
Structural Determinants of Alkyne Reactivity in Copper-Catalyzed Azide-Alkyne Cycloadditions

Abstract
:1. Introduction
2. Results and Discussion
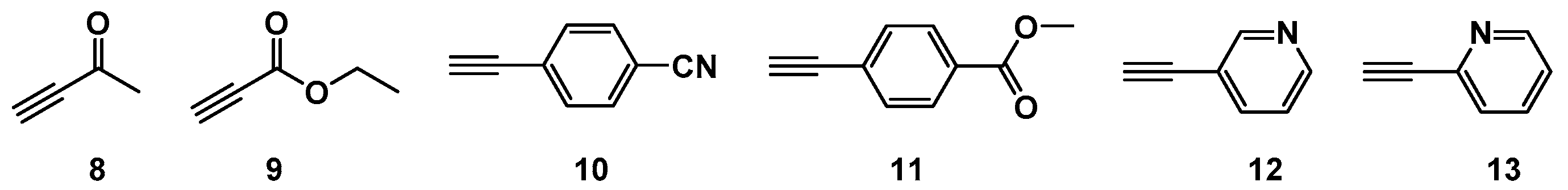
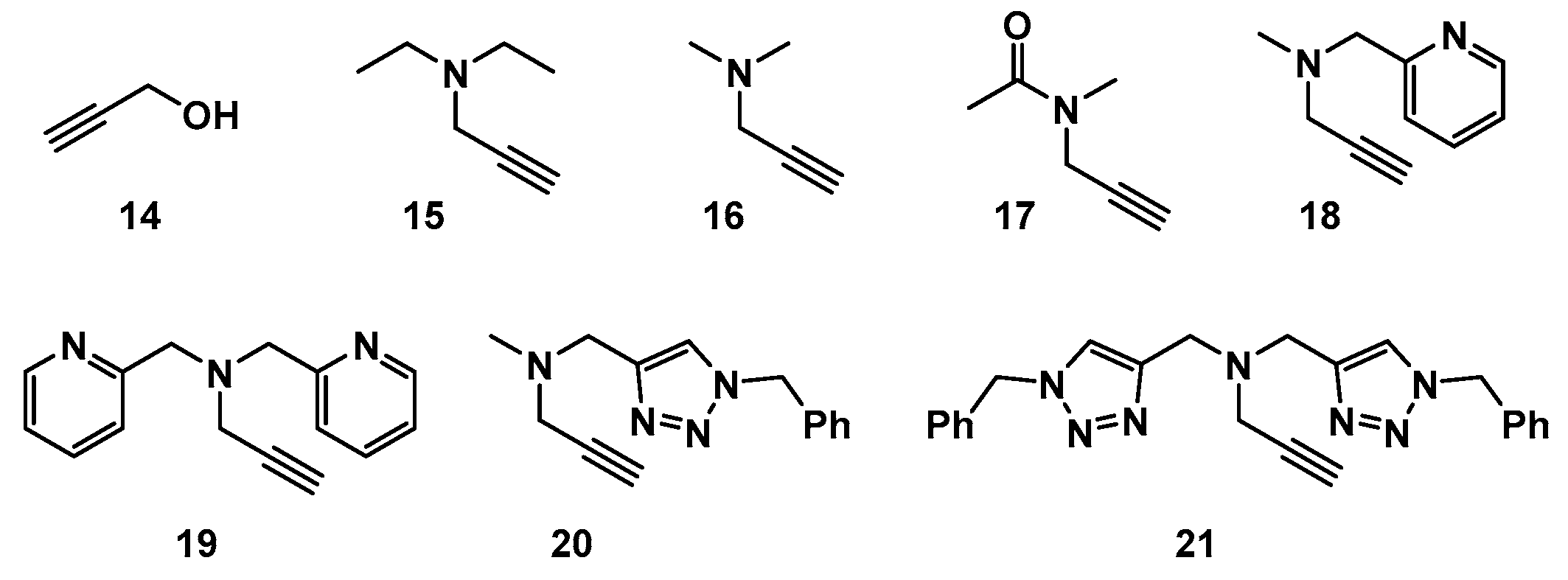
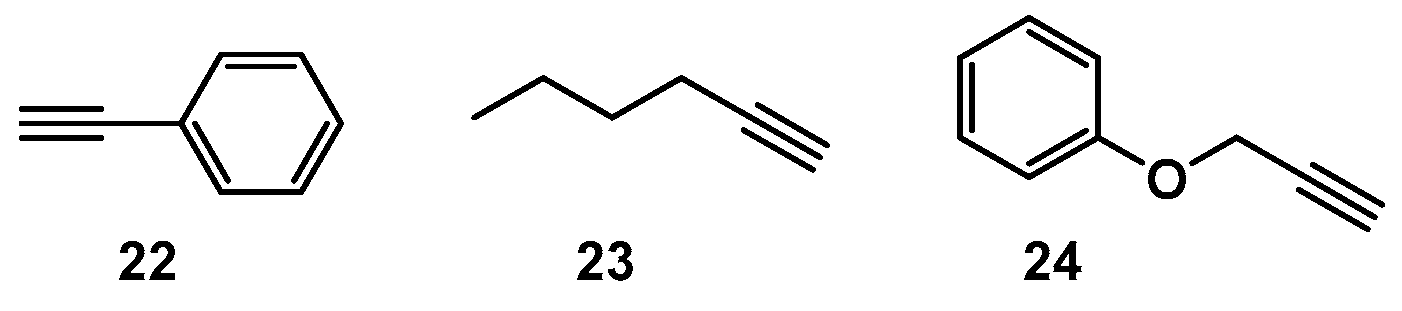
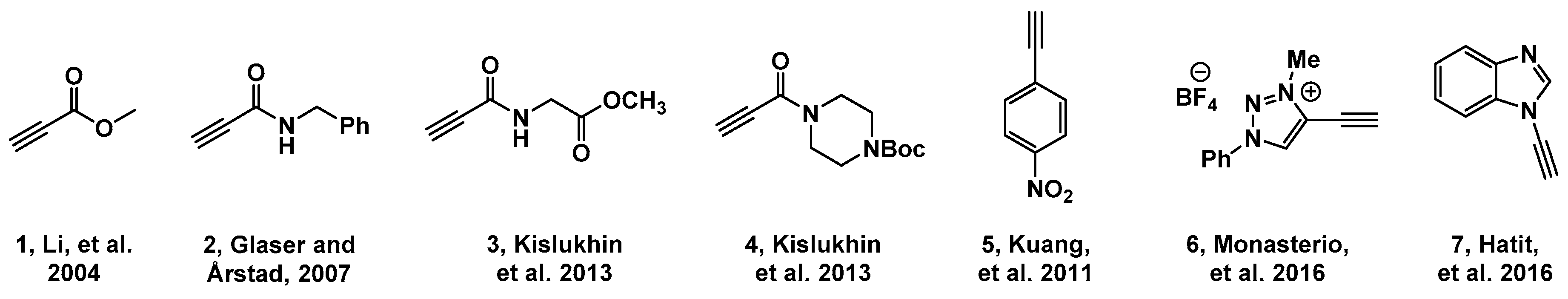
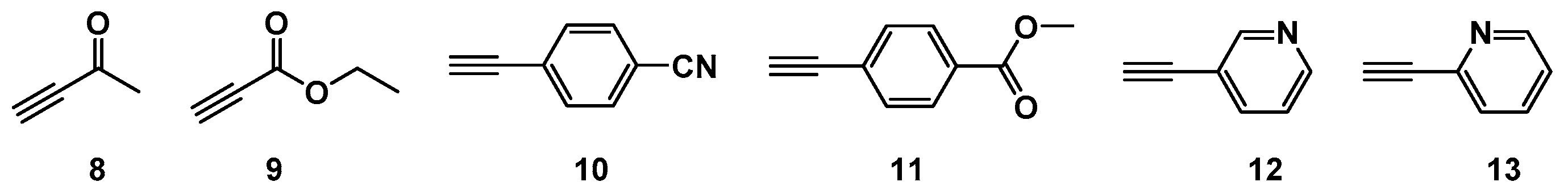
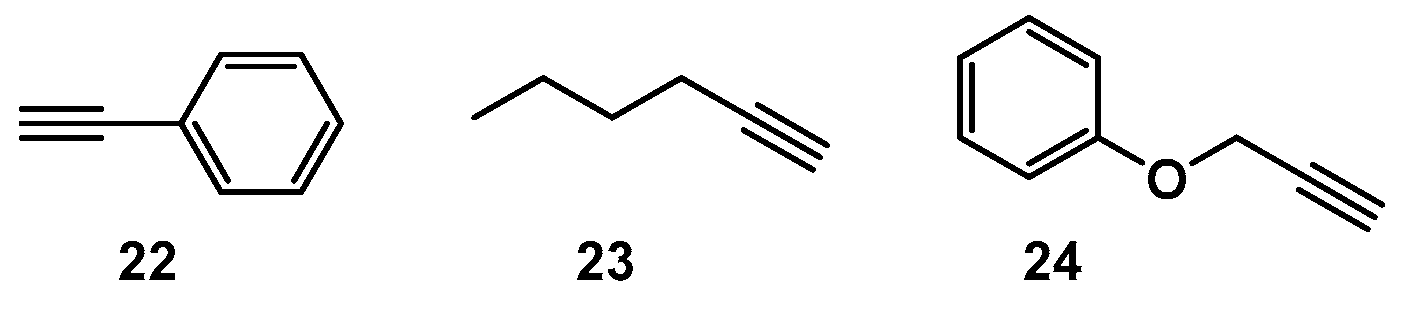
2.1. Choice of Alkynes
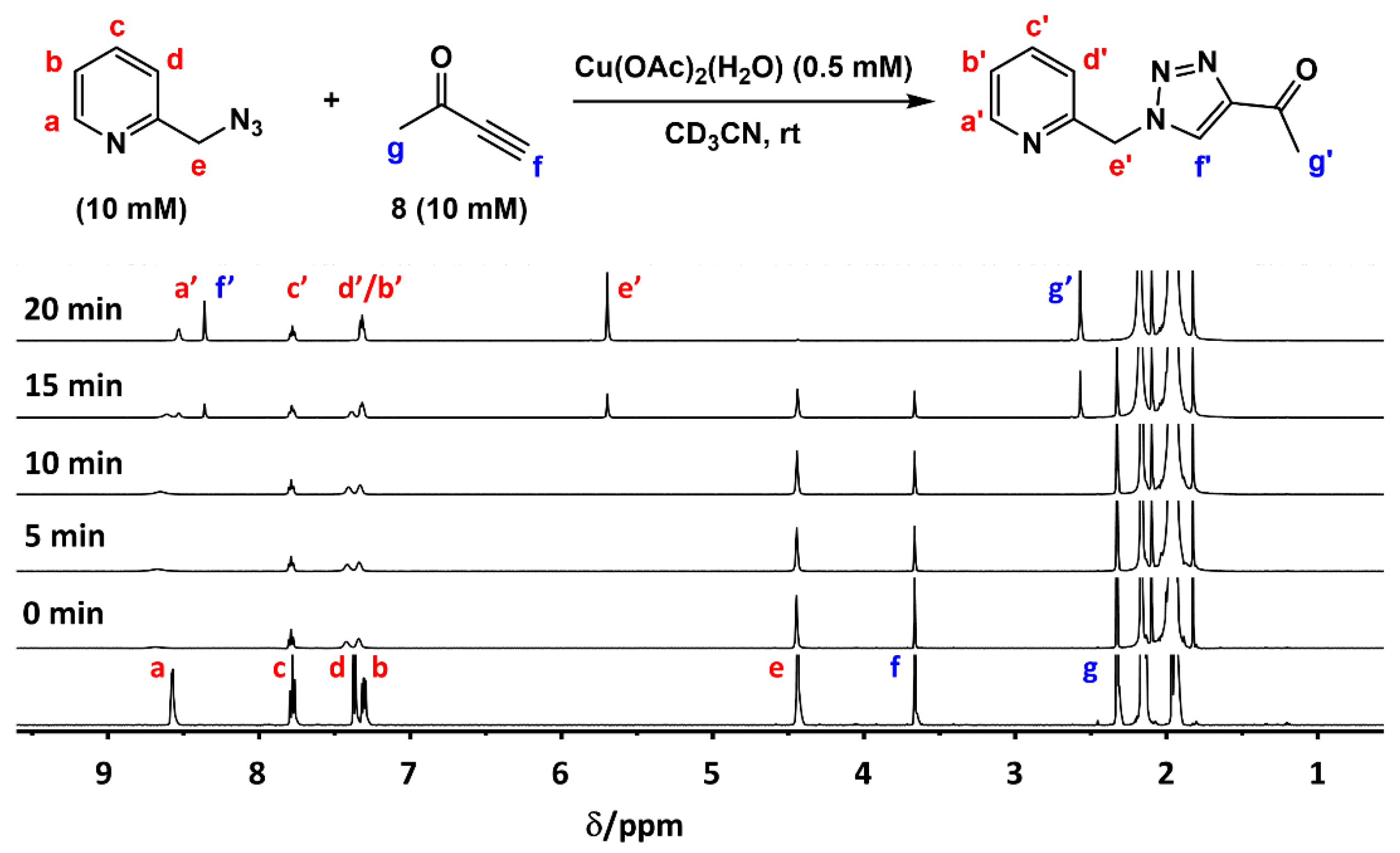
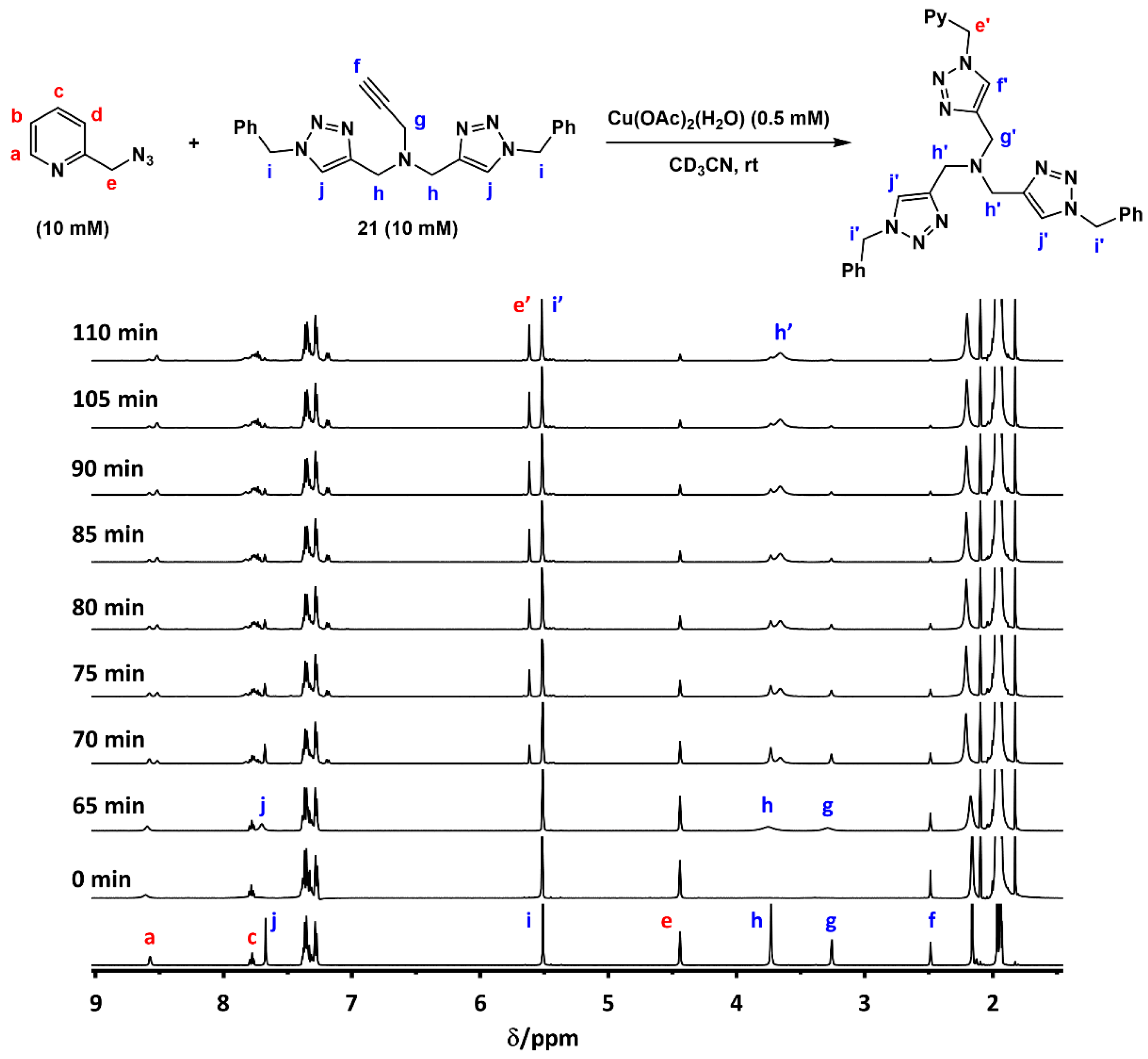
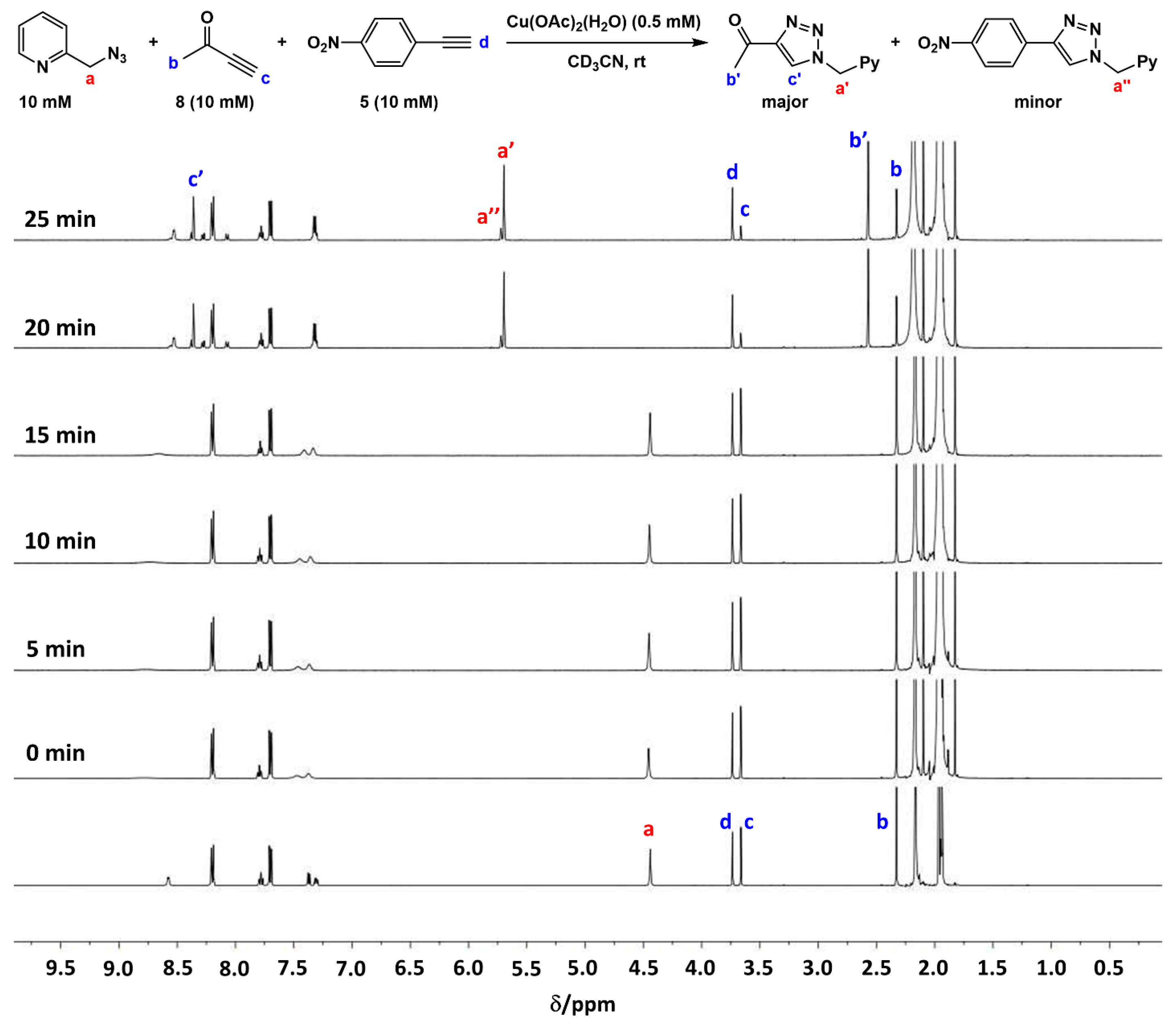
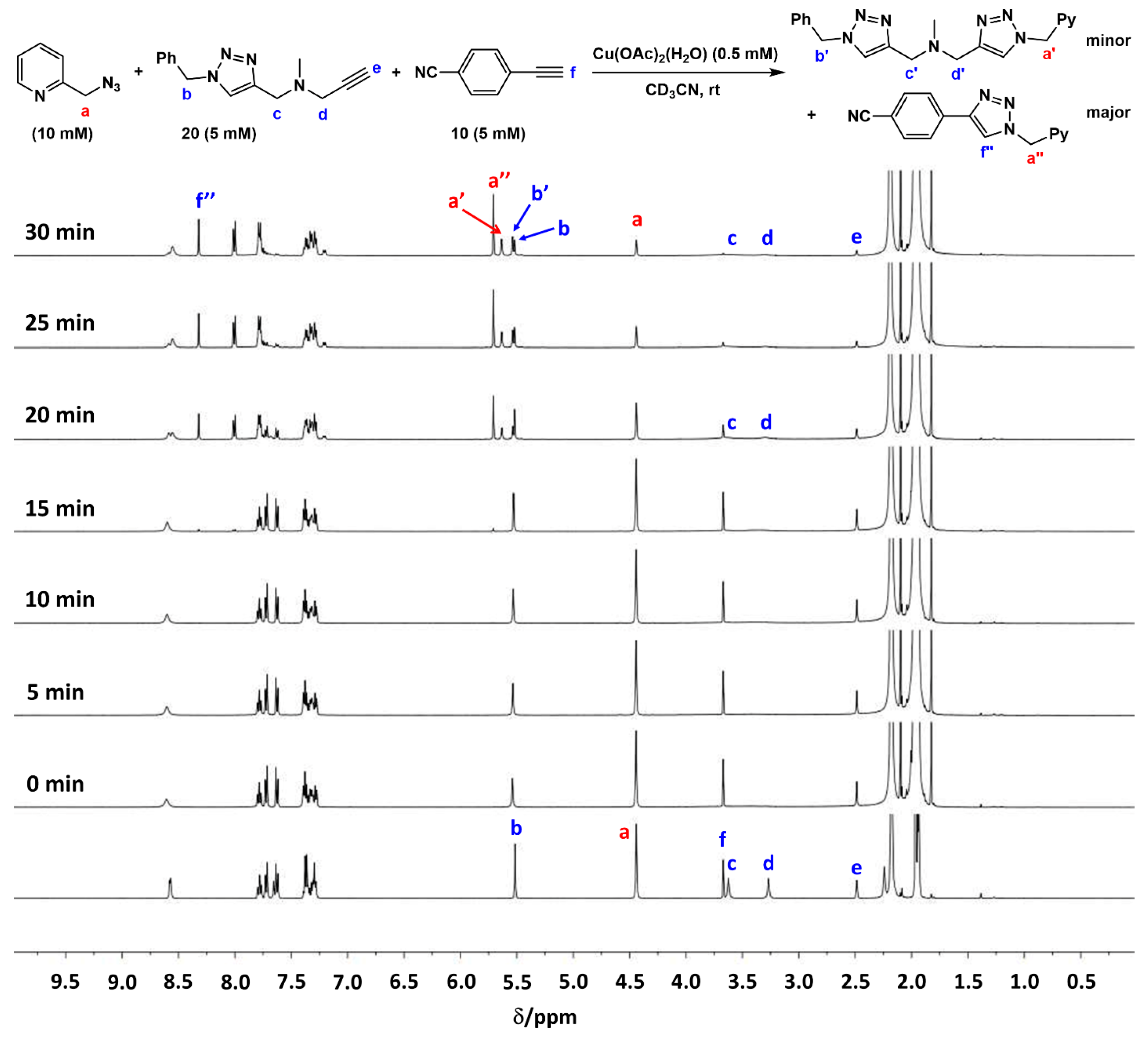
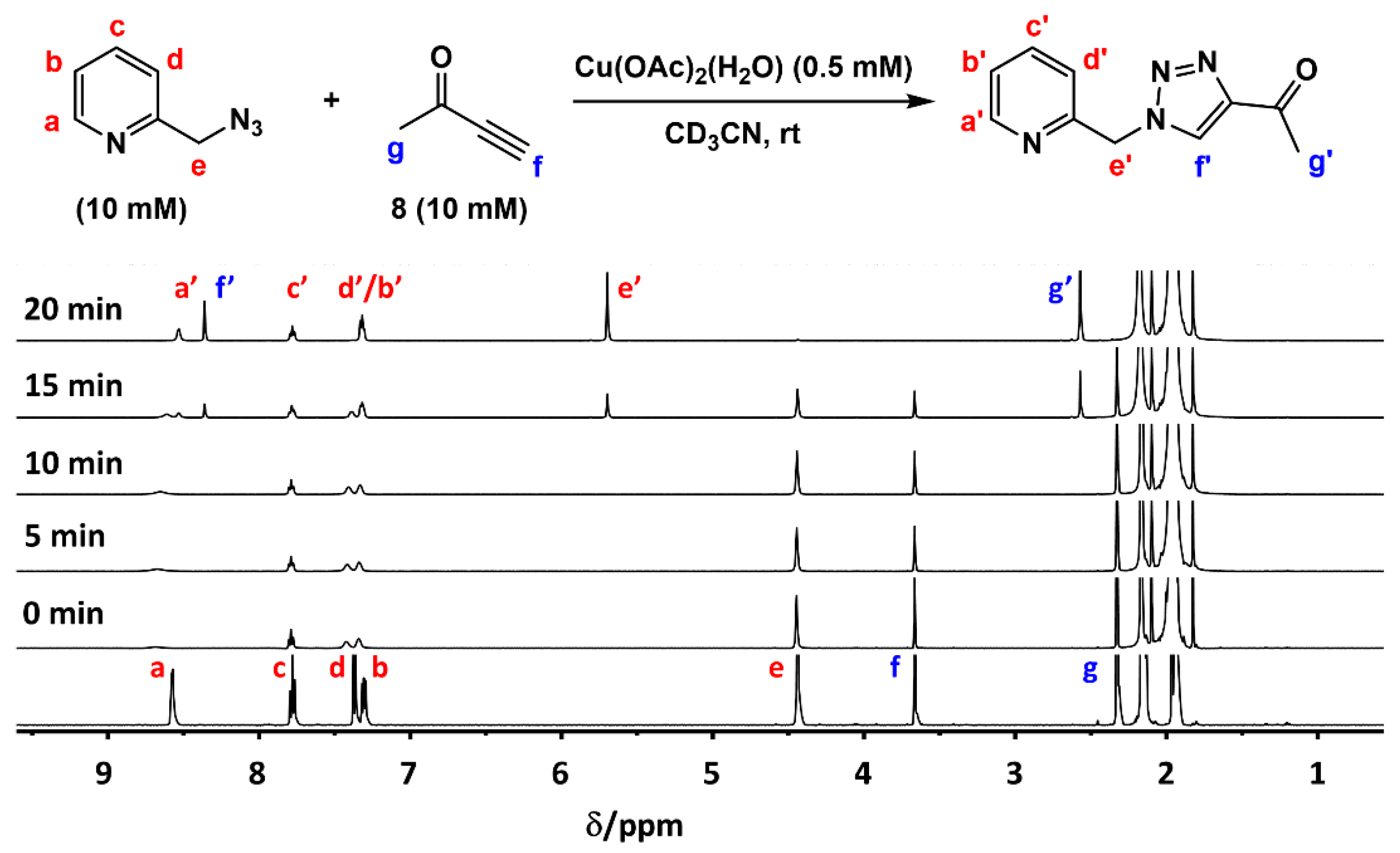
2.2. The 1H-NMR Assay
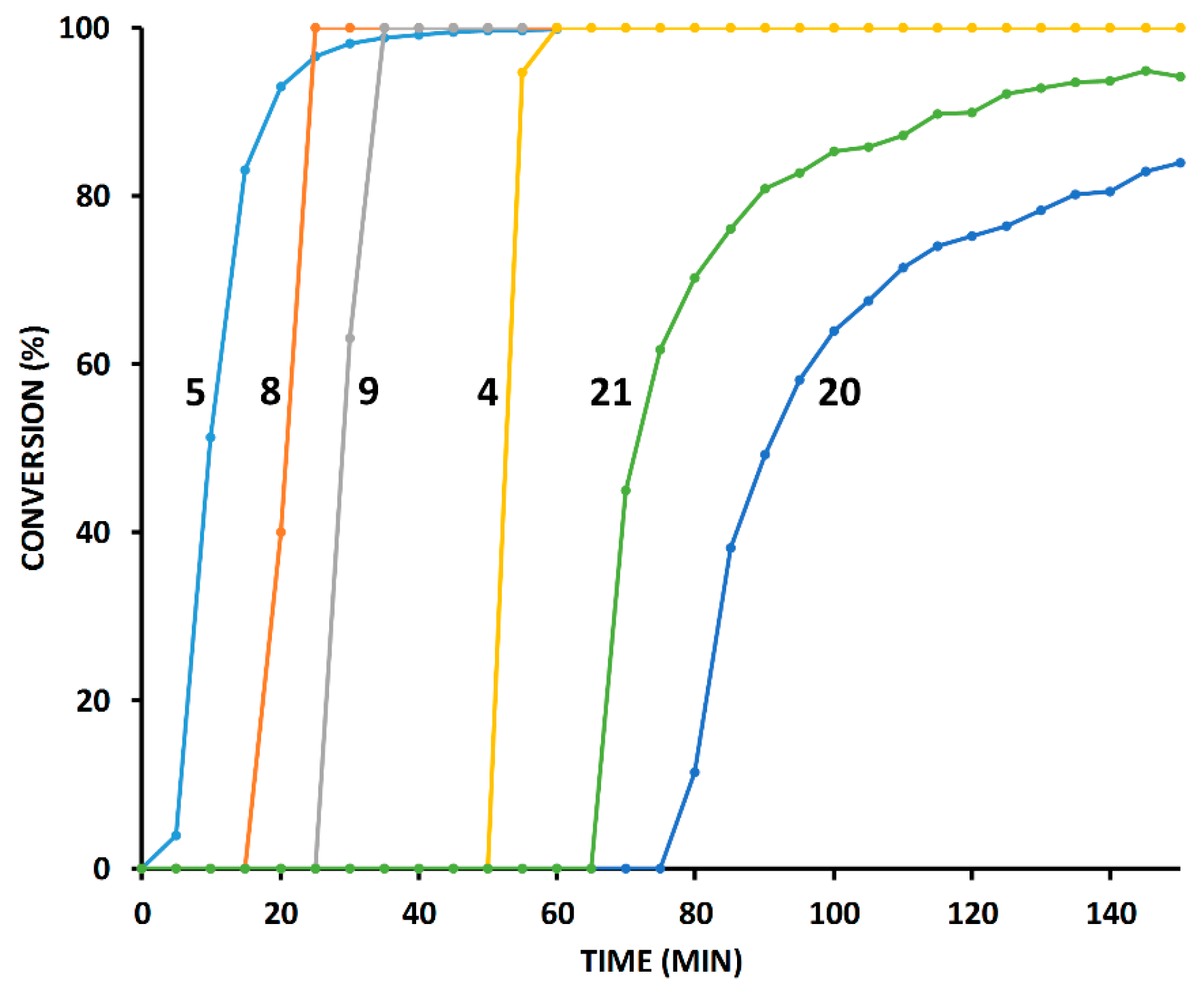
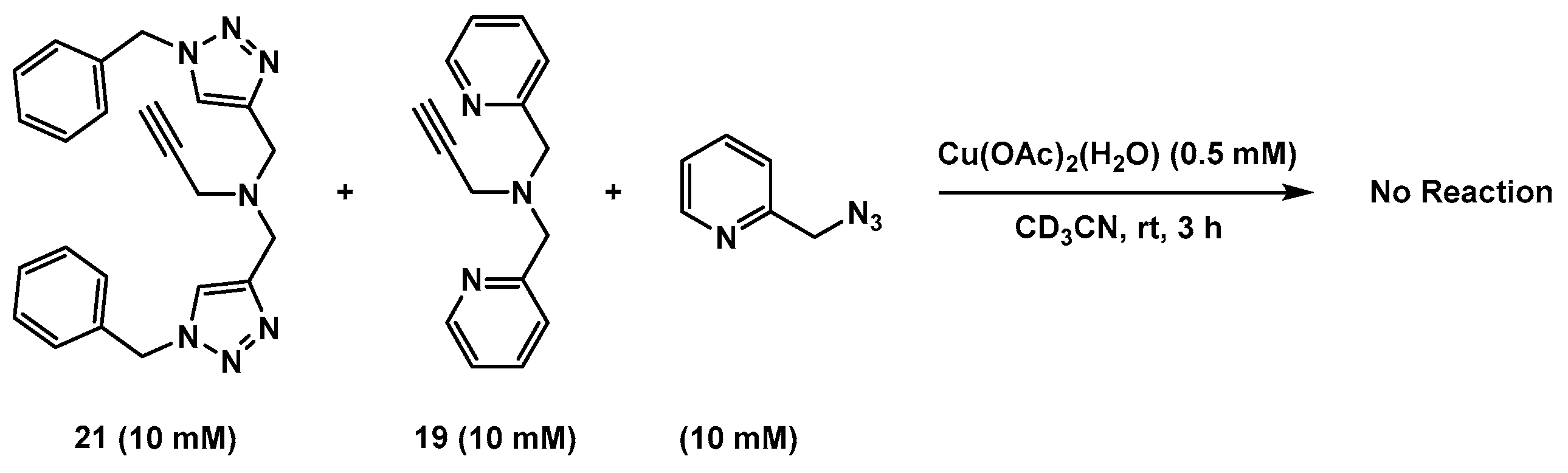
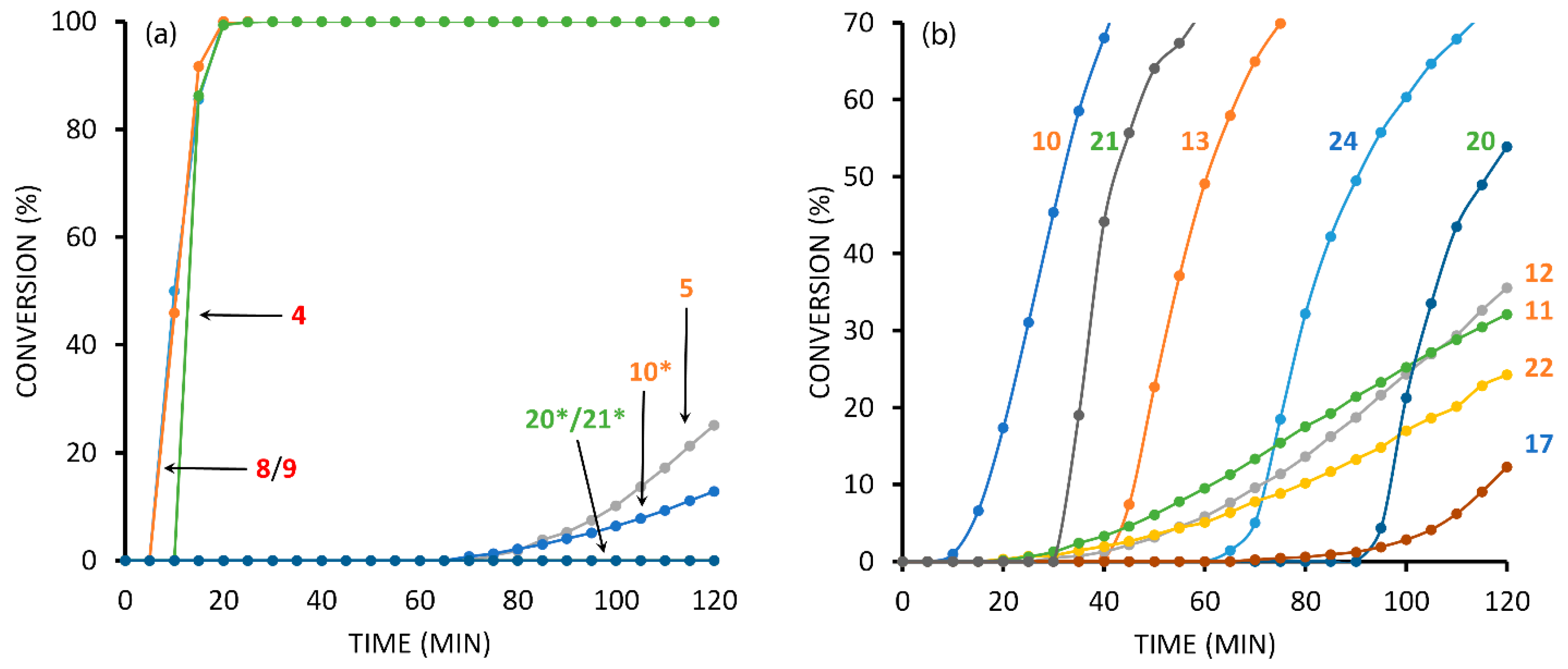
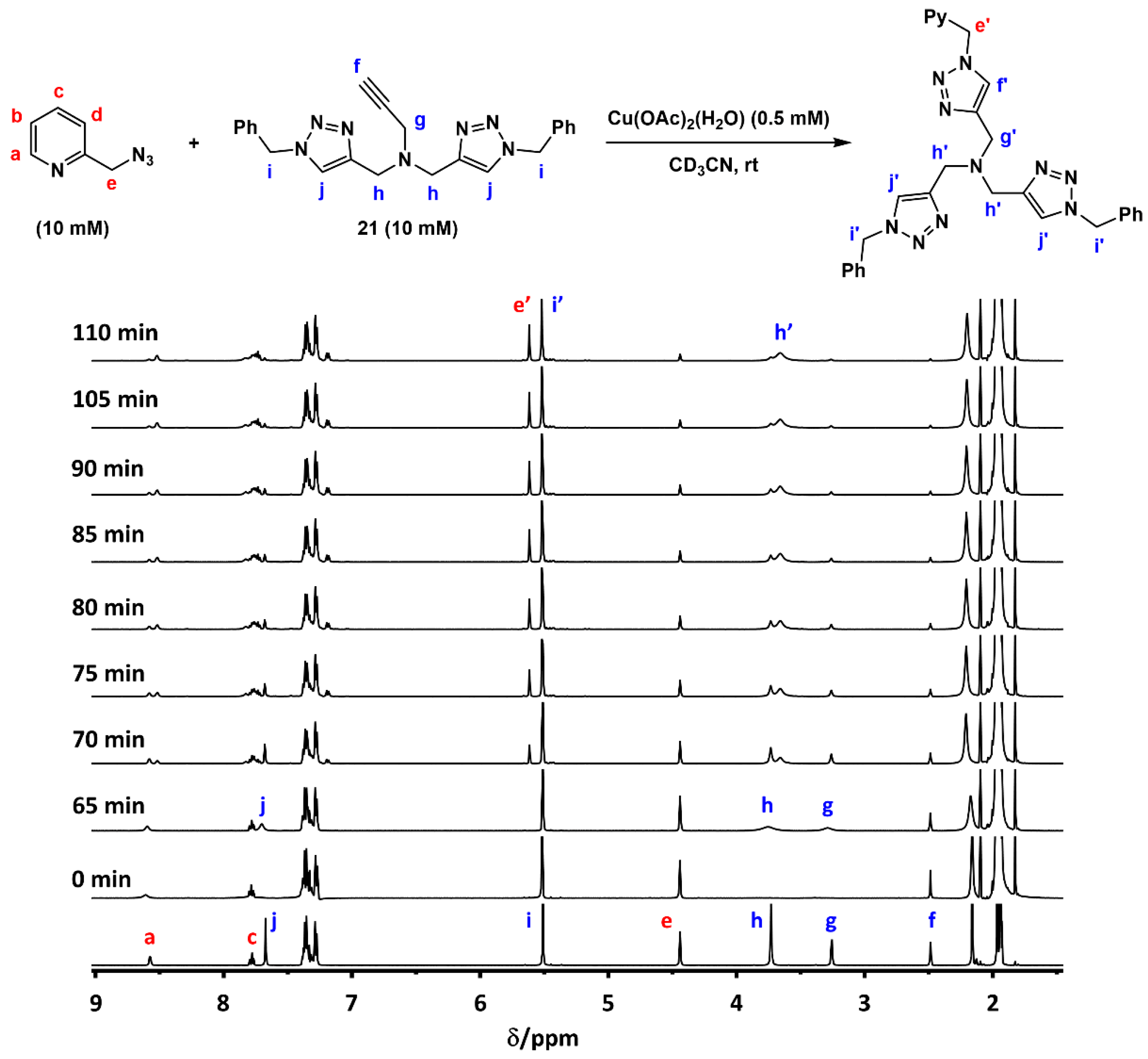
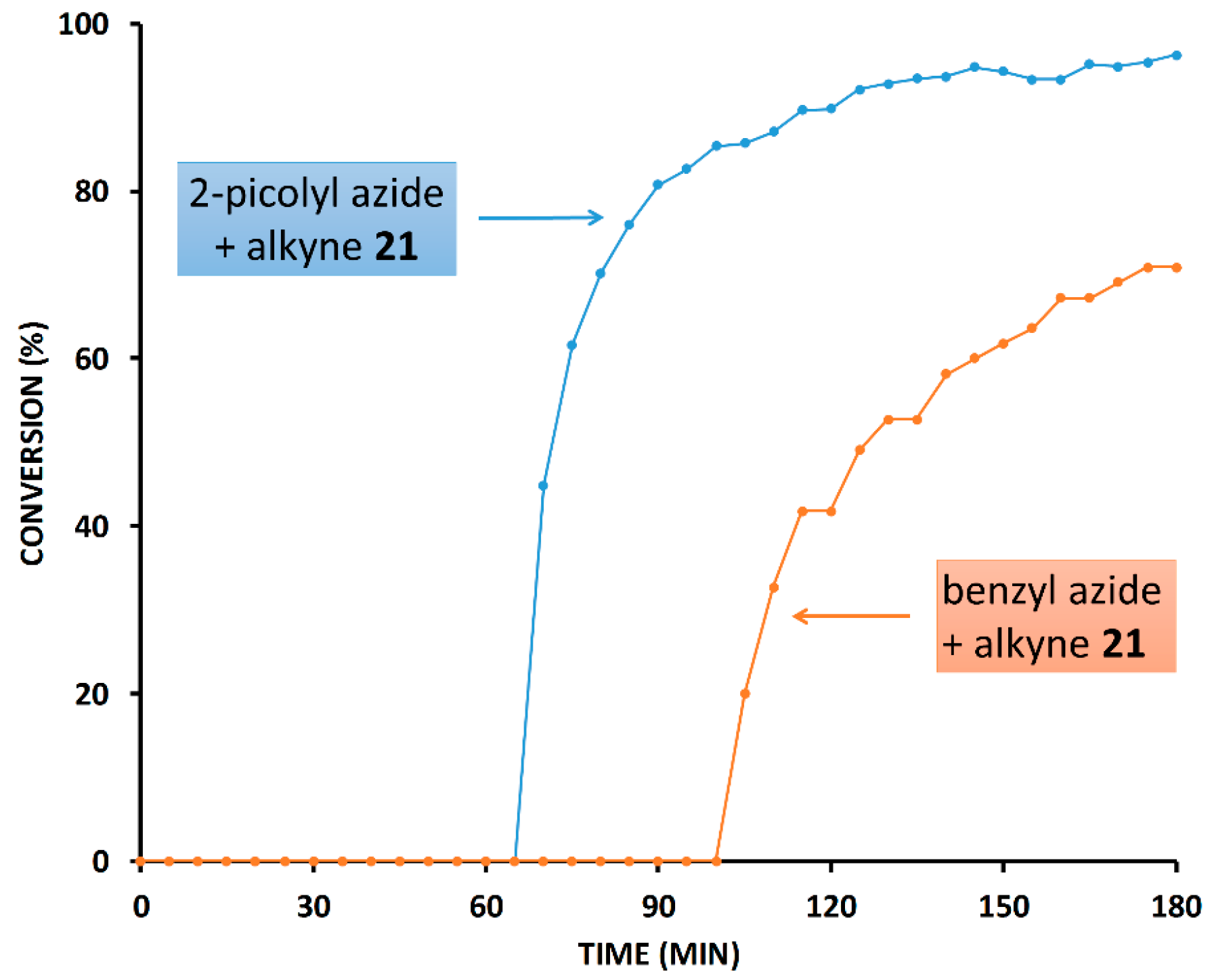
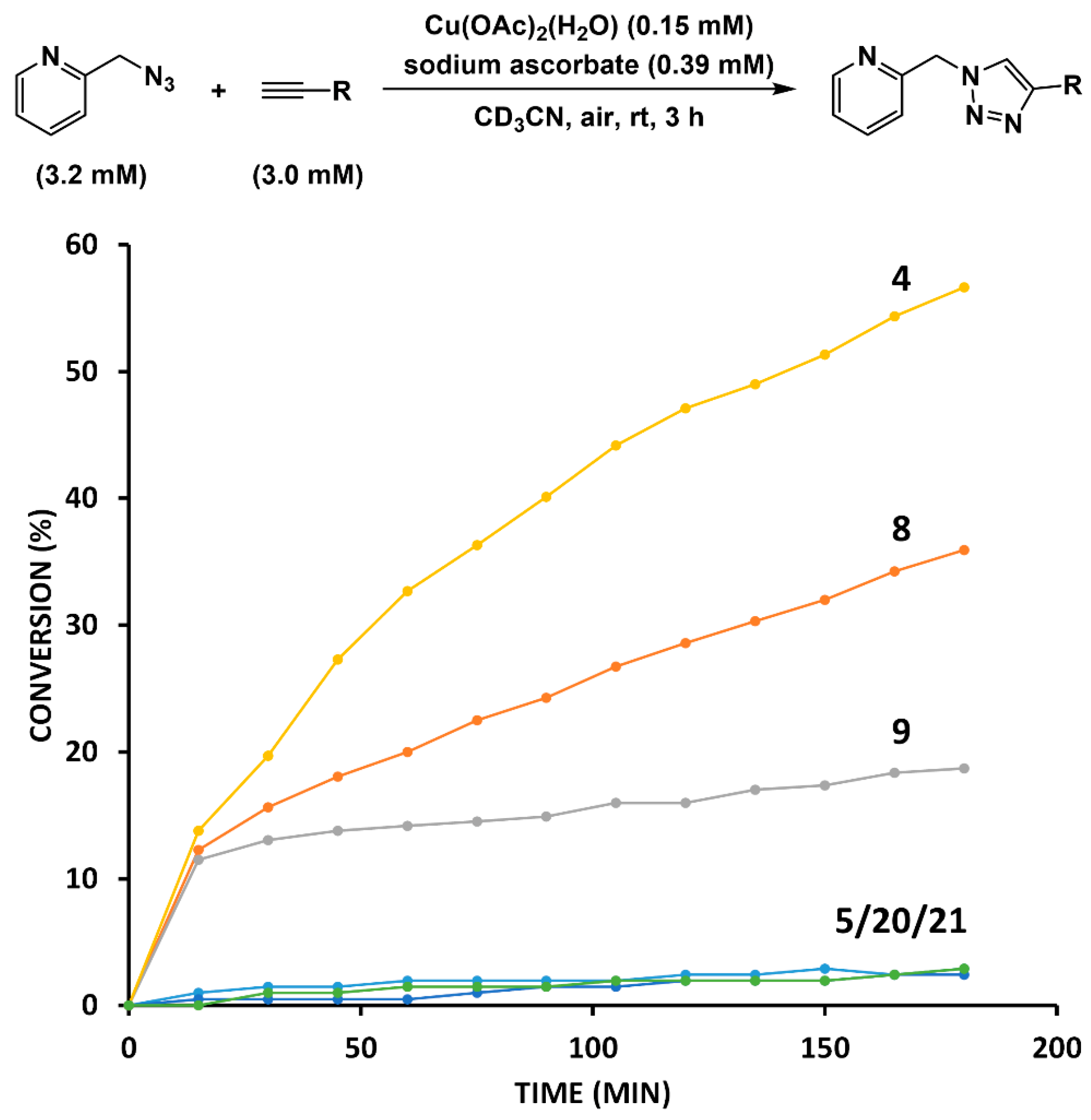
2.3. Reaction with the Non-Chelating Benzyl Azide
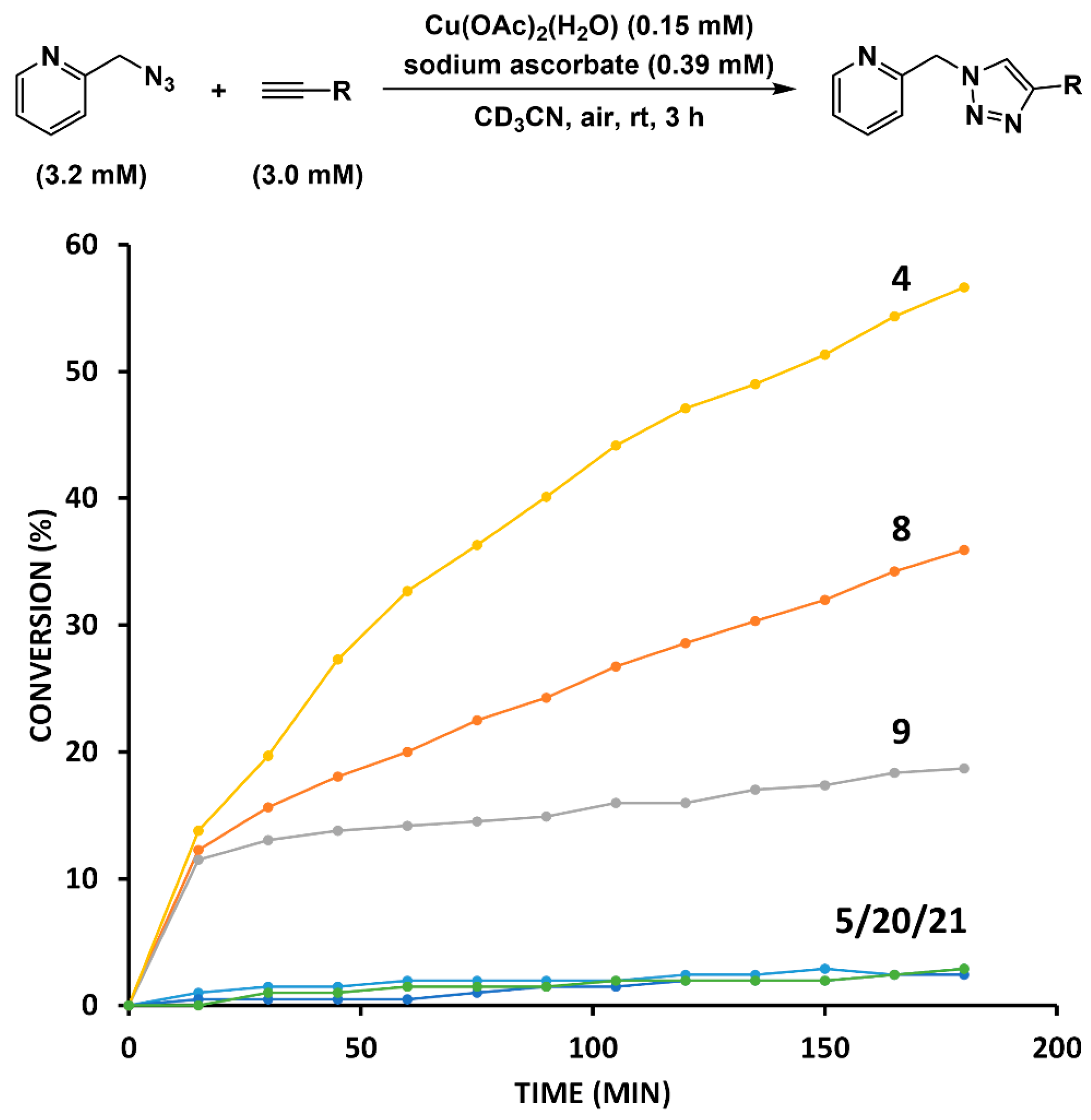
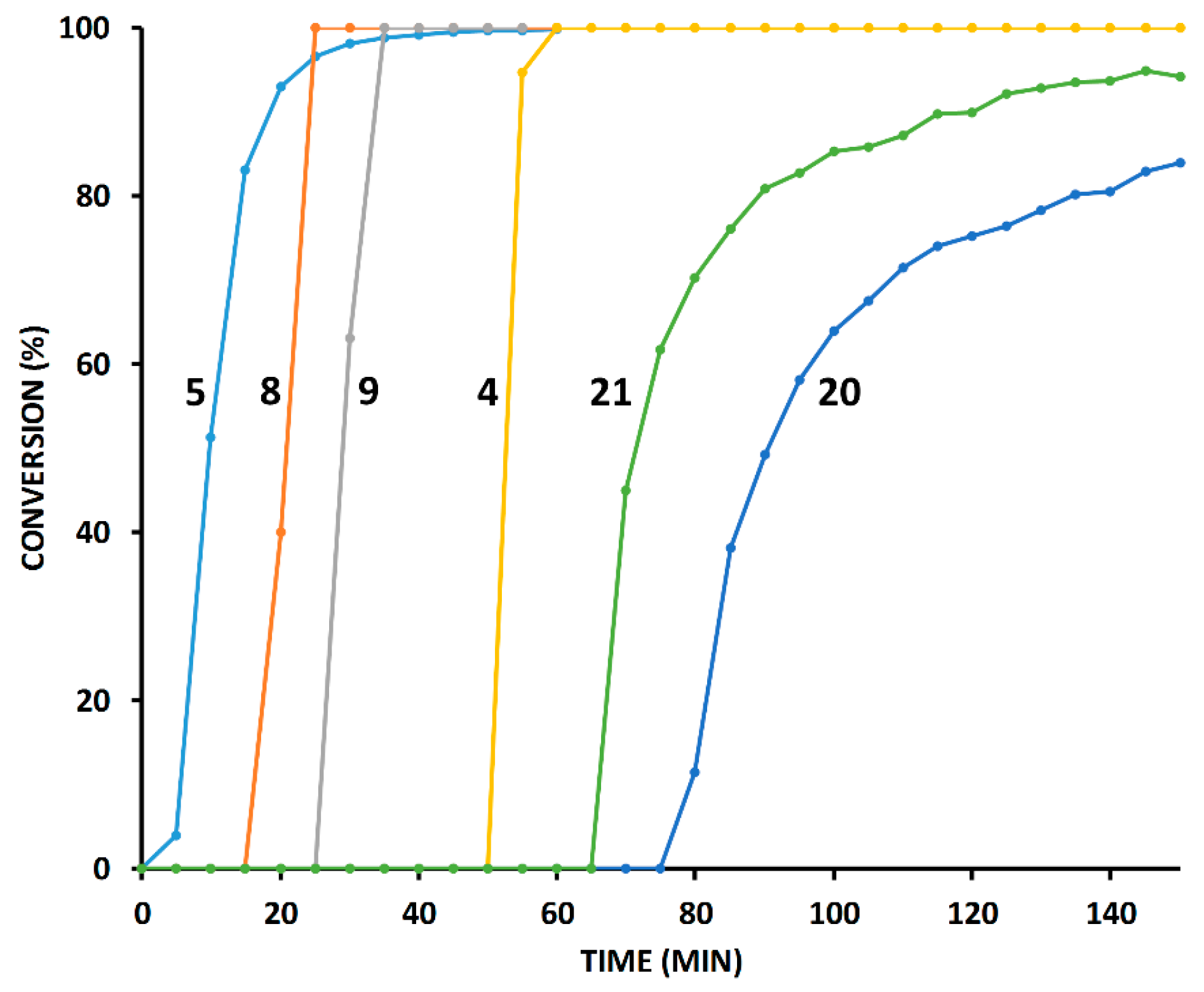
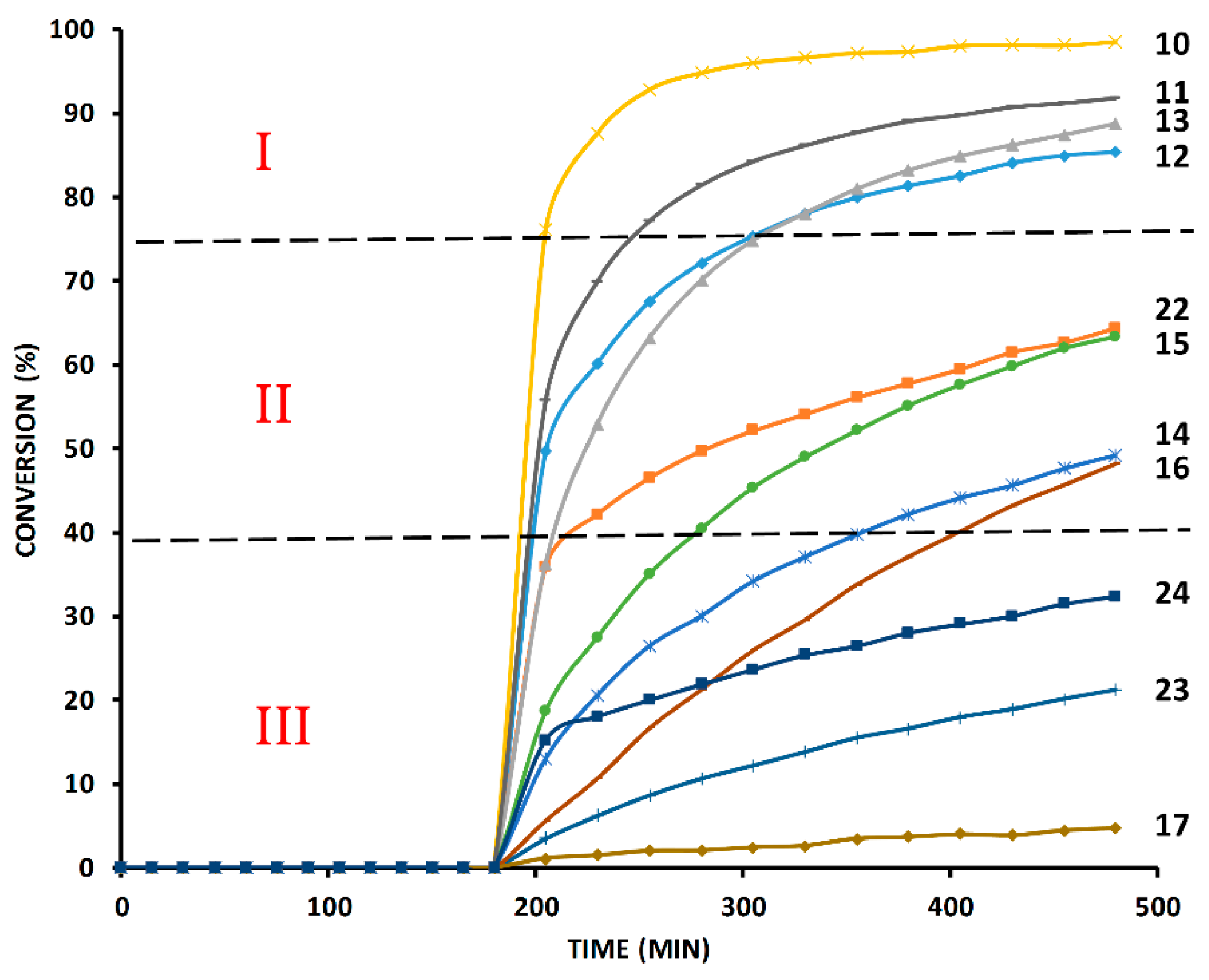
2.4. Comparison of Fast-Reacting Alkynes with a Normalized Amount of Copper(I)
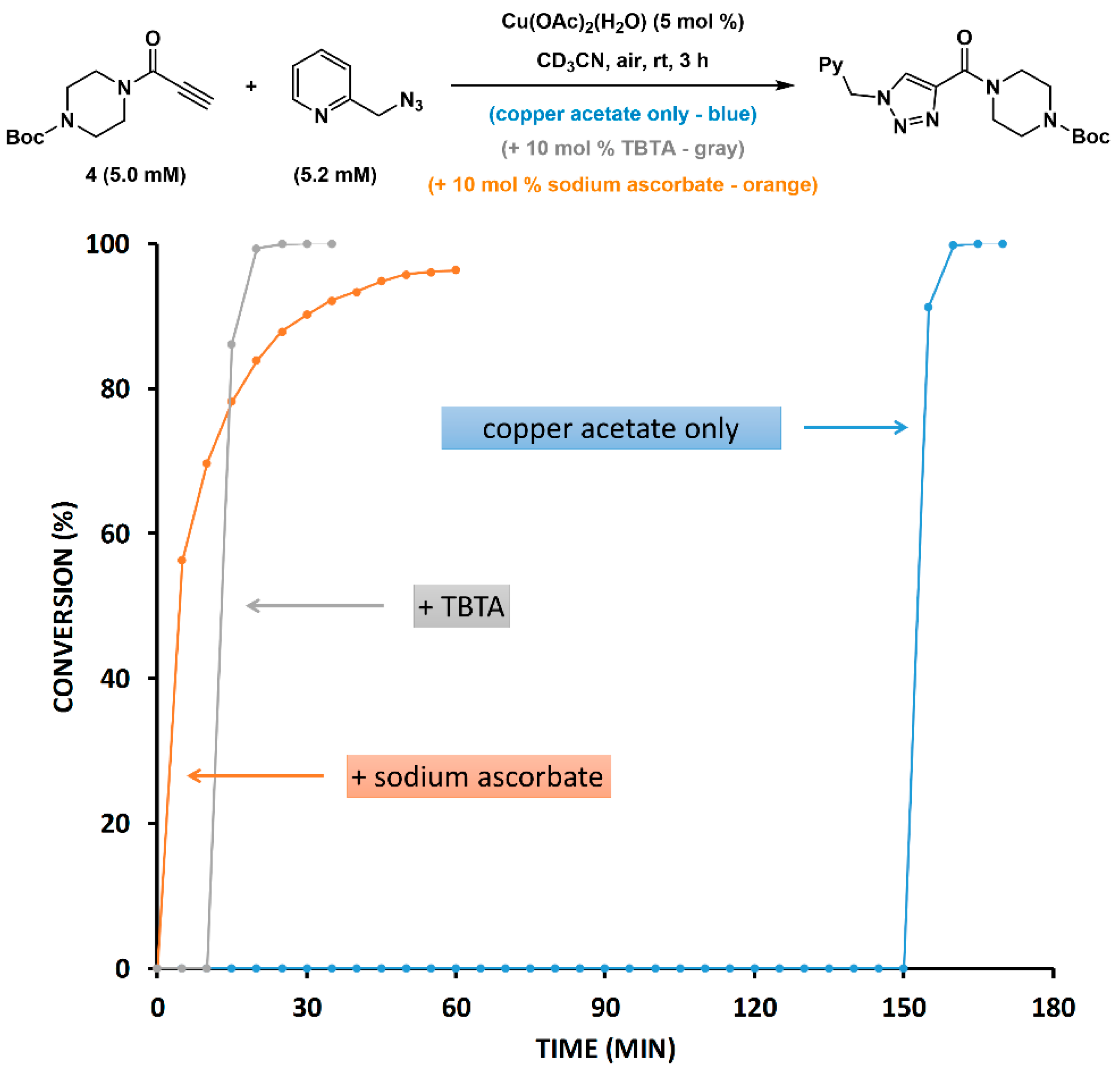
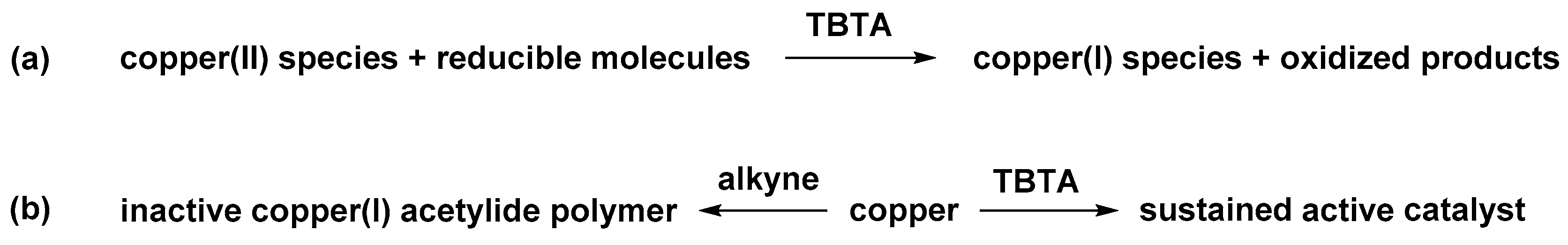
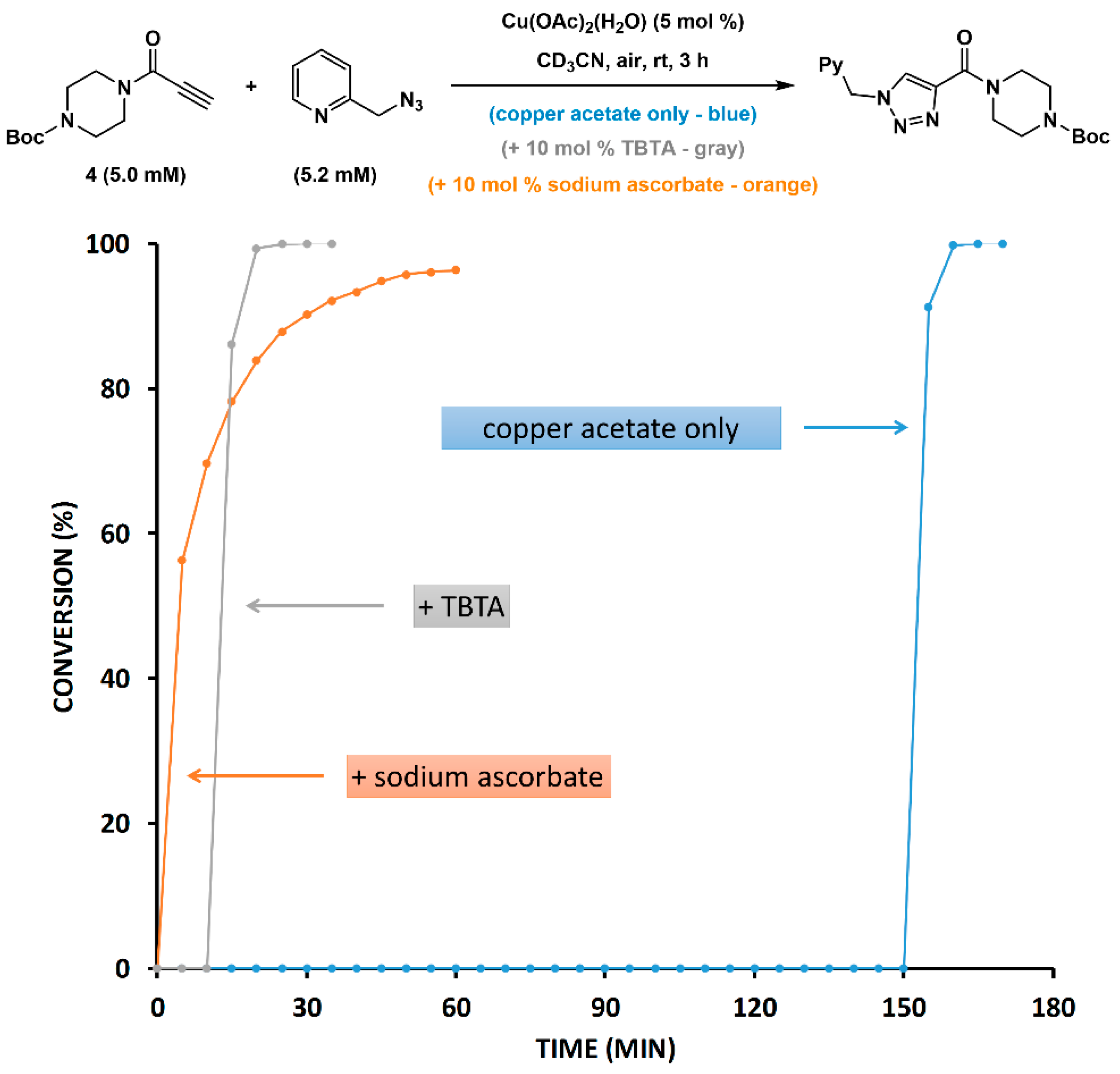
2.5. Reduction of Copper(II) Acetate by Ascorbate Alters the Pathway
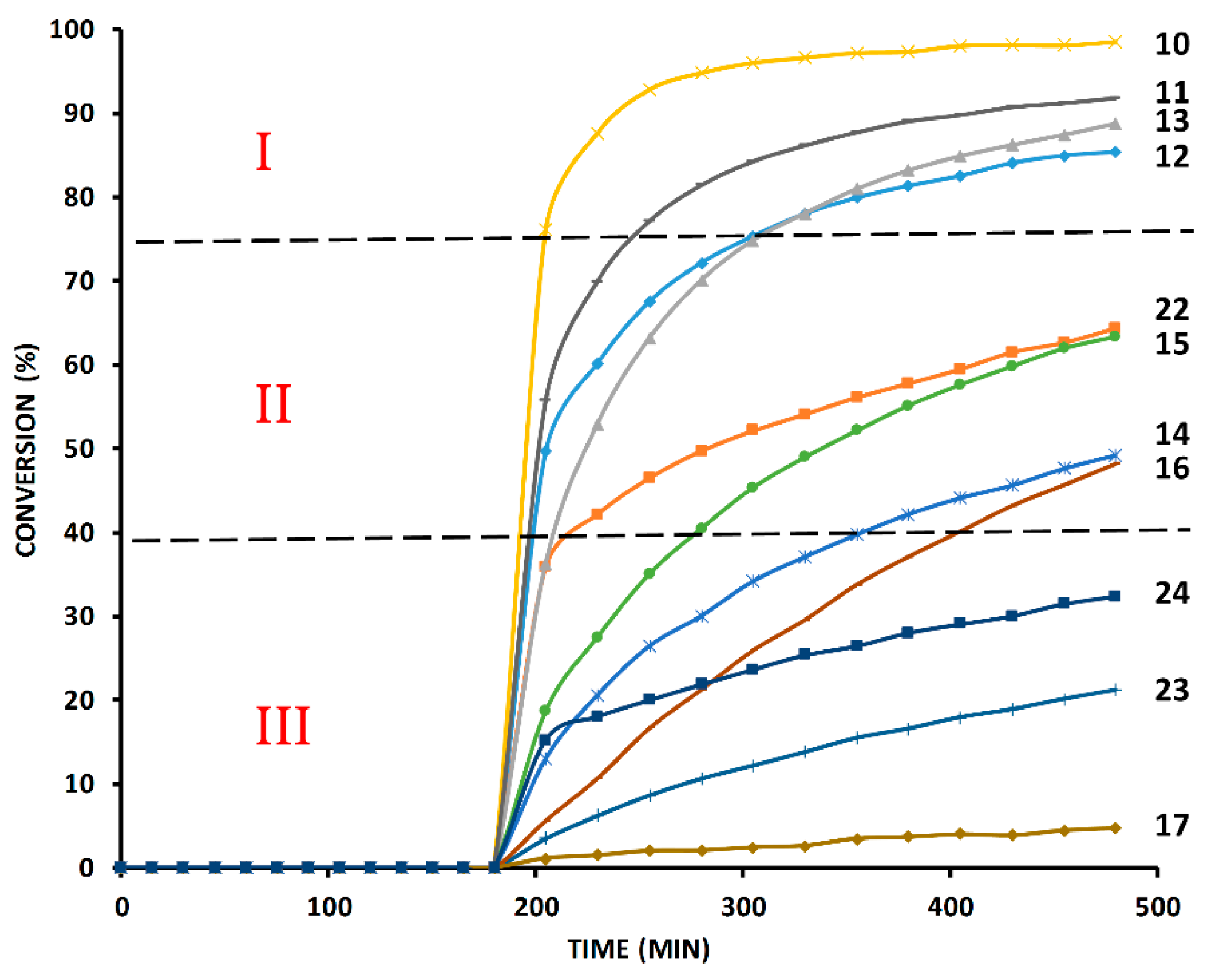
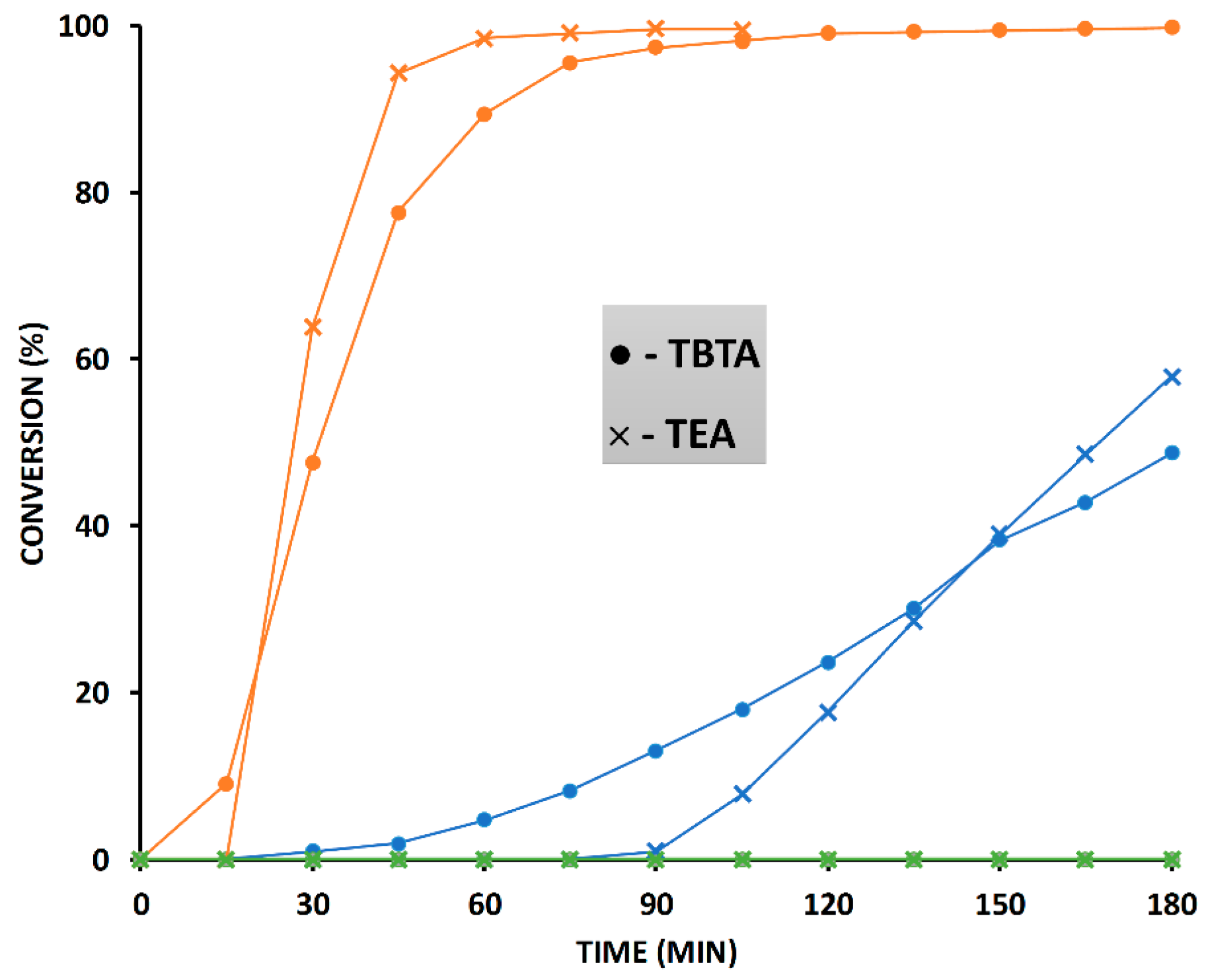
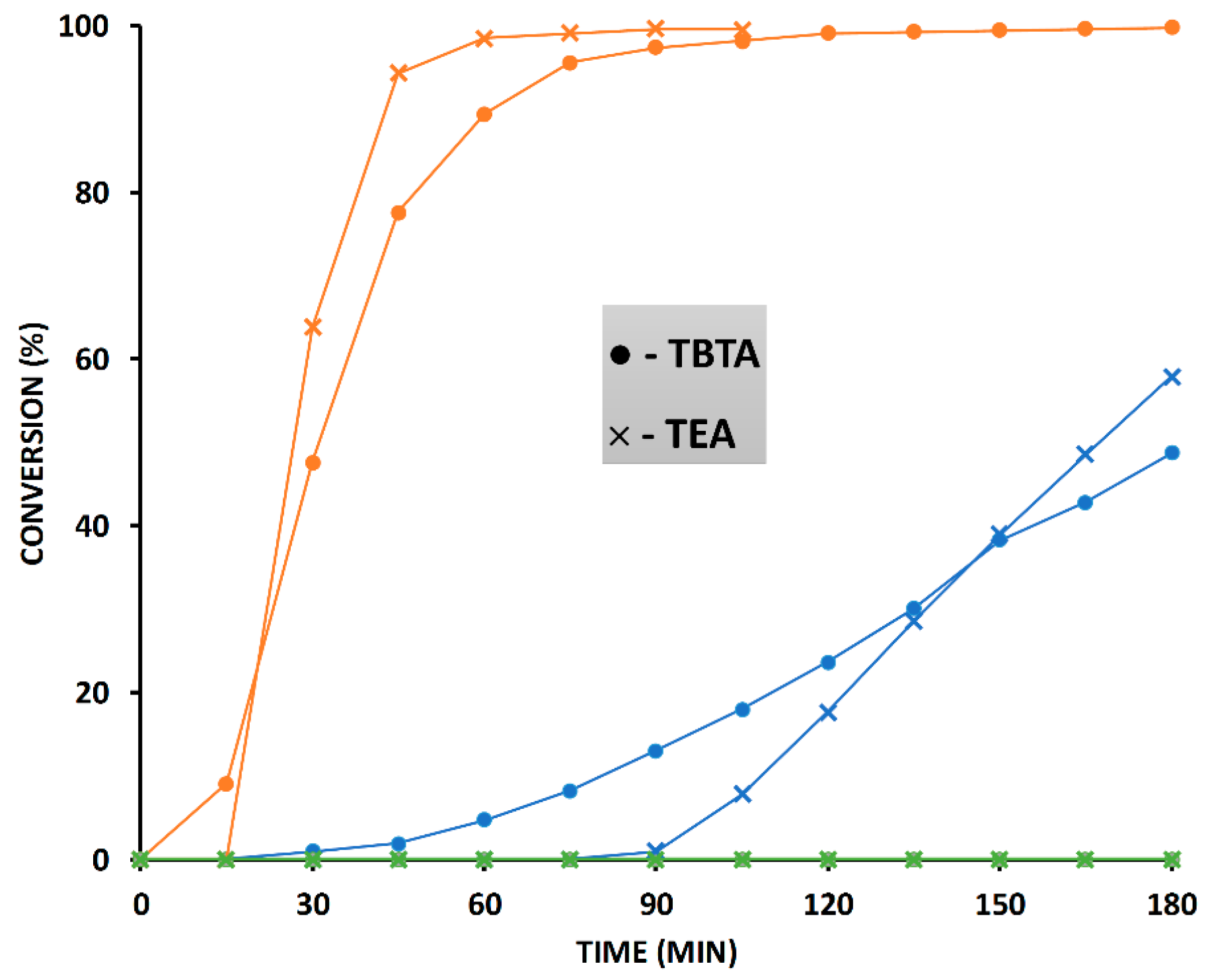
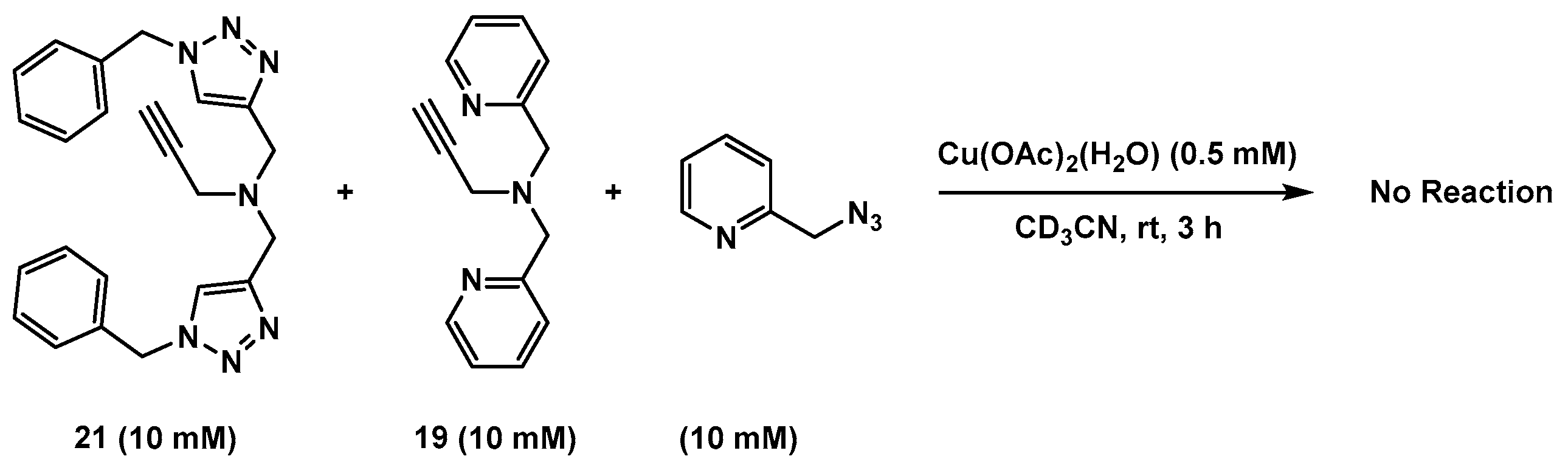
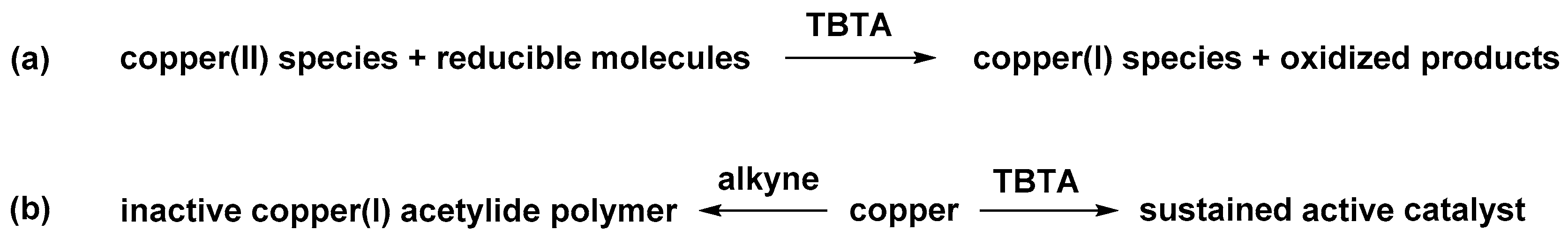
2.6. The Effect of TBTA on the Slow Alkyne Substrates
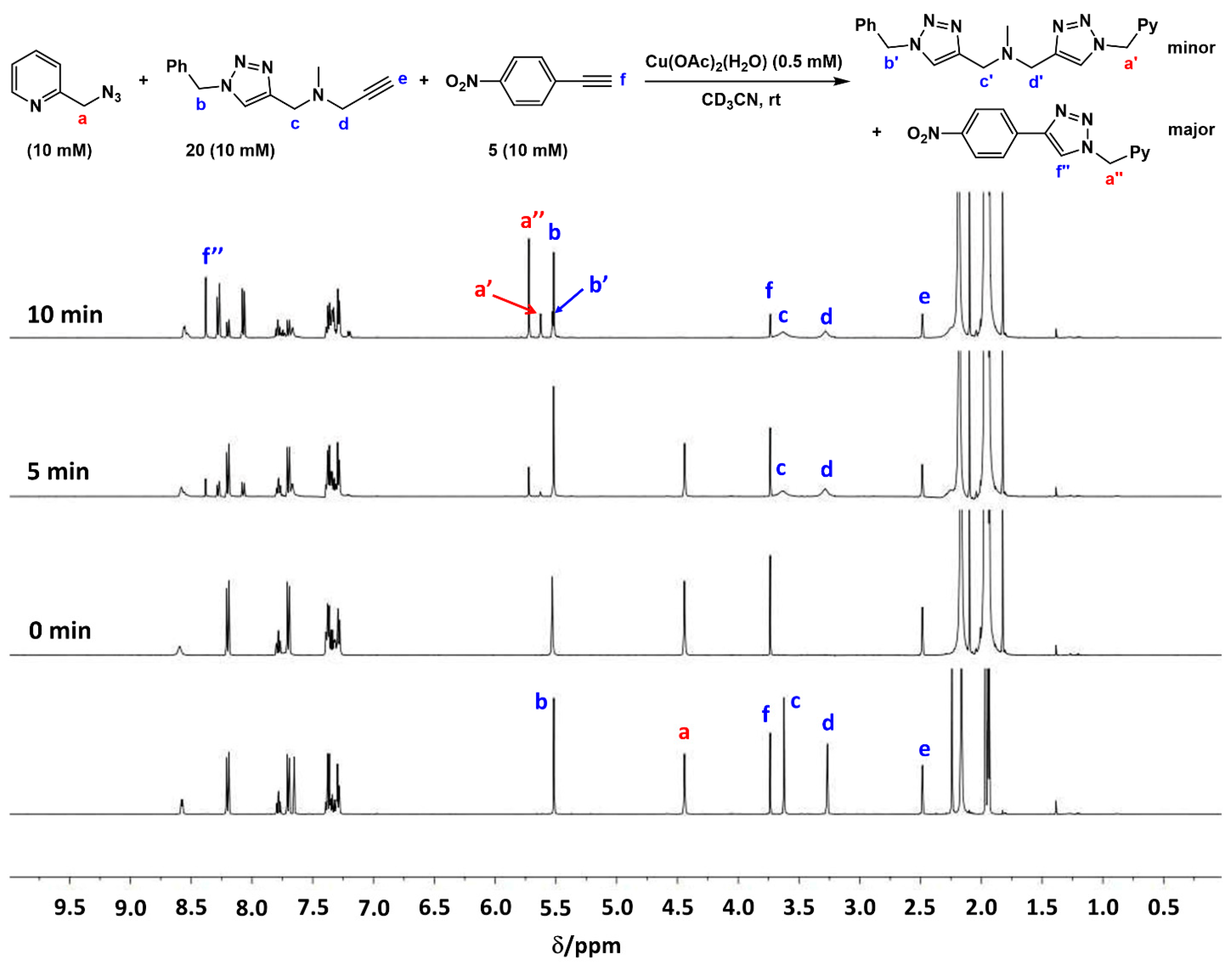
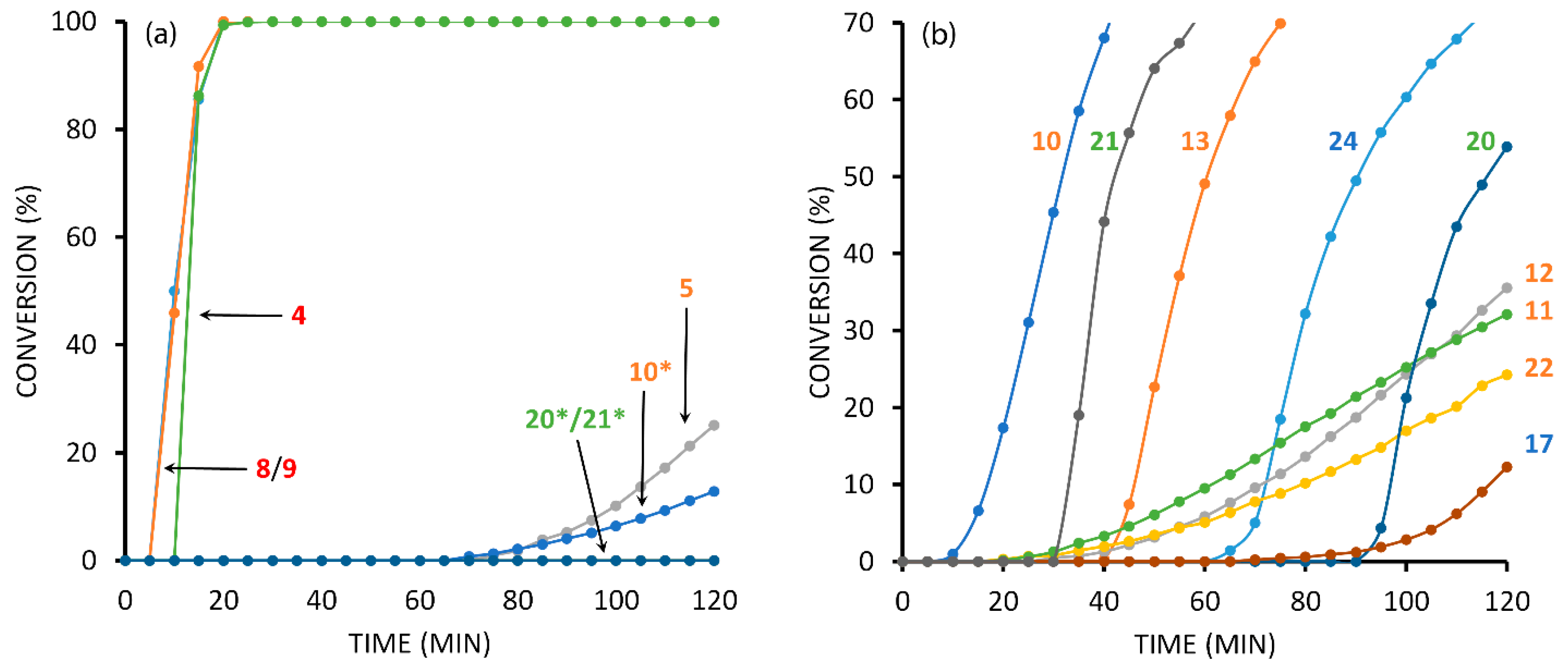
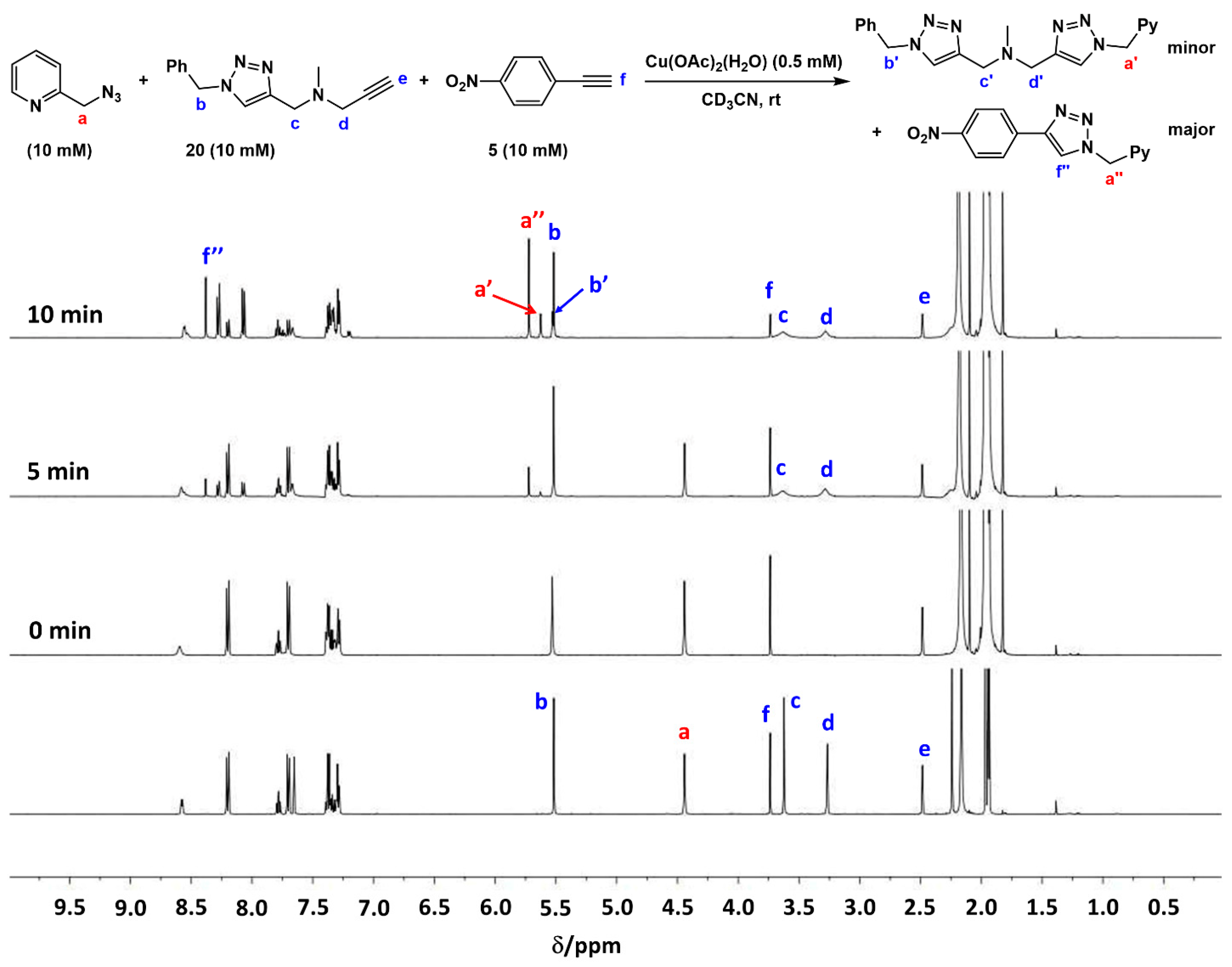
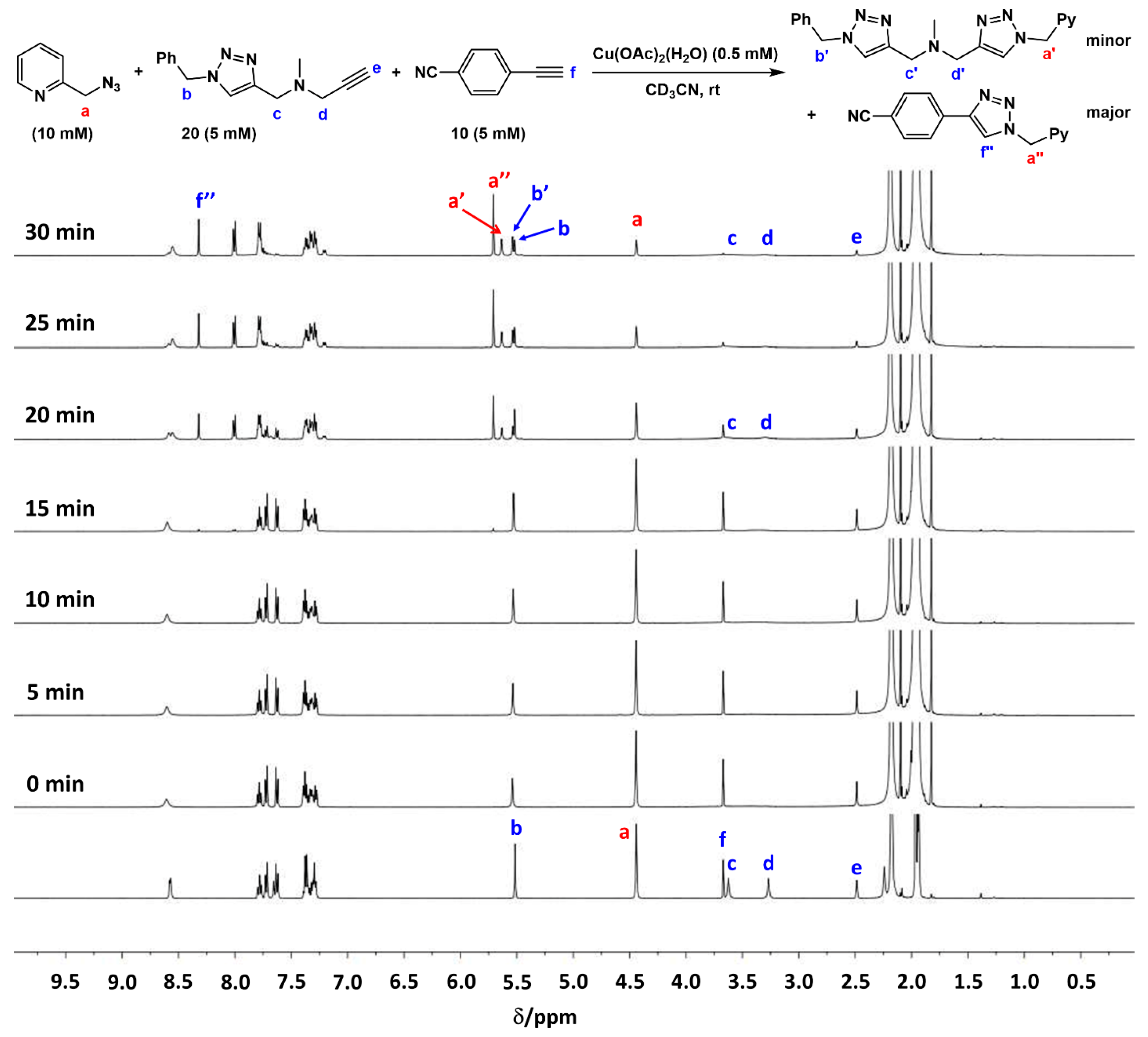
2.7. Competition Experiments
3. Experimental Section
3.1. Materials and General Methods
3.2. Synthesis of Compound 20 (Scheme 4)
3.3. Synthesis of Compound 21 (Scheme 5)
3.4. General Procedure for 1H-NMR Reaction Monitoring Experiments
3.5. Oxidative Homocoupling of Phenylacetylene (Table 1)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Finn, M.G.; Fokin, V.V. Click chemistry: Function follows form. Chem. Soc. Rev. 2010, 39, 1231–1232. [Google Scholar] [CrossRef] [PubMed]
- Košmrlj, J. Click Triazoles; Springer: Berlin/Heidelberg, Germany, 2012; Volume 28. [Google Scholar]
- Ramil, C.P.; Lin, Q. Bioorthogonal chemistry: Strategies and recent developments. Chem. Commun. 2013, 49, 11007–11022. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, J.; Chen, P.R. Transition metal-mediated bioorthogonal protein chemistry in living cells. Chem. Soc. Rev. 2014, 43, 6511–6526. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Chin, J.W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 2014, 9, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Chin, J.W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 2014, 114, 4764–4806. [Google Scholar] [CrossRef] [PubMed]
- Tra, V.N.; Dube, D.H. Glycans in pathogenic bacteria—Potential for targeted covalent therapeutics and imaging agents. Chem. Commun. 2014, 50, 4659–4673. [Google Scholar] [CrossRef] [PubMed]
- McKay, C.S.; Finn, M.G. Click chemistry in complex mixtures: Bioorthogonal bioconjugation. Chem. Biol. 2014, 21, 1075–1101. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.M.; Nazarova, L.A.; Prescher, J.A. Finding the right (bioorthogonal) chemistry. ACS Chem. Biol. 2014, 9, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Boutureira, O.; Bernardes, G.J.L. Advances in chemical protein modification. Chem. Rev. 2015, 115, 2174–2195. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.M.; Prescher, J.A. Orthogonal bioorthogonal chemistries. Curr. Opin. Chem. Biol. 2015, 28, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Mohan, K.; Weiss, G.A. Chemically modifying viruses for diverse applications. ACS Chem. Biol. 2016, 11, 1167–1179. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.C.; McKay, C.S.; Legault, M.C.B.; Danielson, D.C.; Blake, J.A.; Pegoraro, A.F.; Stolow, A.; Mester, Z.; Pezacki, J.P. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 2011, 133, 17993–18001. [Google Scholar] [CrossRef] [PubMed]
- Gary R Abel, J.; Calabrese, Z.; Ayco, J.; Hein, J.E.; Ye, T. Measuring and suppressing the oxidative damage to DNA during Cu(I)-catalyzed azide-alkyne cycloaddition. Bioconjugate Chem. 2016, 27, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cai, H.; He, J.; Chen, H.; Lam, S.; Cai, T.; Zhu, Z.; Bark, S.J.; Cai, C. Extent of the oxidative side reactions to peptides and proteins during the cuaac reaction. Bioconjugate Chem. 2016, 27, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, W.S.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Dalal, N.S.; Zhu, L. Apparent copper(II)-accelerated azide-alkyne cylcoaddition. Org. Lett. 2009, 11, 4954–4957. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.-C.; Michaels, H.A.; Simmons, J.T.; Clark, R.J.; Zhu, L. Chelation-assisted, copper(II) acetate-accelerated azide-alkyne cycloaddition. J. Org. Chem. 2010, 75, 6540–6548. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.-C.; Guha, P.M.; Brotherton, W.S.; Simmons, J.T.; Stankee, L.A.; Nguyen, B.T.; Clark, R.J.; Zhu, L. Experimental investigation on the mechanism of chelation-assisted, copper(II) acetate-accelerated azide-alkyne cycloaddition. J. Am. Chem. Soc. 2011, 133, 13984–14001. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, W.S.; Guha, P.M.; Hoa, P.; Clark, R.J.; Shatruk, M.; Zhu, L. Tridentate complexes of 2,6-bis(1,2,3-triazol-1-ylmethyl)pyridine and its organic azide precursors—An application of the copper(II) acetate-accelerated azide-alkyne cycloaddition. Dalton Trans. 2011, 40, 3655–3665. [Google Scholar] [CrossRef] [PubMed]
- Uttamapinant, C.; Tangpeerachaikul, A.; Grecian, S.; Clarke, S.; Singh, U.; Slade, P.; Gee, K.R.; Ting, A.Y. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed. 2012, 51, 5852–5856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zheng, T.; Lopez-Aguilar, A.; Feng, L.; Kopp, F.; Marlow, F.L.; Wu, P. Monitoring dynamic glycosylation in vivo using supersensitive click chemistry. Bioconjugate Chem. 2014, 25, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, V.; King, M.; Chaumontet, M.; Nothisen, M.; Gabillet, S.; Buisson, D.; Puente, C.; Wanger, A.; Taran, F. Copper-chelating azides for efficient click conjugation reactions in complex media. Angew. Chem. Int. Ed. 2014, 53, 5872–5876. [Google Scholar] [CrossRef] [PubMed]
- Machida, T.; Winssinger, N. One-step derivatization of reducing oligosaccharides for rapid and live-cell-compatible chelation-assisted cuaac conjugation. ChemBioChem 2016, 17, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Uttamapinant, C.; Sanchez, M.I.; Liu, D.S.; Yao, J.Z.; White, K.A.; Grecian, S.; Clark, S.; Gee, K.R.; Ting, A.Y. Site-specific protein labeling using prime and chelation-assisted click chemistry. Nat. Protocols 2013, 8, 1620–1634. [Google Scholar] [CrossRef] [PubMed]
- Hein, J.E.; Fokin, V.V. Copper-catalyzed azide-alkyne cycloaddition (cuaac) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Berg, R.; Straub, B.F. Advancements in the mechanistic understanding of the copper-catalyzed azide–alkyne cycloaddition. Beilstein J. Org. Chem. 2013, 9, 2715–2750. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Seo, T.S.; Ju, J. 1,3-dipolar cycloaddition of azides with electron-deficient alkynes under mild condition in water. Tetrahedron Lett. 2004, 45, 3143–3146. [Google Scholar] [CrossRef]
- Glaser, M.; Årstad, E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjugate Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Kislukhin, A.A.; Hong, V.P.; Breitenkamp, K.E.; Finn, M.G. Relative performance of alkynes in copper-catalyzed azide–alkyne cycloaddition. Bioconjugate Chem. 2013, 24, 684–689. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Tolentino, D.R.; Melaimi, M.; Bertrand, G. Isolation of bis(copper) key intermediates in cu-catalyzed azide-alkyne “click reaction”. Sci. Adv. 2015, 1, e1500304. [Google Scholar] [CrossRef] [PubMed]
- Monasterio, Z.; Sagartzazu-Aizpurua, M.; Miranda, J.I.; Reyes, Y.; Aizpurua, J.M. Cationic 1,2,3-triazolium alkynes: Components to enhance 1,4-regioselective azide−alkyne cycloaddition reactions. Org. Lett. 2016, 18, 788–791. [Google Scholar] [CrossRef] [PubMed]
- Hatit, M.Z.C.; Sadler, J.C.; McLean, L.A.; Whitehurst, B.C.; Seath, C.P.; Humphreys, L.D.; Young, R.J.; Watson, A.J.B.; Burley, G.A. Chemoselective sequential click ligations directed by enhanced reactivity of an aromatic ynamine. Org. Lett. 2016, 18, 1694–1697. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper(i)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef] [PubMed]
- The reactions would proceed to completion under preparative conditions, and many of these alkynes have been reported engage in copper(II) acetate-catalyzed CuAAC reactions. See 19.
- Romary, J.K.; Barger, J.D.; Bunds, J.E. New multidentate alpha-pyridyl ligand. Coordination of bis(2-pyridylmethyl)amine with transition metal ions. Inorg. Chem. 1968, 7, 1142–1145. [Google Scholar] [CrossRef]
- Michaels, H.A.; Zhu, L. Ligand-assisted, copper(II) acetate-accelerated azide–alkyne cycloaddition. Chem. Asian J. 2011, 6, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, H.; Ogata, K.; Fukuzawa, S.-I. 2-ethynylpyridine-promoted rapid copper(I) chloride catalyzed azide-alkyne cycloaddition reaction in water. Synlett 2013, 24, 843–846. [Google Scholar] [CrossRef]
- Donnelly, P.S.; Zanatta, S.D.; Zammit, S.C.; White, J.M.; Williams, S.J. “Click” cycloaddition catalysts: Copper(I) and copper(II) tris(triazolylmethyl)amine complexes. Chem. Commun. 2008, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Brassard, C.J.; Zhang, X.; Guha, P.M.; Clark, R.J. On the mechanism of copper(I)-catalyzed azide-alkyne cycloaddition. Chem. Rec. 2016, 16, 1501–1517. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, B.J.; Baumgold, J.; Paek, R.; Kammula, U.; Zimmet, J.; Jacobson, K.A. Muscarinic receptor binding and activation of second messengers by substituted N-methyl-N-[4-(1-azacycloalkyl)-2-butynyl]acetamides. J. Med. Chem. 1991, 34, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Michaels, H.A.; Murphy, C.S.; Clark, R.J.; Davidson, M.W.; Zhu, L. 2-anthryltriazolyl-containing multidentate ligands: Zinc-coordination mediated photophysical processes and potential in live-cell imaging applications. Inorg. Chem. 2010, 49, 4278–4287. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.T.; Allen, J.R.; Morris, D.R.; Clark, R.J.; Levenson, C.W.; Davidson, M.W.; Zhu, L. Integrated and passive 1,2,3-triazolyl groups in fluorescent indicators for zinc(II) ions—thermodynamic and kinetic evaluations. Inorg. Chem. 2013, 52, 5838–5850. [Google Scholar] [CrossRef] [PubMed]
- Presolski, S.I.; Hong, V.; Cho, S.-H.; Finn, M.G. Tailored ligand acceleration of the cu-catalyzed azide-alkyne cycloaddition reaction: Practical and mechanistic implications. J. Am. Chem. Soc. 2010, 132, 14570–14576. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.R.; Fokin, V.V. Polymer-supported copper(i) catalysts for the experimentally simplified azide–alkyne cycloaddition. QSAR Comb. Sci. 2007, 26, 1274–1279. [Google Scholar] [CrossRef]
- Kamata, K.; Yamaguchi, S.; Kotani, M.; Yamaguchi, K.; Mizuno, N. Efficient oxidative alkyne homocoupling catalyzed by a monomeric dicopper-substituted silicotungstate. Angew. Chem. Int. Ed. 2008, 47, 2407–2410. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.

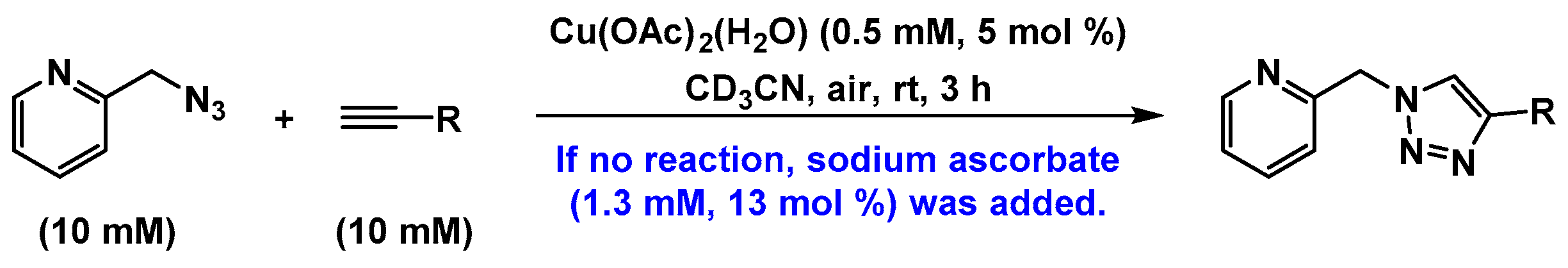


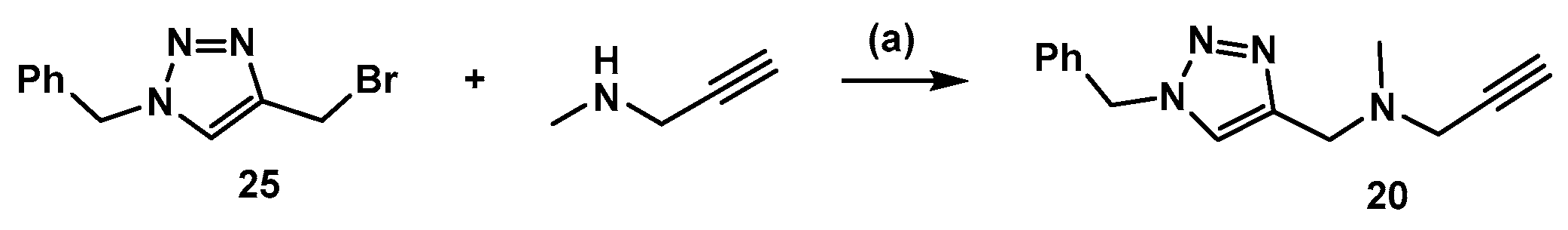
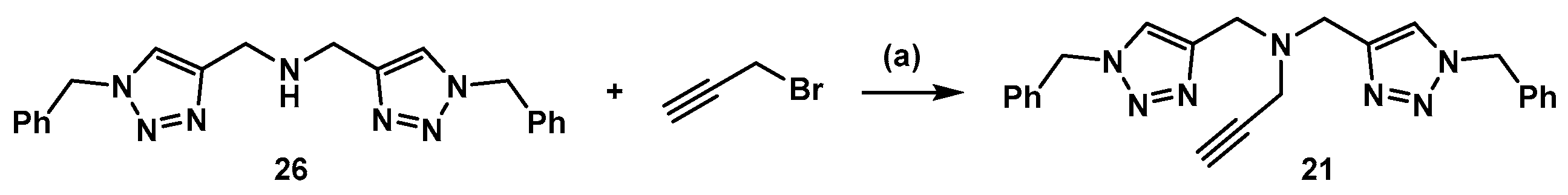

| Entry | Additive | 1H-NMR Yield b |
|---|---|---|
| 1 | TBTA (0.02 mmol) | 57% |
| 2 | None | 0 |
| 3 | 2-picoline (0.02 mmol) | 14% |
| 4 | TEA (0.02 mmol) | 0 |
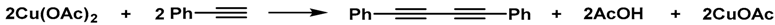
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
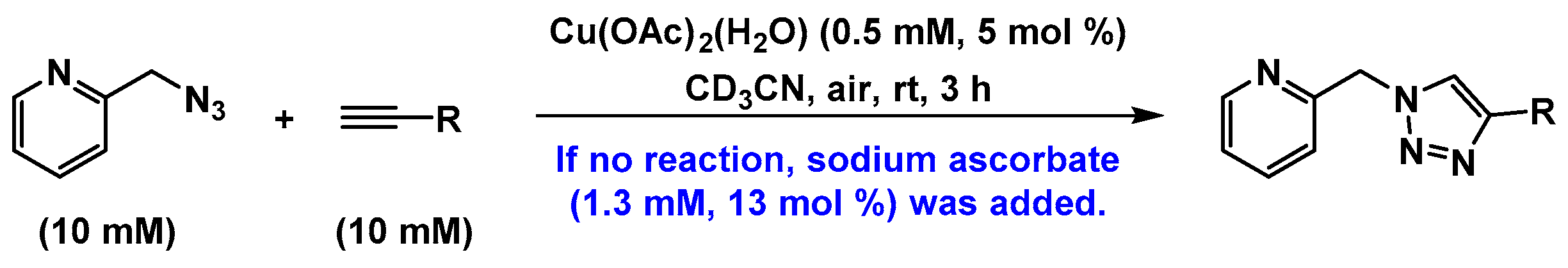{kind=link}
{kind=link}
{kind=link}
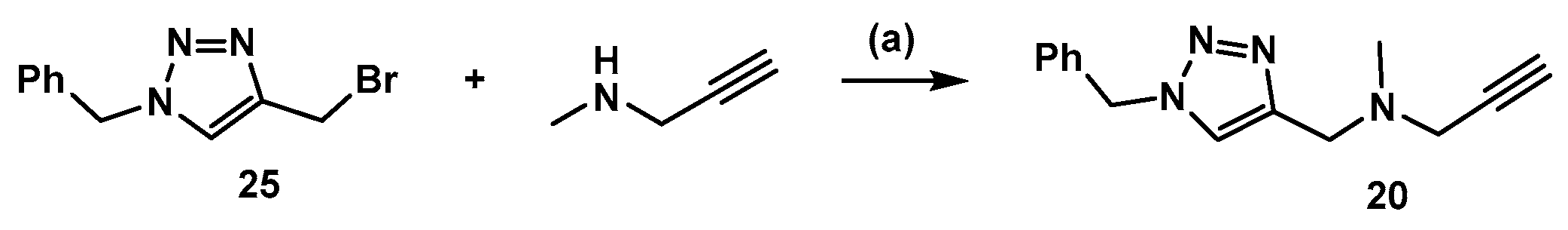{kind=link}
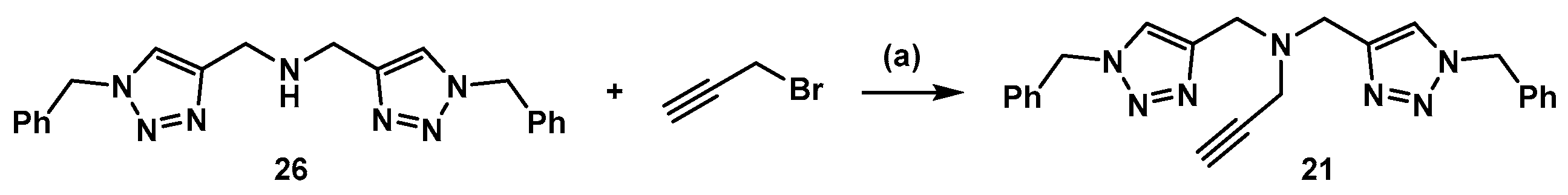{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Liu, P.; Zhu, L. Structural Determinants of Alkyne Reactivity in Copper-Catalyzed Azide-Alkyne Cycloadditions. Molecules 2016, 21, 1697. https://doi.org/10.3390/molecules21121697
Zhang X, Liu P, Zhu L. Structural Determinants of Alkyne Reactivity in Copper-Catalyzed Azide-Alkyne Cycloadditions. Molecules. 2016; 21(12):1697. https://doi.org/10.3390/molecules21121697
Chicago/Turabian StyleZhang, Xiaoguang, Peiye Liu, and Lei Zhu. 2016. "Structural Determinants of Alkyne Reactivity in Copper-Catalyzed Azide-Alkyne Cycloadditions" Molecules 21, no. 12: 1697. https://doi.org/10.3390/molecules21121697
APA StyleZhang, X., Liu, P., & Zhu, L. (2016). Structural Determinants of Alkyne Reactivity in Copper-Catalyzed Azide-Alkyne Cycloadditions. Molecules, 21(12), 1697. https://doi.org/10.3390/molecules21121697