
Antiproliferative Effect of Indole Phytoalexins



Abstract
:1. Introduction
2. Occurrence, Structure, and Biological Activity of Indole Phytoalexins
3. The Antiproliferative Effect of Naturally Occurring Indole Phytoalexins
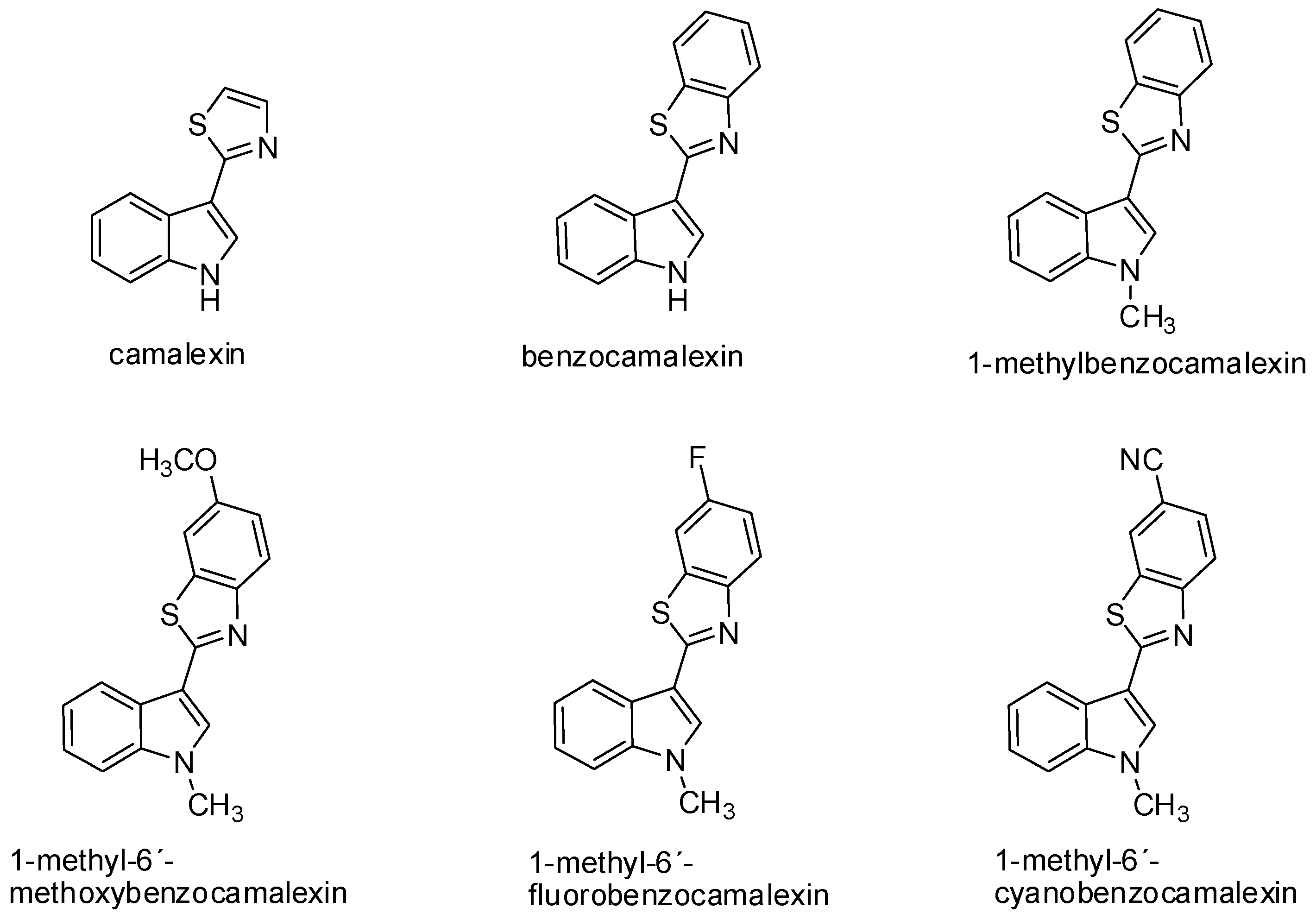
4. Antiproliferative Effect of Synthetic Derivatives of Indole Phytoalexins
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wachtel-Galor, S.; Benzie, I.F.F. Herbal Medicine: An Introduction to Its History, Usage, Regulation, Current Trends, and Research Needs. In Herbal Medicine: Biomolecular and Clinical Aspects, 2nd ed.; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011. [Google Scholar]
- Verhoeven, D.T.H.; Verhagen, H.; Goldbohm, R.A.; van den Brandt, P.A.; van Poppel, G. A review of mechanisms underlying anticarcinogenicity by brassica vegetables. Chem. Biol. Interact. 1997, 103, 79–129. [Google Scholar] [CrossRef]
- Herr, I.; Lozanovski, V.; Houben, P.; Schemmer, P.; Buchler, M.W. Sulforaphane and related mustard oils in focus of cancer prevention and therapy. Wien. Med. Wochenschr. 2013, 163, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Johnson, I.T. Glucosinolates: Bioavailability and importance to health. Int. J. Vitam. Nutr. Res. 2002, 72, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Brooks, J.D.; Paton, V.G.; Vidanes, G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer Epidemiol. Biomark. 2001, 10, 949–954. [Google Scholar]
- Fimognari, C.; Lenzi, M.; Hrelia, P. Interaction of the isothiocyanate sulforaphane with drug disposition and metabolism: Pharmacological and toxicological implications. Curr. Drug Metab. 2008, 9, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, F.; Paredes-Gonzalez, X.; Kong, A.T. Dietary Glucosinolates Sulforaphane, Phenethyl Isothiocyanate, Indole-3-Carbinol/3,3′-Diindolylmethane: Anti-Oxidative Stress/Inflammation, Nrf2, Epigenetics/Epigenomics and In Vivo Cancer Chemopreventive Efficacy. Curr. Pharmacol. Rep. 2015, 1, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Boreddy, S.R.; Sahu, R.P.; Srivastava, S.K. Benzyl isothiocyanate suppresses pancreatic tumor angiogenesis and invasion by inhibiting HIF-α/VEGF/Rho-GTPases: Pivotal role of STAT-3. PLoS ONE 2011, 6, e25799. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.S.; Perkins, S.N.; Hursting, S.D.; Young, H.A.; Kim, Y.S.; Wang, T.C.; Wang, T.T. Inhibition of androgen-responsive LNCaP prostate cancer cell tumor xenograft growth by dietary phenethyl isothiocyanate correlates with decreased angiogenesis and inhibition of cell attachment. Int. J. Oncol. 2012, 40, 1113–1121. [Google Scholar] [PubMed]
- Lai, K.C.; Hsu, S.C.; Kuo, C.L.; Ip, S.W.; Yang, J.S.; Hsu, Y.M.; Huang, H.Y.; Wu, S.H.; Chung, J.G. Phenethyl isothiocyanate inhibited tumor migration and invasion via suppressing multiple signal transduction pathways in human colon cancer HT29 cells. J. Agric. Food Chem. 2010, 58, 11148–11155. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.C.; Lu, C.C.; Tang, Y.J.; Chiang, J.H.; Kuo, D.H.; Chen, F.A.; Chen, I.L.; Yang, J.S. Allyl isothiocyanate inhibits cell metastasis through suppression of the MAPK pathways in epidermal growth factorstimulated HT29 human colorectal adenocarcinoma cells. Oncol. Rep. 2014, 31, 189–196. [Google Scholar] [PubMed]
- Cheung, K.L.; Kong, A.N. Molecular targets of dietary phenethyl isothiocyanate and sulforaphane for cancer chemoprevention. AAPS J. 2010, 12, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Wright, S.E.; Kim, S.H.; Srivastava, S.K. Phenethyl isothiocyanate: A comprehensive review of anti-cancer mechanisms. Biochim. Biophys. Acta 2014, 1846, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.Z.; Zhang, X.; Wu, L.X.; Wen, C.J.; Hu, L.; Lv, Q.L.; Shen, D.Y.; Zhou, H.H. Advances in molecular signaling mechanisms of beta-phenethyl isothiocyanate antitumor effects. J. Agric. Food Chem. 2015, 63, 3311–3322. [Google Scholar] [CrossRef] [PubMed]
- Gross, D. Phytoalexins of the Brassicaceae. J. Plant. Dis. Protect. 1993, 100, 433–442. [Google Scholar]
- Pedras, M.S.; Okanga, F.I.; Zaharia, I.L.; Khan, A.Q. Phytoalexins from crucifers: Synthesis, biosynthesis, and biotransformation. Phytochemistry 2000, 53, 161–176. [Google Scholar] [CrossRef]
- Pedras, M.S.C.; Jha, M.; Ahiahonu, P.W.K. The synthesis and biosynthesis of phytoalexins produced by cruciferous plants. Curr. Org. Chem. 2003, 7, 1635–1647. [Google Scholar] [CrossRef]
- Mezencev, R.; Mojzis, J.; Pilatova, M.; Kutschy, P. Anti-proliferative and cancer chemopreventive activity of phytoalexins: Focus on indole phytoalexins from crucifers. Neoplasma 2003, 50, 239–245. [Google Scholar] [PubMed]
- Pedras, M.S.C.; Zheng, Q.A.; Sarma-Mamillapalle, V.K. The phytoalexins from Brassicaceae: Structure, biological activity, synthesis and biosynthesis. Nat. Prod. Commun. 2007, 2, 319–330. [Google Scholar]
- Pedras, M.S.; Yaya, E.E. Phytoalexins from Brassicaceae: News from the front. Phytochemistry 2010, 71, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Pilatova, M.; Sarissky, M.; Kutschy, P.; Mirossay, A.; Mezencev, R.; Curillova, Z.; Suchy, M.; Monde, K.; Mirossay, L.; Mojzis, J. Cruciferous phytoalexins: Anti-proliferative effects in T-Jurkat leukemic cells. Leuk. Res. 2005, 29, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Pilatova, M.; Ivanova, L.; Kutschy, P.; Varinska, L.; Saxunova, L.; Repovska, M.; Sarissky, M.; Seliga, R.; Mirossay, L.; Mojzis, J. In vitro toxicity of camalexin derivatives in human cancer and non-cancer cells. Toxicol. In Vitro 2013, 27, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Chripkova, M.; Drutovic, D.; Pilatova, M.; Mikes, J.; Budovska, M.; Vaskova, J.; Broggini, M.; Mirossay, L.; Mojzis, J. Brassinin and its derivatives as potential anticancer agents. Toxicol. In Vitro 2014, 28, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Montaut, S.; Suchy, M. Phytoalexins from the crucifer rutabaga: Structures, syntheses, biosyntheses, and antifungal activity. J. Org. Chem. 2004, 69, 4471–4476. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Galizzi, M.; Kutschy, P.; Docampo, R. Trypanosoma cruzi: Anti-proliferative effect of indole phytoalexins on intracellular amastigotes in vitro. Exp. Parasitol. 2009, 122, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.G.; Liu, J.; Constantinou, A.; Thomas, C.F.; Hawthorne, M.; You, M.; Gerhuser, C.; Pezzuto, J.M.; Moon, R.C.; Moriarty, R.M. Cancer chemopreventive activity of brassinin, a phytoalexin from cabbage. Carcinogenesis 1995, 16, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Duhadaway, J.B.; Gaspari, P.; Sutanto-Ward, E.; Munn, D.H.; Mellor, A.L.; Malachowski, W.P.; Prendergast, G.C.; Muller, A.J. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene 2008, 27, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.A.; Mansfield, J.W. Phytoalexins; Blackie: Glasgow, UK, 1982. [Google Scholar]
- Dixon, R.A.; Lamb, C.J. Molecular Communication in Interactions between Plants and Microbial Pathogens. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1990, 41, 339–367. [Google Scholar] [CrossRef]
- Daniel, M.; Purkayastha, R.P. Handbook of Phytoalexin Metabolism and Action; CRC Press: Boca Raton, FL, USA, 1994. [Google Scholar]
- Heil, M.; Bostock, R.M. Induced systemic resistance (ISR) against pathogens in the context of induced plant defences. Ann. Bot. 2002, 89, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.O.; Börger, H. Experimentelle Untersuchungen Über die Phytophthorainfestans—Resistenz der Kartoffel. Arb. Biol. Reichsanst. Land Forstwirtsch 1940, 97, 189–231. [Google Scholar]
- Cruickshank, I.A.; Perrin, D.R. Isolation of a phytoalexin from Pisum sativum L. Nature 1960, 187, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Ahiahonu, P.W. Metabolism and detoxification of phytoalexins and analogs by phytopathogenic fungi. Phytochemistry 2005, 66, 391–411. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Katsui, N.; Shirata, A. Isolation of three Novel Sulfur-Containing Phytoalexins from the Chinese-Cabbage Brassica campestris L. Ssp pekinensis (Cruciferae). J. Chem. Soc. Chem. Commun. 1986, 14, 1077–1078. [Google Scholar] [CrossRef]
- Kutschy, P.; Dzurilla, M.; Takasugi, M.; Sabova, A. Synthesis of some analogs of indole phytoalexins brassinin and methoxybrassenin B and their positional isomers. Collect. Czech. Chem. C 1999, 64, 348–362. [Google Scholar] [CrossRef]
- Mehta, R.G.; Naithani, R.; Huma, L.; Hawthorne, M.; Moriarty, R.M.; McCormick, D.L.; Steele, V.E.; Kopelovich, L. Efficacy of chemopreventive agents in mouse mammary gland organ culture (MMOC) model: A comprehensive review. Curr. Med. Chem. 2008, 15, 2785–2825. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Okinyo, D.P. Remarkable incorporation of the first sulfur containing indole derivative: Another piece in the biosynthetic puzzle of crucifer phytoalexins. Org. Biomol. Chem. 2008, 6, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Chumala, P.B.; Suchy, M. Phytoalexins from Thlaspi arvense, a wild crucifer resistant to virulent Leptosphaeria maculans: Structures, syntheses and antifungal activity. Phytochemistry 2003, 64, 949–956. [Google Scholar] [CrossRef]
- Pedras, M.S.; Suchy, M.; Ahiahonu, P.W. Unprecedented chemical structure and biomimetic synthesis of erucalexin, a phytoalexin from the wild crucifer Erucastrum gallicum. Org. Biomol. Chem. 2006, 4, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Adio, A.M. Phytoalexins and phytoanticipins from the wild crucifers Thellungiella halophila and Arabidopsis thaliana: Rapalexin A, wasalexins and camalexin. Phytochemistry 2008, 69, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Zheng, Q.A.; Sarwar, M.G. Efficient synthesis of brussalexin A, a remarkable phytoalexin from Brussels sprouts. Org. Biomol. Chem. 2007, 5, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Zheng, Q.A.; Strelkov, S. Metabolic changes in roots of the oilseed canola infected with the biotroph Plasmodiophora brassicae: Phytoalexins and phytoanticipins. J. Agric. Food Chem. 2008, 56, 9949–9961. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Monde, K.; Katsui, N.; Shirata, A. Spirobrassinin, a Novel Sulfur-Containing Phytoalexin from the Daikon Rhaphanus sativus L. var. hortensis (Cruciferae). Chem. Lett. 1987, 8, 1631–1632. [Google Scholar] [CrossRef]
- Keplinger, K.; Laus, G.; Wurm, M.; Dierich, M.P.; Teppner, H. Uncaria tomentosa (Willd.) DC.—Ethnomedicinal use and new pharmacological, toxicological and botanical results. J. Ethnopharmacol. 1999, 64, 23–34. [Google Scholar] [CrossRef]
- Falkiewicz, B.; Lukasiak, J. Uncaria tomentosa (Willd) DC. and Uncaria guianensis (Aublet) Gmell.—A review of published scientific literature. Case Rep. Clin. Pract. Rev. 2001, 2, 305–316. [Google Scholar]
- Glawischnig, E. Camalexin. Phytochemistry 2007, 68, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Glawischnig, E. The role of cytochrome P450 enzymes in the biosynthesis of camalexin. Biochem. Soc. Trans. 2006, 34, 1206–1208. [Google Scholar] [CrossRef] [PubMed]
- Leroux, P. Modes of action of agrochemicals against plant pathogenic organisms. C. R. Biol. 2003, 326, 9–21. [Google Scholar] [CrossRef]
- Navneetha, O.; Saritha Jyostna, T. Therapeutic potentials of brassinin and its analogues—A review. Adv. J. Pharm. Life Sci. Res. 2016, 4, 76–82. [Google Scholar]
- Cao, S.L.; Feng, Y.P.; Jiang, Y.Y.; Liu, S.Y.; Ding, G.Y.; Li, R.T. Synthesis and in vitro antitumor activity of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains. Bioorg. Med. Chem. Lett. 2005, 15, 1915–1917. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, F.; Yu, X.; Ding, G.; Xu, P.; Cao, J.; Jiang, Y. The 3D-QSAR analysis of 4(3H)-quinazolinone derivatives with dithiocarbamate side chains on thymidylate synthase. Bioorg. Med. Chem. 2006, 14, 1425–1430. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.C.; Ma, Y.C.; Zhang, E.; Shi, X.J.; Wang, M.M.; Ye, X.W.; Liu, H.M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anti-cancer agents. Eur. J. Med. Chem. 2013, 62, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Navneetha, O.; Anuradha, B.S.; Sandhya, M.S.N.; Sree Kanth, S.; Vijjulatha, M.; Saritha Jyostna, T. Bioisosteres of brassinin: Synthesis, molecular docking and chemotherapeutic activity. Indo Am. J. Pharm. Sci. 2016, 6, 4070–4079. [Google Scholar]
- Xue, W.; Meylan, E.; Oliver, T.G.; Feldser, D.M.; Winslow, M.M.; Bronson, R.; Jacks, T. Response and resistance to NF-κB inhibitors in mouse models of lung adenocarcinoma. Cancer Discov. 2011, 1, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Ghosh, S. Toll-like receptor-mediated NF-κB activation: A phylogenetically conserved paradigm in innate immunity. J. Clin. Investig. 2001, 107, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.S.; Noh, Y.S.; Lee, Y.S.; Cho, Y.W.; Baek, N.I.; Choi, M.S.; Jeong, T.S.; Kang, E.; Chung, H.G.; Lee, K.T. Arvelexin from Brassica rapa suppresses NF-κB-regulated pro-inflammatory gene expression by inhibiting activation of I κB kinase. Br. J. Pharmacol. 2011, 164, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Sabol, M.; Kutschy, P.; Siegfried, L.; Mirossay, A.; Suchy, M.; Hrbkova, H.; Dzurilla, M.; Maruskova, R.; Starkova, J.; Paulikova, E. Cytotoxic effect of cruciferous phytoalexins against murine L1210 leukemia and B16 melanoma. Biologia 2000, 55, 701–707. [Google Scholar]
- Mehta, R.G.; Liu, J.; Constantinou, A.; Hawthorne, M.; Pezzuto, J.M.; Moon, R.C.; Moriarty, R.M. Structure-activity relationships of brassinin in preventing the development of carcinogen-induced mammary lesions in organ culture. Anti Cancer Res. 1994, 14, 1209–1213. [Google Scholar]
- Tempete, C.; Devys, M.; Barbier, M. Growth inhibitions on human cancer cell cultures with the indole sulphur-containing phytoalexins and their analogues. Z. Naturf. C J. Biosci. 1991, 46, 706–707. [Google Scholar]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2887. [Google Scholar] [CrossRef] [PubMed]
- Botting, N.P. Chemistry and neurochemistry of the kynurenine pathway of tryptophan metabolism. Chem. Soc. Rev. 1995, 24, 401. [Google Scholar] [CrossRef]
- Sono, M.; Taniguchi, T.; Watanabe, Y.; Hayaishi, O. Indoleamine 2,3-dioxygenase. Equilibrium studies of the tryptophan binding to the ferric, ferrous, and CO-bound enzymes. J. Biol. Chem. 1980, 255, 1339–1345. [Google Scholar] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, P.; Banerjee, T.; Malachowski, W.P.; Muller, A.J.; Prendergast, G.C.; DuHadaway, J.; Bennett, S.; Donovan, A.M. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J. Med. Chem. 2006, 49, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Izutani, Y.; Yogosawa, S.; Sowa, Y.; Sakai, T. Brassinin induces G1 phase arrest through increase of p21 and p27 by inhibition of the phosphatidylinositol 3-kinase signaling pathway in human colon cancer cells. Int. J. Oncol. 2012, 40, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Park, J.H.; Kim, K.D.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H.; Ahn, K.S. Brassinin induces apoptosis in PC-3 human prostate cancer cells through the suppression of PI3K/Akt/mTOR/S6K1 signaling cascades. Phytother. Res. 2014, 28, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Zhang, Z.; Jiang, B.H.; Shi, X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem. Biophys. Res. Commun. 2003, 310, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386–6405. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N. STATs and MAPKs: Obligate or opportunistic partners in signaling. BioEssays News Rev. Mol. Cell. Dev. Biol. 1996, 18, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Grandis, J.R.; Drenning, S.D.; Chakraborty, A.; Zhou, M.Y.; Zeng, Q.; Pitt, A.S.; Tweardy, D.J. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J. Clin. Investig. 1998, 102, 1385–1392. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernandez-Luna, J.L.; Nunez, G.; et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.M.; Yang-Yen, H.F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar] [PubMed]
- Zimmer, S.; Kahl, P.; Buhl, T.M.; Steiner, S.; Wardelmann, E.; Merkelbach-Bruse, S.; Buettner, R.; Heukamp, L.C. Epidermal growth factor receptor mutations in non-small cell lung cancer influence downstream Akt, MAPK and Stat3 signaling. J. Cancer Res. Clin. Oncol. 2009, 135, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS ONE 2012, 7, e30820. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Oh, E.Y.; Lee, J.H.; Nam, D.; Lee, S.G.; Lee, J.; Kim, S.H.; Shim, B.S.; Ahn, K.S. Brassinin Combined with Capsaicin Enhances Apoptotic and Anti-metastatic Effects in PC-3 Human Prostate Cancer Cells. Phytother. Res. 2015, 29, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Sanchez, M.G.; Malagarie-Cazenave, S.; Olea, N.; Diaz-Laviada, I. Induction of apoptosis in prostate tumor PC-3 cells and inhibition of xenograft prostate tumor growth by the vanilloid capsaicin. Apoptosis 2006, 11, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Kello, M.; Drutovic, D.; Chripkova, M.; Pilatova, M.; Budovska, M.; Kulikova, L.; Urdzik, P.; Mojzis, J. ROS-dependent anti-proliferative effect of brassinin derivative homobrassinin in human colorectal cancer Caco2 cells. Molecules 2014, 19, 10877–10897. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.K.; Lund, E.K.; Parker, M.L.; Clarke, R.G.; Johnson, I.T. Allyl-isothiocyanate causes mitotic block, loss of cell adhesion and disrupted cytoskeletal structure in HT29 cells. Carcinogenesis 2004, 25, 1409–1415. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Mojžiš, J.; Pilátová, M.; Kutschy, P.; Čurillová, Z. Effects of indole phytoalexins from cruciferous plants on the growth of cancer cells.Implications for cancer chemoprevention and chemotherapy. Int. J. Canc. Prev. 2004, 1, 105–112. [Google Scholar]
- Moody, C.J.; Roffey, J.R.; Stephens, M.A.; Stratford, I.J. Synthesis and cytotoxic activity of indolyl thiazoles. Anti Cancer Drugs 1997, 8, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Neal, C.L.; Chetram, M.; Vo, B.; Mezencev, R.; Hinton, C.; Odero-Marah, V.A. The phytoalexin camalexin mediates cytotoxicity towards aggressive prostate cancer cells via reactive oxygen species. J. Nat. Med. 2013, 67, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Updegrove, T.; Kutschy, P.; Repovska, M.; McDonald, J.F. Camalexin induces apoptosis in T-leukemia Jurkat cells by increased concentration of reactive oxygen species and activation of caspase-8 and caspase-9. J. Nat. Med. 2011, 65, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Randle, D.; Mezencev, R.; Thomas, L.; Hinton, C.; Odero-Marah, V. Camalexin-induced apoptosis in prostate cancer cells involves alterations of expression and activity of lysosomal protease cathepsin D. Molecules 2014, 19, 3988–4005. [Google Scholar] [CrossRef] [PubMed]
- Kutschy, P.; Sykora, A.; Curillova, Z.; Repovska, M.; Pilatova, M.; Mojzis, J.; Mezencev, R.; Pazdera, P.; Hromjakova, T. Glyoxyl Analogs of Indole Phytoalexins: Synthesis and Anti-cancer Activity. Collect. Czech. Chem. C 2010, 75, 887–903. [Google Scholar] [CrossRef]
- Kobayashi, J.; Murayama, T.; Ishibashi, M.; Kosuge, S.; Takamatsu, M.; Ohizumi, Y.; Kobayashi, H.; Ohta, T.; Nozoe, S.; Sasaki, T. Hyrtiosin-a and Hyrtiosin-B, New Indole Alkaloids from the Okinawan Marine Sponge Hyrtios erecta. Tetrahedron 1990, 46, 7699–7702. [Google Scholar] [CrossRef]
- Bacher, G.; Nickel, B.; Emig, P.; Vanhoefer, U.; Seeber, S.; Shandra, A.; Klenner, T.; Beckers, T. D-24851, a novel synthetic microtubule inhibitor, exerts curative antitumoral activity in vivo, shows efficacy toward multidrug-resistant tumor cells, and lacks neurotoxicity. Cancer Res. 2001, 61, 392–399. [Google Scholar] [PubMed]
- Kutschy, P.; Salayova, A.; Curillova, Z.; Kozar, T.; Mezencev, R.; Mojzis, J.; Pilatova, M.; Balentova, E.; Pazdera, P.; Sabol, M.; et al. 2-(Substituted phenyl)amino analogs of 1-methoxyspirobrassinol methyl ether: Synthesis and anti-cancer activity. Bioorg. Med. Chem. 2009, 17, 3698–3712. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Kutschy, P.; Salayova, A.; Curillova, Z.; Mojzis, J.; Pilatova, M.; McDonald, J. Anti-cancer properties of 2-piperidyl analogues of the natural indole phytoalexin 1-methoxyspirobrassinol. Chemotherapy 2008, 54, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Mezencev, R.; Kutschy, P.; Salayova, A.; Updegrove, T.; McDonald, J.F. The design, synthesis and anti-cancer activity of new nitrogen mustard derivatives of natural indole phytoalexin 1-methoxyspirobrassinol. Neoplasma 2009, 56, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Budovska, M.; Pilatova, M.; Varinska, L.; Mojzis, J.; Mezencev, R. The synthesis and anti-cancer activity of analogs of the indole phytoalexins brassinin, 1-methoxyspirobrassinol methyl ether and cyclobrassinin. Bioorg. Med. Chem. 2013, 21, 6623–6633. [Google Scholar] [CrossRef] [PubMed]
- Curillova, Z.; Kutschy, P.; Solcaniova, E.; Pilatova, M.; Mojzis, J.; Kovacik, V. Synthesis and anti-proliferative activity of 1-methoxy-, 1-(α-d-ribofuranosyl)- and 1-(β-d-ribofuranosyl)brassenin B. ARKIVOC 2008, 8, 85–104. [Google Scholar]
- Kojiri, K.; Arakawa, H.; Satoh, F.; Kawamura, K.; Okura, A.; Suda, H.; Okanishi, M. New antitumor substances, BE-12406A and BE-12406B, produced by a streptomycete. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J. Antibiot. 1991, 44, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, T.; Ohkubo, M.; Fukasawa, K.; Egashira, S.; Hara, Y.; Matsumoto, M.; Nakai, K.; Arakawa, H.; Morishima, H.; Nishimura, S. Mode of action of a new indolocarbazole anti-cancer agent, J-107088, targeting topoisomerase I. Cancer Res. 1999, 59, 4271–4275. [Google Scholar] [PubMed]
- Arakawa, H.; Morita, M.; Kodera, T.; Okura, A.; Ohkubo, M.; Morishima, H.; Nishimura, S. In vivo anti-tumor activity of a novel indolocarbazole compound, J-107088, on murine and human tumors transplanted into mice. Jpn. J. Cancer Res. 1999, 90, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Csomos, P.; Zupko, I.; Rethy, B.; Fodor, L.; Falkay, G.; Bernath, G. Isobrassinin and its analogues: Novel types of anti-proliferative agents. Bioorg. Med. Chem. Lett. 2006, 16, 6273–6276. [Google Scholar] [CrossRef] [PubMed]
- Monde, K.; Taniguchi, T.; Miura, N.; Kutschy, P.; Curillova, Z.; Pilatova, M.; Mojzis, J. Chiral cruciferous phytoalexins: Preparation, absolute configuration, and biological activity. Bioorg. Med. Chem. 2005, 13, 5206–5212. [Google Scholar] [CrossRef] [PubMed]
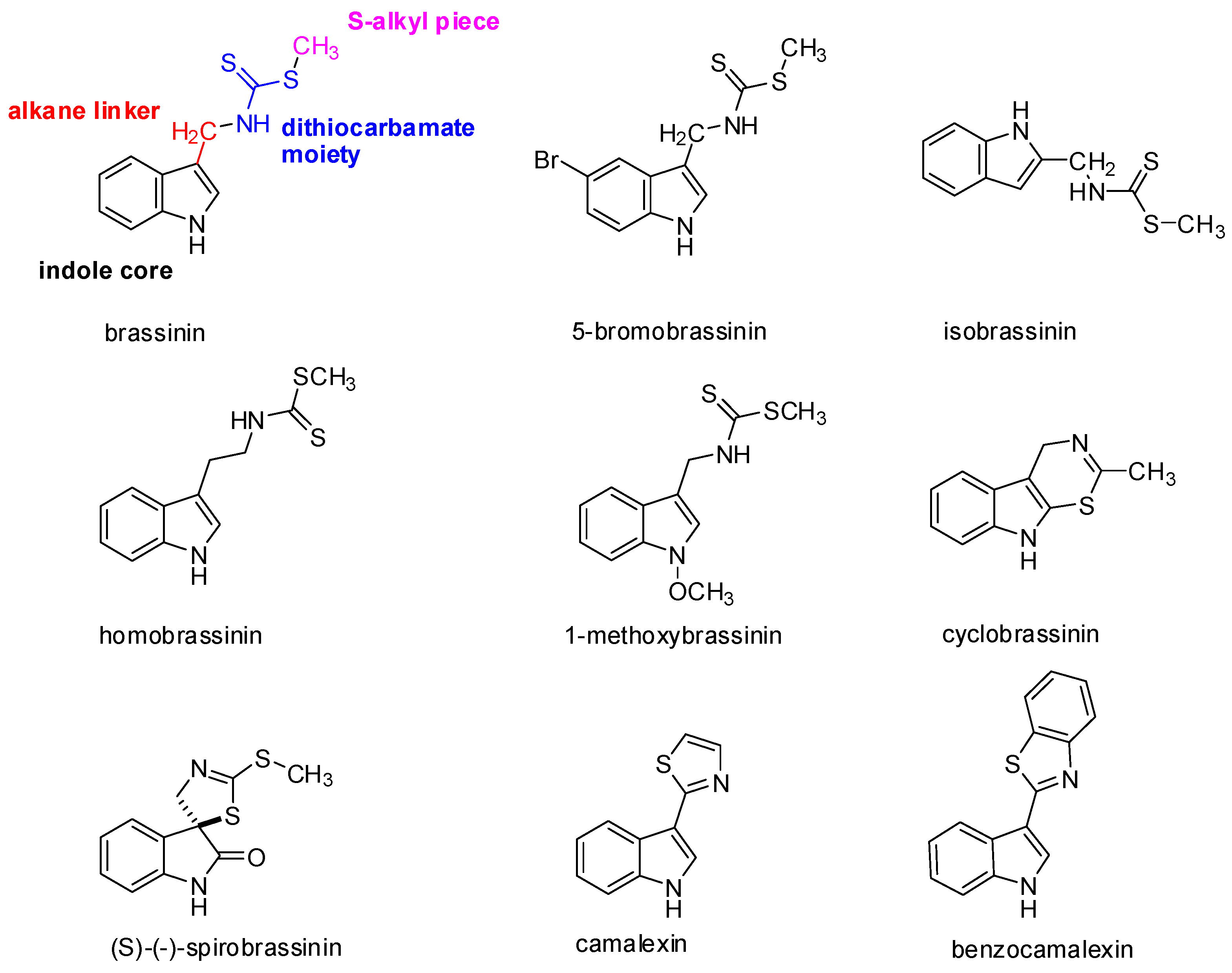

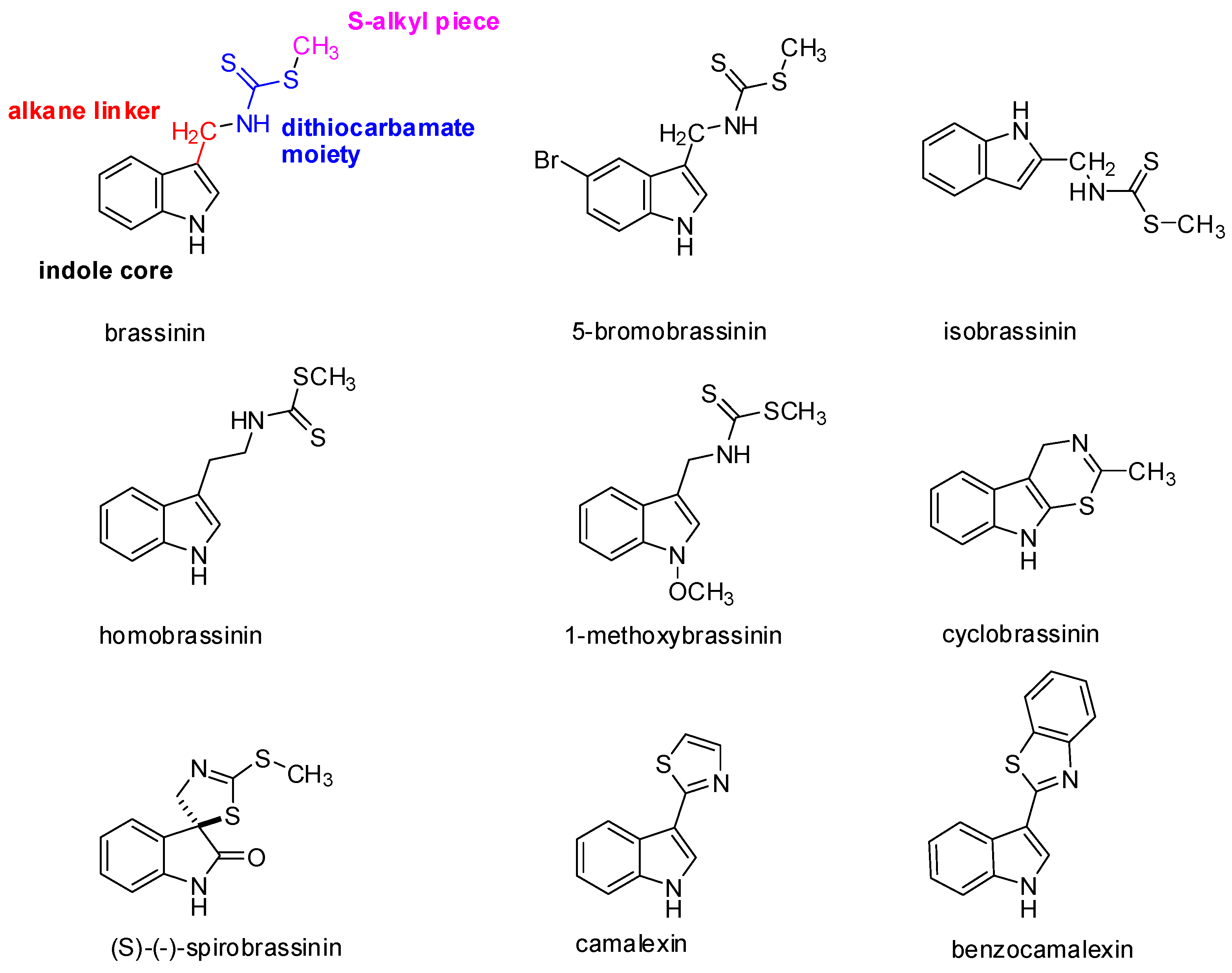{kind=link}
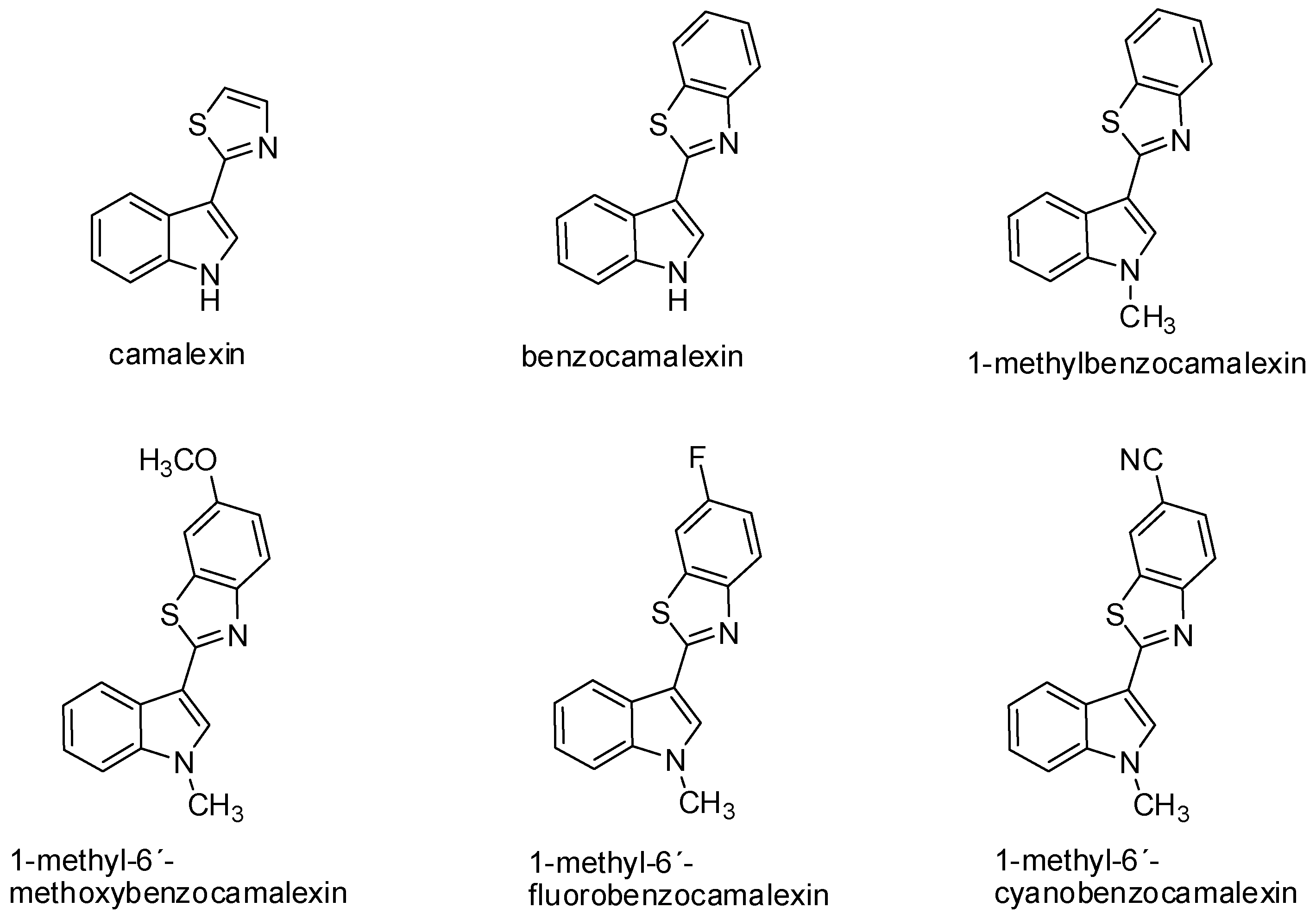{kind=link}
| Indole Phytoalexin | Possible Anti-Cancer Properties | Reference |
|---|---|---|
| Brassinin | Reduces the cell growth of mouse melanoma (B16) and leukemic cancer cell line (L1210) | [59] |
| Exhibits cancer chemopreventive activity: inhibits the formation of preneoplastic mammary lesions in culture | [27] | |
| Induces phase II enzymes that metabolically inactivate chemical carcinogens | ||
| Enhances the effectiveness of tumor immunotherapy by blocking indoleamine 2,3-dioxygenase (IDO), the enzyme that drives immune escape in cancer | [65] | |
| Induces G1 phase arrest through increase of p21 and p27 by inhibition of the PI3K signaling pathway in human colon cancer cells (HT-29) | [67] | |
| Induces apoptosis in human prostate cancer cells (PC-3) through the suppression of PI3K/Akt/mTOR/S6K1 signaling cascades | [69] | |
| Inhibits STAT3 signaling through modulation of PIAS-3 and SOCS-3, thereby reducing tumor cell growthEnhances the antitumor effects of paclitaxel in human lung cancer xenograft in nude mice | [70] | |
| In combination with capsaicin, enhances apoptotic and anti-metastatic effects in human prostate cancer cells (PC-3) | [78] | |
| Potentiates vincristine cytotoxicity to U-87 MG (human glioblastoma astrocytoma) | [82] | |
| Isobrassinin | Antiproliferative effect on cervical carcinoma (HeLa), breast carcinoma (MCF-7), and epidermoid carcinoma (A431) cell lines | [98] |
| 5-Bromobrassinin | Suppresses growth of B16-F10 melanoma xenografts in C57BL/6 mice by inhibiting IDO enzyme | [28] |
| Homobrassinin | Induces mitotic phase arrest via inhibition of microtubule formation (dysregulation of α-tubulin, α1-tubulin, and β5-tubulin expression) in colorectal cancer cells (Caco-2) | [80] |
| Induction of apoptosis in Caco-2 is associated with the loss of mitochondrial membrane potential, caspase-3 activation as well as intracellular reactive oxygen species (ROS) production. | ||
| 1-Methoxybrassinin | Exhibits antiproliferative effects on the human acute T lymphoblastic leukemia cell line (Jurkat) IC50 10 μmol/L | [22] |
Induces apoptosis in Caco-2 cells, which is associated with the:
| [24] | |
| Cyclobrassinin | Exhibits antiproliferative effects on the epidermoid carcinoma cell line (KB) IC50 8 μg/mL | [61] |
| Exhibits cancer chemopreventive activity: inhibits the formation of preneoplastic mammary lesions in culture | [27] | |
| Induces phase II enzymes that metabolically inactivate chemical carcinogens | ||
| Spirobrassinin | Reduces the cell growth of mouse melanoma (B16) and the leukemic cancer cell line (L1210) | [59] |
| Exhibits cancer chemopreventive activity: inhibits the formation of preneoplastic mammary lesions in culture | [27] | |
| Induces phase II enzymes that metabolically inactivate chemical carcinogens | ||
| Potentiates vincristine cytotoxicity to U-87 MG (human glioblastoma astrocytoma) | [82] | |
| Reduces the growth of breast carcinoma cells (MCF-7, MDA-MB-231) | [19] | |
| Camalexin | Antiproliferative activity on the human breast cancer cell line that overexpresses the Her2 (SKBr3) IC50 2.7 μmol/L | [83] |
| Increases expression of topoisomerase IIα in SKBr3 | ||
| Induces apoptosis in prostate cancer cells (PCa) through the generation of ROS | ||
| Induces apoptosis in Jurkat cells by increasing production of ROS and activation of caspase-8 and caspase-9. | [85] | |
| Inhibits the growth of prostate cancer cells (PCa) by increasing activity of the cathepsin lysosomal enzyme (CD) | [86] | |
| Benzocamalexin | The fusion of benzene to thiazole ring of camalexin significantly enhances its cytotoxicity | [23] |
| In comparison with camalexin, significantly decreases survival of all tested cancer cell lines (IC50 ranging from 23.3 to 30.0 μmol/L) | ||
| Induces the mitotic phase arrest via inhibition of microtubule formation (downregulates the expression of α-tubulin, a1-tubulin, β5-tubulin) in Jurkat cells | ||
| Downregulates the expression of anti-apoptotic genes bcl-2, bcl-xL | ||
| Upregulates the expression of pro-apoptotic gene bax | ||
| Minimal toxicity (IC50 > 100.0 μmol/L) in non-cancer cells is observed |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chripkova, M.; Zigo, F.; Mojzis, J. Antiproliferative Effect of Indole Phytoalexins. Molecules 2016, 21, 1626. https://doi.org/10.3390/molecules21121626
Chripkova M, Zigo F, Mojzis J. Antiproliferative Effect of Indole Phytoalexins. Molecules. 2016; 21(12):1626. https://doi.org/10.3390/molecules21121626
Chicago/Turabian StyleChripkova, Martina, Frantisek Zigo, and Jan Mojzis. 2016. "Antiproliferative Effect of Indole Phytoalexins" Molecules 21, no. 12: 1626. https://doi.org/10.3390/molecules21121626
APA StyleChripkova, M., Zigo, F., & Mojzis, J. (2016). Antiproliferative Effect of Indole Phytoalexins. Molecules, 21(12), 1626. https://doi.org/10.3390/molecules21121626