
Native Mass Spectrometry in Fragment-Based Drug Discovery

Abstract
:
1. Introduction
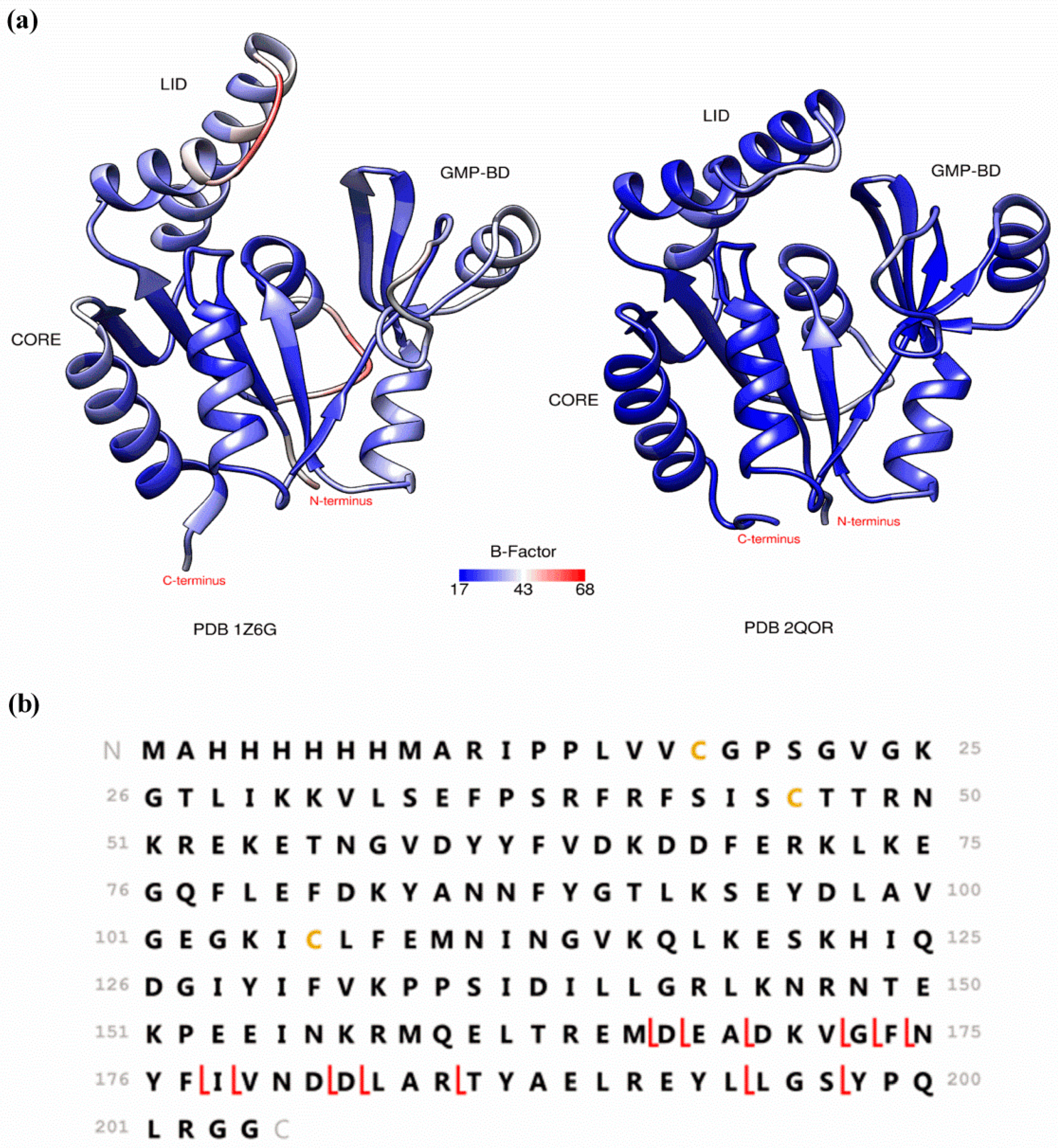
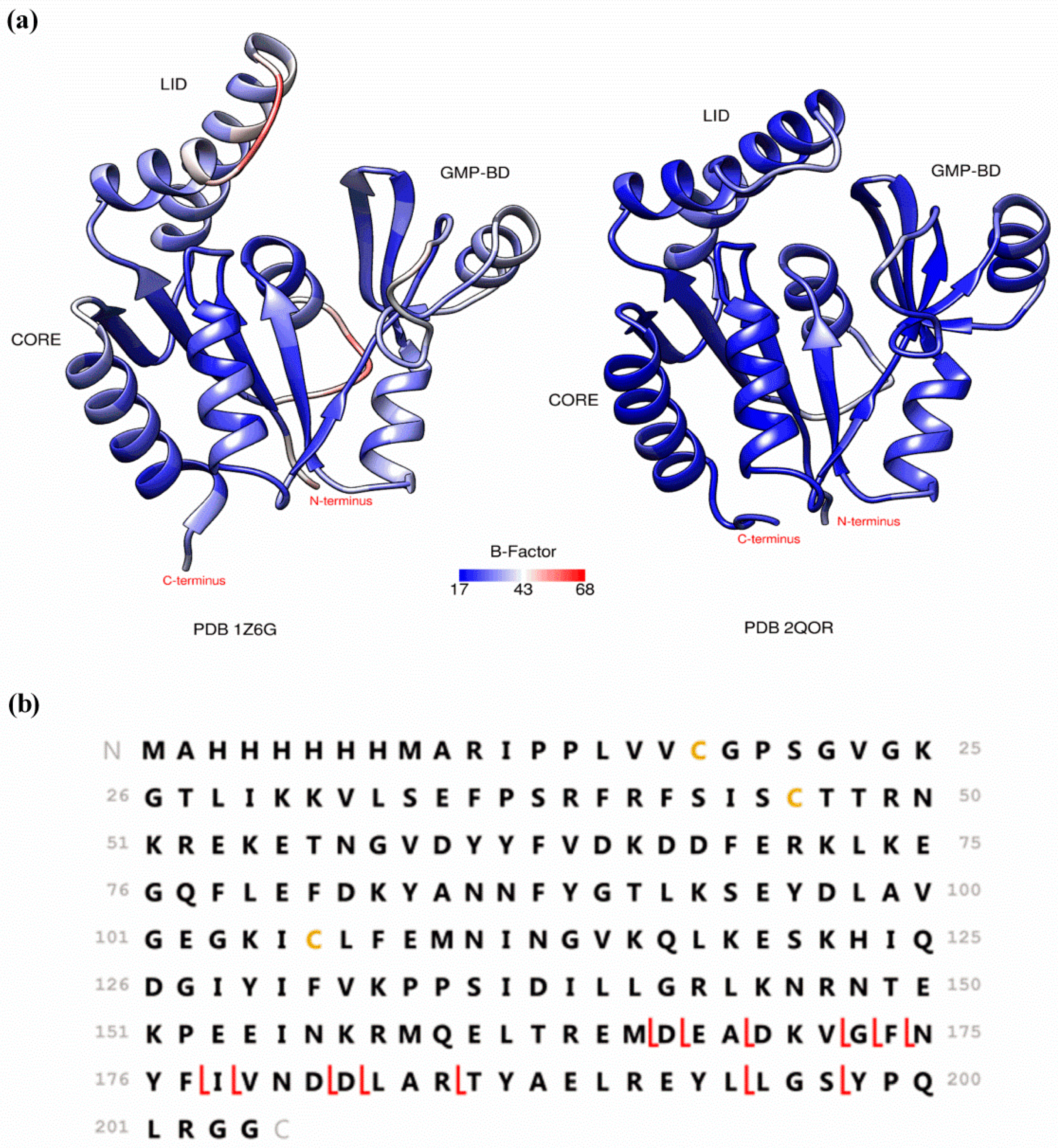
2. Preserving Native or Near-Native Protein Conformations in the Gas-Phase
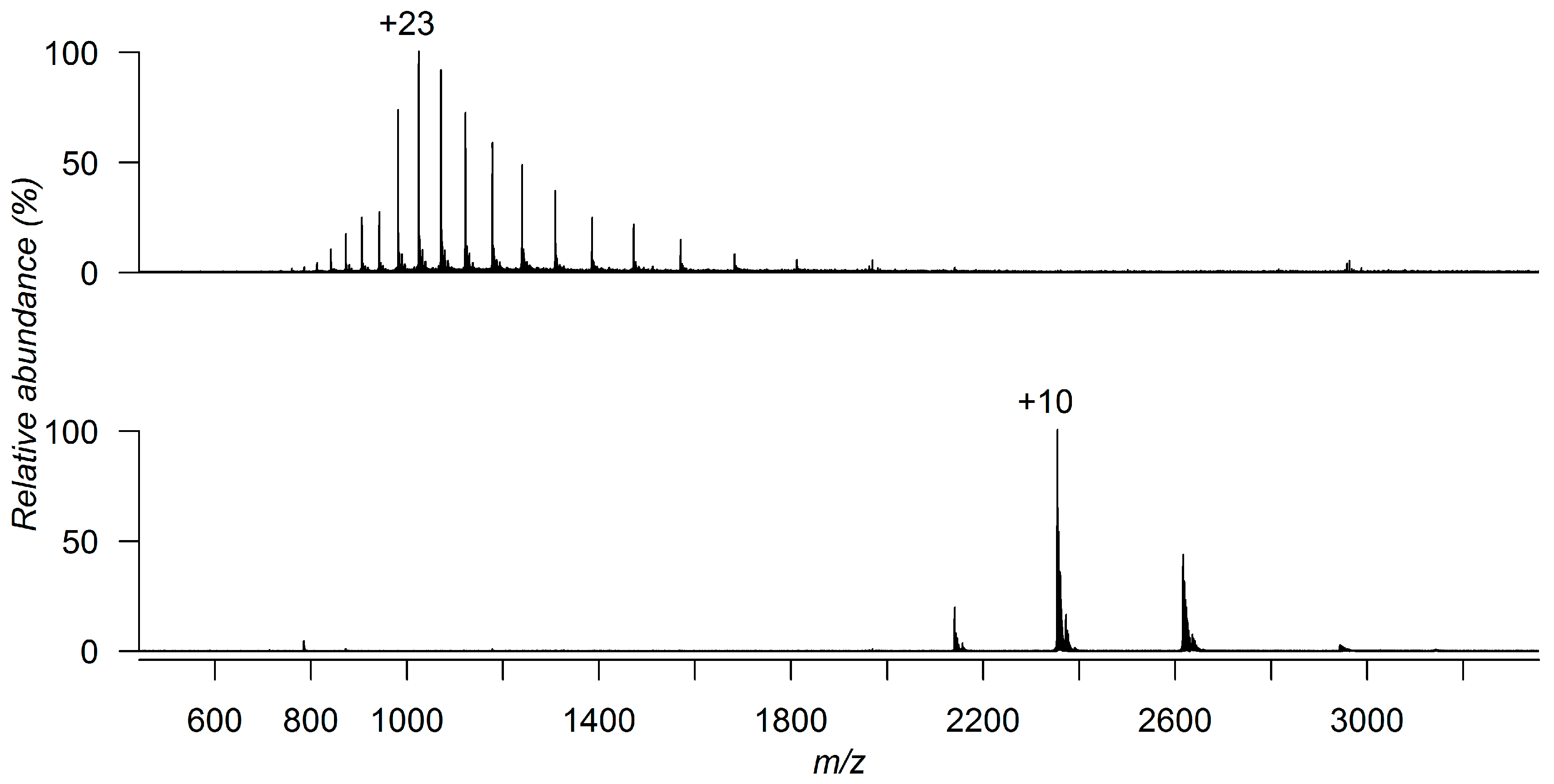
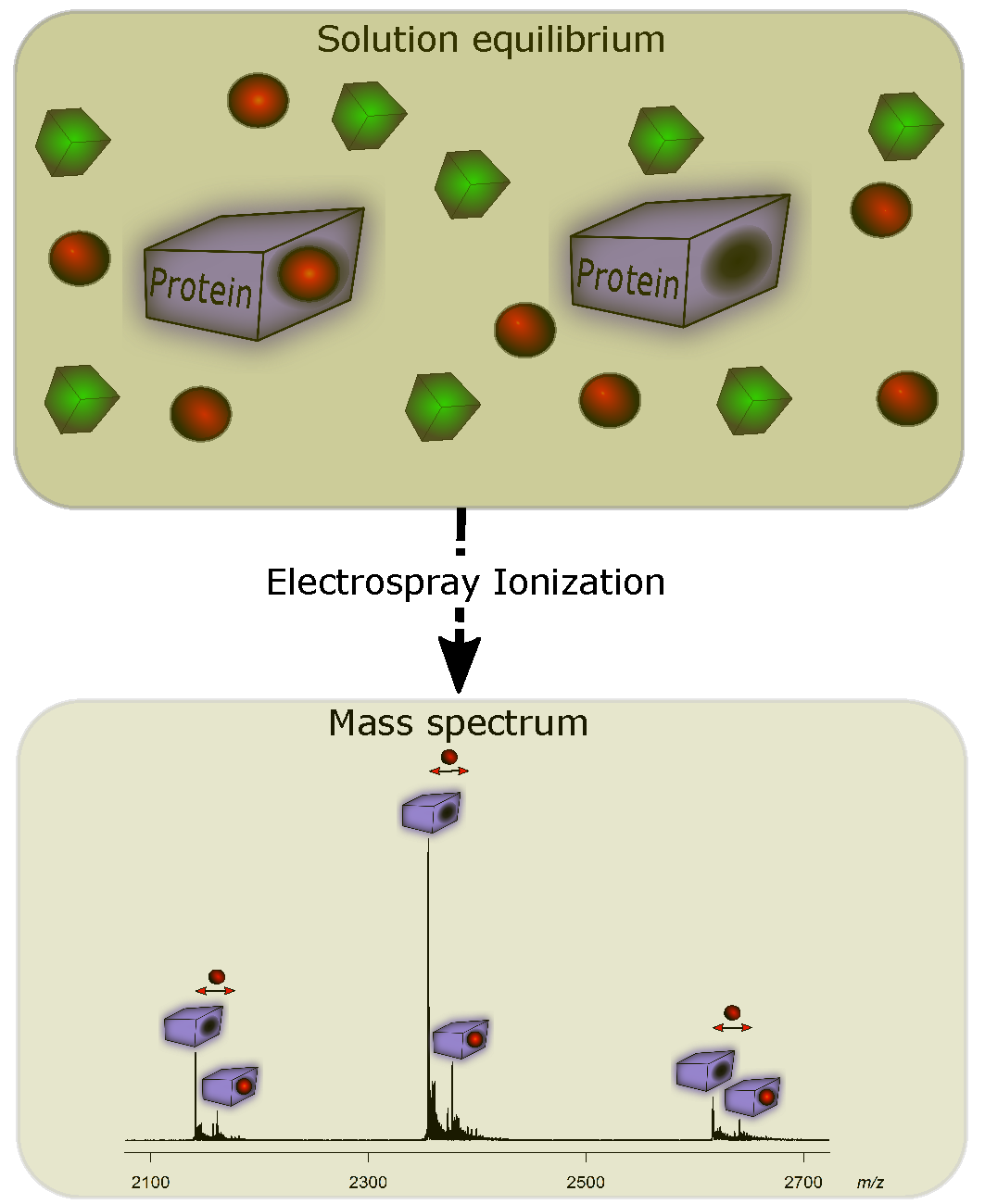
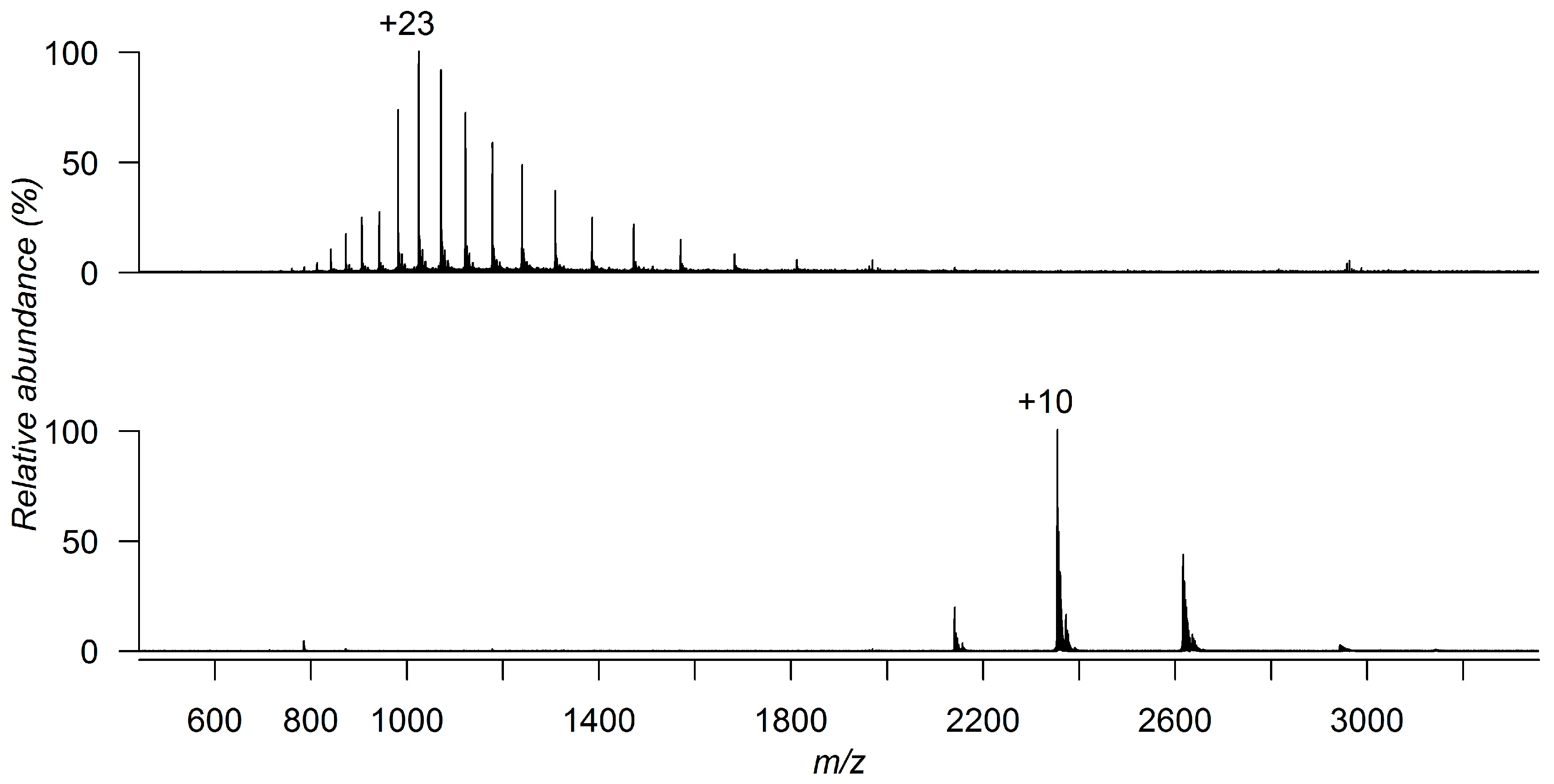
3. Detecting Noncovalent Protein-Fragment Interactions
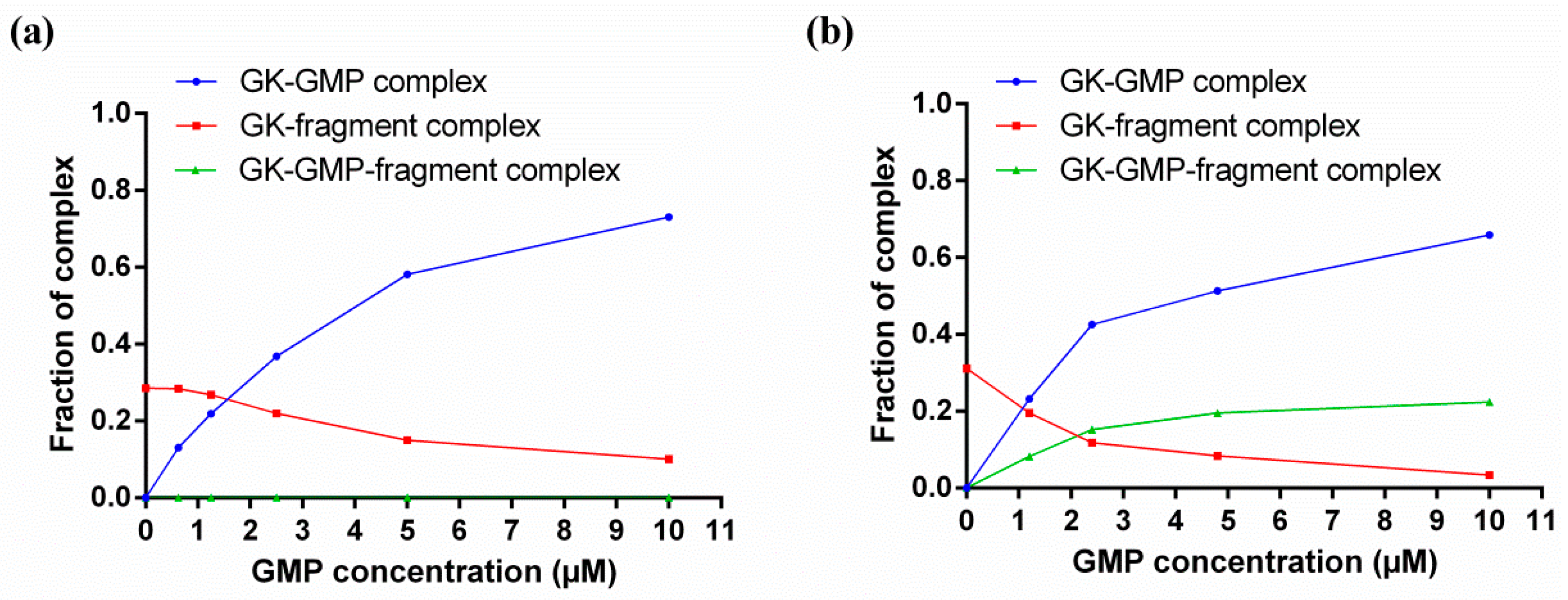
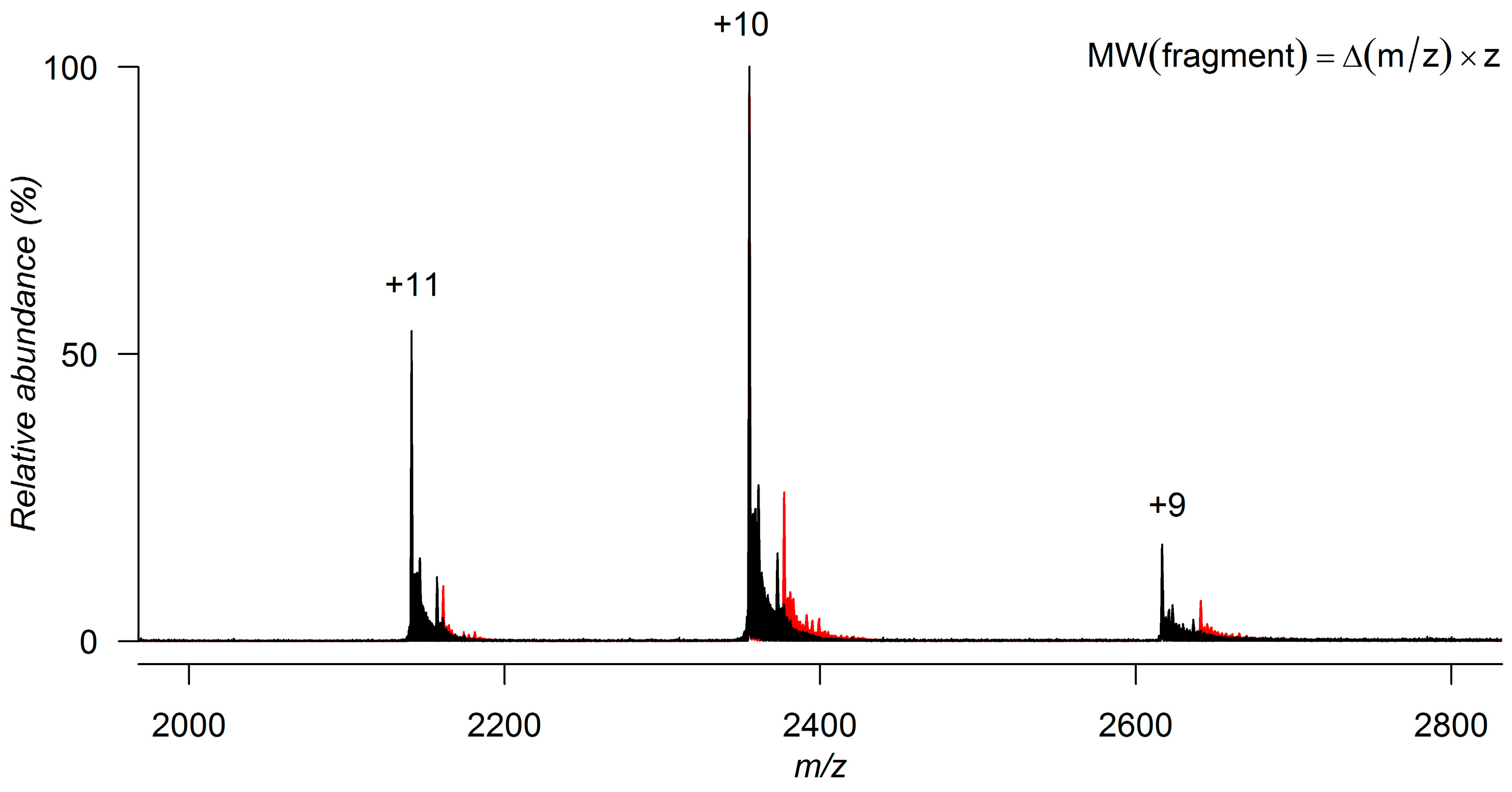
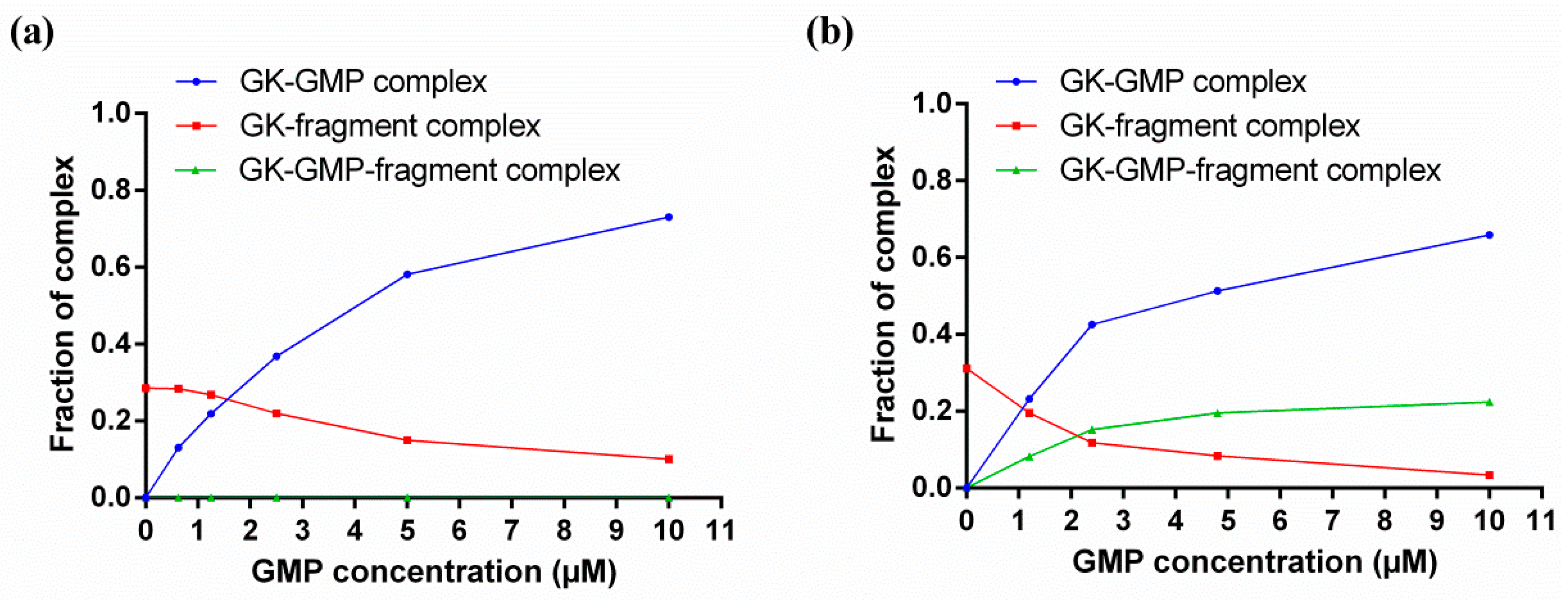
4. Characterizing Noncovalent Protein-Fragment Interactions
5. Native MS in FBDD: Where Are We?
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenn, J.B. Electrospray wings for molecular elephants (nobel lecture). Angew. Chem. Int. Ed. Engl. 2003, 42, 3871–3894. [Google Scholar] [CrossRef] [PubMed]
- Fenn, J.B.; Mann, M.; Meng, C.K.; Wong, S.F.; Whitehouse, C.M. Electrospray ionization for mass spectrometry of large biomolecules. Science 1989, 246, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Bachmann, D.; Bahr, U.; Hillenkamp, F. Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. Ion Process. 1987, 78, 53–68. [Google Scholar] [CrossRef]
- Tanaka, K.; Waki, H.; Ido, Y.; Akita, S.; Yoshida, Y.; Yoshida, T.; Matsuo, T. Protein and polymer analyses up to m/z 100,000 by laser ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 1988, 2, 151–153. [Google Scholar] [CrossRef]
- Chowdhury, S.K.; Katta, V.; Chait, B.T. Probing conformational changes in proteins by mass spectrometry. J. Am. Chem. Soc. 1990, 112, 9012–9013. [Google Scholar] [CrossRef]
- Loo, J.A. Studying noncovalent protein complexes by electrospray ionization mass spectrometry. Mass Spectrom. Rev. 1997, 16, 1–23. [Google Scholar] [CrossRef]
- Erba, E.B.; Zenobi, R. Mass spectrometric studies of dissociation constants of noncovalent complexes. Annu. Rep. Prog. Chem. Sect. C 2011, 107, 199–228. [Google Scholar] [CrossRef]
- Hilton, G.R.; Benesch, J.L.P. Two decades of studying non-covalent biomolecular assemblies by means of electrospray ionization mass spectrometry. J. R. Soc. Interface 2012, 9, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, A.E. Recent developments in electrospray ionisation mass spectrometry: Noncovalently bound protein complexes. Nat. Prod. Rep. 2005, 22, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Hofstadler, S.A.; Sannes-Lowery, K.A. Applications of ESI-MS in drug discovery: Interrogation of noncovalent complexes. Nat. Rev. Drug Discov. 2006, 5, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Ganem, B.; Li, Y.T.; Henion, J.D. Detection of noncovalent receptor ligand complexes by mass-spectrometry. J. Am. Chem. Soc. 1991, 113, 6294–6296. [Google Scholar] [CrossRef]
- Katta, V.; Chait, B.T. Observation of the heme globin complex in native myoglobin by electrospray-ionization mass-spectrometry. J. Am. Chem. Soc. 1991, 113, 8534–8535. [Google Scholar] [CrossRef]
- Ganem, B.; Li, Y.T.; Henion, J.D. Observation of noncovalent enzyme substrate and enzyme product complexes by ion-spray mass-spectrometry. J. Am. Chem. Soc. 1991, 113, 7818–7819. [Google Scholar] [CrossRef]
- Kitova, E.N.; El-Hawiet, A.; Schnier, P.D.; Klassen, J.S. Reliable determinations of protein-ligand interactions by direct ESI-MS measurements. Are we there yet? J. Am. Soc. Mass Spectrom. 2012, 23, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Loo, R.R.O.; Goodlett, D.R.; Smith, R.D.; Loo, J.A. Observation of a noncovalent ribonuclease S-protein S-peptide complex by electrospray ionization mass-spectrometry. J. Am. Chem. Soc. 1993, 115, 4391–4392. [Google Scholar] [CrossRef]
- Cheng, X.H.; Chen, R.D.; Bruce, J.E.; Schwartz, B.L.; Anderson, G.A.; Hofstadler, S.A.; Gale, D.C.; Smith, R.D.; Gao, J.M.; Sigal, G.B.; et al. Using electrospray-ionization fticr mass-spectrometry to study competitive-binding of inhibitors to carbonic-anhydrase. J. Am. Chem. Soc. 1995, 117, 8859–8860. [Google Scholar] [CrossRef]
- Chait, B.T.; Cadene, M.; Olinares, P.D.; Rout, M.P.; Shi, Y. Revealing higher order protein structure using mass spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, D.; Zeng, W.; Hus, J.-C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019. [Google Scholar] [CrossRef] [PubMed]
- Sanglier, S.; Atmanene, C.; Chevreux, G.; van Dorsselaer, A. Nondenaturing mass spectrometry to study noncovalent protein/protein and protein/ligand complexes: Technical aspects and application to the determination of binding stoichiometries. Methods Mol. Biol. 2008, 217–243. [Google Scholar]
- Xie, Y.; Zhang, J.; Yin, S.; Loo, J.A. Top-down ESI-ECD-FT-ICR mass spectrometry localizes noncovalent protein-ligand binding sites. J. Am. Chem. Soc. 2006, 128, 14432–14433. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Loo, J. Elucidating the site of protein-atp binding by top-down mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Loo, J.A. Top-down mass spectrometry of supercharged native protein-ligand complexes. Int. J. Mass Spectrom. 2011, 300, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, M.B.; Thyer, R.; Rosenberg, J.; Ellington, A.; Brodbelt, J.S. Structural characterization of dihydrofolate reductase complexes by top-down ultraviolet photodissociation mass spectrometry. J. Am. Chem. Soc. 2015, 137, 9128–9135. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.M.; Tsybin, Y.O.; Dyson, P.J.; Keppler, B.K.; Hartinger, C.G. Fragmentation methods on the balance: Unambiguous top-down mass spectrometric characterization of oxaliplatin-ubiquitin binding sites. Anal. Bioanal. Chem. 2011, 402, 2655–2662. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wongkongkathep, P.; Orden, S.L.; Ogorzalek Loo, R.R.; Loo, J.A. Revealing ligand binding sites and quantifying subunit variants of noncovalent protein complexes in a single native top-down FTICR MS experiment. J. Am. Soc. Mass Spectrom. 2014, 25, 2060–2068. [Google Scholar] [CrossRef] [PubMed]
- Dyachenko, A.; Gruber, R.; Shimon, L.; Horovitz, A.; Sharon, M. Allosteric mechanisms can be distinguished using structural mass spectrometry. Proc. Natl. Acad. Sci. USA 2013, 110, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Sharon, M.; Horovitz, A. Probing allosteric mechanisms using native mass spectrometry. Curr. Opin. Struct. Biol. 2015, 34, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Cubrilovic, D.; Haap, W.; Barylyuk, K.; Ruf, A.; Badertscher, M.; Gubler, M.; Tetaz, T.; Joseph, C.; Benz, J.; Zenobi, R. Determination of protein-ligand binding constants of a cooperatively regulated tetrameric enzyme using electrospray mass spectrometry. ACS Chem. Biol. 2014, 9, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Daneshfar, R.; Kitova, E.N.; Klassen, J.S. Determination of protein-ligand association thermochemistry using variable-temperature nanoelectrospray mass spectrometry. J. Am. Chem. Soc. 2004, 126, 4786–4787. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, G.K.; Soya, N.; Palcic, M.M.; Klassen, J.S. Temperature-dependent cooperativity in donor-acceptor substrate binding to the human blood group glycosyltransferases. Glycobiology 2008, 18, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Cong, X.; Liu, Y.; Liu, W.; Liang, X.; Russell, D.H.; Laganowsky, A. Determining membrane protein-lipid binding thermodynamics using native mass spectrometry. J. Am. Chem. Soc. 2016, 138, 4346–4349. [Google Scholar] [CrossRef] [PubMed]
- Gülbakan, B.; Barylyuk, K.; Zenobi, R. Determination of thermodynamic and kinetic properties of biomolecules by mass spectrometry. Curr. Opin. Biotechnol. 2015, 31, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Breuker, K.; McLafferty, F.W. Stepwise evolution of protein native structure with electrospray into the gas phase, 10−12 to 102 s. Proc. Natl. Acad. Sci. USA 2008, 105, 18145–18152. [Google Scholar] [CrossRef] [PubMed]
- Frankevich, V.; Barylyuk, K.; Chingin, K.; Nieckarz, R.; Zenobi, R. Native biomolecules in the gas phase? The case of green fluorescent protein. Chemphyschem 2013, 14, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Breuker, K. The study of protein-ligand interactions by mass spectrometry—a personal view. Int. J. Mass Spectrom. 2004, 239, 33–41. [Google Scholar] [CrossRef]
- Loo, R.R.O.; Loo, J.A. Salt bridge rearrangement (sabre) explains the dissociation behavior of noncovalent complexes. J. Am. Soc. Mass Spectrom. 2016, 27, 975–990. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Politis, A.; Davies, R.B.; Liko, I.; Wu, K.-J.; Stewart, A.G.; Stock, D.; Robinson, C.V. Ion mobility-mass spectrometry of a rotary ATPase reveals ATP-induced reduction in conformational flexibility. Nat. Chem. 2014, 6, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Clemmer, D.E.; Hudgins, R.R.; Jarrold, M.F. Naked protein conformations: Cytochrome c in the gas phase. J. Am. Chem. Soc. 1995, 117, 10141–10142. [Google Scholar] [CrossRef]
- Myung, S.; Badman, E.R.; Lee, Y.J.; Clemmer, D.E. Structural transitions of electrosprayed ubiquitin ions stored in an ion trap over ~10 ms to 30 s. J. Phys. Chem. A 2002, 106, 9976–9982. [Google Scholar] [CrossRef]
- Badman, E.R.; Myung, S.; Clemmer, D.E. Evidence for unfolding and refolding of gas-phase cytochrome c ions in a paul trap. J. Am. Soc. Mass Spectrom. 2005, 16, 1493–1497. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, T.; Bowers, M.T. Structural stability from solution to the gas phase: Native solution structure of ubiquitin survives analysis in a solvent-free ion mobility-mass spectrometry environment. J. Phys. Chem. B 2011, 115, 12266–12275. [Google Scholar] [CrossRef] [PubMed]
- Hall, Z.; Robinson, C.V. Do charge state signatures guarantee protein conformations? J. Am. Soc. Mass Spectrom. 2012, 23, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Russell, D.H. How closely related are conformations of protein ions sampled by IM-MS to native solution structures? J. Am. Soc. Mass Spectrom. 2015, 26, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.D.; Chorush, R.A.; Wampler, F.M.; Little, D.P.; O’Connor, P.B.; McLafferty, F.W. Gas-phase folding and unfolding of cytochrome c cations. Proc. Natl. Acad. Sci. USA 1995, 92, 2451–2454. [Google Scholar] [CrossRef] [PubMed]
- McLafferty, F.W.; Guan, Z.; Haupts, U.; Wood, T.D.; Kelleher, N.L. Gaseous conformational structures of cytochrome c. J. Am. Chem. Soc. 1998, 120, 4732–4740. [Google Scholar] [CrossRef]
- Freitas, M.A.; Hendrickson, C.L.; Emmett, M.R.; Marshall, A.G. Gas-phase bovine ubiquitin cation conformations resolved by gas-phase hydrogen/deuterium exchange rate and extent. Int. J. Mass spectrom. 1999, 185, 565–575. [Google Scholar] [CrossRef]
- Evans, S.E.; Lueck, N.; Marzluff, E.M. Gas phase hydrogen/deuterium exchange of proteins in an ion trap mass spectrometer. Int. J. Mass spectrom. 2003, 222, 175–187. [Google Scholar] [CrossRef]
- Zhu, S.; Campbell, J.L.; Chernushevich, I.; le Blanc, J.Y.; Wilson, D.J. Differential mobility spectrometry-hydrogen deuterium exchange (DMS-HDX) as a probe of protein conformation in solution. J. Am. Soc. Mass Spectrom. 2016, 27, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Pringle, S.D.; Murphy, J.P., III; Fadgen, K.E.; Brown, J.; Engen, J.R. Gas-phase hydrogen/deuterium exchange in a traveling wave ion guide for the examination of protein conformations. Anal. Chem. 2009, 81, 10019–10028. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Pringle, S.D.; Morris, M.; Brown, J.M. Site-specific analysis of gas-phase hydrogen/deuterium exchange of peptides and proteins by electron transfer dissociation. Anal. Chem. 2012, 84, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, K. Time-resolved pulsed hydrogen/deuterium exchange mass spectrometry probes gaseous proteins structural kinetics. J. Am. Soc. Mass Spectrom. 2015, 26, 71–82. [Google Scholar] [CrossRef]
- Breuker, K.; Oh, H.; Horn, D.M.; Cerda, B.A.; McLafferty, F.W. Detailed unfolding and folding of gaseous ubiquitin ions characterized by electron capture dissociation. J. Am. Chem. Soc. 2002, 124, 6407–6420. [Google Scholar] [CrossRef]
- Horn, D.M.; Breuker, K.; Frank, A.J.; McLafferty, F.W. Kinetic intermediates in the folding of gaseous protein ions characterized by electron capture dissociation mass spectrometry. J. Am. Chem. Soc. 2001, 123, 9792–9799. [Google Scholar] [CrossRef]
- Breuker, K.; McLafferty, F.W. Native electron capture dissociation for the structural characterization of noncovalent interactions in native cytochrome c. Angew. Chem. Int. Ed. 2003, 42, 4900–4904. [Google Scholar] [CrossRef] [PubMed]
- Breuker, K.; McLafferty, F.W. The thermal unfolding of native cytochrome c in the transition from solution to gas phase probed by native electron capture dissociation. Angew. Chem. Int. Ed. 2005, 44, 4911–4914. [Google Scholar] [CrossRef] [PubMed]
- Breuker, K.; Brüschweiler, S.; Tollinger, M. Electrostatic stabilization of a native protein structure in the gas phase. Angew. Chem. Int. Ed. 2011, 50, 873–877. [Google Scholar] [CrossRef] [PubMed]
- Schennach, M.; Breuker, K. Proteins with highly similar native folds can show vastly dissimilar folding behavior when desolvated. Angew. Chem. Int. Ed. 2014, 53, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Skinner, O.S.; McLafferty, F.W.; Breuker, K. How ubiquitin unfolds after transfer into the gas phase. J. Am. Soc. Mass Spectrom. 2012, 23, 1011–1014. [Google Scholar] [CrossRef] [PubMed]
- Lermyte, F.; Sobott, F. Electron transfer dissociation provides higher-order structural information of native and partially unfolded protein complexes. Proteomics 2015, 15, 2813–2822. [Google Scholar] [CrossRef] [PubMed]
- Schennach, M.; Breuker, K. Probing protein structure and folding in the gas phase by electron capture dissociation. J. Am. Soc. Mass Spectrom. 2015, 26, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cui, W.; Wen, J.; Blankenship, R.E.; Gross, M.L. Native electrospray and electron-capture dissociation fticr mass spectrometry for top-down studies of protein assemblies. Anal. Chem. 2011, 83, 5598–5606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cui, W.; Gross, M.L. Native electrospray ionization and electron-capture dissociation for comparison of protein structure in solution and the gas phase. Int. J. Mass Spectrom. 2013, 354, 288–291. [Google Scholar] [CrossRef] [PubMed]
- Tarun, A.S.; Peng, X.; Dumpit, R.F.; Ogata, Y.; Silva-Rivera, H.; Camargo, N.; Daly, T.M.; Bergman, L.W.; Kappe, S.H.I. A combined transcriptome and proteome survey of malaria parasite liver stages. Proc. Natl. Acad. Sci. USA 2008, 105, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.J.; Napuli, A.J.; Gilligan, J.H.; Gagaring, K.; Borboa, R.; Francek, C.; Chen, Z.; Dagostino, E.F.; Stockmyer, J.B.; Wang, Y.; et al. Identification of inhibitors for putative malaria drug targets among novel antimalarial compounds. Mol. Biochem. Parasit. 2011, 175, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kandeel, M.; Kitade, Y. Binding dynamics and energetic insight into the molecular forces driving nucleotide binding by guanylate kinase. J. Mol. Recognit. 2011, 24, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Delalande, O.; Sacquin-Mora, S.; Baaden, M. Enzyme closure and nucleotide binding structurally lock guanylate kinase. Biophys. J. 2011, 101, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Liu, B.F.; Li, J.; Liu, X. Advances in coupling microfluidic chips to mass spectrometry. Mass Spectrom. Rev. 2015, 34, 535–557. [Google Scholar] [CrossRef] [PubMed]
- Keetch, C.A.; Hernández, H.; Sterling, A.; Baumert, M.; Allen, M.H.; Robinson, C.V. Use of a microchip device coupled with mass spectrometry for ligand screening of a multi-protein target. Anal. Chem. 2003, 75, 4937–4941. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; van Pelt, C.K.; Wilson, D.B. Quantitative determination of noncovalent binding interactions using automated nanoelectrospray mass spectrometry. Anal. Chem. 2003, 75, 3010–3018. [Google Scholar] [CrossRef] [PubMed]
- Benkestock, K.; van Pelt, C.K.; Åkerud, T.; Sterling, A.; Edlund, P.-O.; Roeraade, J. Automated nano-electrospray mass spectrometry for protein-ligand screening by noncovalent interaction applied to human H-FABP and A-FABP. J. Biomol. Screen. 2003, 8, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Bovet, C.; Wortmann, A.; Eiler, S.; Granger, F.; Ruff, M.; Gerrits, B.; Moras, D.; Zenobi, R. Estrogen receptor-ligand complexes measured by chip-based nanoelectrospray mass spectrometry: An approach for the screening of endocrine disruptors. Protein Sci. 2007, 16, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Vu, H.; Lovell, K.M.; Criscione, K.R.; Collins, B.M.; Prisinzano, T.E.; Poulsen, S.A.; McLeish, M.J.; Grunewald, G.L.; Martin, J.L. Fragment-based screening by x-ray crystallography, MS and isothermal titration calorimetry to identify PNMT (phenylethanolamine N-methyltransferase) inhibitors. Biochem. J. 2010, 431, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Vivat Hannah, V.; Atmanene, C.; Zeyer, D.; van Dorsselaer, A.; Sanglier-Cianférani, S. Native MS: An “ESI” way to support structure-and fragment-based drug discovery. Future Med. Chem. 2010, 2, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Maple, H.J.; Garlish, R.A.; Rigau-Roca, L.; Porter, J.; Whitcombe, I.; Prosser, C.E.; Kennedy, J.; Henry, A.J.; Taylor, R.J.; Crump, M.P.; et al. Automated protein-ligand interaction screening by mass spectrometry. J. Med. Chem. 2012, 55, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.A.; Dolezal, O.; Ren, B.; Ryan, J.H.; Peat, T.S.; Poulsen, S.-A. Native state mass spectrometry, surface plasmon resonance, and x-ray crystallography correlate strongly as a fragment screening combination. J. Med. Chem. 2016, 59, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, S.-A. Fragment screening by native state mass spectrometry. Aust. J. Chem. 2014, 66, 1495–1501. [Google Scholar] [CrossRef]
- Daubenfeld, T.; Bouin, A.-P.; van der Rest, G. A deconvolution method for the separation of specific versus nonspecific interactions in noncovalent protein-ligand complexes analyzed by ESI-FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Shimon, L.; Sharon, M.; Horovitz, A. A method for removing effects of nonspecific binding on the distribution of binding stoichiometries: Application to mass spectroscopy data. Biophys. J. 2010, 99, 1645–1649. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Trnka, M.J.; Bushnell, D.A.; Robinson, P.J.; Gestwicki, J.E.; Burlingame, A.L. Deconvolution method for specific and nonspecific binding of ligand to multiprotein complex by native mass spectrometry. Anal. Chem. 2015, 87, 8541–8546. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.V.; Chung, E.W.; Kragelund, B.B.; Knudsen, J.; Aplin, R.T.; Poulsen, F.M.; Dobson, C.M. Probing the nature of noncovalent interactions by mass spectrometry. A study of protein-coa ligand binding and assembly. J. Am. Chem. Soc. 1996, 118, 8646–8653. [Google Scholar] [CrossRef]
- Laskin, J.; Futrell, J.H. Entropy is the major driving force for fragmentation of proteins and protein-ligand complexes in the gas phase. J. Phys. Chem. A 2003, 107, 5836–5839. [Google Scholar] [CrossRef]
- Bich, C.; Baer, S.; Jecklin, M.C.; Zenobi, R. Probing the hydrophobic effect of noncovalent complexes by mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Xie, Y.; Loo, J.A. Mass spectrometry of protein-ligand complexes: Enhanced gas-phase stability of ribonuclease-nucleotide complexes. J. Am. Soc. Mass Spectrom. 2008, 19, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Kutchukian, P.S.; Wassermann, A.M.; Lindvall, M.K.; Wright, S.K.; Ottl, J.; Jacob, J.; Scheufler, C.; Marzinzik, A.; Brooijmans, N.; Glick, M. Large scale meta-analysis of fragment-based screening campaigns privileged fragments and complementary technologies. J. Biomol. Screen. 2015, 20, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Riccardi Sirtori, F.; Caronni, D.; Colombo, M.; Dalvit, C.; Paolucci, M.; Regazzoni, L.; Visco, C.; Fogliatto, G. Establish an automated flow injection ESI-MS method for the screening of fragment based libraries: Application to hsp90. Eur. J. Pharm. Sci. 2015, 76, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Jecklin, M.C.; Touboul, D.; Jain, R.; Toole, E.N.; Tallarico, J.; Drueckes, P.; Ramage, P.; Zenobi, R. Affinity classification of kinase inhibitors by mass spectrometric methods and validation using standard IC50 measurements. Anal. Chem. 2009, 81, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tjernberg, A.; Carnö, S.; Oliv, F.; Benkestock, K.; Edlund, P.-O.; Griffiths, W.J.; Hallén, D. Determination of dissociation constants for protein-ligand complexes by electrospray ionization mass spectrometry. Anal. Chem. 2004, 76, 4325–4331. [Google Scholar] [CrossRef] [PubMed]
- El-Hawiet, A.; Kitova, E.N.; Liu, L.; Klassen, J.S. Quantifying labile protein-ligand interactions using electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2010, 21, 1893–1899. [Google Scholar] [CrossRef] [PubMed]
- Peschke, M.; Verkerk, U.H.; Kebarle, P. Features of the ESI mechanism that affect the observation of multiply charged noncovalent protein complexes and the determination of the association constant by the titration method. J. Am. Soc. Mass Spectrom. 2004, 15, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Jaquillard, L.; Saab, F.; Schoentgen, F.; Cadene, M. Improved accuracy of low affinity protein-ligand equilibrium dissociation constants directly determined by electrospray ionization mass spectrometry. J. Am. Soc. Mass Spectrom. 2012, 23, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kitova, E.N.; Klassen, J.S. Influence of solution and gas phase processes on protein-carbohydrate binding affinities determined by nanoelectrospray fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2003, 75, 4945–4955. [Google Scholar] [CrossRef] [PubMed]
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Swayze, E.E.; Jefferson, E.A.; Sannes-Lowery, K.A.; Blyn, L.B.; Risen, L.M.; Arakawa, S.; Osgood, S.A.; Hofstadler, S.A.; Griffey, R.H. SAR by MS: A ligand based technique for drug lead discovery against structured rna targets. J. Med. Chem. 2002, 45, 3816–3819. [Google Scholar] [CrossRef] [PubMed]
- Ockey, D.A.; Dotson, J.L.; Struble, M.E.; Stults, J.T.; Bourell, J.H.; Clark, K.R.; Gadek, T.R. Structure-activity relationships by mass spectrometry: Identification of novel MMP-3 inhibitors. Bioorg. Med. Chem. 2004, 12, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, S.-A. Direct screening of a dynamic combinatorial library using mass spectrometry. J. Am. Soc. Mass Spectrom. 2006, 17, 1074–1080. [Google Scholar] [CrossRef]
- Moore, C.D.; Wu, H.; Bolaños, B.; Bergqvist, S.; Brooun, A.; Pauly, T.; Nowlin, D. Structural and biophysical characterization of XIAP BIR3 G306E mutant: Insights in protein dynamics and application for fragment-based drug design. Chem. Biol. Drug Des. 2009, 74, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.; Roullier, C.; Campitelli, M.; Trenholme, K.R.; Gardiner, D.L.; Andrews, K.T.; Skinner-Adams, T.; Crowther, G.J.; van Voorhis, W.C.; Quinn, R.J. Plasmodium gametocyte inhibition identified from a natural-product-based fragment library. ACS Chem. Biol. 2013, 8, 2654–2659. [Google Scholar] [CrossRef] [PubMed]
- Lössl, P.; Snijder, J.; Heck, A.J. Boundaries of mass resolution in native mass spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, S.; Allison, T.M.; Robinson, C.V. Mass spectrometry of protein complexes: From origins to applications. Annu. Rev. Phys. Chem. 2015, 66, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Gault, J.; Donlan, J.A.C.; Liko, I.; Hopper, J.T.S.; Gupta, K.; Housden, N.G.; Struwe, W.B.; Marty, M.T.; Mize, T.; Bechara, C.; et al. High-resolution mass spectrometry of small molecules bound to membrane proteins. Nat. Methods 2016, 13, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Morgner, N.; Kleinschroth, T.; Barth, H.-D.; Ludwig, B.; Brutschy, B. A novel approach to analyze membrane proteins by laser mass spectrometry: From protein subunits to the integral complex. J. Am. Soc. Mass Spectrom. 2007, 18, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Morgner, N.; Barth, H.-D.; Brutschy, B. A new way to detect noncovalently bonded complexes of biomolecules from liquid micro-droplets by laser mass spectrometry. Aust. J. Chem. 2006, 59, 109–114. [Google Scholar] [CrossRef]
- Mikhailov, V.A.; Liko, I.; Mize, T.H.; Bush, M.F.; Benesch, J.L.P.; Robinson, C.V. Infrared laser activation of soluble and membrane protein assemblies in the gas phase. Anal. Chem. 2016, 88, 7060–7067. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, J.; Radeva, N.; Köster, H.; Metz, A.; Krotzky, T.; Kuhnert, M.; Diederich, W.E.; Heine, A.; Neumann, L.; Atmanene, C.; et al. One question, multiple answers: Biochemical and biophysical screening methods retrieve deviating fragment hit lists. ChemMedChem 2015, 10, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Schiebel, J.; Radeva, N.; Krimmer, S.G.; Wang, X.; Stieler, M.; Ehrmann, F.R.; Fu, K.; Metz, A.; Huschmann, F.U.; Weiss, M.S.; et al. Six biophysical screening methods miss a large proportion of crystallographically discovered fragment hits: A case study. ACS Chem. Biol. 2016, 11, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-J.; Hubbard, R.E. Lessons for fragment library design: Analysis of output from multiple screening campaigns. J. Comput. Aided Mol. Des. 2009, 23, 603–620. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Target | Library Size | Primary Screening | Hit Validation | Relative Binding Affinty | Absolute Binding Affinity | Binding Specificity | Reference |
|---|---|---|---|---|---|---|---|
| Subdomain of 23S rRNA | NS | × | − | × | − | × | [93] |
| Stromelysin | 15 | × | − | − | × | × | [94] |
| bCA II | 10 | × | − | − | − | − | [95] |
| XIAP | NS | − | × | − | − | × | [96] |
| PNMT | 12 | − | × | − | − | − | [72] |
| Hsp90 | 350 | × | − | − | − | − | [73] |
| Hsp90 | 60 | − | × | − | − | − | [73] |
| Bcl-xL | 157 | × | − | − | × | − | [74] |
| PfdUTPase | 331 | × | − | × | − | − | [97] |
| hCA II | 720 | − | × | × | − | − | [75] |
| hCA II | 70 | × | − | − | − | − | [75] |
| Endothiapepsin | 361 | × | − | − | − | − | [104,105] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedro, L.; Quinn, R.J. Native Mass Spectrometry in Fragment-Based Drug Discovery. Molecules 2016, 21, 984. https://doi.org/10.3390/molecules21080984
Pedro L, Quinn RJ. Native Mass Spectrometry in Fragment-Based Drug Discovery. Molecules. 2016; 21(8):984. https://doi.org/10.3390/molecules21080984
Chicago/Turabian StylePedro, Liliana, and Ronald J. Quinn. 2016. "Native Mass Spectrometry in Fragment-Based Drug Discovery" Molecules 21, no. 8: 984. https://doi.org/10.3390/molecules21080984
APA StylePedro, L., & Quinn, R. J. (2016). Native Mass Spectrometry in Fragment-Based Drug Discovery. Molecules, 21(8), 984. https://doi.org/10.3390/molecules21080984