3.1. Synthesis
3.1.1. General Information
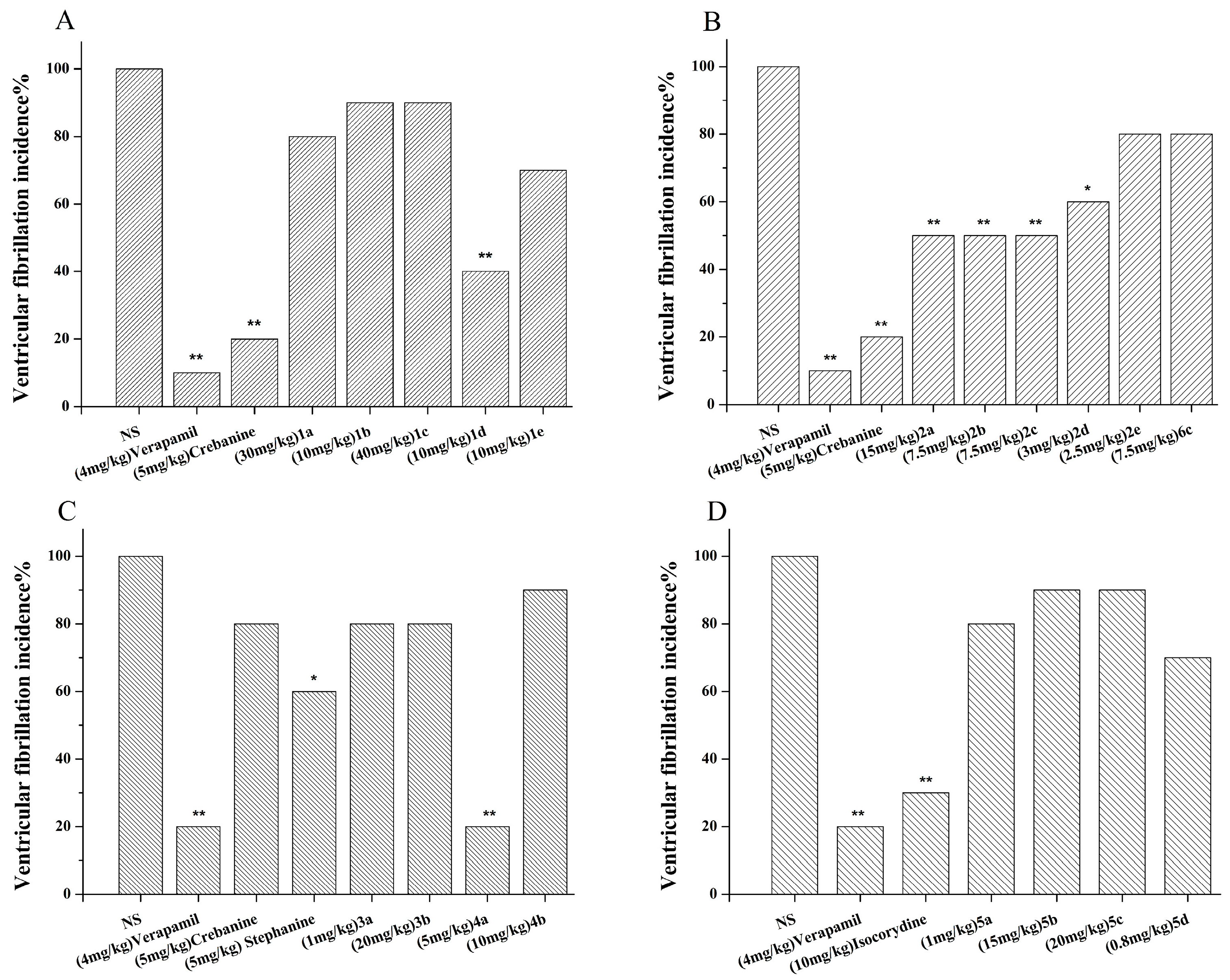
Isocorydine, crebanine, and stephanine were extracted from the plant, identified as
Stephania yunnanensis Lo by Professor Yunshu Ma, Yunnan University of Traditional Chinese Medicine, and as reported previously [
26,
27]. Solvents and reagents used in syntheses were of analytical grade, were purchased from commercial sources, and used without further purification.
All reactions were carried out under an atmosphere of argon, with magnetic stirring, in flame dried or oven-dried glassware. Analytical thin layer chromatography was performed on 0.25-mm silica gel GF 254 plates (Qingdao Haiyang Chemical Co., Ltd., Qingdao, China). Visualization was accomplished with UV light and bismuth potassium iodide solution staining followed by heating. 1H-NMR spectra were recorded on a 400 MHz spectrometer (Amersham Pharmacia Biotech AB Inc., Tokyo, Japan) in CDCl3 or CD3OD at ambient temperature, using the solvent signal as an internal standard. Data are reported as: (br/broad, s/singlet, d/doublet, t/triplet, q/quartet, m/multiplet; integration; coupling constant(s) in hertz). 13C-NMR spectra were recorded on a 400 MHz spectrometer (Amersham Pharmacia Biotech AB Inc., Tokyo, Japan) in CDCl3 at ambient temperature, using the solvent signal as an internal standard. Electrospray ionization mass spectrometry was detected with a Brukerama Zon SL (Bruker Daltonics Inc., Leipzig, Bremen, Germany); HR-MS was detected with 6200 series TOF/6500 series (Agilent Technologies Inc., Santa Clara, UT, USA); rotation was detected with a WZZ-three digital automatic polarimeter (Shanghai Shengguang Technology Co., Ltd., Shanghai, China).
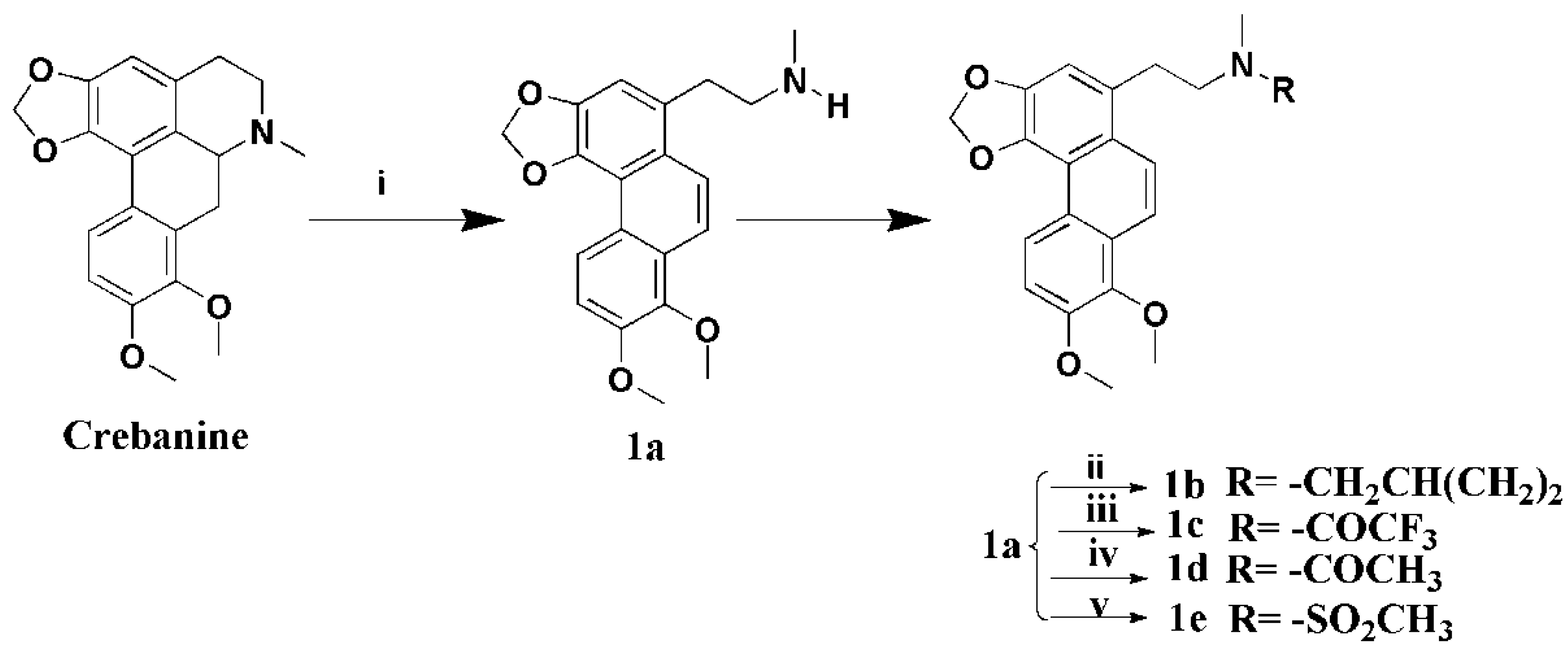
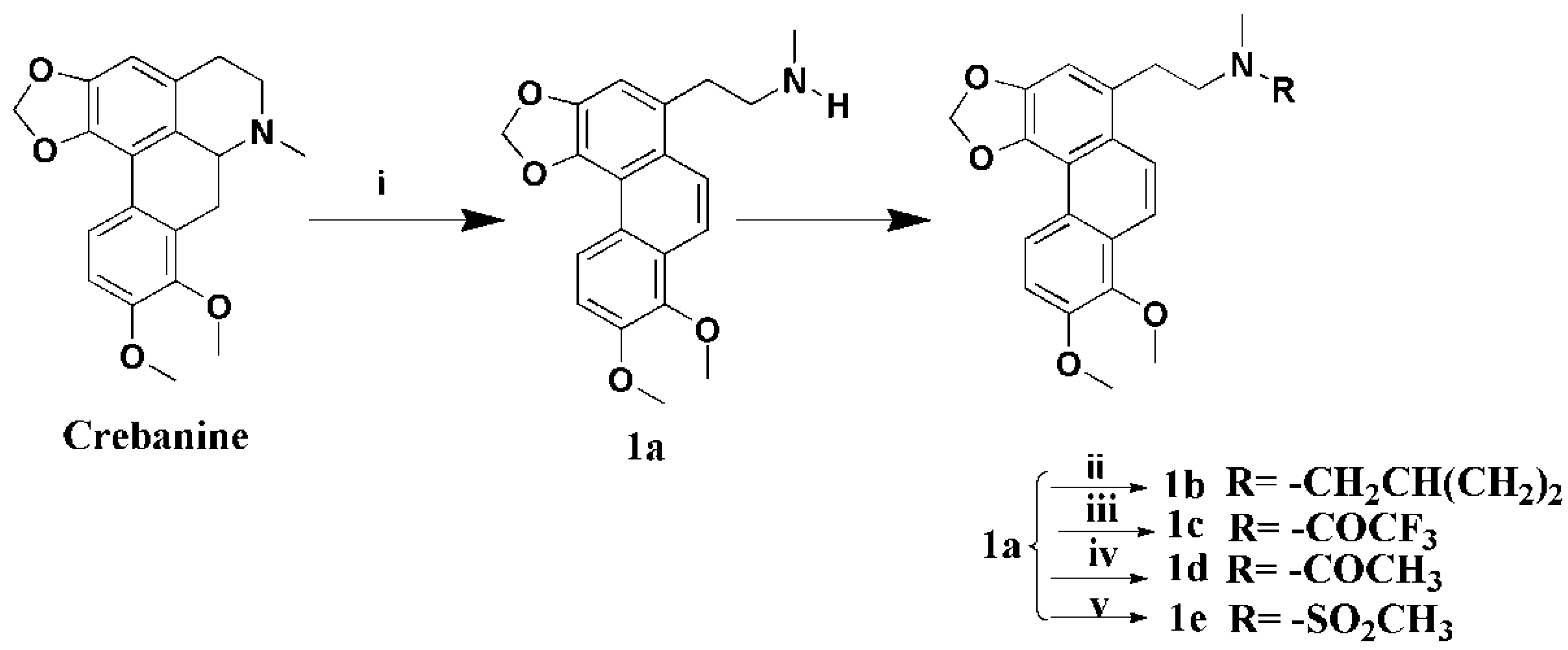
3.1.2. Preparation of Secocrebanine (1a)
A mixture of crebanine (2 g, 5.9 mmol) and K2CO3 (1.7 g, 1.2 mmol) in 1,2-dichloroethane (20 mL) was stirred and 1-chloroethyl chloroformate (1.0 g, 7.1 mmol) was added slowly. The mixture was heated to 85 °C for 6 h, the 1,2-dichloroethane-insoluble material was filtered off, and the filtrate was concentrated to give the residue, which was dissolved in MeOH (20 mL) and five drops of 37% HCl, and refluxed for 2 h. The solution was concentrated and neutralized with saturated Na2CO3, and the residue was extracted with CH2Cl2. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to get a crude product, which was crystallized from EtOAC at 4 °C, yielding compound 1a (1.2 g, 60%) as a pink-white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.83 (d, J = 9.2 HZ, 1H, 5-H), 7.93 (d, J = 9.6 HZ, 1H, 10-H), 7.87 (d, J = 9.6 HZ, 1H, 9-H), 7.29 (d, J = 8.8 HZ, 1H, 6-H), 7.10 (s, 1H, 2-H), 6.22 (s, 2H, -OCH2O-), 4.02 (s, 3H, 8-OCH3), 4.00 (s, 3H, 9-OCH3), 3.28–3.24 (t, J = 7.2 HZ, 2H, -CH2α), 2.97–2.93 (t, J = 7.2 HZ, 2H, -CH2β), 2.47 (s, 3H, -NCH3); 13C-NMR (100 MHZ, CDCl3, δ; ppm) 33.87 (t, -CH2β), 36.43 (q, N-CH3), 53.12 (t, -CH2α), 56.30 (q, 7-OCH3), 61.32 (q, 8-OCH3), 100.95 (t, -OCH2O-), 110.05 (d, C-2), 112.60 (d, C-6), 117.11 (s, C-8a), 118.38 (d, C-5), 123.26 (s, C-4a), 123.64 (d, C-9), 123.79 (d, C-10), 125.19 (s, C-1a), 127.41 (s, C-5a), 130.88 (s, C-1), 141.76 (s, C-4), 143.23 (s, C-7), 144,98 (s, C-3), 149.96 (s, C-8), Positive ESI-MS m/z: 340.2 [M + H]+. HR-MS for C20H21NO4 [M + H]+; calcd. 340.1543, found: 340.1542.
3.1.3. Preparation of N-Cyclopropylmethylsecocrebanine (1b)
Product 1a (200 mg, 0.59 mmol), K2CO3 (163 mg, 1.18 mmol), and cyclopropylmethyl bromide (96 mg, 0.71 mmol) were added to MeCN (20 mL) and heated at 80 °C for 4 h. The reaction mixture was filtered and concentrated to give a residue, which was purified by silica column chromatography (CHCl3/MeOH 80:1), yielding compound 1b (180 mg, 78%) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.81(d, J = 9.2 HZ, 1H, 5-H), 8.04 (d, J = 9.6 HZ, 1H, 10-H), 7.93 (d, J = 9.2 HZ, 1H, 9-H), 7.31 (d, J = 8.4 HZ, 1H, 6-H), 7.16 (s, 1H, 2-H), 6.23 (s, 2H, -OCH2O-), 4.02 (s, 3H, 8-OCH3), 3.99 (s, 3H, 9-OCH3), 3.84–3.78 (m, 1H, CH2α), 3.66–3.60 (m, 1H, -CH2β), 3.47–3.42 (m, 1H, -CH2β), 3.18–3.15 (m, 2H, N-CH2), 2.97 (s, 3H, -NCH3), 2.93–2.89 (m, 1H, CH2α), 1.30–1.25 (m, 1H, -CH), 0.85–0.82 (m, 2H, -CH2), 0.50–0.47 (m, 1H, -CH2). .13C-NMR (100 MHZ, CDCl3, δ; ppm) 100.62 (t, -OCH2O-), 52.82 (t, -CH2β), 29.59 (t, CH2α), 29.26 (t, -CH2), 29.05 (t, -CH2), 61.00 (q, 8-OCH3), 56.02 (q, 8-OCH3), 43.97 (q, N-CH3), 115.77 (d, C-5), 114.88 (d, C-6),62.40(d, -CH), 152.55 (s, C-8), 145.17 (s, C-3), 145.02 (s, C-7), 141.99 (s, C-4), 133.19 (s, C-1),130.69 (s, C-5a), 125.73(s, C-1a), 124.66 (s, C-4a), 114.88(s, C-8a). Positive ESI-MS m/z: 394.2 [M + H]+. HR-MS for C24H27NO4 [M + H]+; calcd. 394.2013, found: 394.2026.
3.1.4. Preparation of N-Trifluoroacetamidesecocrebanine (1c)
A solution of 1a (200 mg, 0.59 mmol) and Et3N (72 mg, 0.71 mmol) in anhydrous CH2Cl2 (30 mL), trifluoroacetic anhydride (149 mg, 0.71 mmol) was slowly added under an ice-bath, then, the solution was stirred for 2 h at room temperature. The mixture was diluted with water (20 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined extracts were dried over anhydrous Na2SO4, filtered and concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 100:1), and yielding compound 1c (210 mg, 82%) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.83 (d, J = 9.2 HZ, 1H, 5-H), 7.99 (d, J = 9.2 HZ, 1H, 10-H), 7.94 (d, J = 9.6 HZ, 1H, 9-H), 7.31 (d, J = 9.2 HZ, 1H, 6-H), 7.07 (s, 1H, 2-H), 6.24 (s, 1H, -OCH2O-), 6.23 (s, 1H, -OCH2O-), 4.03 (s, 3H, 8-OCH3), 4.00 (s, 3H, 9-OCH3), 3.73–3.69 (t, J = 7.4 HZ, 2H, -CH2α), 3.36–3.32 (t, J = 7.2 HZ, 2H, -CH2β), 3.11 (s, 1H, -NCH3), 2.99 (s, 2H, -NCH3). 13C-NMR (100 MHZ, CDCl3, δ; ppm) 30.67 (t, CH2α), 36.04 (q, N-CH3), 51.79 (t, -CH2β ), 56.32 (q, 7-OCH3), 61.33(q, 8-OCH3), 101.21 (t, -OCH2O-), 110.13 (d, C-2), 112.91 (d, C-6), 117.15 (s, C-8a), 119.12 (s, C-4a), 119.28 (d, C-5), 122.08 (s, CF3), 123.77 (d, C-9), 123.81 (d, C-10), 125.39 (C-1a), 127.47 (C-5a), 128.83 (s, C-1), 142.27 (s, C-4), 143.32 (s, C-7), 145.08 (s, C-3), 150.15 (s, C-8). Positive ESI-MS m/z: 458.2 [M + Na]+. HR-MS for C22H20F3NO5 [M + Na]+; calcd. 458.1186, found: 458.1188.
3.1.5. Preparation of N-Acetamidesecocrebanine (1d)
To a stirred solution of 1a (200 mg, 0.59 mmol), 4-dimethylaminopyridine (144 mg, 1.18 mmol), and Et3N (72 mg, 0.71 mmol) in anhydrous CH2Cl2 (20 mL), acetic anhydride (72 mg, 0.71mmol) was slowly added and stirred for 4 h at room temperature. The mixture was diluted with water (10 mL) and extracted with CH2Cl2 (3 × 10mL). The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to get a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 100:1), yielding compound 1d (180 mg, 80%) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.83 (d, J = 8.4 HZ, 1H, 5-H), 8.00 (d, J = 10.0 HZ, 1H, 10-H) 7.96 (d, J = 9.6 HZ, 1H, 9-H), 7.32 (d, J = 9.2 HZ, 1H, 6-H), 7.09 (s, 1H, 2-H), 6.23 (s, 1H, -OCH2O-), 6.22 (s, 1H, -OCH2O-), 4.03 (s, 6H, 8-OCH3), 4.00 (s, 6H, 9-OCH3), 3.67–3.61 (dd, J = 15.6 HZ, J = 8.0 HZ, 2H, -CH2α), 3.31–3.27 (t, J = 7.4 HZ, 2H, -CH2β), 3.00 (s, 1H, -NCH3), 2.81 (s, 2H, -NCH3), 2.09 (s, 2H, -NOCH3), 1.85 (s, 1H, -NOCH3). 13C-NMR (100 MHZ, CDCl3, δ; ppm) 22.03 (q, -NOCH3), 32.57 (t, -CH2α), 37.39 (q, N-CH3), 51.99 (t, -CH2β), 56.33 (q, 7-OCH3), 61.32 (q, 8-OCH3), 100.08 (t, -OCH2O-), 110.28 (d, C-2), 112.88 (d, C-6), 117.17 (s, C-8a), 119.10 (d, C-5),123.76 (d, C-9), 123.81 (d, C-10), 125.55 (s, C-4a), 127.50 (C-1a), 128.72 (C-5a), 130.31 (s, C-1), 142.30 (s, C-4), 143.28 (s, C-7), 145.14 (s, C-3), 150.11 (s, C-8), 170.69 (s, -C=O).Positive ESI-MS m/z: 404.2 [M + Na]+.HR-MS for C22H23NO5 [M + Na]+; calcd. 404.1468, found: 404.1474.
3.1.6. Preparation of N-Methylsulfonamidesecocrebanine (1e)
To a mixture of 1a (200 mg, 0.59 mmol) and Et3N (72 mg, 0.71 mmol) in anhydrous CH2Cl2 (30 mL) under an ice-bath, methanesulfonyl chloride (81 mg, 0.71 mmol) was slowly added and stirred for 2 h at room temperature. The mixture was diluted with water (20 mL) and extracted with CH2Cl2 (3 × 10 mL). The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 100:1), yielding 1e (210 mg, 82%) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.83 (d, J = 9.2 HZ, 1H, 5-H), 7.98 (d, J = 9.6 HZ, 1H, 10-H), 7.85 (d, J = 9.6 HZ, 1H, 9-H), 7.31(d, J = 9.2 HZ, 1H, 6-H), 7.11 (s, 1H, 2-H), 6.23 (s, 2H, -OCH2O-), 4.03 (s, 3H, 8-OCH3), 4.00 (s, 3H, 9-OCH3), 3.46–3.42 (m, 2H, -CH2α), 3.37–3.34 (m, 2H, -CH2β), 2.92 (s, 3H, -NCH3), 2.74 (s, 3H, -NSO2CH3). 13C-NMR (100 MHZ, CDCl3, δ; ppm) 33.26 (t, -CH2α), 35.38 (t, -NSO2CH3), 51.56 (t, -CH2β), 56.31 (q, 7-OCH3), 61.34 (q, 8-OCH3), 101.10 (t, -OCH2O-), 110.30 (d, C-2), 112.76 (d, C-6),117.13 (s, C-8a), 119.06 (d, C-5),122.72 (d, C-9),123.77 (d, C-10),125.21 (s, C-4a),127.40 (C-1a), 128.88 (s, C-1), 142.20 (s, C-4),143.29 (s, C-7), 145.07 (s, C-3),150.07 (s, C-8). Positive ESI-MS m/z: 440.2 [M + Na]+. HR-MS for C21H23NO6S [M + Na]+; calcd. 440.1138, found: 440.1140.
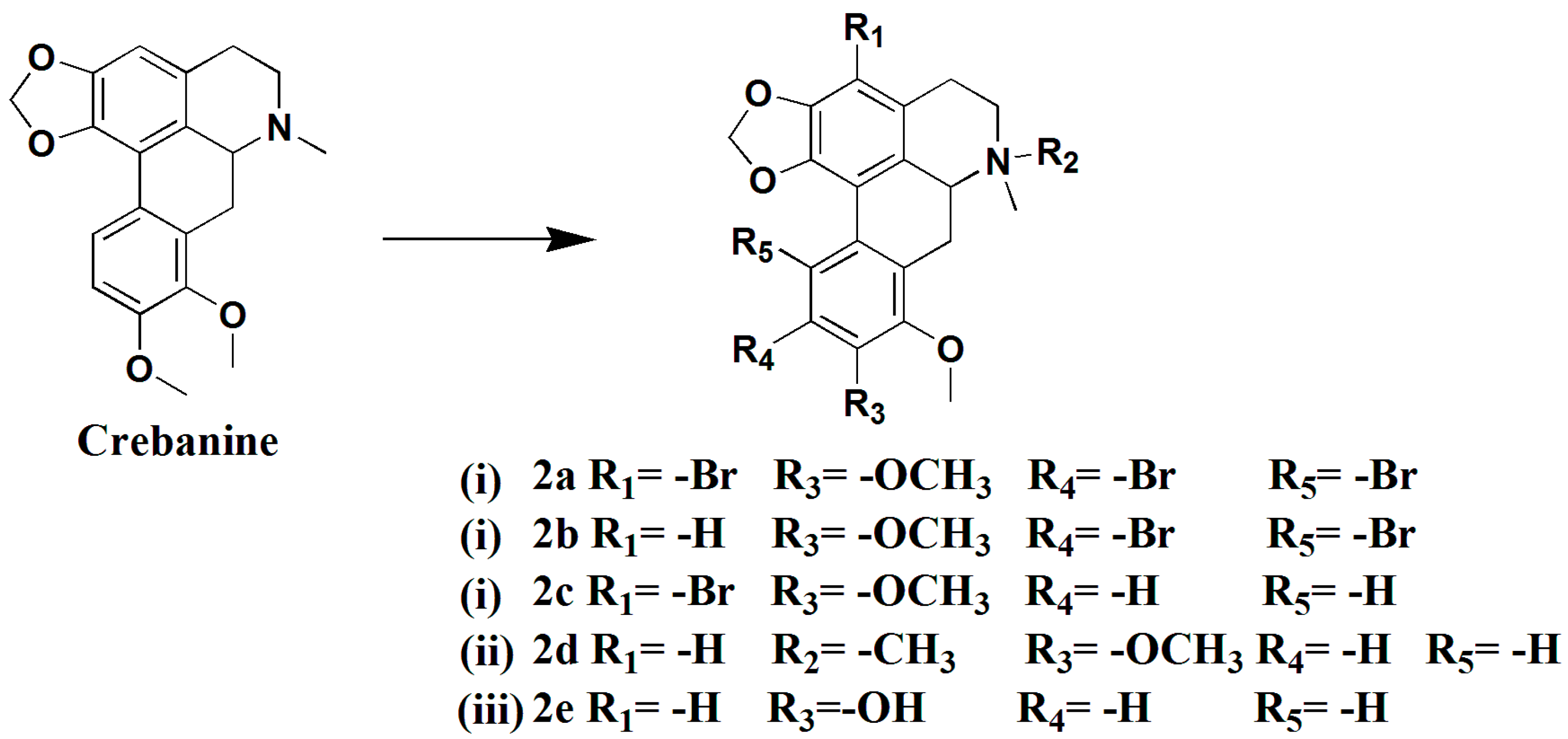
3.1.7. Preparation of 3,10,11-Tribromocrebanine (2a), 10,11-Dibromocrebanine (2b) and 3-Bromocrebanine (2c)
Crebanine (200 mg, 0.59 mmol) and N-Bromosuccinimide (213.6 mg, 1.2 mmol) were added to trifluoroacetic acid (5 mL), the mixture was stirred for 18hat room temperature, diluted with water (10 mL), and neutralized with saturated NaHCO3 and extracted with CH2Cl2 (3 × 10 mL). The combined CH2Cl2 solution was brined with water, dried over anhydrous Na2SO4, filtered, and concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 200:1→100:1), yielding compound 2a (52 mg, 27%), compound 2b (50 mg, 17%), and compound 2c (80 mg, 33%).
Compound 2a, pale yellow solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 6.16 (1H, s, 9, -OCH2O-), 6.06 (1H, s, -OCH2O-), 3.93 (3H, s, 8-OCH3), 3.87 (3H, s, 9-OCH3), 3.54 (1H, d, J = 12.8 HZ, 6a-H), 3.16–3.09 (m, 1H, 7-Ha), 2.99–2.91 (m, 2H, 5-Ha, 7-Hb), 2.77–2.72 (m, 1H, 5-Ha), 2.60 (s, 3H, -NCH3), 2.53–2.50 (m, 1H, 4-Ha), 2.15–2.02 (m, 1H, 4-Hb). 13C-NMR (100 MHZ, CDCl3, δ; ppm), 29.29 (t, C-7), 29.33 (t, C-4), 52.78 (t, C-5), 44.02 (t, -NCH3), 60.82 (q, 9-OCH3), 61.17 (q, 8-OCH3), 62.18 (d, C-6a), 100.77 (t, -OCH2O-),102.96 (s, C-3), 115.29 (s, C-10), 118.63 (s, C-11), 125.78 (s, C-7a), 129.83 (s, C-1b), 130.73 (s, C-3a), 132.67 (s, C-11a), 142.26 (s, C-1), 145.28 (s, C-8), 149.72 (s, C-2), 150.88 (s, C-9a). Positive ESI-MS m/z: 579.2 [M + 3]+. HR-MS for C20H18Br3NO4 [M + H]+; calcd.573.8859, found:573.8853.
Compound 2b, white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.13 (s, 1H, 3-H), 6.14 (s, 1H, -OCH2O-), 6.05 (s, 1H, -OCH2O-), 3.89 (s, 3H, 8-OCH3), 3.79 (s, 3H, 9-OCH3), 3.61–3.57 (m, 1H, 6a-H), 3.11–3.07 (m, 1H, 7-Ha), 2.97–2.87 (m, 2H, 5-Ha, 7-Hb), 2.80–2.66 (m, 1H, 5-Hb), 2.56 (s, 3H, -NCH3), 2.52–2.47 (m, 1H, 4-Ha), 2.11–2.04 (t, J = 13.6 HZ, 1H, 4-Hb). 13C-NMR (100 MHZ, CDCl3, δ; ppm) 100.62 (t, -OCH2O-), 52.82 (t, C-5), 29.26 (t, C-4), 29.05 (t, C-7), 62.40 (d, C-6a), 115.95 (d, C-3), 152,55 (s, C-9), 145.17 (s, C-2), 145.09 (s, C-8), 141.99 (s, C-1), 133.19 (s, C-11a), 130.69 (s, C-3a), 125.73 (s, C-7a), 124.66 (s, C-1a). Positive ESI-MS m/z: 498.0 [M + H]+. HR-MS for C20H19Br2NO4 [M + H]+; calcd. 495.9754, found: 495.9759.
Compound 2c, white solid. Positive ESI-MS m/z: 418 [M]+, 420.0 [M + 2]+. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.76 (d, J = 8.8 HZ, 1H, 11-H), 6.90 (d, J = 8.8 HZ, 1H, 10-H), 6.22 (s, 1H, -OCH2O-), 6.07(s, 1H, -OCH2O-), 3.91 (s, 3H, 8-OCH3), 3.84 (s, 3H, 9-OCH3), 3.82–3.80 (m, 1H, 6a-H), 3.09-3.04 (m, 2H, 5-Ha, 7-Hb), 2.90–2.82 (m, 2H, 5-Ha, 7-Hb), 2.17 (s, 3H, -NCH3), 1.95–1.87 (m, 1H, 4-Ha), 1.79–1.72 (m, 1H, 4-Hb).
3.1.8. Preparation of N-Methylcrebanine (2d)
A mixture of crebanine (200 mg, 0.59 mmol), ether (109 mg, 1.48 mmol) and CH3I (184.5 mg, 1.3 mmol) in MeOH (10 mL) was stirred for 16 h at room temperature. The reaction mixture was concentrated and extracted with CHCl3 (3 × 10 mL). The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 30:1), yielding compound 2d (180 mg, 86%) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.70 (d, J = 8.8 Hz, 1H, 11-H), 7.05 (d, J = 8.8 Hz, 1H, 10-H), 6.78 (s, 1H, 3-H), 6.18 (s, 1H, -OCH2O-), 5.98 (s, 1H, -OCH2O-), 4.74–4.70 (m, 1H, 6a-H), 3.80 (s, 3H, 8-OCH3), 3.76–3.73 (m, 1H, 7-Ha), 3.72 (s, 3H, 9-OCH3), 3.64–3.55 (m, 2H, 7-Hb,5-Ha), 3.33 (s, 3H, -NCH3), 3.18–3.09 (m, 1H, 5-Hb), 2.97 (s, 3H, -NCH3), 2.94–2.89 (m, 1H, 4-Ha), 2.70–2.63 (t, J = 14.2 Hz, 1H, 4Hb). Positive ESI-MS m/z: 354.2 [M]+.
3.1.9. Preparation of Stesakine (2e)
Crebanine (200 mg, 0.59 mmol) in 2mL of anhydrous CH2Cl2 was slowly added into a suspension of AlBr3 (320 mg, 1.2 mmol) in nitrobenzene (5 mL) at 5 °C under argon. The solution was stirred for 18 h, diluted with water (20 mL) and neutralized with saturated NaHCO3. The mixture was extracted with CH2Cl2 (3 × 10 mL), and the combined CH2Cl2 solution was brined with water, dried with anhydrous Na2SO4, filtered, and concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 50:1), yieldingcompound 2e (56 mg, 29%) as a colorless oil. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.63 (d, J = 8.8 HZ, 1H, 11-H), 6.83 (d, J = 8.8 HZ, 1H, 10-H), 6.53 (s, 1H, 3-H), 6.06 (s, 1H, -OCH2O-), 5.92 (s, 1H, -OCH2O-), 5.77 (s, 1H, 9-OH), 3.93 (s, 3H, 8-CH3), 3.69–3.64 (dd, J = 16.0 HZ, J = 4.0 HZ, 1H, 6a-H), 3.19–3.03 (m, 3H, 7-H, 5-Ha), 2.65–2.61 (m, 1H, 5-Hb), 2.60 (s, 3H, -NCH3), 2.56–2.50 (m, 1H, 4-Ha), 2.32–2.25 (t, J = 10.4 HZ, 1H, 4-Hb), 13C-NMR (100 MHZ, CDCl3, δ; ppm) 142.2 (s, C-1), 116.7 (s, C-1a), 126.5 (s, C-1b), 146.6 (s, C-2), 106.7 (d, C-3), 126.7 (s, C-3a), 26.3 (t, C-4), 56 (t, C-5), 61.9 (d, 6a), 29.2 (t, C-7), 124.8 (s, C-7a), 156.4 (s, C-8), 145.8 (s, C-9), 108.5 (d, t-10), 118.7 (d, C-11), 126.6 (s, C-11a), 100.1 (t, -CH2O-), 44.0 (q, -NCH3), 56.0 (q, 8-OCH3). Positive ESI-MS m/z: 326 [M + H]+.
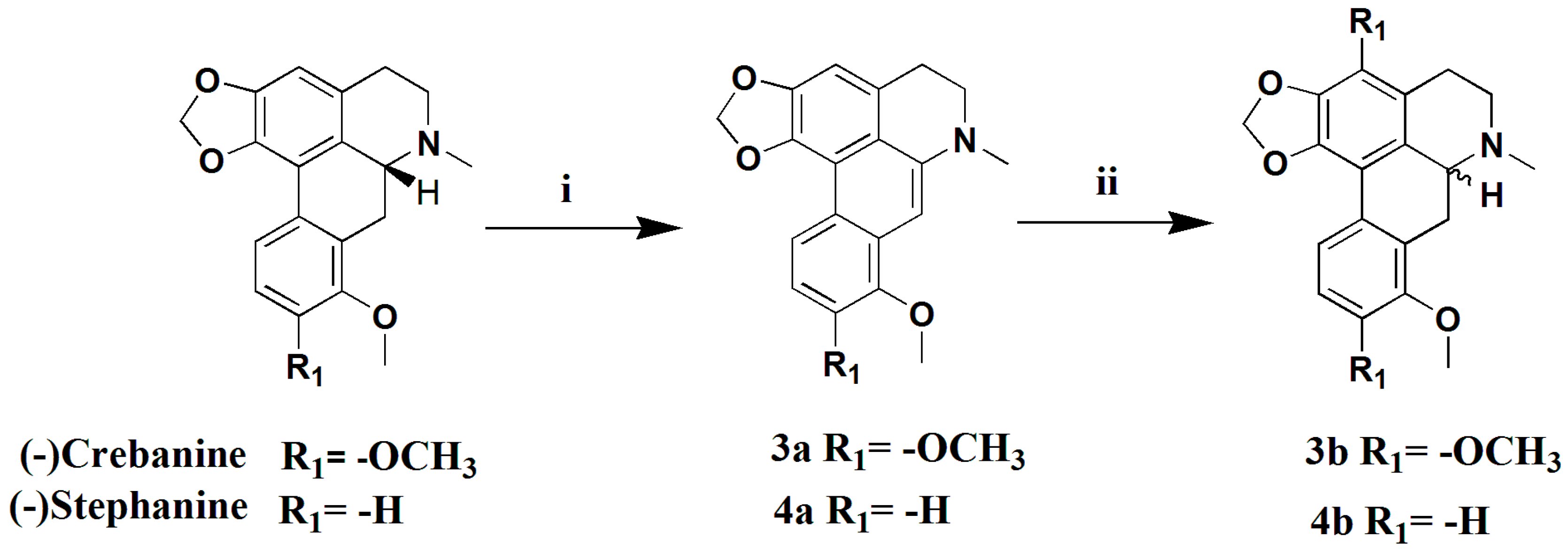
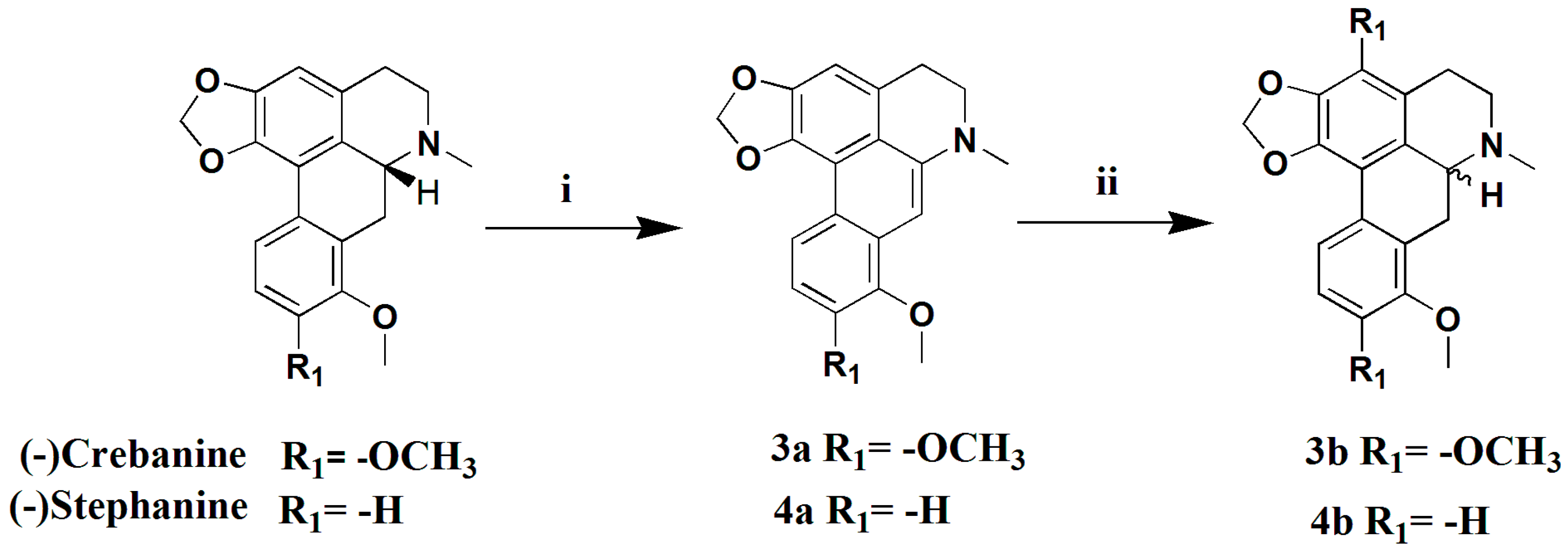
3.1.10. Preparation of Dehydrocrebanine (3a)
Pd/C (10%, 210 mg) was added to a suspension of crebanine (200 mg, 0.59 mmol) in MeCN (20 mL), and the solution was refluxed for 6 h under N2. The mixture reaction solution was filtered and the filtrate was evaporated to give compound 3a (181 mg, 91%) as a yellowish-green solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.66 (d, J = 8.8 Hz, 1H, 11-H), 7.03 (d, J = 9.2 Hz, 1H, 10-H), 6.89 (s, 2H, 3-H, 7-H), 6.19 (s, 2H, -OCH2O-), 3.99 (s, 3H, 8-OCH3), 3.97 (s, 3H, 9-OCH3), 3.34–3.37 (t, J = 5.8 Hz, 2H, 5-H), 3.24–3.31 (t, J = 5.8 Hz, 2H, 4-H), 3.13 (s, 3H, -NCH3). Positive ESI-MS m/z: 348.2 [M + H]+.
3.1.11. Preparation of (±) Crebanine (3b)
A mixture of 3a (150 mg, 0.44 mmol) and NaCNBH3 (36 mg, 0.57 mmol) in absolute EtOH was stirred, and a mixture of EtOH and 2NHCl was added until the pH approached 3.0 (within 4 h). Then, the solution was continuously stirred for 18 h at room temperature. After evaporation of the reaction mixture, the pH was adjusted to 8 with saturated Na2CO3 and the mixture solution was extracted with CH2Cl2 (3 × 10 mL). The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to get a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 50:1), yielding compound 3b (120 mg, 80%) as a yellow oil. : 0° (C 0.2, MeOH). 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.81 (d, J = 8.8 Hz, 1H, 11-H), 6.88 (d, J = 8.8 Hz, 1H, 10-H), 6.53 (s, 1H, 3-H), 6.07 (s, 1H, -OCH3O-), 5.90 (s, 1H, -OCH3O-), 3.90 (s, 3H, 8-OCH3), 3.81 (s, 3H, 9-OCH3), 3.70–3.65 (dd, J = 14.8 Hz, J = 4.4 Hz, 1H, 6a-H), 3.18–3.04 (m, 3H, 7-H, 5-Ha), 2.66–2.61 (m, 1H, 5-Hb), 2.59 (s, 3H, -NCH3), 2.56–2.49 (m, 1H, 4-Ha), 2.33–2.26 (t, J = 14.2 Hz, 1H, 4-Hb). Positive ESI-MS m/z: 340.2 [M + H]+.
3.1.12. Preparation of Dehydrostephanine (4a)
In a similar manner used in the preparation of 3a, stephanine (200 mg, 0.65 mmol) was subjected to a dehydrogenation reaction to give compound 4a (162 mg, 81%) as a yellowish-green solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.56 (d, J = 8.4 HZ, 1H, 11-H), 8.29 (d, J = 8.0 HZ, 10-H), 7.08 (s, 1H, 9-H), 6.96 (s, H, 3-H), 6.93 (s, H, 7-H), 6.21 (s, 2H, -OCH2O-), 4.02 (s, 3H, 8-OCH3), 3.40–3.37 (t, J = 5.6 HZ, 2H, 5-CH2), 3.27–3.24 (m, 2H, 5-CH2), 3.12 (s, 3H). Positive ESI-MS m/z: 308.2 [M + H]+.
3.1.13. Preparation of (±) Stephanine (4b)
In a similar manner used for the preparation of 3b, 4a (150 mg, 0.49 mmol) was subjected to a racemization reaction to give compound 4b (125 mg, 83%) as a yellowish-green solid. : 0° (C 0.2, MeOH). 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.70 (d, J = 8.0 Hz, 1H, 11-H), 7.31–7.26 (t, J = 8.0 Hz, 1H, 10-H), 6.85 (d, J = 8.0 Hz, 1H, 9-H), 6.58 (s, 1H, 3-H), 6.10 (s, 1H, -OCH2O-), 5.95 (s, 1H, -OCH2O-), 3.86 (s, 1H, 8-OCH3), 3.78–3.74 (dd, J = 14.8 Hz, J = 3.6 Hz, 1H, 6a-H), 3.40–3.34 (m, 3H, 7-H, 5a-H), 2.90–2.83 (m, 1H, 5a-H), 2.80 (s, 3H, -NCH3), 2.77–2.72 (m, 1H, 4a-H), 2.58–2.51 (t, J = 14.4 Hz, 1H, 4b-H). Positive ESI-MS m/z: 310.2 [M + H]+.
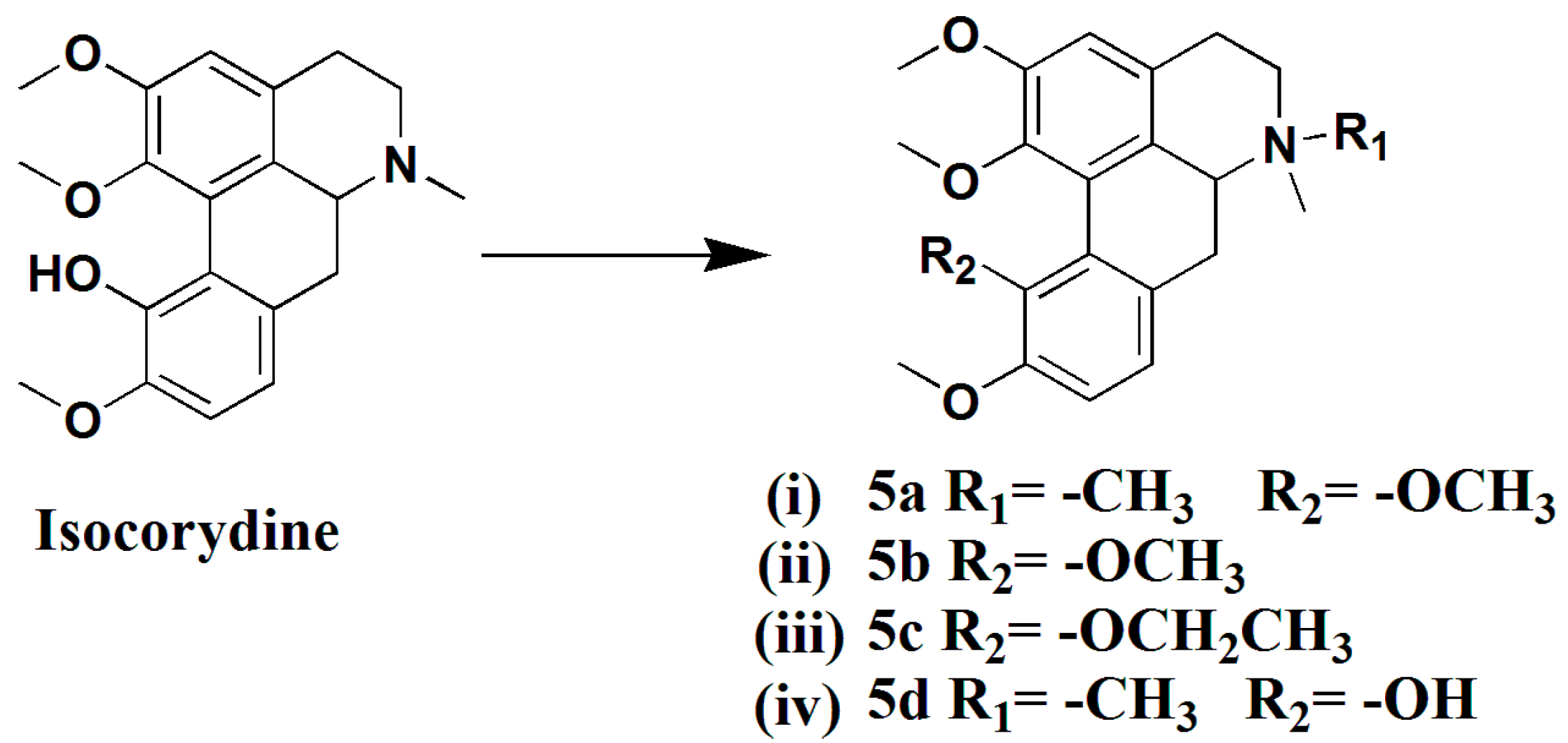
3.1.14. Preparation of 11-Methoxy-N-Methylisocorydine (5a)
To a mixture of isocorydine (200 mg, 0.59 mmol), Tetrabutylammonium bromide (16 mg, 0.03 mmol), and K2CO3 (163 mg, 1.18 mmol) in anhydrous THF (10 mL) CH3I (185 mg, 1.30 mmol) was successively added over a 10-min period and stirred for 8 h at room temperature. The reaction mixture was condensed and extracted with CHCl3. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 50:1), yielding compound 5a (154 mg, 71%) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 6.95 (d, J = 8.0 HZ, 1H, 8-H), 6.84 (d, J = 8.4 HZ, 1H, 9-H), 6.66 (s, 1H, 3-H), 3.88 (d, J = 4.0 HZ, 6H, 1-OCH3,2-OCH3), 3.87 (s, 3H,11-OCH3), 3.72 (s, 3H, 10-OCH3), 3.17–3.10 (m, 1H, 6a-H), 3.04–2.99 (m, 2H, 7-Ha, 5-Ha), 2.87 (d, J = 12.4 HZ, 1H, 7-Hb), 2.69 (d, J = 16.8 HZ, 1H, 5-Hb), 2.53–2.48 (m, 4H, -NCH3, 4-Ha), 2.40–2.33 (t, J = 13.0 HZ, 1H, 4-Hb).13C-NMR (100 MHZ, CDCl3, δ; ppm) 29.73 (t, C-4), 23.03 (t, C-7), 42.94 (q, N-CH3), 53.07 (q, N-CH3), 55.83 (q, 10-OCH3), 55.97(q, 2-OCH3), 60.00 (t, C-5), 60.21 (q, 1-OCH3), 60.40 (q, 11-OCH3), 111.46 (d, C-3), 112.72 (d, C-9), 121.63 (s, C-11a). 122.36 (d, C-8), 123.98 (s, C-1a), 123.96 (s, C-7a), 126.18 (s, C-1b), 146.35 (s, C-1), 147.04 (s, C-10), 152.20 (s, C-11), 152.54 (s, C-20). Positive ESI-MS m/z: 370.2 [M + H]+. HR-MS for C22H28NO4 [M + H]+; calcd. 370.2013, found: 370.2026.
3.1.15. Preparation of 11-Methoxyisocorydine (5b)
A solution of isocorydine (200 mg, 0.59 mmol) in MeCN (5 mL) and MeOH (5 mL) was treated with N,N-diisopropylethylamine (91 mg, 0.71 mmol) and trimethylsilyldiazomethane (0.36 mL, 2M in hexane), the mixture was stirred at room temperature for 15 h, condensed, and extracted with CHCl3. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to give a crude product, which was purified by silica gel column chromatography (CHCl3–MeOH = 80:1), yielding compound 5b (51 mg, 24%) as a colorless oil. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 6.95 (d, J = 8.0 HZ, 1H, 8-H), 6.84 (d, J = 8.4 HZ, 1H, 9-H), 6.66 (s, 1H, 3-H), 3.88 (d, J = 4.0 HZ, 6H, 1-OCH3,2-OCH3), 3.87 (s, 3H,11-OCH3), 3.72 (s, 3H, 10-OCH3), 3.17–3.10 (m, 1H, 6a-H), 3.04–2.99 (m, 2H, 7-Ha, 5-Ha), 2.87 (d, J = 12.4 HZ, 1H, 7-Hb), 2.69 (d, J = 16.8 HZ, 1H, 5-Hb), 2.53–2.48 (m, 4H, -NCH3, 4-Ha), 2.40–2.33 (t, J = 13.0 HZ, 1H, 4-Hb). Positive ESI-MS m/z: 356.2 [M + H]+. HR-MS for C21H25NO4 [M + H]+; calcd. 356.1856, found: 356.1864.
3.1.16. Preparation of 11-Ethoxyisocorydine (5c)
A mixture of isocorydine (200 mg, 0.59 mmol), Tetrabutylammonium bromide (16 mg, 0.03 mmol), and K2CO3 (163 mg, 1.18 mmol) in anhydrous THF (10 mL) was successively added CH3CH2I (203 mg, 1.30 mmol) over a 10-min period and stirred for 8 h at room temperature. The reaction mixture was condensed and extracted with CHCl3. The combined extracts were dried over anhydrous Na2SO4, filtered, and concentrated to get a crude product, which was purified by silica gel column chromatography (CH2Cl2/MeOH 50:1), yielding compound 5c (80 mg, 23%) as a colorless oil. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 6.95 (d, J = 8.0 HZ, 1H, 8-H), 6.83 (d, J = 8.0 HZ,1H, 9-H), 6.66 (s, 1H, 3-H), 3.87 (d, J = 2.8 HZ, 6H, 1-OCH3, 2-OCH3), 3.84–3.79 (m, 1H, 6a-H), 3.66 (s, 3H, 10-OCH3), 3.19–3.14 (m, 1H, 7-Ha), 3.05–2.98 (m, 2H,11-CH2), 2.87–2.83 (m, 1H, 7-Hb), 2.71–2.67 (m 1H, 5-Ha), 2.54 (s, 3H, -NCH3), 2.50–2.47 (m, 4-Ha), 2.40–2.34 (t, 1H, J = 12.8 HZ, 4-Hb), 1.14–1.10 (t, J = 7.0 HZ, 3H, 11-CH3). 13C-NMR (100 MHZ, CDCl3, δ; ppm) 15.79 (q, 11-CH2CH3), 25.87 (t, C-7), 33.34 (t, C-4). 56.21 (q, 10-OCH3), 60.96 (q, 1-OCH3), 61.14 (q, 2-OCH3), 68.89 (t, 11-CH2CH3), 111.41 (d, C-3), 112.72 (d, C-9), 122.04 (d, C-8), 124.00 (C-7a), 125.01 (s, C-1a), 127.00 (s, C-1b), 146 .89 (s, C-1), 147.04 (s, C-10), 152.82 (s, C-11), 153.42 (s, C-2). Positive ESI-MS m/z: 370.2 [M + H]+. HR-MS for C22H27NO4 [M + H]+; calcd. 370.2013, found: 370.2022.
3.1.17. Preparation of N-Methylisocorydine (5d)
In a similar manner used in the preparation of 2d, isocorydine (200 mg, 0.59 mmol) was subjected to a N-methylation reaction to give compound 5d (183 mg, 88% yield) as a white solid. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 8.60 (s, 1H, 11-OH), 7.01 (d, J = 8.0 HZ, 1H, 8-H), 6.89 (d, J = 8.0 Hz, 2H, 9-H), 6.86 (s, 1H, 3-H), 4.77–4.73 (dd, J = 12.0 HZ, J = 5.2 HZ, 1H, 6a-H), 4.24 (d, J = 13.2, 1H, 5a-H), 3.96 (s, 3H, 1-OCH3), 3.89 (s, 3H, 2-OCH3), 3.76 (s, 3H, 10-OCH3), 3.74 (s, 3H, -NCH3), 3.65–3.58 (m, 2H, 7a-H), 3.54–3.41 (m, 2H, 5b-H, 7a-H), 3.33 (s, 3H, -NCH3), 3.11–3.07 (m, 1H, 4-Ha), 2.88–2.81 (t, J = 12.0 HZ, 1H, 4-Hb). Positive ESI-MS m/z: 356.2 [M]+.
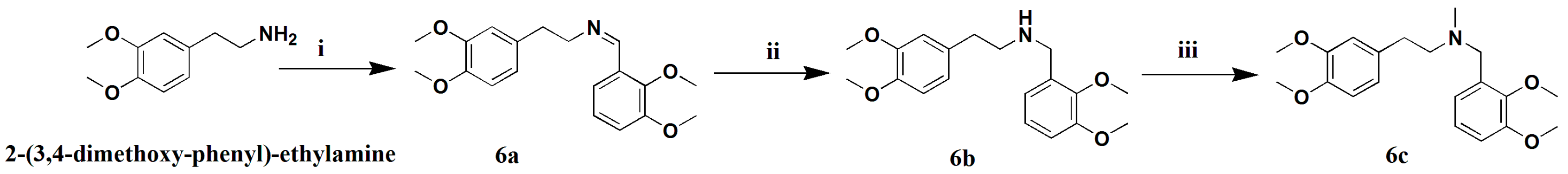
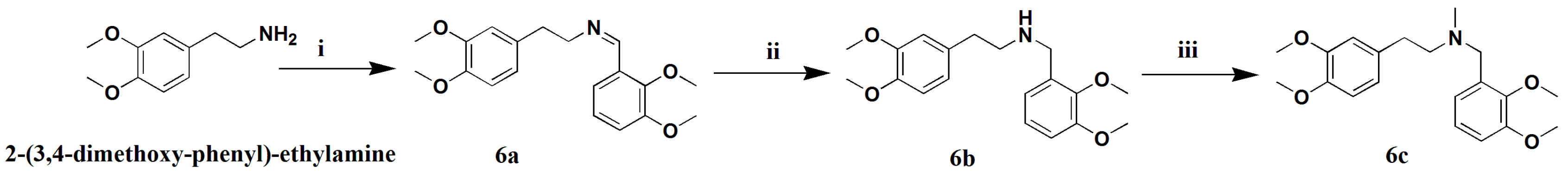
3.1.18. Preparation of (Z)-N-2,3-Dimethoxybenzylidene-3,4-Dimethoxy-2-Phenyl-Ethanamine (6a)
A mixture of 2-(3,4-dimethoxy-phenyl)-ethylamine (1 g, 5.5 mmol) and 2,3-dimethoxy-benzoic acid (1.1 g, 6.1 mmol) in methylbenzene (50 mL) was heated to 120 °C for 8 h. The reaction mixture was condensed and extracted with EtOAC. The combined extracts were washed with water three times, dried over anhydrous Na2SO4, filtered, and concentrated. The crude product was drawn off by a vacuum pump over 4 h to give compound 6a (1.78 g, 92% Purity, 91% yield) as a brown-red oil. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.54 (d, J = 7.6 HZ 1H, N-CH2CH2Ph-2H), 7.09–7.05 (t, J = 8.0 HZ, 1H, N-CH2Ph-4H), 6.96 (d, J = 8.0 HZ, 1H, N-CH2CH2Ph-5H), 6.76 (d, J = 12.0 HZ, 3H, N-CH2CH2Ph-6H, N-CH2Ph-3H, N-CH2Ph-5H), 3.90–3.88 (m, 1H, N-CH2β), 3.87–3.76 (m, 12H, 4×-OCH3), 2.98–2.94 (t, J = 7.0 HZ, 2H, N-CH2α). Positive ESI-MS m/z: 330.2 [M + H]+.
3.1.19. Preparation of N-2,3-Dimethoxybenzyl-3,4-Dimethoxy-2-Phenylethanamine (6b)
Amixture of 6a (1.5 g, 4.6 mmol) and Pd/C(5%, 150 mg) in MeOH (50 mL) was stirred under a hydrogen atmosphere (1 bar) for 8 h. The MeOH-insoluble material was filtered off and the filtrate was evaporated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/petroleum ether 1:1), yielding compound 6b (1.3 g, 87%) as a yellow oil. 1H-NMR (400 MHZ, CDCl3, δ; ppm), 7.01–6.97 (t, J = 8.0 HZ, 1H, N-CH2Ph-4H), 6.83 (d, J = 6.4 HZ, 2H, N-CH2CH2Ph-5H, N-CH2CH2Ph-6H), 6.78 (d, J = 8.0 HZ, 1H, N-CH2CH2Ph-5H), 6.73 (d, 2H, J = 11.6, N-CH2Ph-3H, N-CH2Ph-5H), 3.84–3.77 (m, 12H, 4×-OCH3), 2.87–2.83 (t, J = 7.0 HZ, 2H, N-CH2α), 2.78–2.74 (t, J = 6.8 HZ, 2H, N-CH2β), 1.89 (s, 2H, N-CH2). Positive ESI-MS m/z: 310.2 [M + H]+.
3.1.20. Preparation of N-Methyl-N-2,3-Dimethoxybenzyl-3,4-Dimethoxy-2-Phenyl-Ethanamine (6c)
To a mixture of 6b (1.2 g, 3.6 mmol) and K2CO3 (1.0 g, 7.2 mmol) in anhydrous THF (20 mL),CHI (610.6 mg, 4.3 mmol) was slowly added under an ice-bath over a 10-min period. The solution was stirred for 4 h at room temperature, condensed, and extracted with EtOAC. The combined extracts were dried over anhydrous Na2SO4 and filtered, and the filtrate was concentrated to give a crude product, which was purified by silica gel column chromatography (CH2Cl2/petroleum ether 10:1), yielding compound 6c (950 mg, 76% yield) as a colorless oil. 1H-NMR (400 MHZ, CDCl3, δ; ppm) 7.06–7.00 (dd, 2H, J = 9.6 HZ, J = 4.8 HZ, N-CH2CH2Ph-5H, N-CH2CH2Ph-6H), 6.86 (t, 1H, J = 4.2 HZ, N-CH2Ph-4H), 6.78 (d, 1H, J = 8.4, N-CH2CH2Ph-2H), 6.73 (d, 2H, J = 7.2, N-CH2Ph-5H, N-CH2Ph-3H), 3.86–3.82 (m, 12H, 4×-OCH3), 3.74 (s, 2H, N-CH2), 2.87 (t, 2H, J = 7.2, N-CH2α), 2.77–2.71 (m, 2H, N-CH2β), 2.37 (s, 3H). Positive ESI-MS m/z: 346.2 [M + H]+.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}