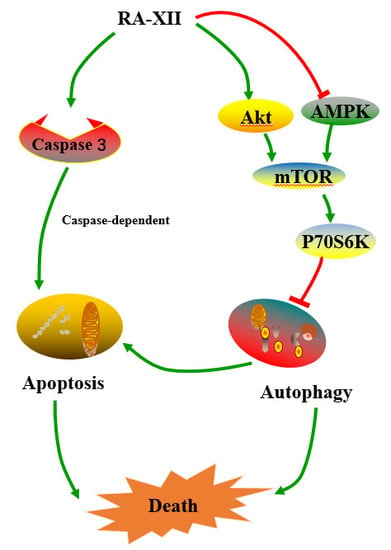
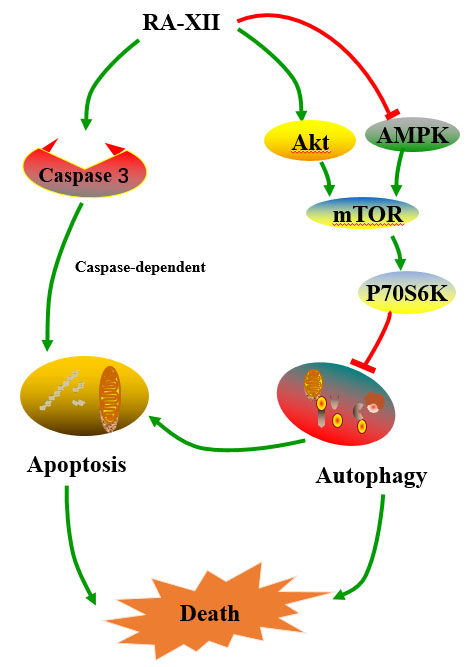
Natural Cyclopeptide RA-XII, a New Autophagy Inhibitor, Suppresses Protective Autophagy for Enhancing Apoptosis through AMPK/mTOR/P70S6K Pathways in HepG2 Cells

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
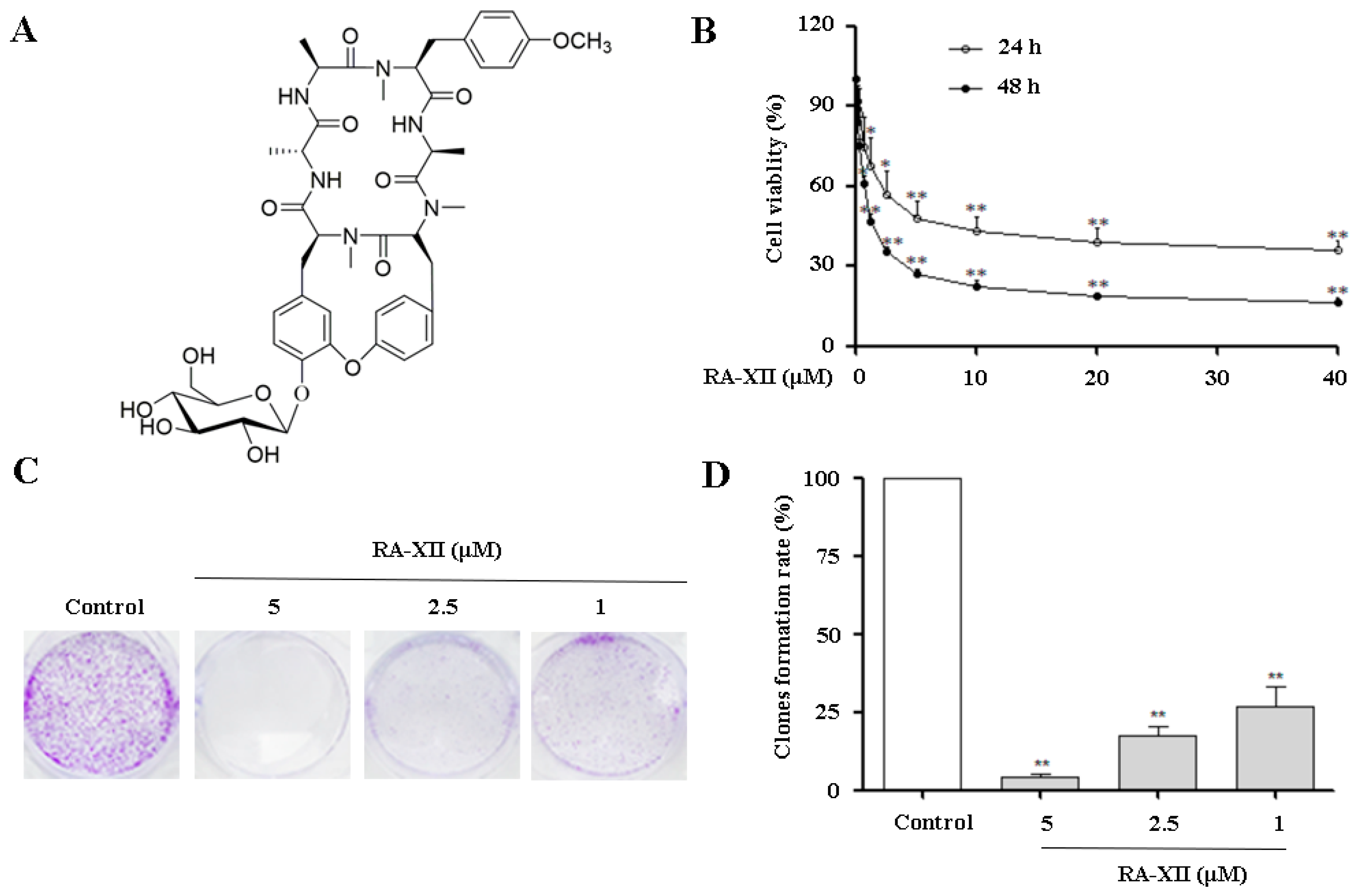
2.1. RA-XII Inhibits Growth and Colony Formation in HepG2 Cells
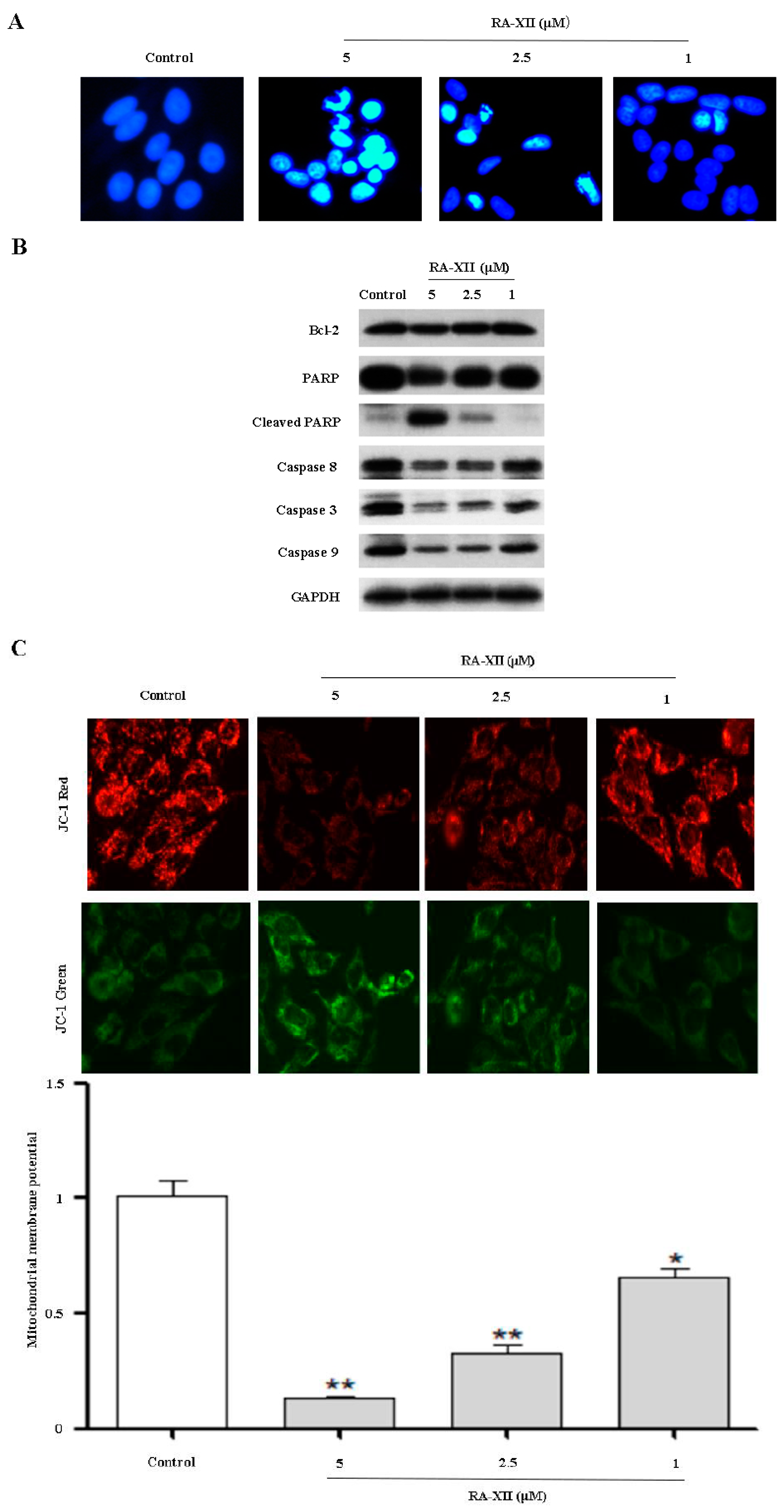
2.2. RA-XII Induces Apoptosis in HepG2 Cells
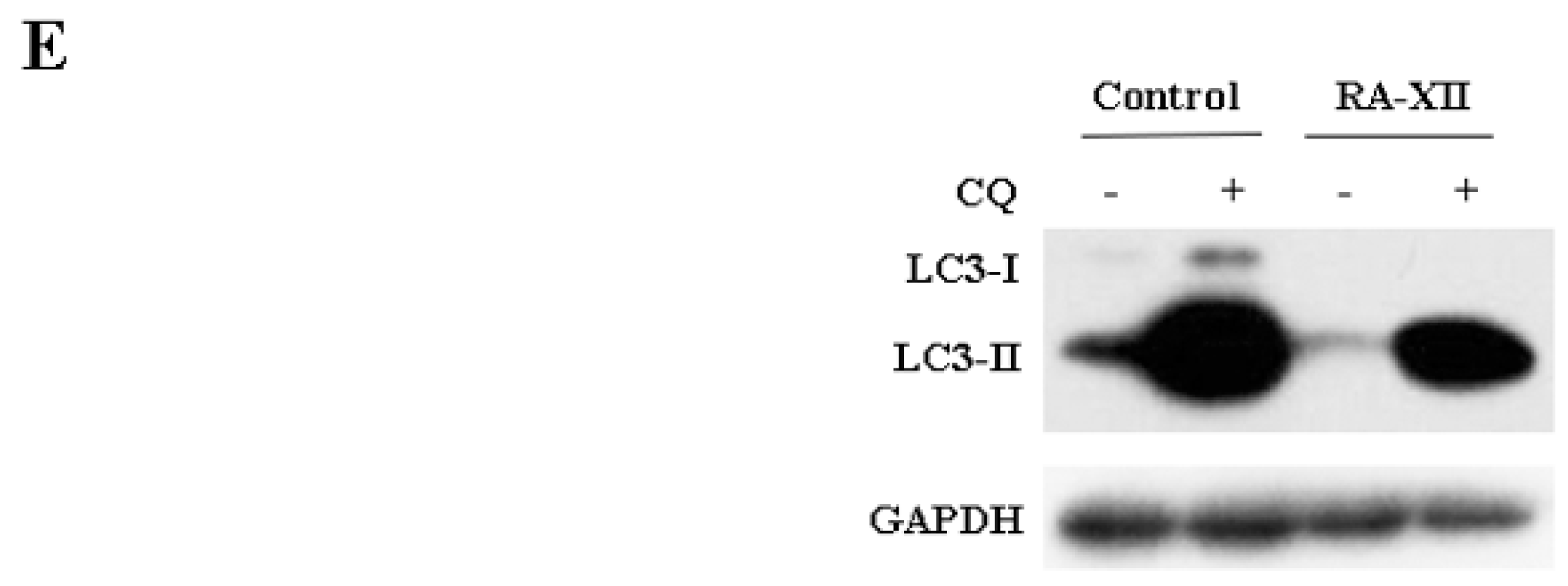
2.3. RA-XII Suppresses Autophagy in HepG2 Cells
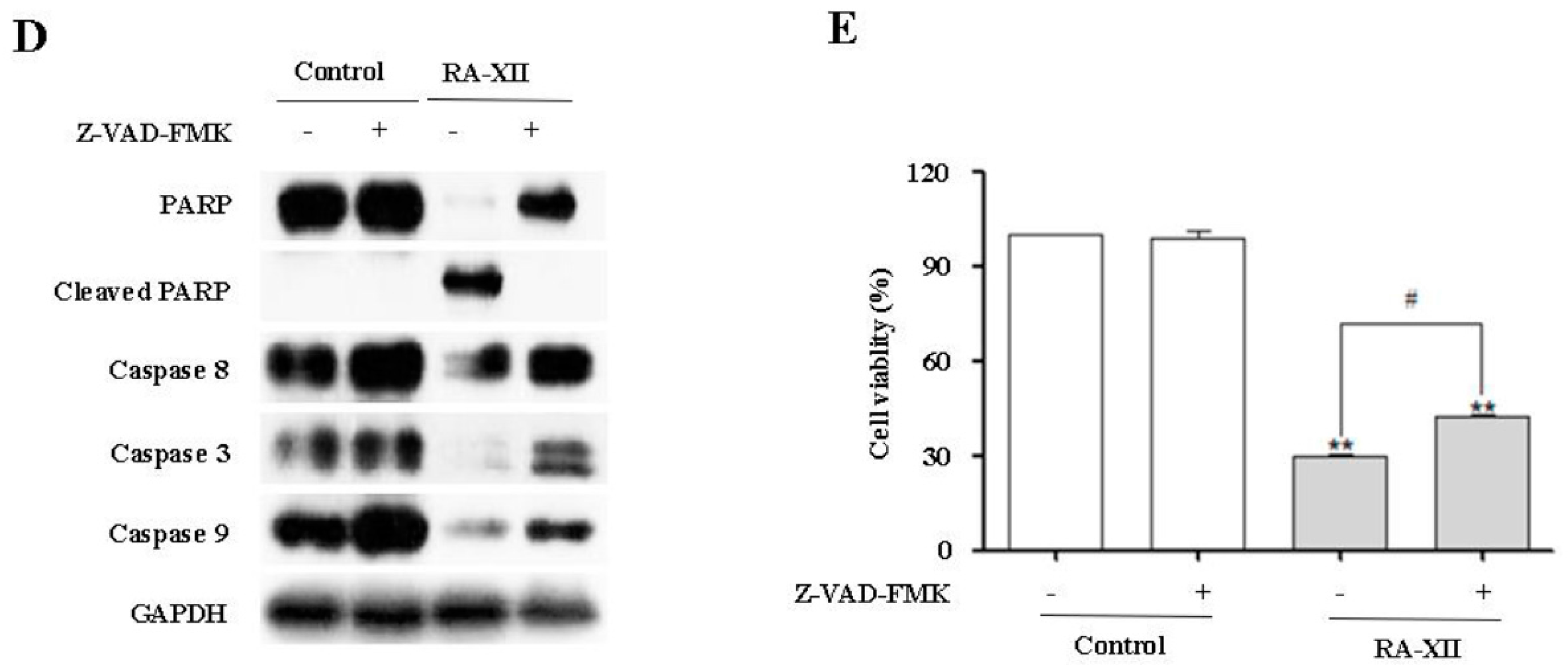
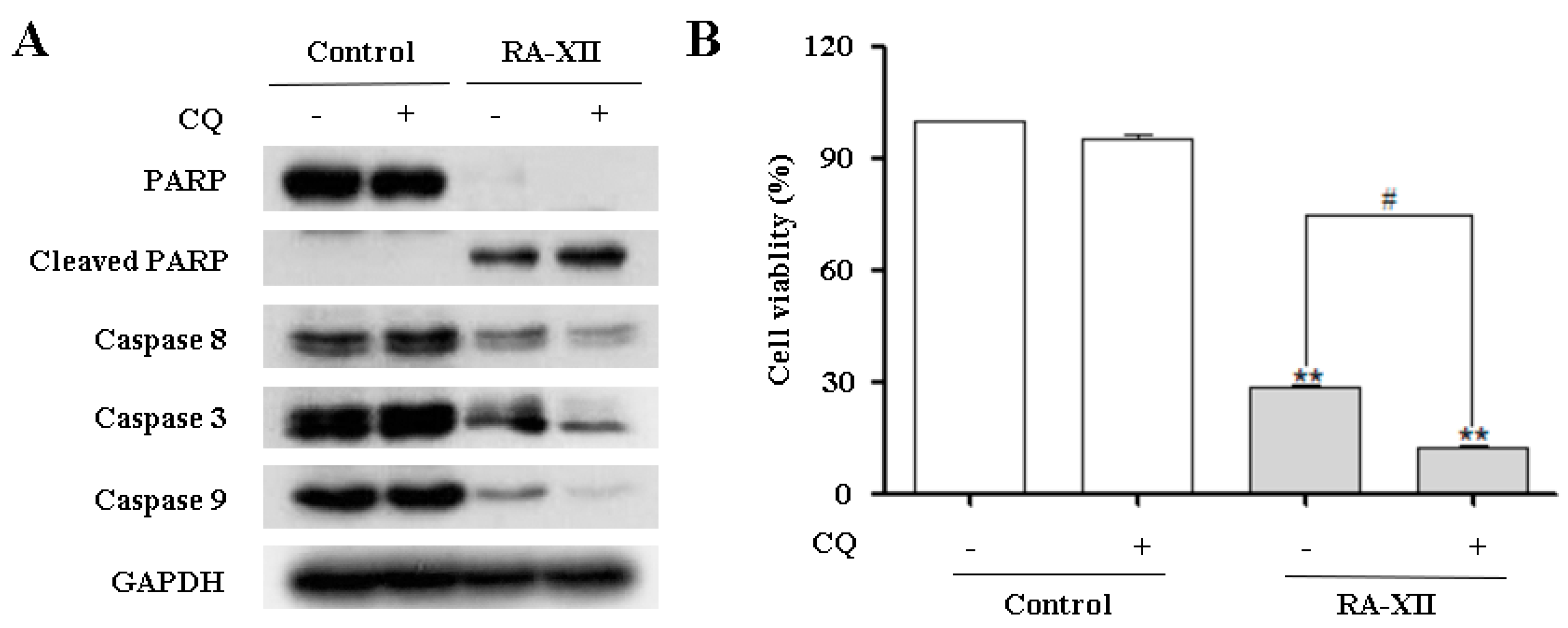
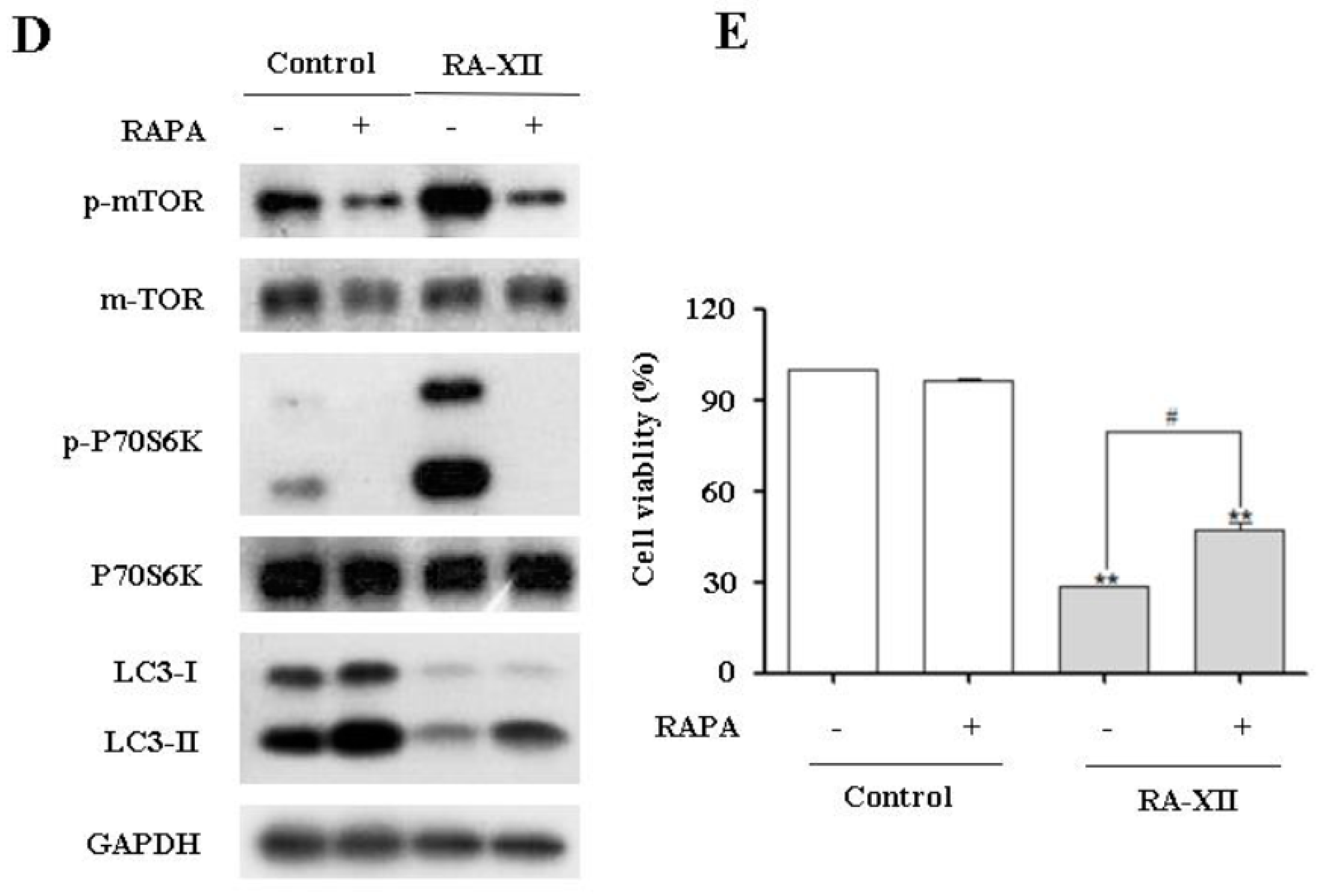
2.4. RA-XII Inhibits Protective Autophagy and Promotes Apoptosis in HepG2 Cells
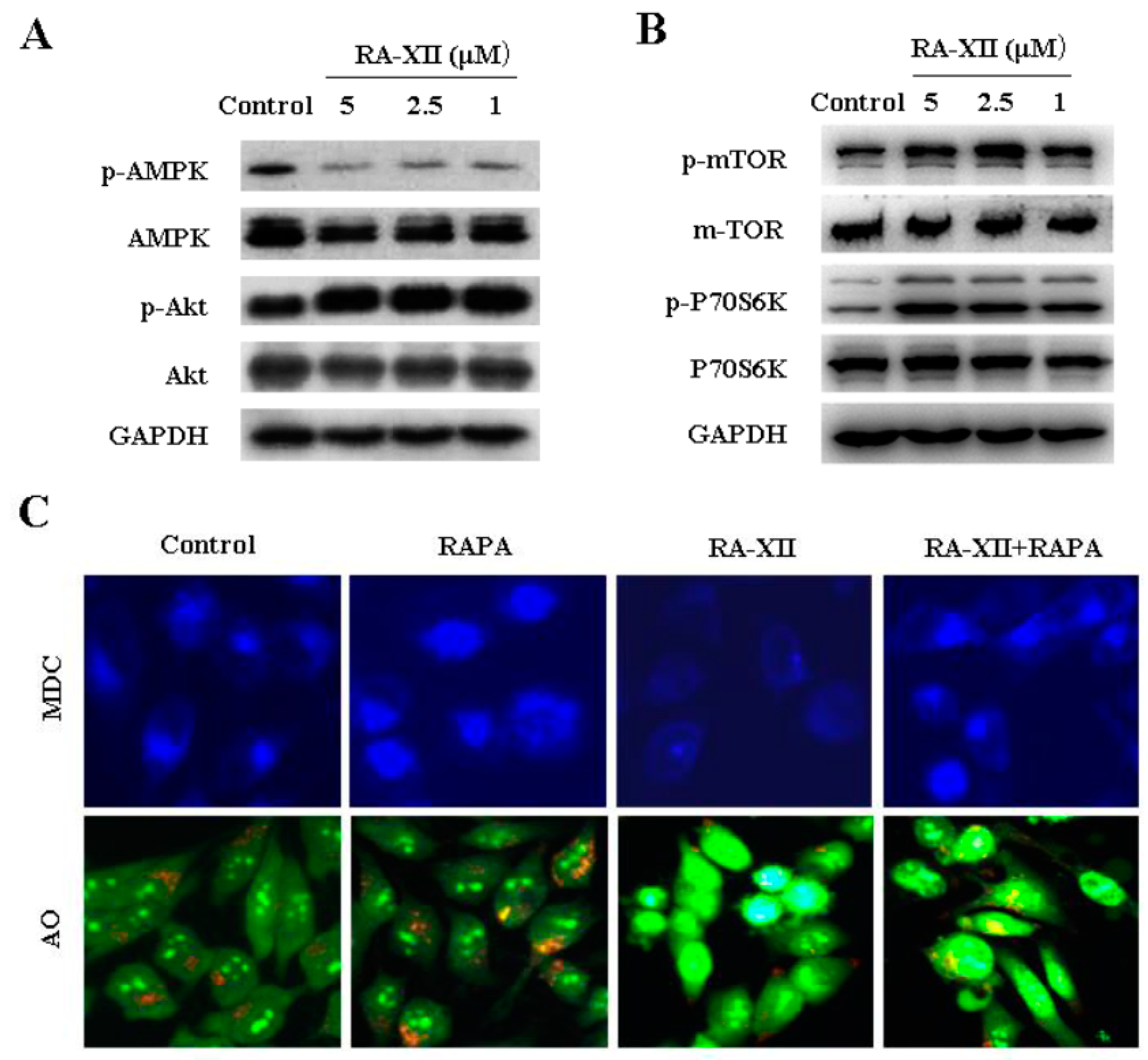
2.5. Akt/AMPK-mTOR Signaling Pathways are Involved in RA-XII-Inhibited Protective Autophagy in HepG2 Cells
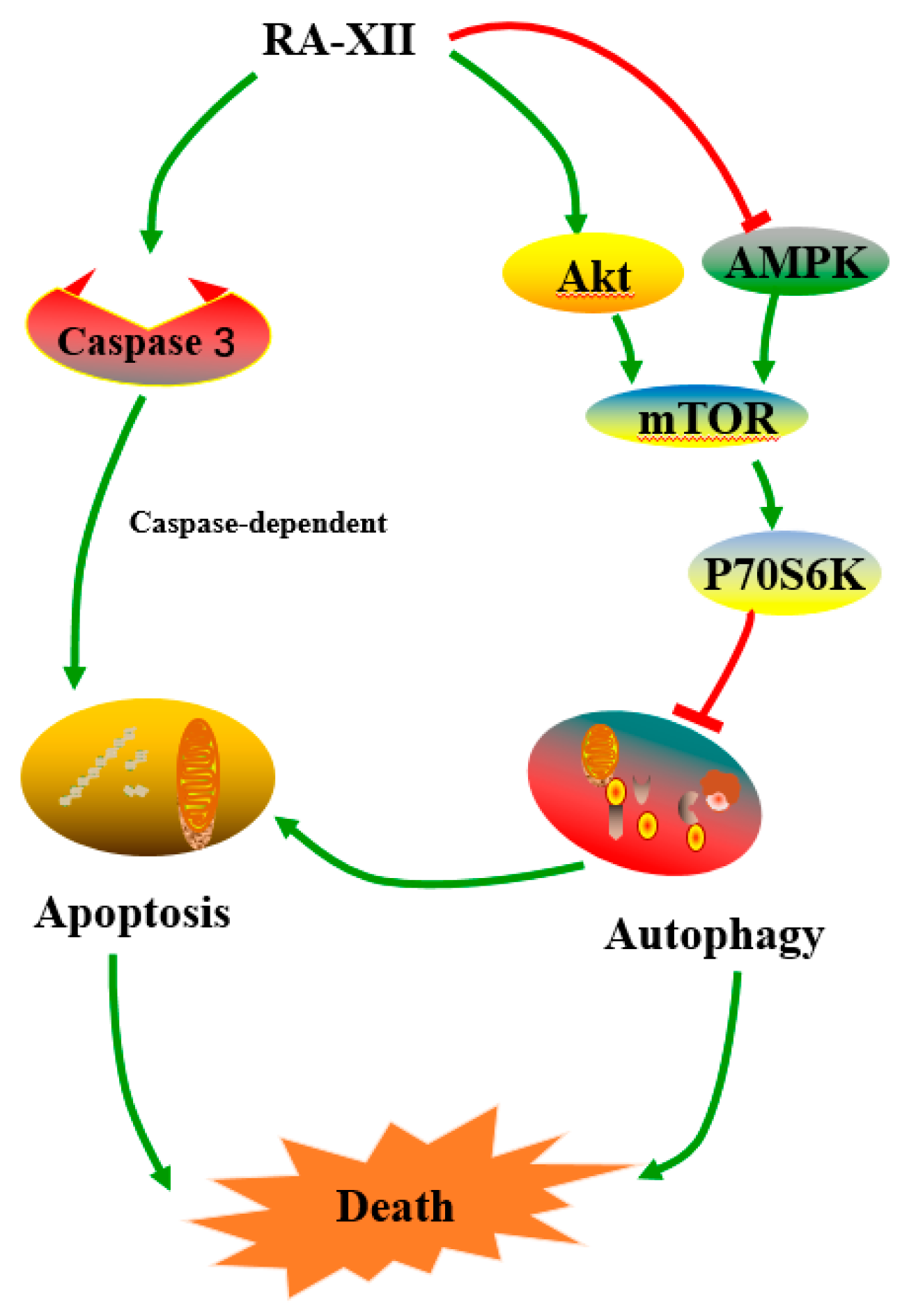
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Reagents and Antibodies
4.3. Cell Viability Assay
4.4. Clonogenic Survival Assay
4.5. Mitochondrial Membrane Potential (JC-1) Assay
4.6. GFP-LC3B Plasmid Transfection
4.7. DAPI Staining
4.8. Staining of Autophagic Vacuoles by Monodansylcadaverine (MDC) and Acridine Orange (AO)
4.9. Immunoblotting Assay
4.10. Quantitative Real-Time PCR
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bertuccio, P.; Turati, F.; Carioli, G.; Rodriguez, T.; La-Vecchia, C.; Malvezzi, M.; Negri, E. Global trends and predictions in hepatocellular carcinoma mortality. J. Hepatol. 2017, 67, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Saran, U.; Humar, B.; Kolly, P.; Dufour, J.F. Hepatocellular carcinoma and lifestyles. J. Hepatol. 2016, 64, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Mahato, R.I. Recent advances in hepatocellular carcinoma therapy. Pharmacol. Ther. 2017, 173, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, H.C.; Thimme, R.; Blum, H.E. Targeted therapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Lalaoui, N.; Lindqvist, L.M.; Sandow, J.J.; Ekert, P.G. The molecular relationships between apoptosis, autophagy and necroptosis. Semin. Cell Dev. Biol. 2015, 39, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.A.; Salmani, J.M.M.; Jiang, Z.; Feng, L.; Song, J.; Jia, X.; Chen, B. Autophagy: An overview and its roles in cancer and obesity. Clin. Chim. Acta 2017, 468, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vicencio, J.M.; Kepp, O.; Tasdemir, E.; Maiuri, M.C.; Kroemer, G. To die or not to die: That is the autophagic question. Curr. Mol. Med. 2008, 8, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Jang, B.K. The role of autophagy in hepatocellular carcinoma. Int. J. Mol. Sci. 2015, 16, 26629–26643. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and interpretation of current autophagy inhibitors and activators. Acta. Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Jarauta, V.; Jaime, P.; Gonzalo, O.; de-Miguel, D.; Ramírez-Labrada, A.; Martínez-Lostao, L.; Anel, A.; Pardo, J.; Marzo, I.; Naval, J. Inhibition of autophagy with chloroquine potentiates carfilzomib-induced apoptosis in myeloma cells in vitro and in vivo. Cancer Lett. 2016, 382, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, C.; Xu, D.; Wang, M.; Xia, Q. AstragalosideII inhibits autophagic flux and enhance chemosensitivity of cisplatin in human cancer cells. Biomed. Pharmacother. 2016, 81, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Sinicrope, F.A. Celecoxib-induced apoptosis is enhanced by ABT-737 and by inhibition of autophagy in human colorectal cancer cells. Autophagy 2010, 6, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Hu, X.; Wu, B.; Jiang, H.; Young, C.Y.; Pang, Y.; Yuan, H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011, 307, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Longo, L.; Platini, F.; Scardino, A.; Alabiso, O.; Vasapollo, G.; Tessitore, L. Autophagy inhibition enhances anthocyanin-induced apoptosis in hepatocellular carcinoma. Mol. Cancer Ther. 2008, 7, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Wahab, A.; Gao, K.; Jia, C.; Zhang, F.; Tian, G.; Murtaza, G.; Chen, J. Significance of resveratrol in clinical management of chronic diseases. Molecules 2017, 18, 22. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Dent, R.; Im, S.A.; Espié, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017. [Google Scholar] [CrossRef]
- Gigliotti, C.L.; Ferrara, B.; Occhipinti, S.; Boggio, E.; Barrera, G.; Pizzimenti, S.; Giovarelli, M.; Fantozzi, R.; Chiocchetti, A.; Argenziano, M.; et al. Enhanced cytotoxic effects of camptothecin nanosponges in anaplastic thyroid cancer cells in vitro and in vivo on orthotopic xenograft tumors. Drug Deliv. 2017, 24, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.Y.; Chen, W.; Fan, J.T.; Song, R.; Wang, L.; Gu, Y.H.; Zeng, G.Z.; Shen, Y.; Wu, X.F.; Tan, N.H.; et al. Plant cyclopeptide RA-V kills human breast cancer cells by inducing mitochondria-mediated apoptosis through blocking PDK1-AKT interaction. Toxicol. Appl. Pharmacol. 2013, 267, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.W.; Wang, Z.; Yue, G.G.; Zhao, S.M.; Lee, J.K.; Fung, K.P.; Leung, P.C.; Lau, C.B.; Tan, N.H. Cyclopeptide RA-V inhibits cell adhesion and invasion in both estrogen receptor positive and negative breast cancer cells via PI3K/AKT and NF-κB signaling pathways. Biochim. Biophys. Acta 2015, 1853, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Yue, G.G.; Fan, J.T.; Lee, J.K.; Zeng, G.Z.; Ho, T.W.; Fung, K.P.; Leung, P.C.; Tan, N.H.; Lau, C.B. Cyclopeptide RA-V inhibits angiogenesis by down-regulating ERK1/2 phosphorylation in HUVEC and HMEC-1 endothelial cells. Br. J. Pharmacol. 2011, 164, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Yamamiya, T.; Takeya, K.; Itokawa, H. New antitumor bicyclic hexapeptides, RA-XI, -XII, -XIII and -XIV from Rubia cordifolia. Chem. Pharm. Bull. 1992, 40, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.T.; Su, J.; Peng, Y.M.; Li, Y.; Li, J.; Zhou, Y.B.; Zeng, G.Z.; Yan, H.; Tan, N.H. Rubiyunnanins C-H, cytotoxic cyclic hexapeptides from Rubia yunnanensis inhibiting nitric oxide production and NF-κB activation. Bioorg. Med. Chem. 2010, 18, 8226–8234. [Google Scholar] [CrossRef] [PubMed]
- Leung, H.W.; Zhao, S.M.; Yue, G.G.; Lee, J.K.; Fung, K.P.; Leung, P.C.; Tan, N.H.; Lau, C.B. RA-XII inhibits tumour growth and metastasis in breast tumour-bearing mice via reducing cell adhesion and invasion and promoting matrix degradation. Sci. Rep. 2015, 5, 16985. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fang, Y.; Yang, Y.; Qin, Y.; Wu, P.; Wang, T.; Lai, H.; Meng, L.; Wang, D.; Zheng, Z.; et al. Elaiophylin, a novel autophagy inhibitor, exerts antitumor activity as a single agent in ovarian cancer cells. Autophagy 2015, 11, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, Z.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016. [Google Scholar] [CrossRef] [PubMed]
- Averous, J.; Proud, C.G. When translation meets transformation: The mTOR story. Oncogene 2006, 25, 6423–6435. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, L.E.; Williamson, L.E.; Chan, E.Y. Advances in autophagy regulatory mechanisms. Cells 2016, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Liu, T.; Kamp, D.W.; Wang, Y.; He, H.; Zhou, X.; Li, D.; Yang, L.; Zhao, B.; Liu, G. AKT/mTOR and c-Jun N-terminal kinase signaling pathways are required for chrysotile asbestos-induced autophagy. Free Radic. Biol. Med. 2014, 72, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gong, H.; Yang, S.; Yang, L.; Fan, Y.; Zhou, Y. Pectic bee pollen polysaccharide from Rosa rugosa alleviates diet-induced hepatic steatosis and insulin resistance via induction of AMPK/mTOR-mediated autophagy. Molecules 2017, 22, 699. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Ferri, K.F.; Kroemer, G. Mammalian target of rapamycin (mTOR): Pro- and anti-apoptotic. Cell Death. Differ. 2002, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nakada, D.; Yilmaz, O.H.; Tothova, Z.; Joseph, N.M.; Lim, M.S.; Gilliland, D.G.; Morrison, S.J. mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 2010, 7, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, B.; Liu, L.; Luo, Y.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Kontos, C.D.; Huang, S. Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death. Free Radic. Biol. Med. 2011, 50, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Wu, Y.T.; Chen, B.; Zhou, J.; Shen, H.M. Impaired autophagy due to constitutive mTOR activation sensitizes TSC2-null cells to cell death under stress. Autophagy 2011, 7, 1173–1186. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds RA-XII is available from the authors. |









© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, L.; Wang, Z.; Wang, Y.; Guo, D.; Yang, J.; Chen, L.; Tan, N. Natural Cyclopeptide RA-XII, a New Autophagy Inhibitor, Suppresses Protective Autophagy for Enhancing Apoptosis through AMPK/mTOR/P70S6K Pathways in HepG2 Cells. Molecules 2017, 22, 1934. https://doi.org/10.3390/molecules22111934
Song L, Wang Z, Wang Y, Guo D, Yang J, Chen L, Tan N. Natural Cyclopeptide RA-XII, a New Autophagy Inhibitor, Suppresses Protective Autophagy for Enhancing Apoptosis through AMPK/mTOR/P70S6K Pathways in HepG2 Cells. Molecules. 2017; 22(11):1934. https://doi.org/10.3390/molecules22111934
Chicago/Turabian StyleSong, Lihua, Zhe Wang, Yurong Wang, Di Guo, Jianhong Yang, Lijuan Chen, and Ninghua Tan. 2017. "Natural Cyclopeptide RA-XII, a New Autophagy Inhibitor, Suppresses Protective Autophagy for Enhancing Apoptosis through AMPK/mTOR/P70S6K Pathways in HepG2 Cells" Molecules 22, no. 11: 1934. https://doi.org/10.3390/molecules22111934
APA StyleSong, L., Wang, Z., Wang, Y., Guo, D., Yang, J., Chen, L., & Tan, N. (2017). Natural Cyclopeptide RA-XII, a New Autophagy Inhibitor, Suppresses Protective Autophagy for Enhancing Apoptosis through AMPK/mTOR/P70S6K Pathways in HepG2 Cells. Molecules, 22(11), 1934. https://doi.org/10.3390/molecules22111934