Current Development Status of MEK Inhibitors


Abstract
:1. Introduction
2. Ras/Raf/MEK/ERK Pathway and MEK Inhibitors
3. MEK Inhibitors Approved by the US Food and Drug Administration (FDA)
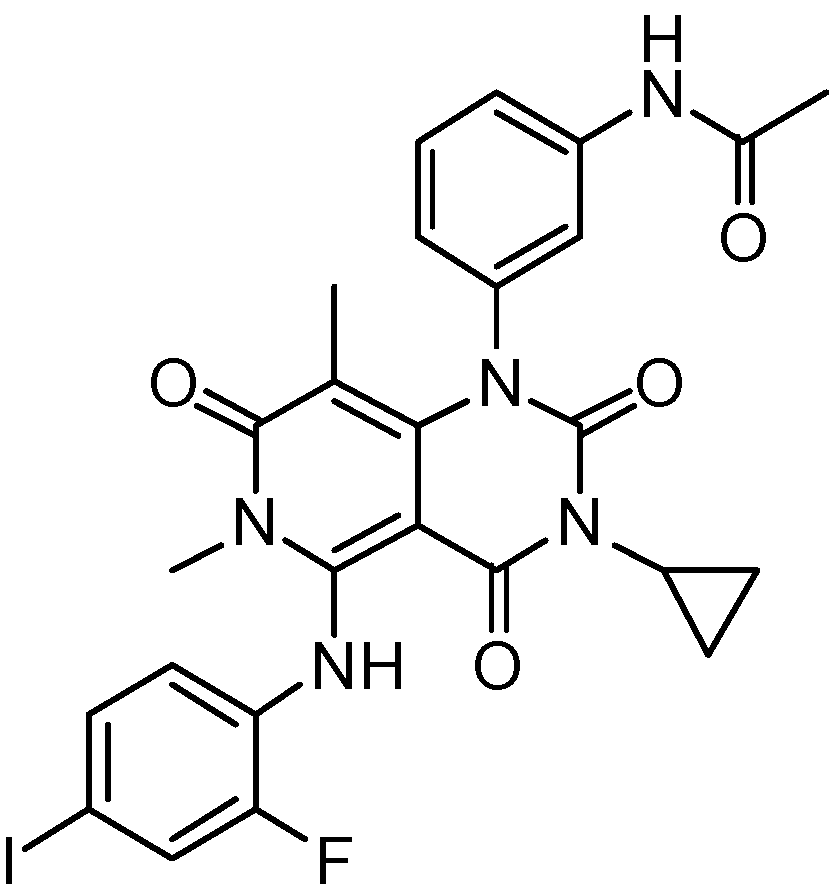
3.1. Trametinib
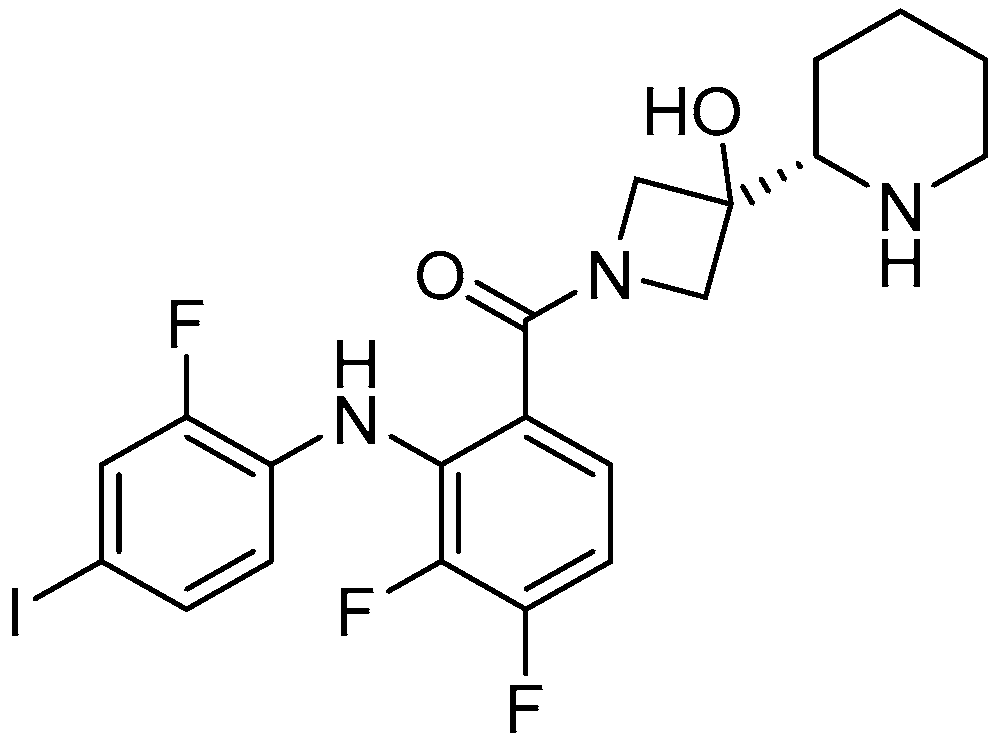
3.2. Cobimetinib
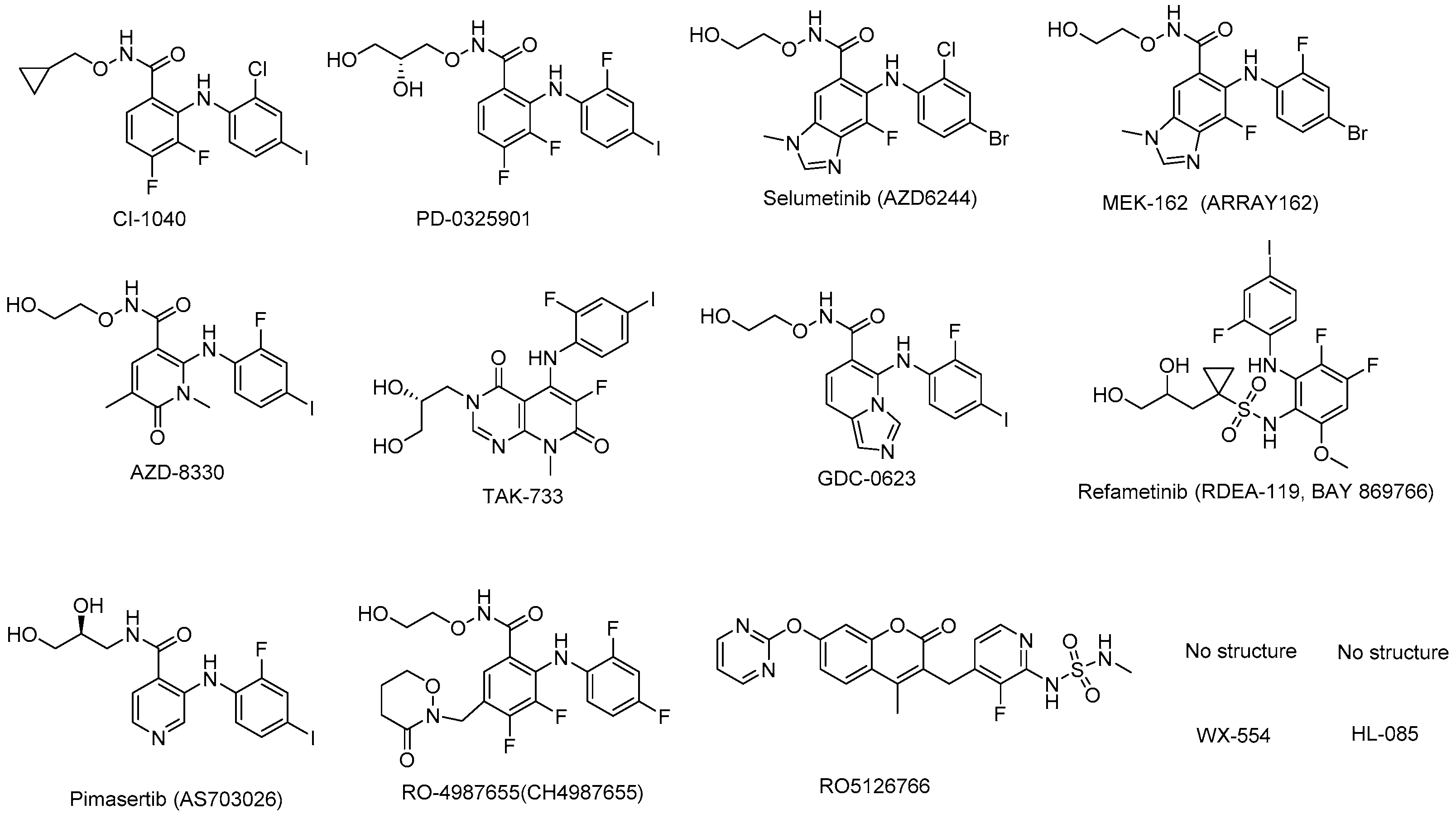
4. MEK Inhibitors under Clinical Development
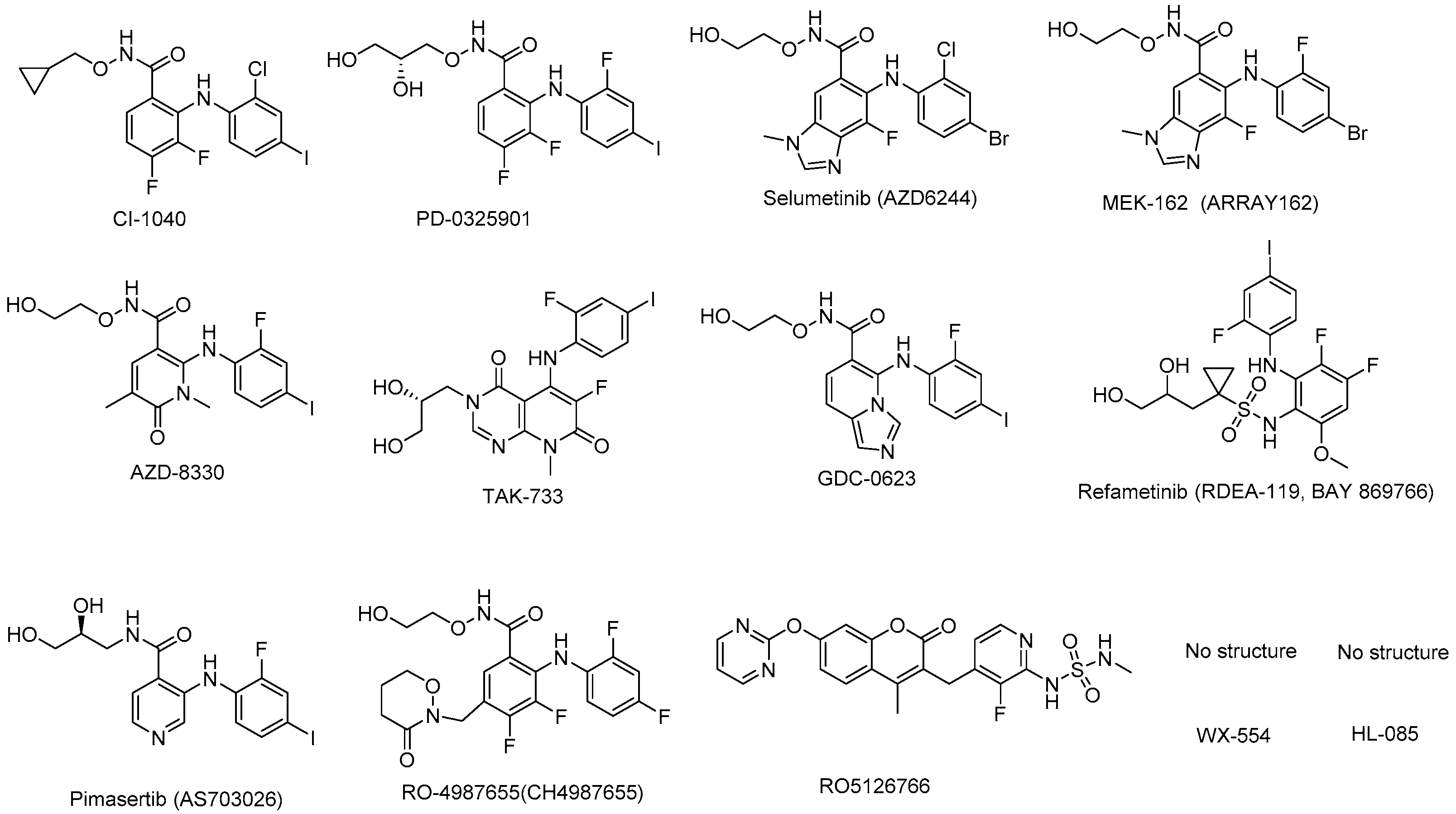
4.1. CI-1040 (PD184352)
4.2. PD-0325901
4.3. Selumetinib (ARRY-142886; AZD6244)
4.4. Binimetinib (MEK162, ARRY-438162)
4.5. AZD-8330 (ARRY-424704)
4.6. TAK-733
4.7. GDC-0623
4.8. Refametinib (RDEA-119, BAY-869766)
4.9. Pimasertib (AS703026)
4.10. RO4987655 (CH4987655)
4.11. RO5126766
4.12. WX-554
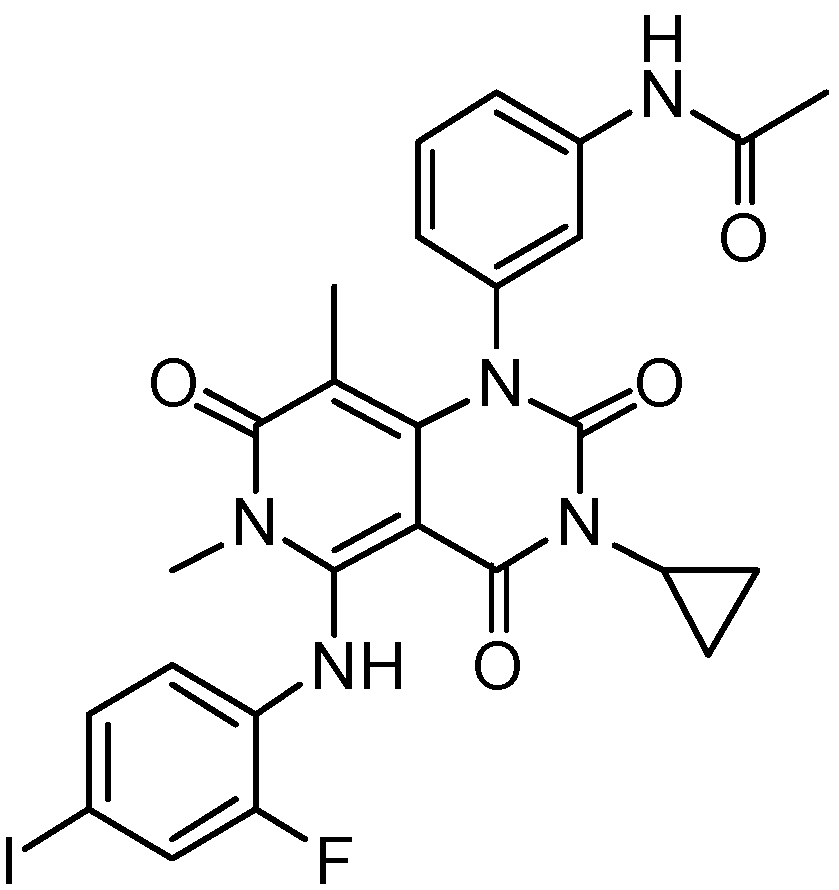
4.13. HL-085
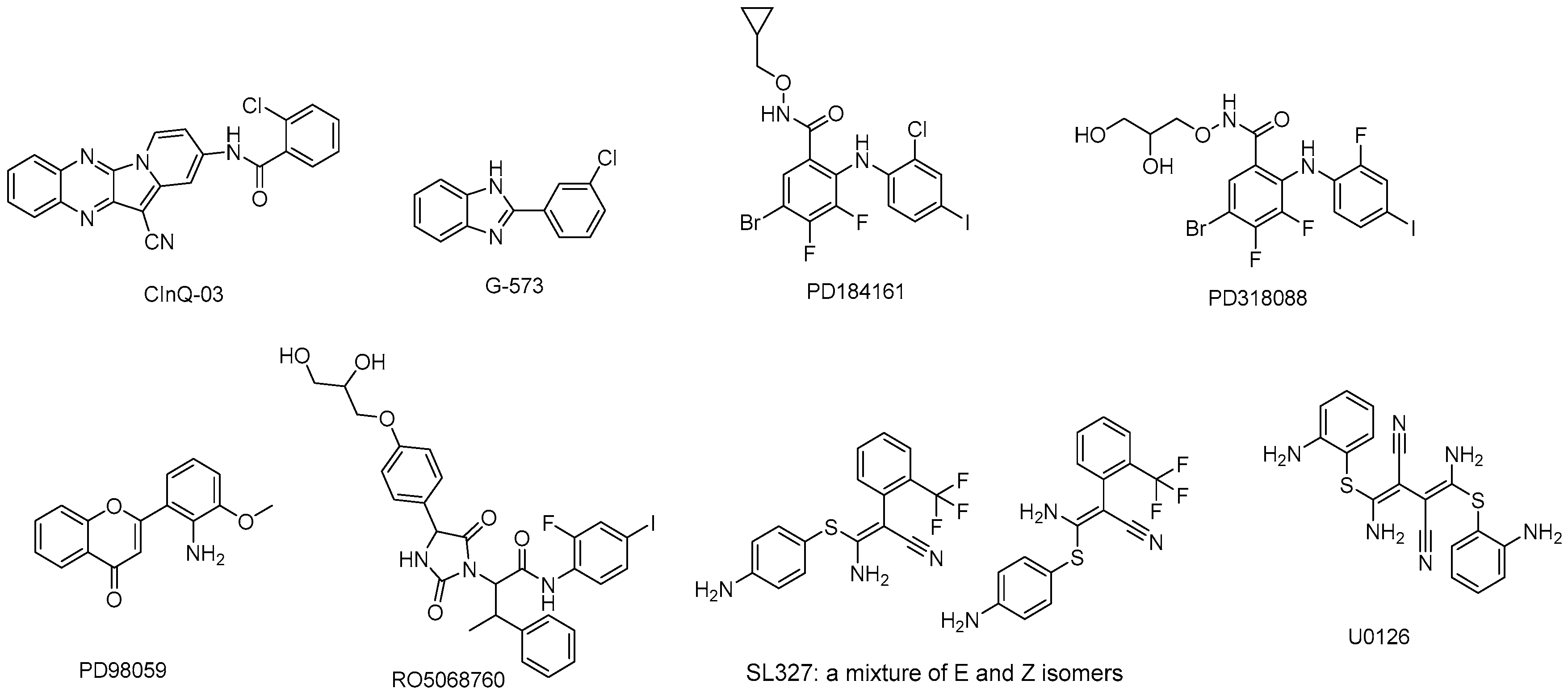
5. MEK Inhibitors in Preclinical Development
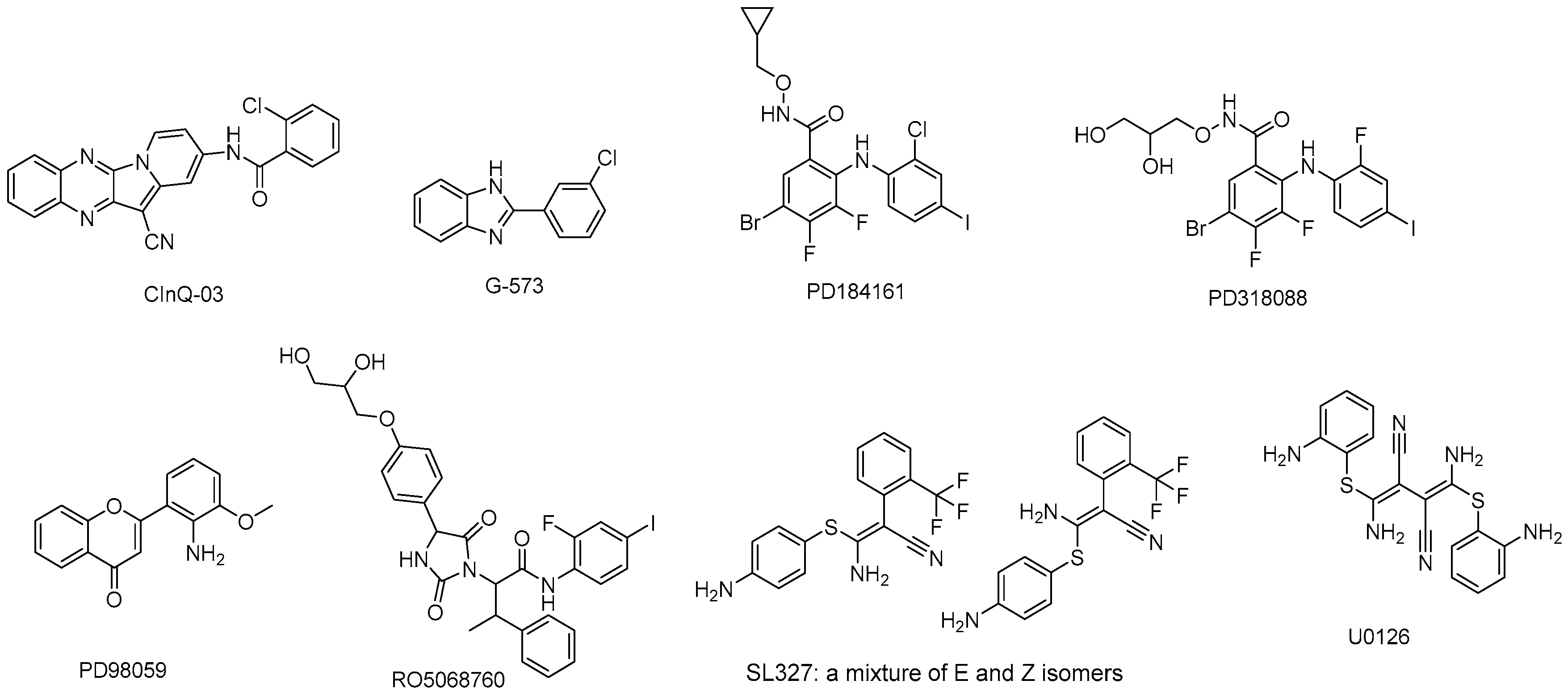
5.1. CInQ-03
5.2. G-573
5.3. PD184161
5.4. PD318088
5.5. PD98059
5.6. RO5068760
5.7. U0126
5.8. SL327
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Karin, L.C.M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar]
- Leonard, J.T.; Raess, P.; Dunlap, J.; Hayes-Lattin, B.; Tyner, J.W.; Traer, E. Functional and genetic screening of acute myeloid leukemia associated with mediastinal germ cell tumor identifies MEK inhibitor as an active clinical agent. J. Hematol. Oncol. 2016, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.; Lyons, J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr. Opin. Pharmacol. 2005, 5, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. MEK inhibitors in oncology: A patent review (2015–Present). Expert Opin. Ther. Pat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Templeton, I.E.; Musib, L. MEK inhibitors beyond monotherapy: Current and future development. Curr. Opin. Pharmacol. 2015, 23, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Martin-Liberal, J.; Lagares-Tena, L.; Larkin, J. Prospects for MEK inhibitors for treating cancer. Expert Opin. Drug Saf. 2014, 13, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alex, A.A. The clinical development of MEK inhibitors. Nat. Rev. 2014, 11, 385–400. [Google Scholar]
- Inamdar, G.S.; Madhunapantula, S.V.; Robertson, G.P. Targeting the MAPK pathway in melanoma: Why some approaches succeed and other fail. Biochem. Pharmacol. 2010, 80, 624–637. [Google Scholar] [CrossRef] [PubMed]
- Antonio, M.G.; Ester, S.; Lucia, F.; Vito, V.; Martina, S.; Paolo, A.A. MEK Inhibitors in the treatment of metastatic melanoma and solid tumors. Am. J. Clin. Dermatol. 2017. [Google Scholar] [CrossRef]
- David, E.U.; Philip, A.H. Recent progress on MAP kinase pathway inhibitors. Bioorg. Med. Chem. Lett. 2015, 19, 4047–4056. [Google Scholar]
- Steve, P. Putative allosteric MEK1 and MEK2 inhibitors. Expert Opin. Ther. Pat. 2008, 18, 603–627. [Google Scholar]
- Juliet, R.; Juan, M.L.; Stefan, D.; James, L. BRAF and MEK inhibition for the treatment of advanced BRAF mutant melanoma. Expert Opin. Pharmacother. 2015, 16, 1285–1297. [Google Scholar]
- Carolina, H.J.; Suzanne, E.B. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar]
- Carsten, H.; Jonathan, L.B. The ups and downs of MEK kinase interactions. Cell. Signal. 2001, 13, 863–875. [Google Scholar]
- Paola, Q.; Virginia, P.; Francesco, S. Combined BRAF and MEK inhibition for the treatment of BRAF-mutated metastatic melanoma. Cancer Treat. Rev. 2015, 41, 519–526. [Google Scholar]
- Neuzillet, C.; Tijeras-Raballand, A.; de Mestier, L.; Cros, J.; Faivre, S.; Raymond, E. MEK in cancer and cancer therapy. Pharmacol. Ther. 2014, 141, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Boerner, S.A.; Winkler, J.D.; LoRusso, P.M. Clinical experience of MEK inhibitors in cancer therapy. Biochim. Biophys. Acta 2007, 1773, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Cuenda, A.; Cohen, P.; Dudley, D.T.; Saltiel, A.R. PD 098059 Is a Specific Inhibitor of the Activation of Mitogen-activated Protein Kinase Kinase in vitro and in vivo. J. Biol. Chem. 1995, 270, 27489–27494. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G.; Rominger, C.M.; Erskine, S.; Fisher, K.E.; Yang, J.; et al. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.D.; Aay, N.; Anand, N.K.; Blazey, C.M.; Bowles, O.J.; Bussenius, J.; Costanzo, S.; Curtis, J.K.; Defina, S.C.; Dubenko, L.; et al. Novel Carboxamide-Based Allosteric MEK Inhibitors: Discovery and Optimization Efforts toward XL518 (GDC-0973). ACS Med. Chem. Lett. 2012, 3, 416–421. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). 2013. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000TOC.cfm (accessed on 29 May 2013).
- U.S. Food and Drug Administration (FDA). 2015. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206192Orig1s000PharmR.pdf (accessed on 10 November 2015).
- U.S. Food and Drug Administration (FDA). 2013. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000ClinPharmR.pdf (accessed on 29 May 2013).
- Takahashi, R.H.; Ma, S.; Yue, Q.; Kim-Kang, H.; Yi, Y.; Ly, J.; Boggs, J.W.; Fettes, A.; McClory, A.; Deng, Y.; et al. Absorption, metabolism and excretion of cobimetinib, an oral MEK inhibitor, in rats and dogs. Xenobiotica 2017, 47, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Rosen, L.S.; LoRusso, P.; Ma, W.W.; Goldman, J.W.; Weise, A.; Colevas, A.D.; Adjei, A.; Yazji, S.; Shen, A.; Johnston, S.; et al. A first-in-human phase I study to evaluate the MEK1/2 inhibitor, cobimetinib, administered daily in patients with advanced solid tumors. Investig. New Drugs 2016, 34, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Kakefuda, R.; Tajima, N.; Sowa, Y.; Sakai, T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 2011, 39, 23–31. [Google Scholar] [PubMed]
- Abe, H.; Kikuchi, S.; Hayakawa, K.; Iida, T.; Nagahashi, N.; Maeda, K.; Sakamoto, J.; Matsumoto, N.; Miura, T.; Matsumura, K.; et al. Discovery of a highly potent and selective MEK inhibitor: GSK1120212 (JTP-74057 DMSO solvate). ACS Med. Chem. Lett. 2011, 2, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Menzies, A.M.; Long, G.V. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2014, 20, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Gandara, D.R.; Leighl, N.; Delord, J.-P.; Barlesi, F.; Bennouna, J.; Zalcman, G.; Infante, J.R.; Reckamp, K.L.; Kelly, K.; Shepherd, F.A.; et al. A phase 1/1b study evaluating trametinib plus docetaxel or pemetrexed in patients with advanced non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.-J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/204114s001lbl.pdf (accessed on 1 January 2014).
- Hsueh, E.C.; Gorantla, K.C. Novel melanoma therapy. Exp. Hematol. Oncol. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). FDA Grants Regular Approval to Dabrafenib and Trametinib Combination for Metastatic NSCLC with BRAF V600E Mutation; FDA: Silver Spring, MD, USA, 2017.
- EMA. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/002643/WC500222159.pdf (accessed on 23 February 2017).
- Planchard, D.; Besse, B.; Groen, H.J.M.; Souquet, P.-J.; Quoix, E.; Baik, C.S.; Barlesi, F.; Kim, T.M.; Mazieres, J.; Novello, S.; et al. Dabrafenib plus trametinib in patients with previously treated BRAFV600E-mutant metastatic non-small cell lung cancer: An open-label, multicentre phase 2 trial. Lancet Oncol. 2016, 17, 984–993. [Google Scholar] [CrossRef]
- Dholaria, B.; Hammond, W.; Shreders, A.; Lou, Y. Emerging therapeutic agents for lung cancer. J. Hematol. Oncol. 2016, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Gilligan, B.M.; Yuan, J.; Li, T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J. Hematol. Oncol. 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Daud, A. Nivolumab plus ipilimumab in the treatment of advanced melanoma. J. Hematol. Oncol. 2015, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Diggs, L.P.; Hsueh, E.C. Utility of PD-L1 immunohistochemistry assays for predicting PD-1/PD-L1 inhibitor response. Biomark. Res. 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Simeone, E.; Grimaldi, A.M.; Festino, L.; Vanella, V.; Palla, M.; Ascierto, P.A. Combination treatment of patients with BRAF-mutant melanoma: A new standard of care. BioDrugs 2017, 31, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.F.; Ng, C.M.; Berry, L.; Belvin, M.; Lewin-Koh, N.; Merchant, M.; Salphati, L. PK-PD modeling of combination efficacy effect from administration of the MEK inhibitor GDC-0973 and PI3K inhibitor GDC-0941 in A2058 xenografts. Cancer Chemother. Pharmacol. 2013, 71, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; McArthur, G.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAFV600-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Miller, W.H.; Kim, T.M.; Lee, C.B.; Flaherty, K.T.; Reddy, S.; Jamal, R.; Chow, L.Q.; Rooney, I.A.; Pitcher, B.; Cha, E.; et al. Atezolizumab (A) + cobimetinib (C) in metastatic melanoma (mel): Updated safety and clinical activity. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Gonzalez, R.; Lewis, K.D.; Hamid, O.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Wallin, J.; Pitcher, B.; Cha, E.; et al. Atezolizumab (A) + Cobimetinib (C) + Vemurafenib (V) in BRAFV600-Mutant Metastatic Melanoma (mel): Updated Safety and Clinical Activity. In Proceedings of the ASCO Annual Meeting, Chicago, IL, USA, 2–6 June 2017; p. 3063. [Google Scholar]
- Johanna, C.B.; Tae, W.K.; Boon, C.G.; Jeffrey, W.; Do-Youn, O.; Sae-Won, H.; Carrie, B.L.; Matthew, D.H.; Jayesh, D.; Jeremy, H.L.; et al. Clinical activity and safety of cobimetinib (cobi) and atezolizumab in colorectal cancer (CRC). J. Clin. Oncol. 2016, 34. [Google Scholar] [CrossRef]
- Barrett, S.D.; Bridges, A.J.; Dudley, D.T.; Saltiel, A.R.; Fergus, J.H.; Flamme, C.M.; Delaney, A.M.; Kaufman, M.; LePage, S.; Leopold, W.R.; et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg. Med. Chem. Lett. 2008, 18, 6501–6504. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Duesbery, N.S. Highly selective MEK inhibitors. Curr. Enzym. Inhib. 2010, 6, 146–157. [Google Scholar] [CrossRef]
- Pheneger, J.; Wallace, E.; Marlow, A.; Hurley, B.; Lyssikatos, J.; Bendele, A.M.; Lee, P.A. Characterization of ARRY-438162, a Potent MEK Inhibitor in Combination with Methotrexate or Ibuprofen in In Vivo Models of Arthritis. In Proceedings of the 2006 Annual Scientific Meeting, Boston, MA, USA, 20–24 October 2006; p. 794. [Google Scholar]
- Wallace, E.; Lyssikatos, J.; Blake, J.; Marlow, A.; Greschuk, J.; Yeh, T.; Callejo, M.; Marsh, V.; Poch, G.; Otten, J.; et al. AZD8330 (ARRY-424704): Preclinical evaluation of a potent, selective MEK 1/2 inhibitor currently in phase I trials. In Proceedings of the AACR, Denver, CO, USA, 18–22 April 2009; Volume 69. [Google Scholar]
- Dong, Q.; Dougan, D.R.; Gong, X.; Halkowycz, P.; Jin, B.; Kanouni, T.; O’Connell, S.M.; Scorah, N.; Shi, L.; Wallace, M.B.; et al. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg. Med. Chem. Lett. 2011, 21, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS-versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Iverson, C.; Larson, G.; Lai, C.; Yeh, L.T.; Dadson, C.; Weingarten, P.; Appleby, T.; Vo, T.; Maderna, A.; Vernier, J.M.; et al. RDEA119/BAY 869766: A potent, selective, allosteric inhibitor of MEK1/2 for the treatment of cancer. Cancer Res. 2009, 69, 6839–6847. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, Y.; Kohchi, Y.; Iikura, H.; Matsubara, Y.; Asoh, K.; Murata, T.; Kohchi, M.; Mizuguchi, E.; Tsujii, S.; Hattori, K.; et al. Design and synthesis of novel allosteric MEK inhibitor CH4987655 as an orally available anticancer agent. Bioorg. Med. Chem. Lett. 2011, 21, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, D.; Griffin, M.J.; Sludden, J.; Drew, Y.; Cresti, N.; Swales, K.; Merriman, M.; Allen, R.; Bevan, P.; Buerkle, M.; et al. A phase I pharmacokinetic and pharmacodynamic study of the oral mitogen-activated protein kinase kinase (MEK) inhibitor, WX-554, in patients with advanced solid tumours. Eur. J. Cancer 2016, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Ji, C.; Liu, C.; Kong, L.; Cheng, Y.; Huang, G.C. Benzoheterocyclic Compounds and Use Thereof. Patent EP 2804855 A4, 25 July 2013. [Google Scholar]
- Wabnitz, P.A.; Mitchell, D.; Wabnitz, D.A. In vitro and in vivo metabolism of the anti-cancer agent CI-1040, a MEK inhibitor, in rat, monkey, and human. Pharm. Res. 2004, 21, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.P.; Carlson, T.C.; Loi, C.M.; Graziano, M.J. Pharmacodynamic and toxicokinetic evaluation of the novel MEK inhibitor, PD0325901, in the rat following oral and intravenous administration. Cancer Chemother. Pharmacol. 2007, 59, 671–679. [Google Scholar] [CrossRef] [PubMed]
- El-Hoss, J.; Kolind, M.; Jackson, M.T.; Deo, N.; Mikulec, K.; McDonald, M.M.; Little, C.B.; Little, D.G.; Schindeler, A. Modulation of endochondral ossification by MEK inhibitors PD0325901 and AZD6244 (Selumetinib). Bone 2014, 59, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Haura, E.B.; Ricart, A.D.; Larson, T.G.; Stella, P.J.; Bazhenova, L.; Miller, V.A.; Cohen, R.B.; Eisenberg, P.D.; Selaru, P.; Wilner, K.D.; et al. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2010, 16, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Boasberg, P.D.; Redfern, C.H.; Daniels, G.A.; Bodkin, D.; Garrett, C.R.; Ricart, A.D. Pilot study of PD-0325901 in previously treated patients with advanced melanoma, breast cancer, and colon cancer. Cancer Chemother. Pharmacol. 2011, 68, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Curran, E.; Iyengar, N.M.; Diaz-Flores, E.; Kunnavakkam, R.; Popplewell, L.; Kirschbaum, M.H.; Karrison, T.; Erba, H.P.; Green, M.; et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: A University of Chicago phase II consortium trial. Clin. Cancer Res. 2014, 20, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.B.; Lui, V.W.; Cheung, C.S.; Lau, C.P.; Ho, K.; Hui, E.P.; Tsui, S.K.; Ng, M.H.; Cheng, S.H.; Ng, P.K.; et al. Activity of the MEK inhibitor selumetinib (AZD6244; ARRY-142886) in nasopharyngeal cancer cell lines. Investig. New Drugs 2013, 31, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Beloueche-Babari, M.; Jamin, Y.; Arunan, V.; Walker-Samuel, S.; Revill, M.; Smith, P.D.; Halliday, J.; Waterton, J.C.; Barjat, H.; Workman, P.; et al. Acute tumour response to the MEK1/2 inhibitor selumetinib (AZD6244, ARRY-142886) evaluated by non-invasive diffusion-weighted MRI. Br. J. Cancer 2013, 109, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.; Van, C.N.; Heng, N.K.; Donald, P.; Su, P.C.; Han, C.T.; Choon, H.T.; Pierce, C.; Hock, S.O.; Alexander, C.; et al. AZD6244 enhances the anti-tumor activity of sorafenib in ectopic and orthotopic models of human hepatocellular carcinoma (HCC). J. Hepatol. 2010, 52, 79–87. [Google Scholar]
- Krishnamurthy, A.; Dasari, A.; Noonan, A.M.; Mehnert, J.M.; Lockhart, A.C.; Stein, M.N.; Sanoff, H.K.; Lee, J.J.; Hansen, A.R.; Malhotra, U.; et al. A phase IB study of the combination of selumetinib (AZD6244, ARRY-142886) and cyclosporin A (CsA) in patients with advanced solid tumors with an expansion cohort in metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Tai, W.M.; Yong, W.P.; Lim, C.; Low, L.S.; Tham, C.K.; Koh, T.S.; Ng, Q.S.; Wang, W.W.; Wang, L.Z.; Hartano, S.; et al. A phase Ib study of selumetinib (AZD6244, ARRY-142886) in combination with sorafenib in advanced hepatocellular carcinoma (HCC). Ann. Oncol. 2016, 27, 2210–2215. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib plus docetaxel compared with docetaxel alone and progression-free survival in patients with KRAS-mutant advanced non–small cell lung cancer. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: A pediatric brain tumor consortium (PBTC) study. Neuro-Oncology 2017, 19, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J.R.; Onar-Thomas, A.; Young-Poussaint, T.; Wu, S.; Ligon, A.H.; Lindeman, N.I.; Banerjee, A.; Packer, R.; Kilburn, L.B.; Pollack, I.; et al. A phase II prospective study of selumetinib in children with recurrent or refractory low-grade glioma (LGG): A Pediatric Brain Tumor Consortium (PBTC) study. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Chappell, W.H.; Russo, S.; Ove, R.; Milella, M.; Tafuri, A.; Lunghi, P.; Bonati, A.; et al. Emerging MEK inhibitors. Expert Opin. Emerg. Drugs 2010, 15, 203–223. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Javle, M.; Bekaii-Saab, T.S.; Finn, R.S.; Wainberg, Z.A.; Laheru, D.A.; Weekes, C.D.; Tan, B.R.; Khan, G.N.; Zalupski, M.M.; et al. A phase 1 dose-escalation and expansion study of binimetinib (MEK162), a potent and selective oral MEK1/2 inhibitor. Br. J. Cancer 2017, 116, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.L.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Marlow, A.L.; Wallace, E.; Seo, J.; Lyssikatos, J.P.; Yang, H.W.; Blake, J.; Storey, R.A.; Booth, R.J.; Pittam, J.D. Heterocyclic Inhibitors of Mek and Methods of Use Thereof. Patent WO2007044084 A3, 19 April 2007. [Google Scholar]
- Haasbach, E.; Hartmayer, C.; Planz, O. Combination of MEK inhibitors and oseltamivir leads to synergistic antiviral effects after influenza A virus infection in vitro. Antivir. Res. 2013, 98, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.B.; Aamdal, S.; Nyakas, M.; Cavallin, M.; Green, D.; Learoyd, M.; Smith, I.; Kurzrock, R. A phase I dose-finding, safety and tolerability study of AZD8330 in patients with advanced malignancies. Eur. J. Cancer 2013, 49, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Adjei, A.A.; LoRusso, P.; Ribas, A.; Sosman, J.A.; Pavlick, A.; Dy, G.K.; Zhou, X.; Gangolli, E.; Kneissl, M.; Faucette, S.; et al. A phase I dose-escalation study of TAK-733, an investigational oral MEK inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.; Belvin, M.; Merchant, M.; Chan, E.; Hollingshead, P.; Orr, C.; Boggs, J.; Plise, E.; Robarge, K.; Zak, M. Preclinical Pharmacokinetics and efficacy assessment of a potent and selective MEK inhibitor, GDC-0623. Eur. J. Cancer 2012, 48, 155. [Google Scholar] [CrossRef]
- Van Laethem, J.L.; Riess, H.; Jassem, J.; Haas, M.; Martens, U.M.; Weekes, C.; Peeters, M.; Ross, P.; Bridgewater, J.; Melichar, B.; et al. Phase I/II study of refametinib (BAY 86-9766) in combination with gemcitabine in advanced pancreatic cancer. Target. Oncol. 2017, 12, 97–109. [Google Scholar] [PubMed]
- Lim, H.Y.; Heo, J.; Choi, H.J.; Lin, C.Y.; Yoon, J.H.; Hsu, C.; Rau, K.M.; Poon, R.T.; Yeo, W.; Park, J.W.; et al. A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86-9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5976–5985. [Google Scholar] [CrossRef] [PubMed]
- Houédé, N.; Delord, J.P.; Awada, A.; Lebbe, C.; Lesimple, T.; Schellens, J.H.M.; Rottey, S.; Kefford, R.; von Richter, O.; Raymond, E. Pharmacokinetics and pharmacodynamics of a selective oral MEK1/2 inhibitor, pimasertib (MSC1936369B/AS703026), in patients with advanced solid tumors. Eur. J. Cancer 2012, 48, 184. [Google Scholar] [CrossRef]
- Naing, A.; Mita, M.; Komarnitsky, P.; Milner, A.; von Richter, O.; Ogden, J.; Piha-Paul, S.; Fu, S.; Asatiani, E.; Kurzrock, R. Phase I dose-escalation trial of a selective oral MEK1/2 inhibitor, pimasertib (MSC1936369B), combined with an mTOR inhibitor, temsirolimus, in patients with advanced solid tumors. Eur. J. Cancer 2012, 48, 187. [Google Scholar] [CrossRef]
- Awada, A.; Delord, J.P.; Houédé, N.; Lebbe, C.; Lesimple, T.; Schellens, J.H.M.; Rottey, S.; Kefford, R.; Rejeb, N.; Raymond, E. Safety and recommended phase II Dose (RP2D) of the selective oral MEK1/2 inhibitor pimasertib (MSC1936369B/AS703026): Results of a phase I trial. Eur. J. Cancer 2012, 48, 185–186. [Google Scholar] [CrossRef]
- Delord, J.P.; Houédé, N.; Awada, A.; Lebbe, C.; Lesimple, T.; Schellens, J.H.M.; Rottey, S.; Kefford, R.; Rejeb, N.; Raymond, E. 616 pimasertib (MSC1936369B/AS703026), a selective oral MEK1/2 inhibitor, shows clinical activity in melanoma. Eur. J. Cancer 2012, 48, 190. [Google Scholar] [CrossRef]
- Kraeber-Bodere, F.; Carlier, T.; Naegelen, V.M.; Shochat, E.; Lumbroso, J.; Trampal, C.; Nagarajah, J.; Chua, S.; Hugonnet, F.; Stokkel, M.; et al. Differences in the biologic activity of 2 novel MEK inhibitors revealed by 18F-FDG PET: Analysis of imaging data from 2 phase I trials. J. Nucl. Med. 2012, 53, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Leijen, S.; Middleton, M.R.; Tresca, P.; Kraeber-Bodere, F.; Dieras, V.; Scheulen, M.E.; Gupta, A.; Lopez-Valverde, V.; Xu, Z.X.; Rueger, R.; et al. Phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of the MEK inhibitor RO4987655 (CH4987655) in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 4794–4805. [Google Scholar] [CrossRef] [PubMed]
- Shinji, N.; Hiroshi, N.; Noboru, Y.; Yasuhide, Y.; Yutaka, F.; Yosuke, T.; Hiroshi, W.; Kazunori, H.; Hidenori, M.; Satoru, K.; et al. Phase I and pharmacokinetics/pharmacodynamics study of the MEK inhibitor RO4987655 in Japanese patients with advanced solid tumors. Investig. New Drugs 2015, 41, 641–651. [Google Scholar]
- Martinez-Garcia, M.; Banerji, U.; Albanell, J.; Bahleda, R.; Dolly, S.; Kraeber-Bodere, F.; Rojo, F.; Routier, E.; Guarin, E.; Xu, Z.X.; et al. First-in-human, phase I dose-escalation study of the safety, pharmacokinetics, and pharmacodynamics of RO5126766, a first-in-class dual MEK/RAF inhibitor in patients with solid tumors. Clin. Cancer Res. 2012, 18, 4806–4819. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Lee, M.H.; Reddy, K.; Li, Y.; Lim, D.Y.; Xie, H.; Lee, S.Y.; Yeom, Y.I.; Bode, A.M.; Dong, Z. CInQ-03, a novel allosteric MEK inhibitor, suppresses cancer growth in vitro and in vivo. Carcinogenesis 2013, 34, 1134–1143. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.F.; Belvin, M.; Chan, J.; Hoeflich, K.; Orr, C.; Robarge, K.; Yang, X.; Zak, M.; Boggs, J. Preclinical disposition and pharmacokinetics-pharmacodynamic modeling of biomarker response and tumour growth inhibition in xenograft mouse models of G-573, a MEK inhibitor. Xenobiotica 2010, 40, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Klein, P.J.; Schmidt, C.M.; Wiesenauer, C.A.; Choi, J.N.; Gage, E.A.; Yip-Schneider, M.T.; Wiebke, E.A.; Wang, Y.; Omer, C.; Sebolt-Leopold, J.S. The effects of a novel MEK inhibitor PD184161 on MEK-ERK signaling and growth in human liver cancer. Neoplasia 2006, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yung, H.W.; Wyttenbach, A.; Tolkovsky, A.M. Aggravation of necrotic death of glucose-deprived cells by the MEK1 inhibitors U0126 and PD184161 through depletion of ATP. Biochem. Pharmacol. 2004, 68, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhou, V.; Pan, S.; Liu, Y.; Hornsby, M.; McMullan, D.; Klock, H.E.; Haugen, J.; Lesley, S.A.; Gray, N.; et al. Identification of coumarin derivatives as a novel class of allosteric MEK1 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 5467–5473. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.T.; Pang, L.; Decker, S.J.; Bridges, A.J.; Saltiel, A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. USA 1995, 92, 7686–7689. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Galuppo, M.; Mazzon, E.; Paterniti, I.; Bramanti, P.; Cuzzocrea, S. PD98059, a specific MAP kinase inhibitor, attenuates multiple organ dysfunction syndrome/failure (MODS) induced by zymosan in mice. Pharmacol. Res. 2010, 61, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Cerioni, L.; Palomba, L.; Cantoni, O. The Raf/MEK inhibitor PD98059 enhances ERK1/2 phosphorylation mediated by peroxynitrite via enforced mitochondrial formation of reactive oxygen species. FEBS Lett. 2003, 547, 92–96. [Google Scholar] [CrossRef]
- Daouti, S.; Higgins, B.; Kolinsky, K.; Packman, K.; Wang, H.; Rizzo, C.; Moliterni, J.; Huby, N.; Fotouhi, N.; Liu, M.; et al. Preclinical in vivo evaluation of efficacy, pharmacokinetics, and pharmacodynamics of a novel MEK1/2 kinase inhibitor RO5068760 in multiple tumor models. Mol. Cancer Ther. 2010, 9, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Niu, H.; Goelzer, P.; Rueger, R.; Deutsch, J.; Busse-Reid, R.; DeSchepper, S.; Blotner, S.; Barrett, J.; Weissgerber, G.; et al. The safety, tolerability, pharmacokinetics, and pharmacodynamics of single oral doses of RO5068760, an MEK inhibitor, in healthy volunteers: Assessment of target suppression. J. Clin. Pharmacol. 2010, 50, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.; Houpt, T.A. Mitogen-activated protein kinase in the amygdala plays a critical role in lithium chloride-induced taste aversion learning. Neurobiol. Learn. Mem. 2012, 97, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Dokladda, K.; Green, K.A.; Pan, D.A.; Hardie, D.G. PD98059 and U0126 activate AMP-activated protein kinase by increasing the cellular AMP: ATP ratio and not via inhibition of the MAP kinase pathway. FEBS Lett. 2005, 579, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, J.; Zhao, L.; Chen, S. Combination of SL327 and Sunitinib Malate leads to an additive anti-cancer effect in doxorubicin resistant thyroid carcinoma cells. Biomed. Pharmacother. 2017, 88, 985–990. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Item | Trametinib | Cobimetinib | |
|---|---|---|---|
| Structure | 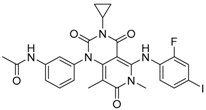 | 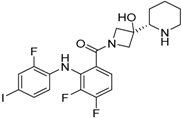 | |
| BCS 1 class | BCS II (high permeability, low solubility) | BCS I (high permeability, high solubility) | |
| Salt form | dimethyl sulfoxide solvate (1:1) | hemifumarate | |
| Molecule weight (free base) | 615.4 | 531.32 | |
| In vitro (enzyme) [19,20] | MEK 2 kinase | MEK1 IC50 3 = 0.7 nM; | MEK1 IC50 = 0.95 nM; |
| MEK2 IC50 = 0.9 nM | MEK2 IC50 = 199 nM | ||
| In vitro (cell potency) [21,22] | A375 | 0.74 nM | 5 nM |
| In vivo efficacy (xenograft) [19,20] | A375 | TGI 4 = 60% @ 0.1 mpk; 14 days; | TGI = 87% @ 3 mg/kg, 21 days, QD 5; |
| TGI = 102% @ 0.3 mpk; 14 days; TGI = 118% @ 3 mpk; 14 days | TGI = 106% @ 5 mg/kg, 21 days, QD | ||
| Pharmacokinetics (rats) [23,24] | Cmax 6 | 2.7 μM (3 mg/kg, mice); | 0.997 μM (30 mg/kg, male rats) |
| 0.47 μM (3 mg/kg, rats) | |||
| Tmax 7 | 4 h (mice, 3 mg/kg, 14 days, repeat); | 2 h (30 mg/kg, male rats) | |
| 4 h (rats, 1 mg/kg, 21days, repeat) | |||
| T1/2 8 | 3.8 h (mice); | 5.56 h (30 mg/kg, male rats) | |
| 5.5 h (rats) | |||
| protein binding rate | 97.40% (human) | 98.8% (5 μM, dog); | |
| 93.5% (5 μM, human) | |||
| Toxicokinetics (rats) [21,22] | Tmax | 4 h (male rats, 0.1667 mg/kg, week 4) | N/A |
| Cmax | 10.1 ng/mL (male rats, 0.1667 mg/kg, week 4) | 39.9 ng/mL (male rats, 3 mg/kg, day 22) | |
| AUC0-t 9 | 188 ng·h/mL (male rats, 0.1667 mg/kg, week 4) | 244 ng·h/mL (male rats, 3 mg/kg, day 22) | |
| Clinical PK [25,26] | MTD 10 | 3 mg/day (QD) | 60 mg/day, QD, 21/7; |
| 100 mg/day, QD, 14/14 | |||
| Tmax | 1.5 h (2 mg, QD) | 2.4 h (60 mg, QD) | |
| Cmax | 22.2 ng/mL (2 mg, QD) | 273 ng/mL (60 mg, QD) | |
| T1/2 | 4–5 d (2 mg, QD) | 43.6 h (60 mg, QD) | |
| CL/F 11 | 5.4 L/h (2 mg, QD) | 13.8 L/h (60 mg, QD) | |
| AUC | 370 ng·h/mL (0-t, day 15, 2 mg, QD) | 4340 ng·h/mL (60 mg, QD) | |
| period/cycle | 21 days/7 days | 21 days/7 days | |
| absolute bioavailability | 72% (2 mg, QD) | 46% (20 mg, QD) | |
| recommended dose | 2 mg, QD | 60 mg, QD | |
| Adverse reactions [25,26] | rash, diarrhea, fatigue, peripheral edema, nausea, and dermatitis acneiform | gastrointestinal disorders, rash, pyrexia, increased blood CPK 12, chorioretinopathy | |
| MEK Inhibitor | Target | IC50 | Indications | Company | Clinical Phase |
|---|---|---|---|---|---|
| CI-1040 (PD184352) [47] | MEK1/2 | 2.3 nM | breast cancer, colorectal cancer, lung cancer, and pancreatic cancer | Pfizer | Phase II |
| PD0325901 [47] | MEK1/2 | 0.33 nM | melanoma, colonic neoplasms, breast neoplasms, carcinoma, NSCLC 1 | Pfizer | Phase II |
| Selumetinib (AZD6244) [48] | MEK1 | 14 nM | melanoma, NSCLC | Array BioPharma and AstraZeneca | Phase III |
| MEK162 [49] | MEK1/2 | 12 nM | BRAF 2 or NRAS 3 mutant melanoma | Array Biopharma/Novartis | Phase III |
| AZD8330 [50] | MEK1/2 | 7 nM | advanced solid tumors | AstraZeneca | Phase I |
| TAK-733 [51] | MEK1/2 | 3.2 nM | advanced non-hematologic malignancies, advanced metastatic melanoma | Millennium Pharmaceutical, Inc./Takeda Pharmaceutical Company Limited | Phase I |
| GDC-0623 [52] | MEK1/2 | 0.13 nM | metastatic solid tumors | Genentech | Phase I |
| Refametinib (RDEA119; BAY 869766) [53] | MEK1/2 | 19 nM/47 nM | hepatocellular cancer, melanoma, colorectal cancer | Ardea Biosciences/Bayer | Phase II |
| Pimasertib (AS703026) | MEK1/2 | 5–11 nM | colorectal cancer, multiple myeloma | Merck and Co. | Phase II |
| RO4987655 (CH4987655) [54] | MEK1 | 42 nM | neoplasms | Hoffman-La Roche | Phase I |
| RO5126766 [54] | RAF/MEK1/2 | 160 nM | neoplasms | Hoffmann-La Roche | Phase I |
| WX-554 [55] | MEK1/2 | 4.7 nM/10.7 nM | advanced solid tumors | Wilex, AG. Germany | Phase I/II (terminated) |
| HL-085 [56] | MEK1 | 1.9–10 nM | no data | Binjiang Pharma | Phase I |
| MEK Inhibitor | Target | IC50 | Current Sponsor | Research Progress |
|---|---|---|---|---|
| CInQ-03 | MEK1/2 | 5/10 μM | No data | in preclinical study |
| G-573 | MEK | No data | Genentech | in preclinical study |
| PD184161 | MEK | 10–100 nM | Pfizer | in preclinical study |
| PD318088 | MEK1 | No data | Pfizer Global Research & Development | in preclinical study |
| PD98059 | MEK1 | 2 μM | No data | in preclinical study |
| RO5068760 | MEK1 | 0.025 ± 0.012 μM | Hoffmann-La Roche, Inc. | in preclinical study |
| U0126 | MEK1/2 | 0.07 μM/0.06 μM | No data | in preclinical study |
| SL327 | MEK1/2 | 0.18 μM/0.22 μM | No data | in preclinical study |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. https://doi.org/10.3390/molecules22101551
Cheng Y, Tian H. Current Development Status of MEK Inhibitors. Molecules. 2017; 22(10):1551. https://doi.org/10.3390/molecules22101551
Chicago/Turabian StyleCheng, Ying, and Hongqi Tian. 2017. "Current Development Status of MEK Inhibitors" Molecules 22, no. 10: 1551. https://doi.org/10.3390/molecules22101551
APA StyleCheng, Y., & Tian, H. (2017). Current Development Status of MEK Inhibitors. Molecules, 22(10), 1551. https://doi.org/10.3390/molecules22101551