
Molecular Dynamics Simulations to Investigate the Binding Mode of the Natural Product Liphagal with Phosphoinositide 3-Kinase α

Abstract
:
1. Introduction
2. Methods
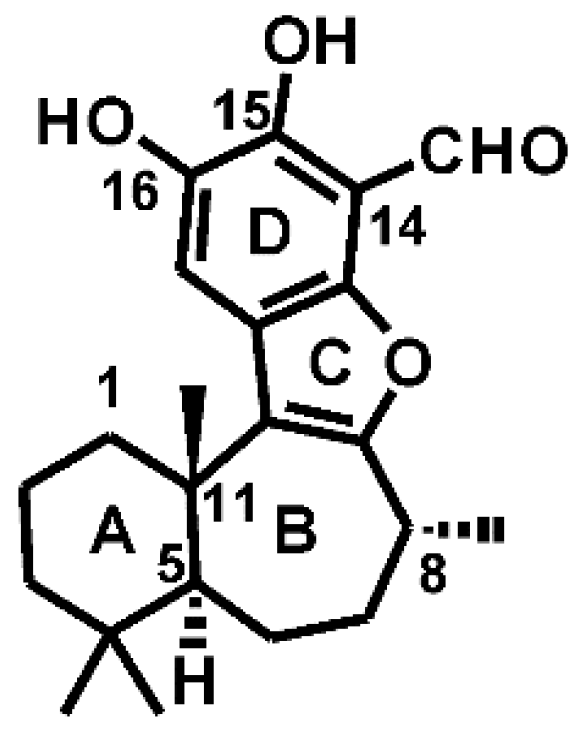
2.1. Preparation of PI3Kα and Liphagal
2.2. Docking Experiments
2.3. MD Simulations of the PI3Kα/Liphagal Complex
2.4. Binding Free Energy Calculations
2.5. Free Energy Decomposition
3. Results and Discussion
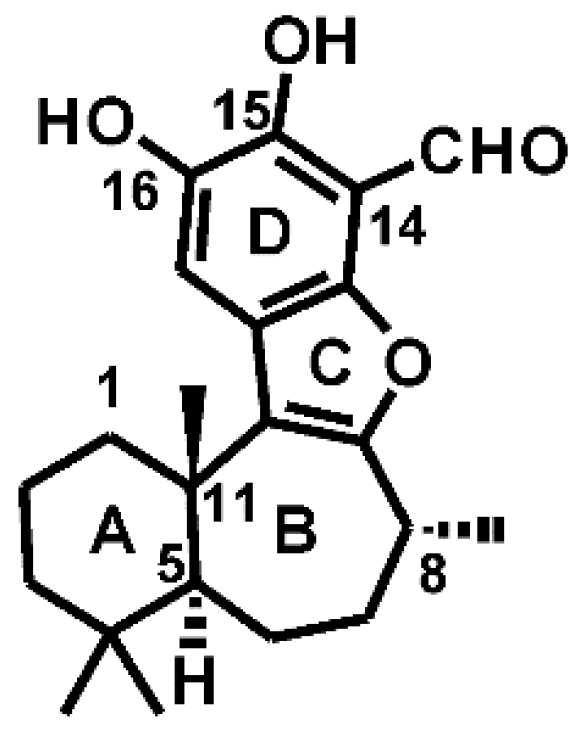
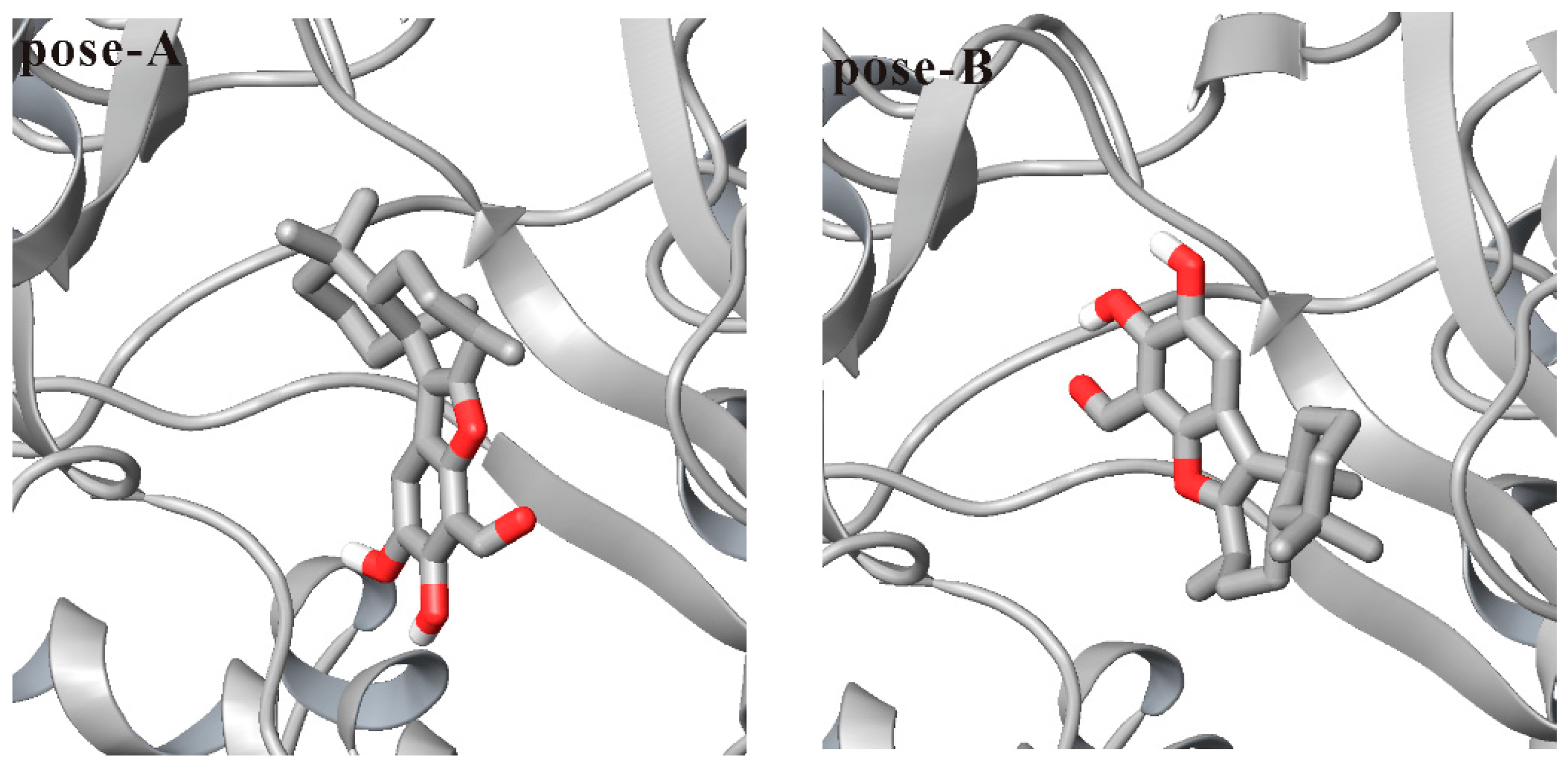
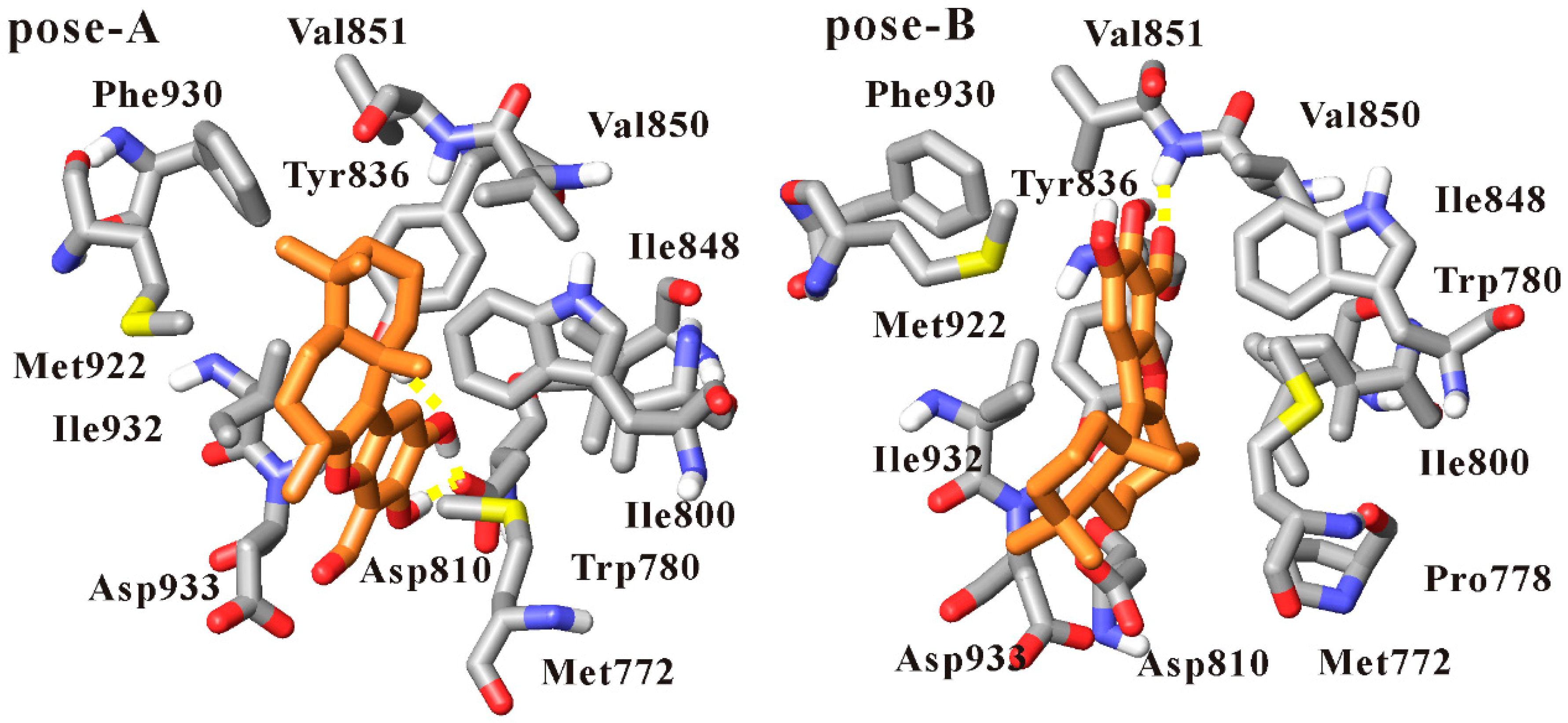
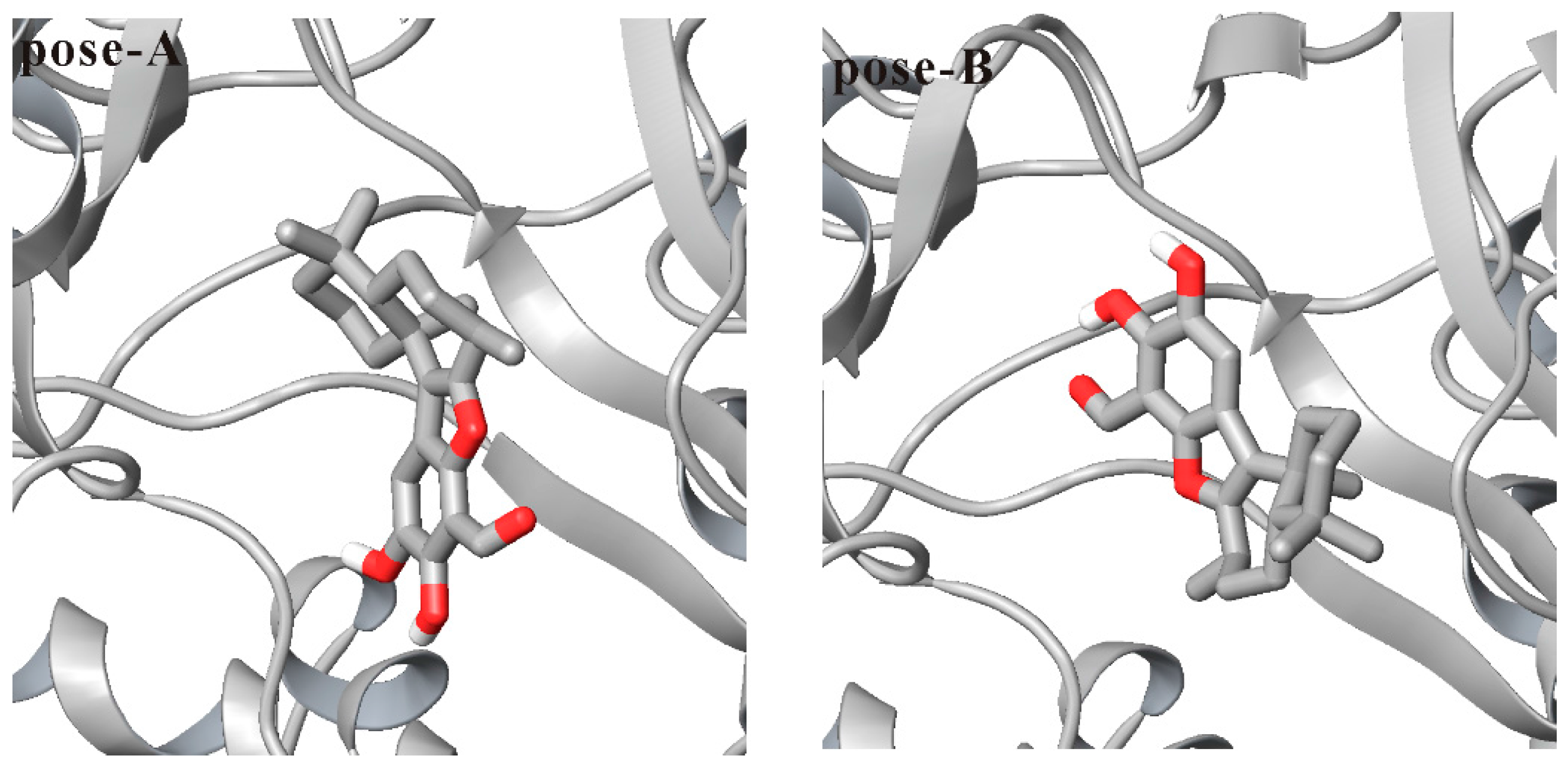
3.1. Docking Liphagal to the Crystal Structure of PI3Kα
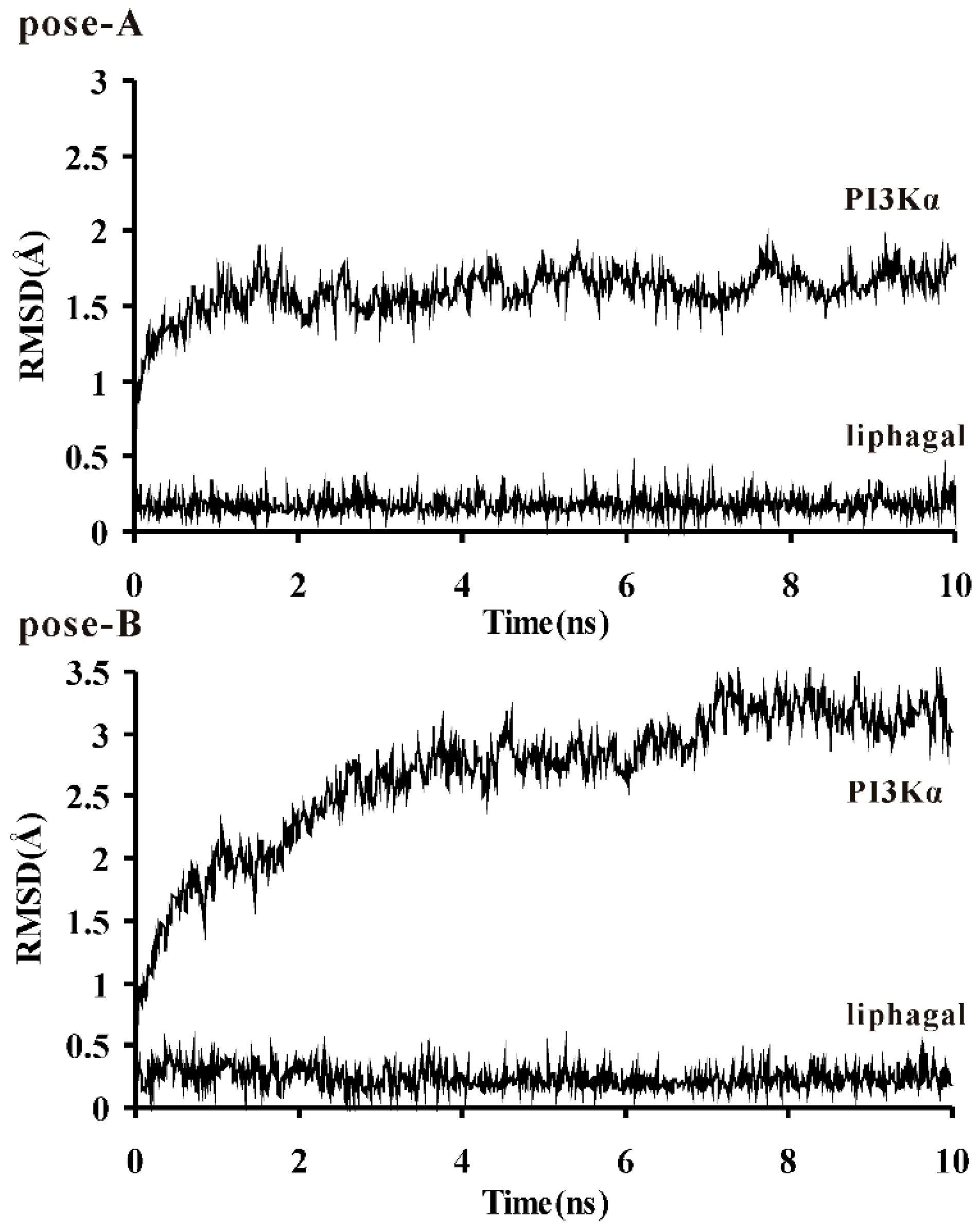
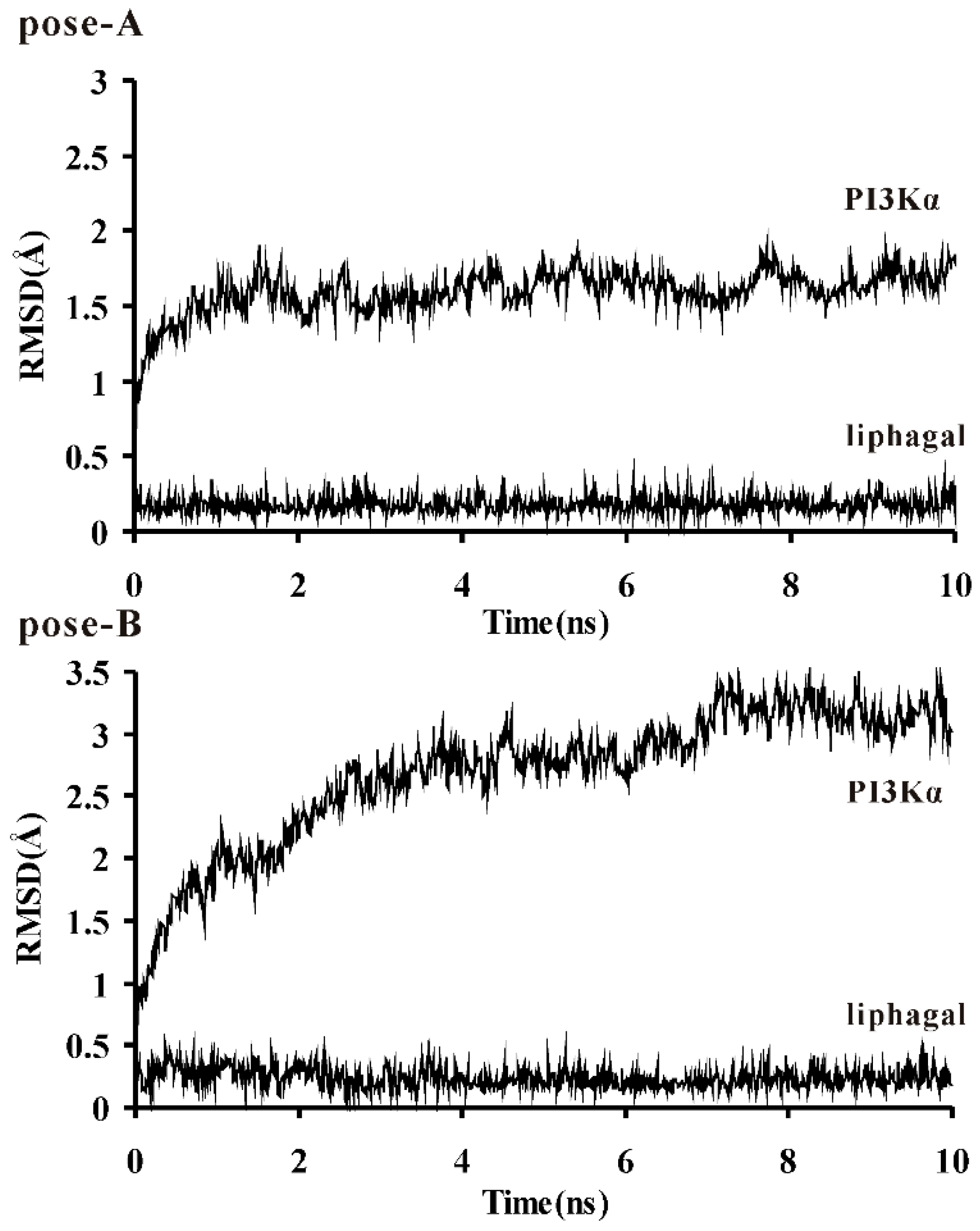
3.2. Molecular Dynamics Simulation of Liphagal-Bound PI3Kα
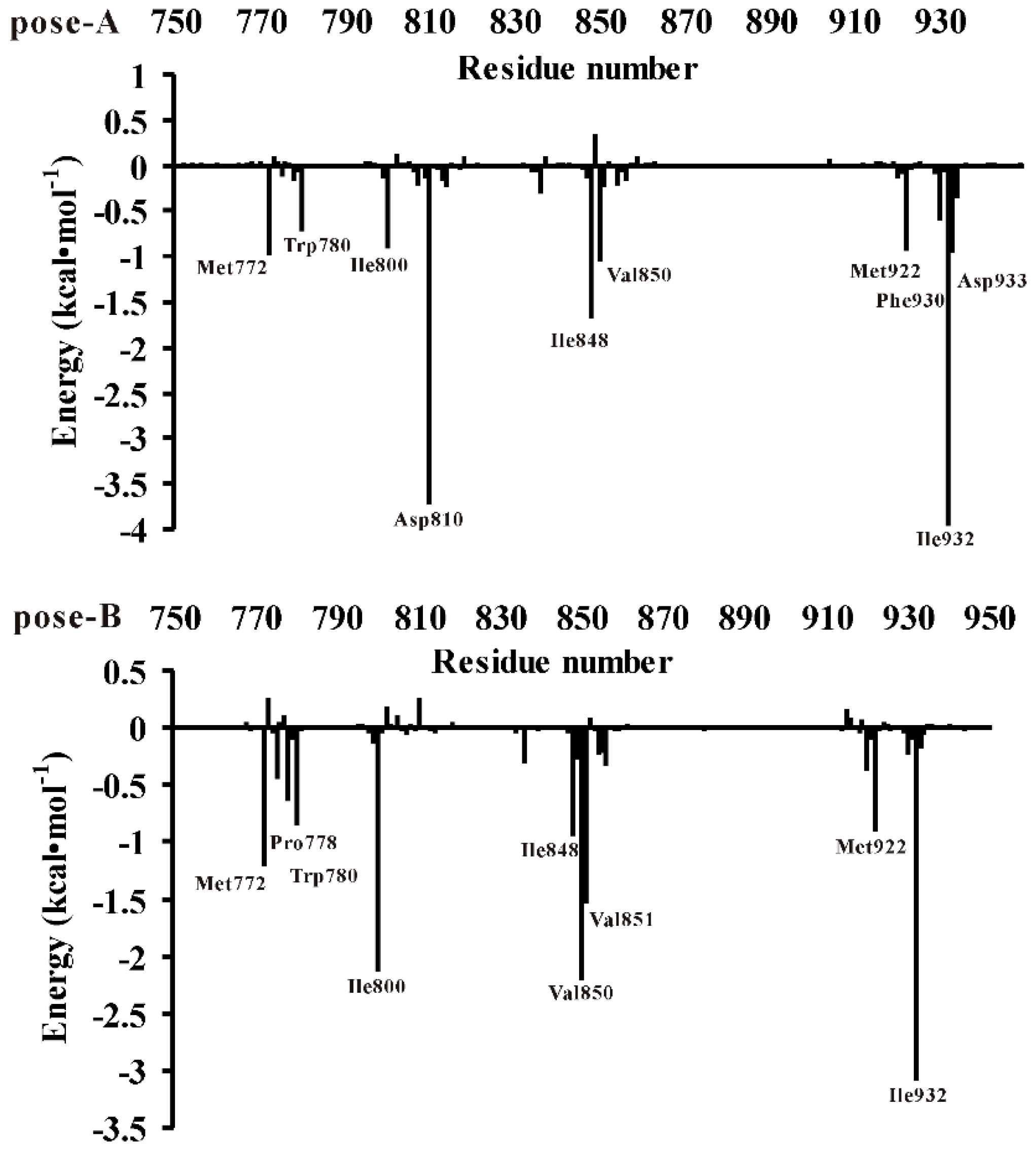
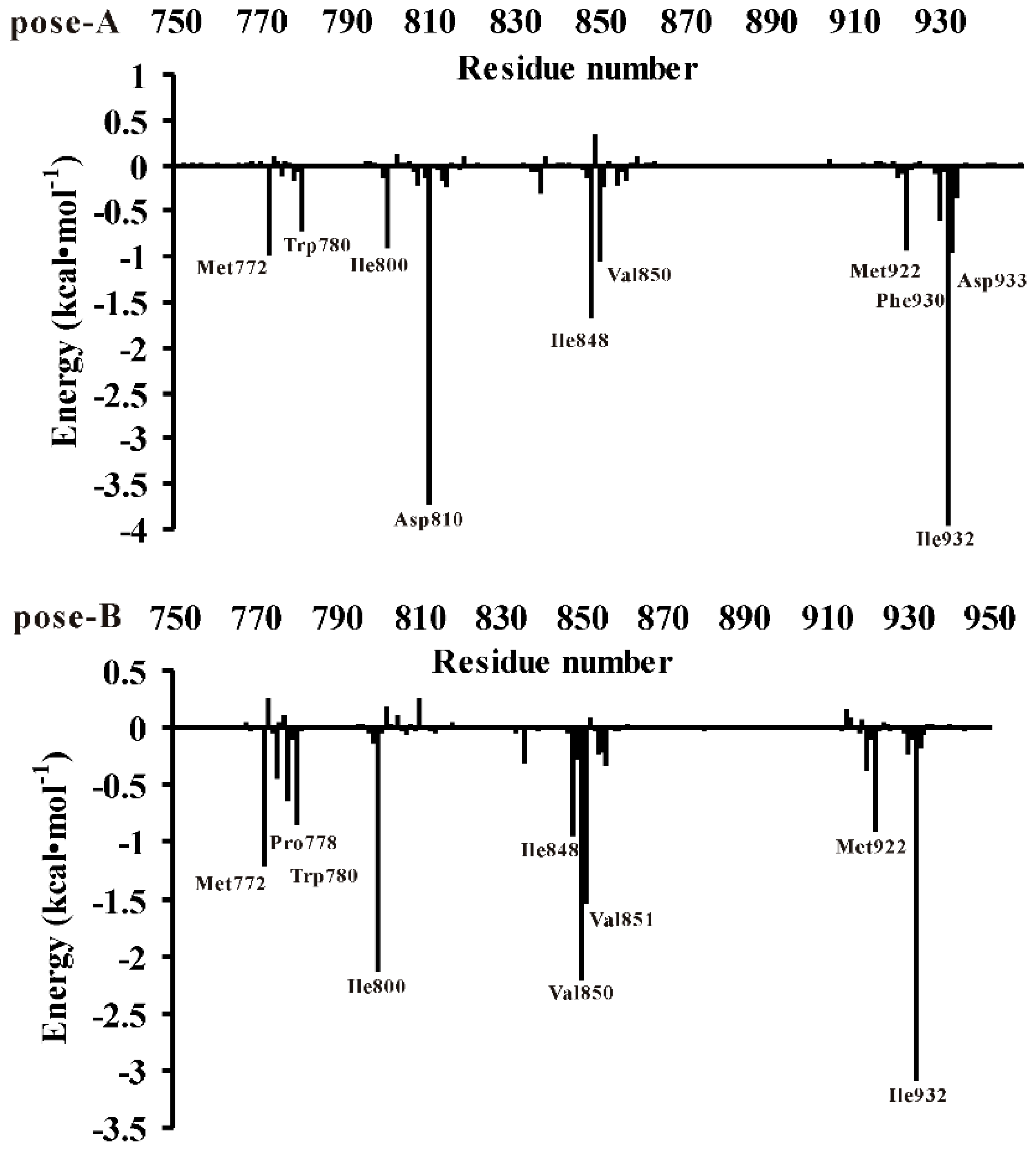
3.3. Decomposition of Binding Energy on a Per-Residue Basis
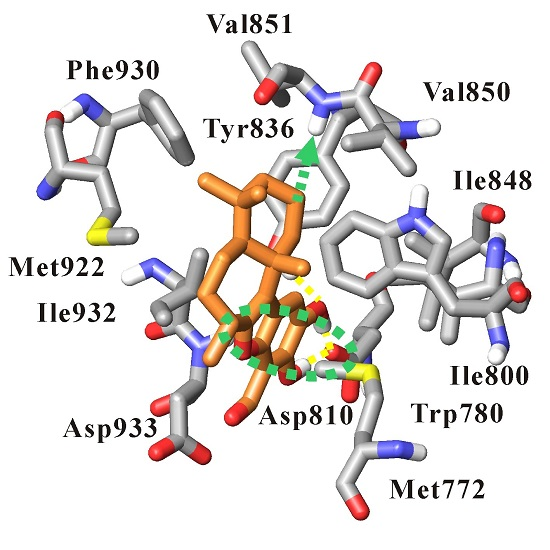
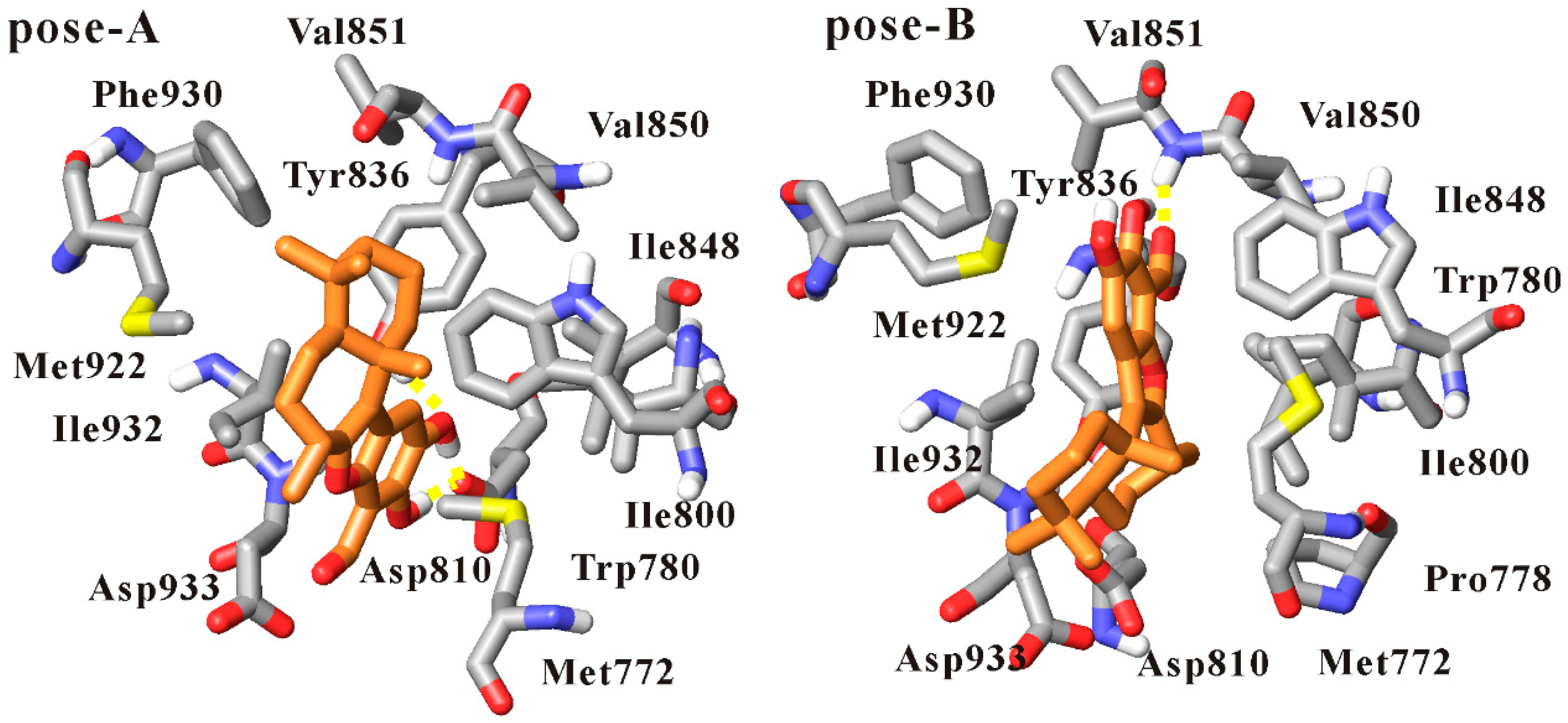
3.4. Dynamics Analysis of the Interactions between PI3Kα and Liphagal
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sundstrom, T.J.; Anderson, A.C.; Wright, D.L. Inhibitors of phosphoinositide-3-kinase: A structure-based approach to understanding potency and selectivity. Org. Biomol. Chem. 2009, 7, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef] [PubMed]
- Marone, R.; Cmiljanovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim. Biophys. Acta 2008, 1784, 159–185. [Google Scholar] [CrossRef] [PubMed]
- Knight, Z.A.; Shokat, K.M. Chemically targeting the PI3K family. Biochem. Soc. Trans. 2007, 35, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, H.; Mills, G.B. Targeting PI3K-AKT pathway for cancer therapy. Rev. Clin. Exp. Hematol. 2003, 7, 205–228. [Google Scholar] [PubMed]
- Wymann, M.P.; Marone, R. Phosphoinositide 3-kinase in disease: Timing, location, and scaffolding. Curr. Opin. Cell Biol. 2005, 17, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Rommel, C.; Camps, M.; Ji, H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat. Immunol. 2007, 7, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Marion, F.; Williams, D.E.; Patrick, B.O.; Hollander, I.; Mallon, R.; Kim, S.C.; Roll, D.M.; Feldberg, L.; van Soest, R.; Andersen, R.J. Liphagal, a Selective inhibitor of PI3 kinase alpha isolated from the sponge akacoralliphaga: Structure elucidation and biomimetic synthesis. Org. Lett. 2006, 8, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Pereira, A.R.; Strangman, W.K.; Marion, F.; Feldberg, L.; Roll, D.; Mallon, R.; Hollander, I.; Andersen, R.J. Synthesis of phosphatidylinositol 3-kinase (PI3K) inhibitory analogues of the sponge meroterpenoid liphagal. J. Med. Chem. 2010, 53, 8523–8533. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, I.; Liu, Y.; Gabelli, S.B.; Amzel, L.M. Oncogenic mutations weaken the interactions that stabilize the p110ɑ-p85ɑ heterodimer in phosphatidylinositol 3-kinase ɑ. FEBS J. 2015, 282, 3528–3542. [Google Scholar] [CrossRef] [PubMed]
- Gkeka, P.; Evangelidis, T.; Pavlaki, M.; Lazani, V.; Christoforidis, S.; Agianian, B.; Cournia, Z. Investigating the structure and dynamics of the PIK3CA wild-type and H1047R oncogenic mutant. PLoS Comput. Biol. 2014, 10, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Gkeka, P.; Papafotika, A.; Christoforidis, S.; Cournia, Z. Exploring a non-ATP pocket for potential allosteric modulation of PI3Kɑ. J. Phys. Chem. B 2015, 119, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H.A. Binding selectivity studies of phosphoinositide 3-kinases using free energy calculations. J. Chem. Inf. Model. 2012, 52, 3213–3224. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, D.A.; Vennerstrom, J.L.; Zhong, H.A. Docking studies on isoform-specific inhibition of phosphoinositide-3-kinases. J. Chem. Inf. Model. 2010, 50, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Do, R.K.; Sali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Mohamadi, F.; Richard, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Model. 1999, 17, 57–61. [Google Scholar] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Darden, T.; Cheatham, T., III; Simmerling, C.; Wang, J.; Duke, R.; Luo, R.; Crowley, M.W.; Zhang, R.C.W.; Merz, K.; et al. AMBER 9; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general Amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Jean-Paul, R.; Giovanni, C.; Herman, J.C.B. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N•log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Case, D.A. Normal mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 1994, 4, 285–290. [Google Scholar] [CrossRef]
- Zhang, Y.; Oblak, E.Z.; Bolstad, E.S.D.; Anderson, A.C.; Jasinski, J.P.; Butcher, R.J.; Wright, D.L. Synthetic and computational studies on liphagal: a natural product inhibitor of PI-3K. Tetrahedron Lett. 2010, 51, 6120–6122. [Google Scholar] [CrossRef]
- Knight, Z.A.; Gonzalez, B.; Feldman, M.E.; Zunder, E.R.; Goldenberg, D.D.; Williams, O.; Loewith, R.; Stokoe, D.; Balla, A.; Toth, B.; et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 2006, 125, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the calculations are available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energies | PI3Kα/Liphagal of Pose-A | PI3Kα/Liphagal of Pose-B | ||
|---|---|---|---|---|
| Mean (kcal·mol−1) | Std (kcal·mol−1) | Mean (kcal·mol−1) | Std (kcal·mol−1) | |
| ΔEele | −50.06 | 4.05 | −7.51 | 1.81 |
| ΔEvdw | −37.74 | 3.58 | −37.96 | 2.31 |
| ΔEMM | −87.80 | 3.86 | −45.47 | 2.57 |
| ΔGpb_sur | −5.65 | 0.22 | −5.94 | 0.22 |
| ΔGpb | 58.84 | 3.05 | 29.82 | 3.55 |
| ΔGpb_sol | 53.19 | 3.02 | 23.88 | 3.47 |
| ΔGgb_sur | −5.65 | 0.22 | −5.94 | 0.22 |
| ΔGgb | 54.95 | 2.44 | 22.66 | 1.71 |
| ΔGgb_sol | 49.30 | 2.43 | 16.72 | 1.64 |
| ΔHpb | −34.61 | 3.61 | −21.59 | 3.06 |
| ΔHgb | −38.50 | 2.96 | −28.75 | 2.03 |
| TΔS | −24.13 | 16.18 | −20.53 | 15.09 |
| ΔGbind(pb) | −10.48 | −1.06 | ||
| ΔGbind(gb) | −14.37 | −8.22 | ||
| Inhibitor | Hydrogen Bond | Occupancy (%) | Distance (Å) | |
|---|---|---|---|---|
| Liphagal of Pose-A | PI3Kα | |||
| Liphagal of pose-A | benzofuran ring 15-OH | O-Asp810 | 89.4 | 2.59 (0.09) |
| benzofuran ring 16-OH | O-Asp810 | 87.4 | 2.59 (0.09) | |
| benzofuran ring 16-O | OH-Tyr836 | 97.2 | 2.85 (0.15) | |
| Liphagal of pose-B | Liphagal of pose-B | PI3Kα | ||
| benzofuran ring 14-formyl-O | NH-Val851 | 70.0 | 3.05 (0.26) | |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Ma, Y.; Yang, G.; Li, Y. Molecular Dynamics Simulations to Investigate the Binding Mode of the Natural Product Liphagal with Phosphoinositide 3-Kinase α. Molecules 2016, 21, 857. https://doi.org/10.3390/molecules21070857
Gao Y, Ma Y, Yang G, Li Y. Molecular Dynamics Simulations to Investigate the Binding Mode of the Natural Product Liphagal with Phosphoinositide 3-Kinase α. Molecules. 2016; 21(7):857. https://doi.org/10.3390/molecules21070857
Chicago/Turabian StyleGao, Yanjuan, Ying Ma, Guangde Yang, and Yiping Li. 2016. "Molecular Dynamics Simulations to Investigate the Binding Mode of the Natural Product Liphagal with Phosphoinositide 3-Kinase α" Molecules 21, no. 7: 857. https://doi.org/10.3390/molecules21070857
APA StyleGao, Y., Ma, Y., Yang, G., & Li, Y. (2016). Molecular Dynamics Simulations to Investigate the Binding Mode of the Natural Product Liphagal with Phosphoinositide 3-Kinase α. Molecules, 21(7), 857. https://doi.org/10.3390/molecules21070857