Abstract
We report single crystal X-ray diffraction (hereafter, SCXRD) analyses of derivatives featuring the electron-donor N-ethylcarbazole or the (4-diphenylamino)phenyl moieties associated with a -CN group attached to a double bond. The compounds are (2Z)-3-(4-(diphenylamino)-phenyl)-2-(pyridin-3-yl)prop-2-enenitrile (I), (2Z)-3-(4-(diphenylamino)phenyl)-2-(pyridin-4-yl)-prop-2-enenitrile (II) and (2Z)-3-(9-ethyl-9H-carbazol-3-yl)-2-(pyridin-2-yl)enenitrile (III). SCXRD analyses reveal that I and III crystallize in the monoclinic space groups P2/c with Z’ = 2 and C2/c with Z’ = 1, respectively. Compound II crystallized in the orthorhombic space group Pbcn with Z’ = 1. The molecular packing analysis was conducted to examine the pyridine core effect, depending on the ortho, meta- and para-positions of the nitrogen atom, with respect to the optical properties and number of independent molecules (Z’). It is found that the double bond bearing a diphenylamino moiety introduced properties to exhibit a strong π-π-interaction in the solid state. The compounds were examined to evaluate the effects of solvent polarity, the role of the molecular structure, and the molecular interactions on their self-assembly behaviors. Compound I crystallized with a cell with two conformers, anti and syn, due to interaction with solvent. DFT calculations indicated the anti and syn structures of I are energetically stable (less than 1 eV). Also electrochemical and photophysical properties of the compounds were investigated, as well as the determination of optimization calculations in gas and different solvent (chloroform, cyclohexane, methanol, ethanol, tetrahydrofuran, dichloromethane and dimethyl sulfoxide) in the Gaussian09 program. The effect of solvent by PCM method was also investigated. The frontier HOMO and LUMO energies and gap energies are reported.
1. Introduction
The study of π-conjugated organic compounds has led to the development of a rich variety of new concepts based on the interplay between their π-electronic structures and their molecular structures. These structures are associated with intense optical properties and thus, the identification and understanding of the structure-property relationships of these molecules is of key importance [1,2,3]. Bulk properties such as luminescence, excitation migration, and carrier mobility depend on the intermolecular dipole coupling, which is determined by the relative positions of adjacent molecules and the directions of their dipole moments [4,5,6,7,8,9,10,11,12,13,14]. Therefore, the design and synthesis of functional molecular solid-state structures or arrangements through tuning the intermolecular interactions remain challenging. The molecular arrangement of a compound in the solid state (i.e., the crystal packing) plays an important role in the performance of organic electronic devices [15,16,17,18].
Of the four main packing motifs in organic solid states [19,20,21,22,23,24,25], the typical herringbone packing, characterized by non-π-π overlapping of neighboring molecules, is unfavorable for charge transport. The large angle between the planes of adjacent molecules along the herringbone diagonal tends to reduce the strength of intermolecular interactions [26,27]. The non-classical herringbone packing with π-π overlap between neighboring molecules, also called slipped π-stacking, lamellar packing with one-dimensional (1-D) π-stacking, and lamellar packing with two-dimensional (2-D) π-stacking represent the other common forms of molecular packing. We have focused our investigations on the synthesis of different α,β-unsaturated acrylonitrile with different derivatives functionalized with electron donors such as diphenylamino-, dimethylamino-, and carbazole attached to the conjugated double bond and with electron acceptors such as the CN and the pyridyl core (in positions 4, 3, 2). Our goal was to observe the effect of substituent position on the optical properties [28,29,30,31]. We reported marked differences in the optical properties of the diphenylamino versus dimethylamine groups, specifically in their fluorescence behaviors, which we compared in the solid state, in single crystals, and in solution in different solvents [28,29]. For some organic materials, the fluorescence of the chromophore is quenched in the solid state, based on the phenomenon of aggregation-caused quenching (ACQ). In contrast, for other compounds, the emission depends on the formation of aggregates, termed aggregation-induced emission (AIE) or aggregation-induced emission enhancement (AIEE) [32,33,34,35,36,37]. The organic materials exhibit either no or weak fluorescence in solution and the aggregated particles (obtained by adding the solution into a poorly solubilizing solvent) exhibit relatively intense fluorescence upon UV irradiation such as for (dimethylamino)acrylonitrile derivatives, specifically the Z-2-(phenyl)-3-(4-dimethylaminophenyl)acrylonitrile [28] and α-cyano-stilbene [38].
Attempts are made to grow adequately sized single crystals in order to investigate that the types of cofacial packing led to different motifs in the herringbone packing. Several research groups have reported [25,26] that in the absence of electrostatic repulsion, organic molecules can be exactly superimposed on top of one another in a perfect co-facial situation, and usually any displacements can occur along the shortest/longest molecular axes between adjacent molecules. These theoretical investigations show that such displacements could strongly affect the intermolecular electronic couplings, in a way that depends on the bonding-antibonding pattern of the frontier molecular orbitals (HOMO, highest occupied molecular orbital, or LUMO, lowest unoccupied molecular orbital) [39]. These studies suggest that changes in the crystal packing were responsible for the crystallochromic property, i.e., changes in the color of the crystal [40,41]. Crystal packing of a given structure is described by the unit cell and the crystallographic symmetry operations of the given space group. In other studies [42,43,44], the packing problem for high Z’ structures (i.e., structures with Z’ > 1) is related to polymorphism for about 9% of crystal structures, although some chemically identical molecules are not related to one another by crystallographic symmetry and occupy distinctly independent positions. An examination of crystal packing is fundamental to investigate properties such as color and emission.
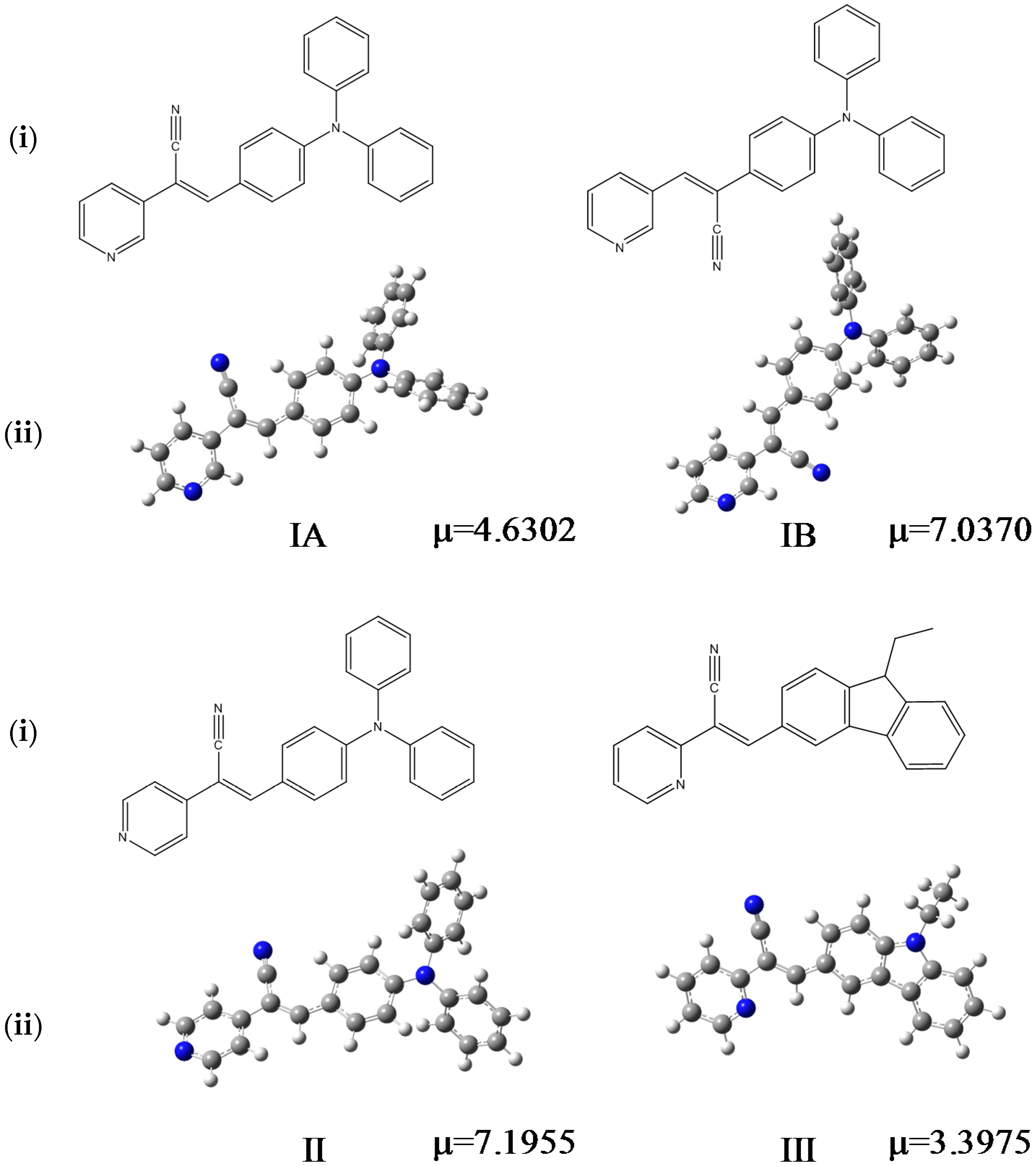
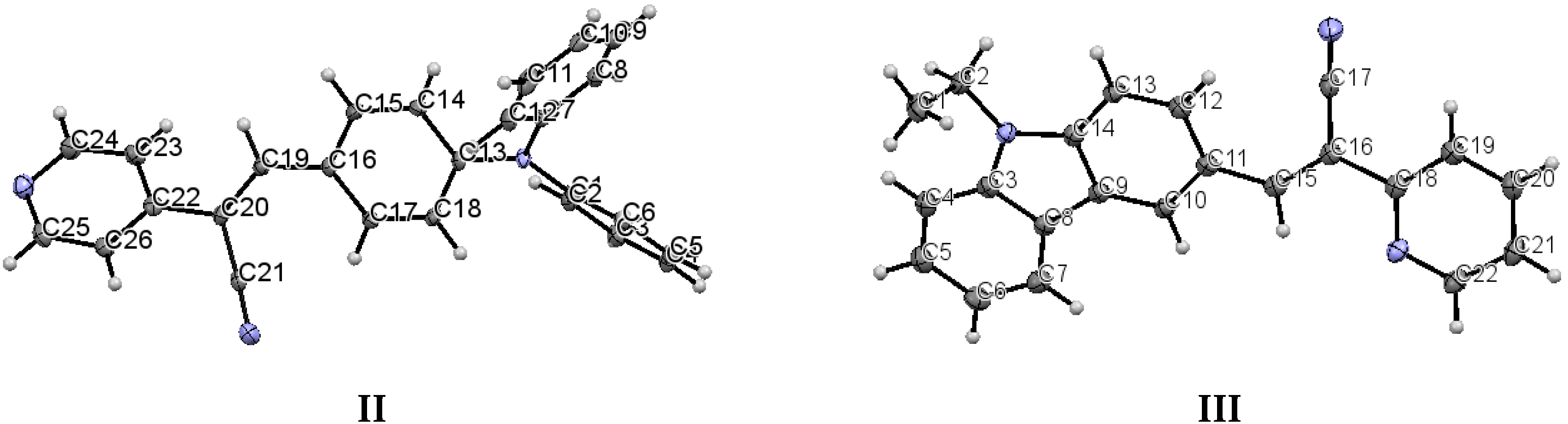
Understanding the nature of the interactions that determine the molecular packing in the solid state and how these interactions affect the optical and electrical properties of these materials is therefore essential for tuning their properties. It is well known that triphenylamine-based materials exhibit a variety of intra- and intermolecular interactions such as Van der Waals interactions, weak hydrogen bonding, π-π stacking, and nitrogen-CN interactions originating from the high polarizability of the CN electrons in the rings. Therefore, in the present study we rely on single crystal X-ray crystallography analyses to investigate different optical properties such as absorbance and fluorescence from derivatives with D-π-A type dyes (D = donor, A = acceptor) and the main packing motifs in organic solid states. The derivatives are (2Z)-3-(4-(diphenylamino)phenyl)-2-(pyridin-3-yl)prop-2-enenitrile, (2Z)-3-(4-(diphenylamino)phenyl)-2-(pyridin-4-yl)prop-2-enenitrile and (2Z)-3-(9-ethyl-9H-carbazol-3-yl)-2-(pyridin-2-yl)enenitrile (Figure 1). The compounds bear an α,β-unsaturated nitrile and a pyridine group in the meta (I), para (II) and ortho (III) positions, respectively. We report the influence of solvent polarity and the roles of the molecular structure and the molecular interactions on their self-assembly behaviors and their optical properties. We also performed theoretical calculations in different solvent (chloroform, cyclohexane, methanol, ethanol, tetrahydrofuran, dichloromethane and dimethyl sulfoxide) using the Gaussian09 program. DFT calculations were made to determine the frontier HOMO and LUMO energies and gap energies.
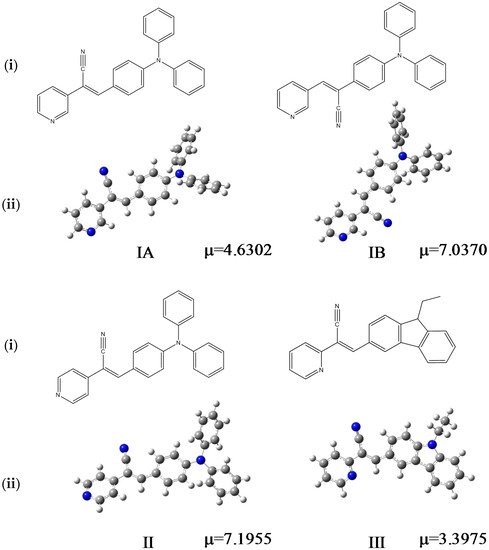
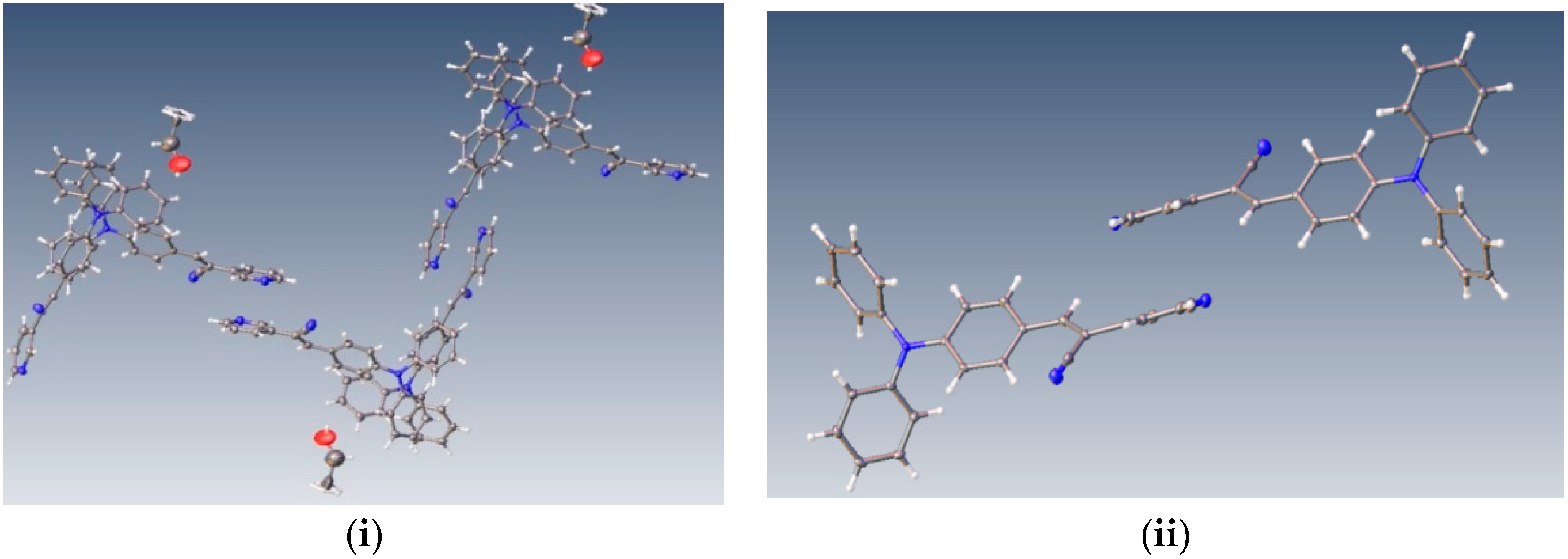
Figure 1.
Chemical formulas of the compounds performed in ChemDraw software (i), optimized structures and µ (dipole moment, Debyes) calculated in gas phase at M06L/cc-pVDZ theory level (ii).
2. Results and Discussion
2.1. X-ray Crystallography and Photolysis Properties of I–III
Crystallographic data for I–III are summarized in Table 1. For compounds I–III the data were collected at 110(2) K after the crystals had been flash-cooled from room temperature. The crystal structure of I (Figure 1) shows that the molecule of the meta position of the pyridyl ring (structure I) belongs to a monoclinic crystal with Z′ > 2 independent molecules of I per asymmetric unit, which could be attributed to a co-crystal with the solvent, (eight per unit cell, Z = 8) and to the P2/c space group. Crystal II belongs to an orthorhombic system (Pbcn, Z = 8). Crystal III also belongs to a monoclinic system with space group C2/c with a Z = 8, indicating an ordered structure. Both II and III display Z′ = 1. Notably, for I, II, and III, the unit cell contained eight molecules.

Table 1.
Summary of Crystallography Data for I–III.
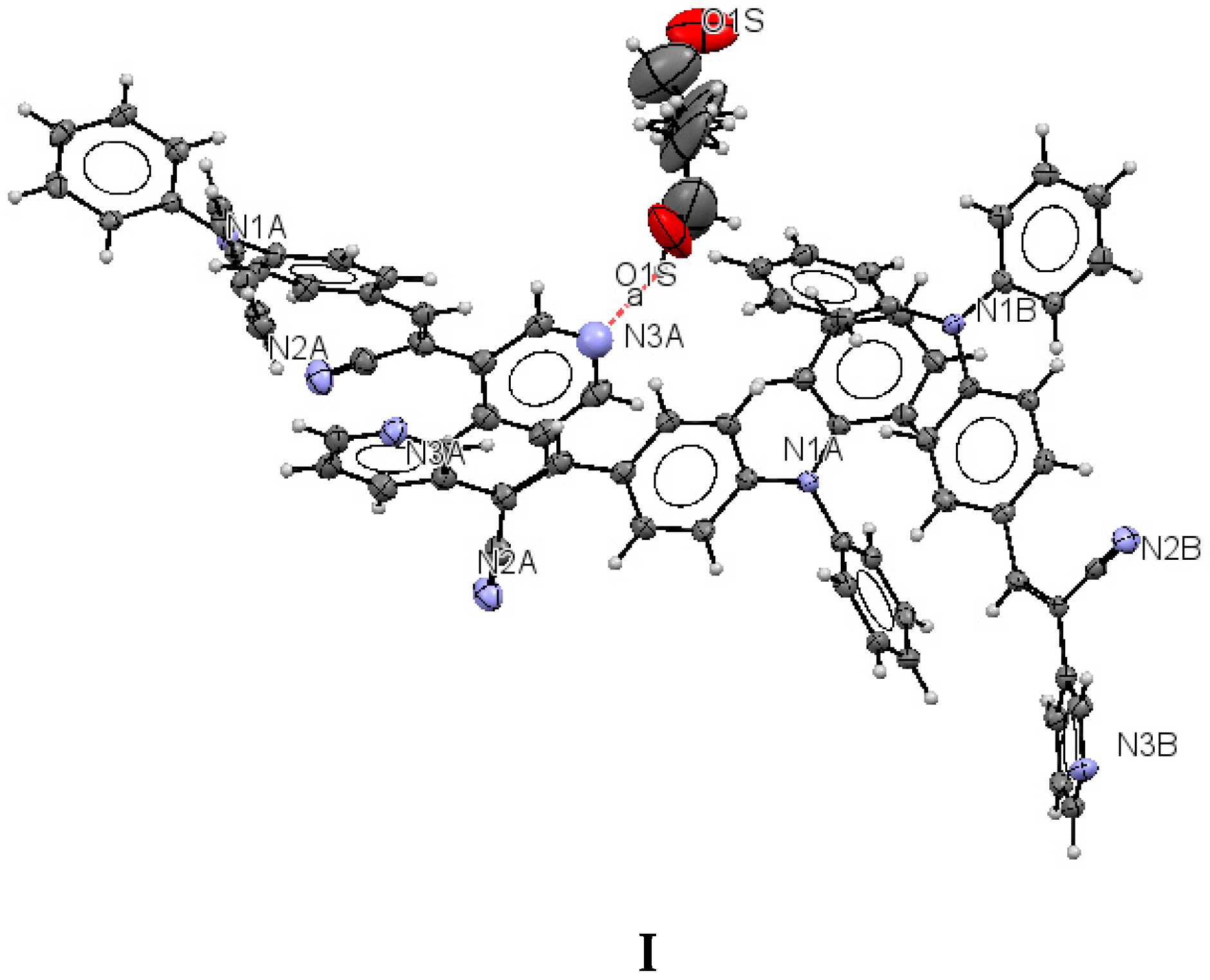
The structure of (I) is modeled as ordered even though the lattice ethanol molecules are likely to be disordered as they are located at sites of two-fold axis symmetry. The elongated ellipsoids found for the lattice solvent molecule (Figure 2) are consistent with this observation. The occupancy factor for the lattice solvent molecule was refined freely, and its value is 0.791(7). It is possible that the void contains a mixture of disordered solvent molecules. The twin relationship corresponded to a two-fold axis found along the c axis. The batch scale factor refined to 0.3680(16). The final refinement was performed using the HKL5 instruction (i.e., the hkl file includes the set of reflections from domain 1 and the set of overlapped reflections from component 2). The two crystallographically independent molecules of the target compound were found to be ordered (Figure 2).
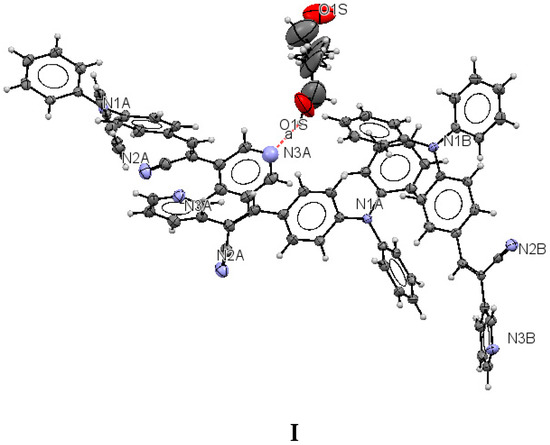
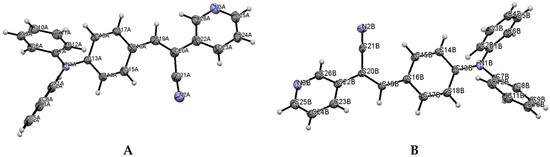
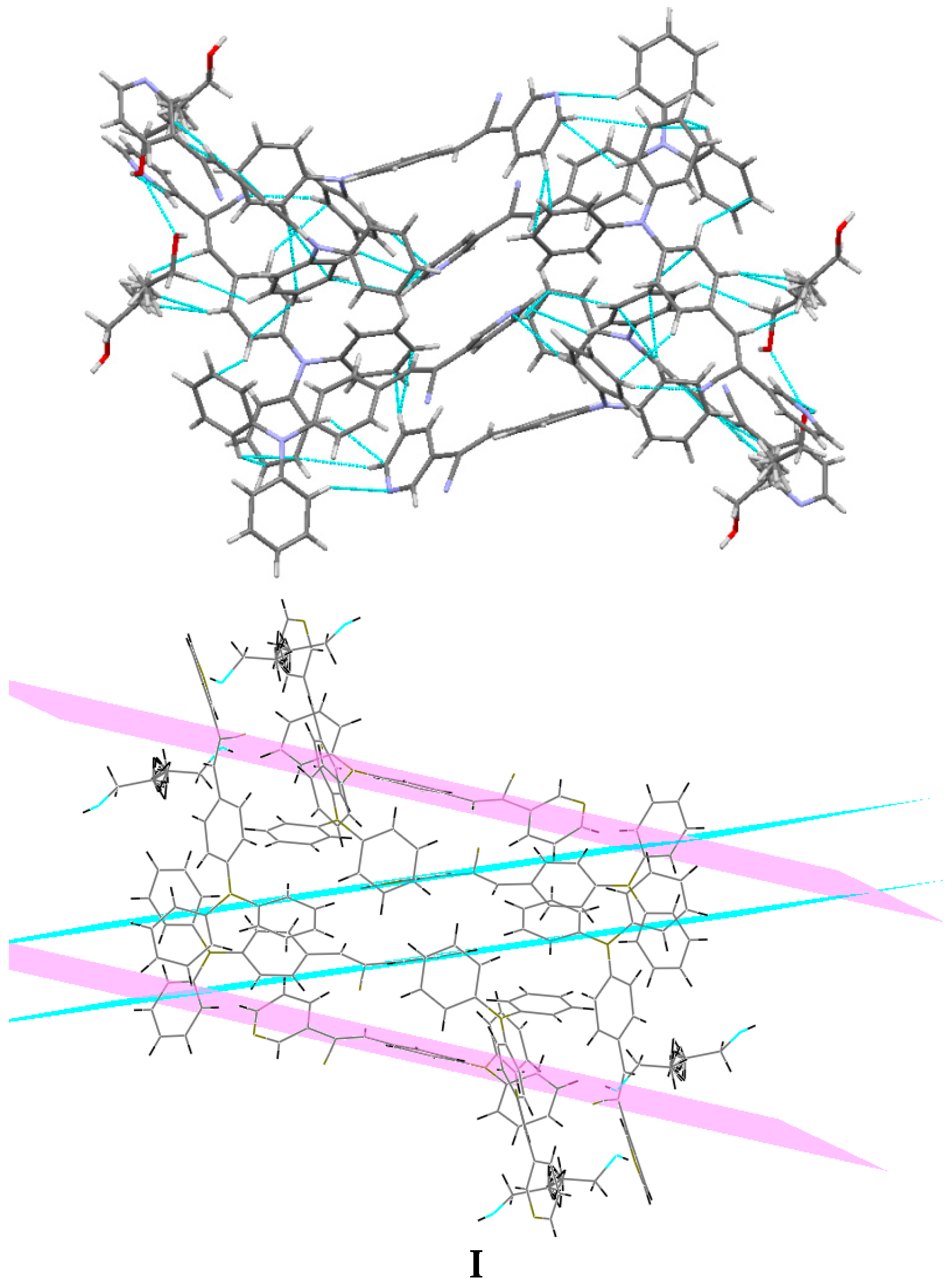
Figure 2.
Displacement ellipsoid plots (50% probability level) of I. The two crystallographically independent molecules A and B are shown separately.
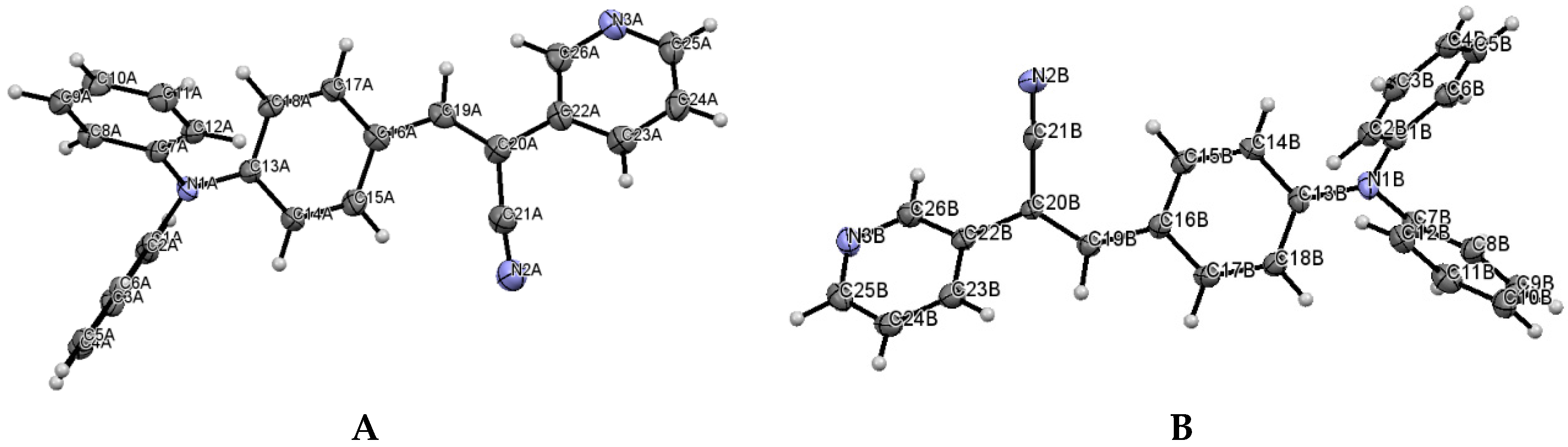
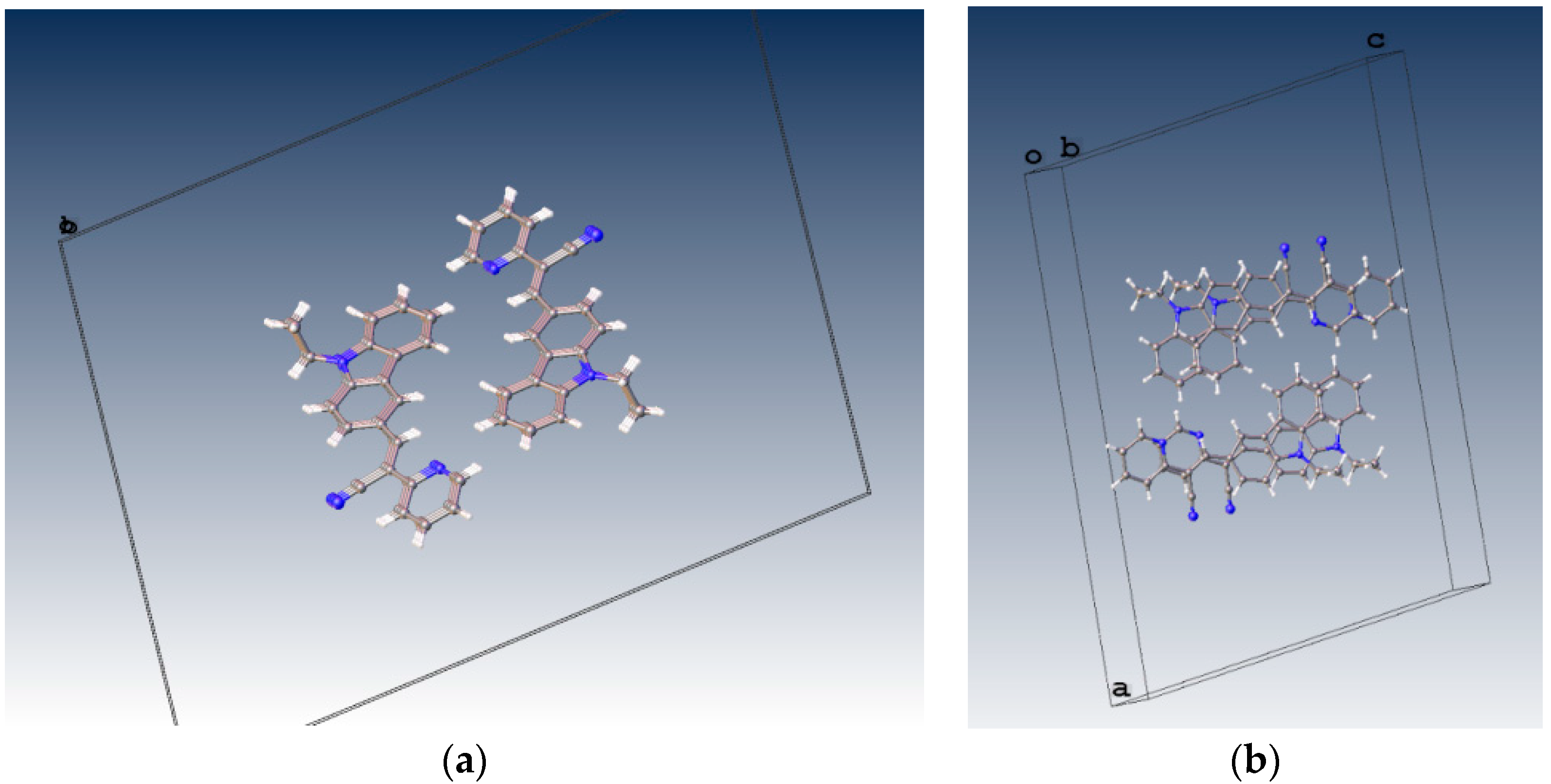
The most interesting feature in the structure of I is concerned with the two independent conformers A and B (Figure 3). For A, there is one O-H(solvent)···N hydrogen bond interaction (Figure 4), and the -C≡N group [C(21A)-N(2A)] is found anti to the pyridine group. For B, there is no such O-H (solvent)···N interaction, and the -C≡N group [C(21B)-N(2B)] is found syn to the pyridine group. The molecules A and B are different conformers along the bond connecting the phenyl, double bond and pyridyl moeties. Table 2 provides a list of torsion angles for molecules A and B, which notable differ in sign. The anti conformation is typically found in different isomeric compounds that have already been reported in the literature [28,29,30], and also in the structures of II and III reported herein.
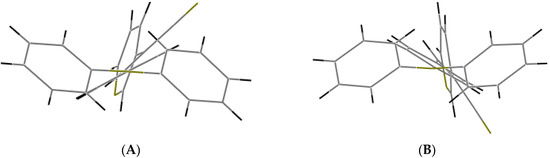
Figure 3.
Two conformers A and B for the structure I.
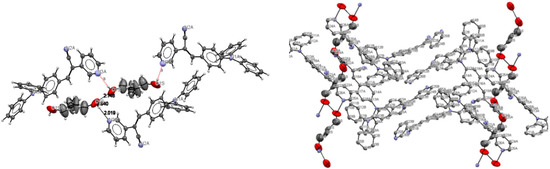
Figure 4.
Intermolecular hydrogen bond interactions between the lattice solvent molecules (donor) and conformers A (acceptor).
Table 2.
Hydrogen-bonds D···H···A (Å) and D−HA angles (°) of compound I.
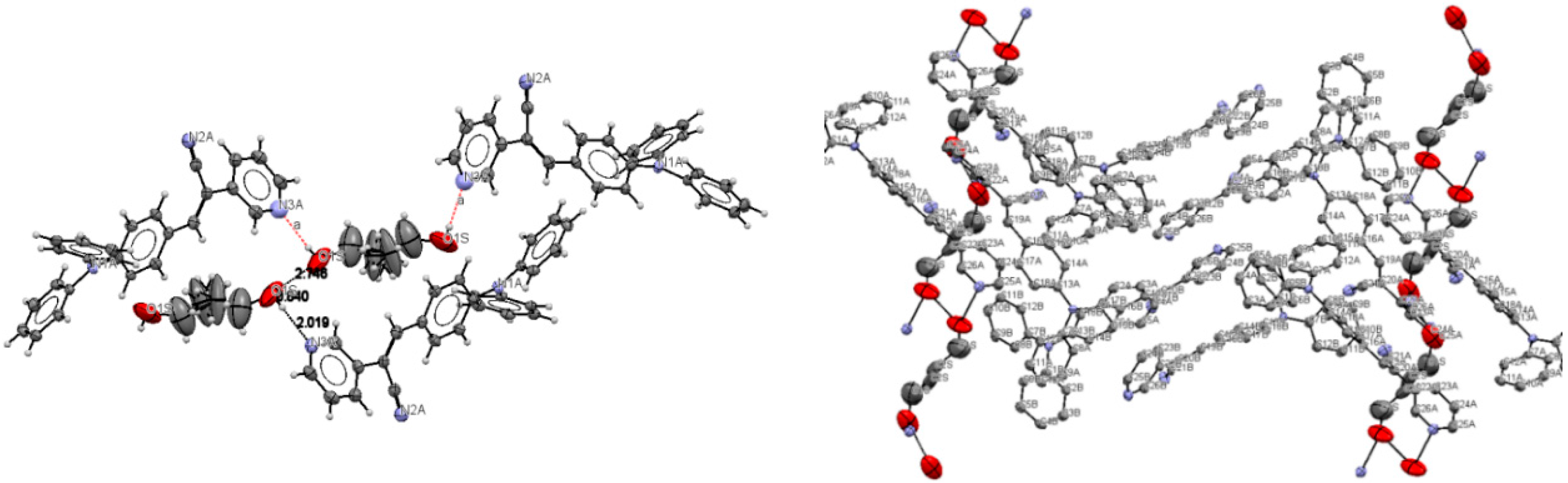
Lattice solvent ethanol molecules act as H-bond donor bridges between molecules of conformers A along the c axis. Table 2 provides a list of H-bond interactions in the structure of I.
The N3A···O1S and O1S···O1S intermolecular distances are ca. 2.77 and 2.75 Å, respectively, which suggests that the H atom from the -OH group of the ethanol molecules is disordered (this is not surprising as the ethanol molecules are most likely disordered) [45]. The presence of lattice solvent molecules in the crystal packing of I might indicate that crystal growth is optimized with O-H(solvent)···N(compound).
Table 3 includes a list of selected bond lengths and torsion angles for conformers A and B. The values for the pyridine ring and the p-(diphenylamino)phenyl group indicate that the N,N-diphenyl substituents are twisted through the single bonds of the N-atom, and those conformers are oppositely twisted. This behavior does not occur for structure II and for similar structures recently reported [28,29,30,31]. Table 3 also provides a list the selected bond lengths for III, as well as, the dihedral angles between the acrylonitrile linkage and the phenyl ring and with the diphenylaminophenyl or carbazole moieties.
Table 3.
Bond lengths (Å) and torsion angles (°) selected in the crystal structures of I and II.
When the nitrogen atom is found at the meta position, the crystal packing significantly differs from those of the analogous structures without nitrogen and those of the structures of II (para position) and III (ortho position). The compounds II and III (Figure 5) have the Z-geometry about the ethylene bridge that links the aromatic rings and heterocyclic groups.
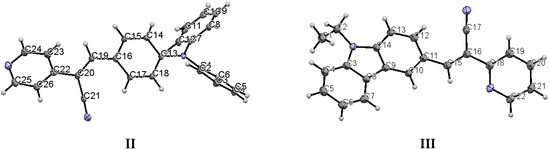
Figure 5.
Displacement ellipsoid plots (50% probability level) II and III. Both compounds have anti conformation.
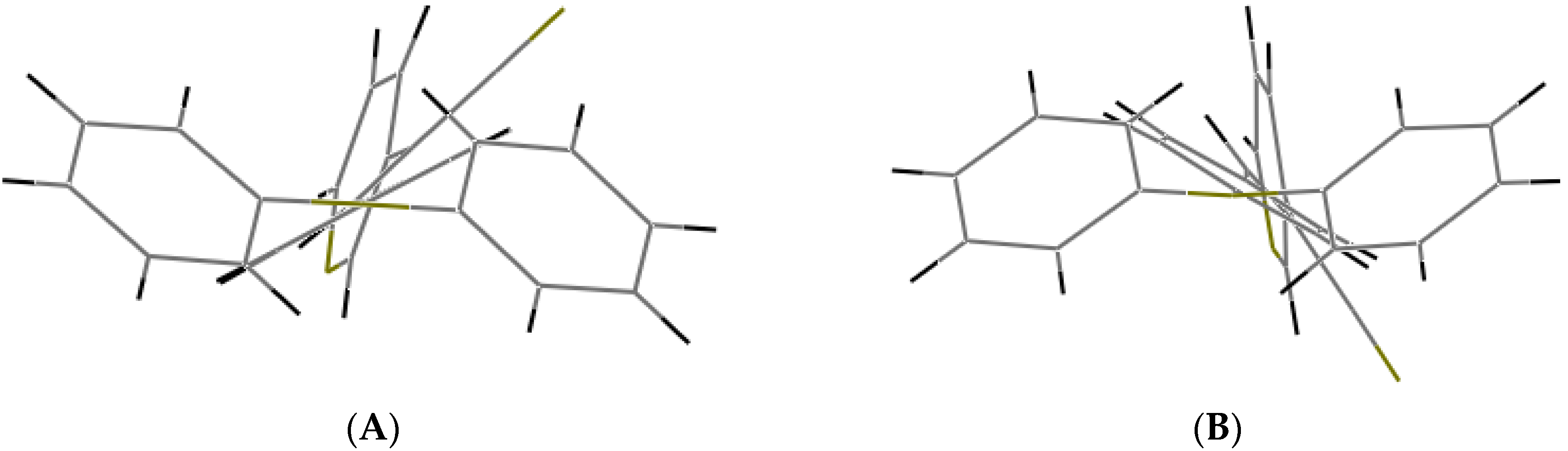
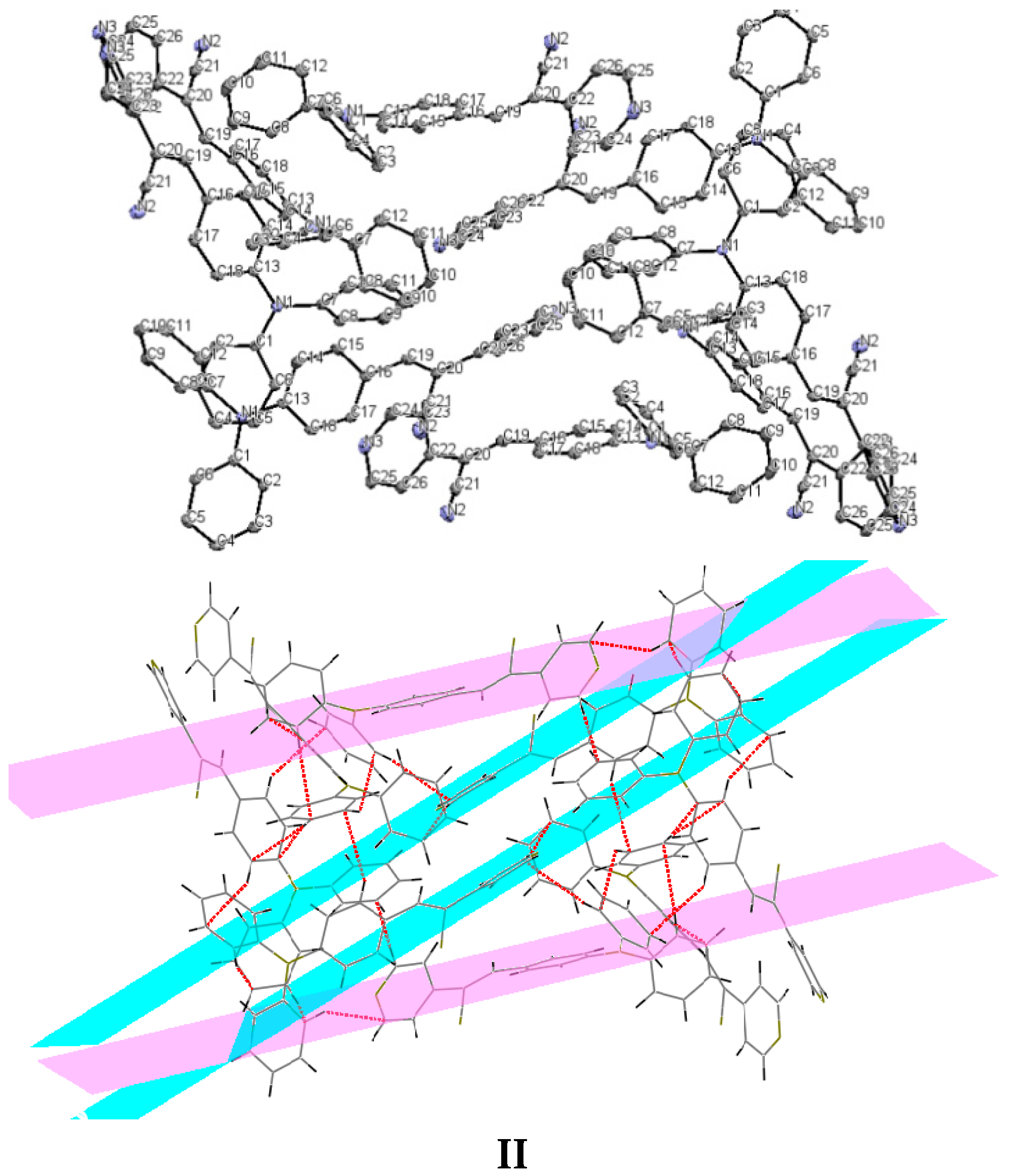
Understanding the different crystal packing modes (e.g., from herringbone to a co-facial π-stacking motif) is of importance to confer good electronic transfer properties. The molecules in I and II showed a tilted face-to-face arrangement (Figure 6) for the aromatic ring constituted of the pyridine ring with a centroid-centroid distance of 3.71 Å, a shift distance of 1.46 Å for I, and 3.84 Å of centroid-centroid distance and a shift distance of 1.843 Å for II. These values for the π-π interactions in the structures were automatically found by the program OLEX 2 [46].
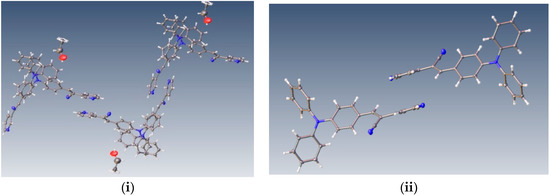
Figure 6.
Perspective view of the packing structure of I (i) and II (ii) showing the strong π-π stacking interactions.
These distances are typical for π-π aromatic face-to-face interactions with centroid distances > 3.65 Å and offsets in the range of 1.6–1.8 Å [47] and they reflect the distribution of electronic density that minimizes the π-electrons. However, for the structure of III, as well as several crystal structure reported with the -CH=CCN- moiety, no obvious π-π interactions are found, even though isomers were present. These packing motifs may be attributed to CH···π, edge-to-face interactions and they tune the packing structure from a herringbone mode toward more of a face-to-face π-stacking motif. Holmes et al. reported that in order to favor face-to-face π-stacking, the CH···π interactions should be minimized [18,22,48,49]. The packing of structures I and II showed that the pyridine unit, the position of the nitrogen atom within the pyridyl core, and the p-(diphenylamino)phenyl- group play important roles as well as the solvent used for crystallization.
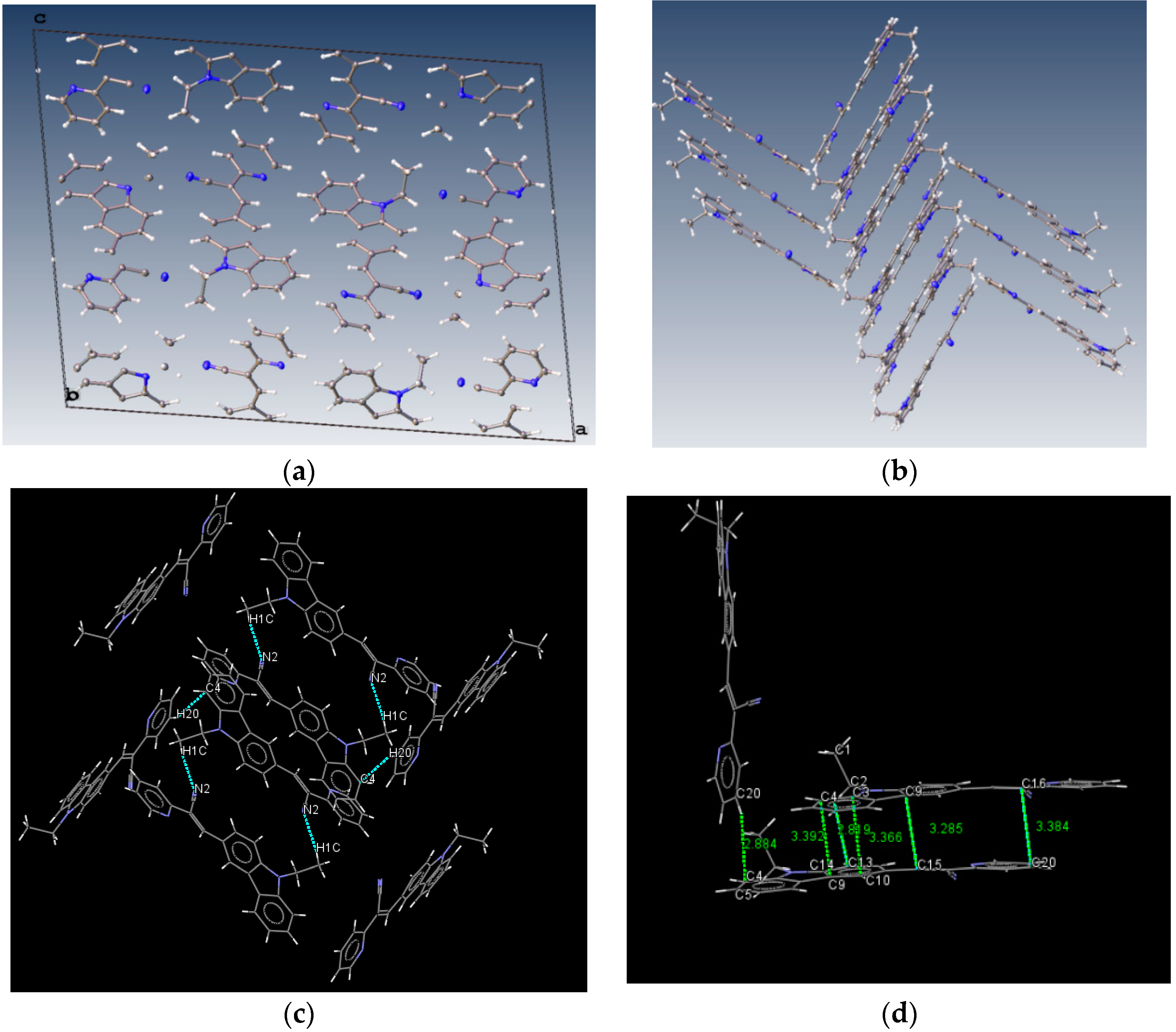
Bao and coworkers reported that tetracene substituted with halogen atoms at the 5 and 11 positions adopted a face-to-face π-stacking motif with enhanced charge transport [22]. In these three packing modes, the charge transport could be three-dimensionally anisotropic and the highest mobility would be observed along the major π-stacking direction (Figure 7). In contrast, the lowest mobility would be found along the directions in which the molecules were insulated from one another. It is worth mentioning that the packing structure of III presents short contacts between the carbazole and the pyridine group. The interactions maintaining the structure of III are CH/EtCz···-C≡N of 2.74Å, and CH/(py)·····CH/π(Cz) of 2.88 Å (Figure 8); however, there are no strong π-π stacking interactions present from centroid-to-centroid, such as in I and II, as it can be seen the short contacts values for III (Figure 8d) calculated by Mercury software [50].
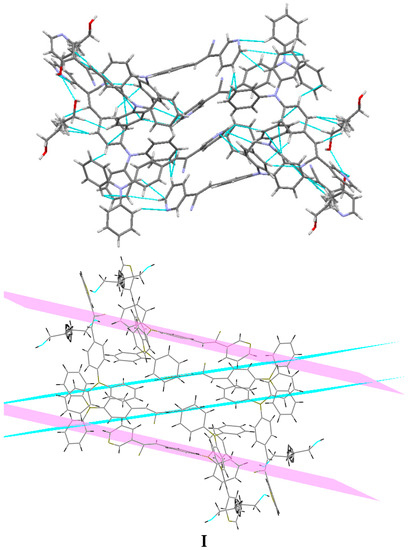
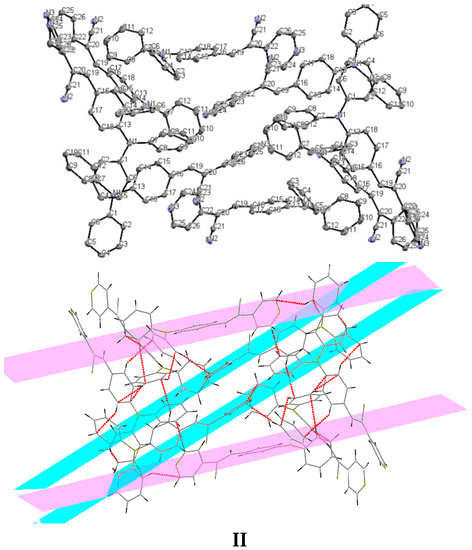
Figure 7.
Crystal packing views of I and II with π-π stacking interactions occurring between neighboring aromatic rings.
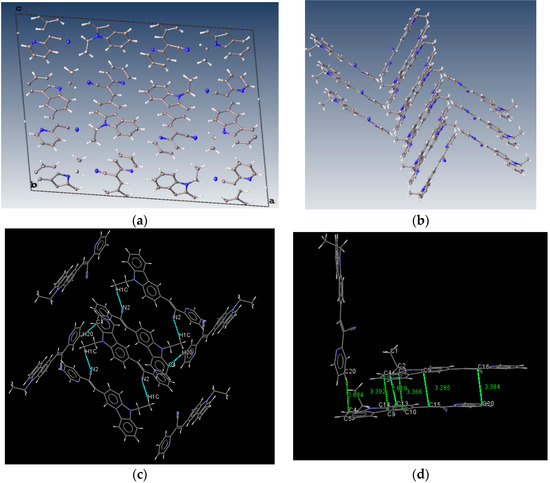
Figure 8.
Molecule arrangement in the unit cell (a) and the herringbone packing motif (b) with the stacking of III, and a view showing the interactions, dashes lines (c) short contacts values along b axis (d).
From the Figure 8d it could be supposed that typical π-π interactions are present. However, in Figure 9 the distance between a pyridine plane and neighboring molecule indicated that there is not a cofacial π-π interaction arising a π-π overlap as it is exhibited in I and II. Consequently, the packing motif seems a classical herringbone packing with a strong slipped π-stacking indicating a weak π-π overlap between neighbor molecules [51].
Figure 9.
Molecular packing of III in the unit cell view from b axis (a) and from c axis (b).
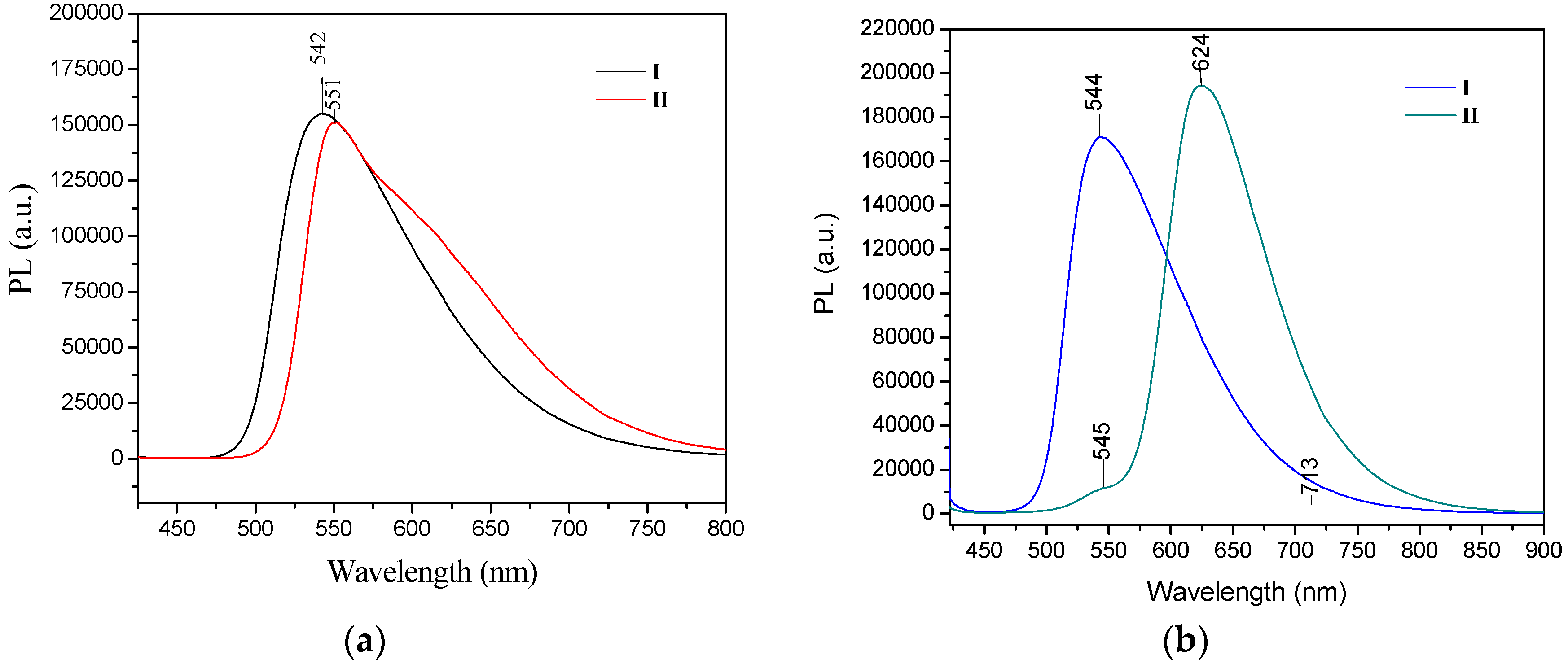
The UV/Vis absorption spectra recorded in chloroform for I, II showed similar maxima of absorption bands, one in the range 416–427 nm in solution with CHCl3 and a smaller absorption band at 297–301 nm. For the solid form, only the change for compound II is reported, which underwent a bathochromic effect of 24 nm (λmax = 451nm) [52]. Only in compound II did the absorption spectrum display an obvious bathochromic shift in the solid state, which some authors suggest is due to the molecules adopting a π-aggregated form in the solid state [18]. The extended delocalization of the nitrogen electron pair onto the two phenyl rings from -N(Ph)2 accounts for the red shift of the absorption band. The emission spectra of I and II in chloroform solution in both the crystal and powder forms showed high fluorescence intensity, both in solution and in the solid state. The photoluminescence spectra of I and II were compared to reported values [52]. Crystals of II showed a strong bathochromic effect of almost 80 nm as crystals, with λem at 624 nm. However, the fluorescence emission maxima for compound I were almost the same for the powder form (535 nm) and the crystals (544 nm) (Figure 10).
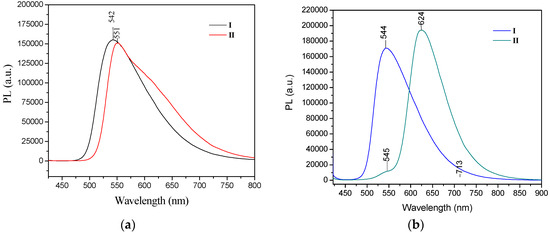
Figure 10.
PL spectra of compounds I–II in (a) powder and (b) in crystal forms. Excitation wavelength for both conditions was 405 nm.
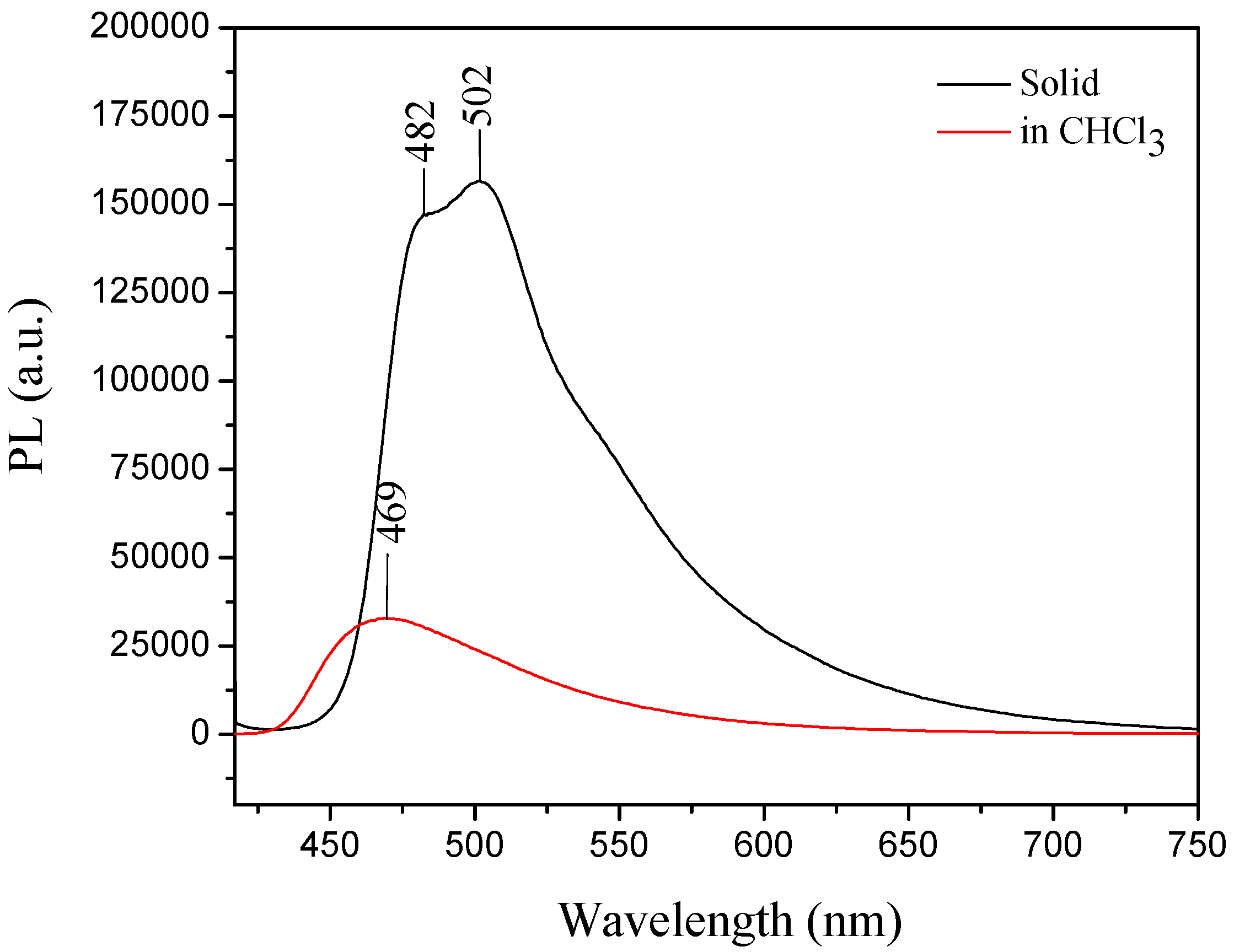
For III, the UV-vis absorption wavelength maximum in solution was at 380 nm, whereas in the solid state its absorption maximum (λmax) was 398 nm (III) [30]. The compound showed a typical AIE effect, because the emission maximum in the solid form was observed at 502 nm and the emission in solution was weak, which could be attributed to the carbazole group effect [30,53] (Figure 11).
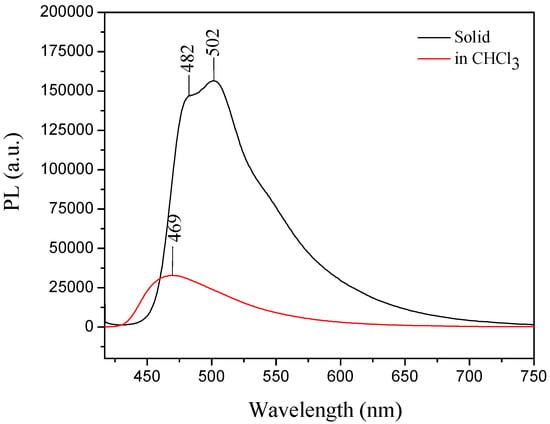
Figure 11.
Emission of compound III in solid form (−) and in solution (−) indicating that III exhibited the AIE property. Excitation wavelength for both conditions was 405 nm.
2.2. Electrochemical Properties.
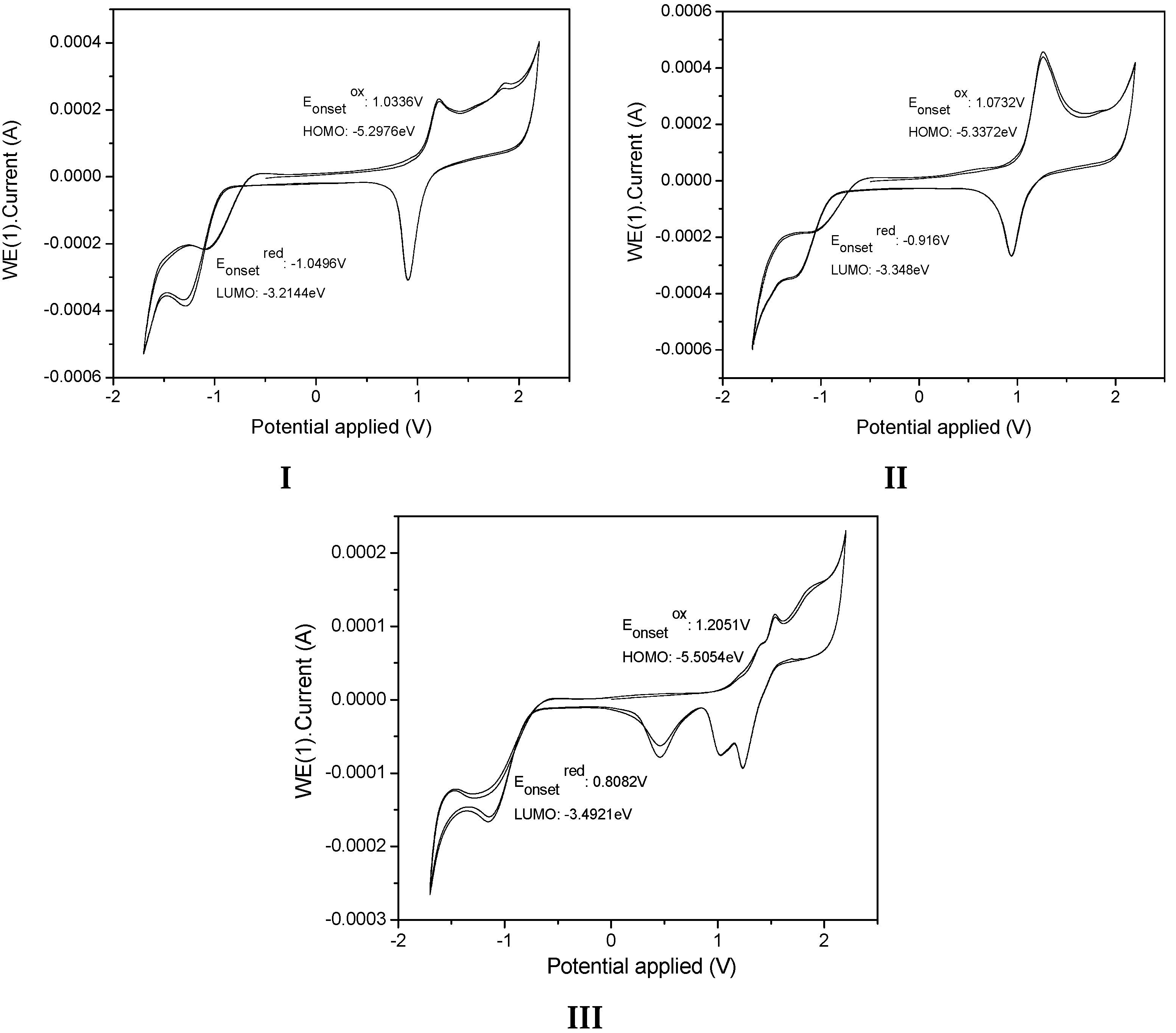
The electrochemical characteristics of the samples I, II, and III were investigated by cyclic voltammetry (CV) (Figure 12). The onset oxidation () estimated from CV curves for I, II, and III are summarized in Table 4. We determined the HOMO and LUMO energy levels of the samples, as calculated from the CV tests applying published equations [54] and using ferrocene as the external standard. All measurements were carried out at room temperature.
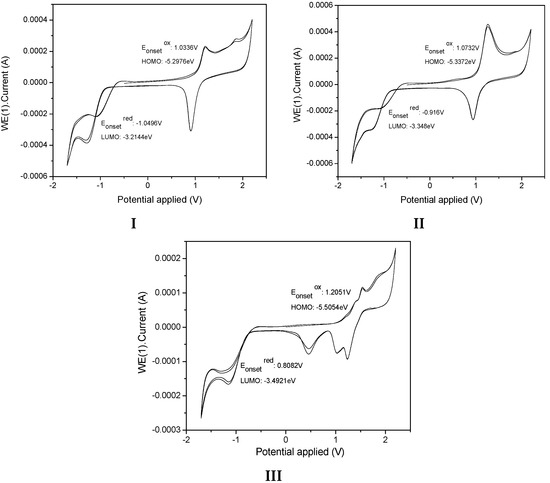
Figure 12.
Cyclic voltammograms of I, II and III in 1 mM in 0.1M TBAFP6/CH2Cl2 solution with Ag/AgCl as reference electrode and a Pt wire as the working electrode. A scan rate of 50 mVs−1 was used.
Table 4.
Electronic properties I, II and III.
The value of III has lower energy HOMO and LUMO levels, suggesting a lower electron injection barrier than observed for I and II, indicating that III had a stronger tendency to donate an electron, whereas the high LUMO energy level of II indicated its strong electron-withdrawing ability. The electronic energy gap values were estimated to be 2.08 eV, 1.99 eV, and 2.01eV for I, II and III, respectively (Table 4). We also determined the energy gap values, as calculated from the absorption spectrum in CHCl3 by using ΔEopt = hc/λ (ΔEopt (eV) =1237.5/λ(nm).
The energy of the lowest gap, which results from a more extended conjugation, was attributed to the (4-diphenylamino) molecule moiety in II. However, the values indicated that the position of substitution of the pyridyl unit reduced the molecule slightly, making it more easily oxidizable. In the structure, the electron withdrawing effect from the pyridine group could also affect the conjugation structure.
2.3. DFT Calculations
We carried out the Gaussian09 program optimization calculations with the M06L method using the cc-pVDZ basis set in the gas and solvent phases.
Geometries with small root-mean-square deviations were obtained with respect to the crystallographic data (the largest 8 degrees in dihedral angles). The frontier HOMO and LUMO energies and gap energies were evaluated in the different solvents (Table 5). The effect of solvent was evaluated by the PCM method. The most energetically stable structures were obtained in solutions of methanol and dimethyl sulfoxide. Also, the highest values of dipole moment (µ) and the lowest were correlate to the solvent polarity used to obtain suitable crystals for X-ray characterization (see Table 6) [55,56,57,58].
Table 5.
Theoretical data of I, II and III in gas and solvent phases at M06L/cc-pVDZ theory level.
Table 6.
Solvent conditions tested to obtain tiny crystals.
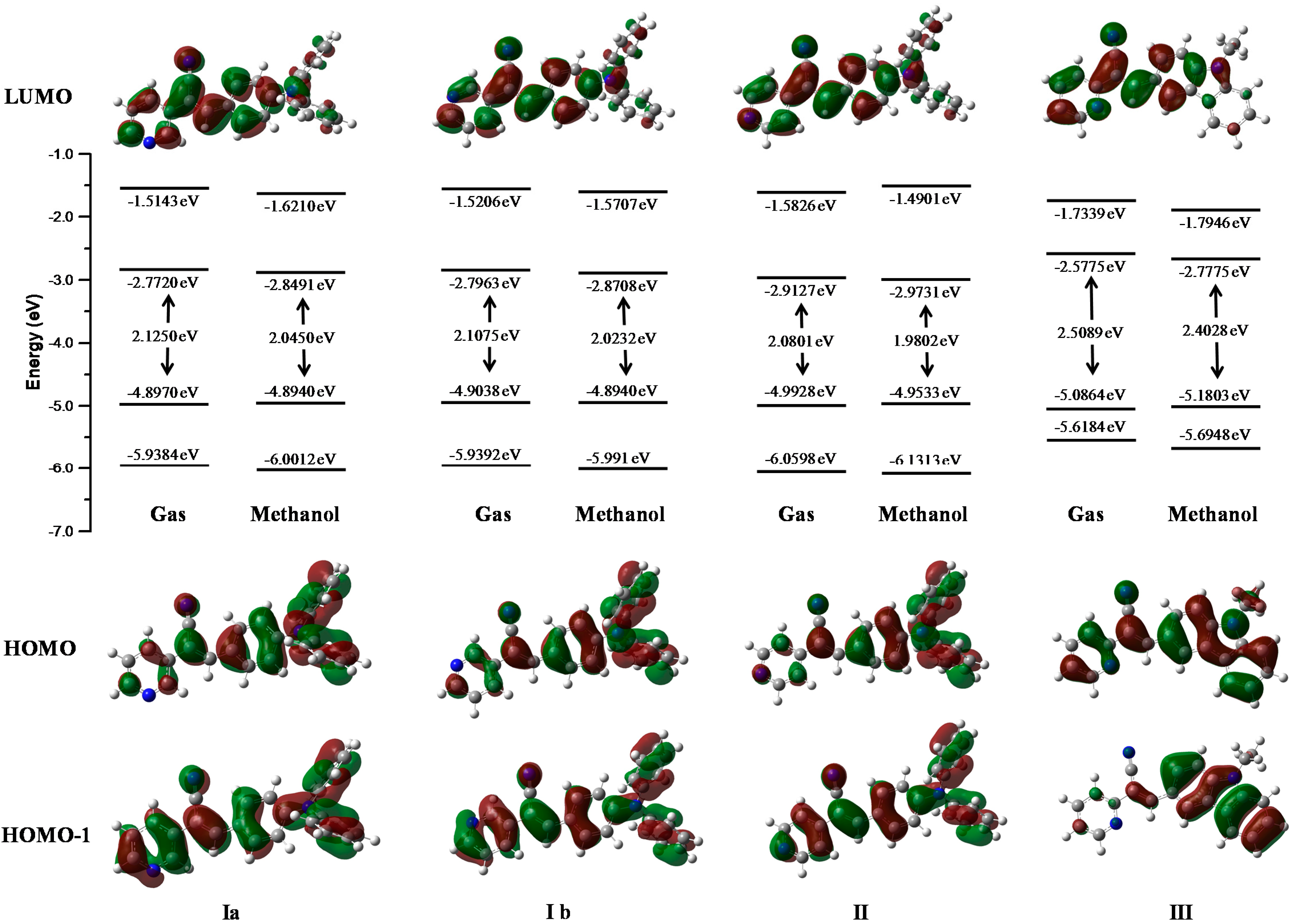
Figure 13 represents the energies of the molecular orbitals LUMO+1, LUMO, HOMO, HOMO-1 in the gas phase and in one of the most stable solvent phase (methanol). Isosurfaces in the diagram represent the maps of HOMO and HOMO-1(bottom) and LUMO (top). The character and energy levels of HOMOs and LUMOs (and the corresponding energy gaps) were determined on fully optimized geometries and compared with electrochemical measurements. The theoretical HOMO energy levels of the three compounds I, II and III in the gas phase were close together, −4.89, −4.99 and −5.08 eV, respectively. I and II p-diphenylamino substituents caused a greater delocalization on the phenyl core than the pyridyl ring. The theoretical calculation showed a delocalization on the pyridine-carbazole backbone for III. Thus, the character of the HOMO of I and II were very different from that of III, for which the HOMO presented a clear pyridyl-phenyl-carbazole delocalization leading to the highest HOMO level (−5.08 eV).
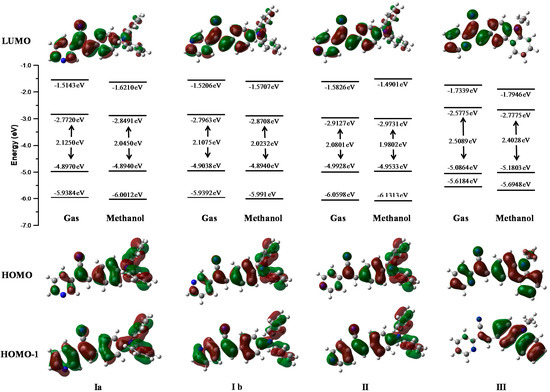
Figure 13.
Molecular orbitals of compounds obtained in gas and methanol and phases at M06L/cc-pVDZ theory level.
The present calculations were almost in accordance with electrochemical data (Table 4), which led us to conclude a better delocalization in I, II and III, therefore diminishing the HOMO level from I, II to III −5.29, −5.33 to −5.50 eV. With respect to the LUMO levels of the pyridine-substituted I, II and III, they were calculated to be between −2.77, 2.91 and −2.57 eV, respectively, presenting a pyridyl character without contribution of the p-diphenylamino and carbazole moieties. The LUMO energy levels of II are 0.34 eV higher than that of III (−2.57eV) showing the less intense withdrawing effect of the pyridyl unit compared to the carbazole unit. The LUMO level of III was calculated at −2.57 eV, lower than that of I and II and the decrease of the LUMO in II emphasized the withdrawing effect of the pyridyl unit and its efficient conjugation with the carbazole unit, rendering this molecule the most easily reducible. The main tendency of the LUMO levels obtained through theoretical calculations did not agree with our electrochemical conclusions, with the lowest LUMO recorded for I. The LUMO calculated for I, II and III are close together (0.44eV), and their LUMO values determined through electrochemical measurements were also very similar (−3.21, −3.34, and −3.39 eV, respectively).
The theoretical energy gaps of I, II and III were close to 2.12, 2.08, and 2.50 eV, respectively, values closer to the energy gap values obtained from the electrochemical measurements (2.08 eV for I, 1.98 for II, and 2.01 for III) showing a good correlation between theoretical calculations. Furthermore, the values showed better fit with the theoretical calculations when the polarity of the solvent was increased.
3. Experimental Section
The compounds I–III were synthesized according to the reported methods [30,59]. We utilized several synthetic procedures to obtain adequate crystals of I–III to permit single crystal characterization (Table 6). In some cases, the crystals were too small to allow diffraction and in other procedures, the compounds were almost insoluble in the solvents. The majority of the isomers were crystallized after reaction or were purified by crystallization.
The optimum crystallization procedures for each compound are indicated below. For I, the solid (0.028 g) was dissolved in ethanol:cyclohexane (12 mL) 58 °C and the vessel was set aside at 4 °C and after 3 days, yellow crystals were obtained. Compound II (0.035 g) was dissolved in dimethylsulfoxide (DMSO) at 90 °C; the vessel was set aside at room temperature and after 9 days, yellow crystals were formed. Compound III (0.020 g) was dissolved in cyclohexane (15 mL) at room temperature, the vessel was set aside at 4 °C and after 4 days, tiny yellow crystals were obtained.
3.1. Single Crystal X-ray Diffraction (SCXRD)
All reflection intensities for I and II were measured at 110(2) K (after the crystals were flash cooled from RT) using a KM4/Xcalibur (detector: Sapphire3) with enhance graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) apparatus under the program CrysAlisPro (Versions 1.171.35.11 or 1.171.36.24, Agilent Technologies 2012, Santa Clara, CA, USA). Data for III were collected at 110(2) K using a SuperNova diffractometer (equipped with Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (Version 1.171.36.28, Agilent Technologies 2013). The same program was used to refine the cell dimensions and for data reduction. The structures were solved with the programs SHELXS-97/SHELXS-2013, and were refined on F2 with SHELXL-97/SHELXL-2013 [60]. Analytical numeric absorption corrections based on a multifaceted crystal model were applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (Oxford Instruments, Abingdon, Oxford, UK). The H atoms were placed at calculated positions using the instructions AFIX 23, AFIX 43, AFIX 123, AFIX 137 or AFIX 147 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C or O atoms.
3.2. Absorbance and Emission (UV-VIS and PL)
The absorbance spectra were acquired on a SD2000 spectrometer (Ocean Optics, Dunedin, FL, USA) equipped with a pulse Xenon light source P-2 (Ocean Optics for the UV region (220–270 nm) and a Cary 300 Spectrometer (Agilent) equipped with a deuterium and halogen lamp. The wavelength detection range was 190–900 nm. For measurements in solution, the solvents were of spectroscopic grade and were preliminarily checked for the absence of absorbing or fluorescent impurities within the scanned spectral ranges. For powder samples, the absorption was measured using pellets prepared with KBr. A UV/Vis DT 1000 CE light source (Analytical Instrument Systems, Inc., Flemington, NJ, USA) was used for measuring absorption. Emission spectra (PL) were acquired from a QE-Pro-FL (Ocean Optics) equipped with a laser diode excitation source at a wavelength of 405 nm.
3.3. Cyclic Voltammetry (CV)
Measurements were carried out with a PGSTAT128N-serial no. AUT85577 potentiostat (Keysight, Santa Rosa, CA, USA) using a three electrode cell assembly comprised of Ag/Ag⁺ in solution of 3 M KCl as the reference electrode, a platinum wire (ϕ = 0.2 mm) as working electrode and counter-electrode, using a solution in CH2Cl2 containing 0.1 M supporting electrolyte of tetrabutylammonium hexafluorophosphate (TBAFP6), and ferrocene was used as external standard. The scanning rate was 50 mV/s.
4. Conclusions
In this work, we have reported three molecular structures of α,β-unsaturated acrylonitriles which crystallized into single crystals with different Z values and according to crystallography data gave information about the arrangement packing that is possible to correlate with the emission with a bathochromic shift. Such emission enhancement in the aggregate state is due to strong π-π molecular stacking in the crystal lattice, which is does not depend on of intermolecular H-bonding interactions between molecule-solvent because it is present in both I and II. The results from crystal structures with Z′ > 1 represent a possible solution to the problem of packing molecules in three dimensions, because the three molecules I, II and III have eight molecules per unit cell, which could be due to the crystallization process, as is particularly well illustrated with I as an example where the compound adopts high Z′ or high symmetry. Also the systems reported for I and II showed stacking via π-π interactions with values closer to those typically found for geometrical parameters of aromatic π-π interactions centroid distances. The data for I represented a rare case with complete hydrogen-bonding of the chains as I, whose structure was all the more remarkable because of the presence of both the syn and anti conformers at the double bond. One of the ultimate goals of solid-state chemistry is the ability to predict or computationally calculate the experimental crystal structure(s) of a compound solely from knowledge of its molecular structure. This factor is a remarkable problem in the computational field because the possible occurrence of more than one symmetry unique molecule greatly complicates the crystal.
Supplementary Materials
Crystallographic data (excluding structure factors) reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1006818, 1006819 and 1006820. Copies of available material can be obtained, free of charge, on application to the CCDC, 12 Union Road. Cambridge CB2 IEZ, UK, (fax: +44-(0)1223-336033 or e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgments
The authors wish to express their gratitude to VIEP-BUAP (projects PEZM-NAT15-G, CHCV-NAT15-G, SOMJ-NAT15-I, CASM-NAT15-I), PROMEP-SEP/103.5/13/2110 (Thematic network of collaboration) and CONACyT (projects 183833 and 157552). Also, the authors acknowledge Laboratorio Nacional de Supercómputo del Sureste (LNS-BUAP) to perform the calculations.
Author Contributions
Conceived and designed the experiments: M.C., G.S.-M. and M.J.P.; Performed the experiments: O.R., M.C., M.E.C. and G.S-M.; Analyzed the data: M.J.P., E.P.-G., G.S.-M. and M.A.S.; Contributed reagents/materials/analysis tools: M.J.P., G.S-M., V.M.C.; Wrote the paper M.J.P. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bader, M.A.; Marowsky, G.; Bahtiar, A.; Koynov, K.; Bubeck, C.; Tillmann, H.; Hörhold, H.H.; Pereira, S. Poly(p-phenylenevinylene) derivatives: New promising materials for nonlinear all-optical waveguide switching. J. Opt. Soc. Am. B 2002, 19, 2250–2262. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Maini, L. The growing world of crystal forms. Chem. Commun. 2010, 46, 6232–6242. [Google Scholar] [CrossRef] [PubMed]
- Schwoerer, M.; Wolf, H.C. Organic Molecular Solids; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2007. [Google Scholar]
- Cornil, J.; Beljonne, D.; Calbert, J.-P.; Brédas, J.-L. Interchain interactions in organic π-conjugated materials: Impact on electronic structure, optical response, and charge transport. Adv. Mater. 2001, 13, 1053–1067. [Google Scholar] [CrossRef]
- Datta, A.; Pati, S.K. Dipolar interactions and hydrogen bonding in supramolecular aggregates: Understanding cooperative phenomena for 1st hyperpolarizability. Chem. Soc. Rev. 2006, 35, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Chow, T.J.; Lin, R.; Ko, C.-W.; Tao, Y.-T. Photo and electroluminescence of 2-anilino-5-phenylpenta-2,4-dienenitrile derivatives. J. Mater. Chem. 2002, 12, 42–46. [Google Scholar]
- Sun, H.; Zhao, Z.; Spano, F.C.; Beljonne, D.; Cornil, J.; Shuai, Z.; Brédas, J.-L. Absorption and emission in quaterthienyl thin films. Adv. Mater. 2003, 15, 818–822. [Google Scholar] [CrossRef]
- Dreuw, A.; Plötner, J.; Lorenz, L.; Wachtveitl, J.; Djanhan, J.E.; Brüning, J.; Metz, T.; Bolte, M.; Schmidt, M.U. Molecular mechanism of the solid-state fluorescence behavior of the organic pigment yellow 101 and its derivatives. Angew. Chem. Int. Ed 2005, 44, 7783–7786. [Google Scholar] [CrossRef] [PubMed]
- Langhals, H.; Potrawa, T.; Nöth, H.; Linti, G. The influence of packing effects on the solid-state fluorescence of diketopyrrolopyrroles. Angew. Chem. Int. Ed. 1989, 28, 478–480. [Google Scholar] [CrossRef]
- Datta, A.; Terenziani, F.; Painelli, A. Cooperative interactions in supramolecular aggregates: Linear and nonlinear responses in calix[4]arenes. Chem. Phys. Chem. 2006, 7, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Swager, T.M. Control of conformational and interpolymer effects in conjugated polymers. Nature 2001, 411, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Jenekhe, S.A.; Osaheni, J.A. Excimers and exciplexes of conjugated polymers. Science 1994, 265, 765–768. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Yang, B.; Li, F.; Cheng, G.; Liu, L.; Yang, G.; Xu, H.; Ye, L.; Hanif, M.; Liu, S.; et al. Cross dipole stacking in the crystal of distyrylbenzene derivative: The approach toward high solid-state luminescence efficiency. J. Am. Chem. Soc. 2005, 127, 14152–14153. [Google Scholar] [CrossRef] [PubMed]
- Mizobe, Y.; Ito, H.; Hisaki, I.; Miyata, M.; Hasegawa, Y.; Tohnai, N. A novel strategy for fluorescence enhancement in the solid-state: Affording rigidity to fluorophores packing. Chem. Commun. 2006, 20, 2126–2128. [Google Scholar]
- Desiraju, G. Crystal Engineering: A holistic view. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, W.; Liu, Y.; Zhu, D. Micro- and nanocrystals of organic semiconductors. Acc. Chem. Res. 2010, 43, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Gsanger, M.; Oh, J.H.; Konemann, M.; Hoffken, H.W.; Krause, A.-M.; Bao, Z.; Wurthner, F. A crystal-engineered hydrogen-bonded octachloroperylene diimide with a twisted core: An n-channel organic semiconductor. Angew. Chem. Int. Ed. 2010, 49, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, X.; Luo, J.; Chi, C.; Chen, H.; Wu, J. A Cruciform 6,6′-dipentacenyl: Synthesis, solid-state packing and applications in thin-film transistors. Chem. Eur. J. 2010, 16, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.D.; Cao, J.; Kampf, J.W. Solid-state packing of conjugated oligomers: From π-stacks to the herringbone structure. J. Am. Chem. Soc. 2004, 126, 4318–4328. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, H.; Li, H.; Zhao, H.; Meng, Q.; Hu, W. Dibenzothiophene Derivatives: From Herringbone to Lamellar Packing Motif. Cryst. Growth Des. 2010, 10, 4155–4160. [Google Scholar] [CrossRef]
- Mattheus, C.C.; de Wijs, G.A.; de Groot, R.A.; Palstra, T.T.M. Modeling the polymorphism of pentacene. J. Am. Chem. Soc. 2003, 125, 6323–6330. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Zeis, R.; Borkent, E.J.; Besnard, C.; Lovinger, A.J.; Siegris, T.; Kloc, C.; Bao, Z.N. Synthesis, Crystal Structure, and Transistor Performance of Tetracene Derivatives. J. Am. Chem. Soc. 2004, 126, 15322–15323. [Google Scholar] [CrossRef] [PubMed]
- Sundar, V.C.; Zaumescil, J.; Podzorov, V.; Menard, E.; Willeett, R.L.; Someya, T.; Gershenson, M.E.; Rogers, J.A. Elastomeric transistor stamps: Reversible probing of charge transport in organic crystals. Science 2004, 303, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, F.; Bacchi, S.; Filippini, G.; Pilati, T.; Gavezzotti, A. Synthesis, X-ray diffraction and computational study of the crystal packing of polycyclic hydrocarbons featuring aromatic and perfluoroaromatic rings condensed in the same molecule: 1,2,3,4-Tetrafluoronaphthalene, -anthracene and -phenanthrene. Chem. Eur. J. 2007, 13, 7177–7184. [Google Scholar] [PubMed]
- Anthony, J.E.; Brooks, J.S.; Eaton, D.J.; Parkin, S.R. Functionalized pentacene: Improved electronic properties from control of solid-state order. J. Am. Chem. Soc. 2001, 123, 9482–9483. [Google Scholar] [CrossRef] [PubMed]
- Coropceanu, V.; Cornil, J.; da Silva, D.A.; Yoann, F.; Silbey, O.R.; Brédas, J.L. Charge transport in organic semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef] [PubMed]
- Valeev, E.F.; Coropceanu, V.; da Silva, D.A.; Salman, S.; Bredas, J.L. Effect of Electronic Polarization on Charge-Transport Parameters in Molecular Organic Semiconductors. J. Am. Chem. Soc. 2006, 128, 9882–9886. [Google Scholar] [CrossRef] [PubMed]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Soriano-Moro, G.; Castro, M.E.; Meléndez, F.J. Fluorescence improvement of pyridylacrylonitrile by dimethylaminophenyl-substitutions: The effect of packing modes of conjugated compounds. J. Mol. Struct. 2013, 1034, 238–248. [Google Scholar] [CrossRef]
- Percino, M.J.; Cerón, M.; Soriano-Moro, G.; Castro, M.E.; Chapela, V.M.; Bonilla, J.; Reyes-Reyes, M.; López-Sandoval, R. The effect of the supramolecular network of (Z)-3-(4-(diphenylamino)phenyl)-2-(pyridin-2-yl)-acrylonitrile on the fluorescence behavior of a single crystal: Experimental and theoretical studies. CrystEngComm 2014, 16, 8591–8604. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Soriano-Moro, G.; Castro, M.E.; Melendez, F.J. Molecular packing and solid-state fluorescence of conjugated compounds of carbazole-acrylonitrile derivatives. Curr. Phys. Chem. 2014, 4, 137–150. [Google Scholar] [CrossRef]
- Percino, M.J.; Cerón, M.; Coca, P.; Soriano, G.; Castro, M.E.; Chapela, V.M.; Bonilla, J.; Reyes-Reyes, M.; López-Sandoval, R.; Siegler, M.A. Important role of molecular packing and intermolecular interactions in two polymorphs of (Z)-2-phenyl-3-(4-(pyridin-2-yl)phenyl)acrylonitrile. Preparation, structures, and optical propertie. J. Mol. Struct. 2014, 1078, 74–82. [Google Scholar] [CrossRef]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 536–5388. [Google Scholar] [CrossRef] [PubMed]
- An, B.-K.; Kwon, S.-K.; Jung, S.-D.; Park, S.Y. Enhanced emission and its switching in fluorescent organic nanoparticles. J. Am. Chem. Soc. 2002, 124, 14401–14415. [Google Scholar] [CrossRef]
- Li, Z.; Dong, Y.Q.; Lam, J.W.Y.; Sun, J.; Qin, A.; Haeussler, M.; Dong, Y.P.; Sung, H.H.Y.; Williams, I.D.; Kwok, H.S.; et al. Functionalized siloles: Versatile synthesis, aggregation-induced emission, and sensory and device applications. Adv. Funct. Mater. 2009, 19, 905–917. [Google Scholar] [CrossRef]
- Zhang, X.; Chi, Z.; Li, H.; Xu, B.; Li, X.; Liu, S.; Zhang, Y.; Xu, J. Synthesis and properties of novel aggregation-induced emission compounds with combined tetraphenylethylene and dicarbazolyl triphenylethylene moieties. J. Mater. Chem. 2011, 21, 1788–1796. [Google Scholar] [CrossRef]
- Zhang, G.-F.; Aldred, M.P.; Gong, W.-L.; Li, C.; Zhu, M.-Q. Utilising tetraphenylethene as a dual activator for intramolecular charge transfer and aggregation induced emission. Chem. Commun. 2012, 48, 7711–7713. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Yoon, S.-J.; An, B.-K.; Park, S.Y. High-contrast on/off fluorescence switching via reversible E–Z isomerization of diphenylstilbene containing the α-cyanostilbenic moiety. J. Phys. Chem. C 2013, 117, 11285–11291. [Google Scholar] [CrossRef]
- Palakollu, V.; Kanvah, S. α-Cyanostilbene based fluorophores: Aggregation-induced enhanced emission, solvatochromism and the pH effect. New J. Chem. 2014, 38, 5736–5746. [Google Scholar] [CrossRef]
- Bredas, J.L.; Calbert, J.P.; da Silva, D.A.; Cornil, J. Organic semiconductors: A theoretical characterization of the basic parameters governing charge transport. Proc. Natl. Acad. Sci. USA 2002, 99, 5804–5809. [Google Scholar] [CrossRef] [PubMed]
- Kazmaier, P.M.; Hoffmann, R. A theoretical study of crystallochromy. Quantum interference effects in the spectra of perylene pigments. J. Am. Chem. Soc. 1994, 116, 9684–9691. [Google Scholar] [CrossRef]
- Wurthner, F. Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures. Chem. Commun. 2004, 1564–1579. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.; Scudder, M.L. Multiple Molecules in the Asymmetric Unit (Z′ > 1) and the Formation of False Conglomerate Crystal Structures. Cryst. Growth Des. 2009, 9, 2890–2894. [Google Scholar] [CrossRef]
- Plötner, J.; Drew, A. Solid state fluorescence of Pigment Yellow 101 and derivatives: A conserved property of the individual molecules. Phys. Chem. Chem. Phys. 2006, 8, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Steed, K.M.; Steed, J.W. Packing problems: High Z' crystal structures and their relationship to cocrystals, inclusion compounds, and polymorphism. Chem. Rev. 2015, 115, 2895–2933. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T. The hydrogen bond in the solid state. Angew. Chem. Int. Ed. 2002, 41, 49–76. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourkos, L.J.; Gilda, R.J.; Howard, J.A.K.; Puschman, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Molčanov, K.; Sabljić, I.; Kojić-Prodić, B. Face-to-face π-stacking in the multicomponent crystals of chloranilic acid, alkali hydrogenchloranilates, and water. CrystEngComm 2011, 13, 4211–4217. [Google Scholar] [CrossRef]
- Li, X.C.; Sirringhaus, H.; Garnier, F.; Holmes, A.B.; Moratti, S.C.; Feeder, M.N.; Clegg, F.W.; Teat, S.J.; Friend, R.H. A highly π-stacked organic semiconductor for thin film transistors based on fused thiophenes. J. Am. Chem. Soc. 1998, 120, 2206–2207. [Google Scholar] [CrossRef]
- Zhang, X.; Cote, A.P.; Matzger, A.J. Synthesis and structure of fused α-oligothiophenes with up to seven rings. J. Am. Chem. Soc. 2005, 127, 10502–10503. [Google Scholar] [CrossRef] [PubMed]
- Mercury 3.5.1. Available online: http://www.ccdc.cam.ac.uk/mercury/ (accessed on 13 December 2014).
- Nagarajan, K.; Rajagopal, S.K.; Hariharan, M. C–H⋯H–C and C–H⋯π contacts aid transformation of dimeric to monomeric anthracene in the solid state. CrystEngComm 2014, 16, 8946–8949. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Cerón, M.; Castro, M.E.; Soriano-Moro, G.; Pérez-Gutiérrez, E.; Meléndez-Bustamante, F. Synthesis and characterization of conjugated pyridine-(N-diphenylamino) acrylonitrile derivatives: Photophysical properties. J. Mater. Res. 2012, 1, 181–192. [Google Scholar]
- Upamali, K.A.N.; Estrada, L.A.; De, P.K.; Cai, X.C.; Krause, J.A.; Neckers, D.C. Carbazole-based cyano-stilbene highly fluorescent microcrystals. Langmuir 2011, 27, 1573–1580. [Google Scholar] [CrossRef] [PubMed]
- Pomrnerehne, J.; Vestweber, H.; Guss, W.; Mahrt, R.F.; Bassler, H.; Porsch, M.; Daub, J. Efficient two layer leds on a polymer blend basis. Adv. Mater. 1995, 7, 551–554. [Google Scholar] [CrossRef]
- Weissbuch, I.; Torbeev, V.Y.; Leiserowitz, L.; Lahav, M. Solvent Effect on Crystal Polymorphism: Why Addition of Methanol or Ethanol to Aqueous Solutions Induces the Precipitation of the Least Stable Beta Form of Glycine. Angew. Chem. Int. Ed. 2005, 44, 3226–3229. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Lee, I.S.; Myerson, A. Factor affecting the polymorphic outcome of gycine crystals constrained on patterned substrates. Chem. Eng. Technol. 2006, 29, 281–285. [Google Scholar] [CrossRef]
- Blagden, N.; Davey, R.J.; Lieberman, H.F.; Williams, L.; Payne, R.; Roberts, R.; Rowe, R.; Docherty, R. Crystal Chemistry and Solvent Effects in Polymorphic Systems Sulfathiazole. J. Chem. Soc. Faraday Trans. 1998, 94, 1035–1044. [Google Scholar] [CrossRef]
- Kitamura, M.; Horimoto, K. Role of Kinetic Process in the Solvent Effect on Crystallization of BPT Propyl Ester Polymorph. J. Cryst. Growth 2013, 373, 151–155. [Google Scholar] [CrossRef]
- Percino, M.J.; Chapela, V.M.; Perez-Gutierrez, E.; Ceron, M.; Soriano, G. Synthesis, optical, and spectroscopic characterisation of substituted 3-phenyl-2-arylacrylonitriles. Chem. Pap. 2011, 65, 42–51. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of compounds I, II and III are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).