
The Protective Effect of Melittin on Renal Fibrosis in an Animal Model of Unilateral Ureteral Obstruction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
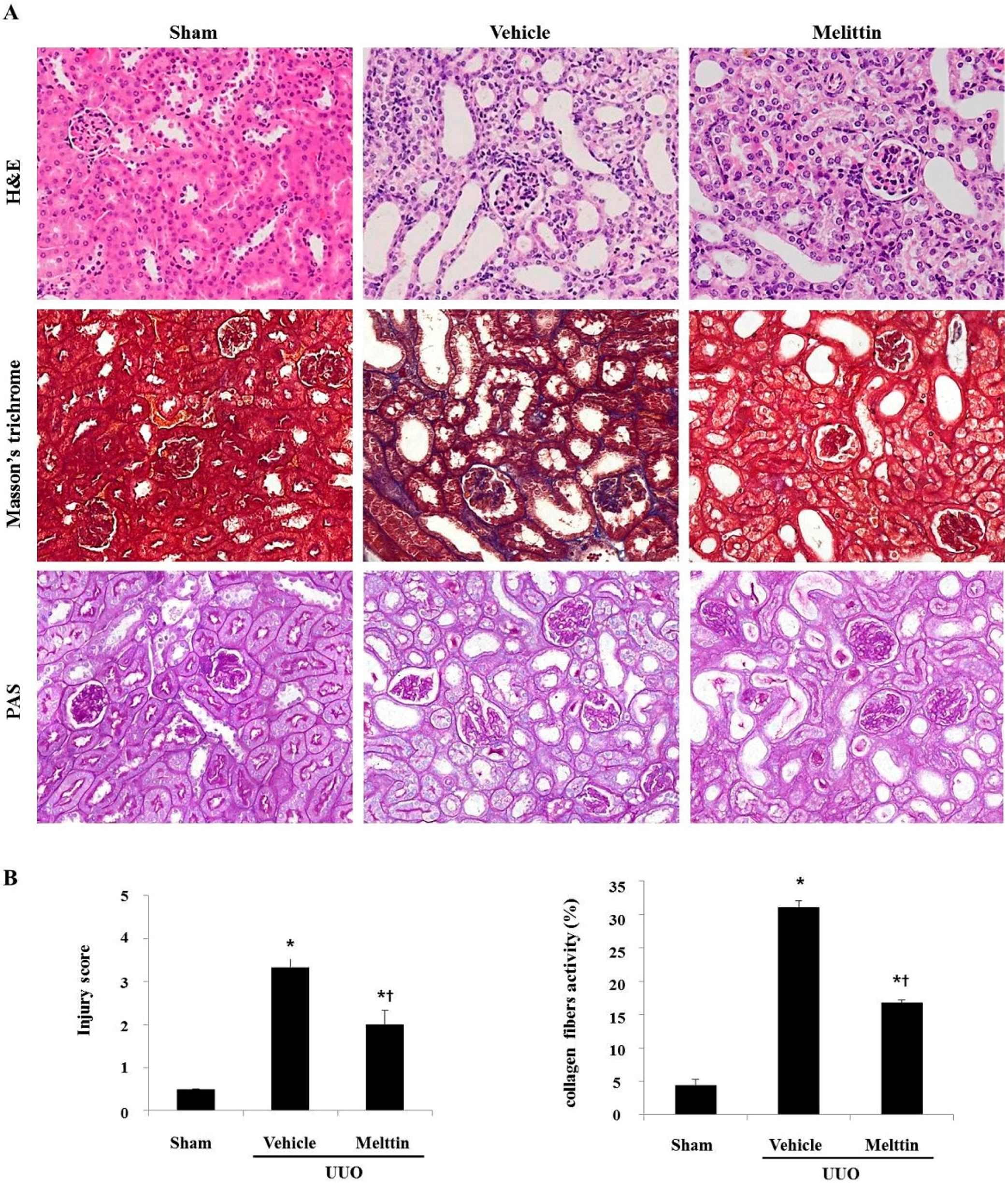
2.1. Melittin Attenuates Renal Interstitial Injury and Fibrosis in UUO Kidneys
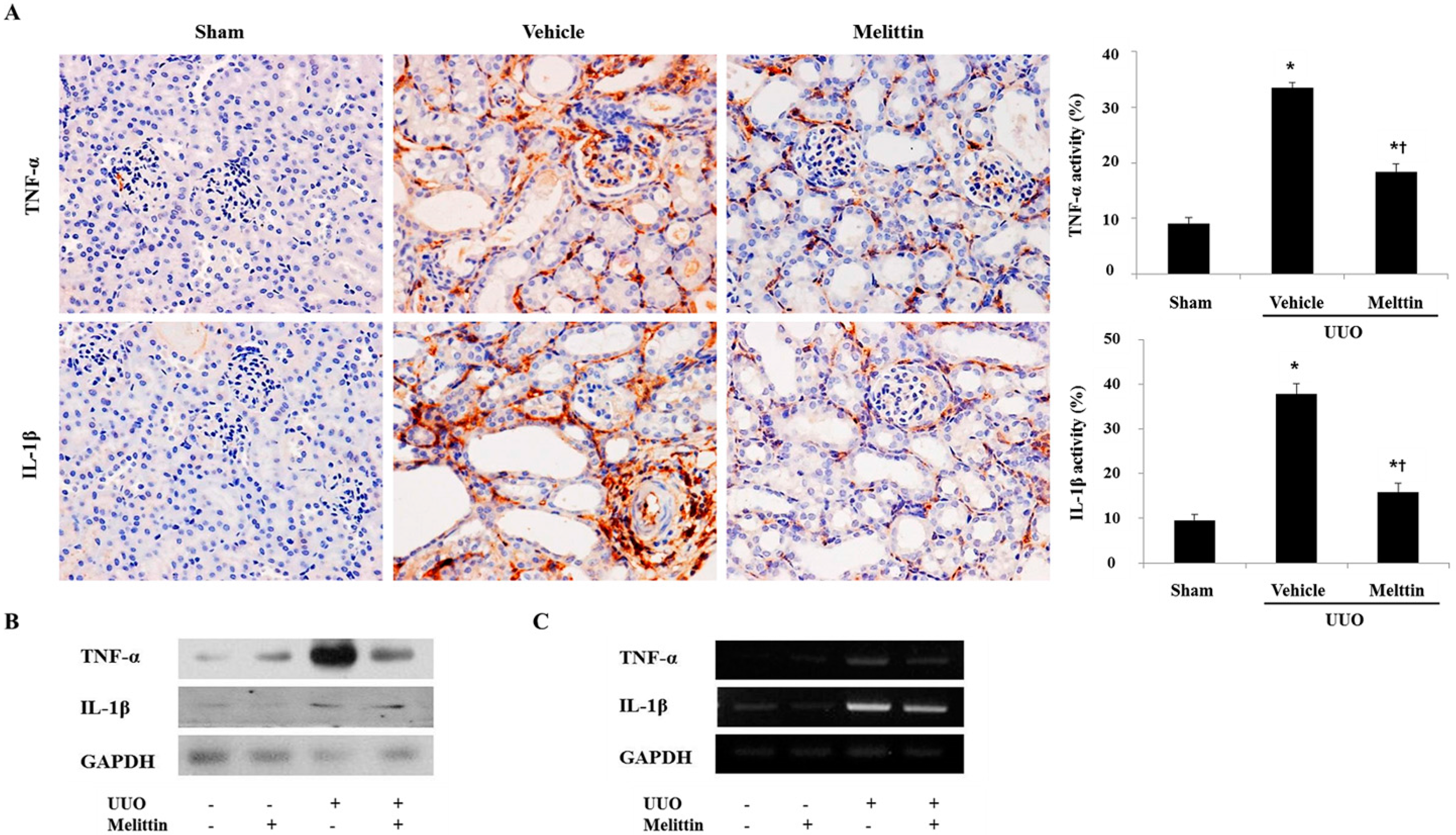
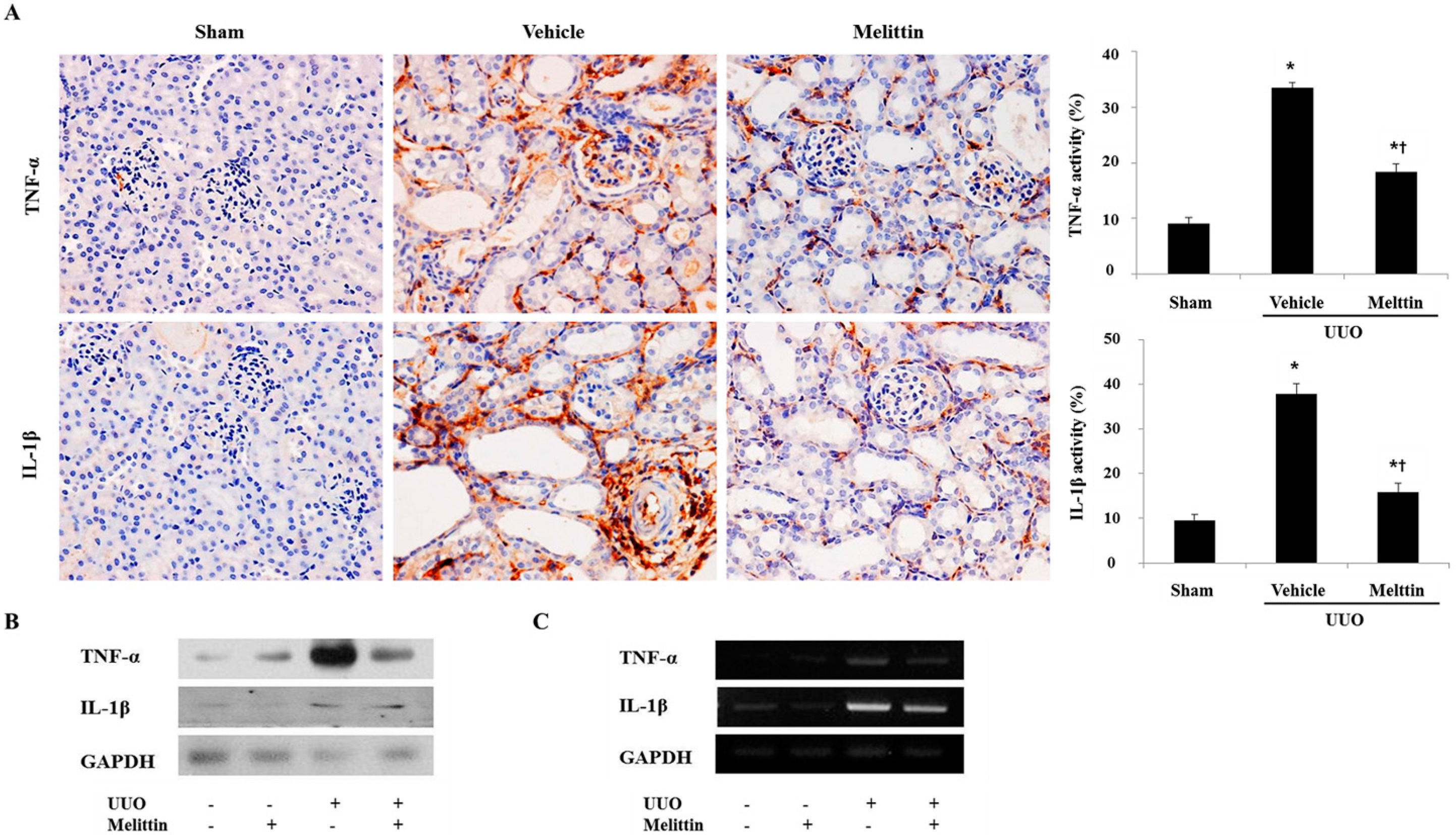
2.2. Melittin Suppresses Pro-Inflammatory Cytokines in Kidneys after UUO
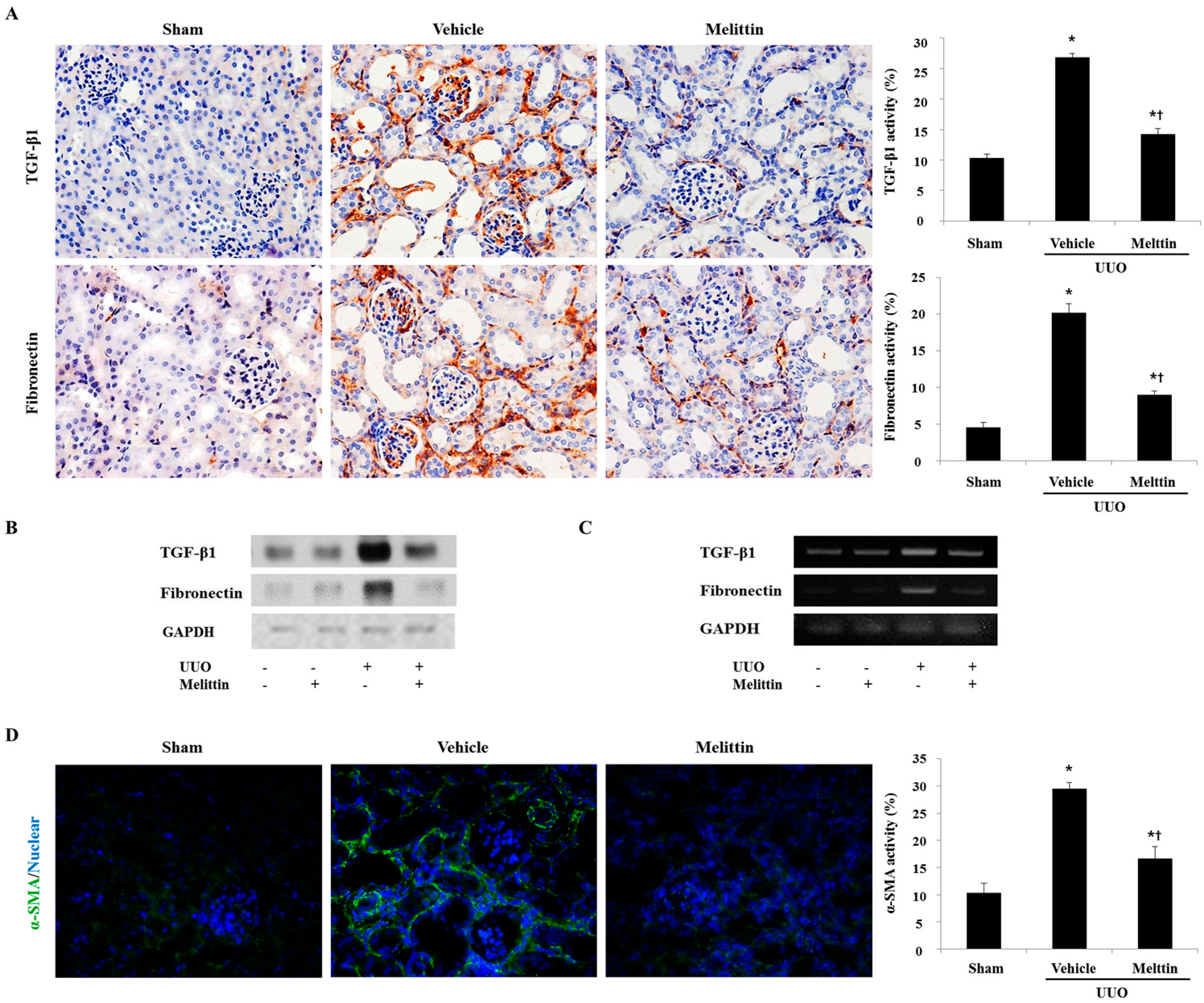
2.3. Melittin Inhibits Fibrotic Gene Expression in the Animal Model of Renal Fibrosis
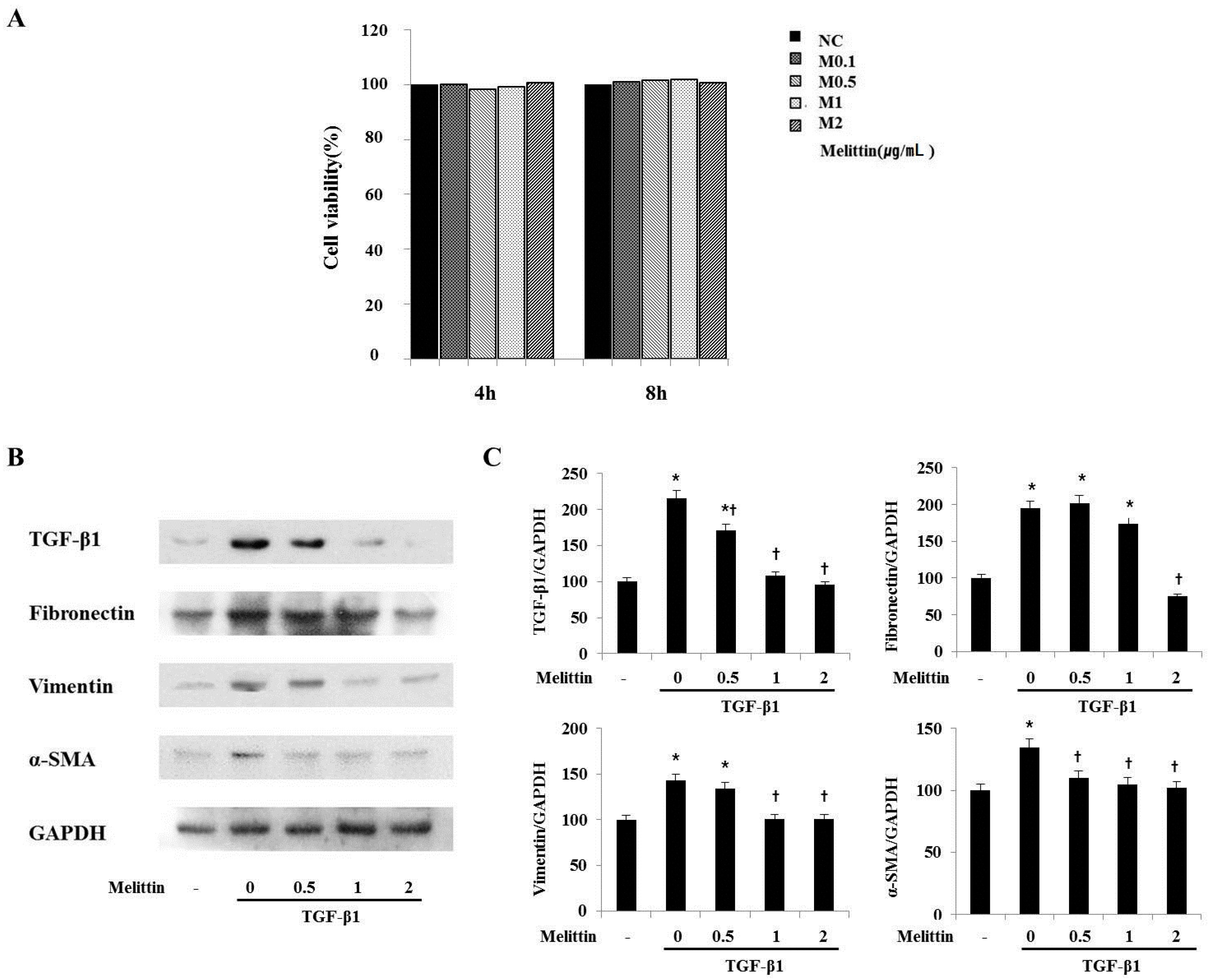
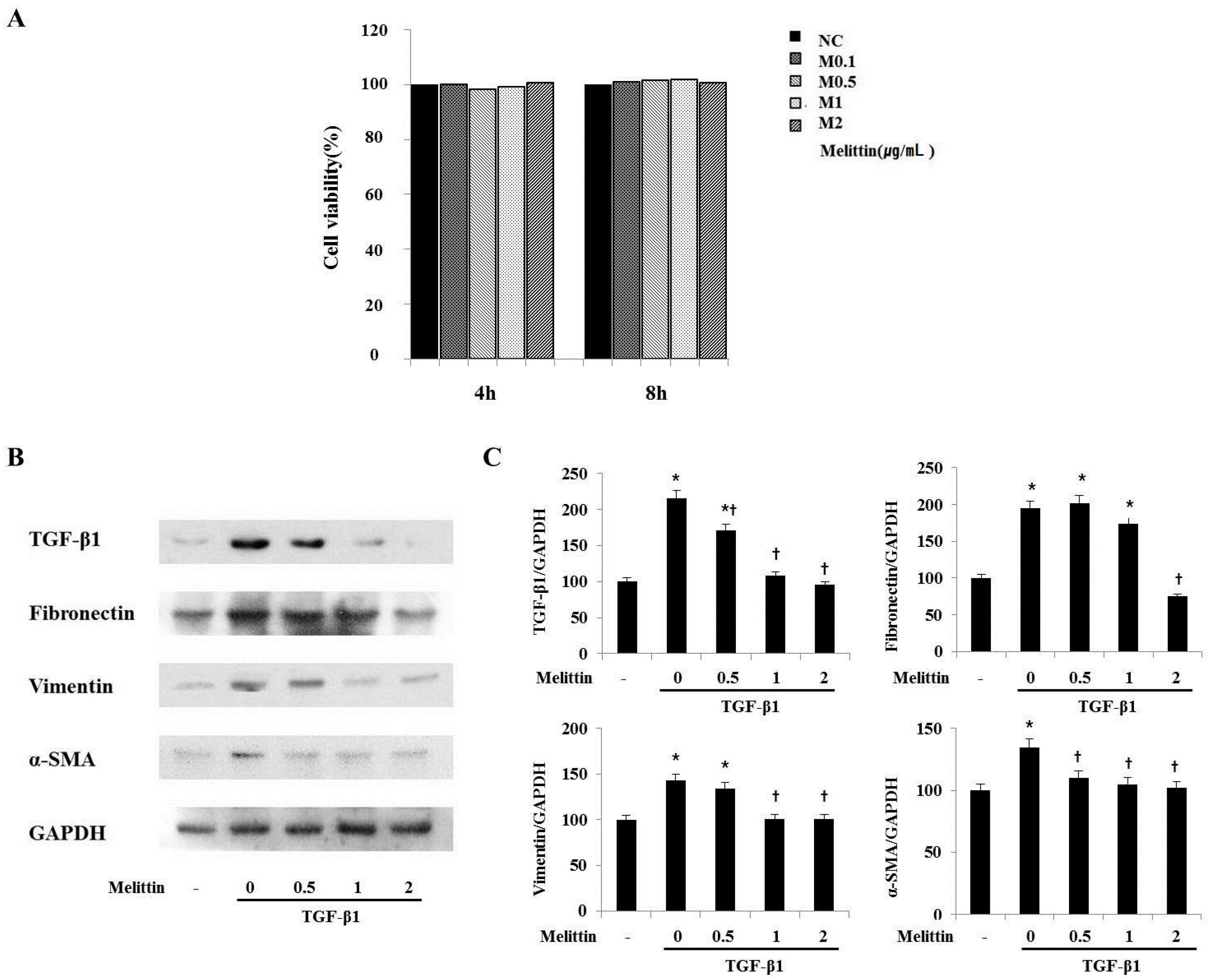
2.4. Anti-Fibrotic Effects of Melittin in Renal Fibroblast Cell
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Histological Analysis
4.3. Immunohistochemical Staining
4.4. Immunofluorescent Staining
4.5. Western Blot Analysis
4.6. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.7. Cell Culture and Viability Assay
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Genovese, F.; Manresa, A.A.; Leeming, D.J.; Karsdal, M.A.; Boor, P. The extracellular matrix in the kidney: A source of novel non-invasive biomarkers of kidney fibrosis? Fibrogen. Tissue Repair 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Kim, Y.J.; Seo, C.S.; Kim, H.T.; Park, S.R.; Lee, M.Y.; Jung, J.Y. Elsholtzia ciliata (Thunb.) hylander attenuates renal inflammation and interstitial fibrosis via regulation of TGF-β and Smad3 expression on unilateral ureteral obstruction rat model. Phytomedicine 2016, 23, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Kawada, N.; Moriyama, T.; Ando, A.; Fukunaga, M.; Miyata, T.; Kurokawa, K.; Imai, E.; Hori, M. Increased oxidative stress in mouse kidneys with unilateral ureteral obstruction. Kidney Int. 1999, 56, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S.; Morrissey, J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. Renal Physiol. 2002, 283, F861–F875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Morrissey, J.; McCracken, R.; Tolley, T.; Liapis, H.; Klahr, S. Contributions of angiotensin ii and tumor necrosis factor-α to the development of renal fibrosis. Am. J. Physiol. Renal Physiol. 2001, 280, F777–F785. [Google Scholar] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, K.K.; Misseri, R.; Metcalfe, P.; Dinarello, C.A.; Hile, K.L.; Meldrum, D.R. TNF-α neutralization ameliorates obstruction-induced renal fibrosis and dysfunction. Am. J. Physiol. Regul., Int. Comp. Physiol. 2007, 292, R1456–R1464. [Google Scholar] [CrossRef] [PubMed]
- Bottinger, E.P.; Bitzer, M. TGF-β signaling in renal disease. J. Am. Soc. Nephrol. 2002, 13, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.Y. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Roberts, I.S.; Burrows, C.; Shanks, J.H.; Venning, M.; McWilliam, L.J. Interstitial myofibroblasts: Predictors of progression in membranous nephropathy. J. Clin. Pathol. 1997, 50, 123–127. [Google Scholar] [CrossRef]
- Billingham, M.E.; Morley, J.; Hanson, J.M.; Shipolini, R.A.; Vernon, C.A. Letter: An anti-inflammatory peptide from bee venom. Nature 1973, 245, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Son, D.J.; Lee, J.W.; Lee, Y.H.; Song, H.S.; Lee, C.K.; Hong, J.T. Therapeutic application of anti-arthritis, pain-releasing, and anti-cancer effects of bee venom and its constituent compounds. Pharmacol. Ther. 2007, 115, 246–270. [Google Scholar] [CrossRef] [PubMed]
- Lariviere, W.R.; Melzack, R. The bee venom test: A new tonic-pain test. Pain 1996, 66, 271–277. [Google Scholar] [CrossRef]
- Saini, S.S.; Chopra, A.K.; Peterson, J.W. Melittin activates endogenous phospholipase d during cytolysis of human monocytic leukemia cells. Toxicon 1999, 37, 1605–1619. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.B.; Lee, J.D.; Lee, H.J.; Han, H.J.; Mar, W.C.; Kang, S.K.; Beitz, A.J.; Lee, J.H. Bee venom injection into an acupuncture point reduces arthritis associated edema and nociceptive responses. Pain 2001, 90, 271–280. [Google Scholar] [CrossRef]
- Perez-Paya, E.; Houghten, R.A.; Blondelle, S.E. The role of amphipathicity in the folding, self-association and biological activity of multiple subunit small proteins. J. Biol. Chem. 1995, 270, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Lee, S.H.; Son, D.J.; Oh, K.W.; Kim, K.H.; Song, H.S.; Kim, G.J.; Oh, G.T.; Yoon, D.Y.; Hong, J.T. Antiarthritic effect of bee venom: Inhibition of inflammation mediator generation by suppression of NF-κb through interaction with the p50 subunit. Arthritis Rheumatism 2004, 50, 3504–3515. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; He, S.; Tolbert, E.; Gong, R.; Bayliss, G.; Zhuang, S. Suramin alleviates glomerular injury and inflammation in the remnant kidney. PloS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Eddy, A.A. Molecular insights into renal interstitial fibrosis. J. Am. Soc. Nephrol. 1996, 7, 2495–2508. [Google Scholar] [PubMed]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.C.; Zuo, Y.; Fogo, A.B. Models of chronic kidney disease. Drug Discovery Today Dis. Models 2010, 7, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L. Obstructive nephropathy: Towards biomarker discovery and gene therapy. Nat. Clin. Pract. Nephrol. 2006, 2, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, E.D., Jr.; Marion, D.; Poppas, D.P.; Felsen, D. Pathophysiology of unilateral ureteral obstruction: Studies from charlottesville to new york. J. Urol. 2004, 172, 2563–2569. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, G.F.; Harris, K.P.; Purkerson, M.L.; Klahr, S. Immunological aspects of acute ureteral obstruction: Immune cell infiltrate in the kidney. Kidney Int. 1988, 34, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Sung, H.J.; Lee, W.R.; An, H.J.; Kim, J.Y.; Pak, S.C.; Han, S.M.; Park, K.K. Effects of melittin treatment in cholangitis and biliary fibrosis in a model of xenobiotic-induced cholestasis in mice. Toxins 2015, 7, 3372–3387. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.H.; Chang, Y.; Reed, N.I.; Sheppard, D. α-Smooth muscle actin is an inconsistent marker of fibroblasts responsible for force-dependent TGFβ activation or collagen production across multiple models of organ fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Kim, H.J.; Yu, M.R.; Kim, W.Y.; Kim, J.; Ryu, J.H.; Kwon, S.H.; Jeon, J.S.; Han, D.C.; Ziyadeh, F. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-beta type ii receptor. Lab. Invest. 2012, 92, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The myofibroblast: Paradigm for a mechanically active cell. J. Biomech. 2010, 43, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, H.-J.; Kim, J.-Y.; Kim, W.-H.; Han, S.-M.; Park, K.-K. The Protective Effect of Melittin on Renal Fibrosis in an Animal Model of Unilateral Ureteral Obstruction. Molecules 2016, 21, 1137. https://doi.org/10.3390/molecules21091137
An H-J, Kim J-Y, Kim W-H, Han S-M, Park K-K. The Protective Effect of Melittin on Renal Fibrosis in an Animal Model of Unilateral Ureteral Obstruction. Molecules. 2016; 21(9):1137. https://doi.org/10.3390/molecules21091137
Chicago/Turabian StyleAn, Hyun-Jin, Jung-Yeon Kim, Woon-Hae Kim, Sang-Mi Han, and Kwan-Kyu Park. 2016. "The Protective Effect of Melittin on Renal Fibrosis in an Animal Model of Unilateral Ureteral Obstruction" Molecules 21, no. 9: 1137. https://doi.org/10.3390/molecules21091137
APA StyleAn, H.-J., Kim, J.-Y., Kim, W.-H., Han, S.-M., & Park, K.-K. (2016). The Protective Effect of Melittin on Renal Fibrosis in an Animal Model of Unilateral Ureteral Obstruction. Molecules, 21(9), 1137. https://doi.org/10.3390/molecules21091137