxicity. Evid. Based Complement. Alternat. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Li, C.; Shan, X.; Liu, H.; Fan, W.; Wang, Z. A Study on isolation of chemical constituents from Sophora flavescens ait. and their anti-glioma effects. Afr. J. Tradit. Complement. Altern. Med. 2014, 11, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Zhu, H.; Li, Z.; Ma, C.; Fan, M.; Sun, Z.; Huang, C. LC-MS characterization of efficacy substances in serum of experimental animals treated with Sophora flavescens extracts. Biomed. Chromatogr. 2007, 21, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C.; Alt, A.; Heiss, E.; Eldeen, A.; Klimo, K.; Knauft, J.; Neumann, I.; Scherf, H.; Frank, N.; Bartsch, H.; et al. Cancer Chemopreventive Activity of Xanthohumol, a Natural Product Derived from Hop. Mol. Cancer Ther. 2002, 1, 959–969. [Google Scholar] [PubMed]
- Ii, T.; Satomi, Y.; Katoh, D.; Shimada, J.; Baba, M.; Okuyama, T.; Nishino, H.; Kitamura, N. Induction of cell cycle arrest and p21CIP1/WAF1 expression in human lung cancer cells by isoliquiritigenin. Cancer Lett. 2004, 207, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Yoon, H.; Kim, K.H.; Jung, S.; Jang, W.; Seo, C.; Lee, Y.M.; Kweon, D.; Hong, J.; Lee, J.; et al. Butein is a novel anti-adipogenic compound. J. Lipid Res. 2013, 54, 1385–1396. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Liu, C.; Tsao, L.; Weng, J.; Ko, H.; Wang, J.; Lin, C. Synthetic chalcones as potential anti-inflammatory and cancer chemopreventive agents. Eur. J. Med. Chem. 2005, 40, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Nowakowska, Z.; Kędzia, B.; Schroeder, G. Synthesis, physicochemical properties and antimicrobial evaluation of new (E)-chalcones. Eur. J. Med. Chem. 2008, 43, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Tomar, V.; Bhattacharjee, G.; Kamaluddin; Kumar, A. Synthesis and antimicrobial evaluation of new chalcones containing piperazine or 2,5-dichlorothiophene moiety. Bioorg. Med. Chem. Lett. 2007, 17, 5321–5324. [Google Scholar] [CrossRef] [PubMed]
- Katsori, A.M.; Hadjipavlou, D. Chalcones in Cancer: Understanding their Role in Terms of QSAR. Curr. Med. Chem. 2009, 16, 1062–1081. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Attaran, B.; Malekzadeh, R.; Graham, D.Y. Furazolidone, an Underutilized Drug for H. pylori Eradication: Lessons from Iran. Dig. Dis. Sci. 2017, 62, 1890–1896. [Google Scholar] [CrossRef] [PubMed]
- Dimmitt, D.C.; Cramer, M.B.; Keung, A.; Arumugham, T.; Weir, S.J. Pharmacokinetics of dolasetron with coadministration of cimetidine or rifampin in healthy subjects. Cancer Chemother. Pharmacol. 1999, 43, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Rachna Pandey, K.V.S.; Madhukar, B. Khetmalas. Indole: A novel signaling molecule and its applications. Indian J. Biotechnol. 2013, 12, 297–310. [Google Scholar]
- James, T.; Maclellan, P.; Burslem, G.M.; Simpson, I.; Grant, J.A.; Warriner, S.; Sridharan, V.; Nelson, A. A modular lead-oriented synthesis of diverse piperazine, 1,4-diazepane and 1,5-diazocane scaffolds. Org. Biomol. Chem. 2014, 12, 2584–2591. [Google Scholar] [CrossRef] [PubMed]
- Matwijczuk, A.; Kluczyk, D.; Górecki, A.; Niewiadomy, A.; Gagos, M. Solvent Effects on Molecular Aggregation in 4-(5-Heptyl-1,3,4-thiadiazol-2-yl)benzene-1,3-diol and 4-(5-Methyl-1,3,4-thiadiazol-2-yl)benzene-1,3-diol. J. Phys. Chem. B 2016, 120, 7958–7969. [Google Scholar] [CrossRef] [PubMed]
- Brancato, G.; Signore, G.; Neyroz, P.; Polli, D.; Cerullo, G.; Abbandonato, G.; Nucara, L.; Barone, V.; Beltram, F.; Bizzarri, R. Dual Fluorescence through Kasha’s Rule Breaking: An Unconventional Photomechanism for Intracellular Probe Design. J. Phys. Chem. B 2015, 119, 6144–6154. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.V.; Siddiqui, S.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Lokwani, D.K.; Nikalje, A.P.G. Microwave-Assisted Facile Synthesis, Anticancer Evaluation and Docking Study of N-((5-(Substituted methylene amino)-1,3,4-thiadiazol-2-yl)methyl) Benzamide Derivatives. Molecules 2017, 22, 995. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Zhao, Y.; Goto, M.; Hsieh, K.; Yang, X.; Morris-Natschke, S.L.; Liu, L.; Zhao, B.; Lee, K. Alkaloids from Oxytropis ochrocephala and Antiproliferative Activity of Sophoridine Derivatives Against Cancer Cell Lines. Bioorg. Med. Chem. Lett. 2016, 26, 1495–1497. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, W.; Jiang, J.; Ren, K.; Du, N.; Li, Y.; Wang, Y.; Bi, C.; Shao, R.; Song, D. Synthesis, structure-activity relationship and biological evaluation of anticancer activity for novel N-substituted sophoridinic acid derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 5251–5254. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Zhang, N.; Yang, P.; Ye, C.; Wang, Y.; Fan, T.; Shao, R.; Deng, H.; Song, D. Synthesis, Biological Evaluation, and Autophagy Mechanism of 12N-Substituted Sophoridinamines as Novel Anticancer Agents. ACS Med. Chem. Lett. 2017, 8, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyana, C.; Narayana, S.H.; Ramasamyc, S.; Vanam, U.; Manivannan, E.; Karunagaran, D.; Trivedi, P. Advances in Chalcones with Anticancer Activities. Recent Pat. Anticancer Drug Discov. 2015, 10, 97–115. [Google Scholar] [CrossRef]
- Wang, L.; You, Y.; Wang, S.; Liu, X.; Liu, B.; Wang, J.; Lin, X.; Chen, M.; Liang, G.; Yang, H. Synthesis, characterization and in vitro anti-tumor activities of matrine derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 4100–4102. [Google Scholar] [CrossRef] [PubMed]
- Galambos, J.; Wágner, G.; Nógrádi, K.; Bielik, A.; Molnár, L.; Bobok, A.; Horváth, A.; Kiss, B.; Kolok, S.; Nagy, J.; et al. Carbamoyloximes as novel non-competitive mGlu5 receptor antagonists. Bioorg. Med. Chem. Lett. 2010, 20, 4371–4375. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 2a–2k, 3a–3f are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R1 | R2 | IC50 (μM) | Clog P a | |
|---|---|---|---|---|---|
| HepG-2 | CNE-2 | ||||
| Sophoridine | 4670 ± 127 | 5379 ± 109 | 1.36 | ||
| 2a | CH2CH2CH2CH3 | / | 82.3 ± 9.3 | >100 | 3.99 |
| 2b | Ph | / | >100 | 87.5 ± 6.8 | 3.73 |
| 2c | PhOCH3-p | / | >100 | >100 | 3.65 |
| 2d | PhCl-p | / | 55.7 ± 5.6 | >100 | 4.44 |
| 2e |  | / | 35.1 ± 2.9 | 38.3 ± 2.1 | 5.56 |
| 2f | 3-naphthyl | / | 75.5 ± 8.3 | 95.3 ± 4.8 | 4.9 |
| 2g | 2-furyl | / | 92.3 ± 7.1 | 89.5 ± 8.3 | 2.91 |
| 2h | 2-thienyl | / | >100 | >100 | 3.38 |
| 2i | 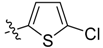 | / | 96.4 ± 5.7 | 89.4 ± 7.8 | 4.13 |
| 2j |  | / | >100 | >100 | 3.87 |
| 2k | 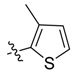 | / | 25.5 ± 3.3 | 33.8 ± 2.3 | 3.87 |
| 3a | / | CH3 | >100 | >100 | 2.41 |
| 3b | / | CH2CH2CH3 | >100 | >100 | 3.47 |
| 3c | / | CH2CH2CH2CH2CH3 | 57.9 ± 6.3 | 83.1 ± 9.2 | 4.53 |
| 3d | / | Ph | >100 | >100 | 3.45 |
| 3e | / | 2-furyl | >100 | >100 | 2.62 |
| 3f | / | 2-thienyl | >100 | >100 | 3.09 |
| Taxo | 15.6 ± 2.3 | 0.18 | |||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Y.; Wu, L.; Dai, H.; Gao, M.; Ur Rashid, H.; Wang, H.; Xie, P.; Liu, X.; Jiang, J.; Wang, L. Novel α, β-Unsaturated Sophoridinic Derivatives: Design, Synthesis, Molecular Docking and Anti-Cancer Activities. Molecules 2017, 22, 1967. https://doi.org/10.3390/molecules22111967
Xu Y, Wu L, Dai H, Gao M, Ur Rashid H, Wang H, Xie P, Liu X, Jiang J, Wang L. Novel α, β-Unsaturated Sophoridinic Derivatives: Design, Synthesis, Molecular Docking and Anti-Cancer Activities. Molecules. 2017; 22(11):1967. https://doi.org/10.3390/molecules22111967
Chicago/Turabian StyleXu, Yiming, Lichuan Wu, Hang Dai, Mingyan Gao, Haroon Ur Rashid, Haodong Wang, Peng Xie, Xu Liu, Jun Jiang, and Lisheng Wang. 2017. "Novel α, β-Unsaturated Sophoridinic Derivatives: Design, Synthesis, Molecular Docking and Anti-Cancer Activities" Molecules 22, no. 11: 1967. https://doi.org/10.3390/molecules22111967
APA StyleXu, Y., Wu, L., Dai, H., Gao, M., Ur Rashid, H., Wang, H., Xie, P., Liu, X., Jiang, J., & Wang, L. (2017). Novel α, β-Unsaturated Sophoridinic Derivatives: Design, Synthesis, Molecular Docking and Anti-Cancer Activities. Molecules, 22(11), 1967. https://doi.org/10.3390/molecules22111967