
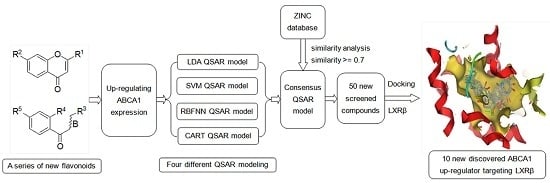
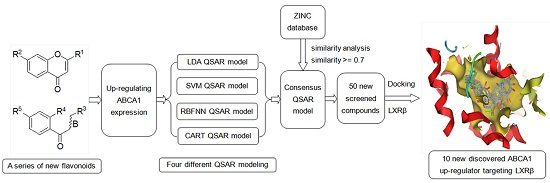
Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRβ Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Results of LDA Model
2.2. Interpretation of Descriptors
2.3. Results of SVM Model
2.4. Results of RBFNN Model
2.5. Results of CART Model
2.6. Comparison of Different Approaches and Consensus Modeling
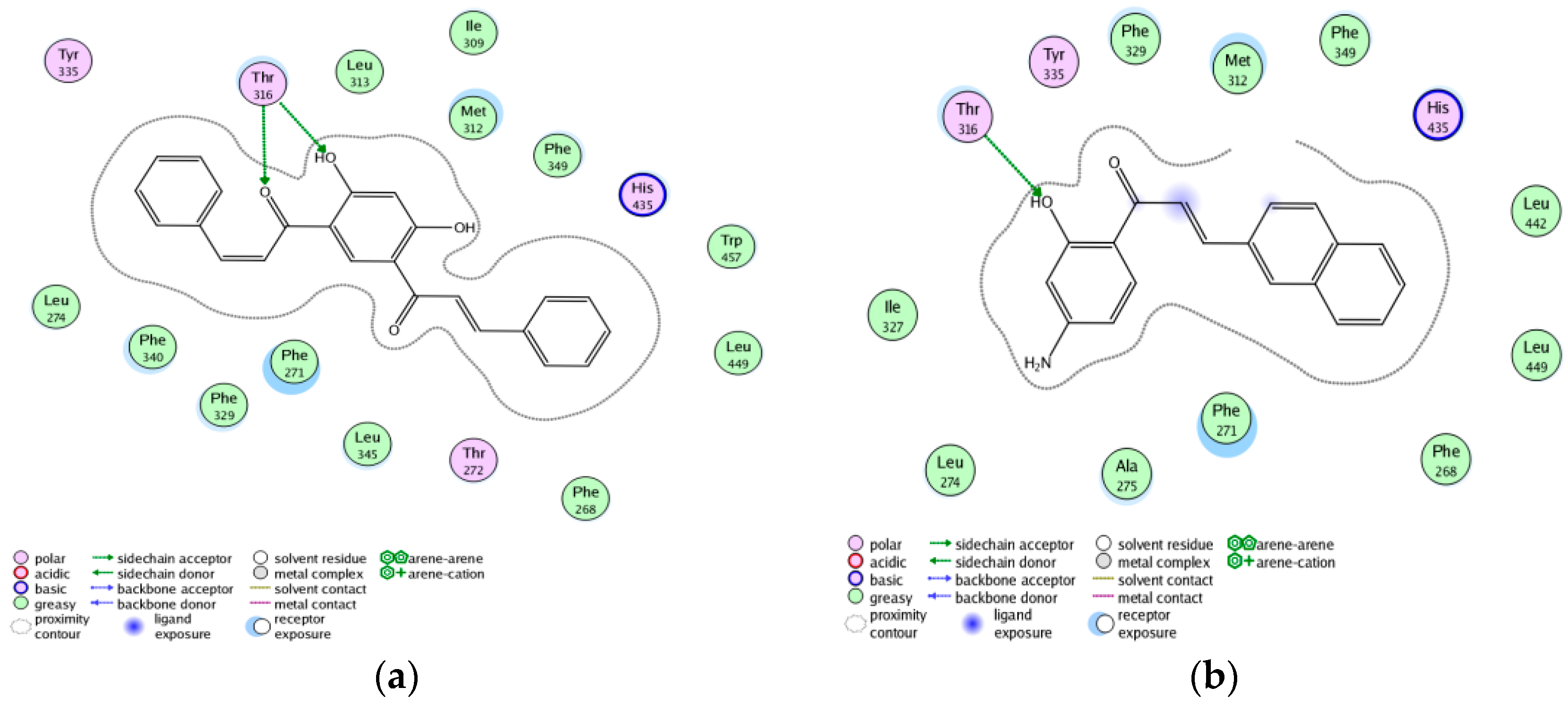
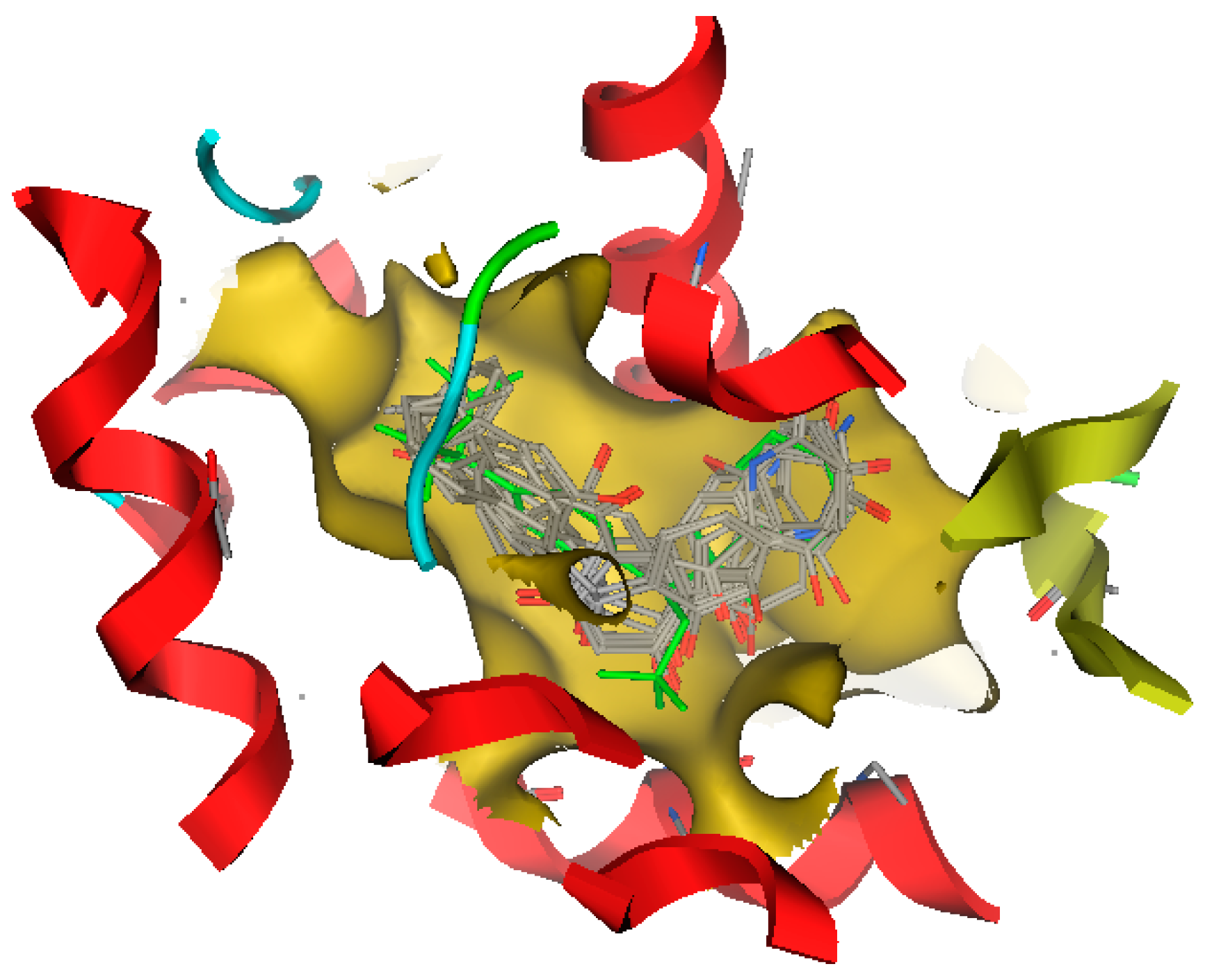
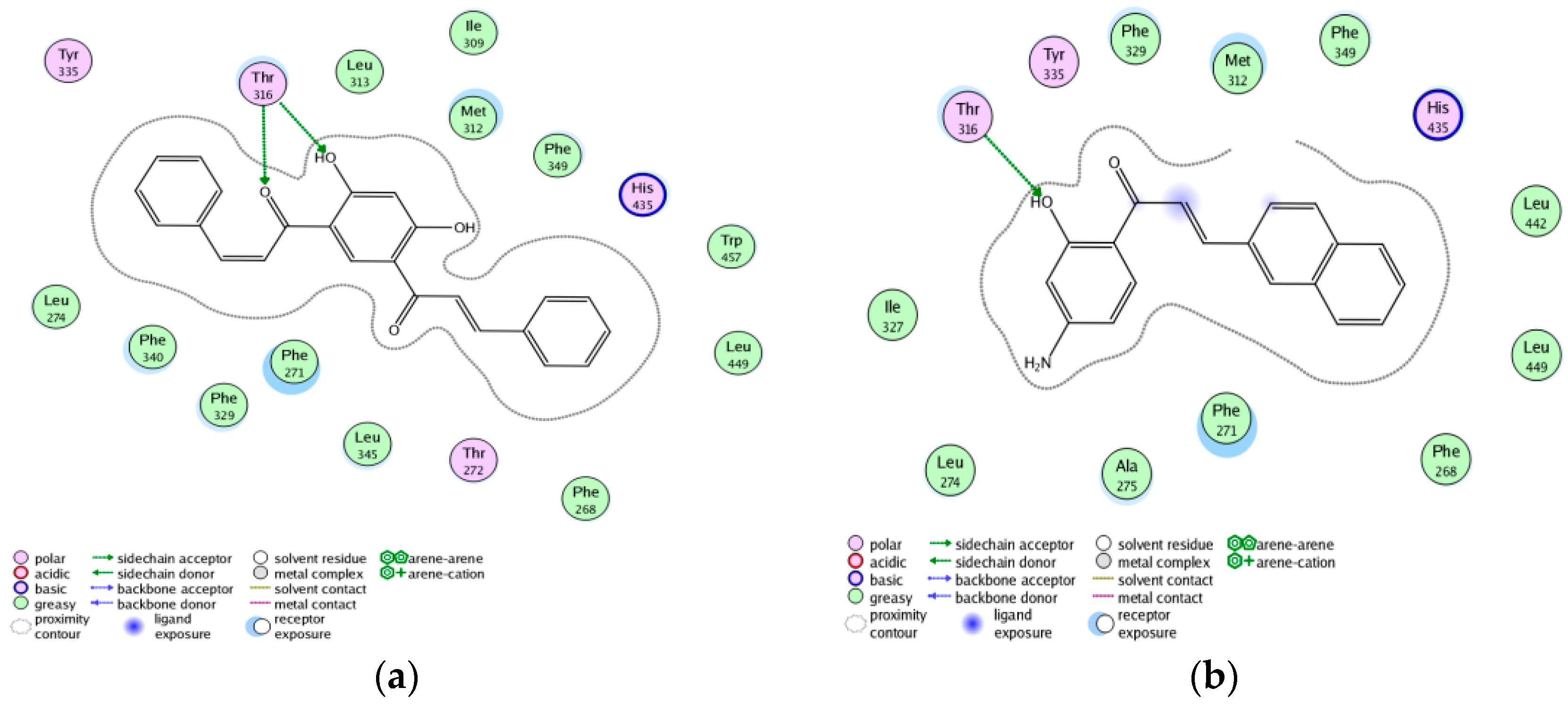
2.7. Screening New Highly-Potent ABCA1 Up-Regulators Targeting LXRβ
3. Materials and Methods
3.1. Dataset
3.2. Descriptor Calculation and Reduction
3.3. QSAR Modeling Approaches
3.3.1. Stepwise Linear Discriminant Analysis (SW-LDA)
3.3.2. Support Vector Machines (SVM)
3.3.3. Radial Basis Function Neural Network (RBFNN)
3.3.4. Classification and Regression Trees (CART)
3.4. Model Validation
3.5. Performance Measures
3.6. Screening New ABCA1 Up-Regulators
3.7. Exploring the Mechanism of ABCA1 Regulator by Molecular Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Oram, F.J.; Vaughan, A.M. ATP-Binding cassette cholesterol transporters and cardiovascular disease. Circ. Res. 2006, 99, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, E.W.; Piehler, A.; Wenzel, J.J. ABCA-subfamily transporters: Structure, function and disease. Biochim. Biophys. Acta 2006, 1762, 510–524. [Google Scholar] [CrossRef] [PubMed]
- Demina, E.P.; Miroshnikova, V.V.; Schwarzman, A.L. Role of the ABC transporters A1 and G1, key reverse cholesterol transport proteins, in atherosclerosis. Mol. Biol. 2016, 50, 193–199. [Google Scholar] [CrossRef]
- Bochem, A.E.; Wijk, D.F.V.; Duivenvoorden, R.; Holleboom, A.G.; de Groot, E.; Kuivenhoven, J.A.; Hovingh, G.K.; Nederveen, A.J.; Stroes, E. Abstract 12617: Mutations in ABCA1 are associated with increased atherosclerosis: A 3.0 tesla MRI study. Circulation 2011, 21, A12617. [Google Scholar]
- Arakawa, R.; Tsujita, M.; Iwamoto, N.; Itoohsumi, C.; Lu, R.; Wu, C.-A.; Shimizu, K.; Aotsuka, T.; Kanazawa, H.; Abe-Dohmae, S.; et al. Pharmacological inhibition of ABCA1 degradation increases HDL biogenesis and exhibits antiatherogenesis. J. Lipid Res. 2009, 50, 2299–2305. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, Y.; Yu, Y.; Wen, G.; Shang, N.; Zhuang, W.; Lu, D.; Zhou, B.; Liang, B.; Yue, X.; et al. Synthesis and identification of new flavonoids targeting liver X receptor β involved pathway as potential facilitators of Aβ clearance with reduced lipid accumulation. J. Med. Chem. 2013, 56, 6033–6053. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jiang, M.; Shui, Y.; Chen, Y.; Wang, Q.; Hu, W.; Ma, X.; Li, X.; Liu, X.; Cao, X.; et al. DNA topoisomerase II inhibitors induce macrophage ABCA1 expression and cholesterol efflux—An LXR-dependent mechanism. BBA Mol. Cell Biol. Lipids 2013, 1831, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Putz, A.M.; Lazea, M.; Ienciu, L.; Chiriac, A. Quantum-SAR extension of the spectral-SAR algorithm. Application to polyphenolic anticancer bioactivity. Int. J. Mol. Sci. 2009, 10, 1193–1214. [Google Scholar] [CrossRef] [PubMed]
- Maggiora, G.; Vogt, M.; Stumpfe, D.; Bajorath, J. Molecular similarity in medicinal chemistry. J. Med. Chem. 2014, 57, 3186–3204. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Dudas, N.A.; Isvoran, A. Double variational binding-(SMILES) conformational analysis by docking mechanisms for anti-HIV pyrimidine ligands. Int. J. Mol. Sci. 2015, 16, 19553–19601. [Google Scholar] [CrossRef] [PubMed]
- Jalali-Heravi, M.; Asadollahi-Baboli, M.; Shahbazikhah, P. QSAR study of heparanase inhibitors activity using artificial neural networks and Levenberg-Marquardt algorithm. Eur. J. Med. Chem. 2008, 43, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, G.; Crivori, P.; Carrupt, P.A.; Testa, B. Molecular fields in quantitative structure-permeation relationships: The VolSurf approach. J. Mol. Struct. THEOCHEM 2000, 503, 17–30. [Google Scholar] [CrossRef]
- Esposito, E.X.; Stouch, T.R.; Wymore, T.; Madura, J.D. Exploring the physicochemical properties of oxime-reactivation therapeutics for cyclosarin, sarin, tabun, and vx inactivated acetylcholinesterase. Chem. Res. Toxicol. 2014, 27, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Thai, K.M.; Nguyen, T.Q.; Ngo, T.D.; Tran, T.D.; Huynh, T.N. A support vector machine classification model for benzo[c]phenathridine analogues with toposiomerase-i inhibitory activity. Molecules 2012, 17, 4560–4582. [Google Scholar] [CrossRef] [PubMed]
- Lei, T.; Li, Y.; Song, Y.; Li, D.; Sun, H.; Hou, T. ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J. Cheminform. 2016, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Makhuri, F.R.; Ghasemi, J.B. Computer-aided scaffold hopping to identify a novel series of casein kinase 1 delta (CK1d) inhibitors for amyotrophic lateral sclerosis. Eur. J. Pharm. Sci. 2015, 78, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Vrontaki, E.; Melagraki, G.; Mavromoustakos, T.; Afantitis, A. Exploiting ChEMBL database to identify indole analogues as HCV replication inhibitors. Methods 2015, 71, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; Rahman, D.E.; Abdel Saleh, D.O.; Abdel Jaleel, G.A. Benzofuran-morpholinomethyl-pyrazoline hybrids as a new class of vasorelaxant agents: Synthesis and quantitative structure-activity relationship study. Chem. Pharm. Bull. 2014, 62, 1238–1251. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.T.; Liu, S.S.; Chen, F.; Xiao, Q.F.; Wu, Q.S. Chemometric model for predicting retention indices of constituents of essential oils. Chemosphere 2013, 90, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanzadeh, M.; Zhang, J.; Andersson, P.L. Binary classification model to predict developmental toxicity of industrial chemicals in zebrafish. J. Chemom. 2016, 30, 298–307. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S. Protein sub-nuclear localization based on effective fusion representations and dimension reduction algorithm LDA. Int. J. Mol. Sci. 2015, 16, 30343–30361. [Google Scholar] [CrossRef] [PubMed]
- Vapnik, V. Statistical Learning Theory; Wiley: New York, NY, USA, 1998. [Google Scholar]
- Cortes-Ciriano, I.; Murrell, D.S.; Westen, G.J.V.; Bender, A.; Malliavin, T.E. Prediction of the potency of mammalian cyclooxygenase inhibitors with ensemble proteochemometric modeling. J. Chemom. 2015, 7, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, Z.; Ghavami, R.; Kompany-Zareh, M. Radial basis function neural networks based on the projection pursuit and principal component analysis approaches: QSAR analysis of fullerene [C60]-based HIV-1 PR inhibitors. Med. Chem. Res. 2016, 25, 19–29. [Google Scholar] [CrossRef]
- Pirhadi, S.; Shiri, F.; Ghasemi, J.B. Multivariate statistical analysis methods in QSAR. RSC Adv. 2015, 5, 104635–104665. [Google Scholar] [CrossRef]
- Malek-Khatabi, A.; Kompany-Zareh, M.; Gholami, S.; Bagheri, S. Replacement based non-linear data reduction in radial basis function networks qsar modeling. Chemometr. Intell. Lab. Syst. 2014, 135, 157–165. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.; Stone, C.J.; Olshen, R.A. Classification and Regression Trees; CRC Press: Boca Raton, FL, USA, 1984. [Google Scholar]
- Kar, S. Development of classification- and regression-based QSAR models and screening of skin sensitisation potential of diverse organic chemicals. Mol. Simul. 2014, 40, 261–274. [Google Scholar]
- Sudhakaran, S.; Calvin, J.; Amy, G.L. QSAR models for the removal of organic micropollutants in four different river water matrices. Chemosphere 2012, 87, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Wegner, J.K.; Fröhlich, H.; Zell, A. Feature selection for descriptor based classification models. 2. Human intestinal absorption (HIA). J. Chem. Inf. Comput. Sci. 2004, 44, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Dejaegher, B.; Dhooghe, L.; Goodarzi, M.; Apers, S.; Pieters, L.; Heyden, Y.V. Classification models for neocryptolepine derivatives as inhibitors of the β-haematin formation. Anal. Chim. Acta 2011, 705, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.P.; Gupta, S.; Rai, P. Predicting acute aquatic toxicity of structurally diverse chemicals in fish using artificial intelligence approaches. Ecotoxicol. Environ. Saf. 2013, 95, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.; Latino, D.A.R.S.; Martins, F. Comparison of multiple linear regressions and neural networks based QSAR models for the design of new antitubercular compounds. Eur. J. Med. Chem. 2013, 70, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Khedr, M.A.; Shehata, T.M.; Mohamed, M.E. Repositioning of 2,4-dichlorophenoxy acetic acid as a potential anti-inflammatory agent: In silico and pharmaceutical formulation study. Eur. J. Pharm. Sci. 2014, 65, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


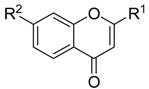
| Descriptor | Chemical Meaning | F to Remove | Wilks’ Lambda | Standardized Coefficient |
|---|---|---|---|---|
| a_nCl | Number of chlorine atoms | 7.391 | 0.479 | 0.707 |
| lip_don | The number of OH and NH atoms | 41.433 | 0.981 | −1.836 |
| vsurf_DD23 | Contact distances of lowest hydrophobic energy | 4.914 | 0.442 | 0.605 |
| vsurf_W2 | Hydrophilic volume | 14.746 | 0.587 | 1.243 |
| Descriptors | a_nCl | lip_don | vsurf_DD23 | vsurf_W2 |
|---|---|---|---|---|
| a_nCl | 1 | 0.150 | −0.078 | −0.047 |
| lip_don | 0.150 | 1 | 0.330 | 0.600 |
| vsurf_DD23 | −0.078 | 0.330 | 1 | 0.096 |
| vsurf_W2 | −0.047 | 0.600 | 0.096 | 1 |
| NO. | R1 | R2 | Fold Activation | Class a Exp. | Predicted Class | |||
|---|---|---|---|---|---|---|---|---|
| LDA | SVM | RBFNN | CART | |||||
| 1 * | quinolin-2-yl | OH | 1.36 | 0 | 0 | 0 | 0 | 0 |
| 2 | naphthalen-2-yl | OH | 1.57 | 0 | 0 | 0 | 0 | 0 |
| 3 | 1H-indol-3-yl | OH | 1.9 | 1 | 1 | 1 | 1 | 1 |
| 4 * | benzo[b]thiophen-3-yl | OH | 1.44 | 0 | 0 | 0 | 0 | 0 |
| 5 | 3-phenoxybenzen-1-yl | OH | 1.17 | 0 | 0 | 0 | 0 | 0 |
| 6 * | 4-carboxybenzen-1-yl | OH | 1.66 | 0 | 0 | 0 | 0 | 0 |
| 7 | (1,1′-biphenyl)-4-yl | OH | 1.15 | 0 | 0 | 0 | 0 | 0 |
| 8 | 5-(4-methoxyphenyl)thiophen-2-yl) | OH | 1.03 | 0 | 0 | 0 | 0 | 0 |
| 9 * | 5-methylfuran-2-yl | NH2 | 1.30 | 0 | 0 | 0 | 0 | 0 |
| 10 | 5-methylthiophen-2-yl | NH2 | 1.47 | 0 | 0 | 0 | 0 | 0 |
| 11 * | 4-isopropylbenzen-1-yl | OH | 1.22 | 0 | 0 | 0 | 0 | 0 |
| 12 | 4-ethoxyphenyl | NH2 | 1.37 | 0 | 0 | 0 | 0 | 0 |
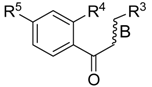
| NO. | R3 | R4 | R5 | B | Fold Activation | Class a Exp. | Predicted Class | |||
|---|---|---|---|---|---|---|---|---|---|---|
| LDA | SVM | RBFNN | CART | |||||||
| 13 | 5-methylfuran-2-yl | OH | NH2 | d | 2.09 | 1 | 1 | 1 | 1 | 1 |
| 14 | 5-methylthiophen-2-yl | OH | NH2 | d | 2.00 | 1 | 1 | 1 | 1 | 1 |
| 15 | 4-ethoxyphenyl | OH | NH2 | d | 1.92 | 1 | 1 | 1 | 1 | 1 |
| 16 | 4-(methylthio)phenyl | OH | NH2 | d | 1.26 | 0 | 0 | 0 | 0 | 0 |
| 17 * | 3-methoxyphenyl | OH | NH2 | d | 2.09 | 1 | 1 | 1 | 1 | 1 |
| 18 | 4-fluorophenyl | OH | NH2 | d | 1.36 | 0 | 0 | 0 | 0 | 0 |
| 19 | 4-chlorophenyl | OH | NH2 | d | 1.16 | 0 | 0 | 0 | 0 | 0 |
| 20 | 3,5-bis(trifluoromethyl)-phenyl | OH | NH2 | d | 1.12 | 0 | 0 | 0 | 0 | 0 |
| 21* | 5-methylfuran-2-yl | OH | NH2 | s | 1.90 | 1 | 1 | 1 | 1 | 1 |
| 22 | 3,4,5-trimethoxyphenyl | OH | NH2 | d | 0.98 | 0 | 1 | 0 | 0 | 0 |
| 23 | 3,4-dimethoxyphenyl | OH | NH2 | d | 1.86 | 1 | 0 | 0 | 0 | 1 |
| 24 | 4-isopropylphenyl | OH | NH2 | d | 2.05 | 1 | 1 | 1 | 1 | 1 |
| 25 | 4-ethylphenyl | OH | NH2 | d | 1.73 | 1 | 1 | 1 | 1 | 1 |
| 26 | (1,1'-biphenyl)-4-yl | OH | NH2 | d | 1.55 | 0 | 0 | 0 | 0 | 0 |
| 27 * | 3-phenoxybenzen-1-yl | OH | NH2 | d | 1.76 | 1 | 1 | 1 | 1 | 1 |
| 28 | 5-(4-methoxyphenyl)-thiophen-2-yl) | OH | NH2 | d | 1.09 | 0 | 0 | 0 | 0 | 0 |
| 29 | benzo[b]thiophen-3-yl | OH | NH2 | d | 2.31 | 1 | 1 | 1 | 1 | 1 |
| 30 * | 1H-indol-3-yl | OH | NH2 | d | 1.76 | 1 | 1 | 1 | 1 | 0 |
| 31 | naphthalen-2-yl | OH | NH2 | d | 2.64 | 1 | 1 | 1 | 1 | 1 |
| 32 | benzo[d][1,3]dioxol-5-yl | OH | NH2 | d | 1.92 | 1 | 1 | 1 | 1 | 1 |
| 33 | 5-methylfuran-2-yl | OCH3 | N(CH3)2 | d | 1.57 | 0 | 0 | 0 | 0 | 0 |
| 34 | naphthalen-2-yl | OCH3 | N(CH3)2 | d | 1.35 | 0 | 0 | 0 | 0 | 0 |
| 35 * | naphthalen-2-yl | OH | NH2 | s | 1.34 | 0 | 1 | 1 | 1 | 1 |
| 36 | naphthalen-2-yl | OCH3 | NH2 | d | 0.88 | 0 | 0 | 0 | 0 | 0 |
| 37 | 5-methylfuran-2-yl | OCH3 | NH2 | d | 1.20 | 0 | 0 | 0 | 0 | 0 |
| 38 | 5-methylthiophen-2-yl | OCH3 | NH2 | d | 1.50 | 0 | 0 | 0 | 0 | 0 |
| 39 | 4-ethoxyphenyl | OCH3 | NH2 | d | 1.06 | 0 | 0 | 0 | 0 | 0 |
| 40 | 5-methylfuran-2-yl | OH | N(CH3)2 | d | 1.02 | 0 | 1 | 0 | 0 | 0 |
| 41 * | 5-methylthiophen-2-yl | OH | CH3CONH | d | 1.08 | 0 | 0 | 0 | 0 | 0 |
{kind=link}
{kind=link}
{kind=link}
| Model | Accuracytrain | Accuracytest | AccuracyLOO | Total Accuracy | Sensitivity | Specificity |
|---|---|---|---|---|---|---|
| LDA | 90% | 90.91% | 83.33% | 90.24% | 90% | 90% |
| SVM | 96.67% | 90.91% | 86.67% | 95.12% | 90% | 100% |
| ANN | 96.67% | 90.91% | 83.33% | 95.12% | 90% | 100% |
| CART | 100% | 81.82% | 83.33% | 95.12% | 100% | 100% |
| Consensus | 96.67% | 90.91% | 83.33% | 95.12% | 90% | 100% |
| NO. | Structure | Docking Score | Similarity |
|---|---|---|---|
| Compound 36 | 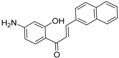 | −10.997 | 1 |
| Compound 19 | 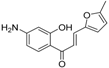 | −8.451 | 0.7 |
| ZINC39949652 | 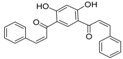 | −13.255 | 0.725 |
| ZINC08665430 | 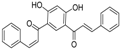 | −14.001 | 0.725 |
| ZINC3250227 | 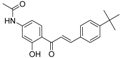 | −11.625 | 0.756 |
| ZINC32502236 | 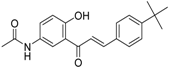 | −11.277 | 0.7 |
| ZINC05777271 | 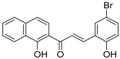 | −11.0604 | 0.723 |
| ZINC32502232 | 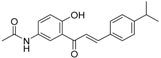 | −10.194 | 0.7 |
| ZINC05173700 | 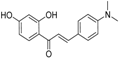 | −10.0822 | 0.8 |
| ZINC05211016 | 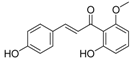 | −9.8112 | 0.707 |
| ZINC32502231 | 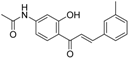 | −9.3668 | 0.756 |
| ZINC32502229 | 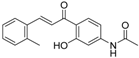 | −9.1047 | 0.747 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, M.; Yang, F.; Kang, J.; Yang, X.; Lai, X.; Gao, Y. Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRβ Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking. Molecules 2016, 21, 1639. https://doi.org/10.3390/molecules21121639
Chen M, Yang F, Kang J, Yang X, Lai X, Gao Y. Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRβ Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking. Molecules. 2016; 21(12):1639. https://doi.org/10.3390/molecules21121639
Chicago/Turabian StyleChen, Meimei, Fafu Yang, Jie Kang, Xuemei Yang, Xinmei Lai, and Yuxing Gao. 2016. "Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRβ Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking" Molecules 21, no. 12: 1639. https://doi.org/10.3390/molecules21121639
APA StyleChen, M., Yang, F., Kang, J., Yang, X., Lai, X., & Gao, Y. (2016). Multi-Layer Identification of Highly-Potent ABCA1 Up-Regulators Targeting LXRβ Using Multiple QSAR Modeling, Structural Similarity Analysis, and Molecular Docking. Molecules, 21(12), 1639. https://doi.org/10.3390/molecules21121639