
Causes of Activation and Deactivation of Modified Nanogold Catalysts during Prolonged Storage and Redox Treatments


 ,
,  , and
, and 
Abstract
:
1. Introduction
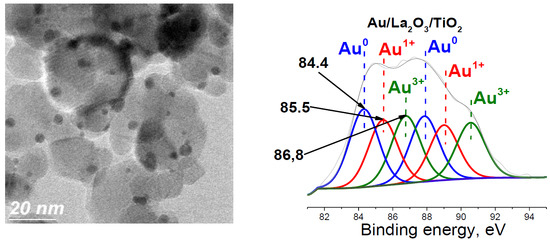
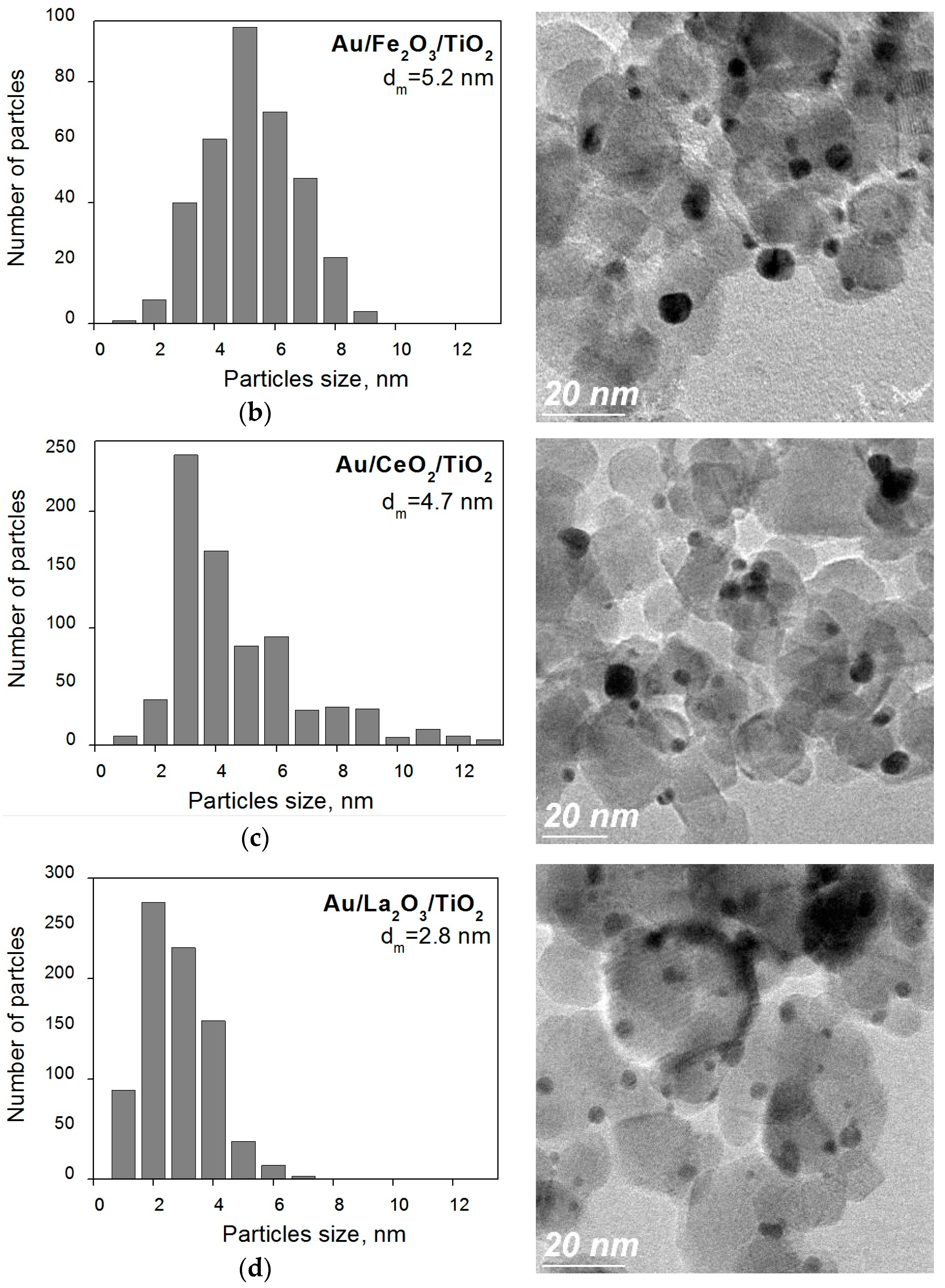
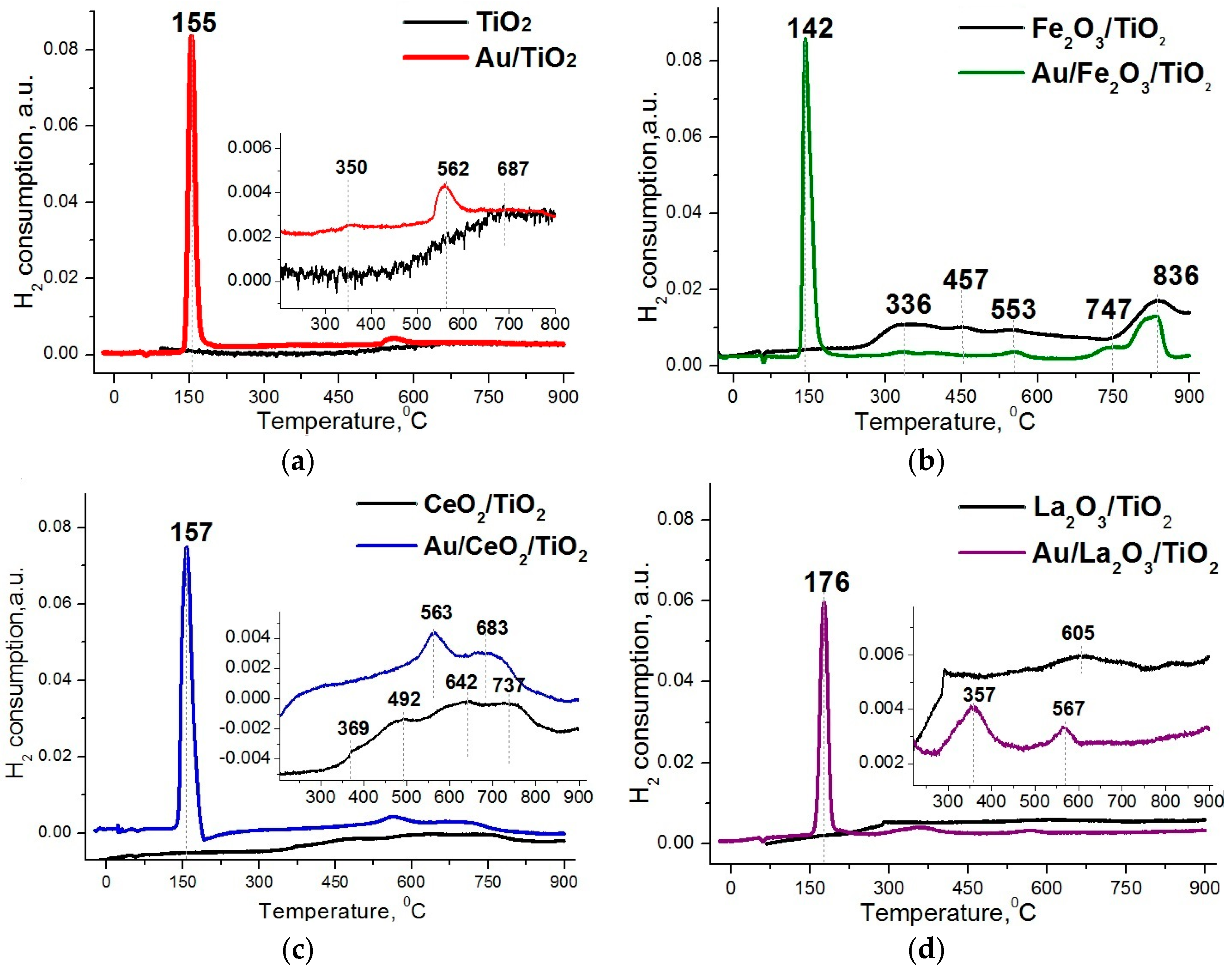
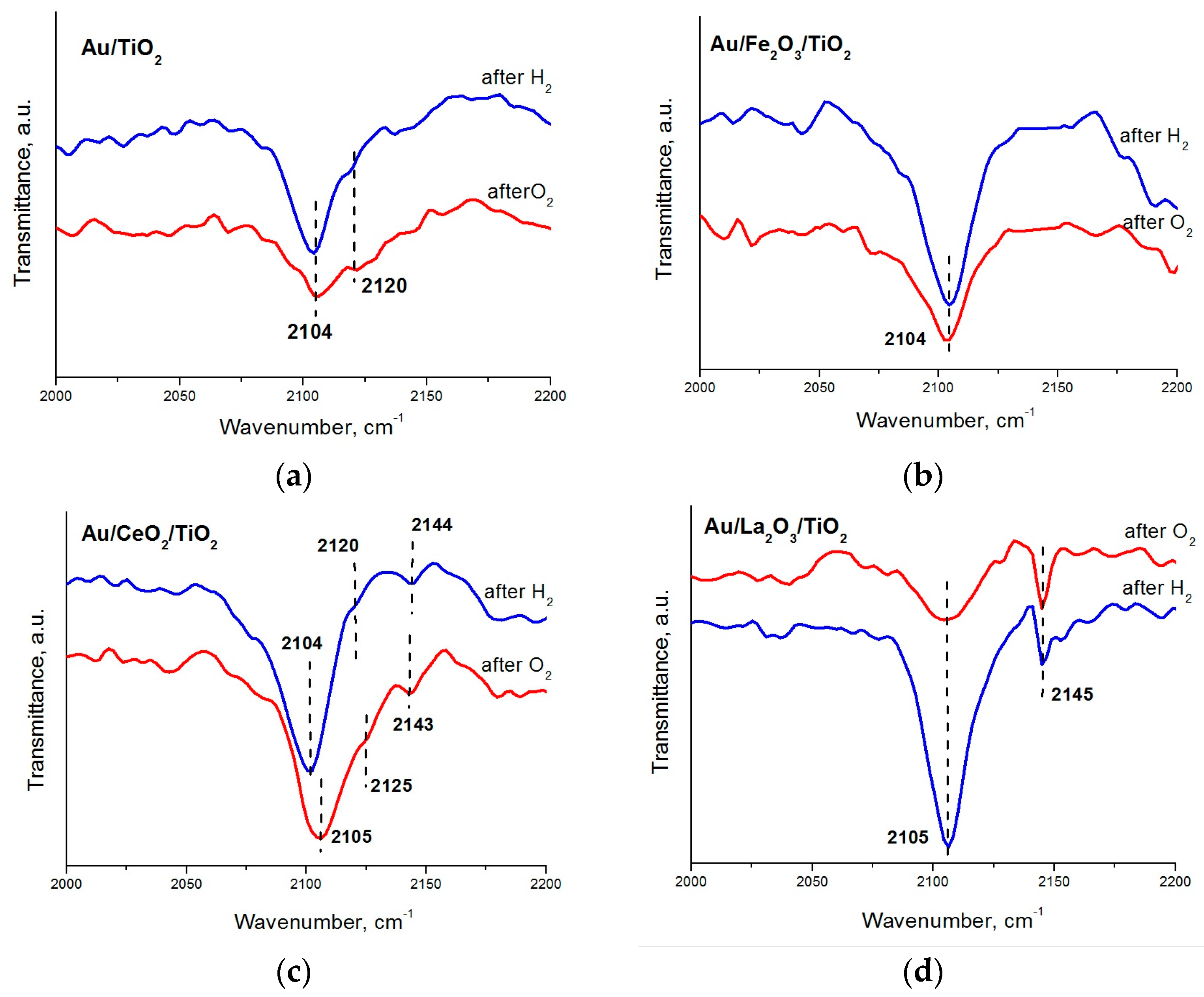
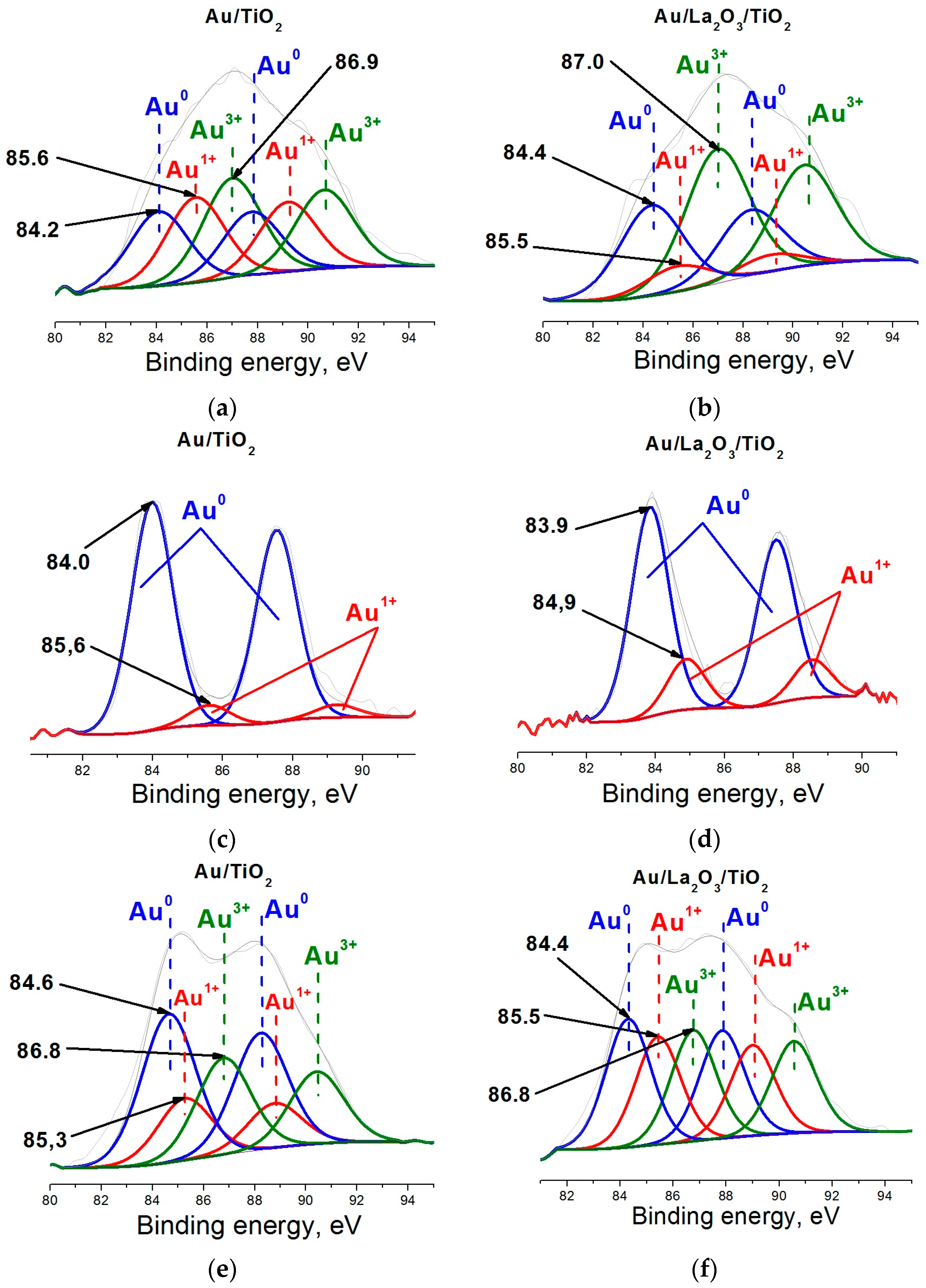
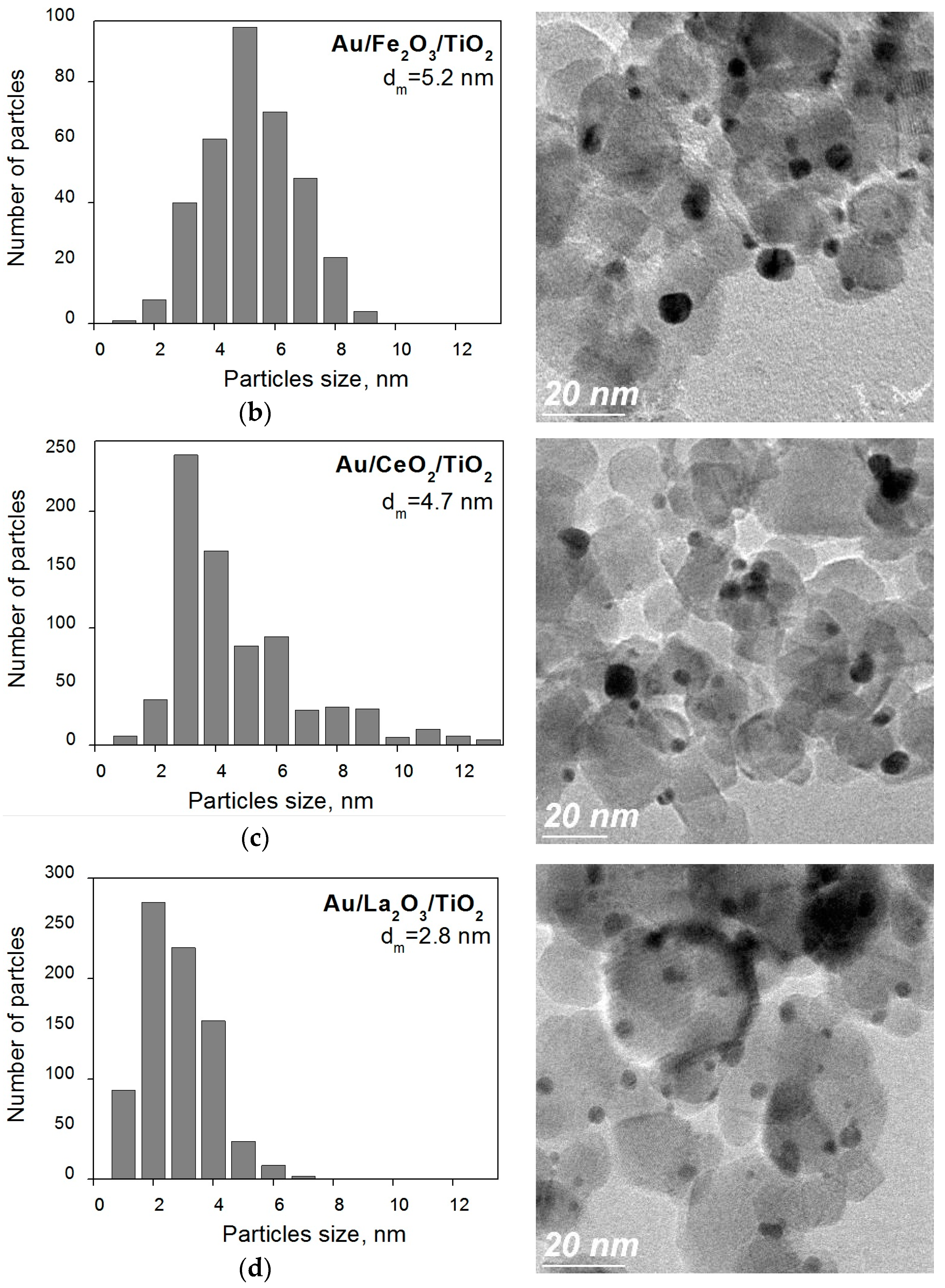
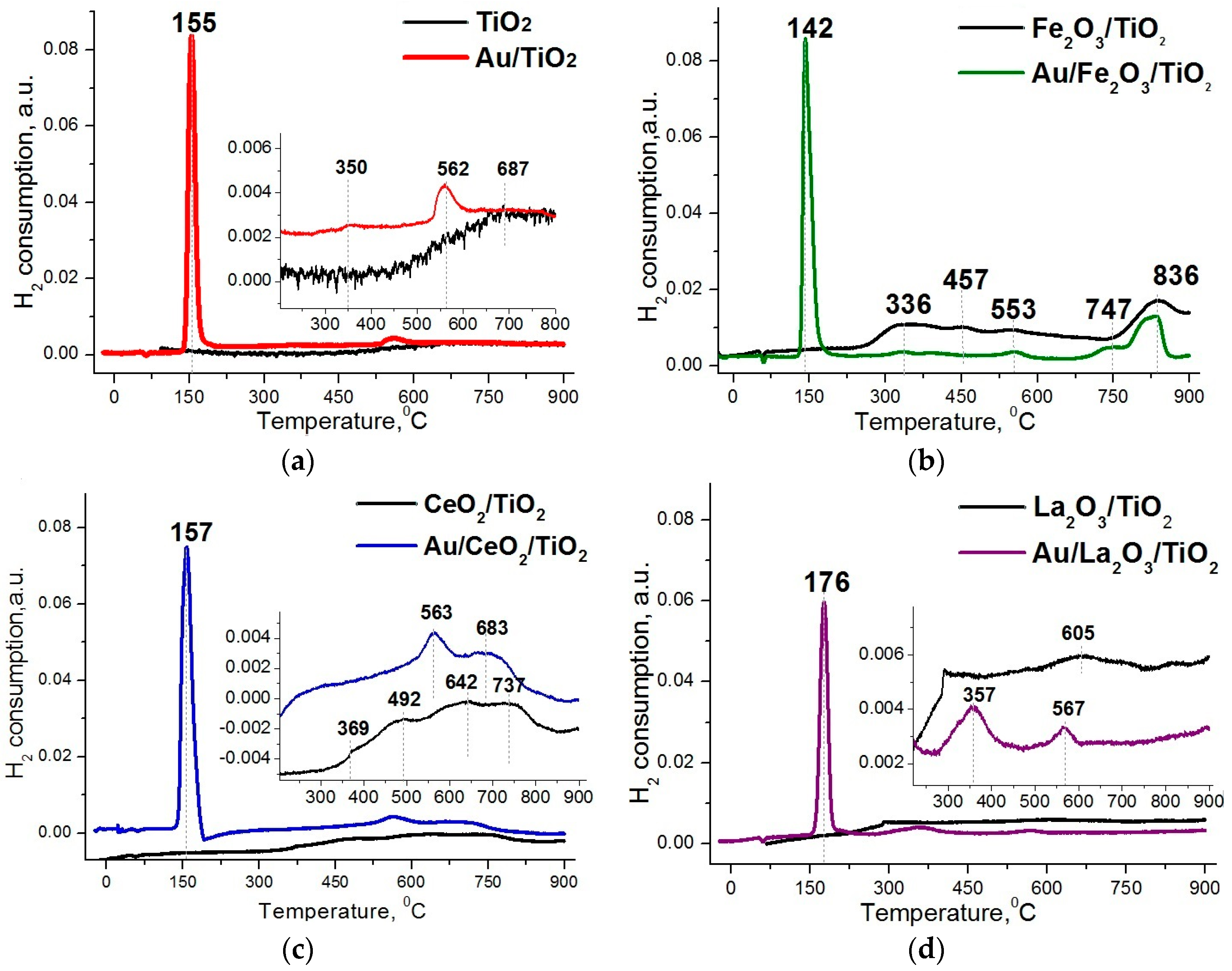
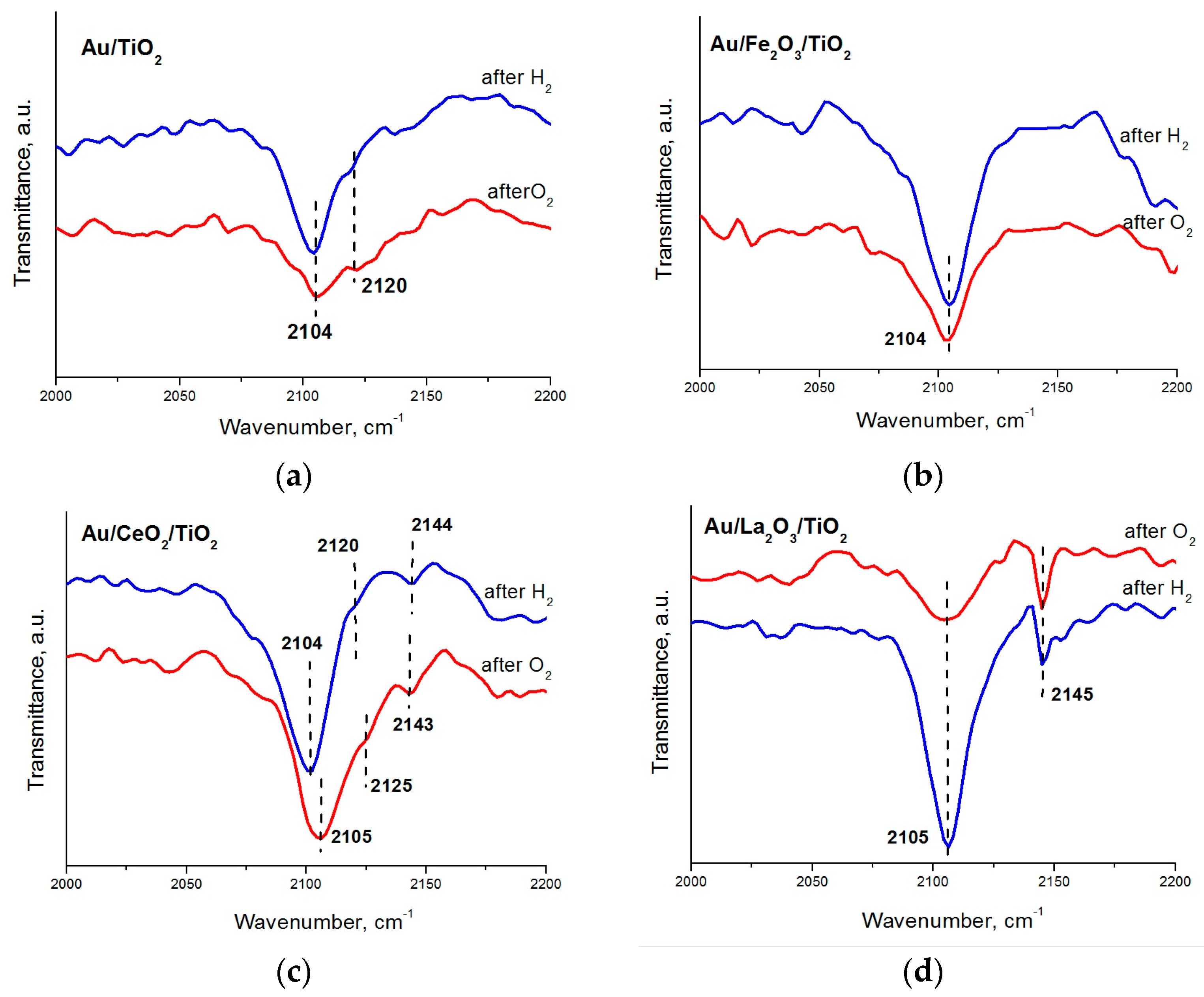
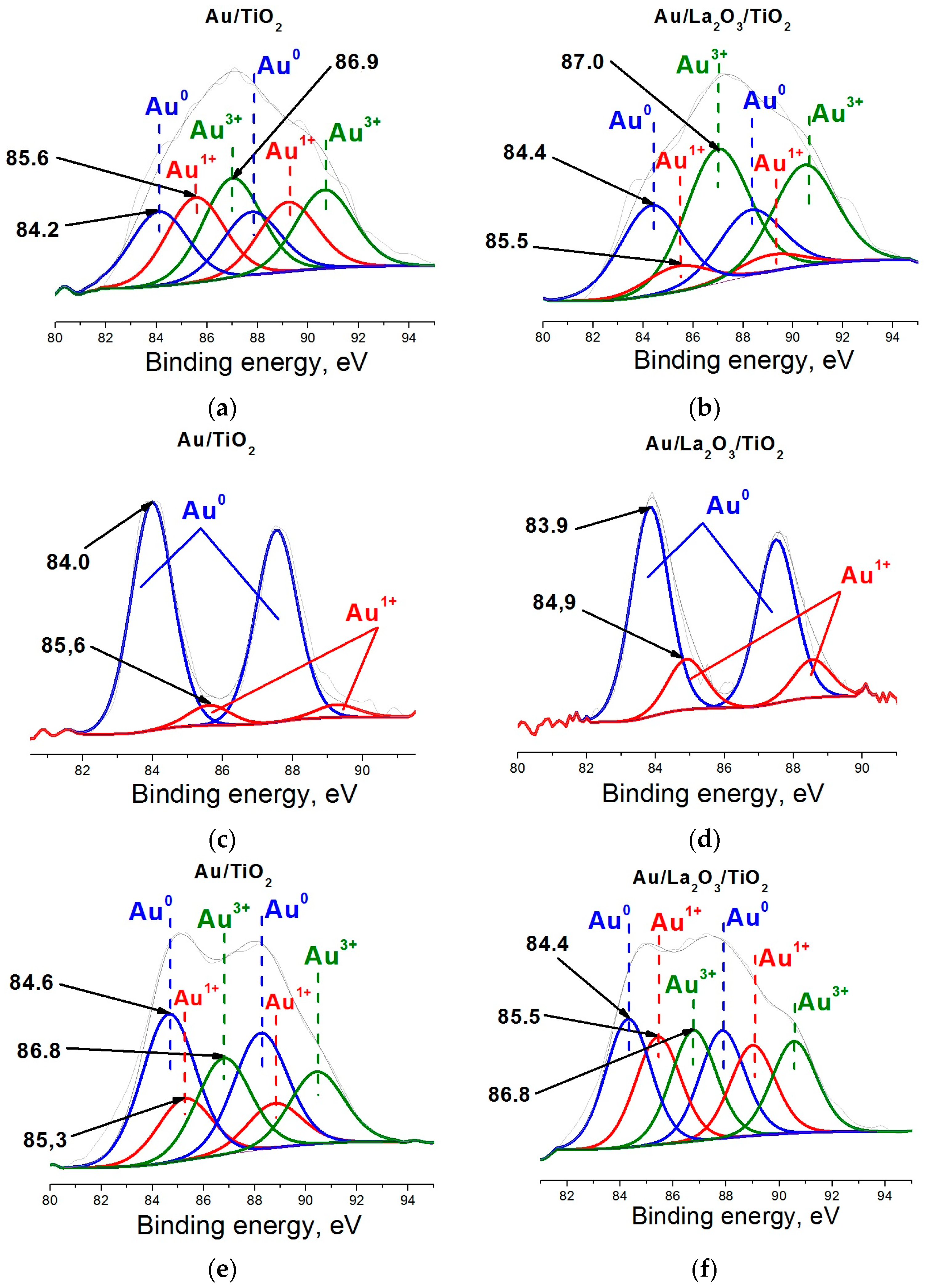
2. Results and Discussion
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Sample Characterization
3.3. Catalytic Testing
4. Conclusions
- Variations in the electronic state of gold are the main cause of the deactivation and activation of the catalysts during redox treatments and after storage.
- The most active catalysts are those with the highest proportion of singly charged ions of gold. Au+ ions seem to be the active sites of the studied catalysts for low-temperature CO oxidation.
- Catalysts with a high content of trivalent gold ions Au3+ and Au0 are inactive at low reaction temperatures.
- The active states of gold in gold-titania catalysts, resistant to the reaction medium and storage conditions, can be stabilized by modification of titanium oxide support with oxides of transition metals.
- Titania modified with lanthanum oxide provides the highest stability and activity after prolonged storage for nanogold catalysts.
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BE | binding energy |
| BET | Brunauer-Emmett-Teller, method of specific surface area measurement |
| EDS | energy dispersive spectroscopy |
| HRTEM | high resolution transmission electron microscopy |
| SEM | scanning electronic microscope |
| TPR | temperature-programmed reduction |
| XPS | X-ray photoelectron spectroscopy |
| FTIR CO | Fourier transformed infrared (FTIR) spectrometry of adsorbed CO |
References
- Park, E.D.; Lee, D.; Lee, H.C. Recent progress in selective CO removal in a H2-rich stream. Catal. Today 2009, 139, 280–290. [Google Scholar] [CrossRef]
- Manzolia, M.; Avgouropoulos, G.; Tabakova, T.; Papavasiliou, J.; Ioannides, T.; Boccuzzi, F. Preferential CO oxidation in H2-rich gas mixtures over Au/doped ceria catalysts. Catal. Today 2008, 138, 239–243. [Google Scholar] [CrossRef]
- McEwan, L.; Julius, M.; Roberts, S.; Fletcher, J.C.Q. A review of the use of gold catalysts in selective hydrogenation reactions. Gold Bull. 2010, 43, 298–306. [Google Scholar]
- Du, X.L.; He, L.; Zhao, S.; Liu, Y.M.; Cao, Y.; He, H.Y.; Fan, K.N. Hydrogen-independent reductive transformation of carbohydrate biomass into γ-valerolactone and pyrrolidone derivatives with supported gold catalysts. Angew. Chem. Int. Ed. 2011, 50, 7815–7819. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.S.; Alvarez, P.J.J.; Fang, Y.-L.; Akёcin, N.; Nutt, M.O.; Miller, J.T.; Heck, K.N. Cleaner water using bimetallic nanoparticle catalysts. J. Chem. Technol. Biotechnol. 2009, 84, 158–166. [Google Scholar] [CrossRef]
- Hernández, W.Y.; Romero-Sarria, F.; Centeno, M.A.; Odriozola, J.A. In situ characterization of the dynamic gold support interaction over ceria modified Eu3+. Influence of the oxygen vacancies on the CO oxidation reaction. J. Phys. Chem. C 2010, 114, 10857–10865. [Google Scholar] [CrossRef]
- Reina, T.R.; Ivanova, S.; Domínguez, M.I.; Centeno, M.A.; Odriozola, J.A. Sub-ambient CO oxidation over Au/MOx/CeO2-Al2O3 (M = Zn or Fe). Appl. Catal. A Gen. 2012, 419–420, 58–66. [Google Scholar] [CrossRef]
- Miah, M.R.; Ohsaka, T. Electrocatalysis of underpotential deposited tin-adatoms-modified gold electrodes toward oxygen reduction reaction in acidic media. J. Electrochem. Soc. 2009, 156, B429–B435. [Google Scholar] [CrossRef]
- Murray, R.W. Nanoelectrochemistry: Metal nanoparticles, nanoelectrodes, and nanopores. Chem. Rev. 2008, 108, 2688–2720. [Google Scholar] [CrossRef] [PubMed]
- Della Pina, C.; Falletta, E.; Prati, L.; Rossi, M. Selective oxidation using gold. Chem. Soc. Rev. 2008, 37, 2077–2095. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Mullins, C.B. Surface science investigations of oxidative chemistry on gold. Acc. Chem. Res. 2009, 42, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Golovko, V.B.; Vaughan, O.P.H.; Abdulkin, P.A.; Berenguer-Murcia, M.S.; Tikhov, M.S.; Johnson, B.F.G.; Lambert, R.M. Selective oxidation with dioxygen by gold nanoparticle catalysts derived from 55-atom clusters. Nature 2008, 454, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.F.; Wang, A.Q.; Wang, Y.L.; Zhang, T.; Li, J. Unusual selectivity of gold catalysts for hydrogenation of 1,3-butadiene toward cis-2-butene: A joint experimental and theoretical investigation. J. Phys. Chem. 2010, C114, 3131–3139. [Google Scholar]
- Bertelsen, S.; Jirgensen, K.A. Organocatalysis—After the gold rush. Chem. Soc. Rev. 2009, 38, 2178–2179. [Google Scholar] [CrossRef] [PubMed]
- Vinod, C.P.; Wilson, K.; Lee, A.F. Recent advances in the heterogeneously catalysed aerobic selective oxidation of alcohols. J. Chem. Technol. Biotechnol. 2011, 86, 161–171. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzynska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Abu Sohel, S.M.; Liu, R.-S. Carbocyclisation of alkynes with external nucleophiles catalysed by gold. Platinum and other electrophilic metals. Chem. Soc. Rev. 2009, 38, 2269–2281. [Google Scholar] [CrossRef] [PubMed]
- Takei, T.; Okuda, I.; Bando, K.K.; Akita, T.; Haruta, M. Gold clusters supported on La(OH)3 for CO oxidation at 193 K. Chem. Phys. Lett. 2010, 493, 207–211. [Google Scholar] [CrossRef]
- Date, M.; Okumura, M.; Tsubota, S.; Haruta, M. Vital role of moisture in the catalytic activity of supported gold nanoparticles. Angew. Chem. Int. Ed. 2004, 43, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.F.; Zhong, Z.Y.; Ramesh, K.; Chen, F.X.; Chen, L.; White, T.; Tay, Q.; Yaakub, S.N.; Wang, Z. Au promotional effects on the synthesis of H2O2 directly from H2 and O2 on supported Pd-Au alloy catalysts. J. Phys. Chem. C 2007, 111, 8410–8413. [Google Scholar] [CrossRef]
- Pestryakov, A.N.; Lunin, V.V.; Bogdanchikova, N.; Temkin, O.N.; Smolentseva, E. Active states of gold in small and big metal particles in CO and methanol selective oxidation. Fuel 2013, 110, 48–53. [Google Scholar] [CrossRef]
- Bogdanchikova, N.; Pestryakov, A.N.; Tuzovskaya, I.; Zepeda, T.A.; Farias, M.H.; Tiznado, H.; Martynyuk, O.A. Effect of redox treatments on activation and deactivation of gold nanospecies supported on mesoporous silica in co oxidation. Fuel 2013, 110, 40–47. [Google Scholar] [CrossRef]
- Bogdanchikova, N.; Pestryakov, A.; Farias, M.H.; Diaz, J.A.; Avalos, M.; Navarrete, J. Formation of TEM- and XRD-undetectable gold clusters accompanying big gold particles on TiO2-SiO2 supports. Solid State Sci. 2008, 10, 908–914. [Google Scholar] [CrossRef]
- Akita, T.; Lu, P.; Ichikawa, S.; Tanaka, K.; Haruta, M. Analytical TEM study on the dispersion of Au nanoparticles in Au/TiO2 catalyst prepared under various temperatures. Surf. Interface Anal. 2001, 31, 73–78. [Google Scholar] [CrossRef]
- Schumacher, B.; Plzak, V.; Kinne, M.; Behm, R.J. Highly active Au/TiO2 catalysts for low-temperature CO oxidation: Preparation, conditioning and stability. Catal. Lett 2003, 89, 109–114. [Google Scholar] [CrossRef]
- Moreau, F.; Bond, G.C. Gold on titania catalysts, influence of some physicochemical parameters on the activity and stability for the oxidation of carbon monoxide. Appl. Catal. A 2006, 302, 110–117. [Google Scholar] [CrossRef]
- Date, M.; Ichihashi, Y.; Yamashita, T.; Chiorino, A.; Boccuzzi, F.; Haruta, M. Performance of Au/TiO2 catalyst under ambient conditions. Catal. Today 2002, 72, 89–94. [Google Scholar] [CrossRef]
- Lee, W.S.; Wan, B.Z.; Kuo, C.N.; Lee, W.C.; Cheng, S. Maintaining catalytic activity of Au/TiO2 during the storage at room temperature. Catal. Commun. 2007, 8, 1604–1608. [Google Scholar] [CrossRef]
- Haruta, M.; Tsubota, S.; Kobayashi, T.; Kageyama, H.; Genet, M.J.; Delmon, B. Low-temperature oxidation of CO over gold supported on TiO2, α-Fe2O3, and Co3O4. J. Catal. 1993, 144, 175–192. [Google Scholar] [CrossRef]
- Zanella, R.; Louis, C. Influence of the conditions of thermal treatments and of storage on the size of the gold particles in Au/TiO2 samples. Catal. Today 2005, 107–108, 768–778. [Google Scholar] [CrossRef]
- Jia, M.; Li, X.; Zhaorigetu; Shen, Y.; Li, Y. Activity and deactivation behavior of Au/LaMnO3 catalysts for CO oxidation. J. Rare Earths 2011, 29, 213–216. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, K.Q.; Yu, J.; Xu, B.Q. A key to the storage stability of Au/TiO2 catalyst. Phys. Chem. Chem. Phys. 2008, 10, 6399–6404. [Google Scholar] [CrossRef] [PubMed]
- Zanella, R.; Delannoy, L.; Louis, C. Mechanism of deposition of gold precursors onto TiO2 during the preparation by cation adsorption and deposition-precipitation with NaOH and urea. Appl. Catal. A Gen. 2005, 291, 62–72. [Google Scholar] [CrossRef]
- Delannoy, L.; Weiher, N.; Tsapatsaris, N.; Beesley, A.M.; Nchari, L.; Schroeder, S.L.M.; Louis, C. Reducibility of supported gold (III) precursors: Influence of the metal oxide support and consequences for CO oxidation activity. Top. Catal. 2007, 44, 263–273. [Google Scholar] [CrossRef]
- Pestryakov, A.N.; Bogdanchikova, N.; Simakov, A.; Tuzovskaya, I.; Jentoft, F.; Farias, M.; Diaz, A. Catalytically active gold clusters and nanoparticles for CO oxidation. Surf. Sci. 2007, 601, 3792–3795. [Google Scholar] [CrossRef]
- Smolentseva, E.; Bogdanchikova, N.; Simakov, A.; Pestryakov, A.; Avalos, M.; Farias, M.H.; Tompos, A.; Gurin, V. Catalytic activity of gold nanoparticles incorporated into modified zeolites. J. Nanosci. Nanotechnol. 2007, 7, 1882–1886. [Google Scholar] [CrossRef]
- Pestryakov, A.; Tuzovskaya, I.; Smolentseva, E.; Bogdanchikova, N.; Jentoft, F.C.; Knop-Gericke, A. Formation of gold nanoparticles in zeolites. Int. J. Modern Phys. B 2005, 19, 2321–2326. [Google Scholar] [CrossRef]
- Pestryakov, A.N.; Lunin, V.V.; Kharlanov, A.N.; Bogdanchikova, N.E.; Tuzovskaya, I.V. Electronic state of gold in supported clusters. Eur. Phys. J. D 2003, 24, 307–309. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Hutchings, G.J. Gold catalysis. Angew. Chem. Int. Ed. 2006, 45, 7896–7936. [Google Scholar] [CrossRef] [PubMed]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; Imperial College Press: London, UK, 2006; p. 384. [Google Scholar]
- Qu, Z.; Huang, W.; Cheng, M.; Bao, X. Restructuring and redispersion of silver on SiO2 under oxidizing/reducing atmospheres and its activity toward CO oxidation. J. Phys. Chem. B 2005, 109, 15842–15848. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, T.; Seshan, K.; Lercher, J.A.; Lefferts, L.; Aika, K. Selective reduction of NO to N2 in the presence of oxygen over supported silver catalysts. Appl. Catal. B Environ. 2002, 37, 205–216. [Google Scholar] [CrossRef]
- Herzing, A.A.; Kiely, C.J.; Carley, A.F.; Landon, P.; Hutchings, G.J. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Martynyuk, O.; Kotolevich, Y.; Vélez, R.; Cabrera Ortega, J.E.; Tiznado, H.; Zepeda, T.; Mota-Morales, J.D.; Pestryakov, A.; Bogdanchikova, N. On the High Sensitivity of the Electronic States of 1 nm Gold Particles to Pretreatments and Modifiers. Molecules 2016, 21, 432. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; De Jesus, J.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Low-content gold-ceria catalysts for the water-gas shift and preferential CO oxidation reactions. Appl. Catal. A Gen. 2005, 291, 126–135. [Google Scholar] [CrossRef]
- Simakov, A.; Tuzovskaya, I.; Pestryakov, A.; Bogdanchikova, N.; Gurin, V.; Avalos, M.; Farías, M.H. On the nature of active gold species in zeolites in CO oxidation. Appl. Catal. A 2007, 331C, 121–128. [Google Scholar] [CrossRef]
- Parida, K.M.; Sahu, N.; Mohapatra, P.; Scurrell, M.S. Low temperature CO oxidation over gold supported mesoporous Fe–TiO2. J. Mol. Cat. A Chem. 2010, 319, 92–97. [Google Scholar] [CrossRef]
- Lenzi, G.G.; Fávero, C.V.B.; Colpini, L.M.S.; Bernabe, H.; Baesso, M.L.; Specchia, S.; Santos, O.A.A. Photocatalytic reduction of Hg(II) on TiO2 and Ag/TiO2 prepared by the sol–gel and impregnation methods. Desalinations 2011, 270, 241–247. [Google Scholar] [CrossRef]
- Biabani-Ravandi, A.; Rezaei, M.; Fattah, Z. Catalytic Performance of Ag/Fe2O3 for the low temperature oxidation of carbon monoxide. Chem. Eng. J. 2013, 219, 124–130. [Google Scholar] [CrossRef]
- Qu, Z.; Yu, F.; Zhang, X.; Wang, Y.; Gao, J. Support effects on the structure and catalytic activity of mesoporous Ag/CeO2 catalysts for CO oxidation. Chem. Eng. J. 2013, 229, 522–532. [Google Scholar] [CrossRef]
- Li, S.; Zhu, H.; Qin, Z.; Wang, G.; Zhang, Y.; Wu, Z.; Li, Z.; Chen, G.; Dong, W.; Wu, Z.; et al. Morphologic effects of nano CeO2–TiO2 on the performance of Au/CeO2–TiO2 catalysts in low-temperature CO oxidation. Appl. Catal. B Environ. 2014, 144, 498–506. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, S.; Cao, A.; Thompson, R.L.; Vesera, G. Au-mixed lanthanum/cerium oxide catalysts for water gas shift. Appl. Catal. B Environ. 2010, 99, 89–95. [Google Scholar] [CrossRef]
- Oros-Ruiz, S.; Zanella, R.; López, R.; Hernández-Gordillo, A.; Gómez, R. Photocatalytic hydrogen production by water/methanol decomposition using Au/TiO2 prepared by deposition–precipitation with urea. J. Hazard. Mater. 2013, 263, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Penkova, A.; Chakarova, K.; Laguna, O.H.; Hadjiivanov, K.; Romero Saria, F.; Centeno, M.A.; Odriozola, J.A. Redox chemistry of gold in a Au/FeOx/CeO2 CO oxidation catalyst. Catal. Commun. 2009, 10, 1196–1202. [Google Scholar] [CrossRef]
- Casaletto, M.P.; Longo, A.; Martorana, A.; Prestianni, A.; Venezia, A.M. XPS study of supported gold catalysts: The role of Au0 and Auδ+ species as active sites. Surf. Interface Anal. 2006, 38, 215–218. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the modified nanogold catalysts are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Temperature for 100% CO Conversion, °C | ||||
|---|---|---|---|---|---|
| 1 * | 2 * | 3 * | 4 * | 5 * | |
| Au/TiO2 | 30 | 30 | 80 | 30 | 30 |
| Au/Fe2O3/TiO2 | 305 | 30 | 80 | 30 | 30 |
| Au/CeO2/TiO2 | 230 | 30 | 80 | 30 | 30 |
| Au/La2O3/TiO2 | 215 | 30 | 30 | 30 | 30 |
| Sample | SBET, m2/g | EDX | |
|---|---|---|---|
| Support | Catalyst | Au Content, wt. % | |
| Au/TiO2 | 55.5 | 45.5 | 4.5 |
| Au/La2O3/TiO2 | 45.3 | 45.2 | 3.6 |
| Au/CeO2/TiO2 | 43.4 | 46.6 | 3.5 |
| Au/Fe2O3/TiO2 | 45.5 | 44.2 | 4.2 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolobova, E.; Kotolevich, Y.; Pakrieva, E.; Mamontov, G.; Farías, M.H.; Bogdanchikova, N.; Cortés Corberán, V.; Pestryakov, A. Causes of Activation and Deactivation of Modified Nanogold Catalysts during Prolonged Storage and Redox Treatments. Molecules 2016, 21, 486. https://doi.org/10.3390/molecules21040486
Kolobova E, Kotolevich Y, Pakrieva E, Mamontov G, Farías MH, Bogdanchikova N, Cortés Corberán V, Pestryakov A. Causes of Activation and Deactivation of Modified Nanogold Catalysts during Prolonged Storage and Redox Treatments. Molecules. 2016; 21(4):486. https://doi.org/10.3390/molecules21040486
Chicago/Turabian StyleKolobova, Ekaterina, Yulia Kotolevich, Ekaterina Pakrieva, Grigory Mamontov, Mario H. Farías, Nina Bogdanchikova, Vicente Cortés Corberán, and Alexey Pestryakov. 2016. "Causes of Activation and Deactivation of Modified Nanogold Catalysts during Prolonged Storage and Redox Treatments" Molecules 21, no. 4: 486. https://doi.org/10.3390/molecules21040486
APA StyleKolobova, E., Kotolevich, Y., Pakrieva, E., Mamontov, G., Farías, M. H., Bogdanchikova, N., Cortés Corberán, V., & Pestryakov, A. (2016). Causes of Activation and Deactivation of Modified Nanogold Catalysts during Prolonged Storage and Redox Treatments. Molecules, 21(4), 486. https://doi.org/10.3390/molecules21040486