Abstract
RNA interference (RNAi) is a potent and specific post-transcriptional gene silencing process. Since its discovery, tremendous efforts have been made to translate RNAi technology into therapeutic applications for the treatment of different human diseases including respiratory diseases, by manipulating the expression of disease-associated gene(s). Similar to other nucleic acid-based therapeutics, the major hurdle of RNAi therapy is delivery. Pulmonary delivery is a promising approach of delivering RNAi therapeutics directly to the airways for treating local conditions and minimizing systemic side effects. It is a non-invasive route of administration that is generally well accepted by patients. However, pulmonary drug delivery is a challenge as the lungs pose a series of anatomical, physiological and immunological barriers to drug delivery. Understanding these barriers is essential for the development an effective RNA delivery system. In this review, the different barriers to pulmonary drug delivery are introduced. The potential of RNAi molecules as new class of therapeutics, and the latest preclinical and clinical studies of using RNAi therapeutics in different respiratory conditions are discussed in details. We hope this review can provide some useful insights for moving inhaled RNAi therapeutics from bench to bedside.
1. Introduction
Lung diseases are among the leading causes of death worldwide. Current treatment of lung diseases such as lung cancer [1], respiratory infections [2,3], inflammatory diseases [4,5] and pulmonary fibrosis [6] have limited efficacy. Almost two decades ago, double-stranded RNAs (dsRNAs) were discovered to play an important role in regulating gene functions by a sequence-specific post-transcriptional gene silencing mechanism called RNA interference (RNAi) [7]. The therapeutic potential of RNAi was soon realized. It presents a new and powerful approach to treat or prevent many diseases including respiratory disorders by modulating gene expression [8,9,10,11,12,13]. RNAi can be mediated by various types of RNA molecules, including long dsRNA, short interfering RNA (siRNA), short hairpin RNA (shRNA) and microRNA (miRNA). The mechanism of RNAi is illustrated in Figure 1, and the general properties of different types of RNAi molecules are summarized in Table 1.
Figure 1.
Mechanism of RNA interference (RNAi) induced gene silencing. Short interfering RNA (siRNA) can be produced inside the cells from double stranded RNA (dsRNA) or expressed from short hairpin RNA (shRNA) vector, or chemically synthesized in the laboratory. Its gene silencing effect is dependent upon fully complementary binding of the target messenger RNA (mRNA). MicroRNA (miRNA) is a naturally occurring molecule. It can also be expressed from miRNA vector or chemically synthesized (miRNA mimic). miRNA mediates gene silencing via the partial complementary binding of the target mRNAs.
Table 1.
Comparison of general properties between long double-stranded RNA (dsRNA), short interfering RNA (siRNA), short hairpin RNA (shRNA) and microRNA (miRNA).
1.1. Long dsRNA and siRNA
Long dsRNAs (around 500–1000 nucleotides in length) have been employed to study gene functions. After the exogenous dsRNA is introduced into the cytoplasm of the cells, it is cleaved by RNase III enzyme Dicer into short dsRNA called siRNA. The siRNA which is 21–23 nucleotides in length is loaded into a protein complex called the RNA-induced silencing complex (RISC). The siRNA is then unwound, and the sense strand (also known as the passenger strand) of the siRNA is degraded, whereas the remaining antisense strand (also known as the guide strand) guides the activated RICS to the target messenger RNA (mRNA) through full complementary binding. The mRNA is then cleaved by Argonaute 2 (Ago2) in the RISC, leading to the silencing of the target gene [14,15]. Since long dsRNA is known to trigger immunostimulatory response through the activation of Dicer-related antiviral pathways and induction of type 1 interferon (IFN) [16], it is less suitable for therapeutic use. In contrast, synthetic siRNA is a more promising gene silencing mediator because of the lower risk of immune response. It is also the most widely investigated RNAi molecule for therapeutic applications, with over 26 clinical trial studies being initiated since 2004 [17].
1.2. shRNA
shRNA is a sequence of RNA transcribed in the nucleus of the cells from a DNA vector by either RNA polymerase II or III. The primary transcript is called primary shRNA (pri-shRNA), which contains a hairpin like stem-loop structure. The pri-shRNA is processed into a 50–70 nucleotides long loop-stem precursor shRNA (pre-shRNA) by a protein complex containing the RNase III nuclease Drosha and the dsRNA binding domain protein DGCR8. It is then transported to the cytoplasm through a specialized nuclear membrane protein, exportin-5 (Exp5). The loop sequence of the pre-shRNA is cleaved by the Dicer to form a double-stranded siRNA. This endogenously produced siRNA is loaded into RISC and induce RNAi through the similar process as the synthetic siRNA [18,19]. Since shRNA expression unit can be incorporated into viral vectors and continuously synthesized by the host cell, it can induce long-lasting gene silencing effect. The RISC-loading process of shRNA is about ten times more efficient than siRNA, implicating that lower dose of shRNA is required to maintain the therapeutic efficacy with less off-target effect [20]. However, the shRNA approach is a DNA-based strategy depending on the expression of shRNA encoding gene which often requires viral vectors. From the delivery perspective, the introduction of synthetic siRNA to the cytoplasm is a more straightforward way to induce RNAi. The use of viral vectors for delivery poses safety concerns in its therapeutic applications [15].
1.3. miRNA
miRNA is a naturally occurring non-coding RNA that plays a key role in regulating gene expression. Primary miRNA (pri-miRNA) is transcribed by RNA polymerase II from endogenous miRNA gene in the nucleus. The hairpin-containing pri-miRNA is structurally similar to the pri-shRNA, therefore the maturation pathway of miRNA is also similar to shRNA [18]. The pri-mRNA is converted by Drosha/DCGR8 complex into precursor miRNA (pre-miRNA), which contains 70–100 nucleotides with interspersed mismatches and adopts a loop structure. The pre-miRNA is subsequently transported to the cytoplasm by Exp5, and is processed by the Dicer into mature miRNA of 18–25 nucleotides in length [21]. As opposed to siRNA, the antisense strand of miRNA is only partially complementary to the target mRNA, leading to gene silencing via translational repression and/or mRNA deadenylation. Position 2 to 7 of the 5′ end of the miRNA is the “seed sequence”, which is essential for target recognition, and the miRNA binding sites of mRNA are located in the 3′ untranslated region (UTR) [22]. There are two major approaches of miRNA-based therapeutics: (i) miRNA inhibition [23,24], which suppresses the action of the endogenous miRNA by antisense oligonucleotide (antimiR); and (ii) miRNA replacement [25,26], which introduces synthetic miRNA (miRNA mimic) to restore the functions of the endogenous miRNA. Only the latter is discussed in this review. Compared to siRNA-based therapeutics, miRNA has the potential to target multiple genes due to the imperfect binding to the target mRNA [27].
2. Pulmonary Delivery of RNAi Therapeutics
RNAi therapeutics offer several advantages over the traditional small molecules and protein-based drugs. They could virtually target any genes with high selectivity, including those “undruggable” targets. The design and synthesis of RNAi molecules are relatively simple because they do not need a cellular expression system, complex protein purification, or refolding schemes [28]. Compared to the antisense oligonucleotides, RNAi is more potent and specific [29]. Despite its huge therapeutic potential, delivery remains to be a major barrier to the clinical application of RNAi therapeutics [30,31]. For the treatment of respiratory disorders, pulmonary route of administration can deliver RNAi molecules directly to the site of action, thereby lowering the dose required while minimizing systemic adverse effects. The RNAi molecules can also avoid the rapid clearance by serum nuclease in the bloodstream. Moreover, inhalation is a non-invasive route of administration that is easily accepted by patients compared to parenteral administration, leading to better patient compliance [14,32]. However, pulmonary drug delivery is a challenge with several key biological barriers, which are highlighted in Figure 2.
Figure 2.
Summary of the major barriers associated with pulmonary delivery of RNAi molecules.
2.1. Extracellular Barriers
Human respiratory tract is divided into the conducting region (nasal cavity, pharynx, trachea, bronchi and bronchioles) and the respiratory region (respiratory bronchioles and alveoli). The former is the pathway for gases conduction whereas the latter is the site of gaseous exchange [14]. The extensive branched structure of the airways with varying length and diameter presents the primary hurdle in pulmonary drug delivery. The mechanisms of inhaled particle deposition in the lungs include inertial impaction, gravitational sedimentation, diffusion, interception and electrostatic precipitation [33]. As illustrated in Figure 3, the first three mechanisms are the major mechanisms which are greatly influenced by the aerodynamic size of the particles [34]. The optimal particle size for lung deposition is between 1 and 5 µm [35,36]. Larger particles are likely to be impacted and trapped on the upper airway wall at bifurcations while the small particles from 0.1 to 1 µm are easily exhaled during normal breathing. Smaller particles under 100 nm may be successfully deposited in the alveolar space because of the increasing diffusional mobility [37]. However, aerosols under 100 nm are difficult to produce, which make their application less favorable.
Figure 3.
The three major mechanisms of particles deposition in the lungs. Inertia impaction occurs when large particles (>5 µm) cannot adjust to the change of airflow direction due to inertia and deposit on the airway walls, typically in the upper respiratory tract. Gravitational sedimentation is the settling of particles due to gravitational force. It mainly affects particles with size of 1–5 µm, which usually deposit in the small airways. Diffusion occurs when submicron-size particles having Brownian motion hit the surface of airway walls.
The presence of fluid layers in the airways, including the mucus and the pulmonary surfactant, also creates a barrier to the delivery of drug molecules to the lungs. Mucus is a gel of three-dimensional network structure. It acts as a sieve that filters out large molecules (>500 nm) [38]. Mucins, which are the major components of mucus, contain the hydrophilic glycosylated residues rich in serine and threonine and the hydrophobic non-glycosylated cysteine-rich domains. This characteristic allows electrostatic, hydrophilic and hydrophobic interactions between mucus and drug particles [39]. Since mucus is continuously produced, shed and replaced, and together with the mucociliary clearance action, the fast turnover rate of mucus leads to the rapid clearance of the entrapped drug molecules, preventing them from reaching the epithelium [40,41]. Moreover, cough clearance that occurs when the mucus reaches the throat is another removal mechanism of RNAi molecules from the airways. On the other hand, the role of pulmonary surfactant in nucleic acid delivery is controversial. Pulmonary surfactant is composed of approximately 90% lipids and 10% proteins [42]. It has been suggested that the lipids and proteins in the lung surfactant interact with the non-viral cationic lipid-based delivery system, leading to the premature release of nucleic acids [43,44]. Conversely, polymer-based delivery systems were found to be more compatible with pulmonary surfactant. Commercial pulmonary surfactant was employed in the preparation of some polymeric nanoparticles to facilitate the cellular uptake of siRNA to improve gene silencing effect [45,46].
Apart from the physical barriers, the RNAi molecules may undergo enzymatic degradation following pulmonary delivery. RNA is extremely susceptible to nuclease activity. Naked siRNA has a short half-life of less than 15 min in serum [47]. Due to the lack of serum in the lungs, the half-life of unmodified siRNA in the airways can reach up to three hours in mouse [48]. RNAi molecules may also be taken up and removed by alveolar macrophages upon lung deposition. The alveolar macrophages are responsible for engulfing and digesting foreign particles in the lower airways via phagocytosis. Unless the target of RNAi molecules is located inside the macrophages, such as Mycobacterium tuberculosis which is the causative agent of tuberculosis that typically reside in the alveolar macrophages [49], otherwise the RNAi molecules are subjected to degradation inside the macrophages before reaching their target sites.
2.2. Intracellular Barriers
Once the RNAi molecules overcome the aforementioned extracellular barriers and reach the target cells, they need to overcome a set of intracellular barriers. The ultimate site of actions depends on the types of RNAi molecules. For synthetic siRNA and miRNA mimic, they need to reach the cytoplasm where the RISC locates, whereas DNA encoding shRNA or miRNA need to enter the nucleus in order for transcription to take place.
Surface adhesion is the first step of cellular entry. There are various endocytic mechanisms involved in the cellular uptake of macromolecules, which subsequently influence their intracellular trafficking and delivery efficiency [50,51,52,53,54,55]. The clathrin-dependent endocytosis is the most common route of cellular entry for macromolecules [56]. Following cell entry, the RNAi molecules are entrapped in the early endosomes where progressive acidification occurs. The late endosomes then fuse with the lysosomes, which contain hydrolases that degrade the RNAi molecules [56,57]. The RNAi molecules must escape from endosome at early stage to exert their biological effect. Strategies of endosomal escape have been reviewed in other literatures [58,59,60,61]. For shRNA or miRNA vectors, nuclear entry is an additional barrier which is particularly challenging due to the presence of nuclear membranes that are impermeable to most substances except small non-polar molecules [62]. Non-viral vectors are inefficient in delivering DNA into the nucleus. Some viruses are evolved to transport their genome into the nucleus; hence, they become attractive DNA carriers to improve transduction efficiency. However, poor safety profile, high production cost and the risk of immunogenicity [63] limited the applications of viral vector mediated RNAi to laboratory tool.
3. Delivery Strategies of RNAi Molecules
A delivery vector or carrier is generally necessary to overcome the aforementioned barriers by promoting cellular uptake and offering protection to the RNAi molecules. RNAi delivery system can be categorized into viral and non-viral vectors according to their nature, and these delivery systems have been extensively reviewed [63,64,65,66,67].
3.1. Viral Vectors
The viral vectors refer to the of use of viruses to deliver genetic materials to the cells. They are extremely efficient in transducing cells and providing either transient or long-term gene expression, depending on the type of viruses employed. Most of the in vivo studies used the adenoviruses, adeno-associated virus (AAV) and retroviruses to express the plasmid DNA encoding shRNA or miRNA to induce RNAi [68]. The most attractive property of viral vectors is that they have the ability to access the nucleus of the cells. Thus, they have high efficacy to express the RNA and subsequently regulate the gene expression [69,70,71,72]. However, the clinical application of viral vectors is limited by the toxicity, insertional mutagenesis (associated with retroviruses) and immunogenicity (associated with adenoviruses) [63]. In addition, the smaller size of AVV limits the amount of therapeutic gene that can be inserted.
3.2. Non-Viral Vectors
Compared to viral vectors, non-viral vectors generally have better safety profile and lower production cost. They have been investigated for the pulmonary delivery of the RNAi molecules. Lipid-based, polymer-based and peptide-based are three major non-viral delivery systems [73]. The lipid-based vectors are the most commonly used vectors for RNAi delivery. They include cationic liposomes, solid lipid nanoparticles, solid nanostructured lipid carriers, lipidoids and pH-responsive lipids [74]. Many commercially available transfection agents (i.e., Lipofectamine, Oligofectamine, TransIT-TKO and DharmaFECT) are lipid-based systems [75,76,77,78]. Despite the promising transfection efficacy, the major challenges of using the lipids are the immune response and cytotoxicity [79]. In addition to the lipids, biocompatible and biodegradable natural polymers such as chitosan and dextran, as well as synthetic polymers such as poly lactic-co-glycolic acid (PLGA), polyethylenimine (PEI) and PAMAM dendrimer are used for the preparation of polymeric nanoparticles for RNA delivery [80,81,82,83,84,85]. The polymers have relatively low toxicity compared to the lipid-based vectors and are more versatile for chemical modification. For peptide-based delivery systems, peptides such poly(l-lysine) (PLL), cell penetrating peptides (CPPs) and pH-responsive peptides have been investigated for the delivery of the RNAi molecules [86,87,88,89,90]. Different types of peptides mediate cellular transfection via different mechanisms such as facilitating cellular uptake and promoting endosomal escape. Peptides can be used to carry the RNAi molecules alone or act as a functional component in other carriers. Currently, efficient peptides with low toxicity and high transfection efficacy are still under development. It is noted that for pulmonary delivery, the reduced nuclease activity in the lungs makes successful delivery of unmodified naked RNA become possible [91,92]. However, the mechanism of how naked siRNA could overcome the extracellular and intracellular barrier following pulmonary administration still remains to be understood.
4. RNAi Therapeutics for the Treatment of Respiratory Diseases
The therapeutic potential of RNAi-based therapy targeting respiratory disorders has been widely explored in the past decade. In the following sections, the pre-clinical and clinical studies that involved the delivery of RNAi molecules to the airways for the treatment of lung diseases are discussed.
4.1. Lung Cancer
Lung cancer is a leading cause of cancer death in the world. The mainstay of treatment is chemotherapy, which is indicated for around 70% of newly diagnosed patients with local and advanced metastatic disease [93]. Other treatment options include surgery and radiotherapy [94]. Angiogenesis inhibitors [95] and epidermal growth factor receptor (EGFR) inhibitors [96,97] are new medications licensed for the treatment of lung cancer. Despite various treatment options, the prognosis of lung cancer is poor. The major problems of chemotherapy are toxicity, lack of specificity and drug resistance [98]. In recent years, pulmonary delivery of RNAi molecules is being explored as a new treatment for lung cancer [99]. Genes that are associated with tumor cell growth and drug resistance are potential targets. Table 2 summarizes some recent in vivo studies of pulmonary RNAi delivery for lung cancer treatment.
Table 2.
Selected in vivo studies of inhaled RNAi therapy on the treatment of lung cancer in the past five years.
4.1.1. Inhibition of Tumor Cell Growth
Genes that are essential for tumor growth and progression, such as Akt1 (RAC-alpha serine/threonine-protein kinase B) and c-myc, are evaluated as RNAi targets. Akt1 is an important mediator of cell growth, proliferation and survival of non-small cell lung cancer (NSCLC). Activation of Akt1 pathway is observed in most NSCLC patients. It is found to reduce chemo- and radiation-induced apoptosis to extend the survival of tumor cells [100,101]. Sorbitol diacrylatepolyethylenimine (SDA-PEI) was employed to deliver shRNA targeting Akt1 (shAkt1) and cDNA encoding programmed cell death protein 4 (Pdcd4) in a dual expression vector via pulmonary route to murine lung cancer model. Simultaneous Akt1 inhibition and Pdcd4 overexpression could induce potent anticancer effect. The total tumor number and the tumor size larger than 1 mm in the lung were significantly reduced after eight doses [101]. Glycerol triacrylate–spermine (GT–SPE) polyspermine [102] and lentivirus [103] were also employed as carriers of shRNA targeting Akt1 signaling pathway for pulmonary delivery. Although repeated aerosol delivery of lentiviral-based shRNA can achieve potent knockdown of the target, inhalation is a procedure that generates large number of viral particles. There are safety concerns regarding the use of viral vectors for inhalation, including the potential mutagenicity, immunogenicity and the risk of aerosol-transmitted infections [104]. Therefore, aerosol delivery of RNAi molecules using viral vectors must go through vigorous biosafety evaluation for clinical use.
C-myc oncogene is a downstream conduit for most oncogenic signals. It is necessary for the growth control, differentiation and apoptosis. Abnormal expression of c-myc is associated with many tumors. De-activation of myc is efficacious for lung tumor therapy [111]. Conde et al. delivered siRNA targeting c-myc to the lung of mouse using RGD-surface-modified gold nanoparticles (AuNPs) by intratracheal instillation. RGD peptide (Arg-Gly-Asp) is a cell adhesion peptide responsible to mediate cell adhesion and proliferation by binding to the integrin avb3 receptor family, a specific marker for angiogenesis and up-regulated in the endothelium. This strategy successfully suppressed c-myc expression level and tumor cell proliferation, resulting in approximately 80% increase in survival of orthograft mouse tumor models [107].
4.1.2. Targeting Multidrug Resistance (MDR)
Genes associated with MDR were also evaluated as target of RNAi, such as ribophorin II (RPN2) [106], B-cell lymphoma 2 (BCL-2) [112] and multidrug resistance protein 1 (MRP1) [110]. RPN2 is known to be associated with the apoptosis in docetaxel-resistant human breast cancer cells [113], and its involvement in NSCLC was investigated recently [114]. A novel RNAi molecule (PnkRNA) targeting RPN2 was developed and delivered to mice bearing lung cancer by intrapulmonary delivery. This new class of RNAi agent has a unique helical structure containing a central stem and two loops, and is claimed to be stable against nuclease degradation. Inhibition of lung tumor growth was observed without any signs of toxicity after treatment. The antitumor effect was triggered by PnkRNA targeting RPN2 without the use of other anticancer drugs. It could be attributed to the multiple activities of RPN2, which is anti-apoptotic and involved in the regulation of tumor survival [106]. However, the mechanism of how the naked the PnkRNA molecules overcame the extracellular and intracellular barriers to initiate RNAi was not elucidated.
Besides the inhibition of MDR associated proteins alone, the co-delivery of anticancer drugs with RNAi therapeutics is as an alternative approach to tackle MDR in various types of cancers [112,115,116,117]. Combination therapy offers the potential advantages of synergistic activity, overcoming the drug resistance and reducing the side effects of chemotherapy. Bcl-2 is a main player of nonpump cellular resistance [118] and MRP1 is responsible for drug efflux in cancer cells [119]. They were investigated in the combination approach. Taratula et al. prepared a lipid-based nanoparticles containing siRNA targeting Bcl-2 and MRP1, as well as anticancer drugs (doxorubicin or paclitaxel). The nanoparticles were delivered to the lung of tumor mouse model using a nose only exposure chamber for inhalation. Almost complete disappearance of lung tumor was observed in animals treated with paclitaxel plus the siRNAs. In contrast, treatment with paclitaxel alone only slowed down the growth of the tumor [110]. Xu et al. developed a polyethylenimine (PEI)-based delivery system that simultaneously delivered siRNA targeting Bcl-2 and doxorubicin to the lungs of mice with metastatic lung cancer. Synergistic antitumor efficacy was achieved compared with the delivery of doxorubicin or siRNA alone [105]. These studies indicated that suppression of resistance gene by RNAi molecules via inhalation could restore the sensitivity of chemotherapeutic drugs, leading to the death of tumor cells.
4.2. Respiratory Infection
Respiratory infection can be caused by a wide range of microorganisms in the airways including bacteria and viruses [120]. It is one of the most common reasons for hospitalization among adults [121]. Due to the rise of antimicrobial resistance, many infectious diseases including respiratory infections have become difficult to treat. RNAi therapeutics appears to be an attractive approach to fight against infections [122]. The pre-clinical studies of RNAi therapeutics to combat against respiratory infections are summarized in Table 3.
Table 3.
Selected in vivo studies of using RNAi therapy on respiratory infections.
Since RNAi was first reported to inhibit respiratory syncytial virus (RSV) in 2001 [131], an increasing number of studies have been initiated to explore the potential of RNAi technology to treat other respiratory viral infections including influenza [86,127,132,133] and severe acute respiratory syndrome (SARS) [134]. RNAi molecules offer several advantages as antiviral therapeutics. They can specifically target the conserved region of mRNA sequence in the viral genome, making it effective against mutated viral strains. In addition, RNAi molecules can be designed rapidly to target viral genes, shortening the lead time of developing new antiviral agents, which is particularly useful in response to an infection outbreak. To achieve effective antiviral effect, the viral targets must be essential for the pathogenesis of viral infection and/or replication cycle. It is also desirable that the target genes share similar conserved sequence among different strains to achieve broad viral inhibition. Furthermore, the viral gene should be substantially different with human gene to minimize any undesirable side effects.
4.2.1. RSV Infection
RSV infection is one of the most common infections in children [2,135]. In adult, RSV infection usually leads to self-limited upper respiratory illness [136]. However, severe RSV infection has been observed in elderly patients with lung diseases or in immunocompromised patients [137]. Current management of RSV includes bronchodilators, corticosteroids, antibiotics and supportive care [2], but their efficacy is not satisfactory. To date, there is no licensed vaccine available to prevent this disease [138]. There is an unmet need to develop effective RSV therapeutics and vaccines in both the pediatric and adult population.
In 2005, Bitko et al. reported the inhibition of RSV and parainfluenza virus (PIV) replication by siRNA targeting viral phosphoprotein (P protein), a crucial subunit of the viral RNA-dependent RNA polymerase. Since RSV replicate primarily in the superficial layer of the respiratory epithelium, local delivery of RNAi molecules to the lungs is a rational approach to inhibit RSV replication [139]. In the study, TransIT-TKO was employed as transfection agent and the siRNA was administered to the lungs of BALB/c mice by intranasal instillation 4 h before viral challenge. The RSV and PIV titer was reduced by over 90% five days after infection. The use of naked siRNA also showed substantial inhibition of infection, with approximately 70%–80% efficiency achieved as compared to complexed siRNA. Interestingly, the administration of siRNA before and concomitant with RSV infection was more effective in reducing viral load than treatment after infection [123]. The prophylactic regimen may not be a clinically attractive approach. To achieve optimal therapeutic benefit, fast detection and early intervention are crucial for the prognosis of RSV infection. It is desirable to initiate the antiviral RNAi therapy as quickly as possible once RSV infection is confirmed in clinical practice.
P protein may not be an ideal target as it is limited by its specificity to a particular viral strain [124]. Another siRNA, ALN-RSV01, was designed to target the highly conserved region of the mRNA encoding viral nucleocapsid protein (N protein). N protein is a core protein in the RNA polymerase and plays a key role in the replication cycle of RSV [140]. After intranasal administration of ALN-RSV01 to the lungs of mouse at 4 h prior to viral infection, 2.5- to 3.0-log-unit reductions in viral load was observed in comparison with the mismatch siRNA control. For the treatment regimen, comparable antiviral efficacy was achieved in multiple daily doses of ALN-RSV01 at day 1, 2 and 3 post-infection [124].
The phase I clinical study of ALN-RSV01 was reported in 2008 [139]. Intranasal administration of ALN-RSV01 was well tolerated and the side effect profile was similar to placebo. Later in 2010, the antiviral effect of ALN-RSV01 was evaluated in a phase II trial. ALN-RSV01 was administered by nasal spray daily to healthy adults for two days before and for three days after RSV inoculation. The group treated with ALN-RSV01 had a significantly lower number of RSV infection compared to the placebo group [140]. Another phase II b clinical trial of ALN-RSV01 in RSV-infected lung transplant patients was also completed [141]. The primary endpoint of the study was to evaluate the effect of ALN-RSV01 on the incidence of new or progressive bronchiolitis obliterans syndrome (BOS) in RSV-infected lung transplant patients. Even though the study marginally missed the primary endpoint statistically (p = 0.058), inhaled ALN-RSV01 treatment induced a clinically meaningful reduction in the incidence of BOS, reinforcing the potential of inhaled RNAi as new antiviral therapeutics.
4.2.2. Influenza
Seasonal influenza viruses, especially influenza type A virus, cause annual epidemics that led to millions of death worldwide [142]. It is one of the major public health problems. Two categories of antiviral drugs are currently approved to treat influenza, including M2 ion-channel protein inhibitors (adamantanes) and neuraminidase inhibitors (zanamivir and oseltamivir). However, some influenza A virus strains have shown to be resistant to these drugs [143,144], making the development of new antiviral drug urgently needed. A number of studies were carried out to investigate the use of RNAi to inhibit viral gene expression and protect cells from influenza infection. The most commonly investigated targets are the nucleocapsid protein (NP), polymerase acidic protein (PA) and polymerase basic protein (PB). These proteins are indispensable to influenza viral replication. More importantly, they are highly conserved across different subtypes of influenza virus strains [132].
The first animal study applying RNAi against influenza was reported by Ge et al. [145]. siRNA targeting NP or PA were complexed with PEI. The complexes were injected intravenously to the mice 3 h before or 5 h after influenza virus (PR8 H1N1) infection. The antiviral effect of siRNA targeting NP was dose-dependent and combination of siRNAs targeting NP and PA enhanced the effect. In the same study, shRNA targeting NP or PB was also investigated. The DNA was mixed with Infasurf, a commercial available pulmonary surfactant. After intranasal instillation, a significant reduction of virus titer was detected 24 h after infection although the effect was less prominent compared with intravenous injection of PEI complexes. Infasurf may play a role as nucleic acid carrier. Little is known about the interaction between DNA and pulmonary surfactant in the airspace. It could be possible that the DNA interacted with the charged lipid components in the pulmonary surfactant leading to the efficient spread and absorption in the airways [146].
Another study reported the administration of siRNA targeting NP and/or PA in lipid carrier to influenza-infected mice by hydrodynamic injection and intranasal administration. The combined siRNA delivery method effectively protected animals against lethal challenge of highly pathogenic avian influenza A viruses, including the H5N1, H7N7 and H9N2 subtypes [126]. Since then, the potential of siRNA for the treatment of influenza virus infection was continuously evaluated in vitro [133,147,148,149] and in vivo [127,128,129]. However, the exploration of RNAi for the treatment of influenza appears to reach the bottleneck. It could be partly attributed to the criticism of the antiviral effect induced by siRNA, which was due to innate immune response rather than viral inhibition through RNAi. Development of chemically modified RNAi molecules with minimal immunostimulatory effect could be helpful to delineate the mechanism of the antiviral effects [150].
4.2.3. Tuberculosis
Tuberculosis (TB) is a bacterial lung infection caused by Mycobacterium tuberculosis (Mtb). Because of the emergence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) TB, the slow development of new anti-TB agents and the inefficiency of vaccination, TB is still a major global health problem. WHO reported that 9.6 million people suffered from TB in 2014 and 1.5 million die annually because of the disease [151]. A new and effective TB therapeutics is highly sought after.
The approach of RNAi-based therapeutics against TB aims to modulate the gene expression of the host instead of the bacillus because bacteria do not contain the requisite machinery for RNAi. Currently, RNAi has been investigated for TB treatment in two directions: (i) targeting the host factors that are essential for the survival of mycobacteria; and (ii) modulating the host immune response to facilitate the elimination of mycobacterium [49]. Autophagy is a host defense mechanism against Mtb. Boosting autophagy could be an effective strategy to promote the eradication of the mycobacteria. Hence, a number of host factors that are involved in the regulation of autophagy were explored as the target of RNAi, including Bfl-1/A1 [152], Rap22a [153], Ras homologue enriched in brain (Rheb) [154] and UV radiation resistance-associated gene (UVRAG) [155]. Down-regulation of these factors promoted autophagosome formation and reduced the bacterial growth. However, the existing studies of using RNAi to suppress host factors are preliminary and in vivo data is lacking. Moreover, it was recently reported that autophagy pathway may not be essential for restricting Mtb growth [156]. Further investigations are required to understand the relationship between Mtb infection and autophagy pathway, and to better position the approach of RNAi for the control of tuberculosis infection.
Another strategy is to modulate host immune response to improve the antimicrobial ability of the host [49]. One of the key characteristics of TB infection is the formation of granuloma, which is a compact, organized aggregate of immune cells. Granuloma is previously considered as a host-protective structure to sequester the infecting mycobacteria, but recent work revealed that the pathogens could take advantage of granuloma formation to facilitate their proliferation and dissemination in the host [157], as it provides a hospitable environment for mycobacteria to survive and replicate over a long period of time. There is a complicated chemokines and cytokines network involved in the granulomatous response. RNAi can be used to target immunosuppressive cytokines in order to inhibit the growth of Mtb. Among them, tumor necrosis factor (TNF)-α and IFN-γ are critical in activating macrophages and triggering the formation of granuloma [158]. XCL1 (lymphotactin) is a chemokine produced by activated CD8+ T cells during chronic TB infection. It was suggested that XCL1 regulates IFN-γ production by CD4+ T cells and contribute to the stability of the granuloma [159]. When a single dose of siRNA targeting XCL1 was delivered to the lungs of mice that were chronically infected with TB, the expression of XCL1 was effectively inhibited. The suppression of XCL1 expression in the lungs was associated with the decreasing number of T lymphocytes, reduction in the IFN-γ response, disorganized granulomatous lesions and higher fibrosis [130]. Although the inhibition effect elicited by siRNA was transient and did not provide significant therapeutic benefit, this proof-of-concept study demonstrated that the local-environment and immunopathology of the lungs can be modulated to favor host response via pulmonary delivery of siRNA.
Other targets of RNAi-based immunotherapy include the transforming growth factor β (TGF-β) and interleukin 10 (IL-10) [13]. Both of them are found to be elevated in TB patients, and their high level of expression is connected with TB reactivation [160]. During chronic infection, these cytokines restrain inflammation through affecting helper T cell 1 (Th1) response and macrophage activation, leading to substantial reduction of their antimicrobial activity [161]. In that study, siRNA targeting TGF-β1 was administered through the intrapulmonary route to IL-10 knockout mice at 60 days post-infection. TGF-β1 gene silencing resulted in the increased expression of the antimicrobial mediators nitric oxide (NO) and inducible nitric oxide synthase (iNOS), and significantly reduced the bacterial load for at least four weeks after treatment. A synergistic effect at improving the antimicrobial activity in the lungs was observed when both IL-10 and TGF-β1 were targeted simultaneously. Although the inhibition of bacterial load in this study was modest, it showed that siRNA immunotherapy is a feasible approach to enhance the host antimicrobial capacity. Moreover, MDR and XDR TB pose huge challenges for the current chemotherapeutic agents. Combined immunotherapy targeting immunosuppressive cytokines and chemotherapy to target drug resistance bacteria population is a direction that is worth future investigation.
4.3. Respiratory Inflammatory Disease
Respiratory inflammatory diseases impose substantial healthcare burden worldwide as many of the patients are inadequately controlled by current therapies [4,5,162]. These diseases are characterized by inflammation in the respiratory tract, which is manifested with increased expression of inflammatory cytokines and chemokines [163,164]. The common lung inflammatory diseases include asthma, chronic obstructive pulmonary disease (COPD) and acute lung injury (ALI). Table 4 lists the major in vivo studies that examine the effects of RNAi molecules in respiratory inflammatory disease.
Table 4.
Selected in vivo studies of using RNAi therapy on respiratory inflammatory diseases.
4.3.1. Asthma
Asthma is characterized by the variable and reversible airflow obstruction, airway hyperresponsiveness (AHR), mucus hypersecretion and chronic inflammation [176]. The most common form of asthma is allergic asthma, which is triggered by environmental stimuli such as house dust and seasonal pollen [177]. According to WHO, it is estimated that around 334 million peoples have asthma worldwide [178]. The mainstay treatments of asthma are corticosteroids, β2-adrenergic receptor agonists and leukotriene receptor antagonists, either alone or in combination. Although these medications can manage the conditions in most of the patients, there are still challenges of poor patient compliance, intolerability and long term adverse effects [179]. For persistent allergic asthmatic patients, omalizumab which is an anti-immunoglobulin E (IgE) monoclonal antibody can be used, but the high cost and parenteral administration make it less favorable [180]. Alternative anti-inflammatory drugs are needed. Many inflammatory mediators are identified to be involved in the pathogenesis of asthma and they are investigated as potential targets of RNAi therapy. These include cytokines/chemokines [181], transcription factors [168], tyrosine kinases/tyrosine kinase receptors [172] and co-stimulatory molecules [182].
In allergic asthma, there is predominant activation of CD4+ T helper 2 (Th2) cell-driven inflammatory response [183]. T cell activation and Th2 cytokine expression are increased in the airways of atopic asthma patients after exposure to allergen [184]. Th2-type cytokines, interleukin-4 (IL-4), IL-5 and IL-13, are the main players in the pathology of allergic airway inflammation, contributing to allergic sensitization, IgE production, eosinophil infiltration and AHR respectively [183,184]. Clinical studies that examine the therapeutic potential of using monoclonal antibodies (mAbs) to neutralize the bioactivity of IL-4 [185], IL-13 [186,187] and IL-5 [188] or target their receptors are in progress. Similarly, RNAi technology can be employed to target these cytokines [169,181].
Targeting suppressor of cytokine signaling (SOCS) 3 is an alternative approach [12]. SOCS proteins are negative regulators of cytokine signaling pathway, which are involved in Th2 cells mediated allergic response via controlling the balance between Th2 and Th1 cells [189]. The expression of SOCS3 is a pathological marker of allergic disease. After siRNA targeting, SOCS3 was intranasally administered to the lungs of chronic asthmatic mouse model [12], the silencing of SOCS3 down-regulated the expression of Th2 cell associated cytokines, IL-4, IL-5 and IL-13, leading to substantial reduction of airway inflammation, AHR as well as IgE production. Reduction of mucus secretion and collagen deposition was also detected in the airways. Besides Th2 cell, other T cells subsets, such as Th9 and Th17 cells, also contribute the inflammatory response in asthma by releasing IL-9 and IL-17A. These molecules may also be the potential targets for RNAi therapy [183]. Furthermore, the combination of inflammatory and antiviral siRNAs is a prospective therapy for patients with virus-induced exacerbation of severe uncontrolled asthma, in view of the report that respiratory viral infection is related to 80% of asthma exacerbation in children and adults [190]. The therapeutic potential of targeting IL-4 and P protein of RSV simultaneously by siRNAs was examined in a mouse model of RSV-induced asthma exacerbation [181]. The intranasal delivery of the two siRNAs was able to: (i) significantly suppress IL-4 mRNA expression and RSV replication in the lungs; (ii) reduce eosinophil and neutrophil infiltration in bronchoalveolar lavage fluid (BALF); (iii) inhibit AHR and inflammation; and (iv) increase interferon (IFN)-γ expression.
Many transcription factors including nuclear factor-κB (NF-κB) [168], signal transducer and activator of transcription factor 6 (STAT6) [166] and GATA-binding protein 3 (GATA3) [170] are involved in the production Th2 cytokine in the airways of patients with asthma. They were investigated as targets for RNAi in the management of asthmatic inflammation. NF-κB plays a key role in the expression of many pro-inflammatory proteins [191]. Inhibition of NF-κB pathway in the lungs was reported to be beneficial in asthma model [176,192]. One approach to inhibit the activation of NF-κB signaling pathway is to target receptor-interacting protein 2 (Rip2), which is a transcriptional product and an activator of NF-κB. Suppression of Rip2 expression was achieved in mice by intratracheal delivery of siRNA, resulting in the inhibition of ovalbumin (OVA)-induced cytokine release, inflammatory cell infiltration, mucus hypersecretion and serum IgE level. This result suggested that Rip2 could be a novel therapeutic target for the treatment of asthma [168]. Consistent with this finding, suppression of STAT6 [10] and GATA3 [170,193] in murine model of allergen-induced asthma by RNAi significantly inhibited the expression of downstream Th2 cytokines. These studies support the hypothesis of using RNAi molecules targeting transcription factors to manage airway inflammation and AHR of asthma.
Tyrosine kinases, which are broadly classified into receptor and non-receptor types, play an important role in activating the transcription factors for inflammatory gene expression upon activation by the inflammatory signals [194,195]. The key receptor tyrosine kinases involved in the inflammatory response in asthma include epidermal growth factor receptor (EGFR), c-kit (a stem cell factor receptor) and vascular endothelial growth factor receptor (VEGFR), while the non-receptor forms are spleen tyrosine kinase (Syk), Src and janus kinase (JAK) [196]. Syk is a pivotal intracellular signaling molecule involved in the activation of several pro-inflammatory transcription factors [197]. Inhibition of Syk expression by antisense oligonucleotides has proven effective in suppression of tracheal contraction and pulmonary inflammation in animal models of OVA-induced asthma [198]. When naked siRNA targeting Syk was administered to the lungs of allergen-induced asthma mice model via nasal instillation, the recruitment of inflammatory cells in the BALF was reduced [194]. Similarly, intranasal administration of siRNA targeting c-kit also induced a promising result in reducing mucus secretion and airway inflammation in experimental asthma mouse model [173]. Moreover, clinical studies have been performed on a siRNA targeting Syk (Excellair™ developed by ZaBeCor Pharmaceuticals, USA) for the treatment of asthma. In the Phase I study, 75% of asthma patients receiving Excellair™ by inhalation for 21 consecutive days showed improvement in breathing or reduced the usage of rescue inhaler compared to placebo. The drug was well tolerated by patients without eliciting serious side effects. A Phase II clinical trial Excellair™ has been initiated in 2009; the results are not yet available during the preparation of this review [199,200].
Targeting dendritic cells (DCs) in the airways by RNAi is another strategy for the treatment of asthma. DCs are antigen presenting cells that could be found in the airways [5,201]. They are actively involved in the differentiation of Th2 cells and hence contribute to the progress of airway inflammation [202]. DCs express co-stimulatory molecules, cluster of differentiation (CD) 80 and 86, on the surface. The binding of CD80/CD86 with CD28 and cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) molecules on T cells is necessary for priming naive T cells into Th2 cells [203]. Blocking CD80 and/or CD86 activity with pharmacological inhibitors can inhibit T cell activation and alleviate AHR and pulmonary inflammation in mice exposed to aerosolized allergen challenge [204]. Likewise, intratracheal administration of siRNA targeting CD86 significantly reduced its expression in the airway mucosa of mouse model of allergic asthma. CD86 siRNA treatment also contributed to the amelioration of OVA-induced airway eosinophilia, hyperresponsiveness, the elevations of IgE in the sera and the production of inflammatory cytokines in BALF [171].
4.3.2. COPD
COPD is a persistent lung disease characterized by irreversible chronic airflow limitation. The disease is usually progressive and associated with an increased chronic inflammatory response in the lungs [205,206]. It is the result of long term exposure to air pollutants and usually associated with cigarette smoking [205]. It affects around 10% of the population in the world. It is the fourth leading cause of death in most of the industrialized countries and expected to become the third by 2020 [207]. Three classes of medications used to manage COPD are bronchodilators (including β2-adrenergic receptor and anticholinergic drugs), corticosteroids and phosphodiesterase-4 inhibitor. They are effective in relieving the symptoms of COPD, but none of them can reverse the progressive deterioration in lung function or cure the disease [4,205]. It is desirable to develop a novel therapy that can simultaneously control the symptoms and improve the long-term prognosis of the disease.
Despite similar clinical features, the mechanisms of underlying airway inflammation in asthma and COPD are different with distinct patterns of inflammatory cells and mediators being involved [163]. The mechanisms and the mediators that drive the progression of chronic inflammation and emphysema in COPD are far from fully understood [208]. The major signaling factors involved in the pathogenesis of COPD include TNF-α, CCL2, CXCL8, TGF-β and VEGF [206]. Many of the cytokines and chemokines secreted in the airways of COPD patients are modulated by the NF-κB pathway [163]. Therefore, RNAi targeting NF-κB pathway can be a strategy for the management of COPD [176,209].
Current research work on RNAi therapy for COPD remains mainly in the in vitro stage, likely due to the insufficient understanding of the highly complex mechanisms underlying the pathology of the disease [120]. Moreover, the degree of airway inflammatory response is determined both by genetic and epigenetic factors. Since multiple inflammatory mediators are involved in the disease process, blocking a single type of molecule is unlikely to exert significant clinical impact [206]. miRNA has the advantage of targeting multiple genes simultaneously. It is involved in the regulation of the inflammatory process of COPD by modulating the transcription and translation of genes. Differentiated expression of several miRNAs, like let-7c, miR-125b, miR-15b [210] and miR-199a-5p [211] were identified in COPD patients compared to subjects without COPD. Modulation the expression of multiple genes using miRNA could be a direction of RNAi therapy for the treatment of COPD.
4.3.3. Acute Lung Injury (ALI)
ALI is a severe form of diffuse lung disease that is commonly caused by acute systemic inflammatory disorders, such as sepsis, pneumonia, trauma and pancreatitis [11,212]. It is a progressive disease, which can lead to acute respiratory distress syndrome (ARDS) or more critically, multiple organ failure and even death [175]. Its pathological changes include the release of pro-inflammatory cytokines and chemokines, and the destruction of the epithelium-capillary interface; the latter can facilitate the extravasation of protein-rich fluid and the infiltration of neutrophils, thereby exacerbating the condition [213,214]. Since the molecular mechanism underlying the pathology of ALI is not well understood, there is no pathophysiologic-driven therapeutic approach available [162].
RNAi targeting the essential inflammatory mediators could offer protection against the progression of the disease. In an in vivo study, systemic delivery of siRNA targeting TNF-α significantly reduced the expression of TNF-α in the lungs and lung injury in a shock-induced ALI mouse model [175]. The findings suggested that pulmonary vascular cells, but not the epithelial cells, contribute to the release of TNF-α leading to lung injury following a systemic inflammatory insult. Although these findings are contradictory to the others who suggest that pulmonary epithelial cells are the major players for causing lung injury induced by a systemic disease [215,216], nevertheless, they provide evidence that support the use of siRNA targeting TNF-α in the management of ALI.
Increase microvascular permeability is a major cause of amplification of the inflammatory responses in ALI, leading to the progression to severe pathological condition [214]. The integrity of pulmonary and systemic endothelial barrier is maintained by the bioactive lipid, sphingosine-1-phosphate (S1P), which can be irreversibly degraded by an enzyme called sphingosine-1-phosphate lyase (S1PLyase) [214]. Therefore, suppressing endothelial S1PLyase expression by siRNA is a potential therapeutic approach to maintain the integrity of the endothelial barrier [217]. Nanoparticles containing siRNA targeting S1PLyase and recombinant high mobility group box-1 A peptide (HMGB1A) were delivered to the lungs of LPS-induced ALI mouse model by intratracheal instillation. HMGB1A peptide is an antagonist of the pro-inflammatory cytokine HMGB1 and provides additional anti-inflammatory effect in ALI. An arginine-rich R3V6 peptide was used as a carrier. S1PLyase level was significantly inhibited in the BALF, leading to a significantly reduction of inflammatory cytokine (TNF-α and IL-6) and inflammatory response in the lungs of the treated animal [11].
4.3.4. Considerations of Managing Respiratory Inflammatory Diseases by RNAi
Although several in vivo studies demonstrated promising results of using RNAi therapeutics for the management of respiratory inflammatory diseases, there are several concerns that need to be taken into consideration with this approach. The distribution of naked siRNA following intranasal and intratracheal administration was investigated. It was shown that the siRNA molecules were predominantly accumulated in the peribronchial epithelial cells within 24 h post-administration [12]. Since the inflammatory mechanism of diseases like asthma appears to be driven by the activated Th2 lymphocytes [218], it is highly desirable for RNAi molecules to target the activated T cells in the airways to achieve therapeutic benefit. However, specific targeting to T cells is extremely difficult [174]. Effective T cells transfection in vitro was achieved by electroporation [219], an approach that is not clinically feasible. Recently, a ligand-polymer conjugate siRNA delivery system, transferrin-polyethylenimine (Tf-PEI), has been developed to target T cells in the lungs [174]. The rational of such a design was based on the increased expression of transferrin receptors on T cells after activation. Biodistribution study showed that Tf-PEI polyplexes can selectively deliver the fluorescently labeled siRNA to the activated T cells in a murine asthma model. However, the therapeutic effect of the system was not evaluated. Therefore, targeting delivery of RNAi therapeutics to specific cell types associated with the disease is a field that certainly required further exploration.
Most of the key signaling molecules involved in the pathogenesis of respiratory inflammatory disorders have multiple functions and participate in normal physiological activities to maintain homeostasis. Short-term regimen (up to four days) was employed in the majority of the animal studies. The duration of treatment may not be sufficient to observe any long-term effect. Since many inflammatory diseases are chronic disorders, long-term therapy is often required to control the disease. Therefore, it is crucial to evaluate the long-term effect of any RNAi therapeutics.
Inhaled corticosteroid is commonly used to control the symptoms of lung inflammatory disorders. Steroid treatment reduces airway inflammation, partially through down-regulation of the key effectors molecules, such as c-kit [220] and GATA3 [221]. For this reason, it is necessary to determine the effect of concurrent medications on the expression of targeting molecules before initiating the RNAi therapy. Given that lung inflammatory diseases are heterogeneous diseases that are categorized into different subtypes according to the inflammatory profiles and responses to treatment, it is desirable to identify the patients’ profile in order to maximize the benefit of RNAi therapy.
4.4. Pulmonary Fibrosis
Pulmonary fibrosis is a respiratory disorder characterized by the progressive and irreversible destruction of lung architecture, which ultimately leads to organ malfunction, disruption of gas exchange and even death [222]. It is initiated by lung tissue damage resulting from autoimmune disorders or exposure to external irritants. The damage in turn triggers inflammatory reactions for tissue repair in the attempt to restore the normal tissue architecture and functions [222]. Under pathological conditions, extracellular matrix (ECM) components, which are released by the activated myofibroblasts during the wound healing process, are excessively produced and accumulate at the site of tissue injury, resulting in fibrosis [223]. There are many forms of pulmonary fibrosis. All of them are lethal diseases with high mortality rate but with very limited therapeutic options [223]. Thus pulmonary fibrosis is a lung disease with huge unmet medical need.
RNAi has been exploited to suppress the production of key molecules involved in the fibrotic process, such as TGF-β [92,224], amphiregulin (AR) and connective transforming growth factor (CTGF) [8,225] (Table 5). Clinical trials examining the effect of neutralized antibodies to block the activity of TGF-β are currently ongoing [14]. Since lung epithelial cells are the key cell types in the pathogenesis of pulmonary fibrosis, local administration of RNAi targeting TGF-β to the lungs can ensure high local concentration of RNAi molecules to produce therapeutic effects. Intratracheal delivery of siRNA targeting TGF-β1 could effectively inhibit pulmonary fibrosis, improved lung function, and prolonged survival in human TGF-β1 transgenic mice [224]. In another in vivo study, intranasal delivery of miRNA-326 mimic, which was designed to suppresses the expression of TGF-β and other profibrotic genes, significantly reduced TGF-β1 expression level and further alleviated the fibrotic feature in the bleomycin-induced pulmonary fibrosis [92]. The results suggest the potential of targeting TGF-β or its signaling pathway using RNAi technology for the management of lung fibrosis.
Table 5.
Selected in vivo studies of using RNAi therapy on pulmonary fibrosis.
One complication of using TGF-β as the molecular target of RNAi is related to its physiological role in maintaining immune and cellular homeostasis. It has a role in tumor suppression as suggested by the finding that specific inhibition of TGF-β in mouse stromal fibroblasts results in spontaneous carcinoma formation in the adjacent epithelium [226]. As a result, targeting the downstream effectors of TGF-β may offer the therapeutic benefit of inhibiting the TGF-β-mediated fibrotic response while preserving the major physiological function of the profibrotic cytokine [227]. CTGF is a key molecule that regulates fibroblast mitosis and promotes collagen deposition in pulmonary fibrosis induced by TGF-β. A non-viral delivery system consists of poly(dimethylamino)ethylmethacrylate (PDMAEMA) and its copolymer with poly(methylether-methacrylate-ethyleneglycol) [PMAPEG] was used to incorporate and form complexes with siRNA targeting CTGF. After intratracheal administration, significant reduction of CTGF expression, collagen deposition and inflammatory cytokines (IL-6, TNF-α and TGF-β) production were observed in bleomycin-induced lung fibrosis animal model. More importantly, the survival rate of the animals treated with siRNA targeting CTGF was significantly increased in comparison with the group treated with scrambled siRNA [225].
Another downstream mediator of TGF-β is amphiregulin (AR), which is an epidermal growth factor receptor (EGFR) ligand primarily induced by TGF-β1 [228]. siRNAs targeting AR and CTGF were delivered to the lung of pulmonary fibrosis mouse model via either intratracheal or intravenous administration. To improve the efficiency and cell-specific delivery, a modified self-assembled micelle interfering RNA nanoparticles (SAMiRNA) was developed. The nanoparticles contain hydrophilic polymer and hydrophobic lipid on each ends of siRNA and can spontaneously form micelle in solution. After administration of AR or CTGF SAMiRNAs, the expression of AR and CTGF, and collagen accumulation in the lung of pulmonary fibrosis model were significantly reduced compared to control SAMiRNA [8].
5. Clinical Translation and Future Perspectives
RNAi technology has been under rapid development for the last decade. The therapeutic potential of RNAi molecules in airway diseases has been demonstrated in several clinical studies (Table 6), and they could be the solution to some unmet medical needs. No RNAi therapeutics has gained approval by regulatory authority at the time this manuscript was prepared. While delivery is generally considered as the major obstacle to RNAi therapeutics development, it is interesting to notice that siRNAs or miRNA mimics do not need a carrier to generate therapeutic effects following pulmonary delivery. The cellular uptake mechanism of naked RNA in the airways is not clear, but this is certainly an attractive feature that allows simple formulation without the need of delivery vectors. Chemically modified siRNA or miRNA is necessary to improve the stability of the RNA molecules. On the other hand, the delivery of shRNA or miRNA encoding plasmids requires viral vectors to ensure efficient transduction and the subsequent expression of RNAi molecules. However, the use of viral vectors could raise safety concerns for clinical applications, especially when they are used for pulmonary delivery. The employment of liposomal or polymer based nanoparticle delivery technology could be a promising approach. Whether the nanoparticles or naked RNA are used, it is important to evaluate the pharmacokinetic profile of each delivery system to ensure the RNAi therapeutics are localized at the site of action but not quickly distributed to other organs in the body. In terms of formulation of RNAi therapeutics, dry powder aerosol is preferred over the liquid aerosol due to higher stability and the dry powder inhalers are easier to operate with better lung deposition [229]. This would be the direction for RNAi formulation development.
Table 6.
Summary of clinical trials of RNAi therapeutics in respiratory diseases.
Among the different types of RNAi molecules for therapeutic development, siRNA has made significant progress, with a couple of clinical studies investigate the pulmonary delivery of siRNA to treat respiratory diseases. There are also a growing number of clinical pipelines involving the use of siRNA and other RNAi molecules. Several key questions are raised that need to be answered. For infectious diseases, as demonstrated in several in vivo studies, it appears that RNAi is more effective in prophylactic regimen than in therapeutic regimen. How can we improve the therapeutic efficacy of RNAi in combating infections to make it clinically relevant? For inflammatory diseases, long-term therapy is expected. How can we minimize the undesirable effects when cytokines/chemokines are being targeted? When the respiratory functions are compromised in patients with lung disease, is pulmonary delivery a suitable route for administration? As complicated signaling pathway is involved in the pathogenesis of different lung disorders, a thorough investigation of the underlying pathological mechanisms of the disease is a prerequisite for identifying the target of RNAi. Despite the challenges and obstacles, we believe that the pulmonary delivery of RNAi molecules targeting airway diseases is promising.
Acknowledgments
The work was supported by the Research Grant Council of Hong Kong (17110414) and the Health and Medical Research Fund, Hong Kong (11123011, 15140962).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pilkington, G.; Boland, A.; Brown, T.; Oyee, J.; Bagust, A.; Dickson, R. A systematic review of the clinical effectiveness of first-line chemotherapy for adult patients with locally advanced or metastatic non-small cell lung cancer. Thorax 2015. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Chang, C.; Gershwin, M.E.; Gershwin, L.J. Respiratory syncytial virus-a comprehensive review. Clin. Rev. Allrgy Immunol. 2013, 45, 331–379. [Google Scholar] [CrossRef] [PubMed]
- Olaru, I.D.; von Groote-Bidlingmaier, F.; Heyckendorf, J.; Yew, W.W.; Lange, C.; Chang, K.C. Novel drugs against tuberculosis: A clinician’s perspective. Eur. Respir. J. 2015, 45, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Mushtaq, Y. The COPD pipeline. Nat. Rev. Drug Discov. 2014, 13, 253–254. [Google Scholar] [CrossRef] [PubMed]
- Olin, J.T.; Wechsler, M.E. Asthma: Pathogenesis and novel drugs for treatment. BMJ 2014, 349, g5517. [Google Scholar] [CrossRef] [PubMed]
- Rafii, R.; Juarez, M.M.; Albertson, T.E.; Chan, A.L. A review of current and novel therapies for idiopathic pulmonary fibrosis. J. Thorac. Dis. 2013, 5, 48–73. [Google Scholar] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Yoon, P.O.; Park, J.W.; Lee, C.M.; Kim, S.H.; Kim, H.N.; Ko, Y.; Bae, S.J.; Yun, S.; Park, J.H.; Kwon, T.; et al. Self-assembled micelle interfering RNA for effective and safe targeting of dysregulated genes in pulmonary fibrosis. J. Biol. Chem. 2016, 291, 6433–6446. [Google Scholar] [CrossRef] [PubMed]
- Ihara, D.; Hattori, N.; Horimasu, Y.; Masuda, T.; Nakashima, T.; Senoo, T.; Iwamoto, H.; Fujitaka, K.; Okamoto, H.; Kohno, N. Histological quantification of gene silencing by intratracheal administration of dry powdered small-interfering RNA/chitosan complexes in the murine lung. Pharm. Res. 2015, 32, 3877–3885. [Google Scholar] [CrossRef] [PubMed]
- Healey, G.D.; Lockridge, J.A.; Zinnen, S.; Hopkin, J.M.; Richards, I.; Walker, W. Development of pre-clinical models for evaluating the therapeutic potential of candidate siRNA targeting STAT6. PLoS ONE 2014, 9, e90338. [Google Scholar] [CrossRef] [PubMed]
- Oh, B.; Lee, M. Combined delivery of HMGB-1 box A peptide and S1PLyase siRNA in animal models of acute lung injury. J. Control. Release 2014, 175, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Zafra, M.P.; Mazzeo, C.; Gámez, C.; Marco, A.R.; de Zulueta, A.; Sanz, V.; Bilbao, I.; Ruiz-Cabello, J.; Zubeldia, J.M.; del Pozo, V. Gene silencing of SOCS3 by siRNA intranasal delivery inhibits asthma phenotype in mice. PLoS ONE 2014, 9, e91996. [Google Scholar]
- Rosas-Taraco, A.G.; Higgins, D.M.; Sánchez-Campillo, J.; Lee, E.J.; Orme, I.M.; González-Juarrero, M. Local pulmonary immunotherapy with siRNA targeting TGFβ1 enhances antimicrobial capacity in Mycobacterium tuberculosis infected mice. Tuberculosis 2011, 91, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Liang, W.; Chan, H.K. Pulmonary delivery of therapeutic siRNA. Adv. Drug Deliv. Rev. 2012, 64, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Karpala, A.J.; Doran, T.J.; Bean, A.G. Immune responses to dsRNA: Implications for gene silencing technologies. Immunol. Cell Biol. 2005, 83, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, G.; Ozpolat, B.; Coleman, R.L.; Sood, A.K.; Lopez-Berestein, G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015, 87, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Rathi, M.; Baradia, D.; Trehan, S.; Misra, A. In vivo delivery aspects of miRNA, shRNA and siRNA. Crit. Rev. Ther. Drug Carr. Syst. 2012, 29, 487–527. [Google Scholar] [CrossRef]
- Singh, S.; Narang, A.S.; Mahato, R.I. Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm. Res. 2011, 28, 2996–3015. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA versus miRNA as therapeutics for gene silencing. Mol. Ther. Nucl. Acids 2015, 4, e252. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Shu, D.; Li, H.; Shu, Y.; Xiong, G.; Carson, W.E., III; Haque, F.; Xu, R.; Guo, P. Systemic delivery of anti-miRNA for suppression of triple negative breast cancer utilizing RNA nanotechnology. ACS Nano 2015, 9, 9731–9740. [Google Scholar] [CrossRef] [PubMed]
- McLendon, J.M.; Joshi, S.R.; Sparks, J.; Matar, M.; Fewell, J.G.; Abe, K.; Oka, M.; McMurtry, I.F.; Gerthoffer, W.T. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J. Control. Release 2015, 210, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Ai, C.; Jiang, R.; Fu, L.; Chen, Y. MicroRNA-495 mimics delivery inhibits lung tumor progression. Tumour Biol. 2015, 36, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Lu, H.; Qu, Y.; Fu, W.; Ma, Z. Developing an effective therapeutic by delivery of synthetic microRNA-520e in lung cancer treatment. Biomed. Pharmacother. 2015, 69, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Kuwano, K.; Ochiya, T. Development of small RNA delivery systems for lung cancer therapy. Int. J. Mol. Sci. 2015, 16, 5254–5270. [Google Scholar] [CrossRef] [PubMed]
- Birmingham, A.; Anderson, E.; Sullivan, K.; Reynolds, A.; Boese, Q.; Leake, D.; Karpilow, J.; Khvorova, A. A protocol for designing siRNAs with high functionality and specificity. Nat. Protoc. 2007, 2, 2068–2078. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, J.R.; Pottier, M.; Vekris, A.; Opolon, P.; Maksimenko, A.; Malvy, C. Comparison of antisense oligonucleotides and siRNAs in cell culture and in vivo. Biochem. Biophys. Res. Commun. 2002, 296, 1000–1004. [Google Scholar] [CrossRef]
- Bobbin, M.L.; Rossi, J.J. RNA interference (RNAi)-based therapeutics: Delivering on the promise? Annu. Rev. Pharmacol. Toxicol. 2016, 56, 103–122. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. RNA-based drugs and vaccines. Expert Rev. Vaccines 2015, 14, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Günther, M.; Lipka, J.; Malek, A.; Gutsch, D.; Kreyling, W.; Aigner, A. Polyethylenimines for RNAi-mediated gene targeting in vivo and siRNA delivery to the lung. Eur. J. Pharm. Biopharm. 2011, 77, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, M.; Müller-Goymann, C.C. Nanoparticle-mediated pulmonary drug delivery: A review. Int. J. Mol. Sci. 2014, 15, 5852–5873. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.C.; Peters, J.I.; Williams, R.O. Influence of particle size on regional lung deposition-what evidence is there? Int. J. Pharm. 2011, 406, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, M. In vivo, in vitro and ex vivo models to assess pulmonary absorption and disposition of inhaled therapeutics for systemic delivery. Adv. Drug Deliv. Rev. 2006, 58, 1030–1060. [Google Scholar] [CrossRef] [PubMed]
- Patton, J.S.; Byron, P.R. Inhaling medicines: Delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 2007, 6, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Merkel, O.M.; Rubinstein, I.; Kissel, T. siRNA delivery to the lung: What’s new? Adv. Drug Deliv. Rev. 2014, 75, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [PubMed]
- Thornton, D.J.; Rousseau, K.; McGuckin, M.A. Structure and function of the polymeric mucins in airways mucus. Annu. Rev. Physiol. 2008, 70, 459–486. [Google Scholar] [CrossRef] [PubMed]
- Salathe, M. Regulation of mammalian ciliary beating. Annu. Rev. Physiol. 2007, 69, 401–422. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.K.; Wang, Y.Y.; Hanes, J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv. Drug Deliv. Rev. 2009, 61, 158–171. [Google Scholar] [CrossRef] [PubMed]
- Parra, E.; Perez-Gil, J. Composition, structure and mechanical properties define performance of pulmonary surfactant membranes and films. Chem. Phys. Lipids 2015, 185, 153–175. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.E.; Whitsett, J.A.; Horowitz, A.D. Pulmonary surfactant inhibits cationic liposome-mediated gene delivery to respiratory epithelial cells in vitro. Hum. Gene Ther. 1997, 8, 431–438. [Google Scholar] [CrossRef] [PubMed]
- De Backer, L.; Braeckmans, K.; Demeester, J.; de Smedt, S.C.; Raemdonck, K. The influence of natural pulmonary surfactant on the efficacy of siRNA-loaded dextran nanogels. Nanomedicine 2013, 8, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Benfer, M.; Kissel, T. Cellular uptake mechanism and knockdown activity of siRNA-loaded biodegradable DEAPA-PVA-g-PLGA nanoparticles. Eur. J. Pharm. Biopharm. 2012, 80, 247–256. [Google Scholar] [CrossRef] [PubMed]
- De Backer, L.; Braeckmans, K.; Stuart, M.C.; Demeester, J.; De Smedt, S.C.; Raemdonck, K. Bio-inspired pulmonary surfactant-modified nanogels: A promising siRNA delivery system. J. Control. Release 2015, 206, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Haupenthal, J.; Baehr, C.; Kiermayer, S.; Zeuzem, S.; Piiper, A. Inhibition of RNAse A family enzymes prevents degradation and loss of silencing activity of siRNAs in serum. Biochem. Pharmacol. 2006, 71, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Lipka, J.; Semmler-Behnke, M.; Wenk, A.; Burkhardt, J.; Aigner, A.; Kreyling, W. Biokinetic studies of non-complexed siRNA versus nano-sized PEI F25-LMW/siRNA polyplexes following intratracheal instillation into mice. Int. J. Pharm. 2016, 500, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Man, D.K.; Chow, M.Y.; Casettari, L.; Gonzalez-Juarrero, M.; Lam, J.K. Potential and development of inhaled RNAi therapeutics for the treatment of pulmonary tuberculosis. Adv. Drug Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. Cold Spring Harb. Perspect. Biol. 2014, 6, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Harush-Frenkel, O.; Debotton, N.; Benita, S.; Altschuler, Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem. Biophys. Res. Commun. 2007, 353, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Scales, S.J.; Reilly, D.E. DNA internalized via caveolae requires microtubule-dependent, Rab7-independent transport to the late endocytic pathway for delivery to the nucleus. J. Biol. Chem. 2007, 282, 22953–22963. [Google Scholar] [CrossRef] [PubMed]
- Khalil, I.A.; Kogure, K.; Futaki, S.; Harashima, H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J. Biol. Chem. 2006, 281, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, D.; Piest, M.; van der Aa, L.J.; al Soraj, M.; Jones, A.T.; Engbersen, J.F.; de Smedt, S.C.; Braeckmans, K. Flotillin-dependent endocytosis and a phagocytosis-like mechanism for cellular internalization of disulfide-based poly(amido amine)/DNA polyplexes. Biomaterials 2011, 32, 3072–3084. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, D.; Rejman, J.; Martens, T.F.; Demeester, J.; de Smedt, S.C.; Braeckmans, K. On the cellular processing of non-viral nanomedicines for nucleic acid delivery: Mechanisms and methods. J. Control. Release 2012, 161, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Zaki, N.M.; Tirelli, N. Gateways for the intracellular access of nanocarriers: A review of receptor-mediated endocytosis mechanisms and of strategies in receptor targeting. Expert Opin. Drug Deliv. 2010, 7, 895–913. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stoter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: SiRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Ma, D. Enhancing endosomal escape for nanoparticle mediated siRNA delivery. Nanoscale 2014, 6, 6415–6425. [Google Scholar] [CrossRef] [PubMed]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Lam, J.K. Endosomal Escape Pathways for Non-Viral Nucleic Acid Delivery Systems. In Molecular Regulation of Endocytosis; Chapter 17; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Dean, D.; Strong, D.; Zimmer, W. Nuclear entry of nonviral vectors. Gene Ther. 2005, 12, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, M.; Frijlink, H.; Hinrichs, W. Pulmonary administration of small interfering RNA: The route to go? J. Control. Release 2016, 235, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Youngren-Ortiz, S.R.; Gandhi, N.S.; España-Serrano, L.; Chougule, M.B. Aerosol delivery of siRNA to the lungs. Part 2: Nanocarrier-based delivery systems. Kona 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Satterlee, A.; Huang, L. In vivo gene delivery by nonviral vectors: Overcoming hurdles & quest. Mol. Ther. 2012, 20, 1298–1304. [Google Scholar] [PubMed]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Couto, L.B.; High, K.A. Viral vector-mediated RNA interference. Curr. Opin. Pharmacol. 2010, 10, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Tang, Q.; Gruntman, A.; Blomenkamp, K.; Teckman, J.; Song, L.; Zamore, P.D.; Flotte, T.R. Sustained miRNA-mediated knockdown of mutant AAT with simultaneous augmentation of wild-type AAT has minimal effect on global liver miRNA profiles. Mol. Ther. 2012, 20, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.J.; Li, W.M.; Liu, D.; Zhnag, W. Construction of sh-rpS6 lentivirus vectors and its effect on proliferation in lung adenocarcinoma A549 cell lines. Sichuan Da Xue Xue Bao Yi Xue Ban 2014, 45, 293–298. [Google Scholar] [PubMed]
- Liu, X.; Li, G.; Zhang, B.; Wang, L.; Li, X.H.; Li, X.P. Influence of suppression of Epstein-Barr Virus-encoded latent membrane protein 1 by rAAV vector mediated RNA interference on metastatic ability of nasopharyngeal cancer cells in vivo. Zhonghua Zhong Liu Za Zhi 2009, 31, 324–329. [Google Scholar] [PubMed]
- Wu, C.J.; Huang, W.C.; Chen, L.C.; Shen, C.R.; Kuo, M.L. Pseudotyped adeno-associated virus 2/9-delivered CCL11 shRNA alleviates lung inflammation in an allergen-sensitized mouse model. Hum. Gene Ther. 2012, 23, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Youngren-Ortiz, S.R.; Gandhi, N.S.; España-Serrano, L.; Chougule, M.B. Aerosol delivery of siRNA to the lungs. Part 1: Rationale for gene delivery systems. Kona 2016, 33, 63–85. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhi, D.; Huang, L. Lipid-based vectors for siRNA delivery. J. Drug Target. 2012, 20, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-C.; Lai, S.; Guo, X.; Zhang, X.; de Crombrugghe, B.; Sonnylal, S.; Arnett, F.C.; Zhou, X. Attenuation of fibrosis in vitro and in vivo with SPARC siRNA. Arthritis Res. Ther. 2010, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Dalby, B.; Cates, S.; Harris, A.; Ohki, E.C.; Tilkins, M.L.; Price, P.J.; Ciccarone, V.C. Advanced transfection with Lipofectamine 2000 reagent: Primary neurons, siRNA, and high-throughput applications. Methods 2004, 33, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Reich, S.J.; Fosnot, J.; Kuroki, A.; Tang, W.; Yang, X.; Maguire, A.M.; Bennett, J.; Tolentino, M.J. Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. Mol. Vis. 2003, 9, 210–216. [Google Scholar] [PubMed]
- Vornlocher, H.-P. Antibody-directed cell-type-specific delivery of siRNA. Trends Mol. Med. 2006, 12, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Neuberg, P.; Kichler, A. Recent developments in nucleic acid delivery with polyethylenimines. Adv. Genet. 2014, 88, 263–288. [Google Scholar] [PubMed]
- Höbel, S.; Aigner, A. Polyethylenimines for siRNA and miRNA delivery in vivo. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 484–501. [Google Scholar] [PubMed]
- Nafee, N.; Taetz, S.; Schneider, M.; Schaefer, U.F.; Lehr, C.-M. Chitosan-coated PLGA nanoparticles for DNA/RNA delivery: Effect of the formulation parameters on complexation and transfection of antisense oligonucleotides. Nanomedicine 2007, 3, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, A.; Wang, H.; Pu, P.; Jiang, X.; Kang, C.; Chang, J. Tat-BMPs-PAMAM conjugates enhance therapeutic effect of small interference RNA on U251 glioma cells in vitro and in vivo. Hum. Gene Ther. 2010, 21, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Raemdonck, K.; Naeye, B.; Buyens, K.; Vandenbroucke, R.E.; Høgset, A.; Demeester, J.; de Smedt, S.C. Biodegradable dextran nanogels for RNA interference: Focusing on endosomal escape and intracellular siRNA delivery. Adv. Funct. Mater. 2009, 19, 1406–1415. [Google Scholar] [CrossRef]
- Howard, K.A.; Rahbek, U.L.; Liu, X.; Damgaard, C.K.; Glud, S.Z.; Andersen, M.Ø.; Hovgaard, M.B.; Schmitz, A.; Nyengaard, J.R.; Besenbacher, F. RNA interference in vitro and in vivo using a chitosan/siRNA nanoparticle system. Mol. Ther. 2006, 14, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Kwok, P.C.; Chow, M.Y.; Tang, P.; Mason, A.J.; Chan, H.-K.; Lam, J.K. Formulation of pH responsive peptides as inhalable dry powders for pulmonary delivery of nucleic acids. Eur. J. Pharm. Biopharm. 2014, 86, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.S.; Qin, B.; Cheng, K. Peptides used in the delivery of small noncoding RNA. Mol. Pharm. 2014, 11, 3395–3408. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tsui, T.Y.; Ma, W. Intracellular delivery of molecular cargo using cell-penetrating peptides and the combination strategies. Int. J. Mol. Sci. 2015, 16, 19518–19536. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Choi, Y.H.; Kim, S.W. Polyester Analogue of Poly-l-Lysine as a Soluble, Biodegradable Gene Delivery Carrier. U.S. Patent 6,217,912, 17 April 2001. [Google Scholar]
- Hartono, S.B.; Gu, W.; Kleitz, F.; Liu, J.; He, L.; Middelberg, A.P.; Yu, C.; Lu, G.Q.; Qiao, S.Z. Poly-l-lysine functionalized large pore cubic mesostructured silica nanoparticles as biocompatible carriers for gene delivery. ACS Nano 2012, 6, 2104–2117. [Google Scholar] [CrossRef] [PubMed]
- Del Sorbo, L.; Costamagna, A.; Muraca, G.; Rotondo, G.; Civiletti, F.; Vizio, B.; Bosco, O.; Martin Conte, E.L.; Frati, G.; Delsedime, L.; et al. Intratracheal administration of small interfering RNA targeting fas reduces lung ischemia-reperfusion injury. Crit. Care Med. 2016, 44, e604–e613. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Kumar, M.; Negi, V.; Pattnaik, B.; Prakash, Y.S.; Agrawal, A.; Ghosh, B. MicroRNA-326 regulates profibrotic functions of transforming growth factor-β in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2014, 50, 882–892. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Takeshita, F.; Kuwano, K.; Ochiya, T. RNAi therapeutic platforms for lung diseases. Pharmaceuticals 2013, 6, 223–250. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Popat, S.; Reinmuth, N.; de Ruysscher, D.; Kerr, K.; Peters, S. Metastatic non-small-cell lung cancer (NSCLC): ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Gootenberg, J.; Keegan, P.; Pazdur, R. FDA drug approval summary: Bevacizumab (Avastin®) plus carboplatin and paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist 2007, 12, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Johnson, J.R.; Chattopadhyay, S.; Tang, S.; Justice, R.; Sridhara, R.; Pazdur, R. Approval summary: Erlotinib maintenance therapy of advanced/metastatic non-small cell lung cancer (NSCLC). Oncologist 2010, 15, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA drug approval summary: Gefitinib (ZD1839)(Iressa®) tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Landesman-Milo, D.; Ramishetti, S.; Peer, D. Nanomedicine as an emerging platform for metastatic lung cancer therapy. Cancer Metastasis Rev. 2015, 34, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Park, S.-J.; Lee, S.; Cho, C.S.; Cho, M.-H. Aerosol gene delivery using viral vectors and cationic carriers for in vivo lung cancer therapy. Expert Opin. Drug Deliv. 2015, 12, 977–991. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Clark, A.S.; Ni, Y.; Dennis, P.A. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar] [PubMed]
- Hong, S.H.; Lee, J.H.; Jiang, H.L.; Kim, J.E.; Lee, A.Y.; Kim, S.; Cho, C.S.; Cho, M.H. Dual expression of shAkt1 and Pdcd4 suppresses lung tumorigenesis in K-rasLA1 mice. Anticancer Res. 2015, 35, 2015–2019. [Google Scholar] [PubMed]
- Hong, S.-H.; Kim, J.-E.; Kim, Y.-K.; Minai-Tehrani, A.; Shin, J.-Y.; Kang, B.; Kim, H.-J.; Cho, C.-S.; Chae, C.; Jiang, H.-L. Suppression of lung cancer progression by biocompatible glycerol triacrylate-spermine-mediated delivery of shAkt1. Int. J. Nanomed. 2012, 7, 2293–2306. [Google Scholar]
- Hwang, S.-K.; Chang, S.-H.; Minai-Tehrani, A.; Kim, Y.-S.; Cho, M.-H. Lentivirus-AIMP2-DX2 shRNA suppresses cell proliferation by regulating Akt1 signaling pathway in the lungs of AIMP2+/–Mice. J. Aerosol. Med. Pulm. Drug Deliv. 2013, 26, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Li, Y.; Eames, I.; Chan, P.; Ridgway, G. Factors involved in the aerosol transmission of infection and control of ventilation in healthcare premises. J. Hosp. Infect. 2006, 64, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, P.; Zhang, J.; Tian, H.; Park, K.; Chen, X. Pulmonary codelivery of doxorubicin and siRNA by pH-sensitive nanoparticles for therapy of metastatic lung cancer. Small 2015, 11, 4321–4333. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Takeshita, F.; Mizutani, T.; Ohgi, T.; Kuwano, K.; Ochiya, T. A novel platform to enable inhaled naked RNAi medicine for lung cancer. Sci. Rep. 2013, 3, 3325. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Tian, F.; Hernández, Y.; Bao, C.; Cui, D.; Janssen, K.-P.; Ibarra, M.R.; Baptista, P.V.; Stoeger, T.; Jesús, M. In vivo tumor targeting via nanoparticle-mediated therapeutic siRNA coupled to inflammatory response in lung cancer mouse models. Biomaterials 2013, 34, 7744–7753. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-H.; Minai-Tehrani, A.; Chang, S.-H.; Jiang, H.-L.; Lee, S.; Lee, A.-Y.; Seo, H.W.; Chae, C.; Beck, G.R., Jr.; Cho, M.-H. Knockdown of the sodium-dependent phosphate co-transporter 2b (NPT2b) suppresses lung tumorigenesis. PLoS ONE 2013, 8, e77121. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Kito, D.; Oiwa, A.; Fukushima, M.; Hira, D.; Okamoto, H. Gene silencing in a mouse lung metastasis model by an inhalable dry small interfering RNA powder prepared using the supercritical carbon dioxide technique. Biol. Pharm. Bull. 2013, 36, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Taratula, O.; Kuzmov, A.; Shah, M.; Garbuzenko, O.B.; Minko, T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J. Control. Release 2013, 171, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.M.; Zhang, M.; Wei, D.; Stueber, D.; Taratula, O.; Minko, T.; He, H. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small 2009, 5, 2673–2677. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Iwao-Koizumi, K.; Takeshita, F.; Yamamoto, Y.; Yoshida, T.; Nishio, K.; Nagahara, S.; Kato, K.; Ochiya, T. RPN2 gene confers docetaxel resistance in breast cancer. Nat. Med. 2008, 14, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Yagishita, S.; Takeshita, F.; Yamamoto, Y.; Kuwano, K.; Ochiya, T. Prognostic and therapeutic impact of RPN2-mediated tumor malignancy in non-small-cell lung cancer. Oncotarget 2015, 6, 3335. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Oh, C.; Kim, I.-S.; Kwon, I.C.; Kim, S. Co-delivery of chemosensitizing siRNA and an anticancer agent via multiple monocomplexation-induced hydrophobic association. J. Control. Release 2015, 210, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, M.; Gong, P.; Deng, J.; Yi, H.; Zhang, P.; Zhang, Y.; Liu, P.; Ma, Y.; Cai, L. Polypeptide cationic micelles mediated co-delivery of docetaxel and siRNA for synergistic tumor therapy. Biomaterials 2013, 34, 3431–3438. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gu, W.; Chen, J.; Chen, W.; Xu, Z.P. Co-delivery of siRNAs and anti-cancer drugs using layered double hydroxide nanoparticles. Biomaterials 2014, 35, 3331–3339. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Reynolds, C.P. Bcl-2 inhibitors: Targeting mitochondrial apoptotic pathways in cancer therapy. Clin. Cancer Res. 2009, 15, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Young, L.C.; Campling, B.G.; Cole, S.P.; Deeley, R.G.; Gerlach, J.H. Multidrug resistance proteins MRP3, MRP1, and MRP2 in lung cancer correlation of protein levels with drug response and messenger RNA levels. Clin. Cancer Res. 2001, 7, 1798–1804. [Google Scholar] [PubMed]
- Koli, U.; Krishnan, R.A.; Pofali, P.; Jain, R.; Dandekar, P. siRNA-based therapies for pulmonary diseases. J. Biomed. Nanotechnol. 2014, 10, 1953–1997. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Becker, K.L.; Swinburne, A.J.; Nylen, E.S.; Formica, M.A.; Hennessey, P.A.; Criddle, M.M.; Peterson, D.R.; Baran, A.; Walsh, E.E. Bacterial complications of respiratory tract viral illness: A comprehensive evaluation. J. Infect. Dis. 2013, 208, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fraga, M.; Wright, N.; Jimenez, A. RNA interference-based therapeutics: New strategies to fight infectious disease. Infect. Disord. Drug Targets 2008, 8, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Musiyenko, A.; Shulyayeva, O.; Barik, S. Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 2005, 11, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, R.; Elbashir, S.; Borland, T.; Toudjarska, I.; Hadwiger, P.; John, M.; Roehl, I.; Morskaya, S.S.; Martinello, R.; Kahn, J. RNA interference-mediated silencing of the respiratory syncytial virus nucleocapsid defines a potent antiviral strategy. Antimicrob. Agents Chemother. 2009, 53, 3952–3962. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Filip, L.; Bai, A.; Nguyen, T.; Eisen, H.N.; Chen, J. Inhibition of influenza virus production in virus-infected mice by RNA interference. Proc. Natl. Acad. Sci. USA 2004, 101, 8676–8681. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, S.M.; Lo, C.Y.; Tumpey, T.M.; Epstein, S.L. Protection against lethal influenza virus challenge by RNA interference in vivo. Proc. Natl. Acad. Sci. USA 2004, 101, 8682–8686. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Lu, J.J.; Ge, Q.; Zhang, C.; Chen, J.; Klibanov, A.M. Full deacylation of polyethylenimine dramatically boosts its gene delivery efficiency and specificity to mouse lung. Proc. Natl. Acad. Sci. USA 2005, 102, 5679–5684. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Jin, M.; Yu, Z.; Xu, X.; Peng, Y.; Wu, H.; Liu, J.; Liu, H.; Cao, S.; Chen, H. Effective small interfering RNAs targeting matrix and nucleocapsid protein gene inhibit influenza A virus replication in cells and mice. Antiviral Res. 2007, 76, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; He, H.; Wu, Y.; Duan, M. RNA interference of avian influenza virus H5N1 by inhibiting viral mRNA with siRNA expression plasmids. J. Biotechnol. 2008, 135, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Taraco, A.G.; Higgins, D.M.; Sánchez-Campillo, J.; Lee, E.J.; Orme, I.M.; González-Juarrero, M. Intrapulmonary delivery of XCL1-targeting small interfering RNA in mice chronically infected with Mycobacterium tuberculosis. Am. J. Respir. Cell Mol. 2009, 41, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Bitko, V.; Barik, S. Phenotypic silencing of cytoplasmic genes using sequence-specific double-stranded short interfering RNA and its application in the reverse genetics of wild type negative-strand RNA viruses. BMC Microbiol. 2001, 1, 34. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Eisen, H.N.; Chen, J. Use of siRNAs to prevent and treat influenza virus infection. Virus Res. 2004, 102, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Stoppani, E.; Bassi, I.; Dotti, S.; Lizier, M.; Ferrari, M.; Lucchini, F. Expression of a single siRNA against a conserved region of NP gene strongly inhibits in vitro replication of different Influenza A virus strains of avian and swine origin. Antiviral Res. 2015, 120, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Li, B.-J.; Tang, Q.; Cheng, D.; Qin, C.; Xie, F.Y.; Wei, Q.; Xu, J.; Liu, Y.; Zheng, B.-J.; Woodle, M.C. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat. Med. 2005, 11, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Ralston, S.L.; Lieberthal, A.S.; Meissner, H.C.; Alverson, B.K.; Baley, J.E.; Gadomski, A.M.; Johnson, D.W.; Light, M.J.; Maraqa, N.F.; Mendonca, E.A. Clinical practice guideline: The diagnosis, management, and prevention of bronchiolitis. Pediatrics 2014, 134, e1474–e1502. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Hennessey, P.A.; Formica, M.A.; Cox, C.; Walsh, E.E. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005, 352, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.; Dormitzer, P.; Nokes, D.J.; Rappuoli, R.; Roca, A.; Graham, B. Strategic priorities for respiratory syncytial virus (RSV) vaccine development. Vaccine 2013, 31, B209–B215. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.; Cehelsky, J.E.; Alvarez, R.; Elbashir, S.; Harborth, J.; Toudjarska, I.; Nechev, L.; Murugaiah, V.; van Vliet, A.; Vaishnaw, A.K. Evaluation of the safety, tolerability and pharmacokinetics of ALN-RSV01, a novel RNAi antiviral therapeutic directed against respiratory syncytial virus (RSV). Antiviral Res. 2008, 77, 225–231. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2010, 107, 8800–8805. [Google Scholar] [CrossRef] [PubMed]
- Alnylam Pharmaceuticals, Inc. Complete Results of Our ALN-RSV01 Phase IIb Study. Available online: http://www.alnylam.com/capella/presentations/complete-results-of-our-aln-rsv01-phase-iib-study/ (accessed on 9 April 2012).
- Medina, R.A.; García-Sastre, A. Influenza A viruses: New research developments. Nat. Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Peng, C.; Luo, J.; Wang, C.; Han, L.; Wu, B.; Ji, G.; He, H. Adamantane-resistant influenza a viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS ONE 2015, 10, e0119115. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Pizzorno, A.; Abed, Y.; Boivin, G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res. 2013, 98, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; McManus, M.T.; Nguyen, T.; Shen, C.-H.; Sharp, P.A.; Eisen, H.N.; Chen, J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc. Natl. Acad. Sci. USA 2003, 100, 2718–2723. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, A.; Cruz, A.; Pérez-Gil, J. Barrier or carrier? Pulmonary surfactant and drug delivery. Eur. J. Pharm. Biopharm. 2015, 95, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Khantasup, K.; Kopermsub, P.; Chaichoun, K.; Dharakul, T. Targeted small interfering RNA-immunoliposomes as a promising therapeutic agent against highly pathogenic Avian Influenza A (H5N1) virus infection. Antimicrob. Agents Chemother. 2014, 58, 2816–2824. [Google Scholar] [CrossRef] [PubMed]
- Sui, H.-Y.; Zhao, G.-Y.; Huang, J.-D.; Jin, D.-Y.; Yuen, K.-Y.; Zheng, B.-J. Small interfering RNA targeting M2 gene induces effective and long term inhibition of influenza A virus replication. PLoS ONE 2009, 4, e5671. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Chow, M.Y.; Lau, P.N.; Zhou, Q.T.; Kwok, P.C.; Leung, G.P.; Mason, A.J.; Chan, H.-K.; Poon, L.L.; Lam, J.K. Inhalable dry powder formulations of siRNA and pH-responsive peptides with antiviral activity against H1N1 influenza virus. Mol. Pharm. 2015, 12, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Robbins, M.; Judge, A.; Ambegia, E.; Choi, C.; Yaworski, E.; Palmer, L.; McClintock, K.; MacLachlan, I. Misinterpreting the therapeutic effects of small interfering RNA caused by immune stimulation. Hum. Gene Ther. 2008, 19, 991–999. [Google Scholar] [CrossRef] [PubMed]
- WHO World Health Organization: Tuberculosis—Global TB Programme Core Functions. Available online: http://www.who.int/tb/publications/GTBcorporate_factsheet.pdf?ua=1 (accessed on 1 November 2015).
- Kathania, M.; Raje, C.I.; Raje, M.; Dutta, R.K.; Majumdar, S. Bfl-1/A1 acts as a negative regulator of autophagy in mycobacteria infected macrophages. Int. J. Biochem. Cell Biol. 2011, 43, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.A.; Chua, J.; Kyei, G.B.; Deretic, V. Higher order Rab programming in phagolysosome biogenesis. J. Cell Biol. 2006, 174, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, K.; Zhou, L.; Wu, Y.; Zhu, M.; Lai, X.; Chen, T.; Feng, L.; Li, M.; Huang, C. MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PLoS Pathog. 2013, 9, e1003697. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Yuk, J.-M.; Kim, S.Y.; Kim, T.S.; Jin, H.S.; Yang, C.-S.; Jo, E.-K. MicroRNA-125a inhibits autophagy activation and antimicrobial responses during mycobacterial infection. J. Immunol. 2015, 194, 5355–5365. [Google Scholar] [CrossRef] [PubMed]
- Behar, S.M.; Baehrecke, E.H. Tuberculosis: Autophagy is not the answer. Nature 2015, 528, 482–483. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, L. Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 2012, 12, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Silva Miranda, M.; Breiman, A.; Allain, S.; Deknuydt, F.; Altare, F. The tuberculous granuloma: An unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clin. Dev. Immunol. 2012, 139127. [Google Scholar] [CrossRef] [PubMed]
- Ordway, D.; Higgins, D.M.; Sanchez-Campillo, J.; Spencer, J.S.; Henao-Tamayo, M.; Harton, M.; Orme, I.M.; Juarrero, M.G. XCL1 (lymphotactin) chemokine produced by activated CD8 T cells during the chronic stage of infection with Mycobacterium tuberculosis negatively affects production of IFN-γ by CD4 T cells and participates in granuloma stability. J. Leukoc. Biol. 2007, 82, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Toossi, Z.; Gogate, P.; Shiratsuchi, H.; Young, T.; Ellner, J.J. Enhanced production of TGF-beta by blood monocytes from patients with active tuberculosis and presence of TGF-beta in tuberculous granulomatous lung lesions. J. Immunol. 1995, 154, 465–473. [Google Scholar] [PubMed]
- Bonecini-Almeida, M.G.; Ho, J.L.; Boéchat, N.; Huard, R.C.; Chitale, S.; Doo, H.; Geng, J.; Rego, L.; Lazzarini, L.C.O.; Kritski, A.L. Down-modulation of lung immune responses by interleukin-10 and transforming growth factor β (TGF-β) and analysis of TGF-β receptors I and II in active tuberculosis. Infect. Immun. 2004, 72, 2628–2634. [Google Scholar] [CrossRef] [PubMed]
- Perl, M.; Lomas-Neira, J.; Venet, F.; Chung, C.-S.; Ayala, A. Pathogenesis of indirect (secondary) acute lung injury. Expert Rev. Respir. Med. 2011, 5, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Immunology of asthma and chronic obstructive pulmonary disease. Nat. Rev. Immunol. 2008, 8, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.; Dancer, R.C.; Thickett, D.R. Acute lung injury. Clin. Med. 2011, 11, 615–618. [Google Scholar] [CrossRef]
- Khaitov, M.R.; Shilovskiy, I.P.; Nikonova, A.A.; Shershakova, N.N.; Kamyshnikov, O.Y.; Babakhin, A.A.; Zverev, V.V.; Johnston, S.L.; Khaitov, R.M. Small interfering RNAs targeted to Interleukin-4 and Respiratory Syncytial Virus reduce airway inflammation in a mouse model of virus-induced asthma exacerbation. Hum. Gene Ther. 2014, 25, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Darcan-Nicolaisen, Y.; Meinicke, H.; Fels, G.; Hegend, O.; Haberland, A.; Kühl, A.; Loddenkemper, C.; Witzenrath, M.; Kube, S.; Henke, W. Small interfering RNA against transcription factor STAT6 inhibits allergic airway inflammation and hyperreactivity in mice. J. Immunol. 2009, 182, 7501–7508. [Google Scholar] [CrossRef] [PubMed]
- Takyar, S.; Vasavada, H.; Zhang, J.-G.; Ahangari, F.; Niu, N.; Liu, Q.; Lee, C.G.; Cohn, L.; Elias, J.A. VEGF controls lung Th2 inflammation via the miR-1-Mpl (myeloproliferative leukemia virus oncogene)-P-selectin axis. J. Exp. Med. 2013, 210, 1993–2010. [Google Scholar] [CrossRef] [PubMed]
- Goh, F.Y.; Cook, K.L.; Upton, N.; Tao, L.; Lah, L.C.; Leung, B.P.; Wong, W.F. Receptor-interacting protein 2 gene silencing attenuates allergic airway inflammation. J. Immunol. 2013, 191, 2691–2699. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Ahmad, T.; Sharma, A.; Mabalirajan, U.; Kulshreshtha, A.; Agrawal, A.; Ghosh, B. Let-7 microRNA-mediated regulation of IL-13 and allergic airway inflammation. J. Allergy Clin. Immunol. 2011, 128, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-C.; Huang, H.-Y.; Chiang, B.-L. Lentiviral-mediated GATA-3 RNAi decreases allergic airway inflammation and hyperresponsiveness. Mol. Ther. 2008, 16, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Asai-Tajiri, Y.; Matsumoto, K.; Fukuyama, S.; Kan-o, K.; Nakano, T.; Tonai, K.; Ohno, T.; Azuma, M.; Inoue, H.; Nakanishi, Y. Small interfering RNA against CD86 during allergen challenge blocks experimental allergic asthma. Respir. Res. 2014, 15, 132. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-Y.; Kim, M.-K.; Kim-Han, T.-H.; Indik, Z.K.; Schreiber, A.D. Effect of locally administered Syk siRNA on allergen-induced arthritis and asthma. Mol. Immunol. 2013, 53, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chen, H.; Li, Y.-M.; Wang, S.-Y.; Diao, X.; Liu, K.-G. Intranasal sirna targeting c-kit reduces airway inflammation in experimental allergic asthma. Int. J. Clin. Exp. Pathol. 2014, 7, 5505–5514. [Google Scholar] [PubMed]
- Xie, Y.; Kim, N.H.; Nadithe, V.; Schalk, D.; Thakur, A.; Kılıç, A.; Lum, L.G.; Bassett, D.J.; Merkel, O.M. Targeted delivery of siRNA to activated T cells via transferrin-polyethylenimine (Tf-PEI) as a potential therapy of asthma. J. Control. Release 2016, 229, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Lomas-Neira, J.; Perl, M.; Venet, F.; Chung, C.S.; Ayala, A. The role and source of tumor necrosis factor-α in hemorrhage-induced priming for septic lung injury. Shock 2012, 37, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Bartlett, N.W.; Clarke, D.; Birrell, M.; Belvisi, M.; Johnston, S.L. Targeting the NF-κB pathway in asthma and chronic obstructive pulmonary disease. Pharmacol. Ther. 2009, 121, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.E. Asthma: Defining of the persistent adult phenotypes. Lancet 2006, 368, 804–813. [Google Scholar] [CrossRef]
- WHO. The Global Asthma Report 2014. Available online: http://www.globalasthmareport.org/ (accessed on 30 October 2014).
- Chung, K.F.; Wenzel, S.E.; Brozek, J.L.; Bush, A.; Castro, M.; Sterk, P.J.; Adcock, I.M.; Bateman, E.D.; Bel, E.H.; Bleecker, E.R. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur. Respir. J. 2013, 43, 343–373. [Google Scholar] [CrossRef] [PubMed]
- Likhar, N.; Kanukula, R.; Mothe, R.; Vsn, M.; Dang, A. Investigating the value of omalizumab in the treatment of severe persistent allergic asthma: A systematic review of cost-effectiveness studies. Value Health 2016, 19, A115. [Google Scholar] [CrossRef]
- Khaitov, M.; Shilovskiy, I.; Nikonova, A.; Shershakova, N.; Kamyshnikov, O.; Babakhin, A.; Zverev, V.; Johnston, S.; Khaitov, R. siRNAs targeted to IL-4 and RSV reduce airway inflammation in a mouse model of virus-induced asthma exacerbation. Hum. Gene Ther. 2014, 25, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Merkel, O.M. Pulmonary delivery of siRNA via polymeric vectors as therapies of asthma. Arch. Pharm. 2015, 348, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.M.; Hessel, E.M. Functions of T cells in asthma: More than just TH2 cells. Nat. Rev. Immunol. 2010, 10, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Larché, M.; Robinson, D.S.; Kay, A.B. The role of T lymphocytes in the pathogenesis of asthma. J. Allergy Clin. Immunol. 2003, 111, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Ford, L.; Pearlman, D.; Spector, S.; Sher, L.; Skobieranda, F.; Wang, L.; Kirkesseli, S.; Rocklin, R.; Bock, B. Dupilumab in persistent asthma with elevated eosinophil levels. N. Engl. J. Med. 2013, 368, 2455–2466. [Google Scholar] [CrossRef] [PubMed]
- Hodsman, P.; Ashman, C.; Cahn, A.; de Boever, E.; Locantore, N.; Serone, A.; Pouliquen, I. A phase 1, randomized, placebo-controlled, dose-escalation study of an anti-IL-13 monoclonal antibody in healthy subjects and mild asthmatics. Br. J. Clin. Pharmacol. 2013, 75, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Brightling, C.E.; Chanez, P.; Leigh, R.; O’Byrne, P.M.; Korn, S.; She, D.; May, R.D.; Streicher, K.; Ranade, K.; Piper, E. Efficacy and safety of tralokinumab in patients with severe uncontrolled asthma: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 692–701. [Google Scholar] [CrossRef]
- Castro, M.; Wenzel, S.E.; Bleecker, E.R.; Pizzichini, E.; Kuna, P.; Busse, W.W.; Gossage, D.L.; Ward, C.K.; Wu, Y.; Wang, B. Benralizumab, an anti-interleukin 5 receptor α monoclonal antibody, versus placebo for uncontrolled eosinophilic asthma: A phase 2b randomised dose-ranging study. Lancet Respir. Med. 2014, 2, 879–890. [Google Scholar] [CrossRef]
- Kubo, M.; Inoue, H. Suppressor of cytokine signaling 3 (SOCS3) in Th2 cells evokes Th2 cytokines, IgE, and eosinophilia. Curr. Allergy Asthma Rep. 2006, 6, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W.; Lemanske, R.F.; Gern, J.E. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet 2010, 376, 826–834. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Gagliardo, R.; Chanez, P.; Mathieu, M.; Bruno, A.; Costanzo, G.; Gougat, C.; Vachier, I.; Bousquet, J.; Bonsignore, G.; Vignola, A.M. Persistent activation of nuclear factor–κB signaling pathway in severe uncontrolled asthma. Am. J. Respir. Crit. Care Med. 2003, 168, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Finotto, S.; de Sanctis, G.T.; Lehr, H.A.; Herz, U.; Buerke, M.; Schipp, M.; Bartsch, B.; Atreya, R.; Schmitt, E.; Galle, P.R. Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J. Exp. Med. 2001, 193, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.F. Inhibitors of the tyrosine kinase signaling cascade for asthma. Curr. Opin. Pharmacol. 2005, 5, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.F.; Leong, K.P. Tyrosine kinase inhibitors: A new approach for asthma. Biochim. Biophys. Acta Proteins Proteom. 2004, 1697, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Guntur, V.P.; Reinero, C.R. The potential use of tyrosine kinase inhibitors in severe asthma. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, S.; Koya, T.; Takeda, K.; Joetham, A.; Miyahara, N.; Pine, P.; Masuda, E.S.; Swasey, C.H.; Gelfand, E.W. Syk activation in dendritic cells is essential for airway hyperresponsiveness and inflammation. Am. J. Respir. Cell Mol. 2006, 34, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Stenton, G.R.; Ulanova, M.; Déry, R.E.; Merani, S.; Kim, M.-K.; Gilchrist, M.; Puttagunta, L.; Musat-Marcu, S.; James, D.; Schreiber, A.D. Inhibition of allergic inflammation in the airways using aerosolized antisense to Syk kinase. J. Immunol. 2002, 169, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.K.; Corey, D.R. Clinical status of duplex RNA. Bioorgan. Med. Chem. Lett. 2010, 20, 3203–3207. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.C.; Rossi, J.J.; Tiemann, K. Current progress of siRNA/shRNA therapeutics in clinical trials. Biotechnol. J. 2011, 6, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Hammad, H.; Lambrecht, B.N. Dendritic cells and epithelial cells: Linking innate and adaptive immunity in asthma. Nat. Rev. Immunol. 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Van Rijt, L.S.; Vos, N.; Willart, M.; KleinJan, A.; Coyle, A.J.; Hoogsteden, H.C.; Lambrecht, B.N. Essential role of dendritic cell CD80/CD86 costimulation in the induction, but not reactivation, of Th2 effector responses in a mouse model of asthma. J. Allergy Clin. Immunol. 2004, 114, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Nadithe, V.; Elsayed, M.; Merkel, O. Tracking and treating activated T cells. J. Drug Deliv. Sci. Technol. 2013, 23, 17–21. [Google Scholar] [CrossRef]
- Jaffar, Z.H.; Stanciu, L.; Pandit, A.; Lordan, J.; Holgate, S.T.; Roberts, K. Essential role for both CD80 and CD86 costimulation, but not CD40 interactions, in allergen-induced Th2 cytokine production from asthmatic bronchial tissue: Role for αβ, but not γδ, T cells. J. Immunol. 1999, 163, 6283–6291. [Google Scholar] [PubMed]
- Vestbo, J.; Hurd, S.S.; Agustí, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin. Chest Med. 2014, 35, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic aspects of chronic obstructive pulmonary disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S. Recent advances in pre-clinical mouse models of COPD. Clin. Sci. 2014, 126, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Velichko, S.; Thai, P.; Hung, L.-Y.; Huang, F.; Wu, R. Regulation of airway MUC5AC expression by IL-1β and IL-17A; the NF-κB paradigm. J. Immunol. 2009, 183, 6236–6243. [Google Scholar] [CrossRef] [PubMed]
- Ezzie, M.E.; Crawford, M.; Cho, J.-H.; Orellana, R.; Zhang, S.; Gelinas, R.; Batte, K.; Yu, L.; Nuovo, G.; Galas, D. Gene expression networks in COPD: MicroRNA and mRNA regulation. Thorax 2012, 67, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Bogaard, H.J.; Gomez-Arroyo, J.; Alhussaini, A.; Kraskauskas, D.; Cool, C.D.; Voelkel, N.F. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1α expression in lungs from patients with COPD. Chest 2012, 142, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; King, L.S.; Aggarwal, N.R.; de Gorordo, A.; D’Alessio, F.R.; Kubo, K. Acute lung injury review. Intern. Med. 2009, 48, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Perl, M.; Chung, C.-S.; Lomas-Neira, J.; Rachel, T.-M.; Biffl, W.L.; Cioffi, W.G.; Ayala, A. Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am. J. Pathol. 2005, 167, 1545–1559. [Google Scholar] [CrossRef]
- Patel, B.V.; Wilson, M.R.; O’Dea, K.P.; Takata, M. TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J. Immunol. 2013, 190, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Gorshkova, I.A.; Berdyshev, E.; He, D.; Fu, P.; Ma, W.; Su, Y.; Usatyuk, P.V.; Pendyala, S.; Oskouian, B. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am. J. Respir. Cell Mol. Biol. 2011, 45, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Holgate, S.T. Innate and adaptive immune responses in asthma. Nat. Med. 2012, 18, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zheng, Z.; Cohen, C.J.; Gattinoni, L.; Palmer, D.C.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. High-efficiency transfection of primary human and mouse T lymphocytes using RNA electroporation. Mol. Ther. 2006, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.; da Silva, C.A.; Frossard, N. Stem cell factor and its receptor c-Kit as targets for inflammatory diseases. Eur. J. Pharmacol. 2006, 533, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Maneechotesuwan, K.; Yao, X.; Ito, K.; Jazrawi, E.; Usmani, O.S.; Adcock, I.M.; Barnes, P.J. Suppression of GATA-3 nuclear import and phosphorylation: A novel mechanism of corticosteroid action in allergic disease. PLoS Med. 2009, 6, e1000076. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 2011, 208, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.; Wynn, T. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro-Gabazza, C.N.; Kobayashi, T.; Boveda-Ruiz, D.; Takagi, T.; Toda, M.; Gil-Bernabe, P.; Miyake, Y.; Yasukawa, A.; Matsuda, Y.; Suzuki, N.; et al. Development and preclinical efficacy of novel transforming growth factor-β1 short interfering RNAs for pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 46, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Sung, D.K.; Kong, W.H.; Park, K.; Kim, J.H.; Kim, M.Y.; Kim, H.; Hahn, S.K. Noncovalenly PEGylated CTGF siRNA/PDMAEMA complex for pulmonary treatment of bleomycin-induced lung fibrosis. Biomaterials 2013, 34, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-ß signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-M.; Park, J.W.; Cho, W.-K.; Zhou, Y.; Han, B.; Yoon, P.O.; Chae, J.; Elias, J.A.; Lee, C.G. Modifiers of TGF-β1 effector function as novel therapeutic targets of pulmonary fibrosis. Korean J. Intern. Med. 2014, 29, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lee, J.-Y.; Lee, C.-M.; Cho, W.-K.; Kang, M.-J.; Koff, J.L.; Yoon, P.-O.; Chae, J.; Park, H.-O.; Elias, J.A. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J. Biol. Chem. 2012, 287, 41991–42000. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.Y.T.; Lam, J.K.W. Dry powder formulation of plasmid DNA and siRNA for inhalation. Curr. Pharm. Des. 2015, 21, 3854–3866. [Google Scholar] [CrossRef] [PubMed]
- DeVincenzo, J.P. The promise, pitfalls and progress of RNA-interference-based antiviral therapy for respiratory viruses. Antivir. Ther. 2012, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Zamora, M.R.; Budev, M.; Rolfe, M.; Gottlieb, J.; Humar, A.; DeVincenzo, J.; Vaishnaw, A.; Cehelsky, J.; Albert, G.; Nochur, S. RNA interference therapy in lung transplant patients infected with respiratory syncytial virus. Am. Respir. Crit. Care Med. 2011, 183, 531–538. [Google Scholar] [CrossRef] [PubMed]
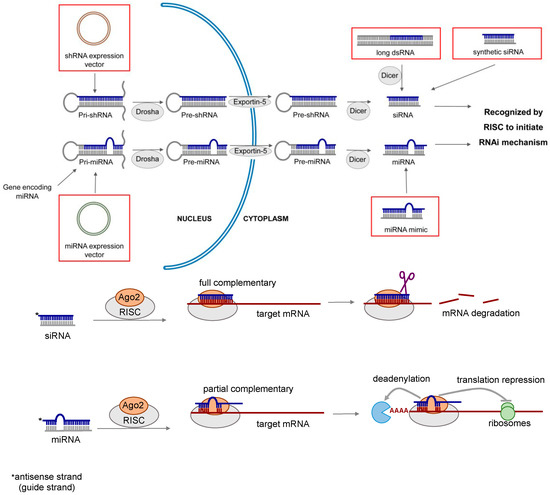
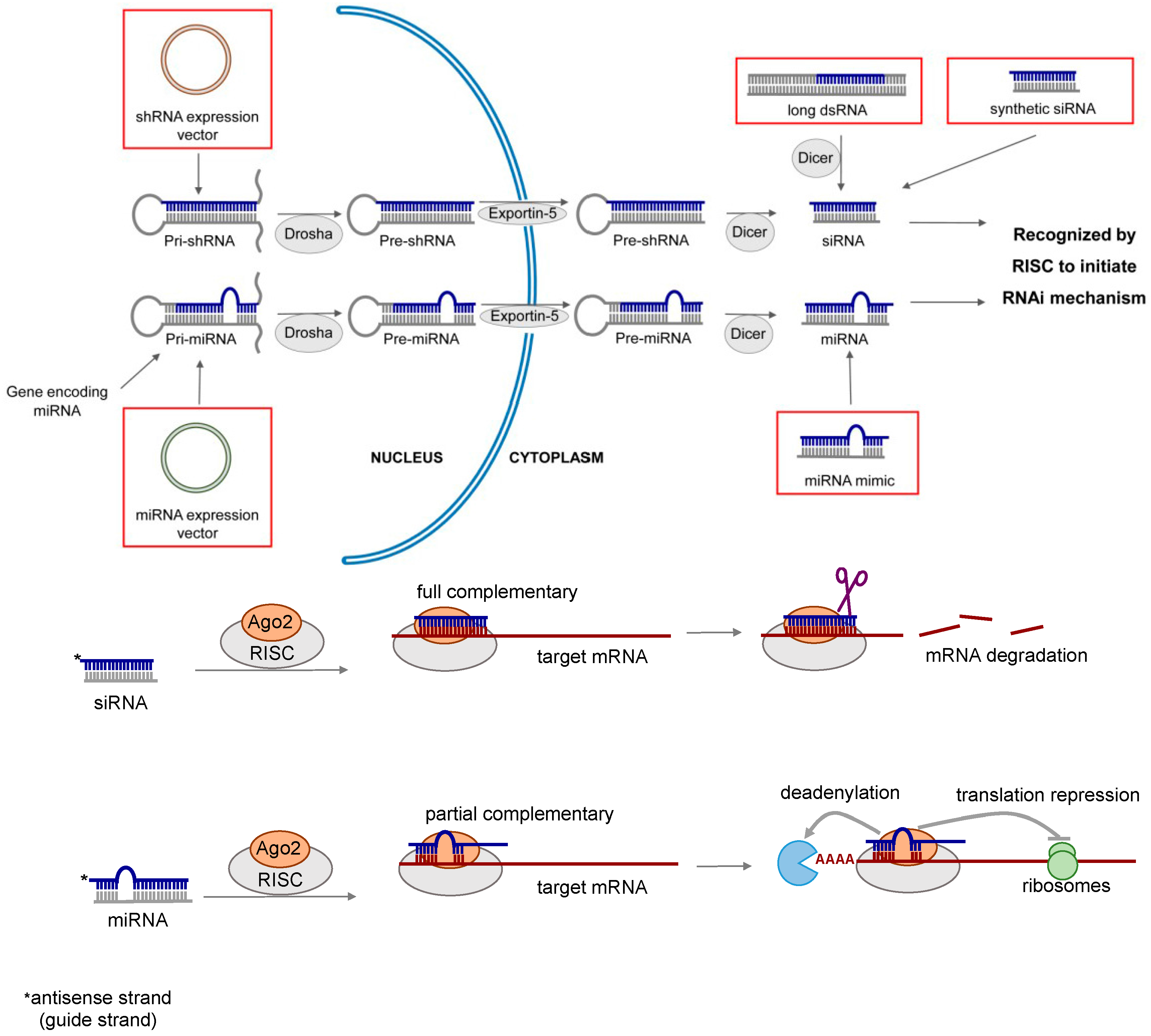
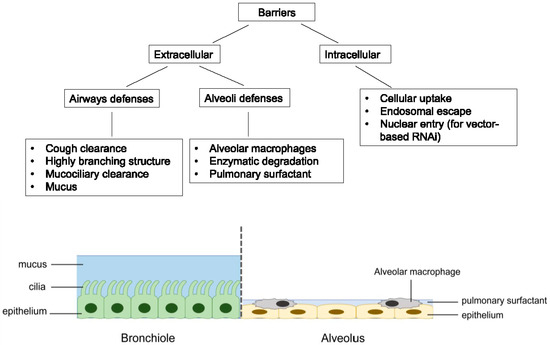
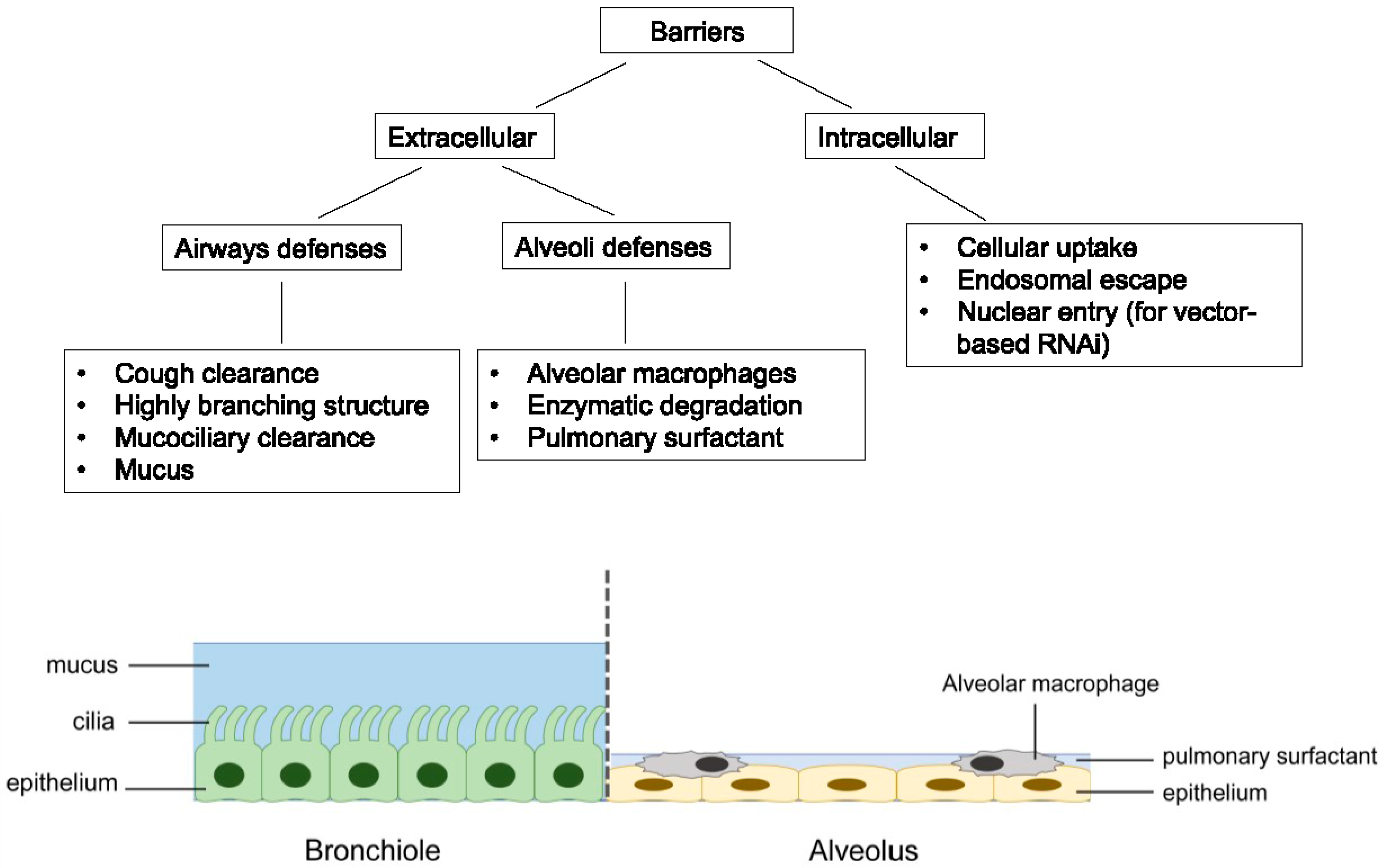
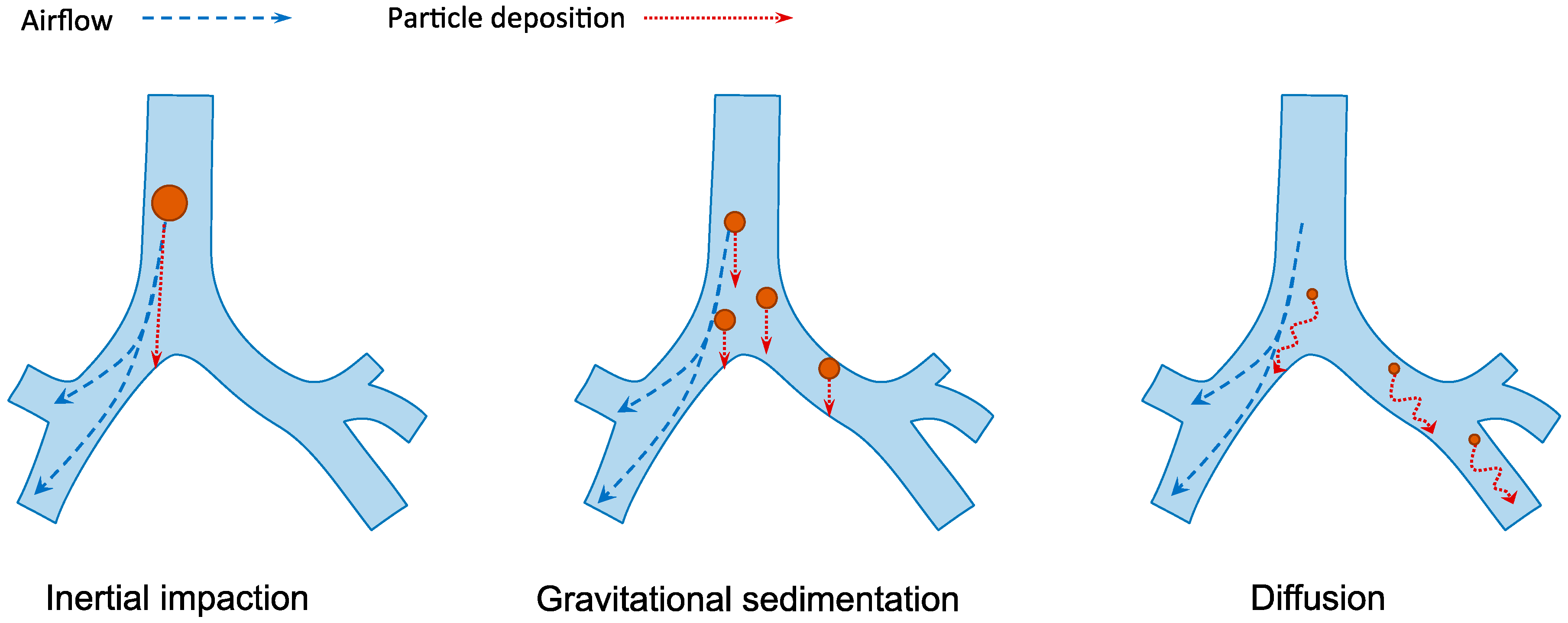

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).