
σ-Bond Electron Delocalization in Oligosilanes as Function of Substitution Pattern, Chain Length, and Spatial Orientation



 and
and
Abstract
:
1. Introduction
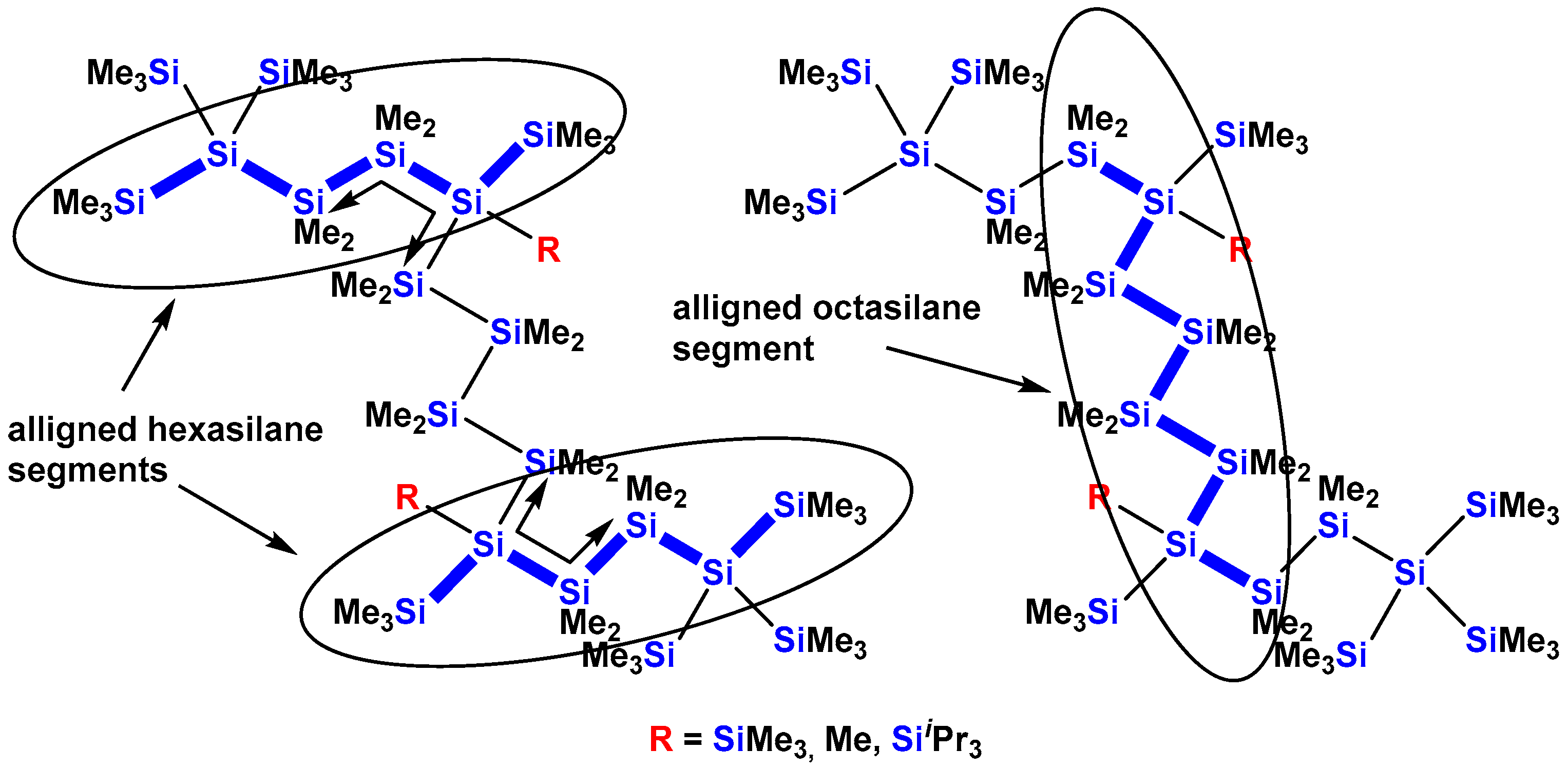
2. Results
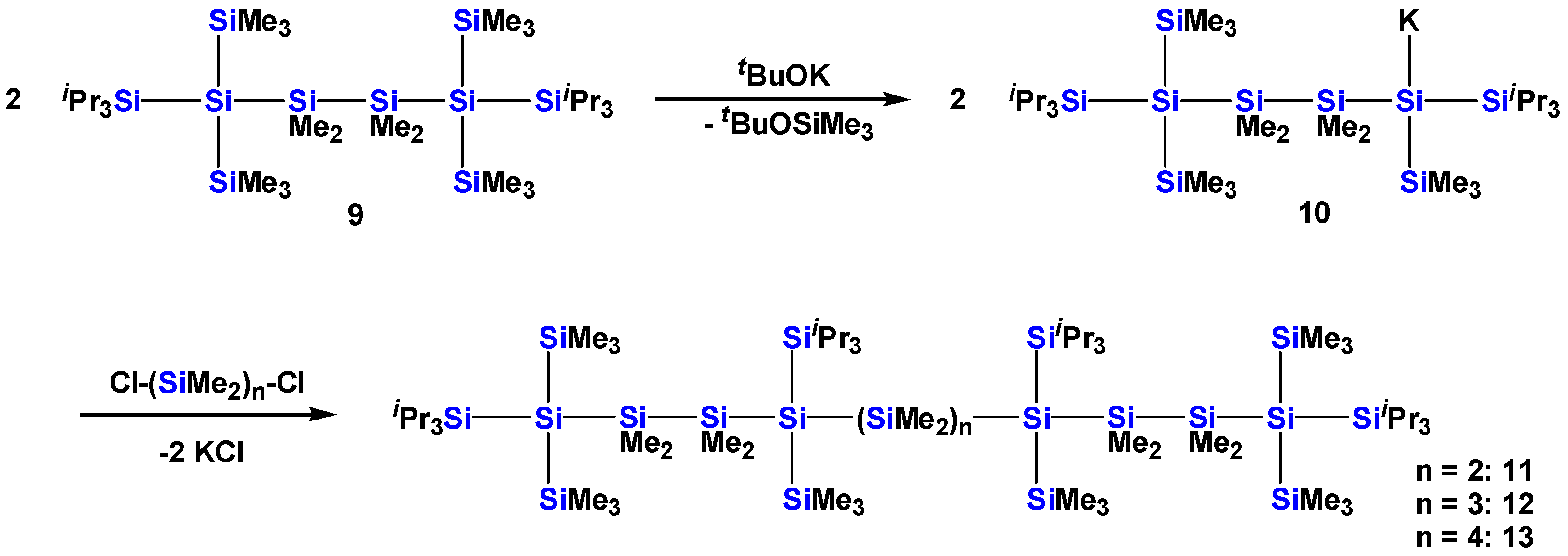
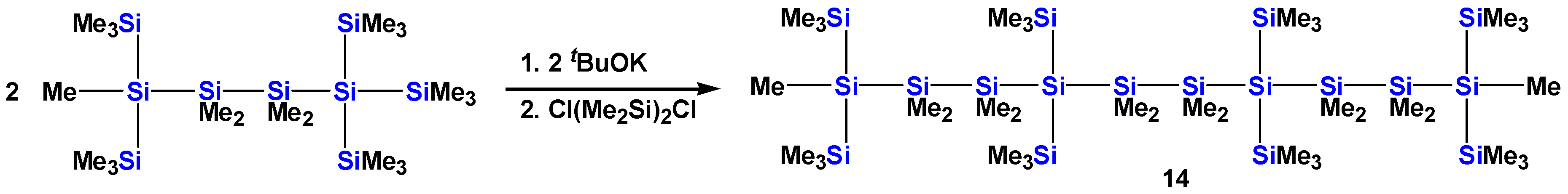
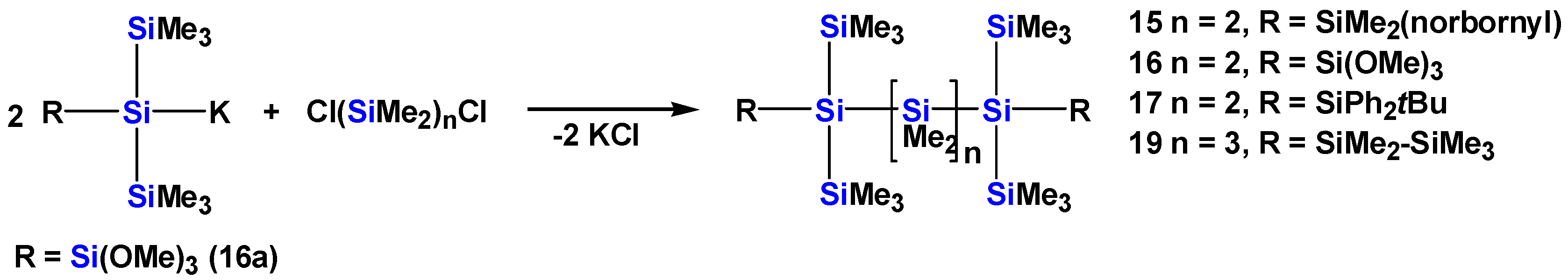
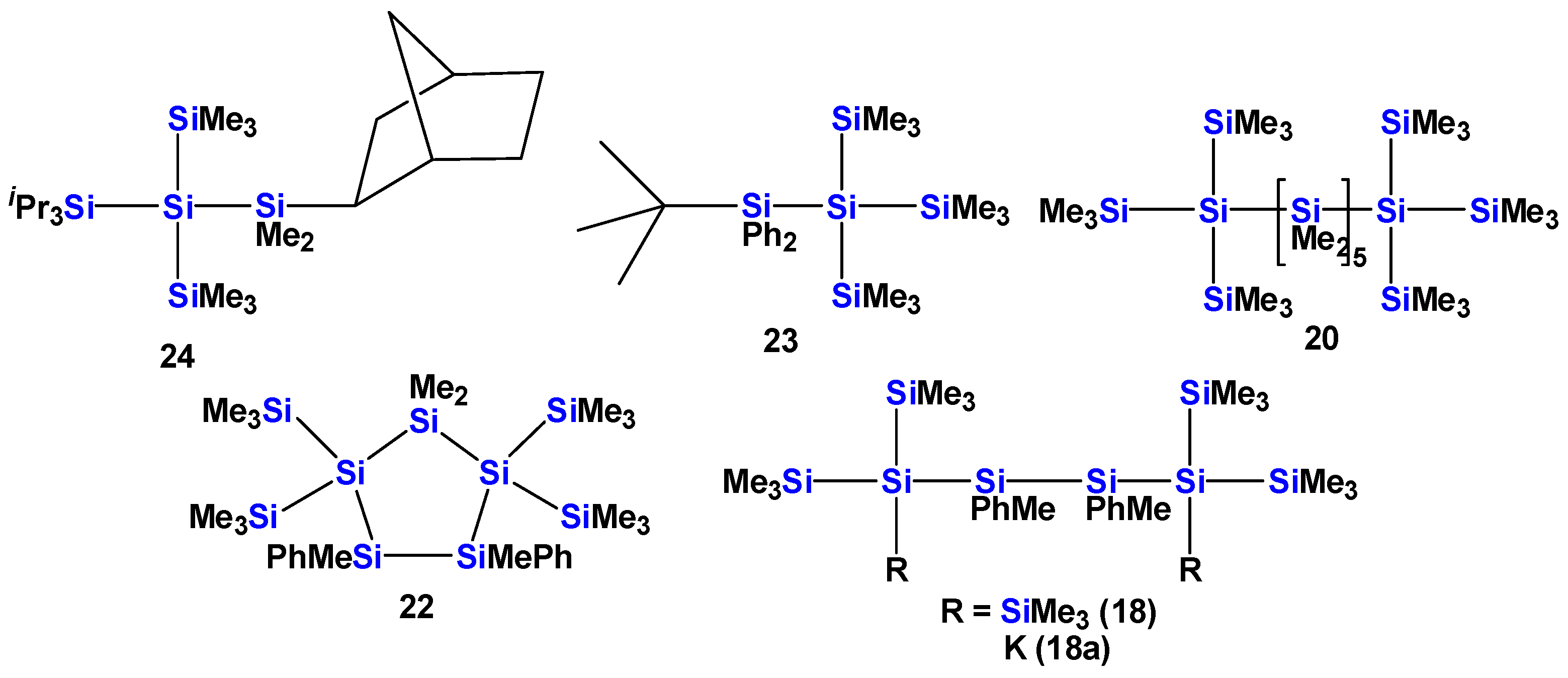
2.1. Synthesis of Oligosilanes
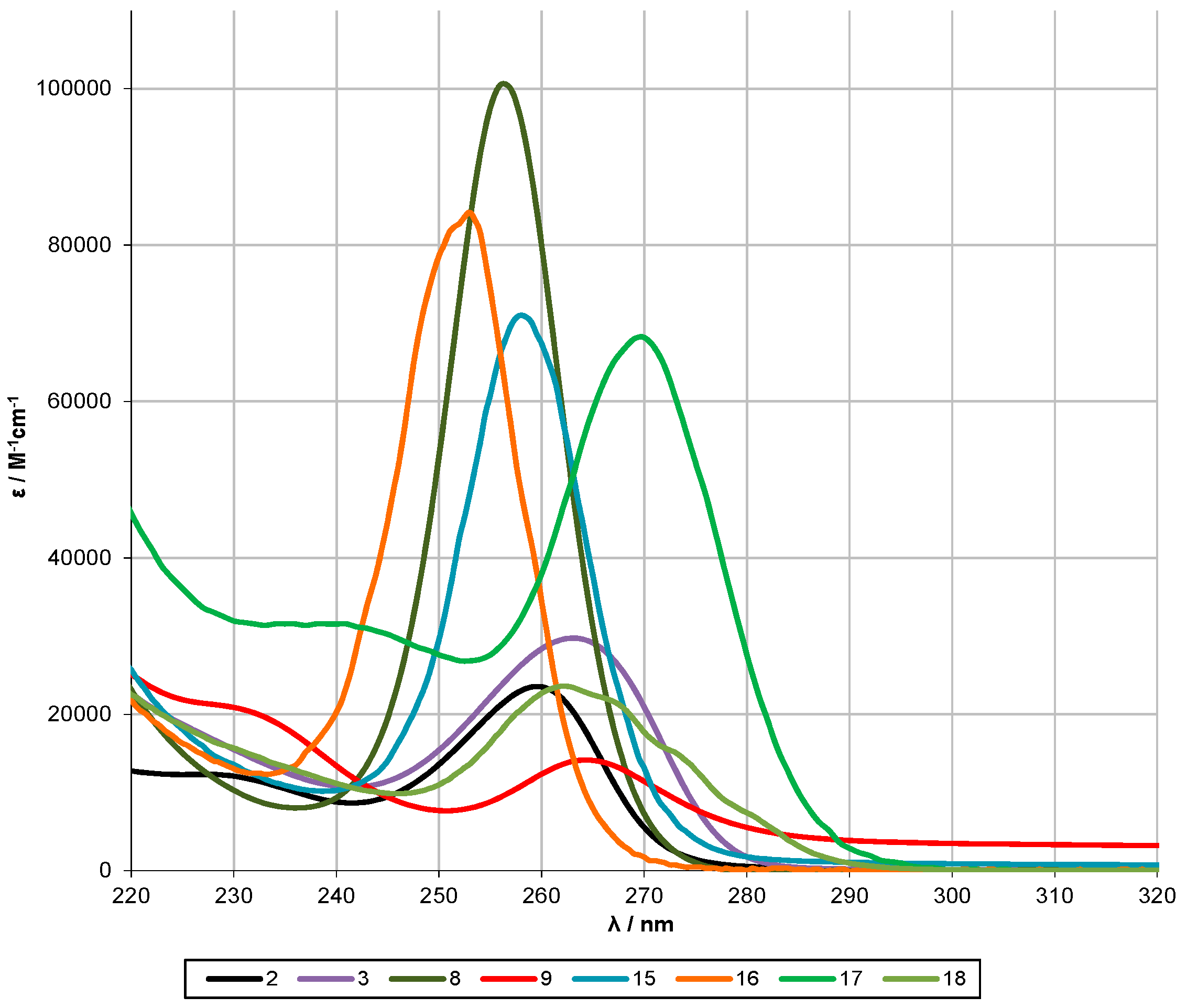
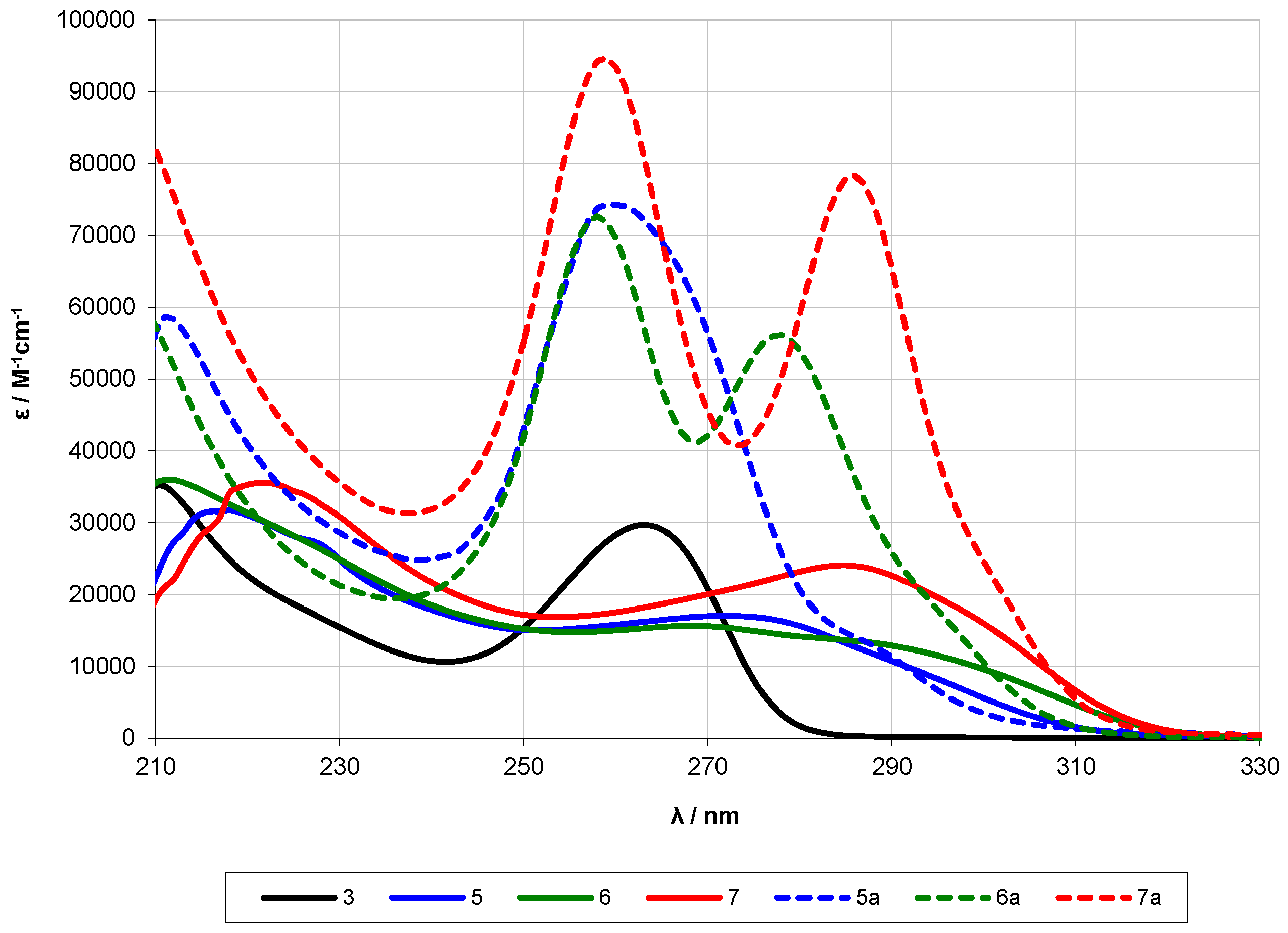
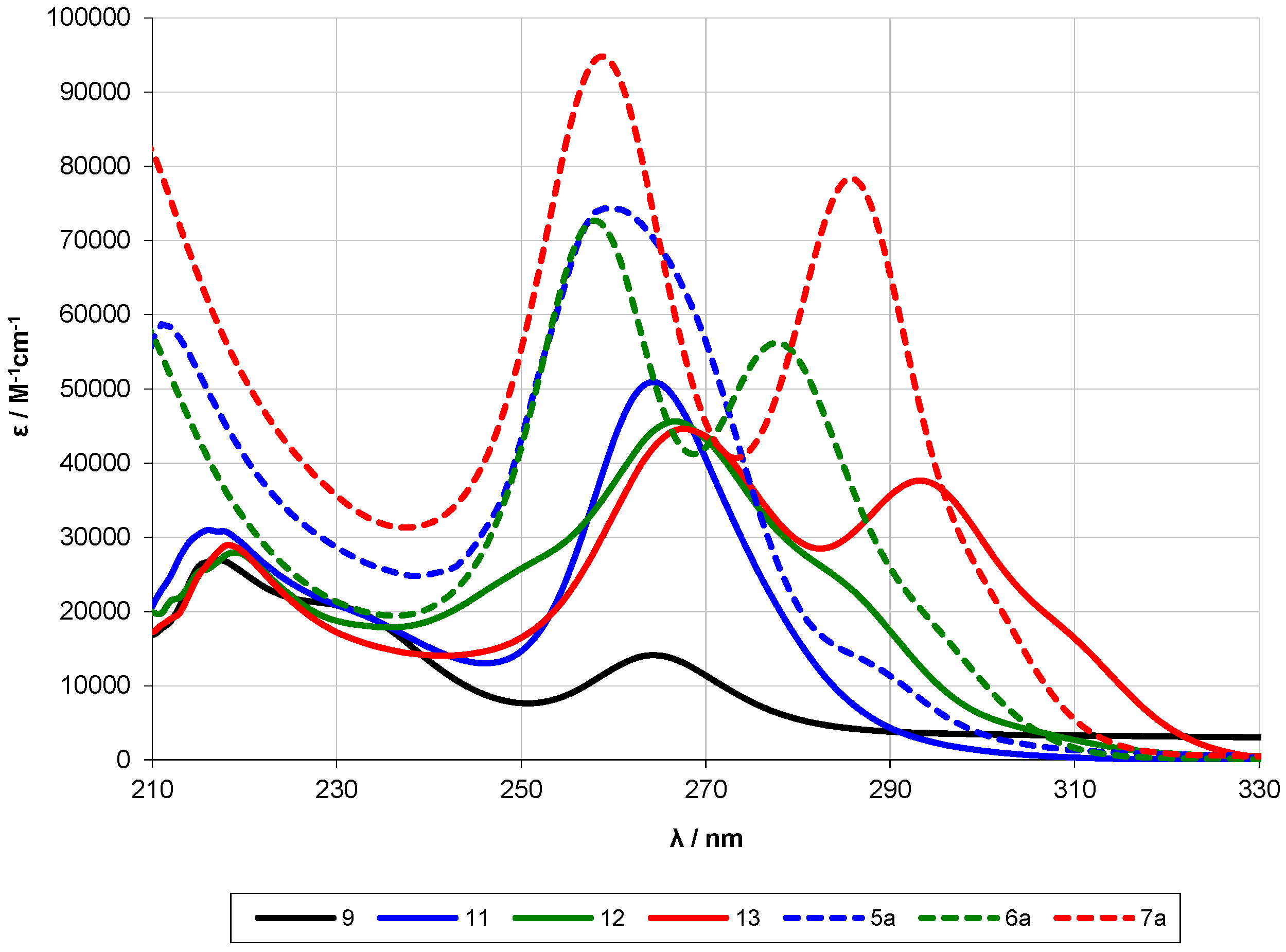
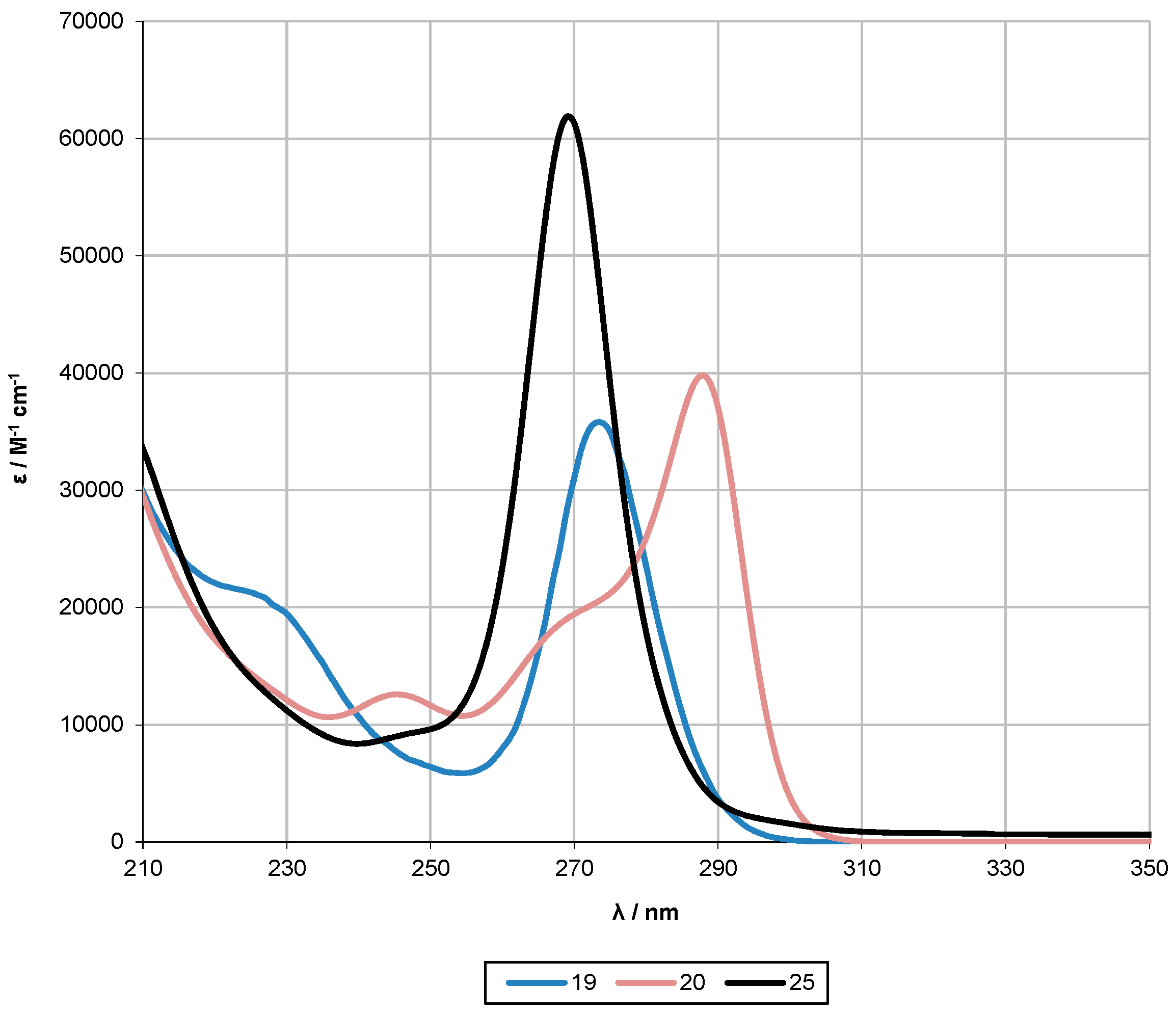
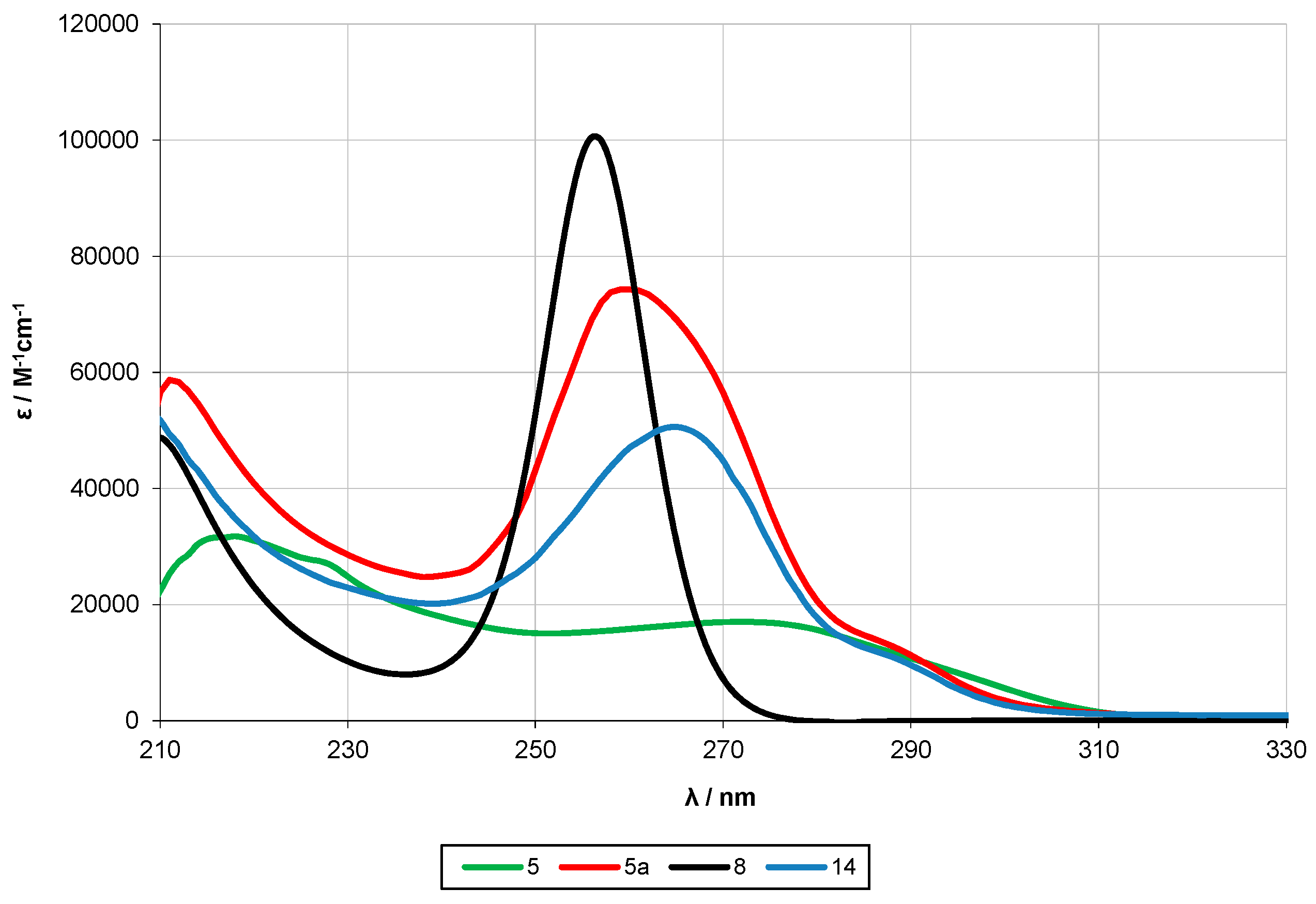
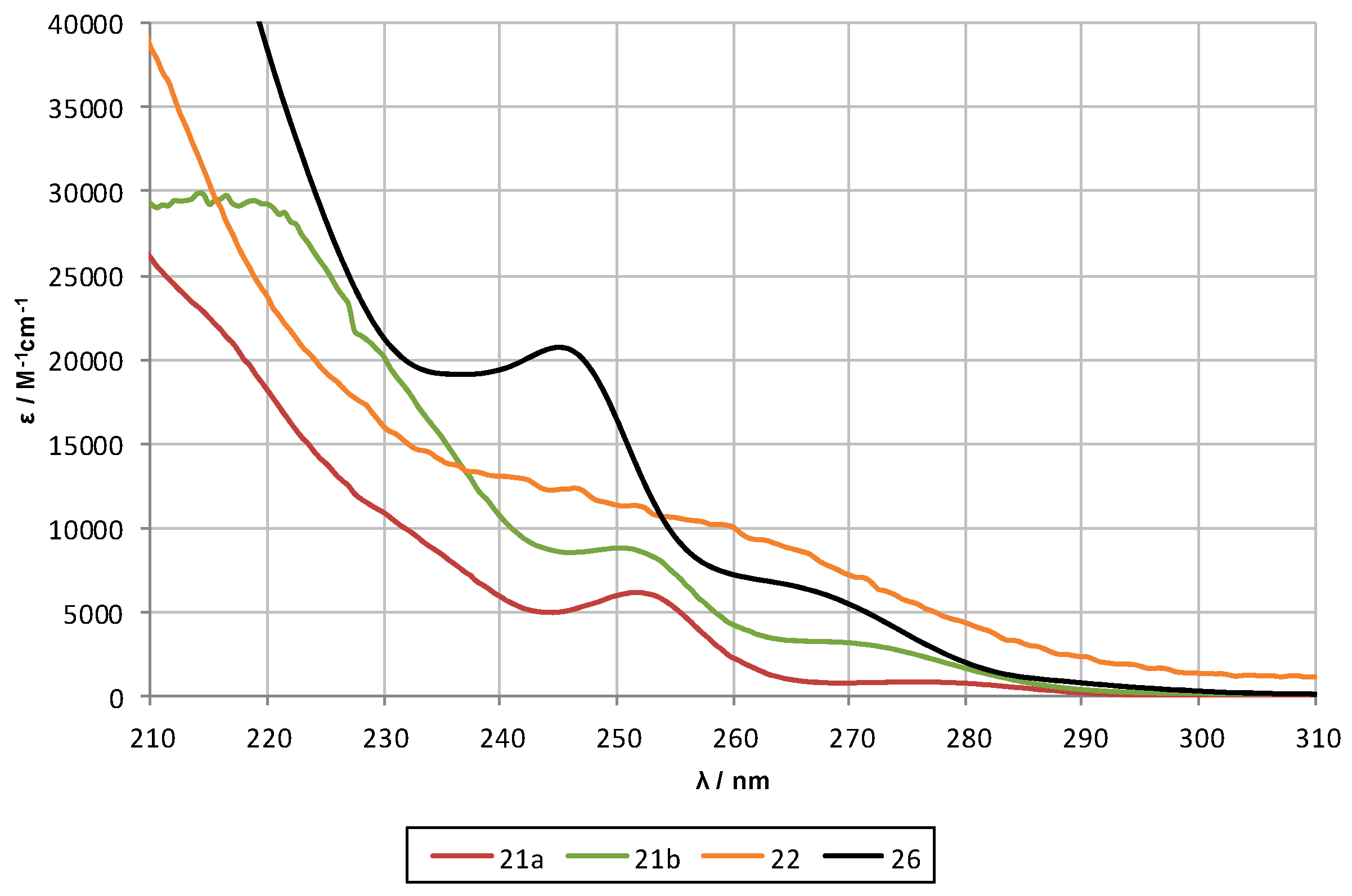
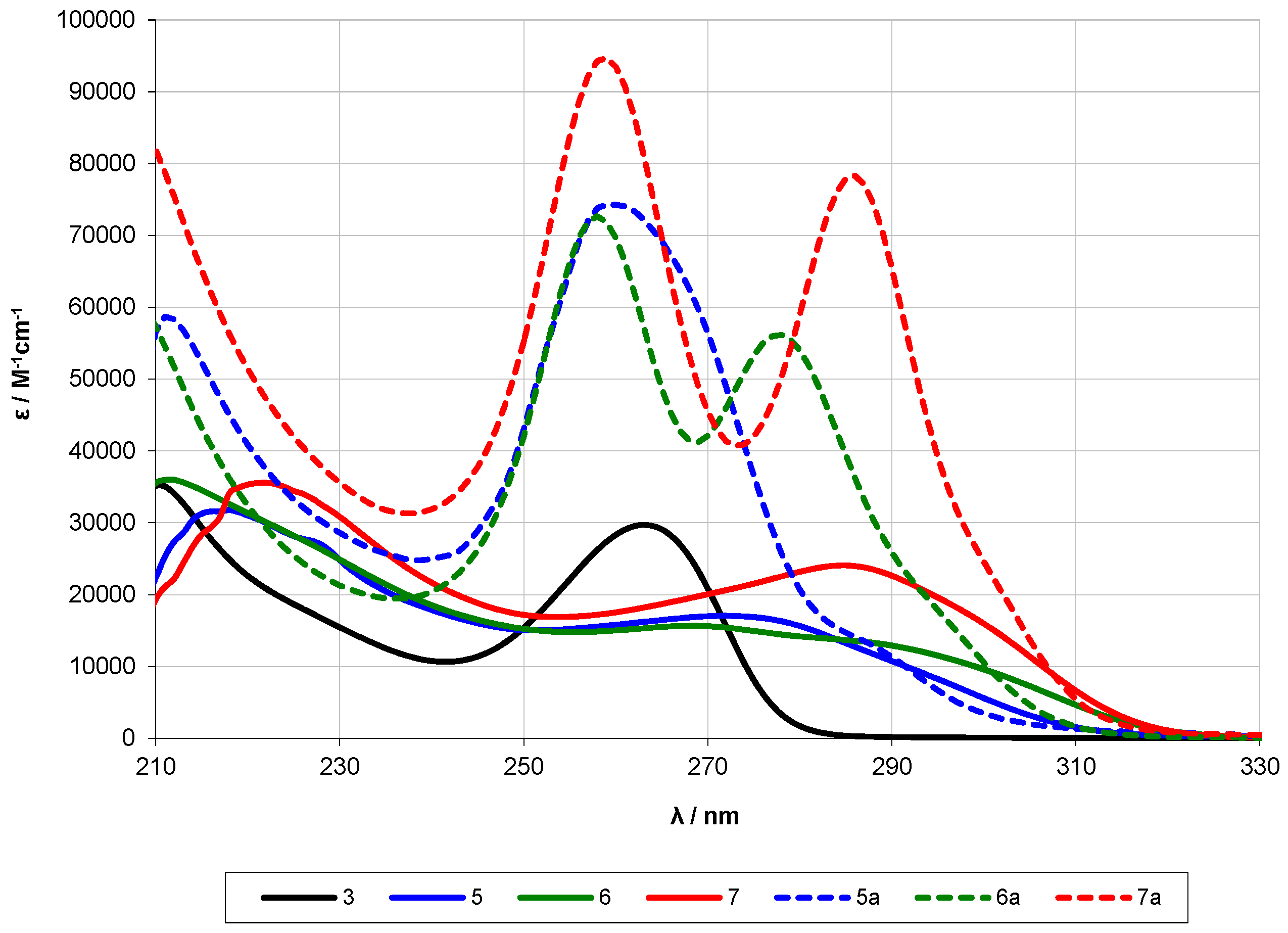
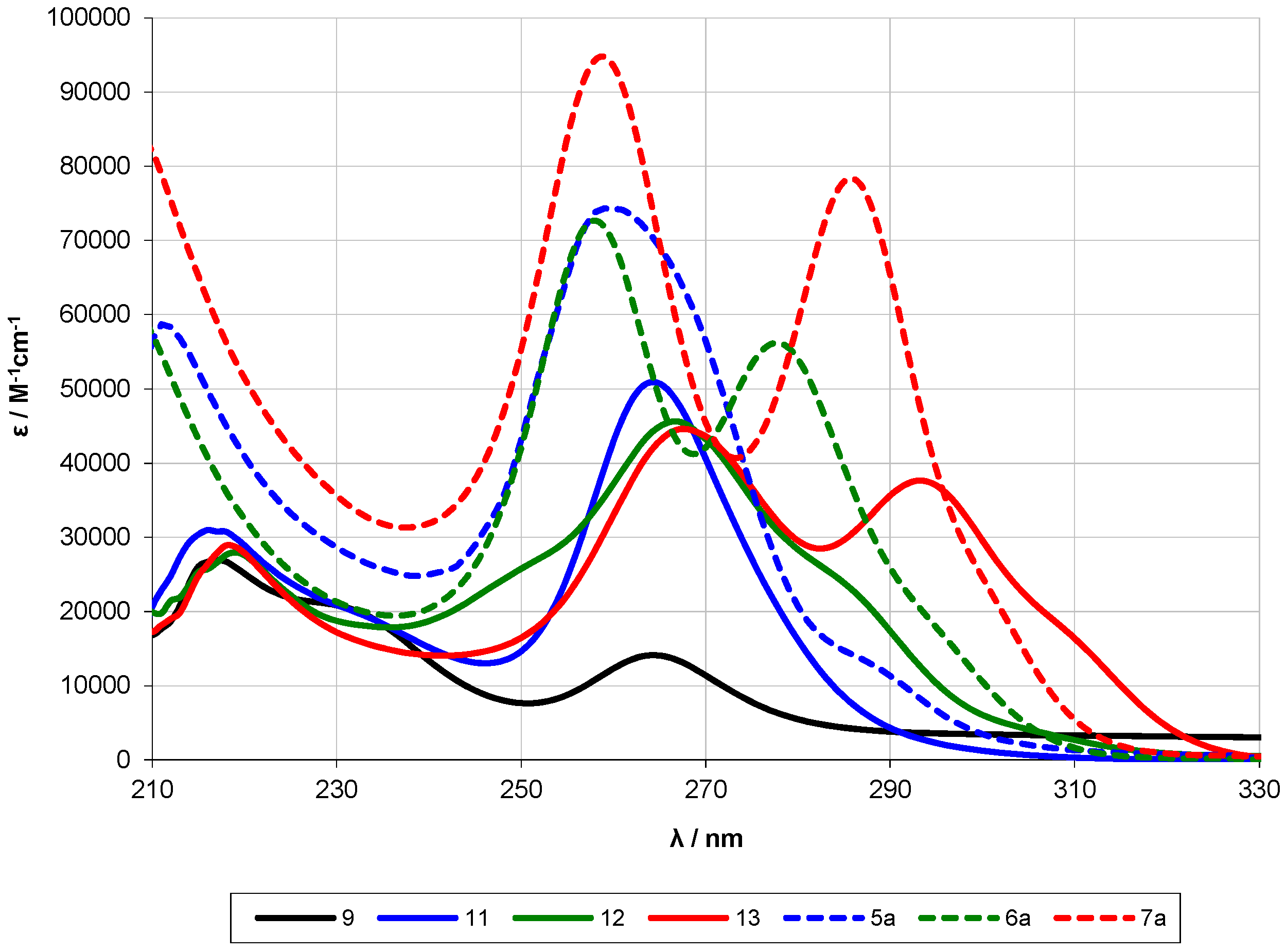
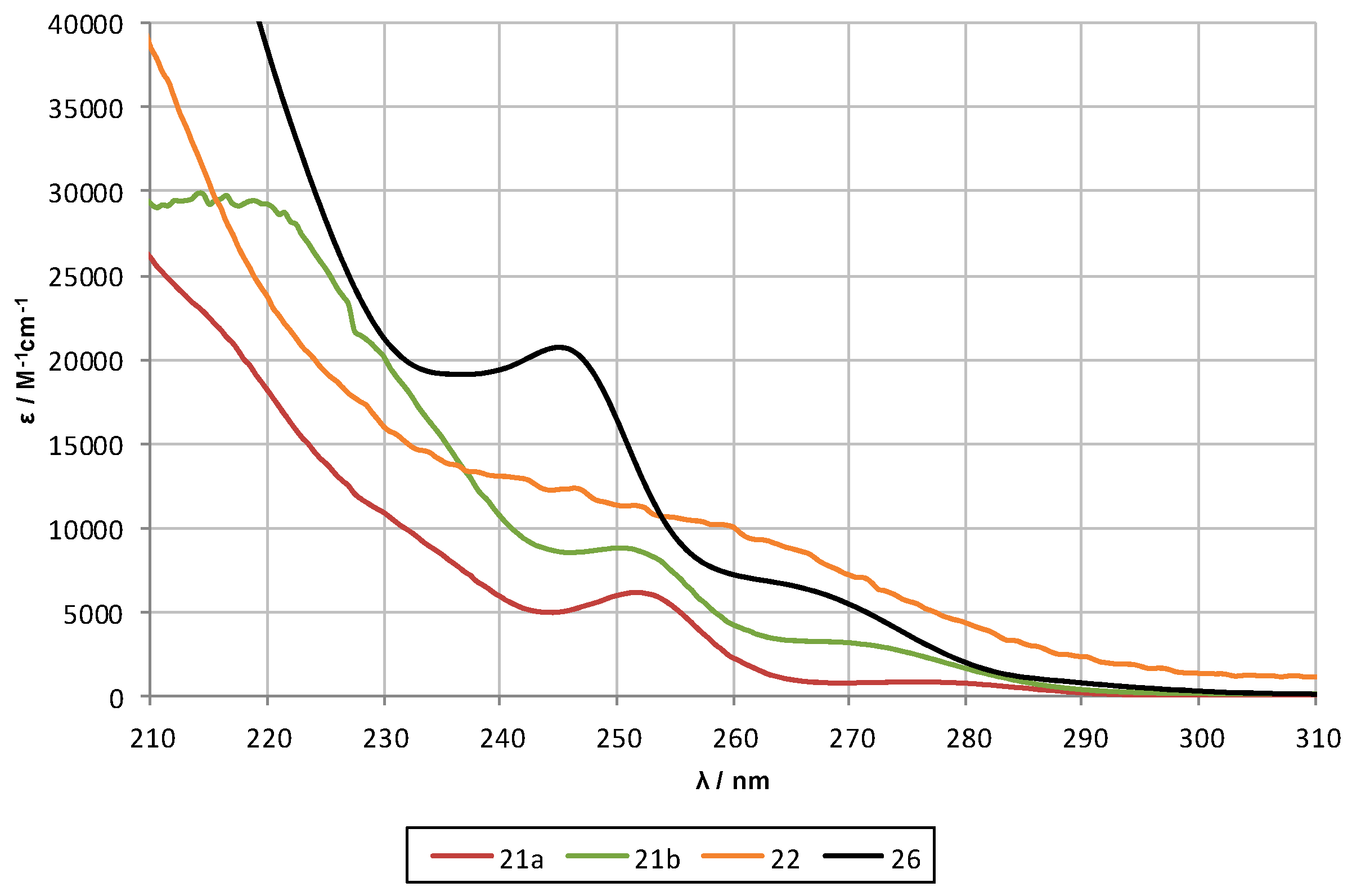
2.2. UV-Spectroscopy Studies
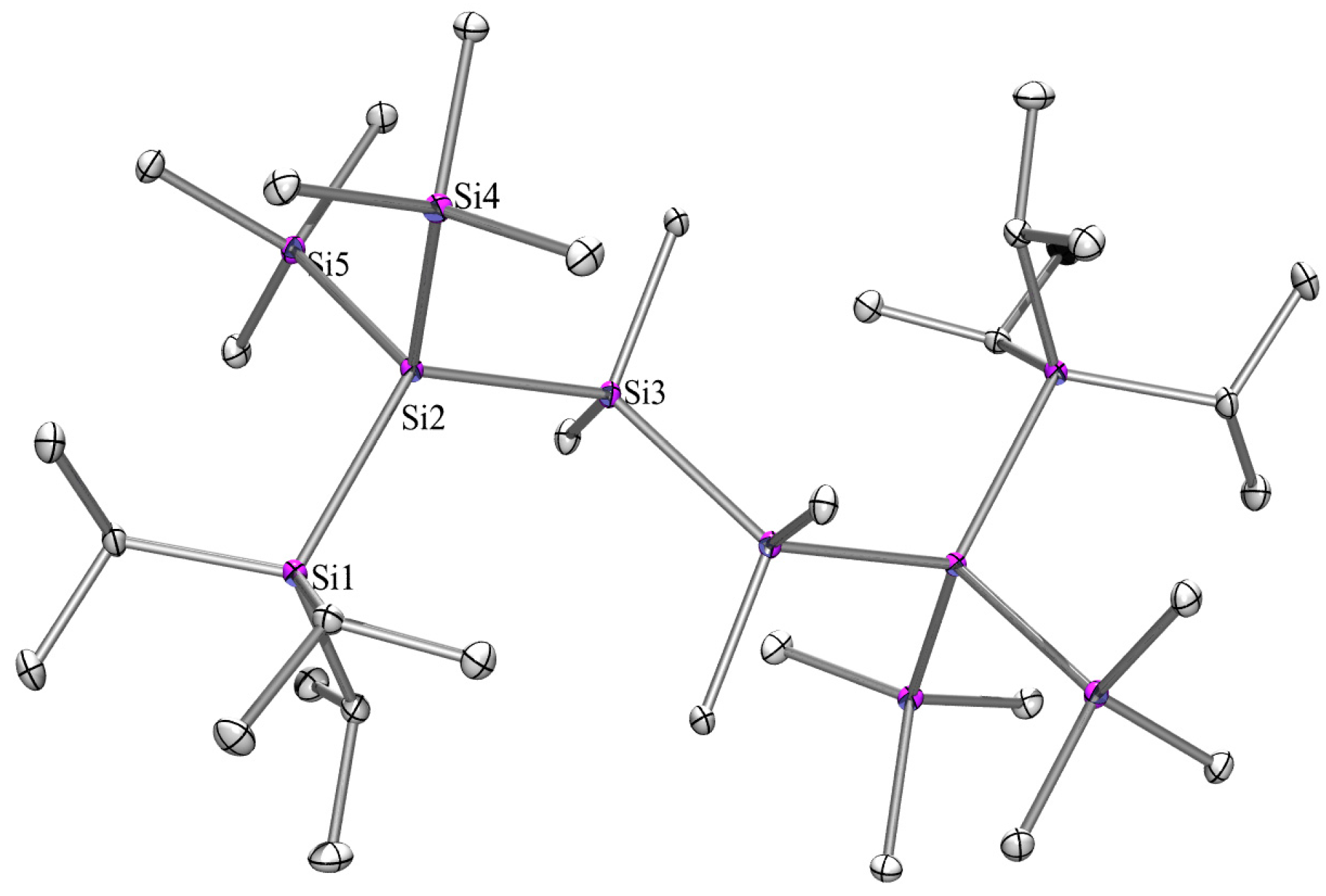
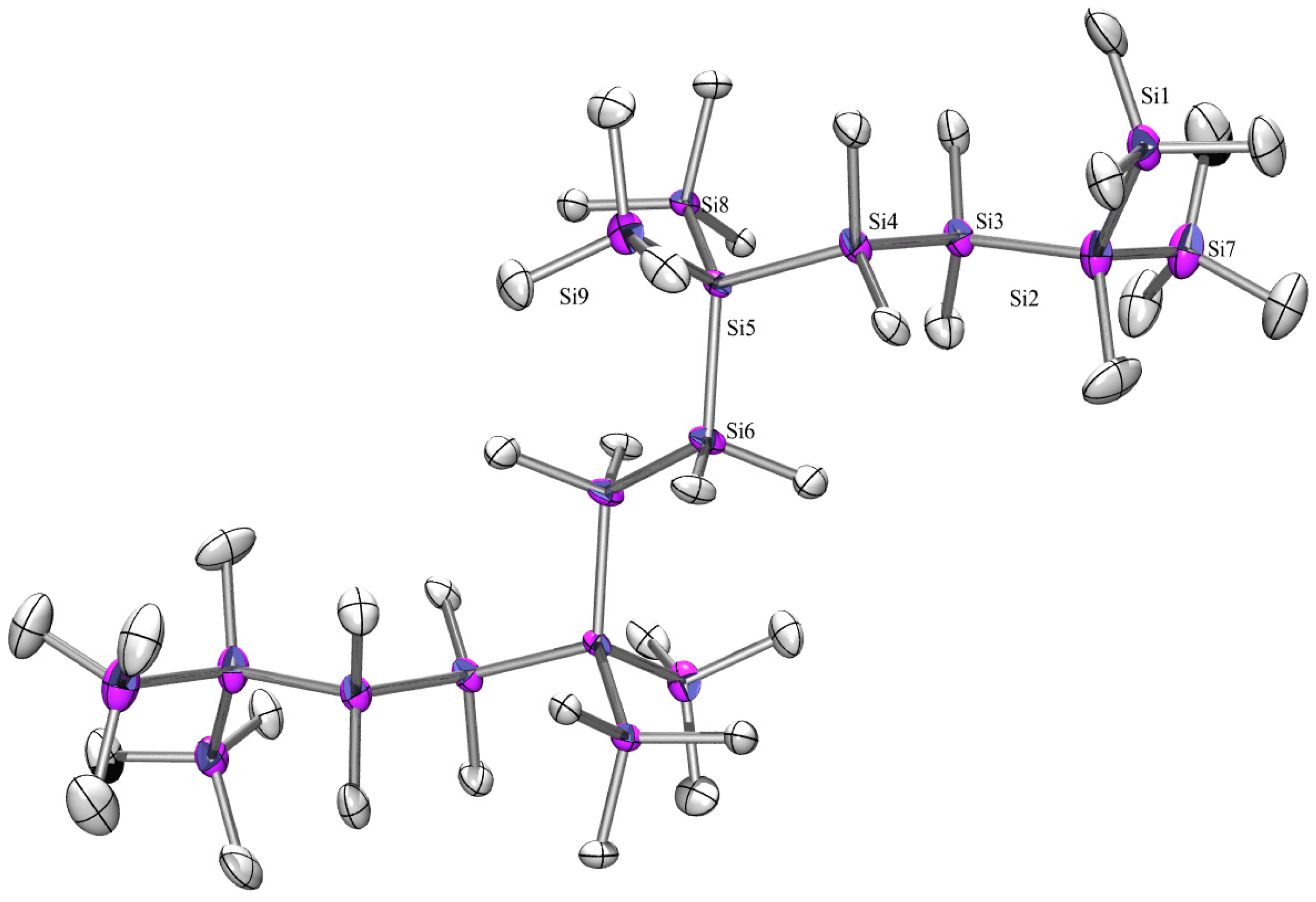
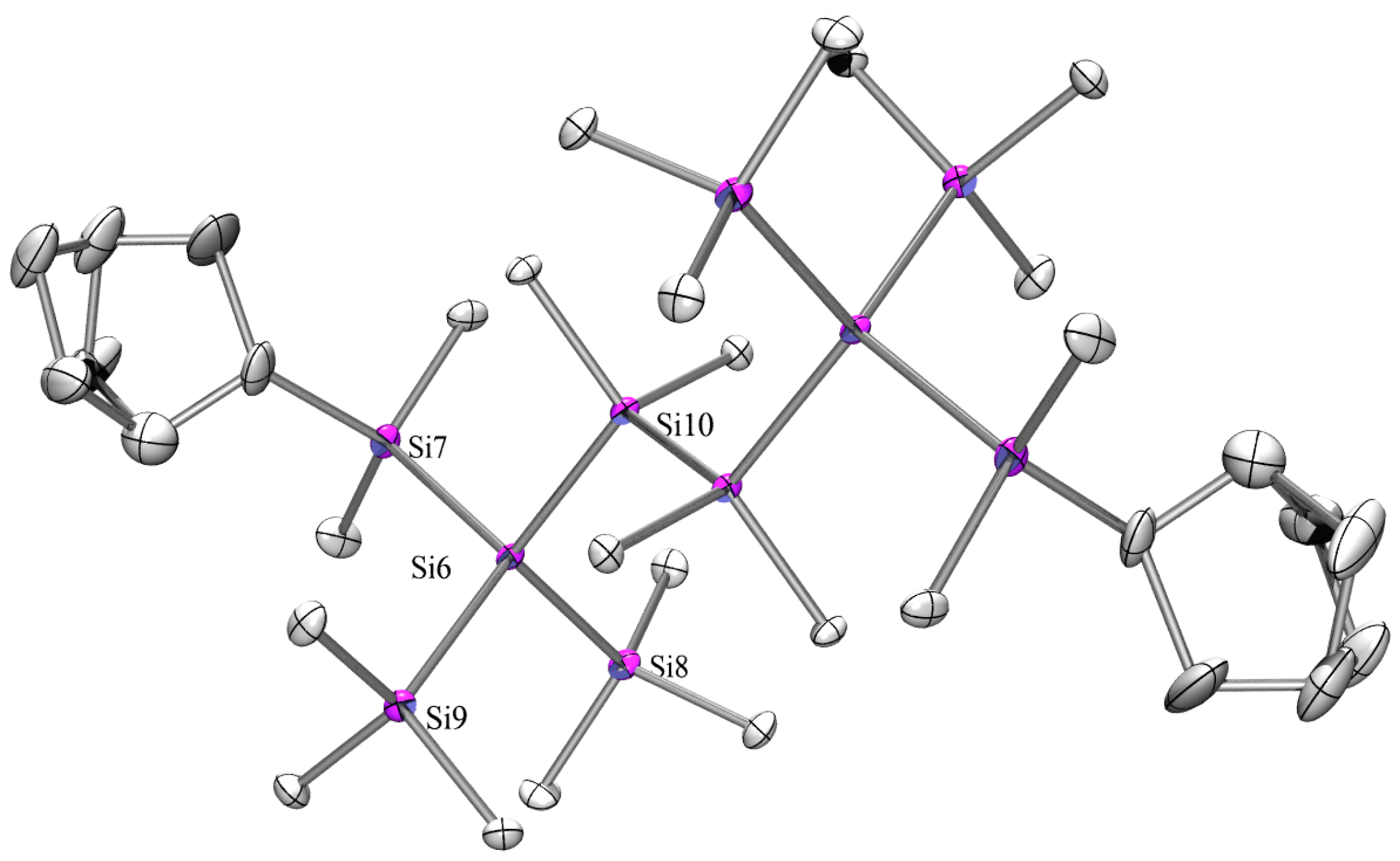
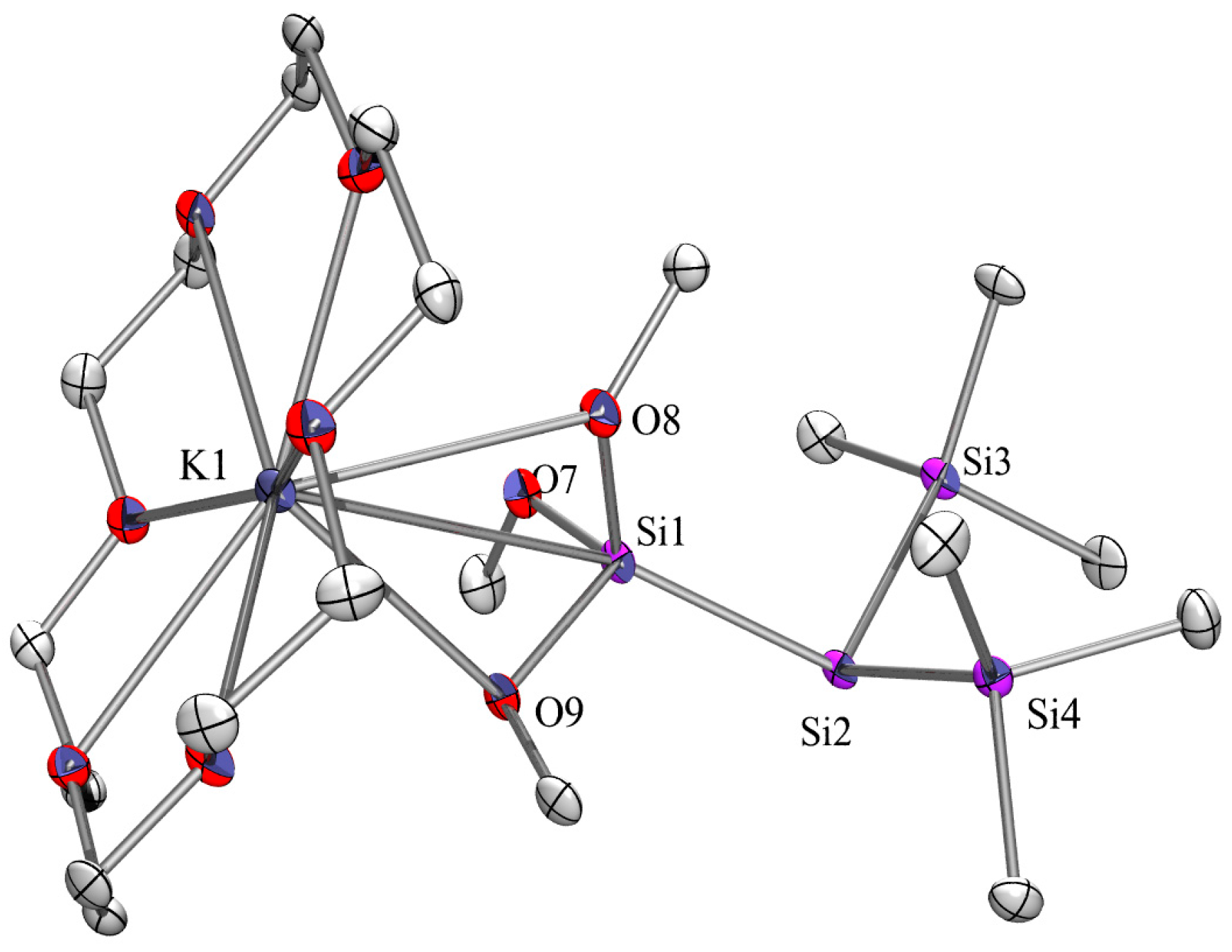
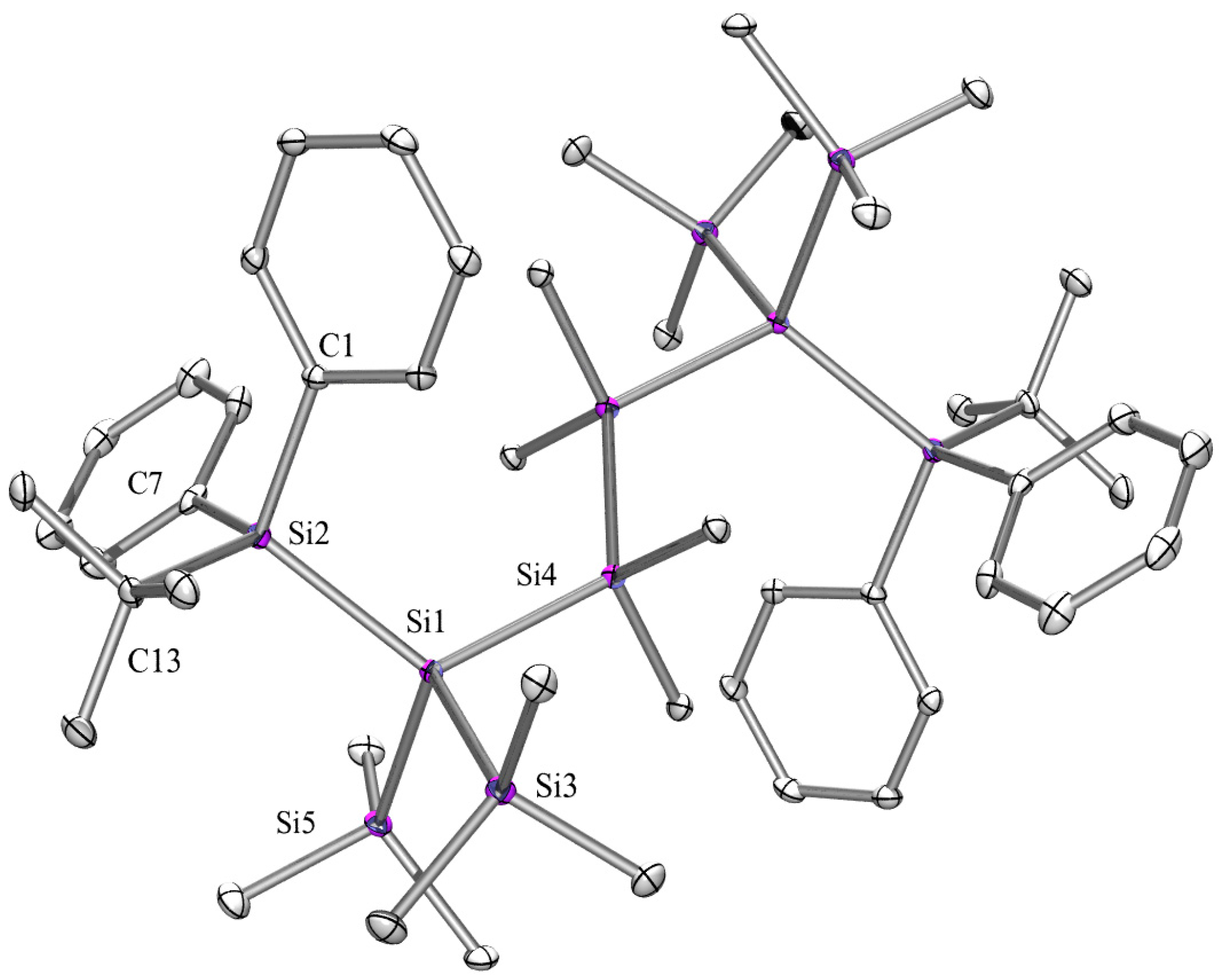
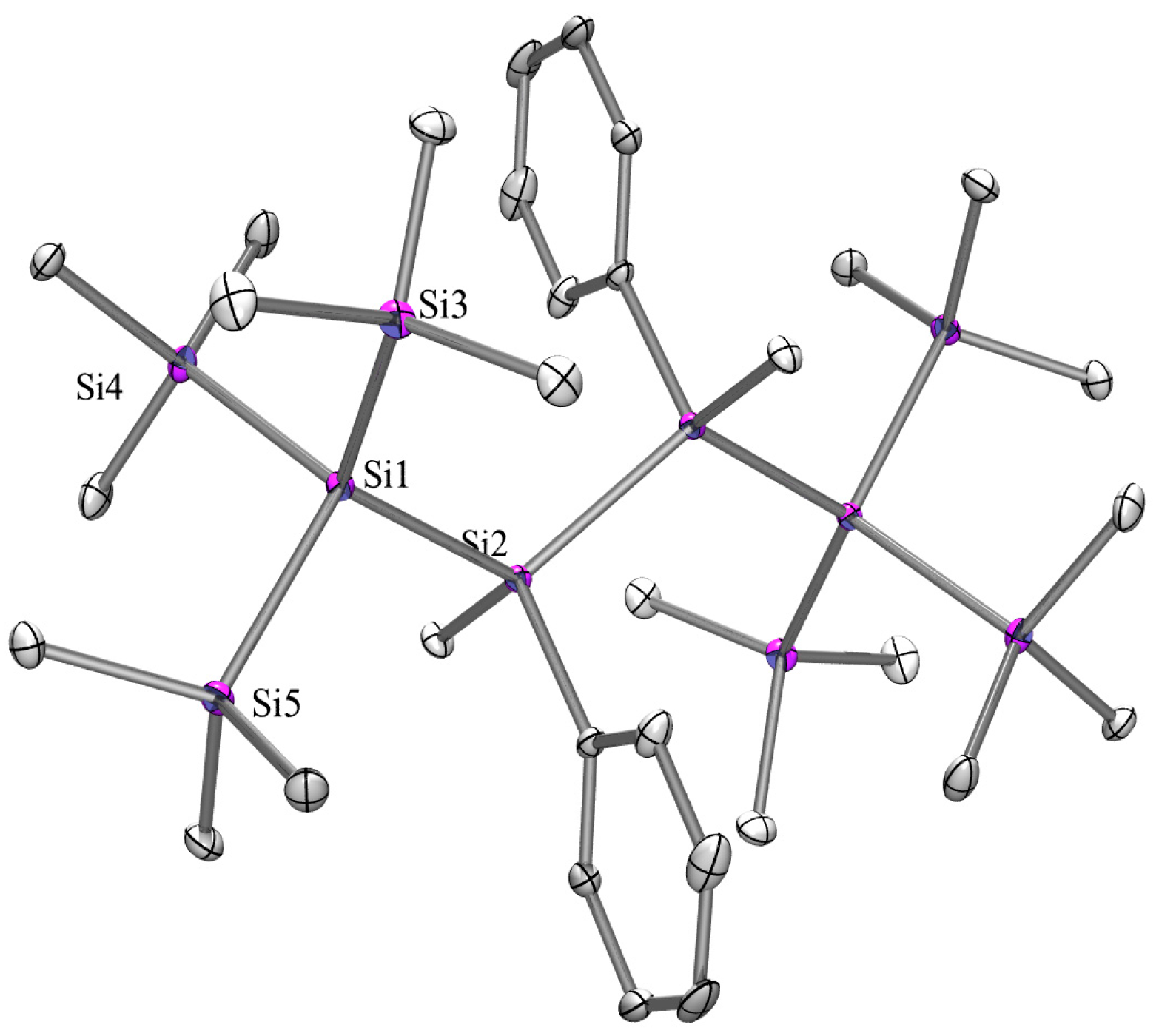
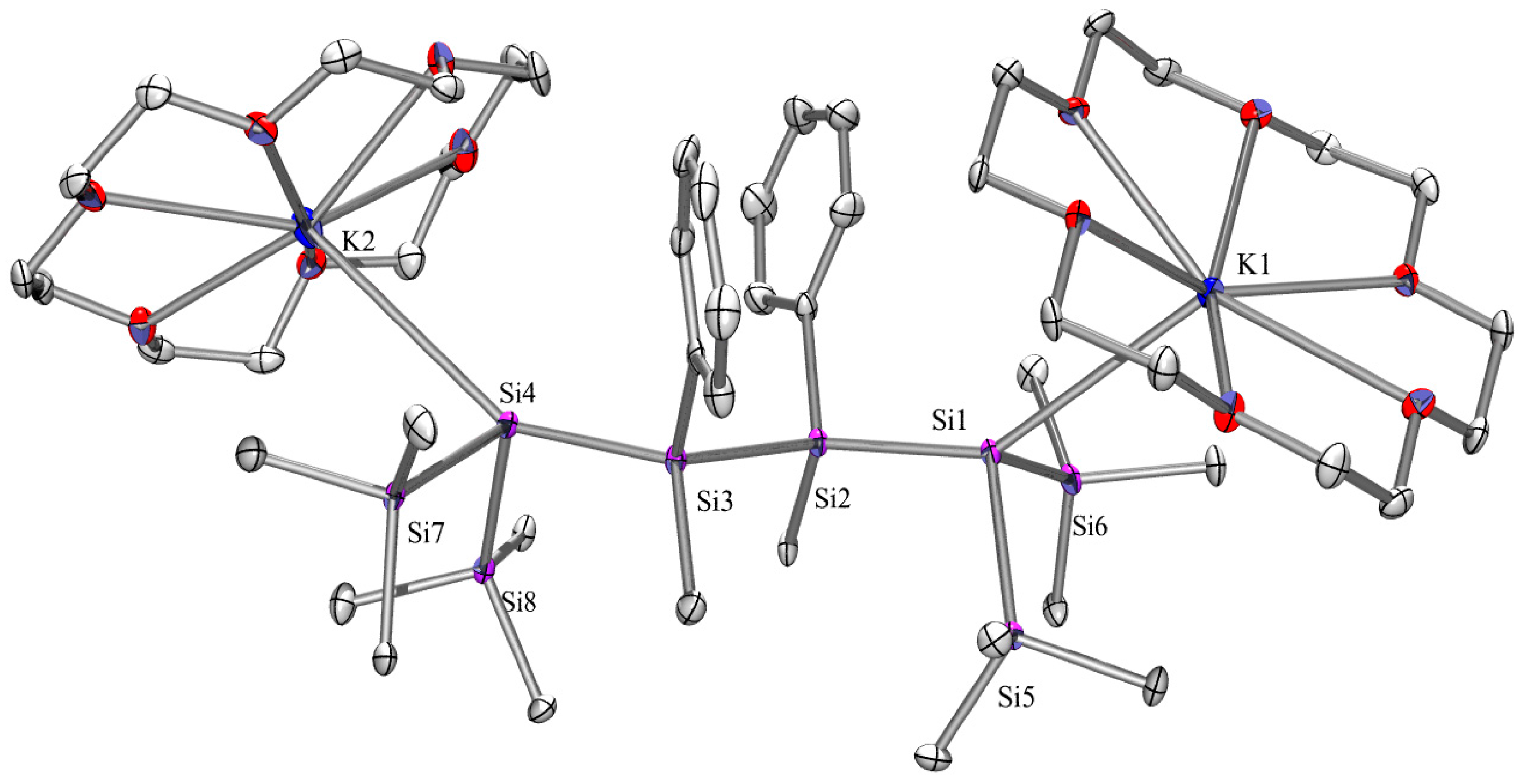
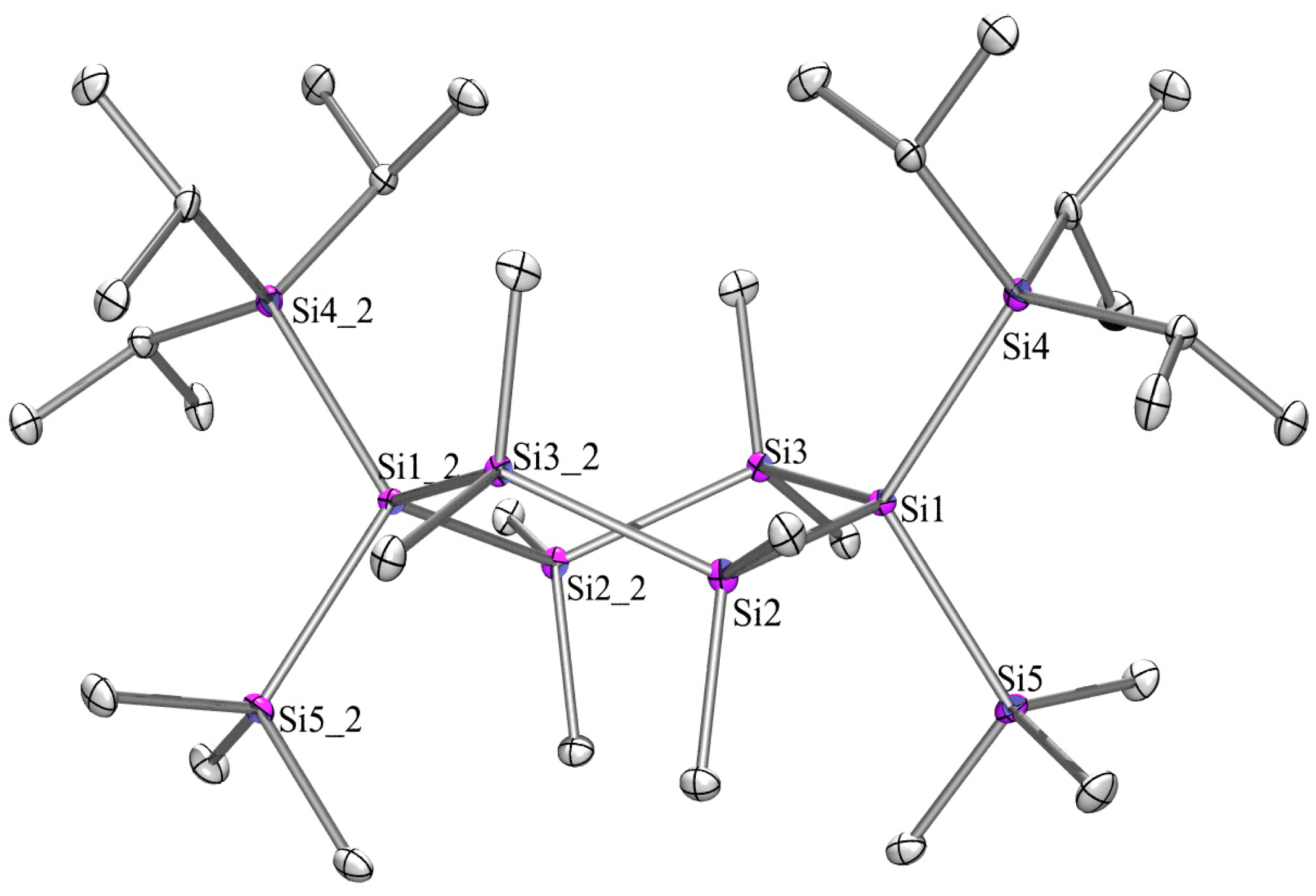
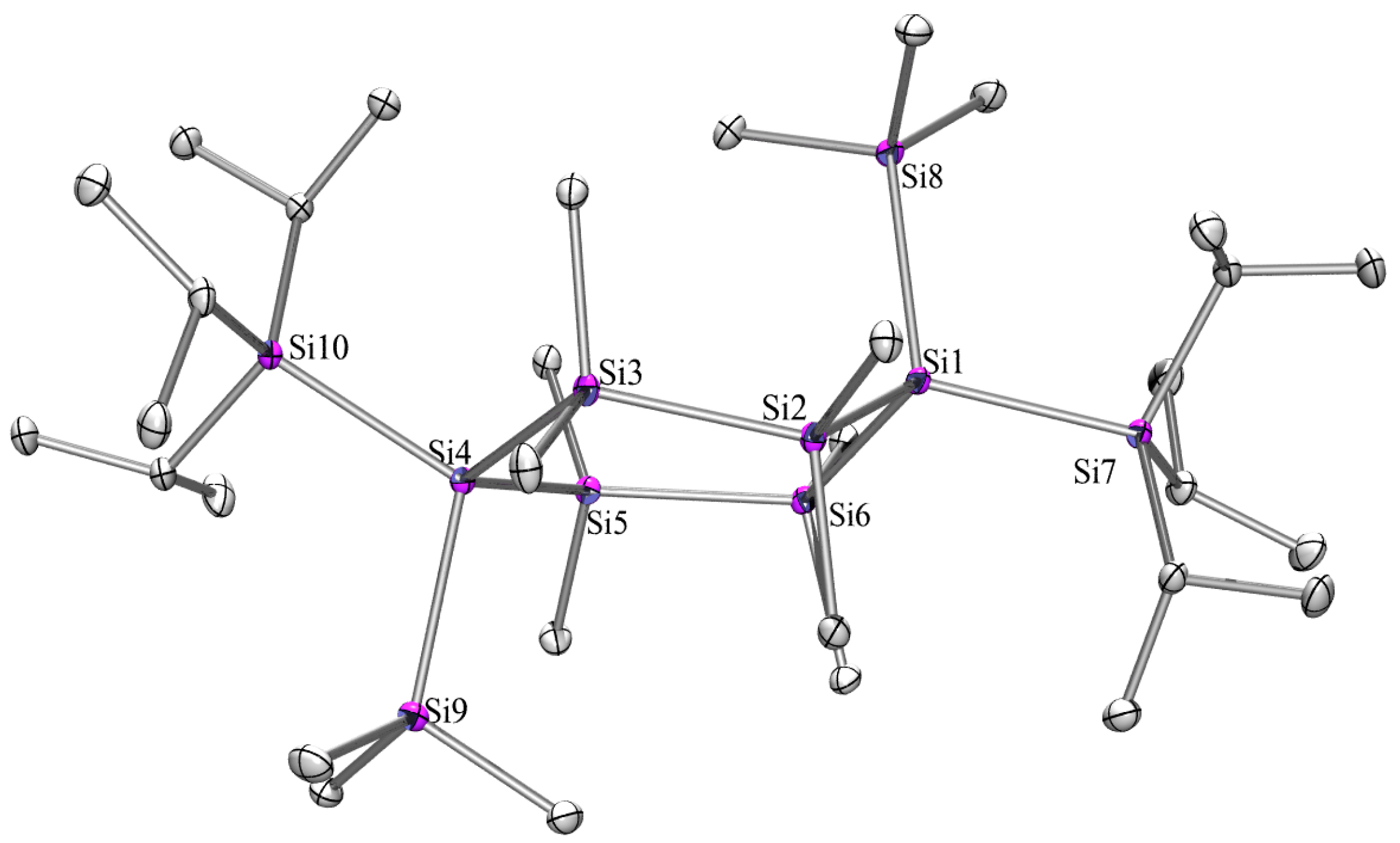
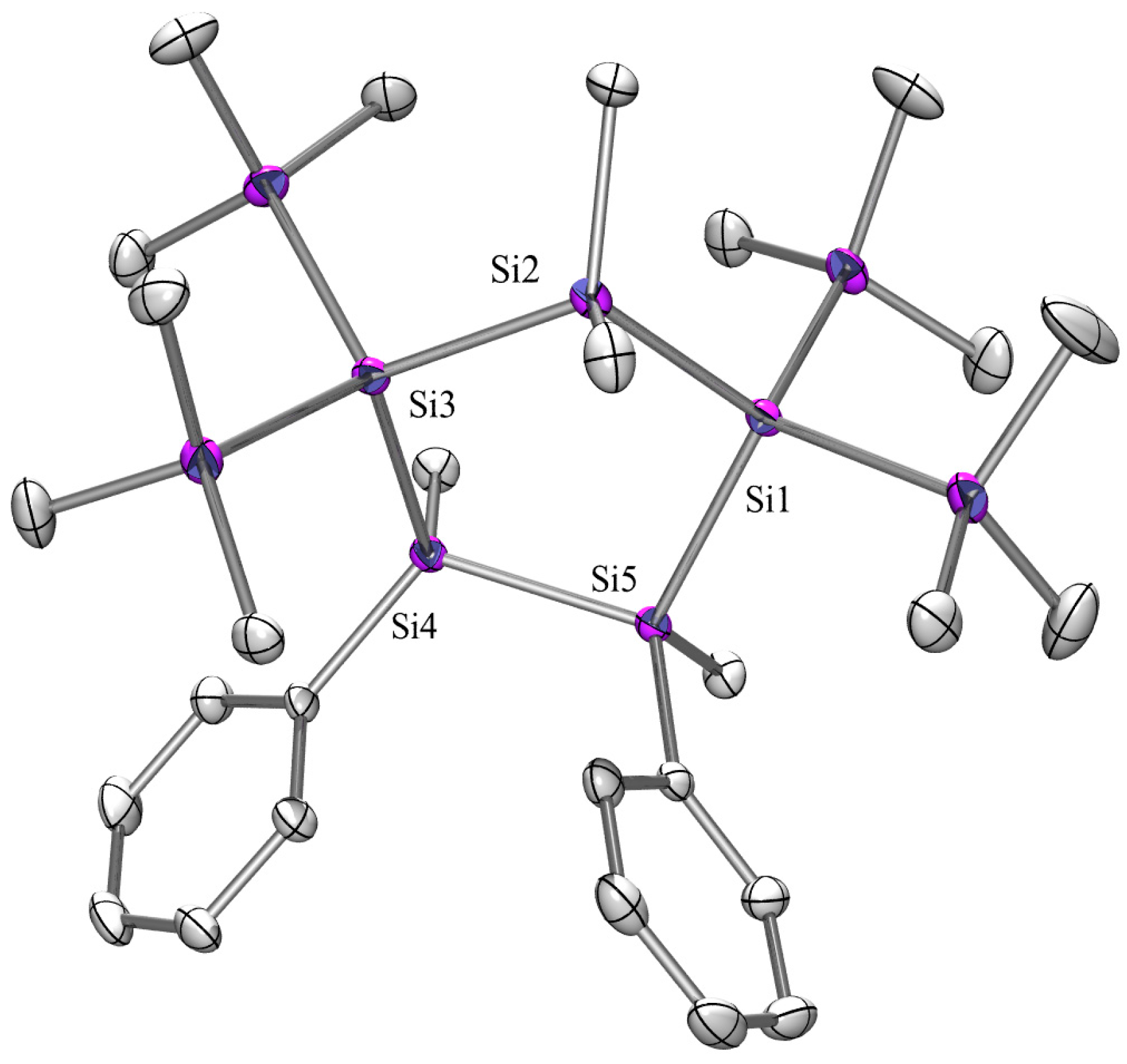
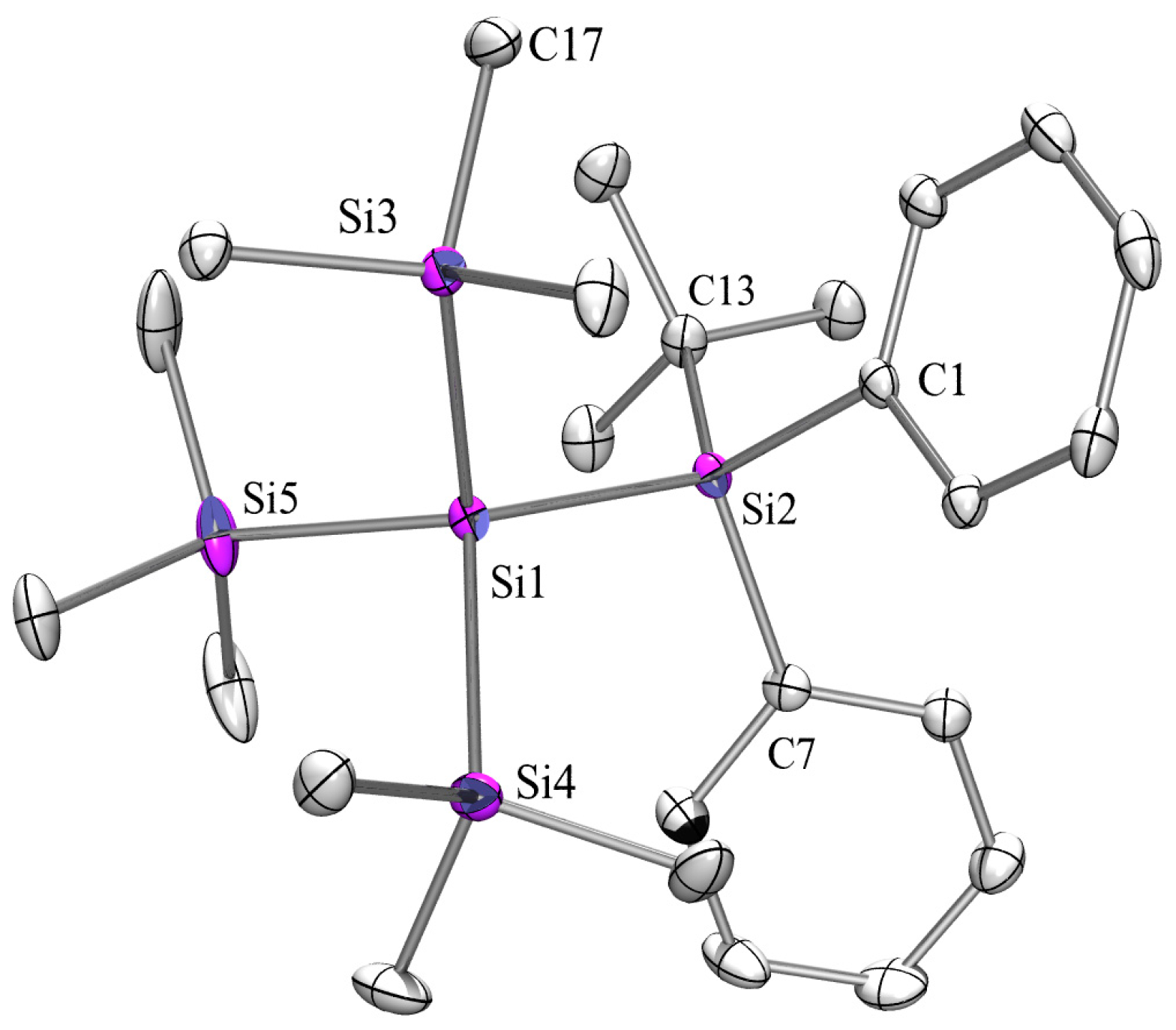
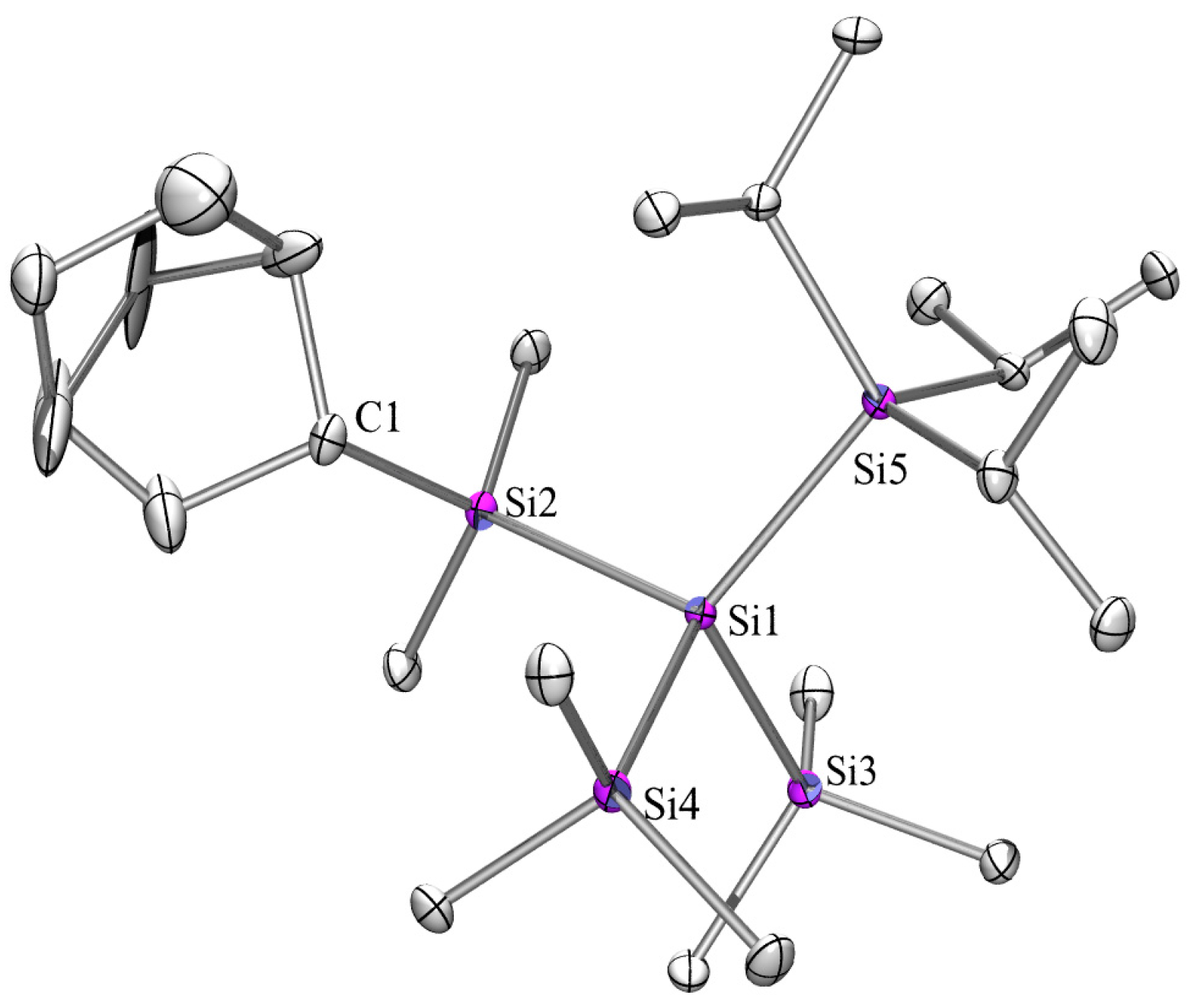
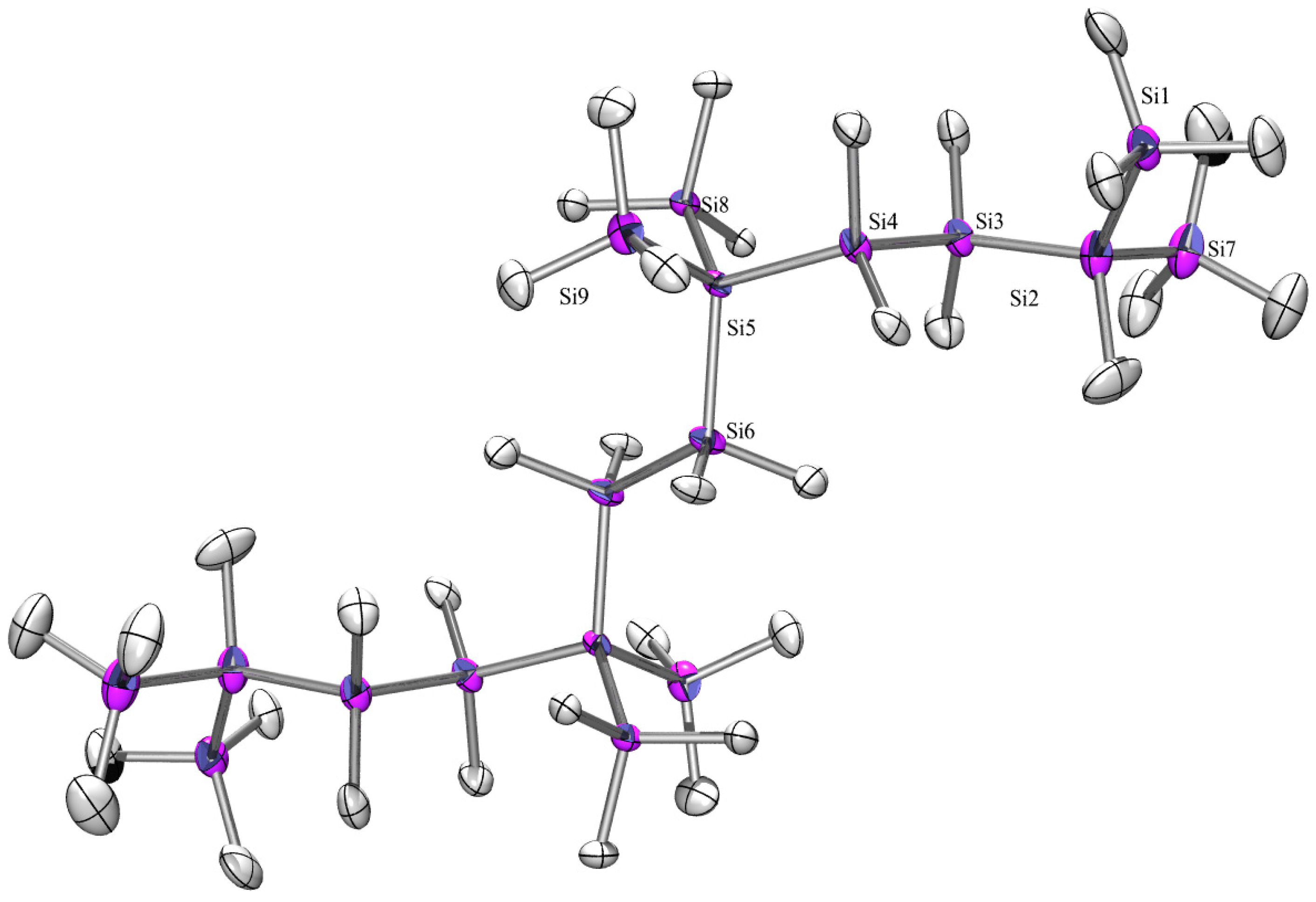
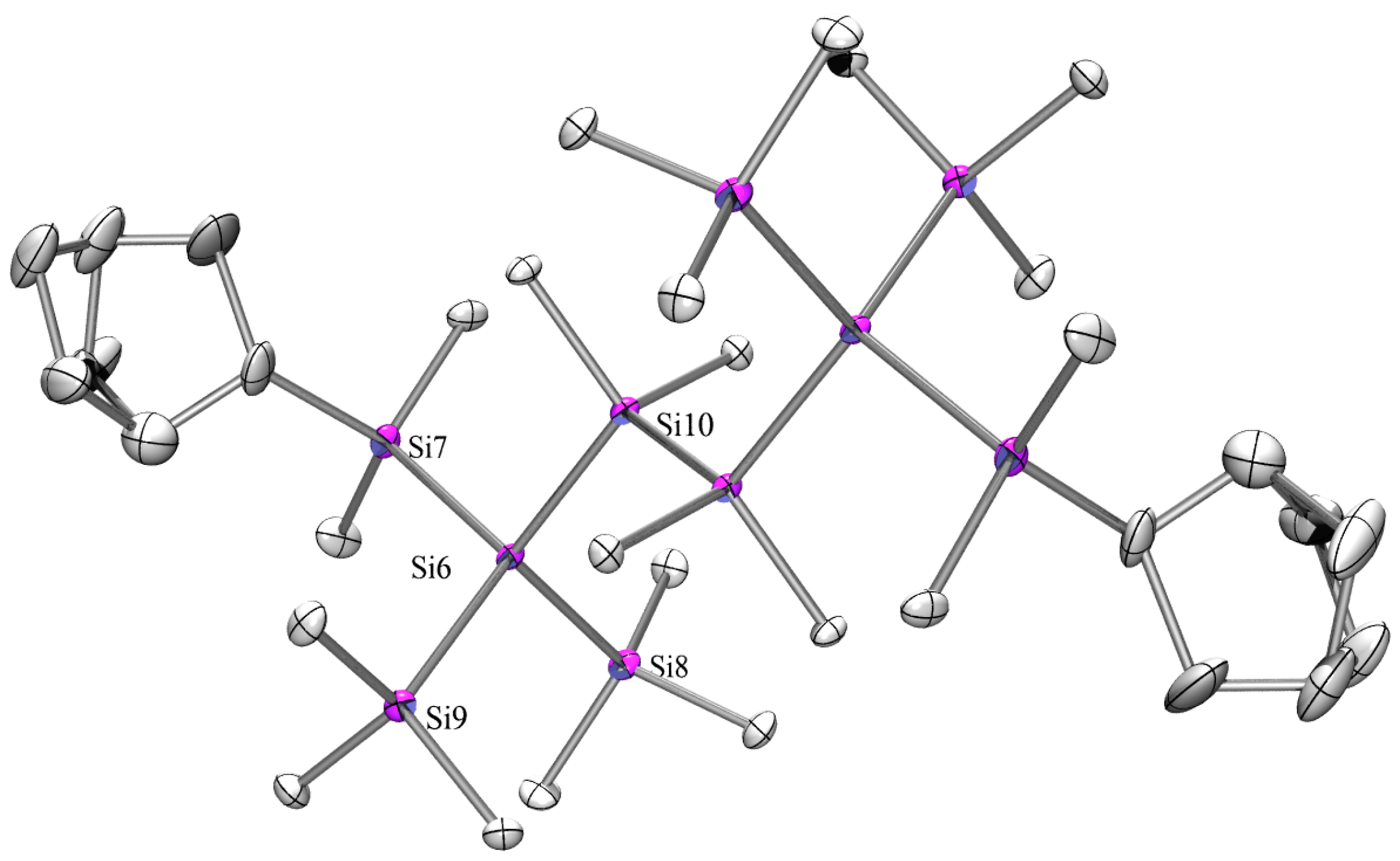
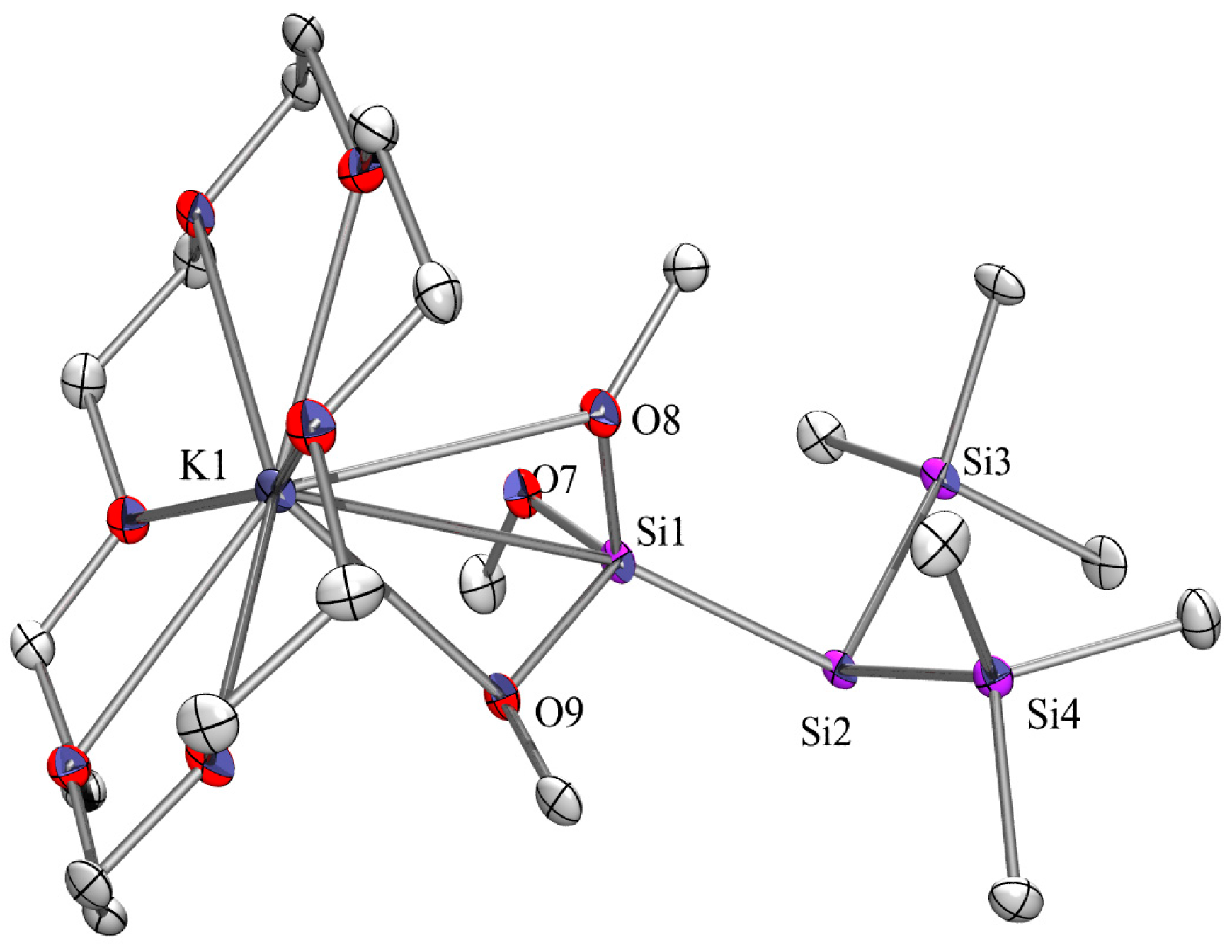
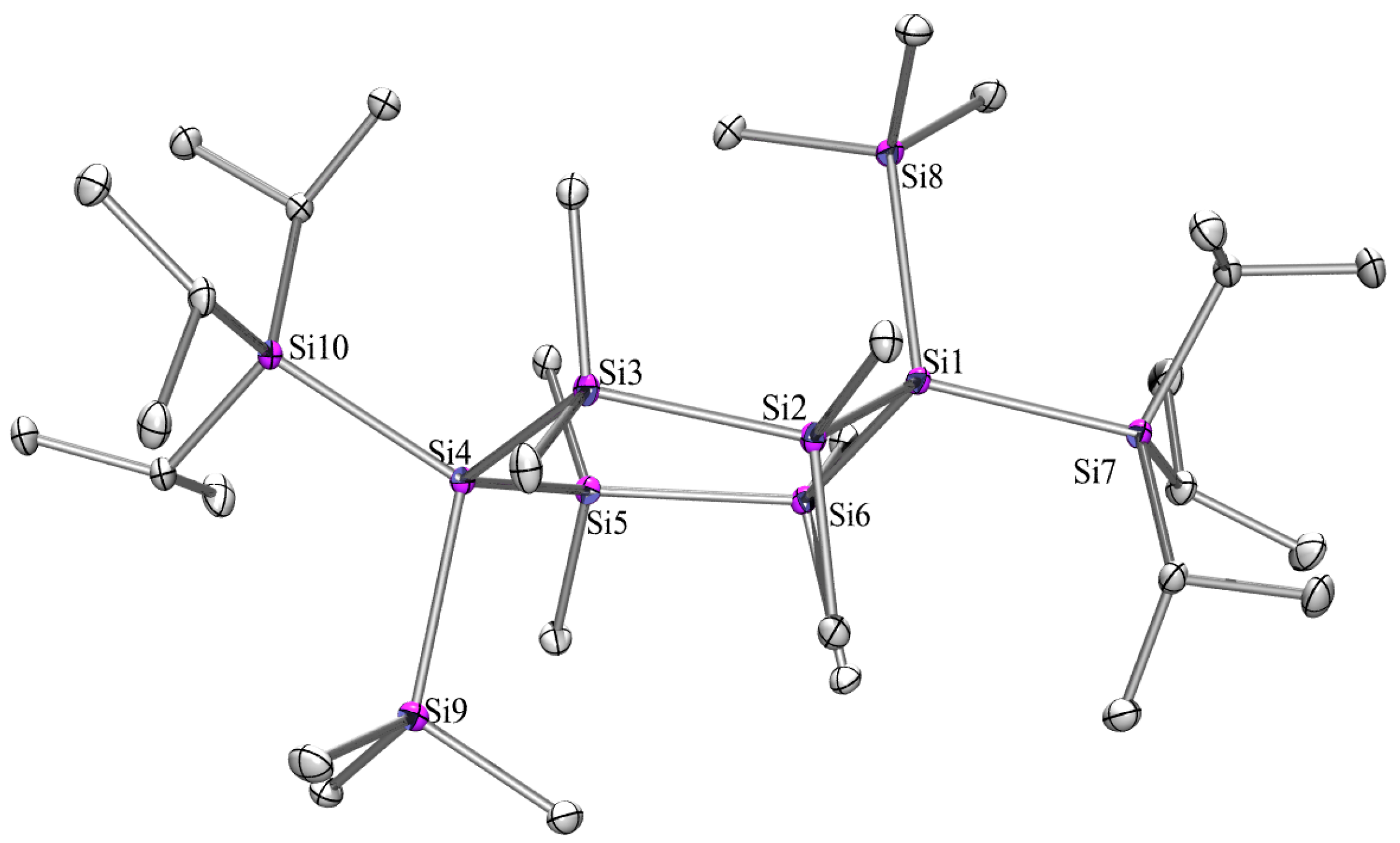
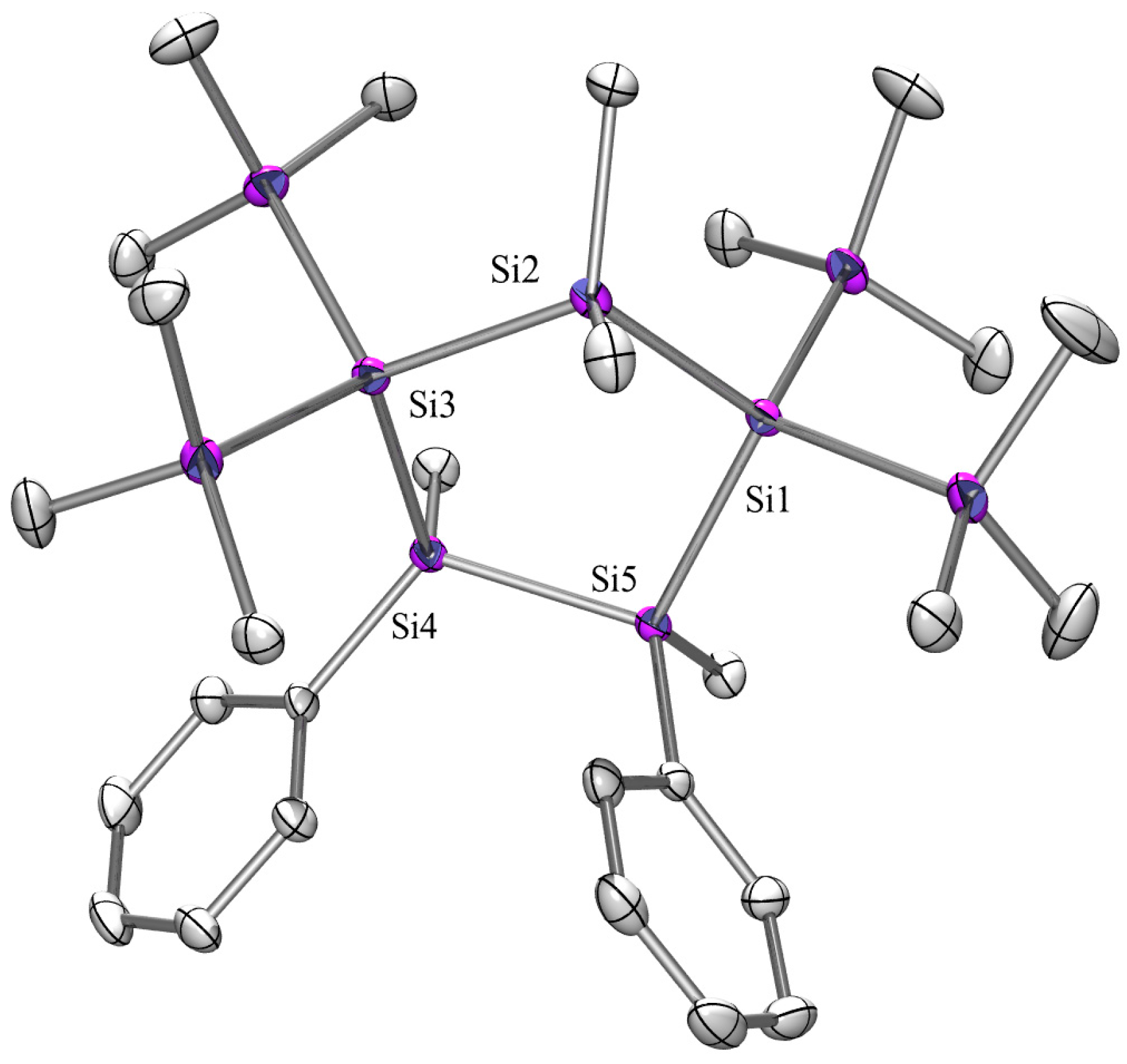
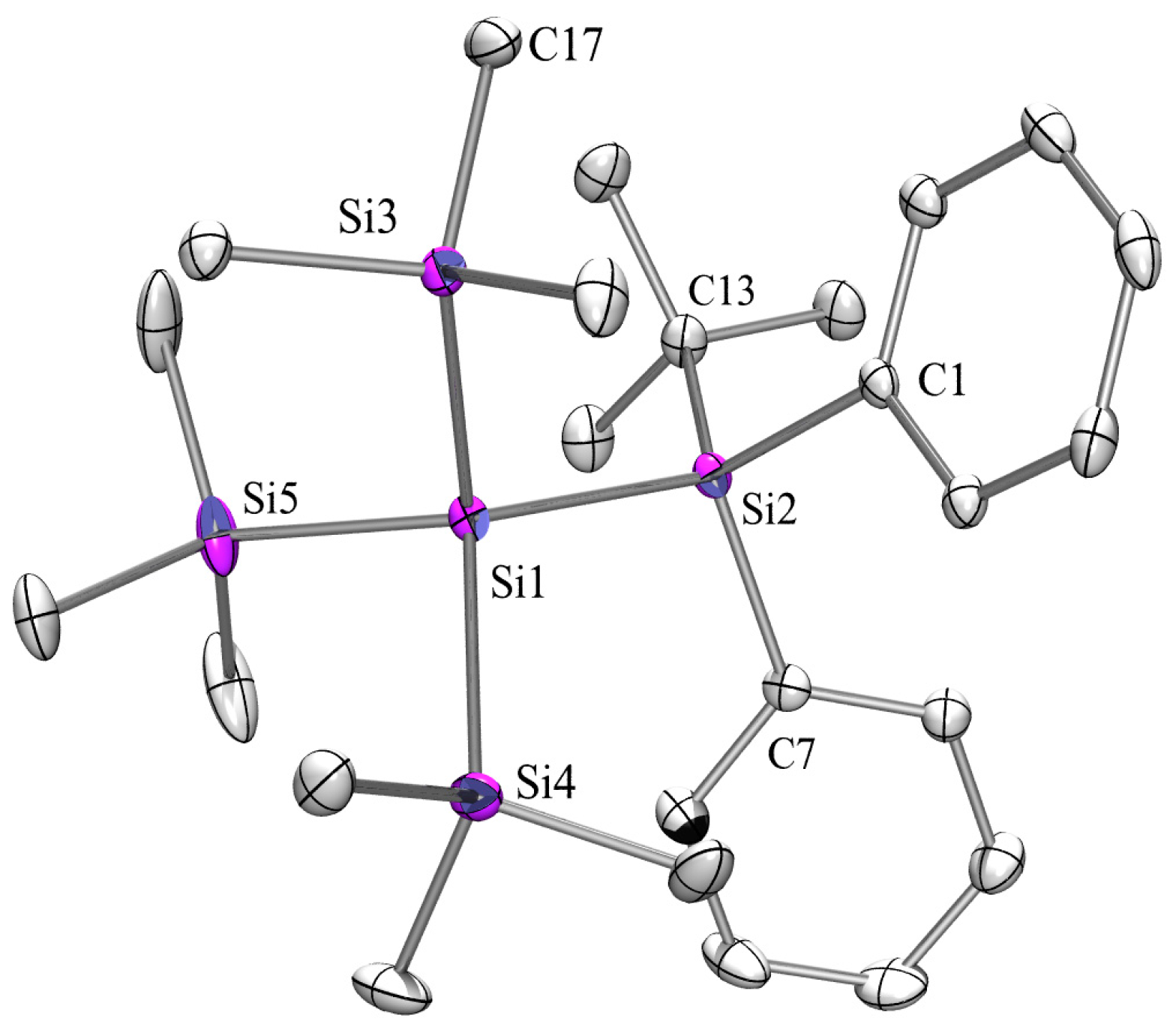
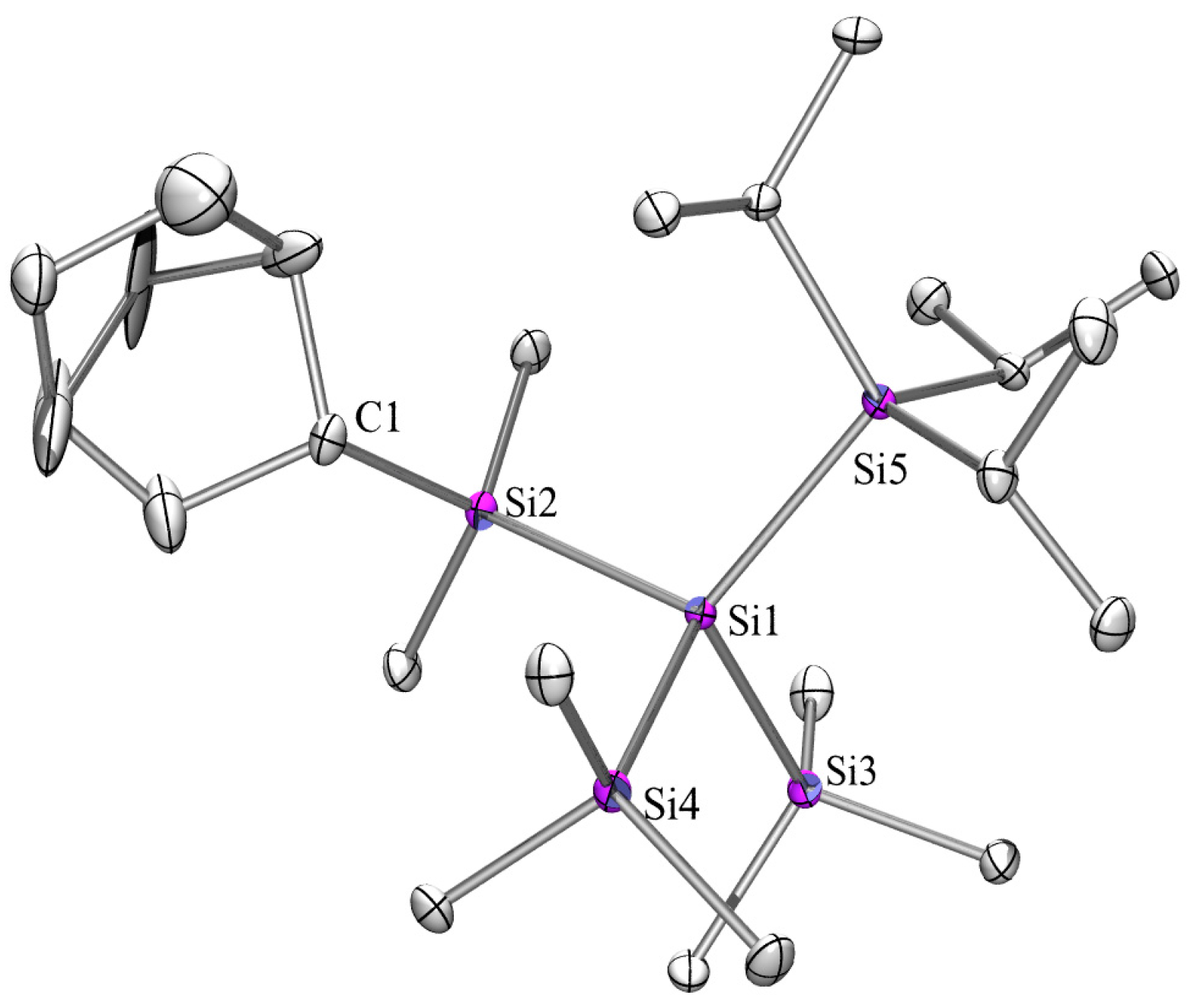
2.3. X-ray Crystallography
3. Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- West, R. Polysilanes. In Organic Silicon Compounds (1989); Patai, S., Rappoport, Z., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1989; Volume 1, pp. 1207–1240. [Google Scholar]
- Michl, J.; West, R. Conformations of Linear Chains. Systematics and Suggestions for Nomenclature. Acc. Chem. Res. 2000, 33, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Bande, A.; Michl, J. Conformational Dependence of σ-Electron Delocalization in Linear Chains: Permethylated Oligosilanes. Chem. Eur. J. 2009, 15, 8504–8517. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Kumada, M. The Ultraviolet Spectra of Some Polysilanes. Bull. Chem. Soc. Jpn. 1964, 37, 1894–1895. [Google Scholar] [CrossRef]
- Sakurai, H.; Yamamori, H.; Kumada, M. The Substituent Effect on the Ultraviolet Spectrum of 1,2-Diphenyltetramethyldisilane. Bull. Chem. Soc. Jpn. 1965, 38, 2024–2024. [Google Scholar] [CrossRef]
- Kumada, M.; Tamao, K. Aliphatic Polysilanes. Adv. Organomet. Chem. 1968, 6, 19–117. [Google Scholar]
- Gilman, H.; Atwell, W.H.; Schwebke, G.L. Ultraviolet properties of compounds containing the silicon-silicon bond. J. Organomet. Chem. 1964, 2, 369–371. [Google Scholar] [CrossRef]
- Gilman, H.; Atwell, W.H. The ultraviolet properties of some perphenylated linear and cyclic polysilanes. J. Organomet. Chem. 1965, 4, 176–178. [Google Scholar] [CrossRef]
- Miller, R.D.; Michl, J. Polysilane high polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- Marschner, C.; Baumgartner, J.; Wallner, A. Structurally and conformationally defined small methyl polysilanes. Dalton Trans. 2006, 5667–5674. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, J.; Fischer, R.; Fischer, J.; Wallner, A.; Marschner, C.; Flörke, U. Structural Aspects of Trimethylsilylated Branched Group 14 Compounds. Organometallics 2005, 24, 6450–6457. [Google Scholar] [CrossRef]
- Wallner, A.; Hoelbling, M.; Baumgartner, J.; Marschner, C. Structural and spectroscopic studies of silylated cyclo- and bicyclosilanes. Silicon Chem. 2007, 3, 175–185. [Google Scholar] [CrossRef]
- Wallner, A.; Wagner, H.; Baumgartner, J.; Marschner, C.; Rohm, H.W.; Kockerling, M.; Krempner, C. Structure, Conformation, and UV Absorption Behavior of Partially Trimethylsilylated Oligosilane Chains. Organometallics 2008, 27, 5221–5229. [Google Scholar] [CrossRef]
- Wallner, A.; Hlina, J.; Konopa, T.; Wagner, H.; Baumgartner, J.; Marschner, C.; Flörke, U. Cyclic and Bicyclic Methylpolysilanes and Some Oligosilanylene-Bridged Derivatives. Organometallics 2010, 29, 2660–2675. [Google Scholar] [CrossRef]
- Wallner, A.; Hlina, J.; Wagner, H.; Baumgartner, J.; Marschner, C. Conformational Control of Polysilanes: The Use of CH2-Spacers in the Silicon Backbone. Organometallics 2011, 30, 3930–3938. [Google Scholar] [CrossRef] [PubMed]
- Wallner, A.; Emanuelsson, R.; Baumgartner, J.; Marschner, C.; Ottosson, H. Coupling of Disilane and Trisilane Segments Through Zero, One, Two, and Three Disilanyl Bridges in Cyclic and Bicyclic Saturated Carbosilanes. Organometallics 2013, 32, 396–405. [Google Scholar] [CrossRef]
- Hlina, J.; Zitz, R.; Wagner, H.; Stella, F.; Baumgartner, J.; Marschner, C. σ-Bond electron delocalization of branched oligogermanes and germanium containing oligosilanes. Inorg. Chim. Acta 2014, 422, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Marschner, C. Preparation and Reactions of Polysilanyl Anions and Dianions. Organometallics 2006, 25, 2110–2125. [Google Scholar] [CrossRef]
- Kayser, C.; Fischer, R.; Baumgartner, J.; Marschner, C. Tailor-made Oligosilyl Potassium Compounds. Organometallics 2002, 21, 1023–1030. [Google Scholar] [CrossRef]
- Marschner, C. A New and Easy Route to Polysilanylpotassium Compounds. Eur. J. Inorg. Chem. 1998, 221–226. [Google Scholar] [CrossRef]
- Wagner, H.; Baumgartner, J.; Marschner, C.; Poelt, P. Rearrangement/Fragmentation Reactions of Oligosilanes with Aluminum Chloride. Organometallics 2011, 30, 3939–3954. [Google Scholar] [CrossRef] [PubMed]
- Rooklin, D.W.; Schepers, T.; Raymond-Johansson, M.K.; Michl, J. Time-dependent density functional theory treatment of the first UV absorption band in all-transoid permethyloligosilanes SinMe2n+2 (n = 2–8, 10). Photochem. Photobiol. Sci. 2003, 2, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, A.; Tsuji, H.; Tamao, K. all-anti-Octasilane: Conformation Control of Silicon Chains Using the Bicyclic Trisilane as a Building Block. J. Am. Chem. Soc. 2006, 128, 6800–6801. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Fukazawa, A.; Yamaguchi, S.; Toshimitsu, A.; Tamao, K. all-anti-Pentasilane: Conformation Control of Oligosilanes Based on the Bis(tetramethylene)-Tethered Trisilane Unit. Organometallics 2004, 23, 3375–3377. [Google Scholar] [CrossRef]
- Mallesha, H.; Tsuji, H.; Tamao, K. UV Absorption and Mass Spectra of n-Alkylsilyl End-Capped Anti, Cisoid-Alternating Oligosilanes up to Docosasilane (Si22). Organometallics 2004, 23, 1639–1642. [Google Scholar] [CrossRef]
- Tsuji, H.; Terada, M.; Toshimitsu, A.; Tamao, K. σσ* Transition in anti,cisoid Alternating Oligosilanes: Clear-Cut Evidence for Suppression of Conjugation Effect by a cisoid Turn. J. Am. Chem. Soc. 2003, 125, 7486–7487. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, H.; Michl, J.; Tamao, K. Recent experimental and theoretical aspects of the conformational dependence of UV absorption of short chain peralkylated oligosilanes. J. Organomet. Chem. 2003, 685, 9–14. [Google Scholar] [CrossRef]
- Suto, S.; Suzuki, H.; Ono, R.; Shimizu, M.; Goto, T.; Watanabe, A.; Matsuda, M. Exciton Dynamics and Lattice Relaxation in Oligosilanes. Int. J. Mod. Phys. B 2001, 15, 4025–4028. [Google Scholar] [CrossRef]
- Krempner, C.; Chtchian, S.; Reinke, H. First synthesis of a dihydrido functionalized double-cored oligosilane dendrimer. Inorg. Chim. Acta 2004, 357, 3733–3738. [Google Scholar] [CrossRef]
- Apeloig, Y.; Bravo-Zhivotovskii, D.; Yuzefovich, M.; Bendikov, M.; Shames, A.I. Polysilyl radicals: EPR study of the formation and decomposition of star polysilanes. Appl. Magn. Reson. 2000, 18, 425–434. [Google Scholar] [CrossRef]
- West, R. Electron delocalization and “aromatic” behavior in cyclic polysilanes. Pure Appl. Chem. 1982, 54, 1041–1050. [Google Scholar] [CrossRef]
- Zirngast, M.; Baumgartner, J.; Marschner, C. Synthesis of Cyclic and Bicyclic Polysilanes of Variable Ring Sizes. Organometallics 2008, 27, 6472–6478. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Ully, S.; Baumgartner, J.; Marschner, C. Route Si6 revisited. J. Organomet. Chem. 2003, 685, 79–92. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Ully, S.; Wallner, A.; Baumgartner, J.; Marschner, C. Preparation and structural studies on cyclohexasilane compounds. In Organosilicon Chemistry VI; Auner, N., Weis, J., Eds.; Wiley-VCH: Weinheim, Germany, 2005; pp. 355–360. [Google Scholar]
- Lambert, J.B.; Pflug, J.L.; Allgeier, A.M.; Campbell, D.J.; Higgins, T.B.; Singewald, E.T.; Stern, C.L. A Branched Polysilane. Acta Cryst. C 1995, 51, 713–715. [Google Scholar] [CrossRef]
- Wagner, H.; Baumgartner, J.; Müller, T.; Marschner, C. Shuttling Germanium Atoms into Branched Polysilanes. J. Am. Chem. Soc. 2009, 131, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.M.; Brun, M.-C.; Cervantes-Lee, F.; Pannell, K.H. Synthesis, structure, and reactivity of the permethylated decasilane (Me3Si)3SiSiMe2SiMe2SiMe3)3. J. Organomet. Chem. 1995, 499, 247–252. [Google Scholar] [CrossRef]
- Zirngast, M.; Flock, M.; Baumgartner, J.; Marschner, C. Formation of Formal Disilene Fluoride Adducts. J. Am. Chem. Soc. 2008, 130, 17460–17470. [Google Scholar] [CrossRef] [PubMed]
- Wagner, H.; Wallner, A.; Fischer, J.; Flock, M.; Baumgartner, J.; Marschner, C. Rearrangement of Cyclic Silanes with Aluminum Trichloride. Organometallics 2007, 26, 6704–6717. [Google Scholar] [CrossRef]
- Stüger, H.; Fuerpass, G.; Renger, K.; Baumgartner, J. Synthesis, Structures, and Unusual Photoluminescence of O- and N-Functional Cyclohexasilanes. Organometallics 2005, 24, 6374–6381. [Google Scholar] [CrossRef]
- Dinnebier, R.E.; Dollase, W.A.; Helluy, X.; Kümmerlen, J.; Sebald, A.; Schmidt, M.U.; Pagola, S.; Stephens, P.W.; van Smaalen, S. Order–disorder phenomena determined by high-resolution powder diffraction: The structures of tetrakis(trimethylsilyl)methane C[Si(CH3)3]4 and tetrakis(trimethylsilyl)silane Si[Si(CH3)3]4. Acta Cryst. B 1999, 55, 1014–1029. [Google Scholar] [CrossRef]
- Fischer, R.; Frank, D.; Gaderbauer, W.; Kayser, C.; Mechtler, C.; Baumgartner, J.; Marschner, C. α,ω-Oligosilyl Dianions and Their Application in the Synthesis of Homo- and Heterocyclosilanes. Organometallics 2003, 22, 3723–3731. [Google Scholar] [CrossRef]
- Fischer, R.; Konopa, T.; Baumgartner, J.; Marschner, C. Small Cyclosilanes: Syntheses and Reactions toward Mono- and Dianions. Organometallics 2004, 23, 1899–1907. [Google Scholar] [CrossRef]
- Aghazadeh Meshgi, M.; Baumgartner, J.; Marschner, C. Oligosilanylsilatranes. Organometallics 2015, 34, 3721–3731. [Google Scholar] [CrossRef] [PubMed]
- Krempner, C.; Chisholm, M.H.; Gallucci, J. The multidentate ligand (MeOMe2Si)3Si-: Unusual coordination modes in alkali metal silanides. Angew. Chem. Int. Ed. 2008, 47, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hope-Weeks, L.J.; Krempner, C. A supramolecular approach to zwitterionic alkaline metal silanides and formation of heterobimetallic silanides. Chem. Commun. 2011, 4117–4119. [Google Scholar] [CrossRef] [PubMed]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and Convenient Procedure for Solvent Purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Gilman, H.; Inoue, S. The Preparation of Some α,ω-Dichloro Permethylated Polysilanes. J. Org. Chem. 1964, 29, 3418–3419. [Google Scholar] [CrossRef]
- Marschner, C.; Baumgartner, J. 4.4.5 Product Subclass 5: Disilanes and Oligosilanes. In Science of Synthesis: Houben-Weyl Methods of Molecular Transformations; Oestreich, M., Ed.; Thieme: Stuttgart, Germany, 2013. [Google Scholar]
- Gilman, H.; Smith, C.L. Tetrakis(trimethylsilyl)silane. J. Organomet. Chem. 1967, 8, 245–253. [Google Scholar] [CrossRef]
- Kayser, C.; Kickelbick, G.; Marschner, C. Simple Synthesis of Oligosilyl-α,ω-dipotassium Compounds. Angew. Chem. Int. Ed. 2002, 41, 989–992. [Google Scholar] [CrossRef]
- Baumgartner, J.; Frank, D.; Kayser, C.; Marschner, C. Comparative Study of Structural Aspects of Branched Oligosilanes. Organometallics 2005, 24, 750–761. [Google Scholar] [CrossRef]
- Derouiche, Y.; Lickiss, P.D. Preparation and reactions of tris(trimethylsilyl)silyl silicon derivatives and related tetrasilylsilanes. J. Organomet. Chem. 1991, 407, 41–9. [Google Scholar] [CrossRef]
- Gilman, H.; Shiina, K.; Aoki, D.; Gaj, B.J.; Wittenberg, D.; Brennan, T. Hexasubstituted disilanes from chlorosilanes and lithium in tetrahydrofuran. J. Organomet. Chem. 1968, 13, 323–328. [Google Scholar] [CrossRef]
- Morris, G.A.; Freeman, R. Enhancement of nuclear magnetic resonance signals by polarization transfer. J. Am. Chem. Soc. 1979, 101, 760–762. [Google Scholar] [CrossRef]
- Helmer, B.J.; West, R. Enhancement of silicon-29 NMR signals by proton polarization transfer. Organometallics 1982, 1, 877–879. [Google Scholar] [CrossRef]
- SAINTPLUS: Software Reference Manual, version 6.45; Bruker-AXS: Madison, WI, USA; pp. 1997–2003.
- Blessing, R.H. An empirical correction for absorption anisotropy. Acta Cryst. A 1995, 51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, version 2.10; Bruker AXS Inc.: Madison, WI, USA, 2003. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- POVRAY 3.6; Persistence of Vision Pty. Ltd.: Williamstown, Victoria, Australia, 2004; Available online: http://www.povray.org/download/ (accessed on 9 July 2008).
- Sample Availability: Not available.
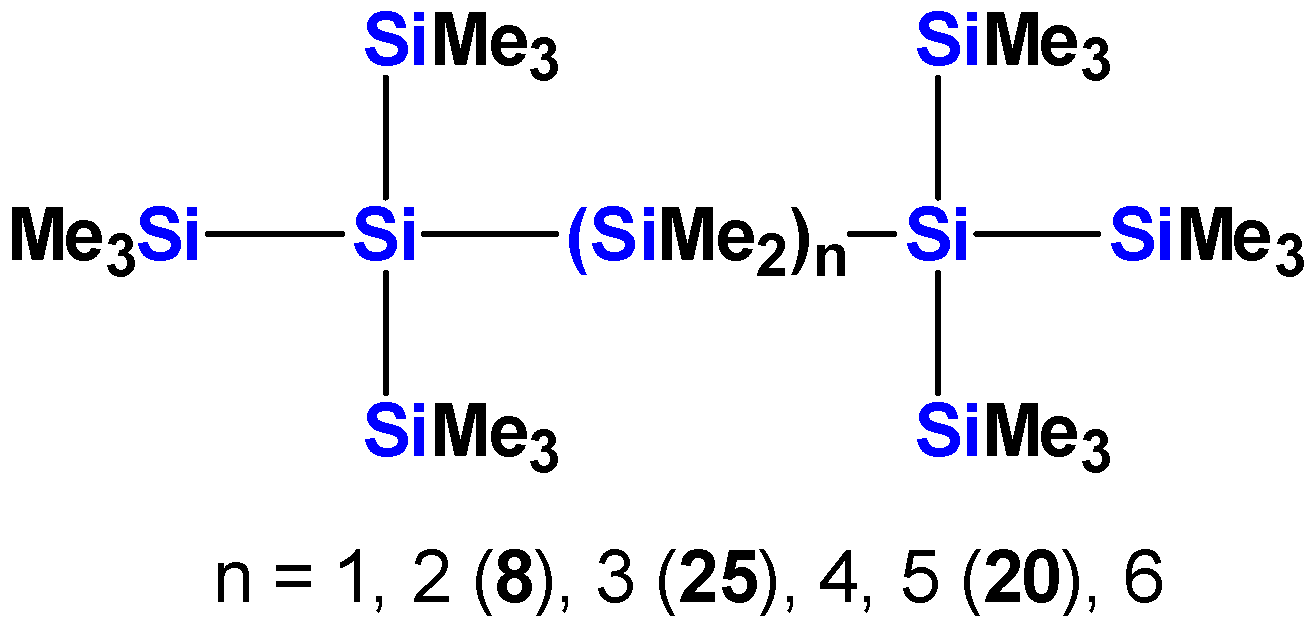

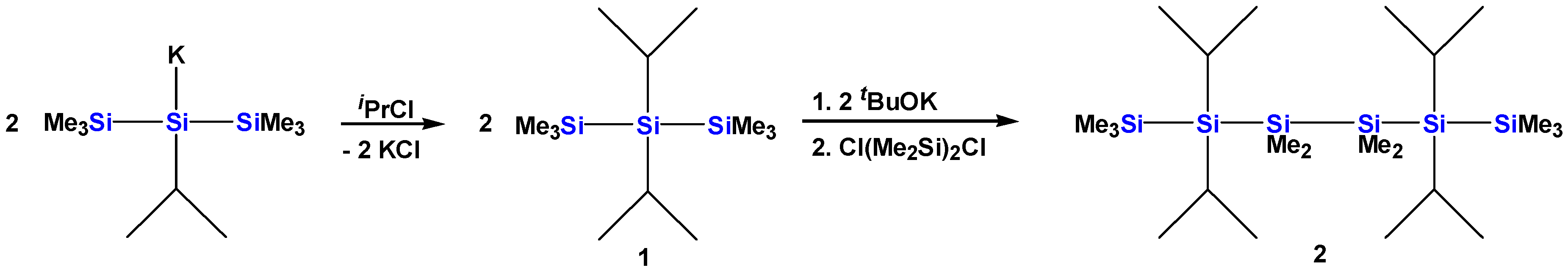
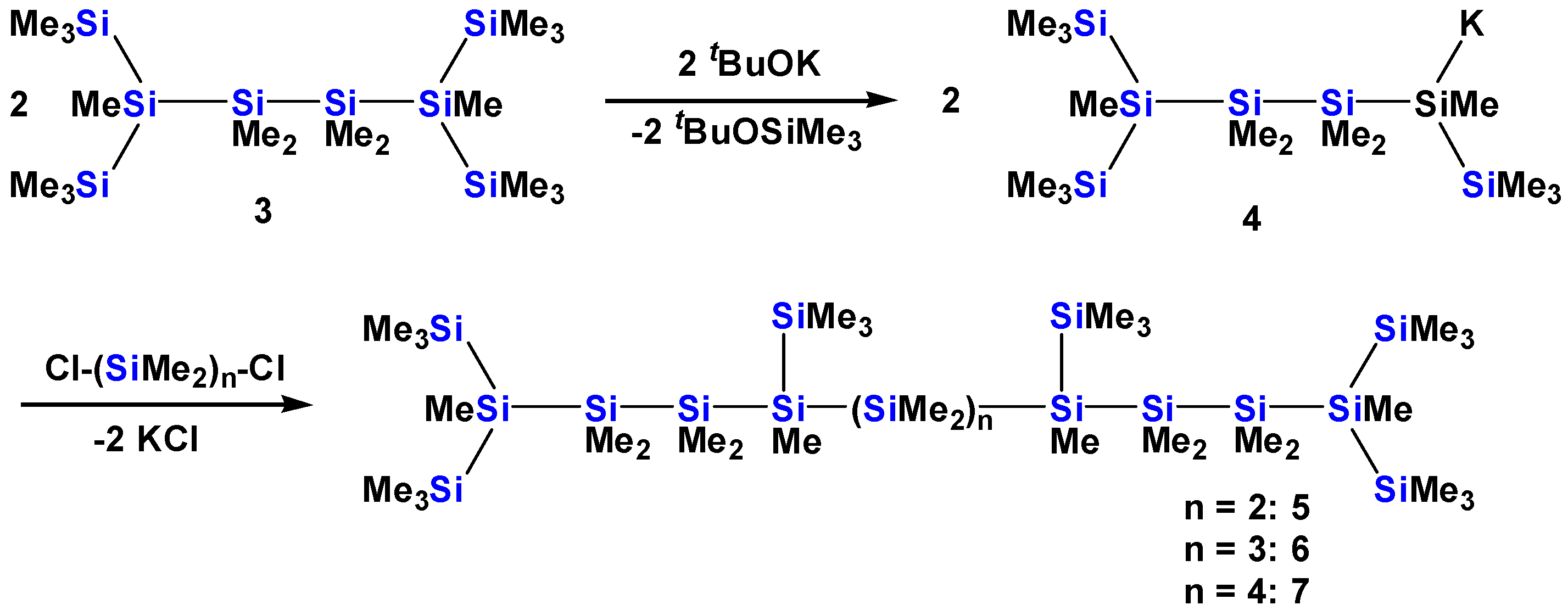
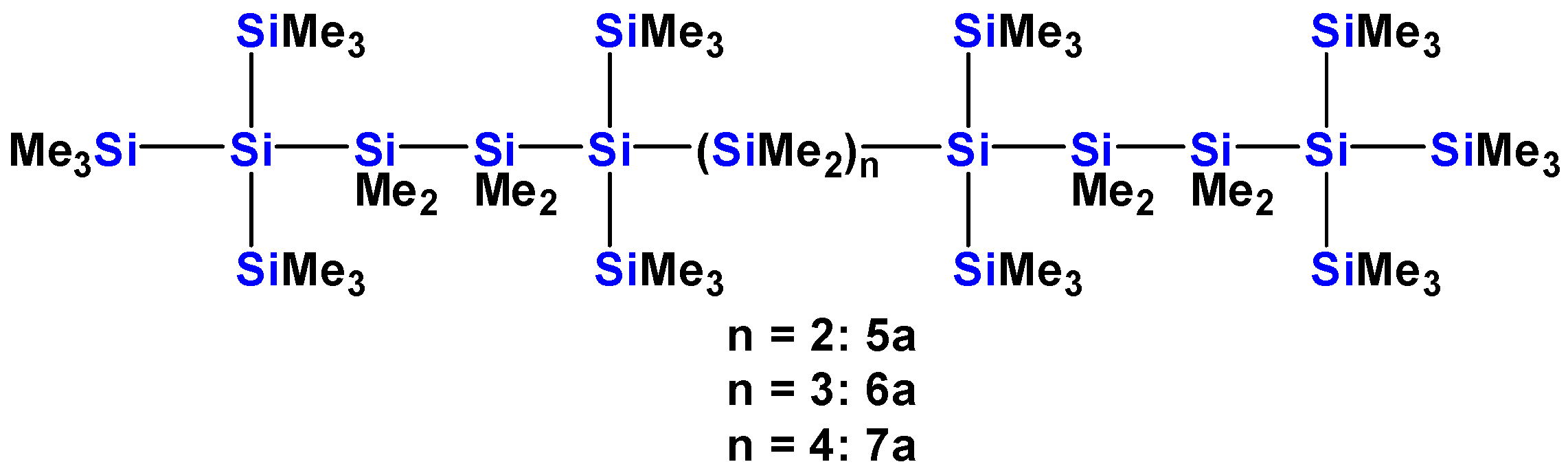
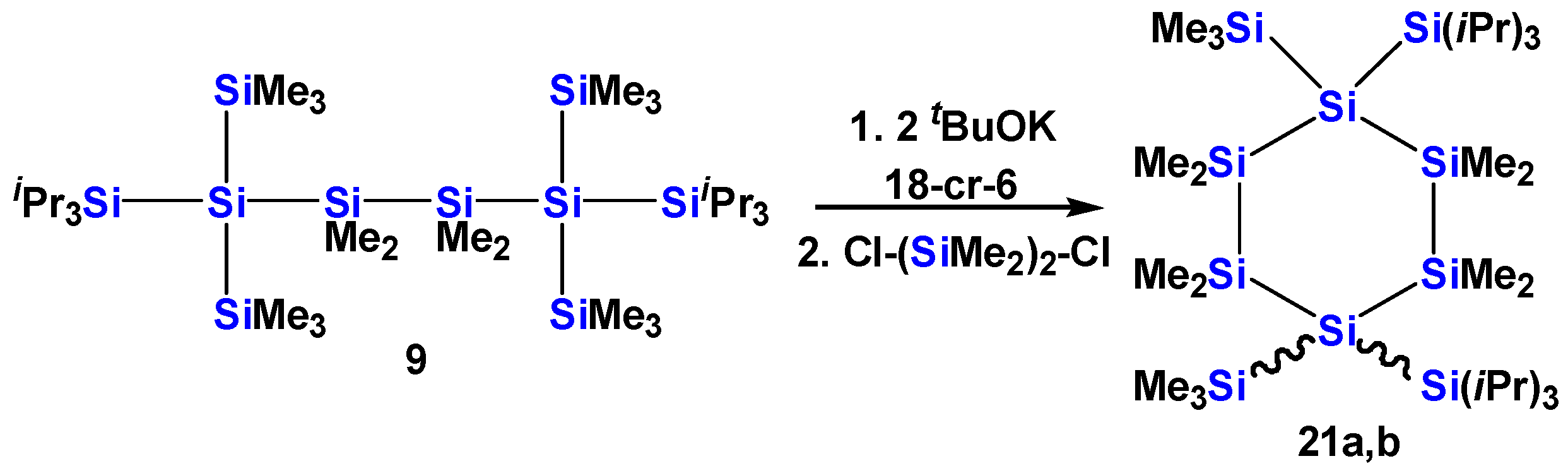

{kind=link}
{kind=link}
{kind=link}
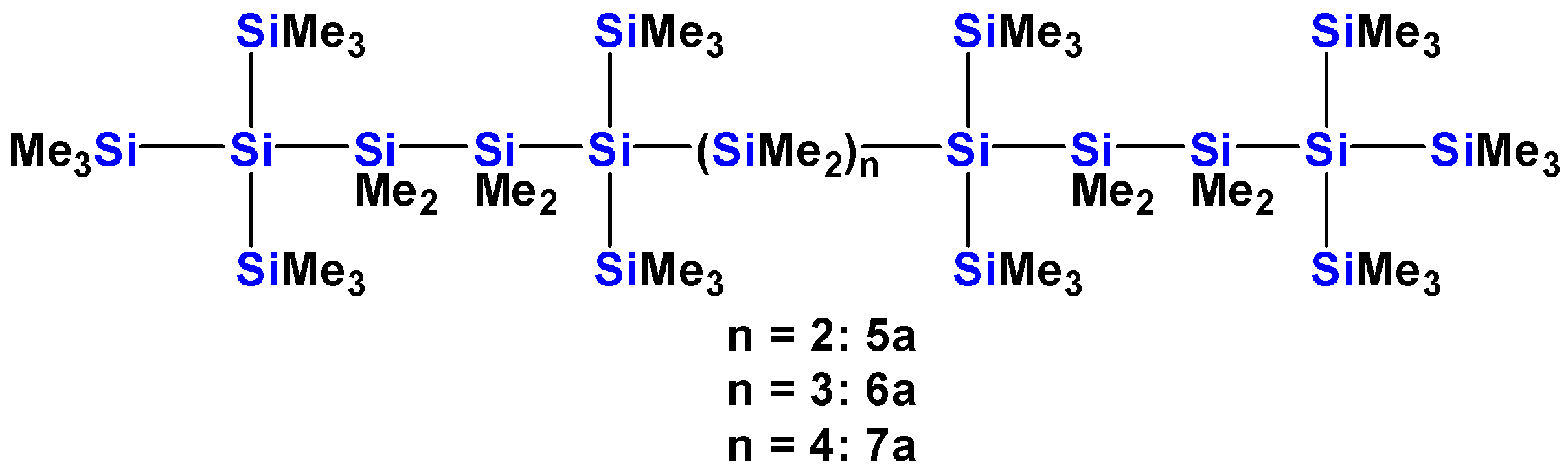{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
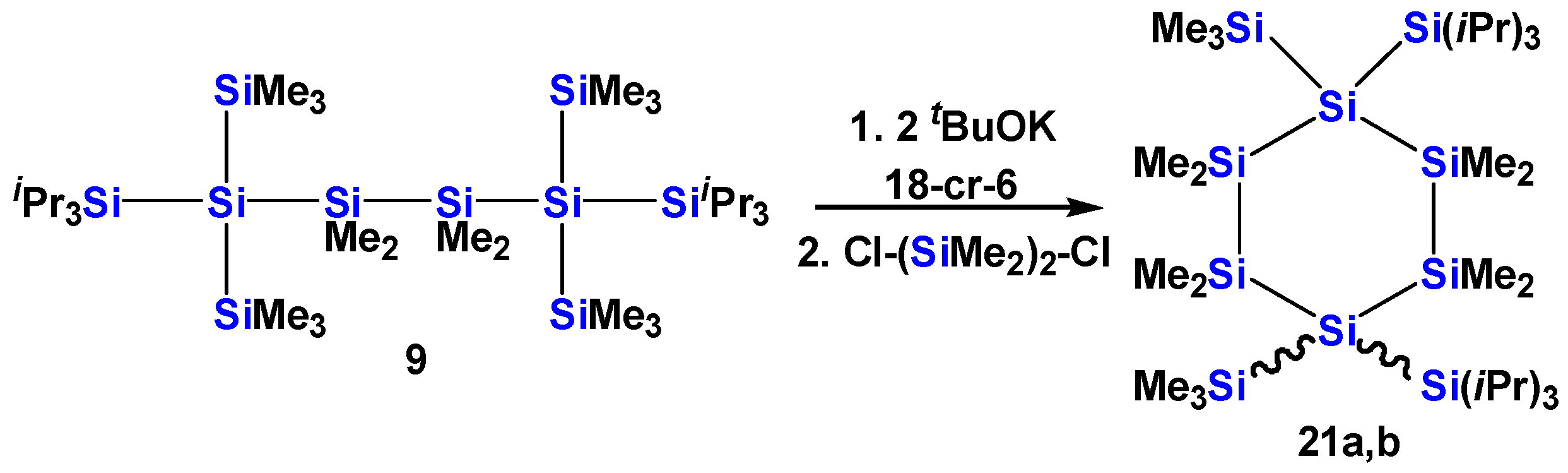{kind=link}
| Compound | Oligosilane Chain Length | λ1 [nm] | ε1 [M−1·cm−1] | λ2 [nm] | ε2 [M−1·cm−1] | λ3 [nm] | ε3 [M−1·cm−1] |
|---|---|---|---|---|---|---|---|
| n-Si6Me14 [6] | 6 | 260 | 1.4 × 104 | ||||
| 8 [10] | 6 | 257 | 6.6 × 104 | - | - | - | - |
| 2 | 6 | 260 | 2.4 × 104 | ||||
| 3 [29] | 6 | 264 | 3.0 × 104 | ||||
| 9 | 6 | 264 | 1.4 × 104 | ||||
| 15 | 6 | 258 | 7.1 × 104 | ||||
| 16 | 6 | 253 | 8.4 × 104 | ||||
| 18 | 6 | 262 | 2.4 × 104 | ||||
| 17 | 6 | 270 | 6.8 × 104 | 240 | 3.2 × 104 | ||
| 25 [10] | 7 | 269 | 6.2 × 104 | - | - | - | - |
| n-Si12Me26 [28] | 12 | 290 | |||||
| 5a [13] | 12 | ~289 (sh) | 2.3 × 104 | 259 | 7.4 × 104 | ||
| 5 | 12 | 268 | 1.6 × 104 | 218 | 3.2 × 104 | ||
| 11 | 12 | 264 | 5.1 × 104 | 232 | 216 | 3.1 × 104 | |
| 14 | 12 | 265 | 5.0 × 104 | ||||
| 6a [13] | 13 | ~295 (sh) | 1.8 × 104 | 278 | 5.6 × 104 | 258 | 7.3 × 104 |
| 6 | 13 | ~287 (sh) | 1.3 × 104 | 272 | 1.7 × 104 | 212 | 2.7 × 104 |
| 12 | 13 | ~307 (sh) | 3.1 × 103 | ~283 (sh) | 2.6 × 104 | 267 | 4.6 × 104 |
| 7a [13] | 14 | ~303 (sh) | 1.8 × 104 | 286 | 7.8 × 104 | 259 | 9.5 × 104 |
| 7 | 14 | 285 | 2.4 × 104 | 222 | 3.6 × 104 | ||
| 13 | 14 | ~311 (sh) | 1.5 × 104 | 293 | 3.8 × 104 | 268 | 4.5 × 104 |
| 27 [13] | 9 | ~284 (sh) | 1.2 × 104 | 262 | 9.2 × 104 | ||
| 19 | 9 | 273 | 3.6 × 104 | 230 | 1.9 × 104 | 226 | 2.1 × 104 |
| 20 | 9 | 288 | 4.0 × 104 | 271 | 2.7 × 104 | 246 | 1.7 × 104 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hlina, J.; Stella, F.; Aghazadeh Meshgi, M.; Marschner, C.; Baumgartner, J. σ-Bond Electron Delocalization in Oligosilanes as Function of Substitution Pattern, Chain Length, and Spatial Orientation. Molecules 2016, 21, 1079. https://doi.org/10.3390/molecules21081079
Hlina J, Stella F, Aghazadeh Meshgi M, Marschner C, Baumgartner J. σ-Bond Electron Delocalization in Oligosilanes as Function of Substitution Pattern, Chain Length, and Spatial Orientation. Molecules. 2016; 21(8):1079. https://doi.org/10.3390/molecules21081079
Chicago/Turabian StyleHlina, Johann, Filippo Stella, Mohammad Aghazadeh Meshgi, Christoph Marschner, and Judith Baumgartner. 2016. "σ-Bond Electron Delocalization in Oligosilanes as Function of Substitution Pattern, Chain Length, and Spatial Orientation" Molecules 21, no. 8: 1079. https://doi.org/10.3390/molecules21081079
APA StyleHlina, J., Stella, F., Aghazadeh Meshgi, M., Marschner, C., & Baumgartner, J. (2016). σ-Bond Electron Delocalization in Oligosilanes as Function of Substitution Pattern, Chain Length, and Spatial Orientation. Molecules, 21(8), 1079. https://doi.org/10.3390/molecules21081079