1. Introduction
One of the most important goals in cancer therapy is the formulation of alternative systems for pharmacological treatments. Nanotechnologies have the potential to revolutionize the existing therapies, through innovative strategies able to deliver molecules to desired sites in the body, thus improving the therapeutic index of drugs. The use of nanoparticles (NPs) could increase the bioavailability of water-insoluble drugs allowing them to pass physiological barriers; moreover, they can carry large amount of cargo [
1]. Tumor targeting approaches have been developed to enhance the effectiveness and reduce side effects by regulating the random cancer drugs biodistribution in the body [
2].
It is well established that solid tumors are often characterized by the over-expression of specific molecules like antigens or receptors on their cell surface. Therefore, passive nanoparticles accumulation by the enhanced permeability and retention (EPR) effect can be associated with an active targeting, since nanoparticles can be functionalized with specific compounds able to link tumor cell surface molecules [
3]. For instance, small molecules have received considerable attention as potential targeting ligands, due to their low molecular weight, ease of conjugation with nanoparticles and low production costs [
4]. Folate, which is essential in many metabolic processes involved in cell survival, shows high specificity for its receptor that is over-expressed in many types of tumor cells. Indeed, it can be conjugated to NPs for a specific targeting in drug delivery systems [
5]. For example, folate-functionalized liposomes were used for acute myelogenous leukemia treatment, while folate-conjugated methotrexate-loaded PAMAM(G5) dendrimer induced a tenfold reduction in cervical tumor size and exhibited less systemic toxicity, compared with free methotrexate [
6,
7]. Furthermore, nanocomplexes can be optimized in order to obtain a controlled drug release. Recently, various “smart nanosystems” have been developed to deliver drugs or biological molecules in response to an internal or external stimulus such as redox, temperature or pH variations: in this way it is possible to improve both in vitro and/or in vivo drug release profiles [
4,
8,
9]. Indeed, the in depth knowledge of the target site (like tumor tissue) can be exploited to perform a specific drug controlled release nanosystem. For example, reduced glutathione (GSH) is well known to be implicated in the maintenance of cellular homeostasis in biological systems, acting as detoxifying agent toward reactive oxygen and xenobiotic species [
10,
11]. Indeed, GSH acts by inhibiting the toxic action of metabolites produced during normal metabolic processes or in stress condition that it typical of the tumor cells. The presence of a pendant thiol group (−SH) in GSH makes it a very efficient reducing agent present at different concentrations in intracellular and extracellular compartments [
8]. It is well-known that the intracellular environment is different from extracellular in pH and redox states. In particular, intracellular glutathione concentration (~1–10 mM) is higher with respect to the extracellular GSH concentration (~2–20 μM). In addition, the cytosolic GSH level in cells with enhanced oxidative stress (like tumor cells) is 7–10 fold higher than normal cells [
12]. In this view, a GSH-controlled release strategy is based on this largely difference of the redox potential between intracellular and extracellular compartments as well as the further elevated GSH concentration in tumor cells, compared to the healthy cells [
9,
10,
12].
Herein, we have developed carboxyl functionalized poly(vinylpyrrolidone) (PVP) nanogels (NGs), produced by pulsed electron-beam irradiation. The choice of nanogels as substrate is motivated by the peculiar properties of these nanoparticles; their high colloidal stability when dispersed in aqueous media in a wide pH range and their shape change ability, provided that the crosslinking density is not too high. Their flexibility and shape changing ability can facilitate the bypass of biological barriers, ensure protection of the payload and enable interaction of the attached ligand with its receptors [
13,
14,
15].
The synthetic methodology chosen for the production of nanogels utilizes ionizing radiation to induce inter- and intra-molecular crosslinking and functionalization of polymer chains in aqueous solutions. The distinctive advantage of this approach is that no surfactants are required to control the locus of reaction and that chemically stable polymers, such as PVP, can be used as a starting material. As a result, purification is simpler and products are non-toxic. Radiation doses within the sterilization dose range are applied to the systems.
The reactions constituting the basis for nanogel formation and the approaches to control particle size and functionality have been discussed in two recent reviews [
16,
17]. Briefly, N
2O-saturated aqueous solutions of PVP are irradiated to short pulses of accelerated electrons. The ionizing irradiation interacts mainly with water producing the following species:
In N
2O-saturated aqueous solutions electrons are converted in hydroxyl radicals.
•OH abstracts hydrogen atoms from the polymer forming macroradicals and can also recombine at low polymer concentration. The polymer carbon center radicals can undergo several possible reactions, depending on polymer concentration and dose rate, as reported in
Scheme 1.
In the absence of oxygen, the main reactions of the polymer radicals are inter- and intra-molecular crosslinking. However, disproportionation reactions can also occur to less extent, leading to the formation of a double bond on one chain (when intermolecular) or on one site of the polymer chain (when intramolecular).
While the inter-molecular processes take place mainly between the lower molecular weight polymer molecules in the system, due to the lower number of free radicals per chain and their higher chain mobility, the intramolecular processes occur preferentially in the higher molecular weight chains, due to the higher number of radicals per chain and lower chain mobility. These two simultaneous processes lead to narrower molecular weight and particle size distributions, hence lower polydispersity values [
18,
19].
Under our experimental conditions, at high doses, molecular oxygen can be produced via the following reactions:
and, in turn, react with the polymer radicals forming peroxyl radicals.
The peroxyl radicals can undergo further reactions, producing ketones and carboxyl groups. The most interesting chemical modification of PVP nanogels for the purpose of their application as biomedical nanocarriers is the formation of carboxyl groups and primary amino groups [
18,
19].
The addition of a vinyl monomer to the polymer solutions introduces a competitive reaction between
•OH with the double bond, and
•OH with the polymer. This decreases the concentration of formed macroradicals. It is also expected that the monomer free radicals would react with the disproportionally-produced double bonds in the backbone of the polymer chain through addition reaction (grafting). The grafting of the monomer onto the polymer can also take place through the combination reactions of
with polymer radicals (see
Scheme 2).
Since grafting competes with inter-molecular crosslinking reactions, it leads to comparatively smaller particles [
20].
By varying the initial concentrations of polymer and monomer, dose-rate and total absorbed dose, one can enhance the probability of certain reactions taking place over others. As a result, PVP-co-AA nanogels with various molecular weights, hydrodynamic diameters and average concentration of functional groups per nanoparticle can be produced.
In particular, four PVP-co-AA systems were synthetized by varying the polymer and acrylic acid concentrations, and characterized for their hydrodynamic size, molecular weight, surface charge density and concentration of functional groups (carboxyl groups and primary amino groups).
The available primary amino groups of the nanoparticles were covalently bonded to the carboxyl group of folic acid. The ability of the folate-conjugated nanoparticles to recognize tumor cells was tested using Hela cells (characterized by high expression of folate receptor) and NIH3T3 cells (a fibroblastic cells line used as negative control).
Additionally, a selected nanogel system was conjugated to Doxorubicin or Bcl-2 siRNA through a redox-sensitive spacer in order to develop GSH-responsive nanosystems. The resulting nanosystems were characterized in vitro, measuring the drug release kinetics under reducing conditions. Moreover, their effects were evaluated both in tumor and healthy cells. The results show the DOX release from nanogels, triggered by higher intracellular GSH concentration, which resulted in enhanced inhibition of the cellular proliferation after internalization. Proof-of-principle experiment has also demonstrated the gene silencing ability of siRNA-loaded-nanogels on Hela cells under reducing conditions, providing evidence of their potential as nanomedicine carrier for the treatment of cancer.
3. Discussion
In the recent years, active tumor-targeting drug delivery systems have attracted particular interest in cancer treatment, since the conventional chemotherapy lacks specificity against cancer cells and is associated with significant side effects. Functionalized nanoparticles have been shown to have a potential to recognize cancer and accumulate drugs in tumor cells, by conjugating specific molecules to realize this objective [
18]. In previous works, we described a physico-chemical and biological characterization of PVP nanogels like size, shape, biocompatibility, oxidative stress induction, the ability to cross biological membranes, genotoxicity and the easy surface-decoration, qualifying them as potential therapeutic nanocarriers [
13,
14,
15,
16,
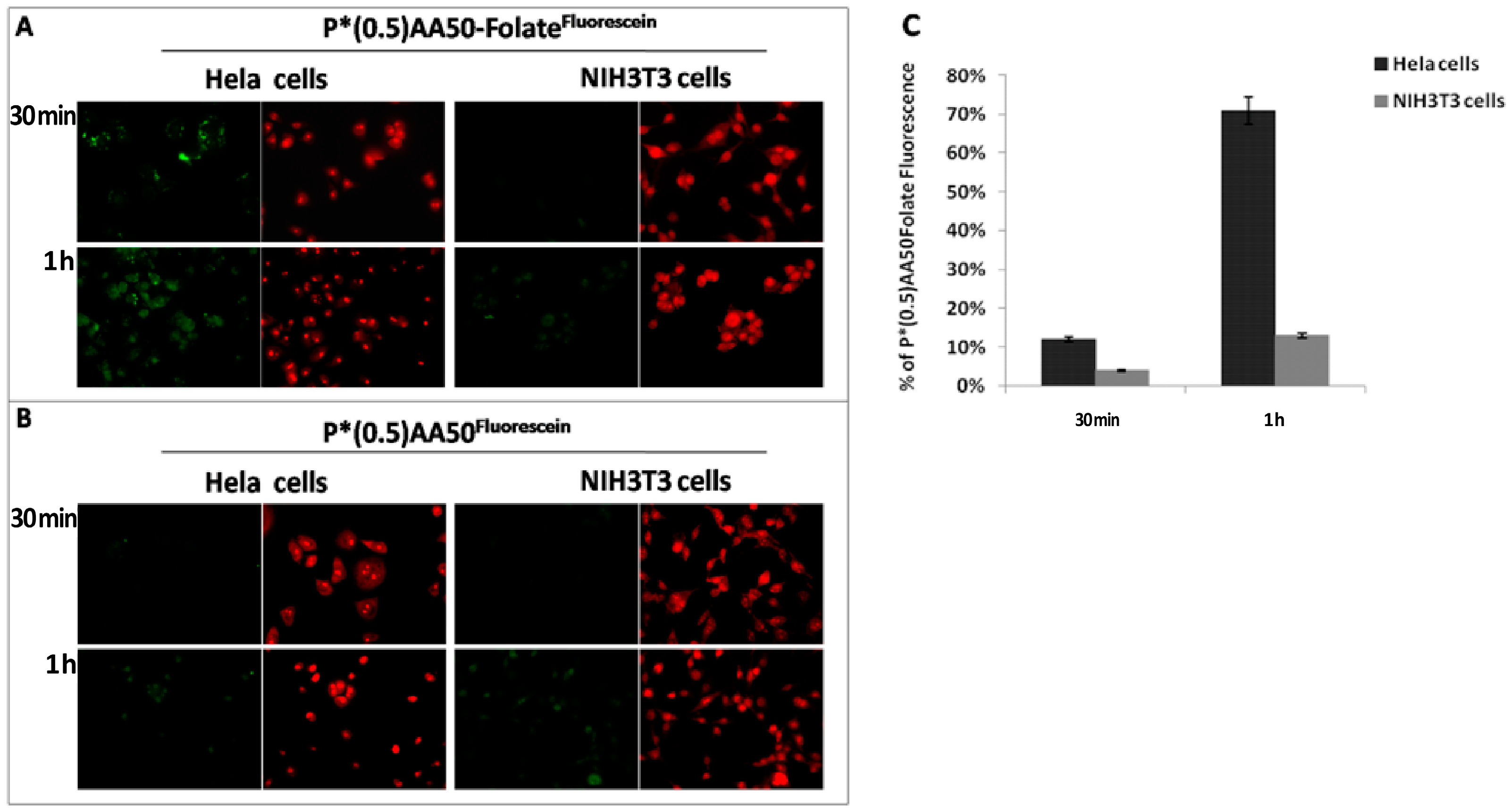
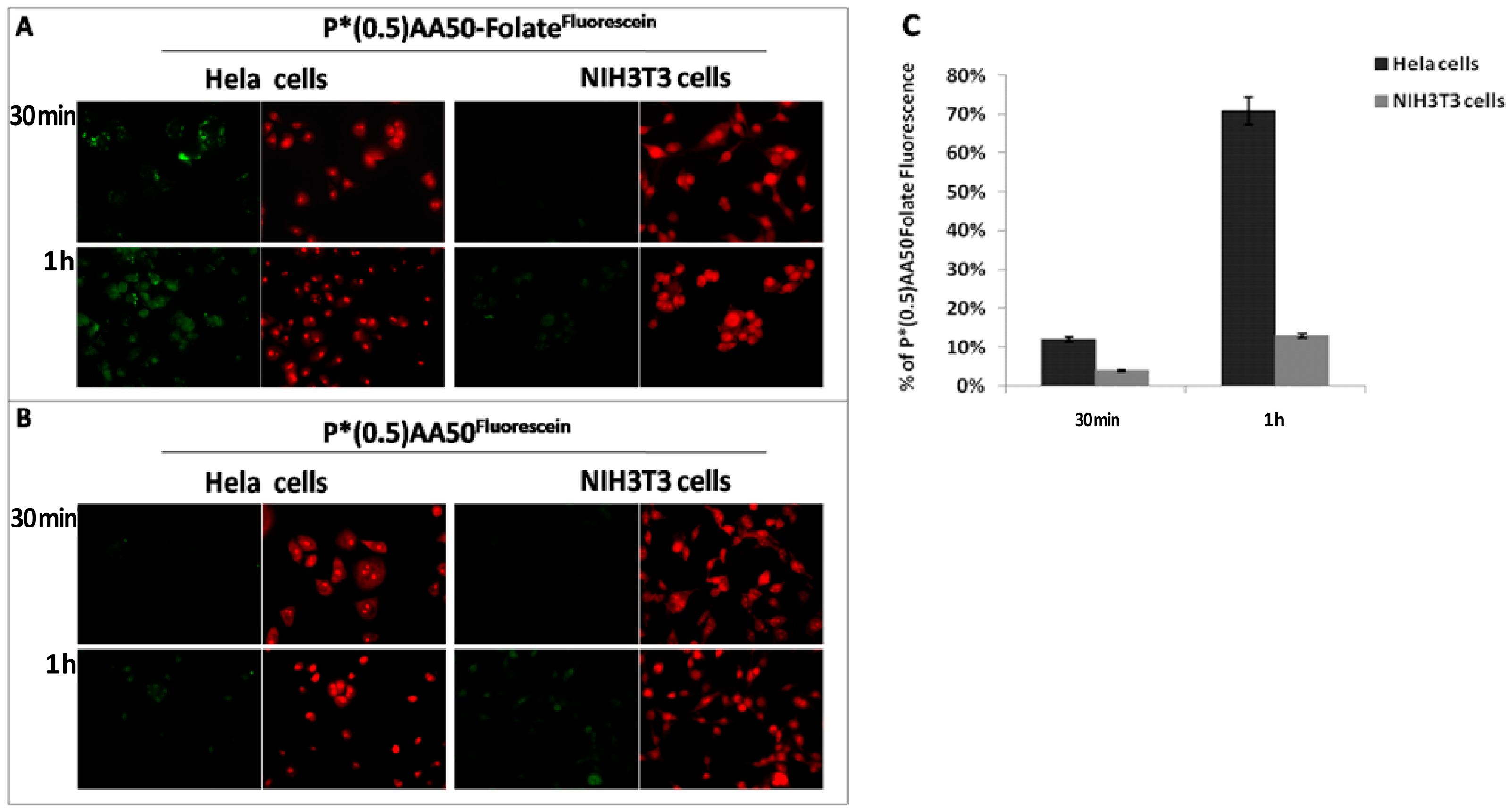
17]. In this study, we initially investigated if the conjugation of NGs with folic acid could improve its tumor targeting ability. Cancer cells are highly dependent on folate for DNA synthesis and cell division, and they express high levels of FR, compared to normal cells. We exploited this differential expression of folic acid receptors on cancer and normal cells in a co-culture system of Hela and NIH3T3 cells respectively, demonstrating that the conjugation of nanogels with folic acid increase the cellular uptake in cancer cells (
Figure 3). Even flow cytometric analysis suggested a fundamental folate role for tumor specific targeting; indeed, it increased NGs affinity for Hela cells through a specific ligand-receptor recognition. One of the mechanisms for the higher efficacy might be upper intracellular accumulation of chemotherapeutics linked to nanoparticles, exploiting a controlled release. Therefore, once established the nanosystems targeting ability, we focused our attention to perform a drug controlled release mechanism, basing on specific tumor cell characteristics like redox state determinate by glutathione (GSH) concentration [
10]. The proposed action mechanism provides the creation of a “stimulus-responsive” nanodrug, sensitive to redox environment changes: at normal reducing condition, such as the extracellular microenvironment or in the body stream, the drug remains hooked to the nanogels. Conversely, when nanogels cross the cellular membrane, in presence of high glutathione levels, as observed in cancer cells, the breaking of the sulfide bridge occurs and drug is release into the cytosol [
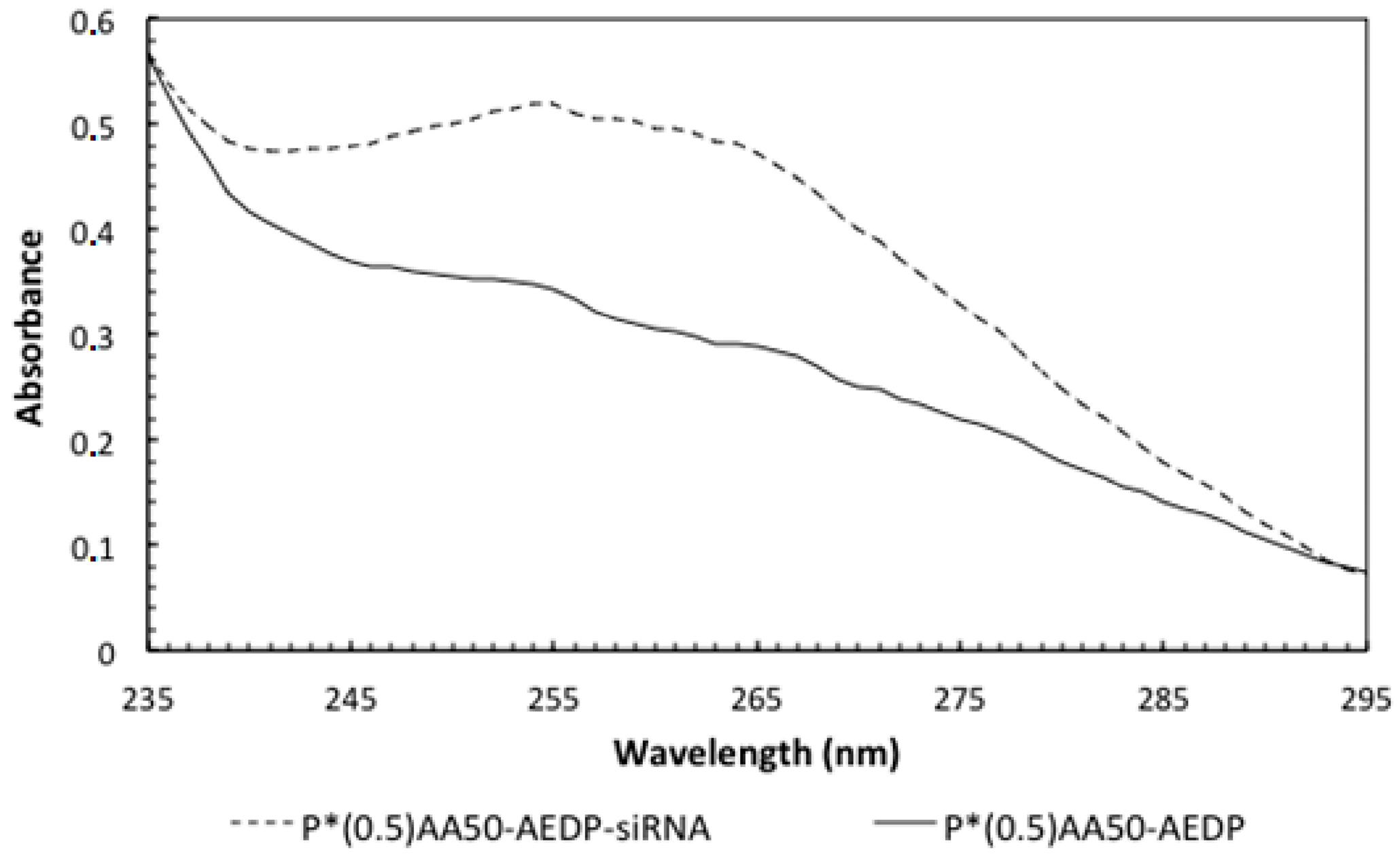
12]. In this view, we selected chemotherapeutic drug doxorubicin as model because of its significant anticancer activity, its chemical structure that allows both the bind with nanogels and follow its trend by red autofluorescence. To develop a glutathione-mediated delivery, drug was linked to nanogels through AEDP spacer containing a cleavable disulfide bridge. P*(0.5)AA50-AEDP-DOX system was prepared and characterized by size and drug loading.
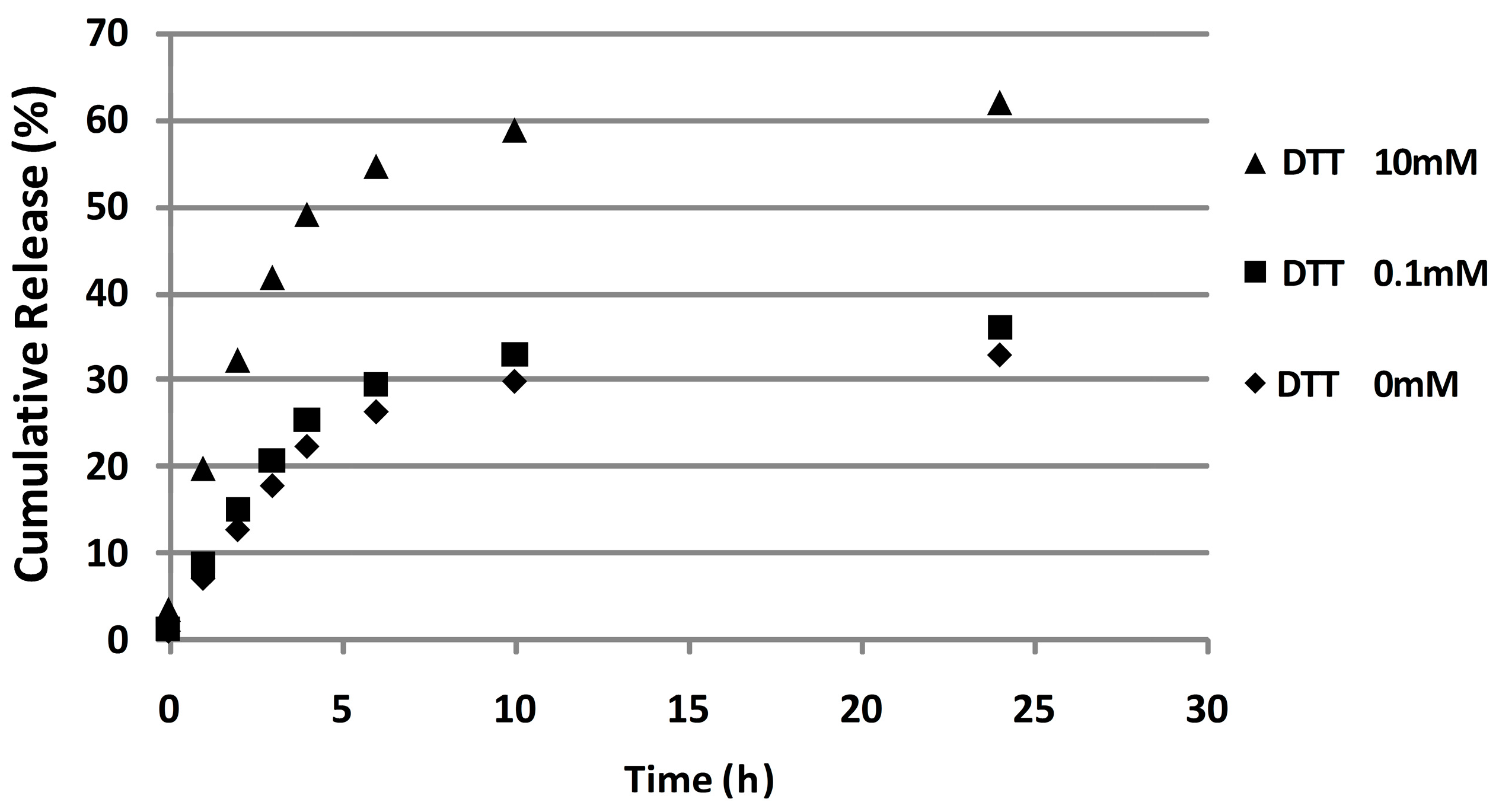
To investigate whether the disulfide bridge is degradable in reducing environments, DOX release profiles were determined in the presence of different concentrations of reducing agent. In particular, 10 mM and 0.1 mM of DTT solution were used to mime the cytoplasm reductive environment of cancer cells and physiological extracellular fluid, respectively. P*(0.5)AA50-AEDP-DOX was able to release twice as much doxorubicin under the reductive stimulus which matches the intracellular cancer GSH concentration (10 mM), by inducing the sulfhydryl bond reduction of the spacer. As expected, in vitro DOX release was highest in reducing condition, which matches with the intracellular cancer GSH concentration (10 mM).
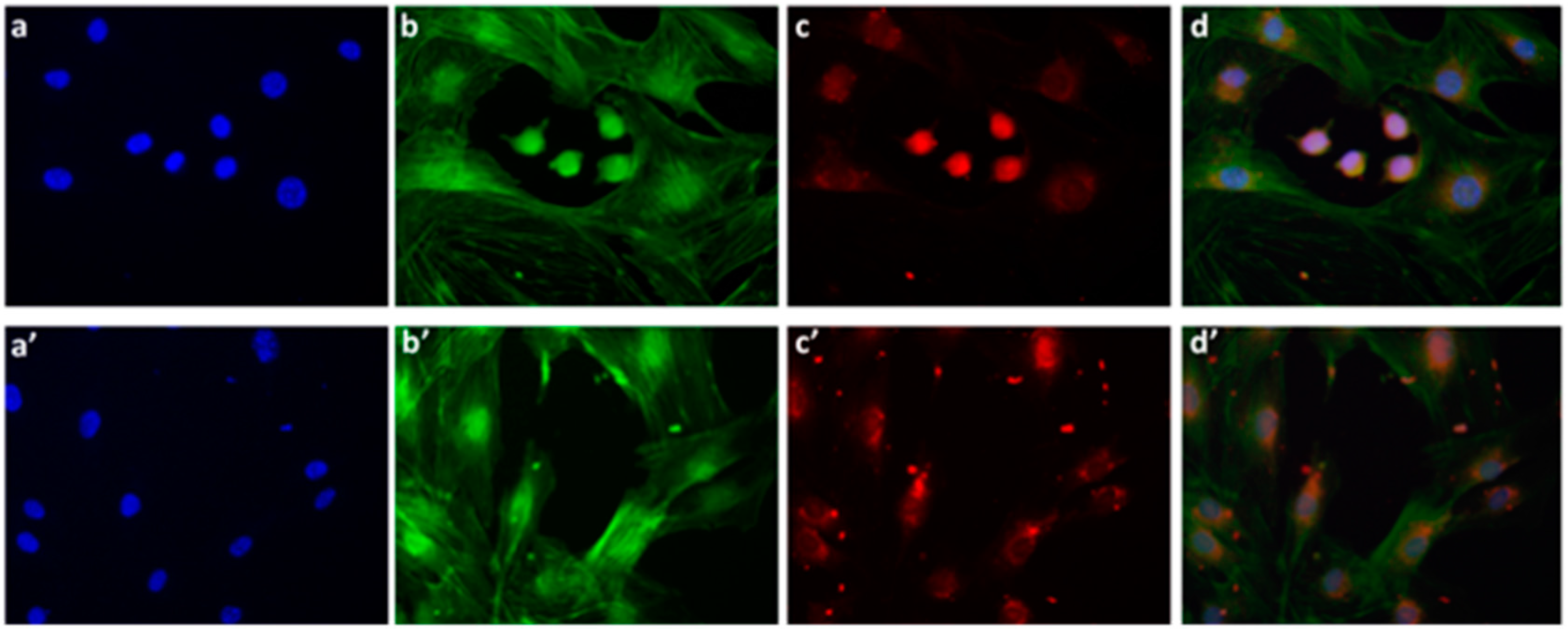
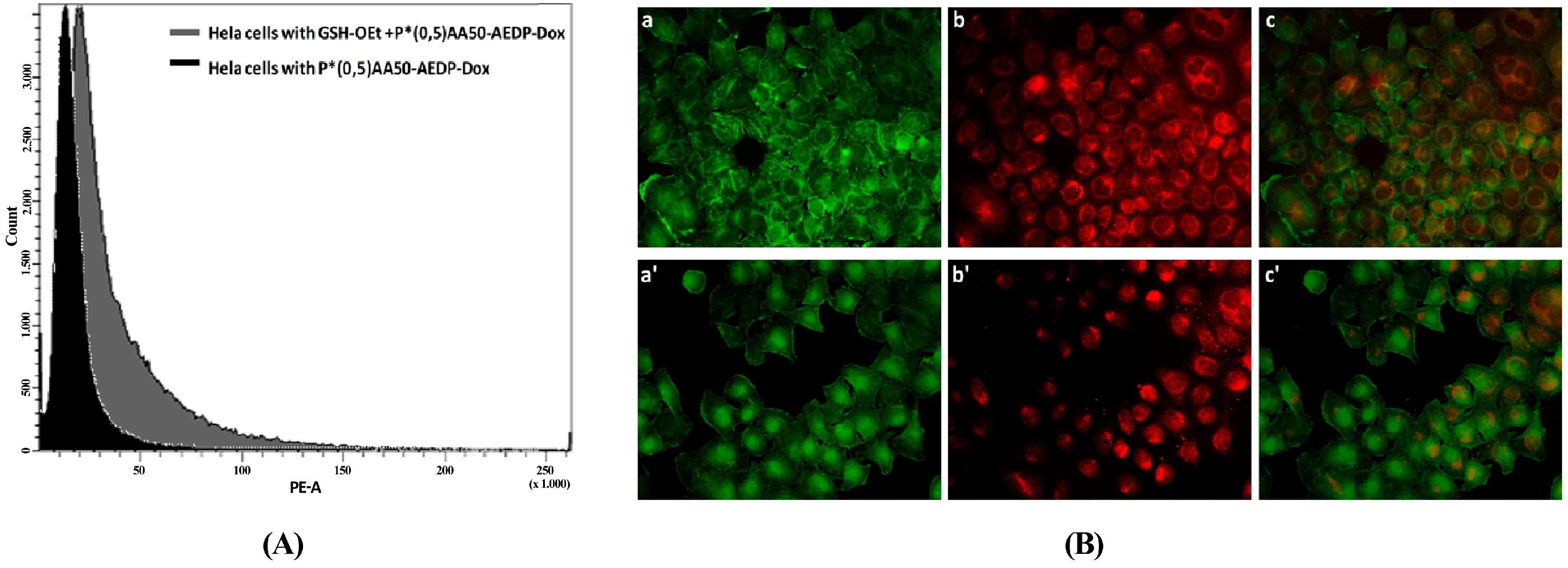
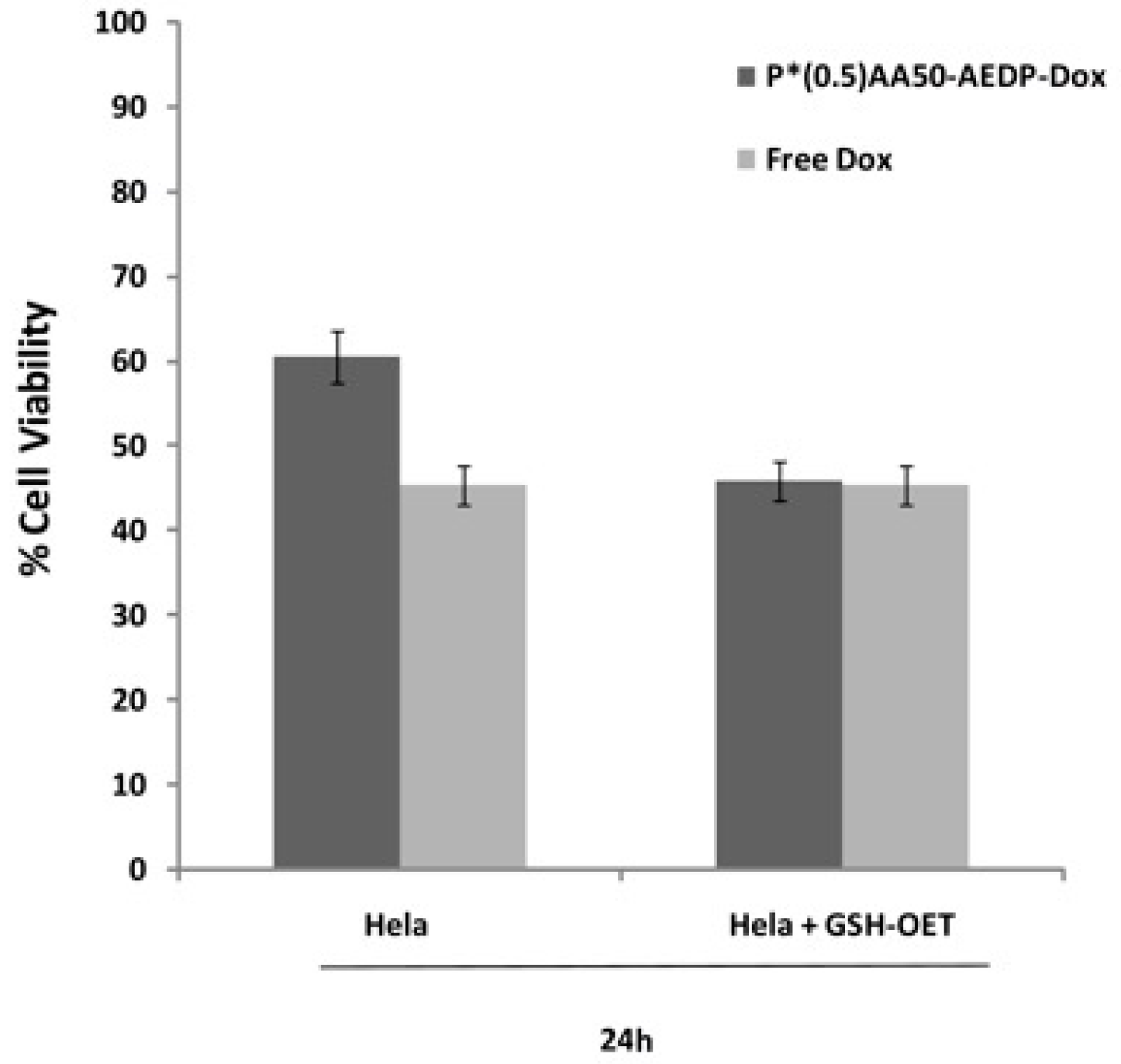
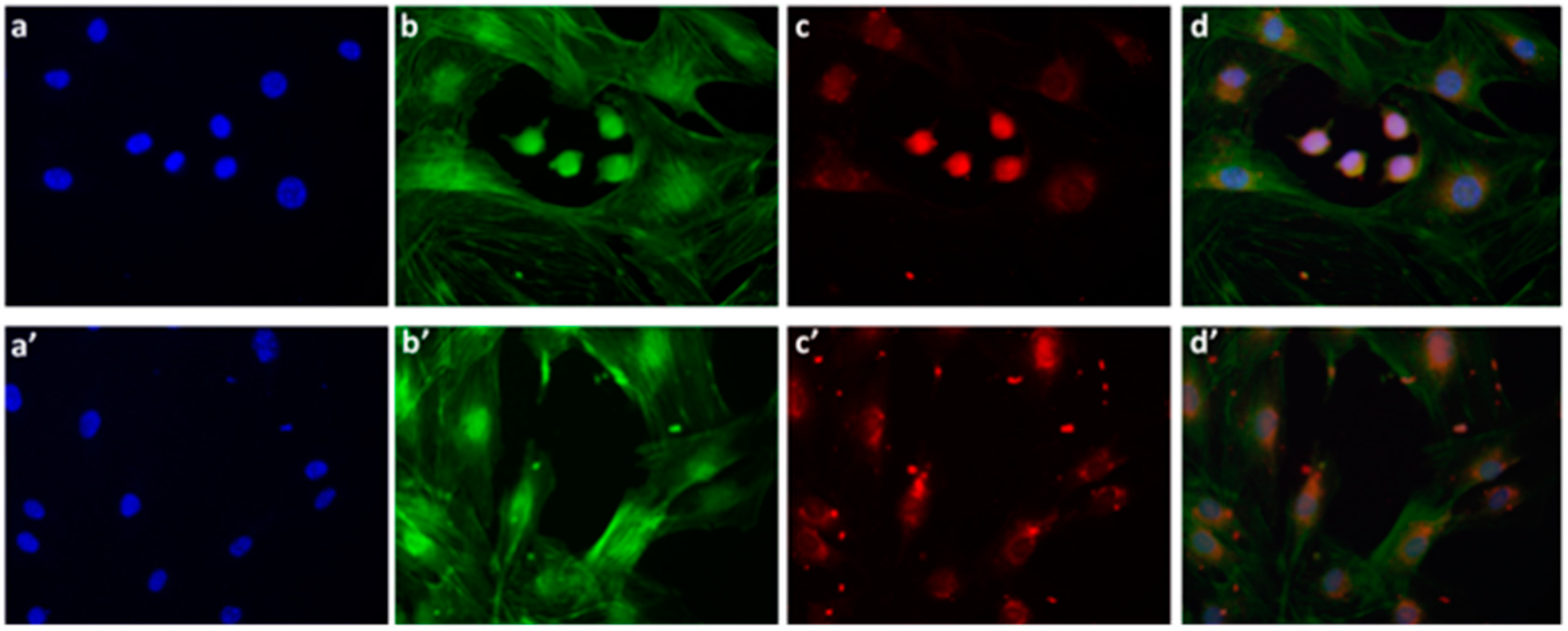
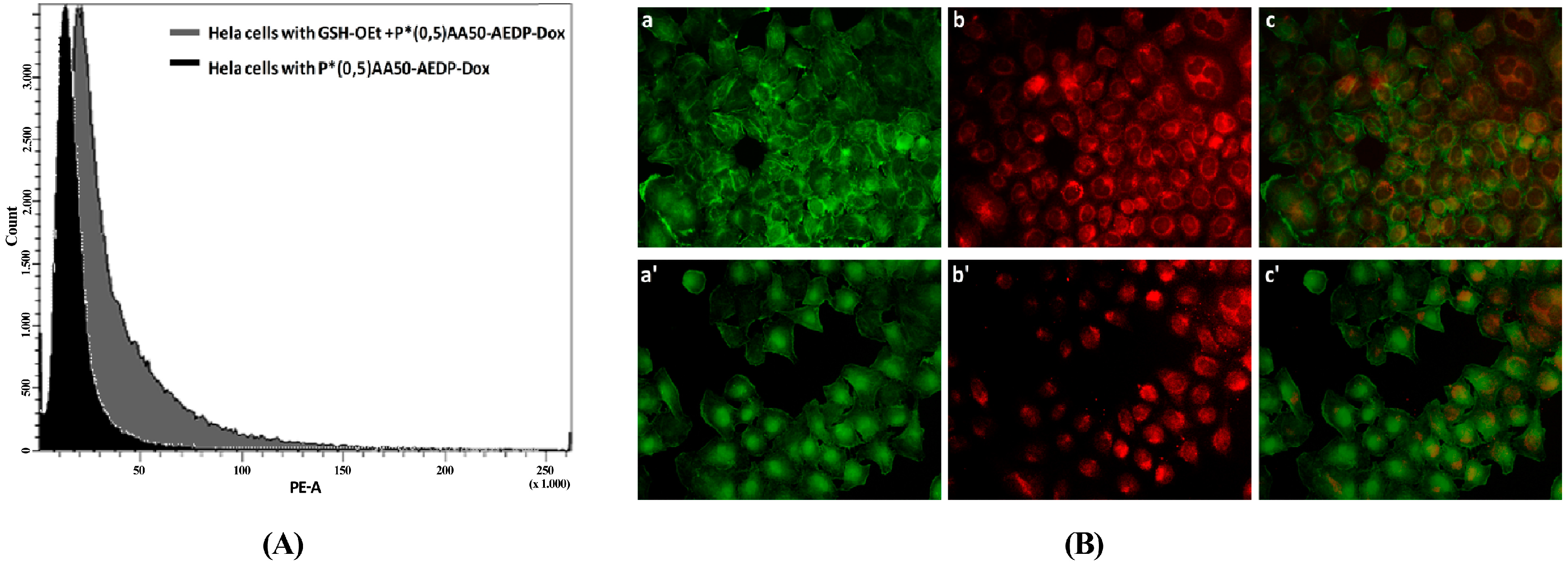
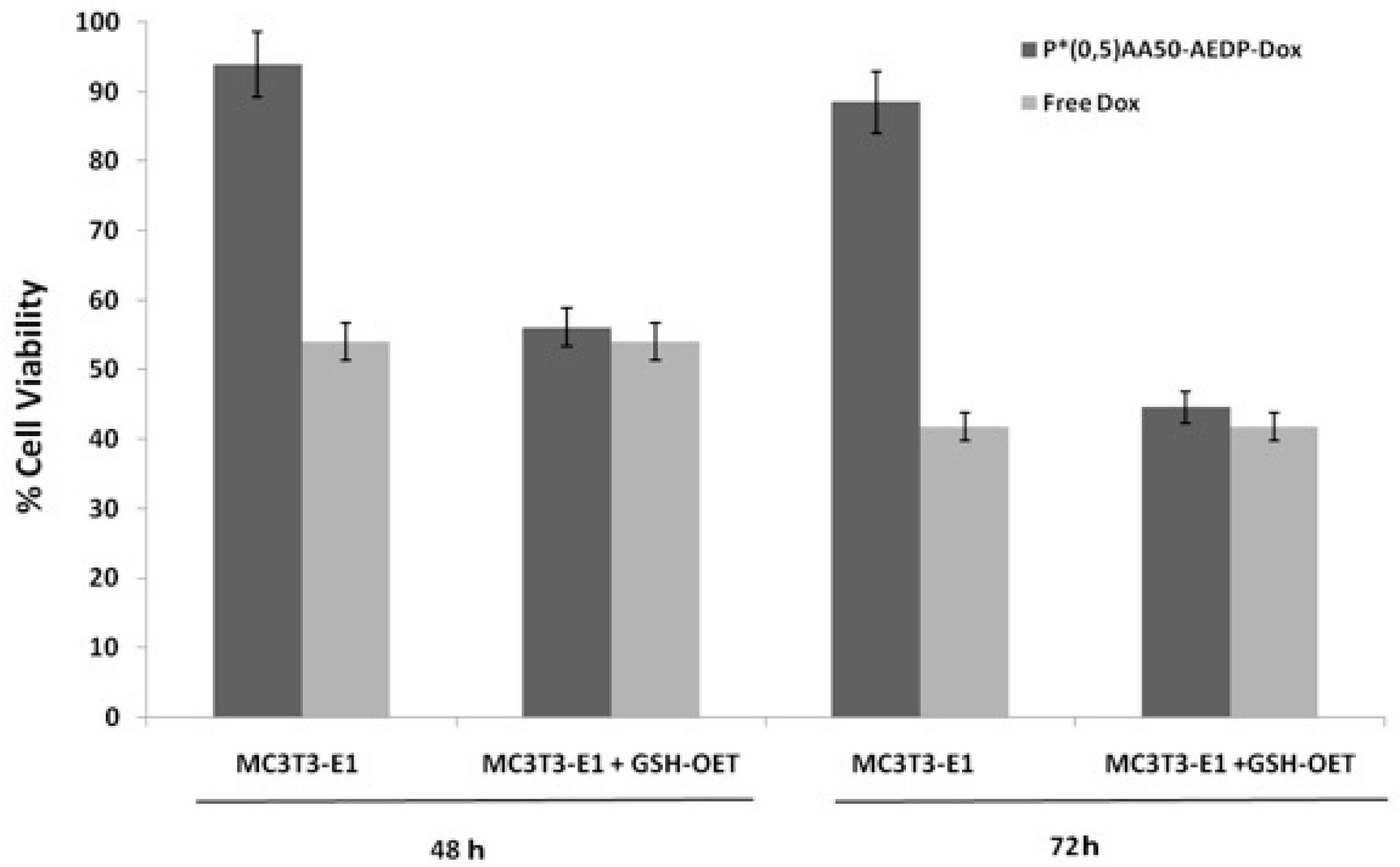
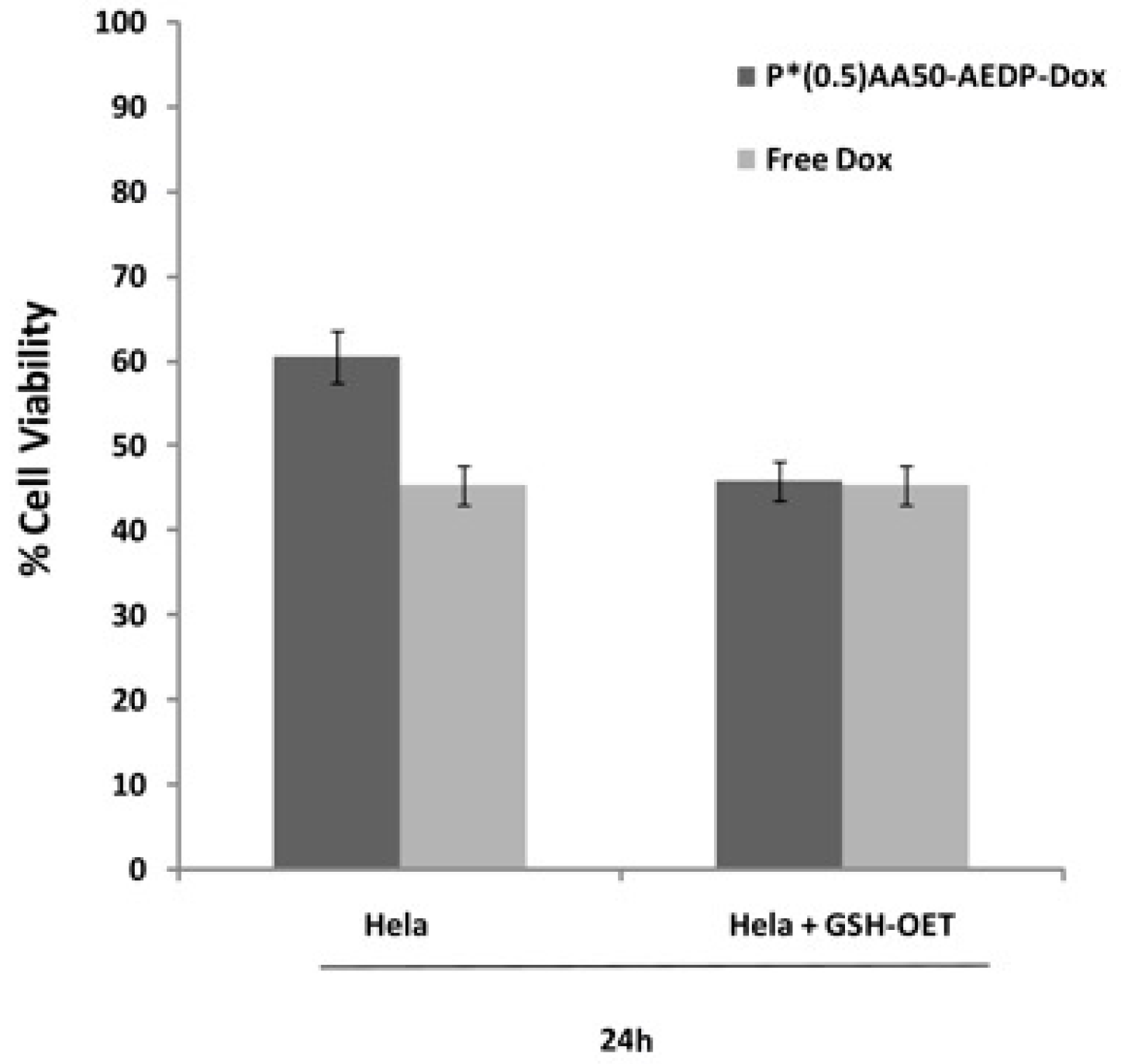
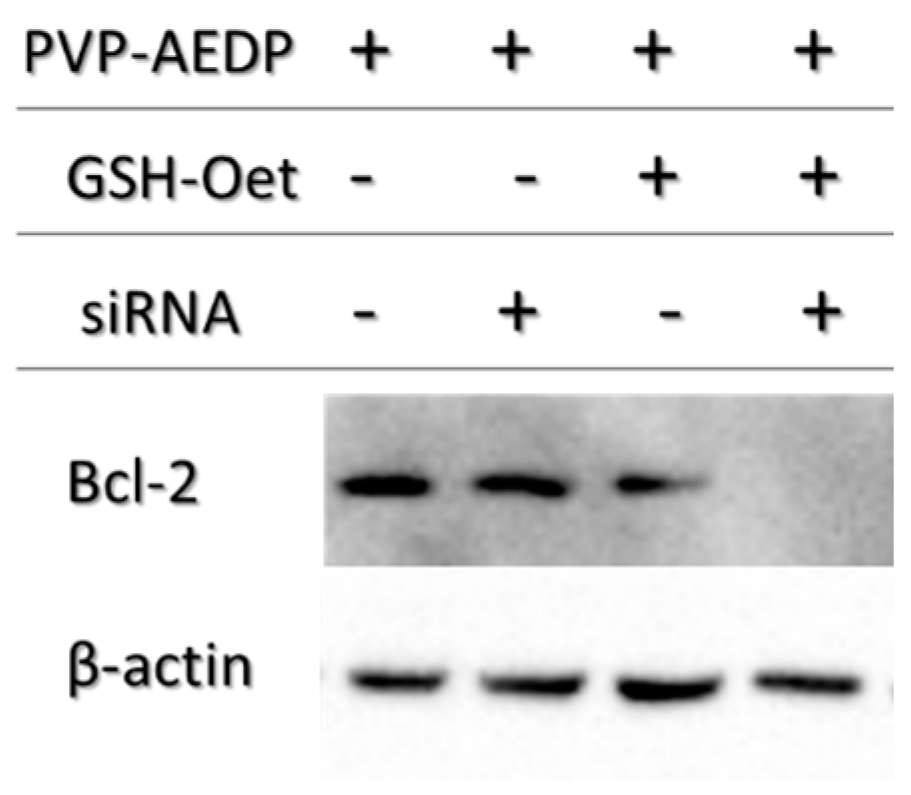
We also examined the P*(0.5)AA50-AEDP-DOX intracellular release in normal and cancer cells (MC3T3-E1 and Hela cells) and the enhanced intracellular DOX release was observed in glutathione monoester (GSH-OEt) pretreated cells. These data suggested that the cytoplasmic glutathione increase could induce disulfide bridge degradation of AEDP linker and consequently the intracellular DOX release from P*(0.5)AA50-AEDP-DOX, as indicated by doxorubicin presence in nuclei and therefore its intercalation into DNA (
Figure 6). By contrast, MC3T3-E1 cells without GSH-OET pre-treatment emitted weak and spotted red DOX fluorescence in the cytoplasm surrounding the nuclei and no evidences of apoptotic events related morphology were observed; thus, it could be hypothesized that DOX remained linked to nanogels, due to the low physiological reductive environment. Moreover, even if glutathione concentration in tumor cells is higher respect to normal ones, several reports also describe GSH-OET pretreatment of cancer cells to enhance cellular GSH levels [
21]. Effectively, confocal microscopy and flow cytometric analysis shown a different drug release profile by comparing pretreated or not tumor cells. Furthermore, the redox responsive drug release also induces higher DOX efficiency as shown by higher cytotoxicity in GSH-OET pretreated Hela and MC3T3E1 cell lines. In relation to cytotoxicity data, it is possible to suppose that the disulfide bridges are stable at physiological condition, but respond to high reductive conditions (GSH) via reversible cleavage into free thiols, triggering a fast DOX release in cytoplasm. Therefore, the glutathione-mediated drug delivery of P*(0.5)AA50-AEDP-DOX is able to realize the rapid intracellular drug release and enhance the growth inhibition to tumor cells. The possibility to easily functionalize these kinds of nanoparticles with molecules of different nature (like folate, aminofluorescein, doxorubicin, and small oligonucleotides) make them optimal candidate for siRNA delivery. Small oligonucleotides, like siRNA, are very unstable and can be easily degraded in bloodstream or in the extracellular matrix. Therefore, their conjugation with PVP nanogels could protect them and target to the specific site [
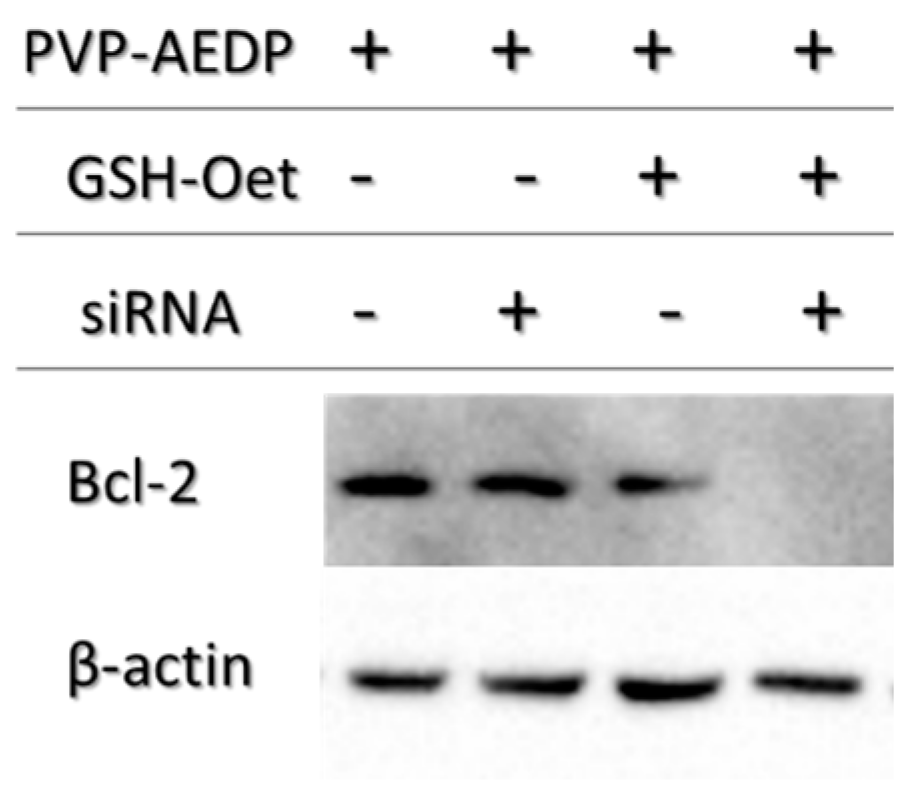
14]. For this purpose, we choose a siRNA for Bcl-2, the first anti-death gene discovered, implicated in tumor progression. RNAi preliminary studies had shown P*(0.5)AA50-AEDP-siRNA propensity as siRNA delivery system; indeed, they are able to link modified siRNA without any alteration of their silencing functionality after has been released, as indicated by Western blot analysis. All these data suggest the possibility to develop an innovative therapeutic approach for tumor therapy. Evidently, the progress in the development of these smart nanogels and their prodrugs version will keep by in vivo studies in order to better validate their properties. The introduction of stimuli-responsive drug delivery system should be considered a good way to improve nanomedicine progress. Moreover, the opportunity to obtain a specific tumor targeting and to trigger drug release systems can be considered an innovative nanomedicine device. The future of our current work will be directed to identify new therapeutic targets to extend the use of these nano-systems in various human diseases observing their behavior in vivo, using mouse model. The simultaneous conjugation with different bio-molecules could improve their efficiency not only concerning tumor therapy, but also neurodegenerative pathologies.
4. Materials and Methods
4.1. Materials
Aminofluorescein (AF), Folic Acid (FolAc), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC),
N-Hydroxysulfosuccinimide (Sulfo-NHs), 2-(
N-morpholino)ethanesulfonic acid (MES), glutathione (GSH), Nickel(II) chloride hexahydrate, 4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), Pyrocatechol Violet (PV) were supplied by Aldrich and were used without further purification. 3-(2-aminoethyl) dithiopropionicacid (AEDP) was supplied by VWR. Poly(
N-vinyl pyrrolidone)-co-acrylic acid nanogels (NG) were synthetized by electron beam irradiation and used as substrates [
20]. Systems are named as P*(X)AAY, where X is the weight percent of polymer in solution and Y is the molar ratio between the PVP repeating unit in the polymer and the acrylic acid in the irradiated solution (nominal concentration of carboxyl groups in the system). The actual concentration of carboxyl groups either formed by radiation-induced oxidation of PVP or grafted with AA was determined by colorimetric method that is based on Nickel complexation [
19]. The primary amino groups present in the nanogels were estimated were titrated upon reaction with fluorescamine [
19].
4.2. Preparation of NG-Fluorescein, NG-Folic Acid, NG-Doxorubicin and NG-siRNA Variants
NGs were labeled with a fluorescent probe, AF, using an EDC/Sulfo-NHS protocol in order to activate the carboxyl groups of the nanogels [
20]. A given volume of NG aqueous dispersion was mixed with EDC and Sulfo-NHS aqueous solution (MES pH 5.5) for 30 min, then a given amount of AF was added, keeping constant the molar ratio between the nominal concentration of carboxyl groups in the system and the probe added to the reactor feed (0.25 and 10). The reaction was conducted for four hours under gentle stirring at room temperature in dark conditions. The determination of the amount of probe conjugated to the nanogel was carried out by spectrophotometer analysis. While bare NGs do not show any absorption band in the wavelength range where AF absorbs, the AF-conjugated systems show the absorption band of AF, with a red-shift (+5 nm). Since AF can also be physically entrapped into the hydrophobic pockets created by crosslinking, nanogels incubated with AF in the same conditions as for the conjugation, but without EDC/sulfo-NHS, were prepared as a control and subjected to the same release experiment. While all entrapped AF was released after 48 h by the control, a percentage of AF was retained by the conjugated systems. The amount of conjugated AF was calculated as the difference between the amount of probe in the uptake solution and the amount of probe released after 48 h. Samples for biological analysis were dialyzed for longer than 48 h in dark conditions.
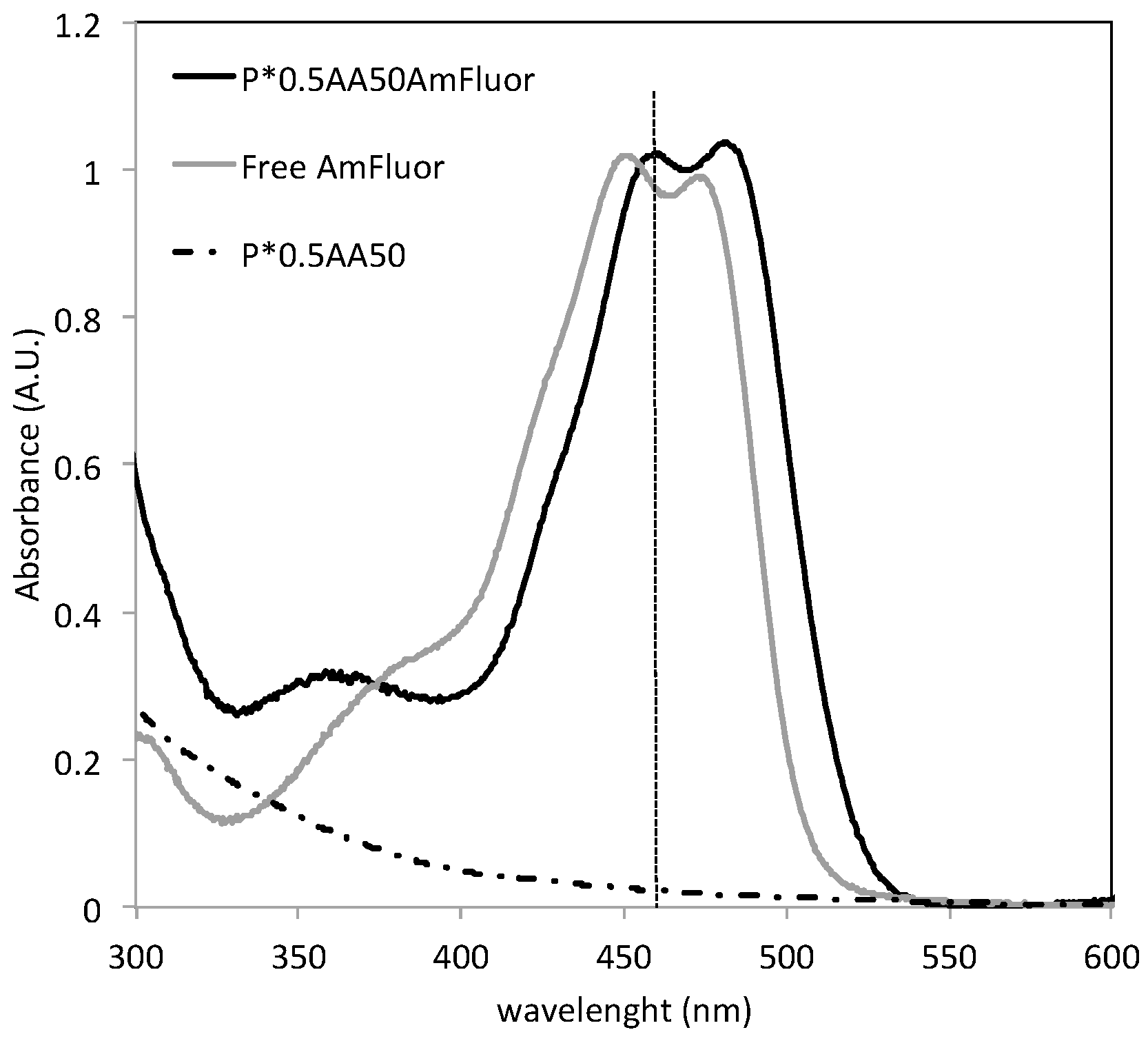
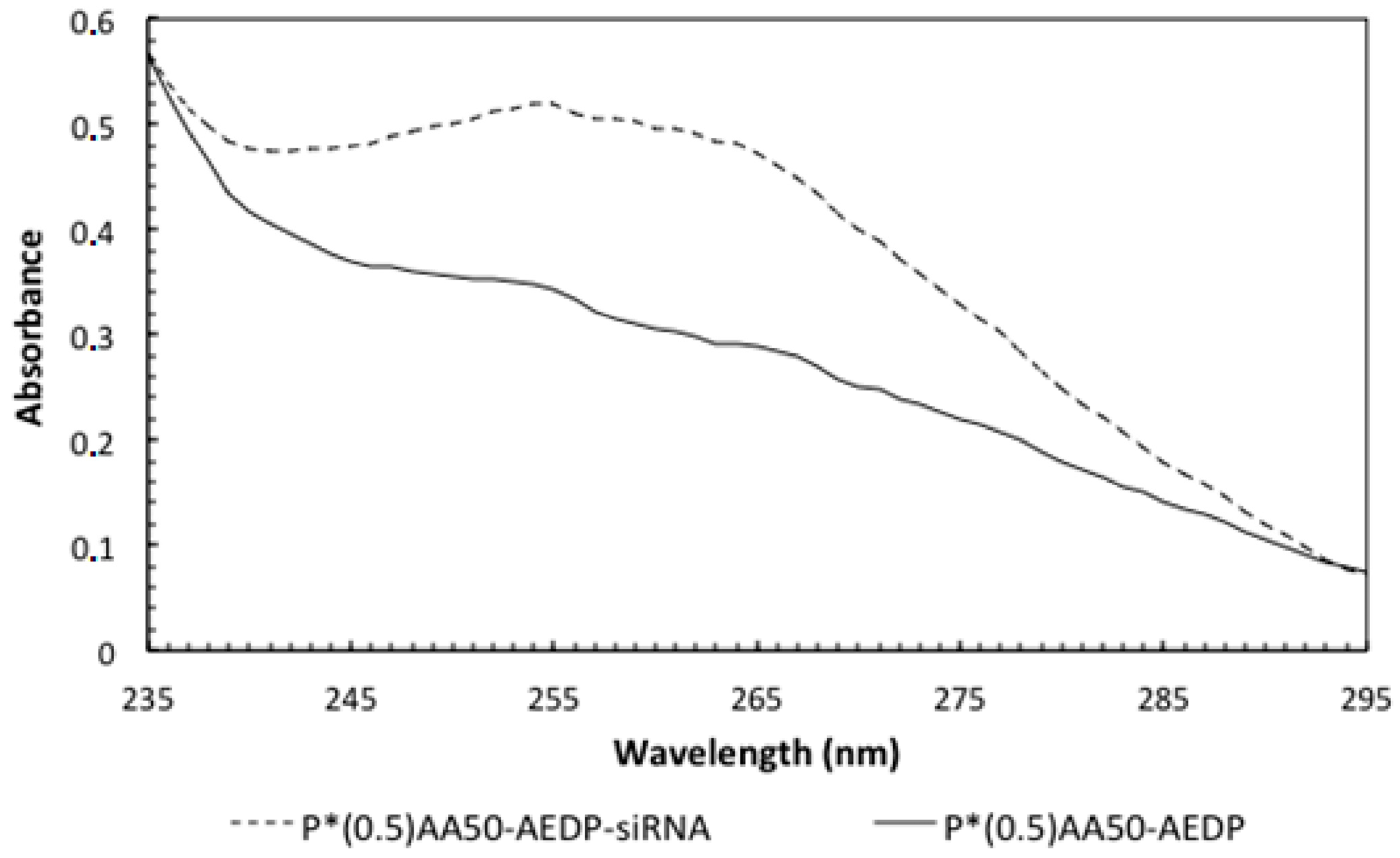
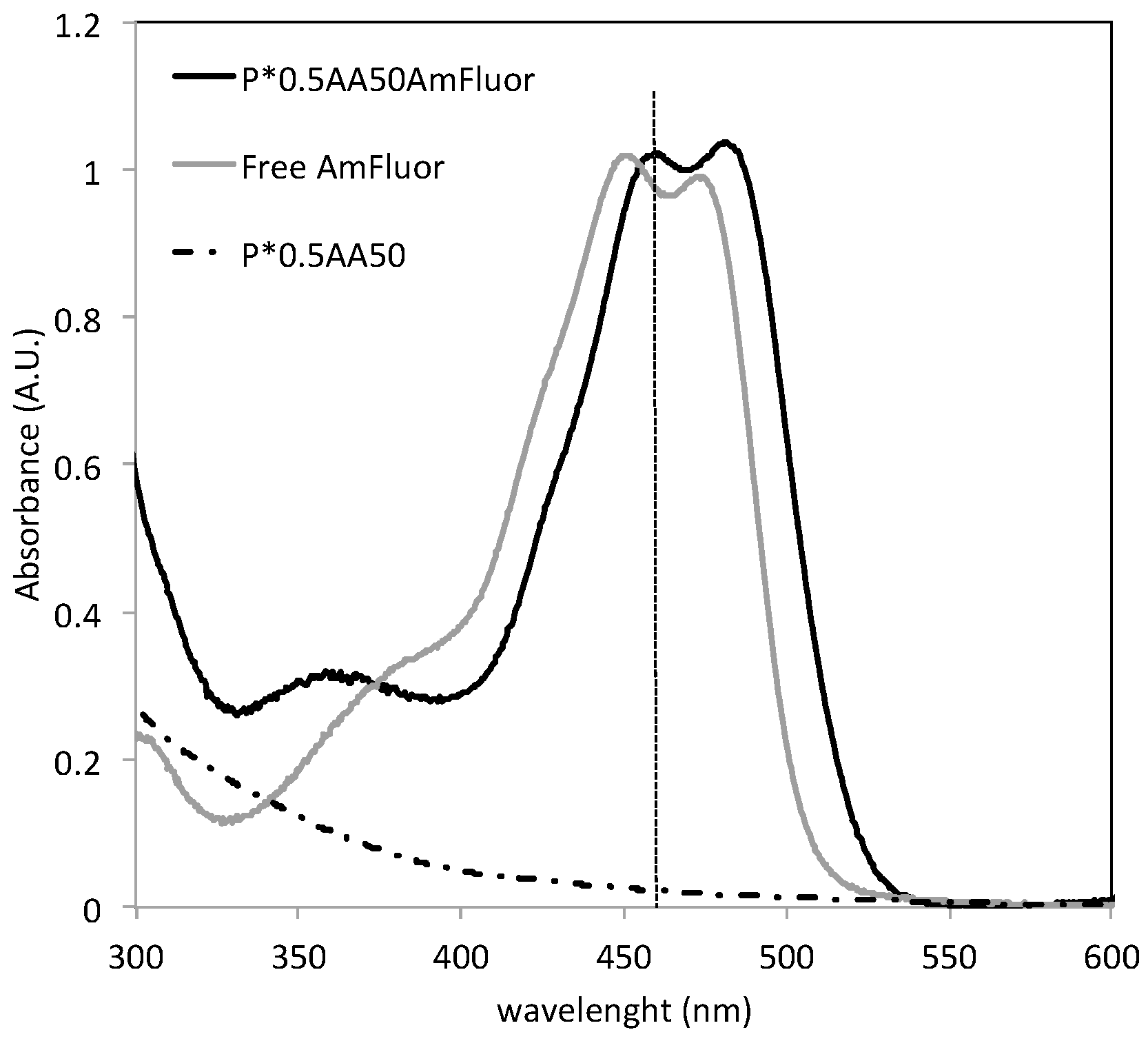
Conjugation with folic acid was performed using the same EDC/Sulfo-NHS protocol used for AF In consideration of the fact that the NGs have primary amino groups, folic acid is likely to be coupled to the amino groups of the nanogels via reaction of one the carboxyl groups [
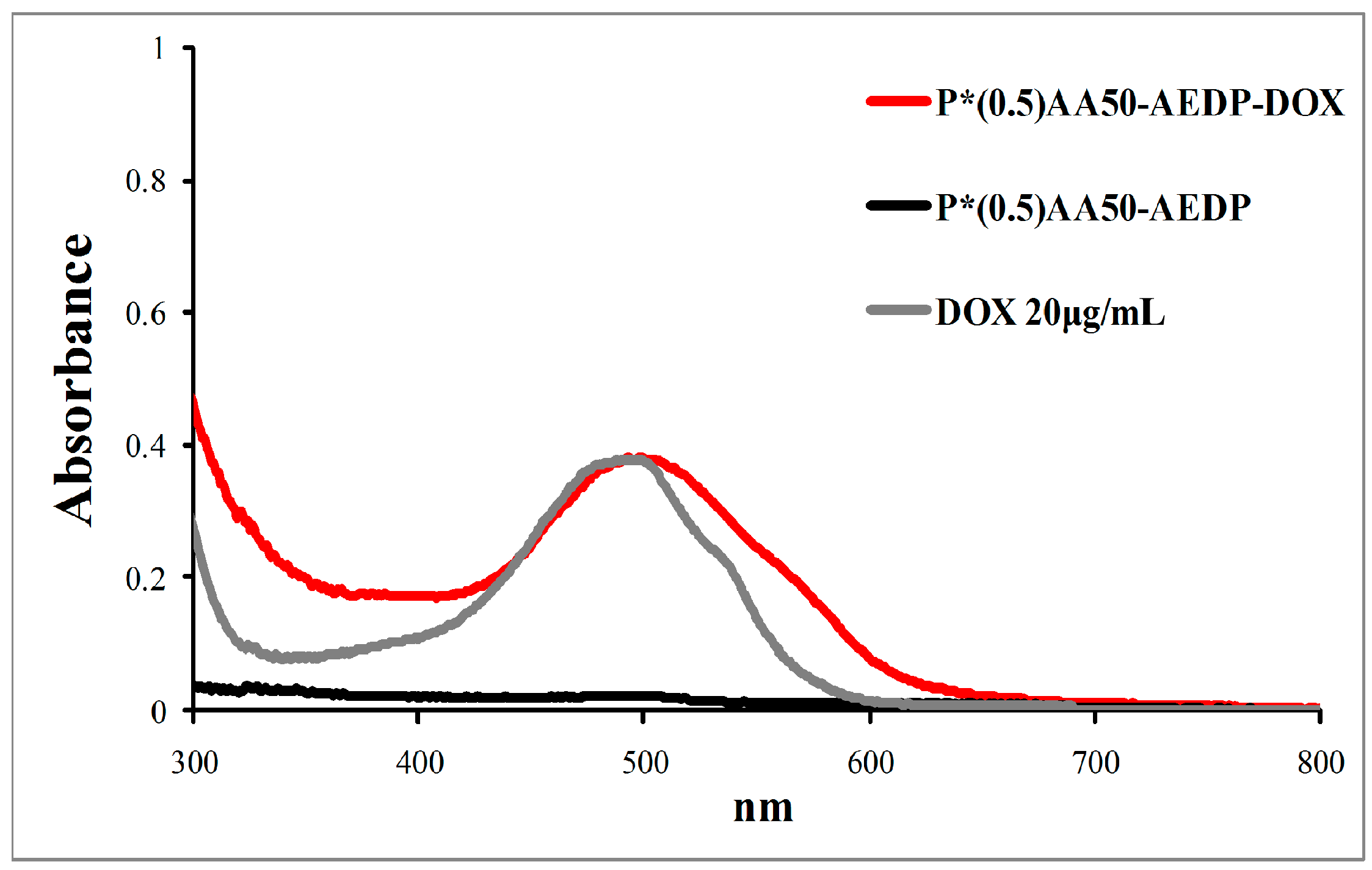
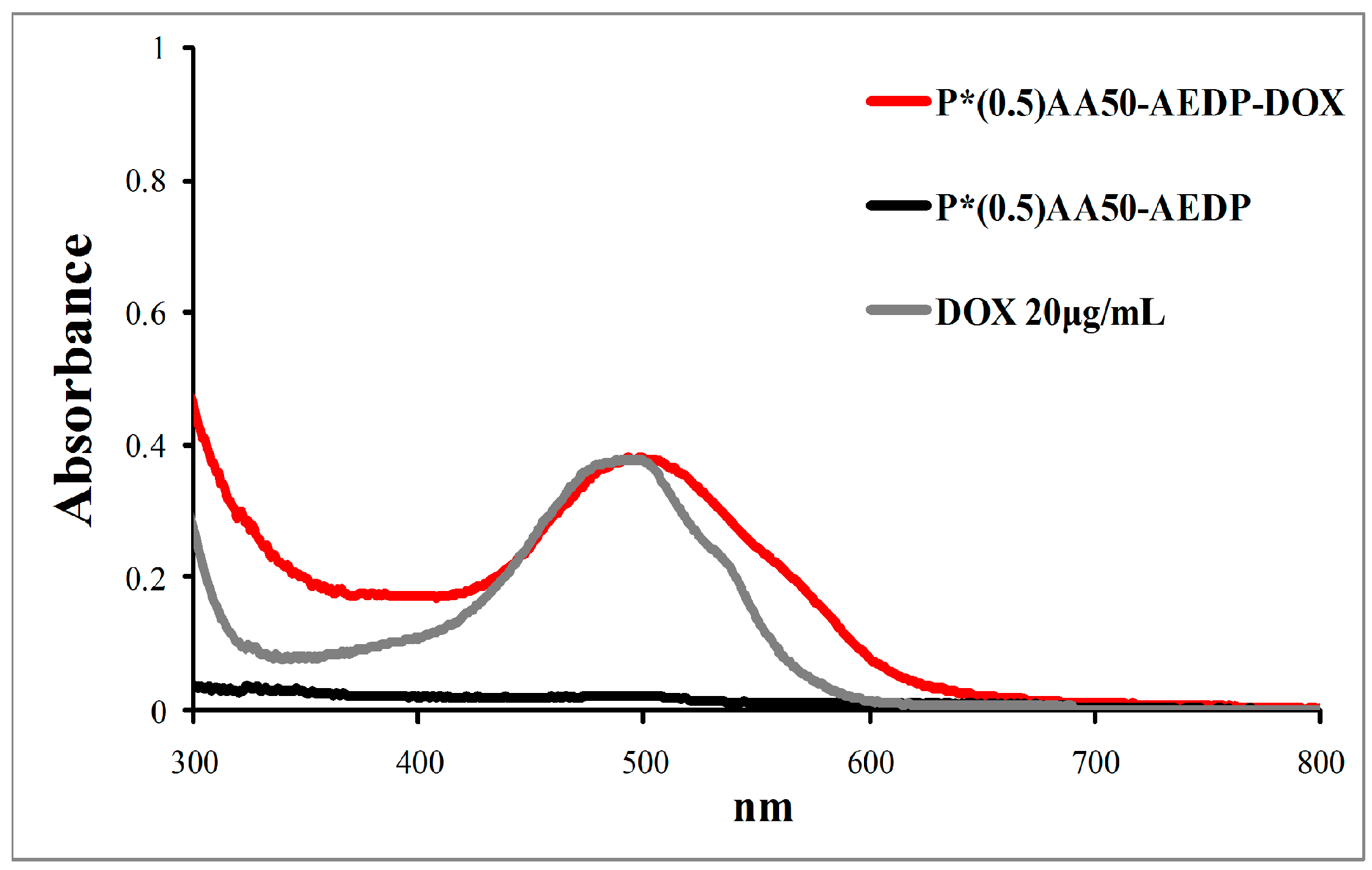
22]. The product was purified by dialysis against water, using 100 kDa cutoff dialysis tubes. The conjugation degree was determined by spectrophotometric measurements of the dialyzed product (
Figure 12).
For the synthesis of doxorubicin-conjugated variants, P*0.5AA50 NGs were selected and conjugated first with AEDP and then with doxorubicin hydrochloride (DOX, Ebewe pharma). For both conjugation reactions, the standard EDC/Sulfo-NHS protocol was used. Reaction and purification conditions after each modification step were the same as those used for AF conjugation.
For the first step, the amount of AEDP was scaled down in order to minimize interparticle bridging. In these conditions, AEDP homopolymerization was tested to be negligible.
For siRNA-conjugation, P*(0.5)AA50 nanogels were dialyzed (14 kDa) against NaOH 0.2 M for 24 h in order to deactivate Ribonuclease (RNAse) that could degrade the siRNA, and then dialyzed against H2O RNAse free. The standard EDC/Sulfo-NHS conjugation protocol was adopted. The blend was incubated for 30 min in agitation at room temperature. Therefore, 720 pmol of 5′-AminoC6linker Bcl-2 siRNA (Qiagen, Hilden, Germany) were mixed and incubated for 3 h in agitation at 4 °C and then dialyzed (14 kDa) against H2O RNAse free to eliminate the excess of unlinked siRNA.
Conjugation degrees of the various ligands were estimated by UV-visible absorption measurements at room temperature with Shimadzu 2401-PC spectrofluorimeter (Shimadzu Corporation, Kyoto, Japan) (scan speed 100 nm/min, integration time 2 s, bandwidth 1 nm) and for NGs conjugated with doxorubicin also with a DU-730 Life Science spectrophotometer, Beckman Coulter (Brea, CA, USA). For RNAi experiments, the samples were then dialyzed against the appropriate cell colure medium. The amount of siRNA conjugated to NGs was determined by reading the absorbance values at 260 nm employing the NanoDrop 1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). Different concentrations of free siRNA were used for a standard assay.
4.3. Physico-Chemical Characterizations
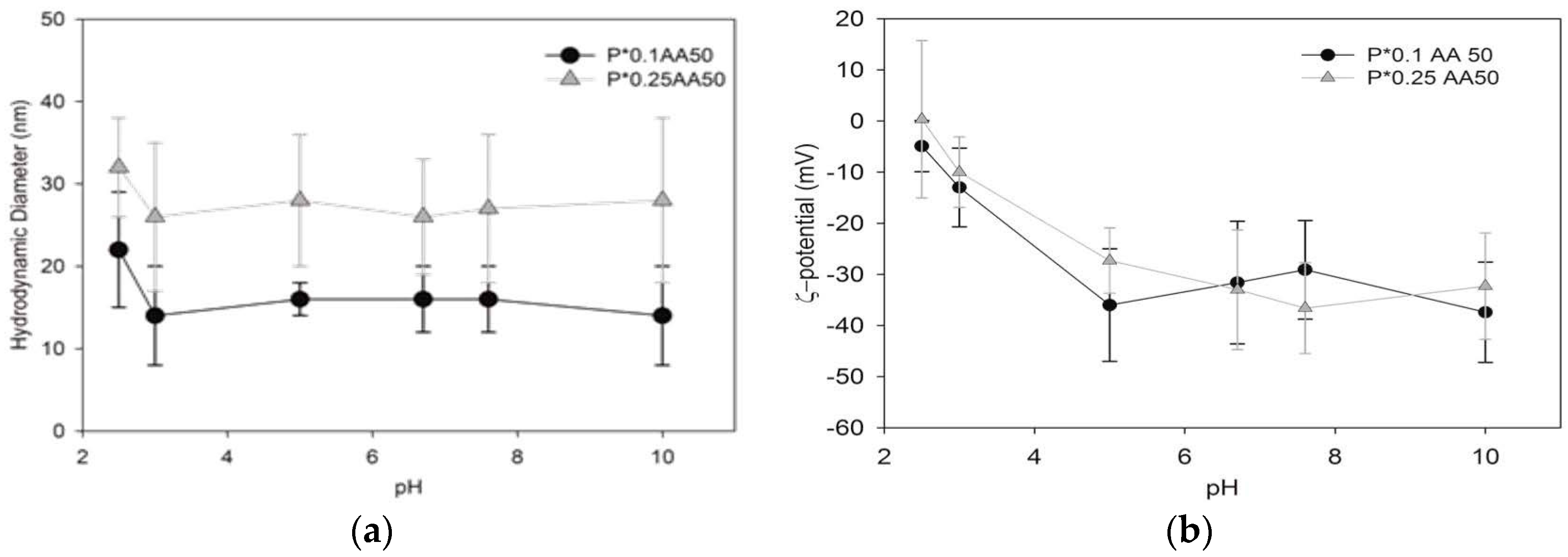
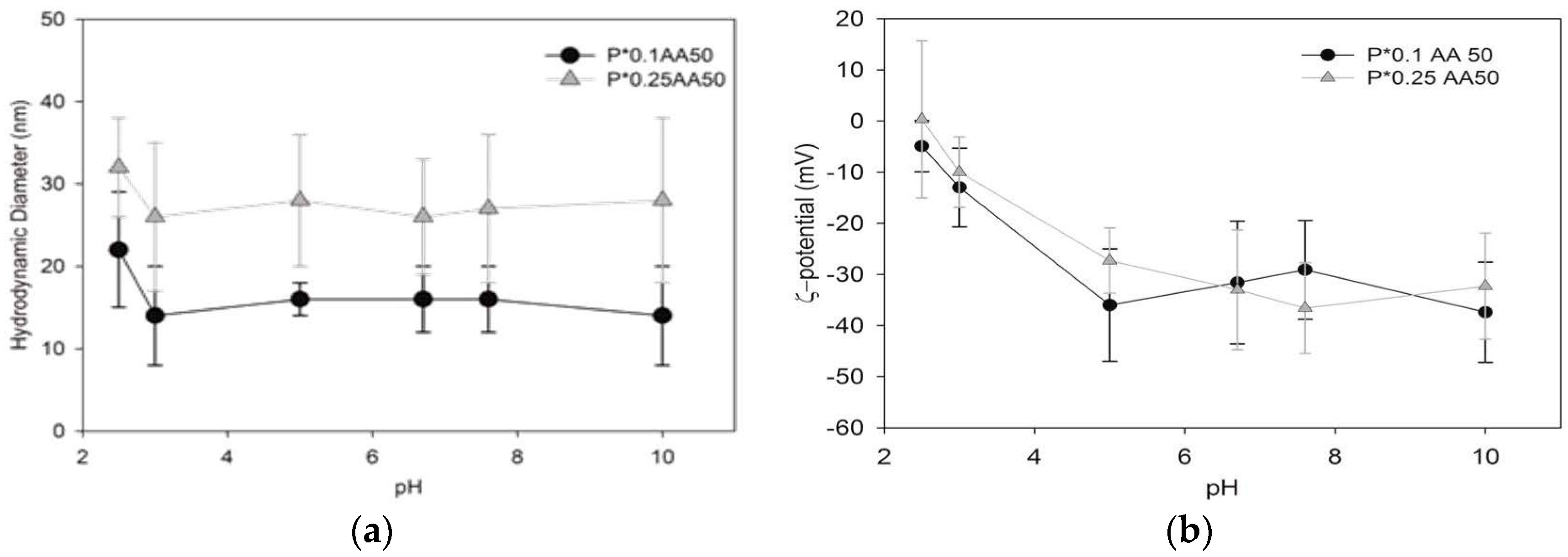
The hydrodynamic dimensions of the differently conjugated NG variants were measured by dynamic light scattering (DLS) using a Brookhaven Instruments BI200-SM goniometer (Brookhaven Instruments Corporation, Holtsville, NY, USA) equipped with a 50 mW He-Ne laser tuned at λ = 632.8 nm. Measurements were carried out placing samples in a quartz cell within the thermostated cell compartment of the instrument at 25 ± 0.1 °C and at 90° scattering angle after filtration (pore size 0.45 μm). In consideration of their monomodal size distribution, DLS data were analyzed according to the method of cumulants [
17,
23,
24,
25]. Ζ-potential measurements were acquired at 25 °C using a Malvern Zetasizer Nano-ZS (Malvern instrument Ltd., Malvern, UK) equipped with a He–Ne laser at a power of 4.0 mW. Systems are characterized by a monomodal ζ-potential distribution; the mean ζ-potential and the relative distribution width are reported in
Table 1. For zeta-potential measurements systems were dialyzed against water. Atomic force microscopy (AFM) was performed on a selected aqueous system (5 ng/mL) deposited on mica substrates modified with 3-aminopropyl triethoxysilane (APTES) [
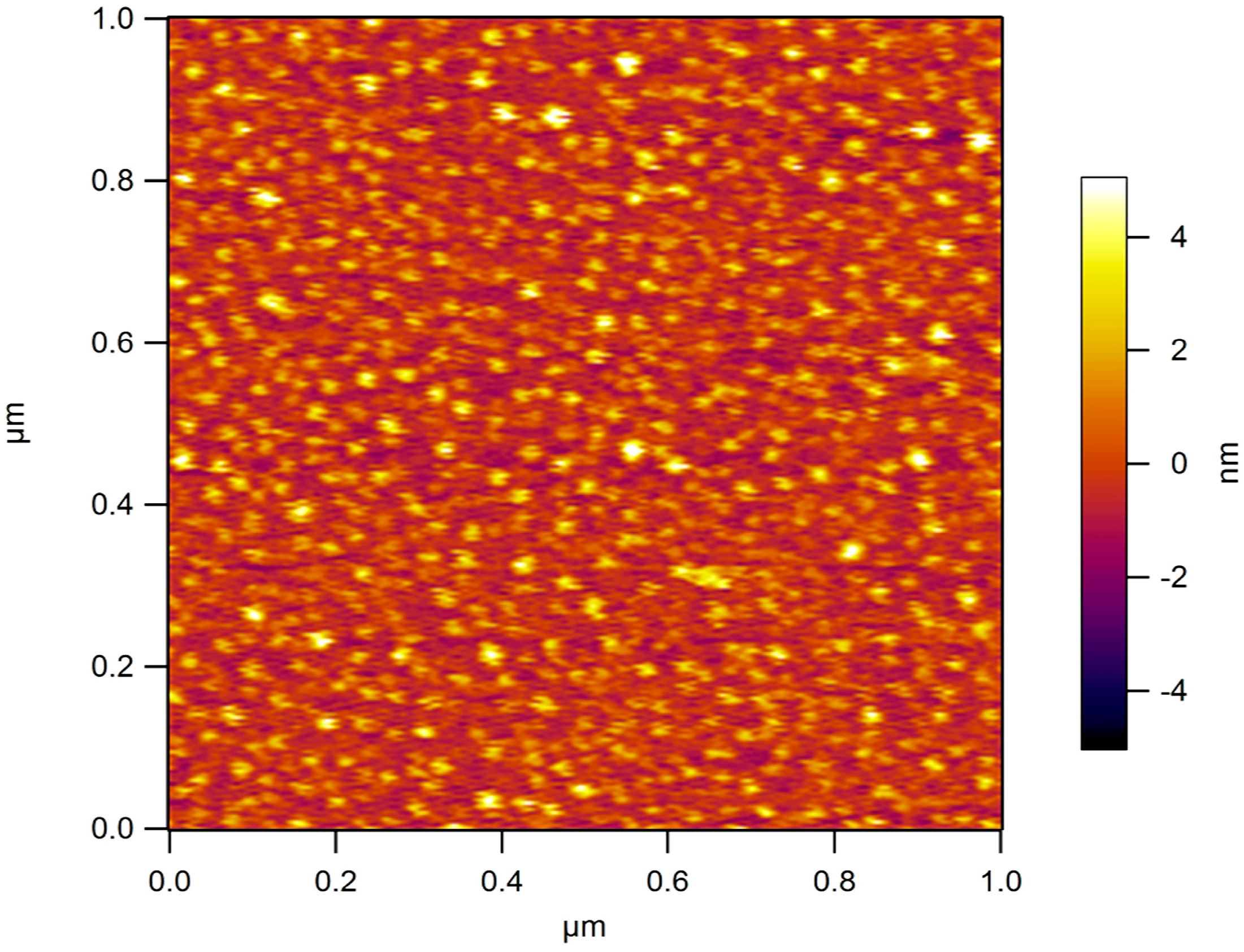
15]. Samples have been examined under water in tapping mode at 2 Hz scanning rate using a MMFP3D AFM (Park Systems Corporate Headquarters, Suwon, Korea) and MSNL D tips (Bruker Nano Inc., Billerica, MA, USA) with nominal resonance frequency of 10–20 kHz and spring constant k ~30 pN/nm.
4.4. Co-Culture Experiment
Human cervical cancer (Hela) and mouse embryo fibroblast (NIH3T3) cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS, EuroClone, Milan, Italy), 100 units per mL penicillin G, 100 µg·mL−1 streptomycin (EuroClone) and 2 mM l-glutamine (EuroClone) at a density of 5 × 103 cells/well into 12 well/plates containing sterile glass coverlips, for 24 h at 37 °C in a humidified atmosphere of 5% CO2. Following, the two slides, everyone with each cell line seeded on, were placed in the same culture environment and incubated with P*(0.5)AA50-Folatefluorescein nanogels (45 μg/mL) for 30 min or 1 h. Equivalent conditions were used as control: both cell lines were treated with the same amount of nanocomplex without folate (P*(0.5)AA50fluorescein). After the established incubation times, cells were washed twice with complete phosphate buffer saline (PBS, EuroClone), fixed with 3.7% formaldehyde for 15 min and again washed twice with PBS. Afterwards, cells were stained with ethidium bromide (1:1000 in PBS) for 1 min at room temperature. Fluorescence within the cells was evaluated by means of epifluorescence microscopy (Leica DFC3000 G, Wetzlar, Germany).
4.5. Internalization Studies by Flow Cytometry
Hela and NIH3T3 cells were cultured in a 6-well plates in complete DMEM medium until 70% of confluence. After, both cell lines were incubated with P*(0.5)AA50-Folatefluorescein nanogels (45 μg/mL), for 30 min and 1 h. After washing with PBS without Ca2+ and Mg2+, cells were detached by Trypsin-EDTA 1× in PBS (EuroClone) and collected by centrifugation at 1000 rpm for 5 min. Therefore, cellpellets were resuspended in 0.5 mL of PBS buffer and analyzed by flow cytometric analysis, using a FACS-Calibur (Becton Dickinson, Heidelberg, Germany). For each sample, data of 1 × 104 gated events were collected and analysis was performed by BD FACS diva software.
4.6. In Vitro Drug Release Measurement
P*(0.5)AA50-AEDP-DOXnanogels (5 mL) were placed into dialysis membranes (MWCO 12 kDa) and each tube was dipped in 50 mL of buffer (1 mM Tris-HCl pH 7.4) alone ( as control) or containing 10 or 0.1 mM of DTT (Dithiothreitol, Sigma Aldrich, Saint Louis, MO, USA).Every system was maintained at 37 °C under continuous shaking condition (200 rpm). At predetermined time (0, 1, 2, 4, 6, 10, and 24 h), 10 mL of external buffer solution was withdrawn and replaced with 10 mL of fresh buffer. Doxorubicin percentage released was determined by using spectrophotometer measurements (490 nm) (Glomax®-Multi Detection System, Promega, Madison, AL, USA).
4.7. Intracellular DOX Release Studies
Intracellular drug release experiments were performed by flow cytometry and analyzed by confocal and epifluorescence microscopy.
4.7.1. FACS analysis
Hela cells were seeded in 6-well plates, until the confluence state. Subsequently, cells were treated with glutathione reduced ethyl ester (GSH-OET, Sigma Aldrich) (10 mM) for 2 h [
26]. After washing with PBS, cells were incubated at 37 °C for 3 h with P*(0.5)AA50-AEDP-DOXnanogels at final DOX concentration of 1 μg/mL in complete DMEM medium. GSH-OET untreated cells were used as control. Next, cells were washed twice with PBS, detached by Trypsin-EDTA 1×, collected by centrifugation at 1000 rpm for 5 min order to obtain cell pellet that was re-suspended into 0.5 mL of PBS. For each sample were collected 1 × 10
4 events investigated by FACS-Calibur (Becton Dickinson) using BD FACS Diva software.
4.7.2. Fluorescence observation
Mouse Osteoblastic cell line (MC3T3-E1) and Hela cells were seeded, at a density of 12 × 104 cells per well, in a 12-multiwell slides containing sterile covers lip in complete DMEM. After 24 h of growth at 37 °C, a sample of each type of cells was treated with 10 mM GSH-OET for 2 h. Cells were washed by PBS and incubated for 3 h at 37 °C with the P*(0.5)AA50-A EDP-DOX nanogels, at final DOX concentration of 1 μg/mL in complete DMEM. Cells without GSH-OET treatment were used as a control. Following, both cell lines on the slides were fixed with 3.6% formaldehyde for 15 min and rinsed with PBS for three times. The actin cytoskeleton was labeled with phalloidinFITC (1:500 in PBS) for 30 min at room temperature. After 3 washes with PBS, MC3T3-E1 cells were stained with DAPI (1:1000 in PBS) for 30 s, at room temperature for marking the nuclei in blue. Finally, the MC3T3-E1 slides were mounted and observed by epifluorescence microscope (Leica, Wetzlar, Germany), while the Hela slided with confocal microscopy (CLSM, Olympus IX70 laser system with MellesGriot, Shinjuku, Tokyo, Japan).
4.8. Cytotoxicity Assay
P*(0.5)AA50-AEDP-DOX cytotoxic effect was evaluated on MC3T3-E1 and Hela cells by Alamar Blue assay (Sigma Aldrich). Both cell lines were seeded into a 96-multiwell at 6 × 103 cells per well, in complete DMEM for 24 h and then treated with 10 mM GSH-OET for 2 h at 37 °C. Therefore, Hela cells were incubated with P*(0.5)AA50-AEDP-DOX nanogels or free DOX as control at the final DOX concentration of 1 μg/mL for 24 and 48 h; while MC3T3-E1 cells were treated with drug alone (as control) or conjugated with NGs at the final DOX concentration of 0.3 μg/mL for 48 and 72 h. Cells GSH-OET untreated and incubated with NGs were used as another control. After treatment, cells were incubated with Alamar Blue solution (10% in culture medium), a reagent for evaluating cellular health, for 4 h at 37 °C. Fluorescence intensity (λexc 530/25 nm and λemm 590/35 nm) was read through spectrofluorometer (SinergyHT, BioTek, Winooski, VT, USA), which changes according to the degree of cell viability. The results were expressed as the percentage ratio of the fluorescence between the treated sample with respect to untreated cells, used as negative control.
4.9. Treatment with NG-AEDP-siRNA Complex
Hela cells were seeded in 6-well plates and grown at 37 °C with 5% CO2 until the confluence state. Some samples were incubated for 2 h at 37 °C with 10 mM of GSH-OET in complete medium. After the opportune washes with PBS, P*(0.5)AA50-AEDP or P*(0.5)AA50-AEDP-siRNA (300 pmol of siRNA) complex were added for 48 h.
4.10. Extraction of Soluble Proteins and Western Blot Analysis
Hela cells were seeded in 6-well plates maintained at 37 °C in a humidified atmosphere of 5% CO2. After the appropriate treatment, the samples were washed with PBS without Ca2+ and Mg2+, detached by Trypsin-EDTA 1× in PBS and collected by centrifugation at 1000 rpm for 5 min. The pellets were re-suspended in an appropriate volume (depending of the pellet size) of RIPA Buffer 1× (50 mM Tris-HCl pH 7.5; 0.5% sodium deoxycholate; 150 mM NaCl; 1% Triton X-100) supplemented of proteases inhibitors (1 mM of phenylmethylsulfonyl fluoride (PMSF); 1 µM of Pepstatin A; 100 µM Leupeptin; 10 mM Ethylenediaminetetraacetic acid (EDTA)). Therefore, samples were incubated in ice for 15 min and constantly subject to micro vortex mixer treatments and then centrifuged at 10,000 rpm for 20 min. The amount of proteins extracted presented into the supernatant were calculated by the Bradford assay (Bradford Reagent, Sigma Aldrich), using different concentrations of bovine serum albumin (BSA) as Standard Assay. Twenty micrograms of proteins were mixed with sample buffer 1× (62.5 mM Tris-HCl pH 6.8; 2.5% SDS; 0.002% Bromophenol Blue; 0.7135 M (5%) β-mercaptoethanol; 10% glycerol) and incubated at 100 °C for 5 min and immediately situated in ice. The proteins were separate by 15% sodium dodecyl sulfate Polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred from the gel to a nitrocellulose membrane (Hybond, Amersham) through electroblotting at 100 V and 300 mA for 90 min at low temperature, in transfer buffer (20% methanol; 10% Running buffer 1×: Tris base 3 g/L-Glycine 14.4 g/L). Blocking of non-specific binding was achieved by placing the membrane in a solution of non-fat dry milk (3%) overnight a 4 °C. Therefore, Bcl-2 antibody (1:1000, Sigma Aldrich) was incubated overnight at 4 °C; after washing with TBS-T solution (0.6% Trizma base; 0.87% NaCl; 0.05% Tween 20), the peroxidase-linked anti-mouse secondary antibody (0.04 µg/mL, Sigma Aldrich) was incubated for 2 h at room temperature. After other TBS-T washes, the presence of Bcl-2 protein was identify using “SuperSignal West Femto Maximum Sensitivity Substrate” (Thermo Scientific) and the chemiluminescence signal was reveal using the ChemiDoc XRS (Biorad, Hercules, CA, USA). β-actin (antibody anti-β-actin 0.4 µg/mL, Sigma Aldrich) was used to normalize each sample.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
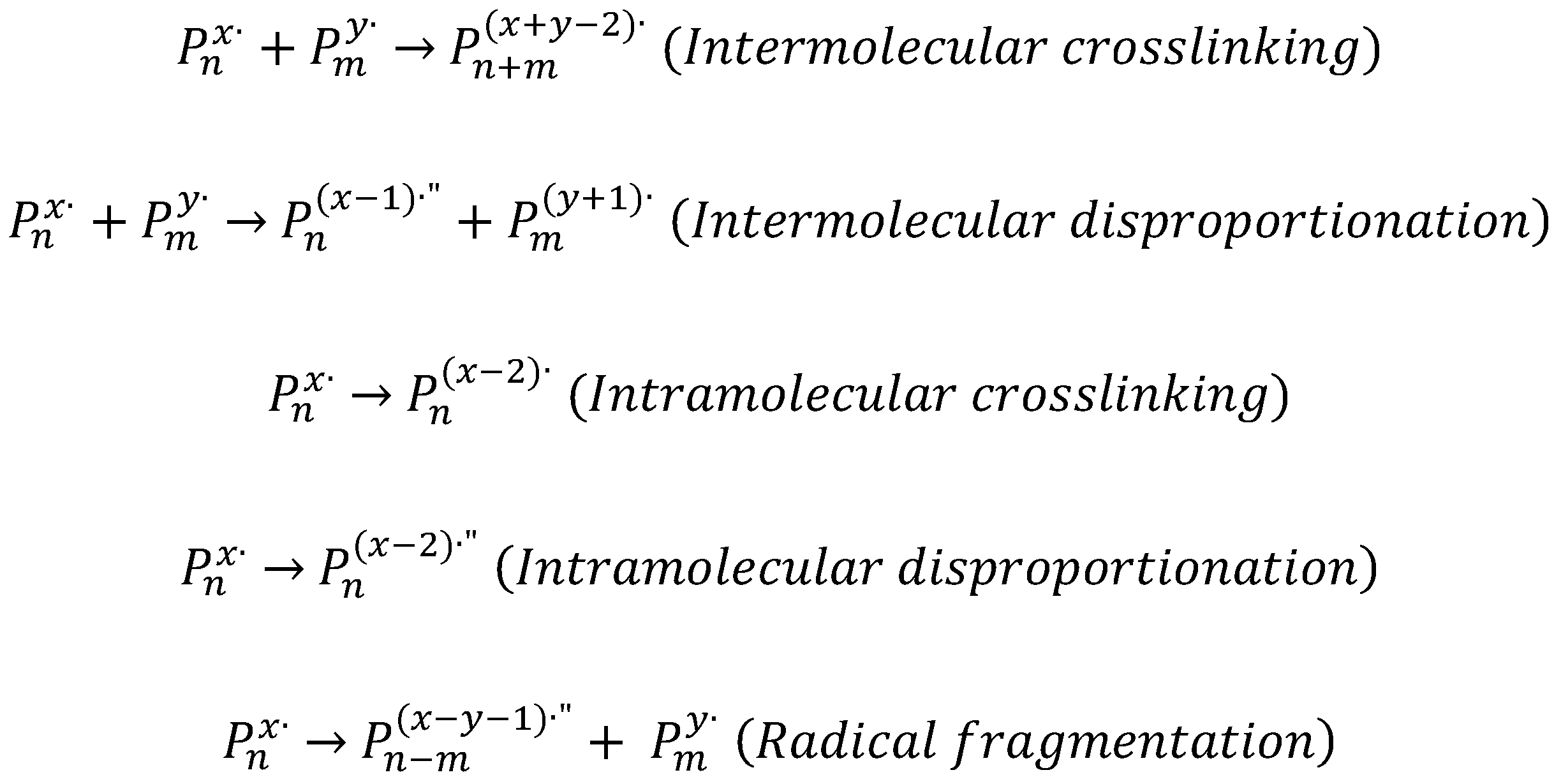{kind=link}
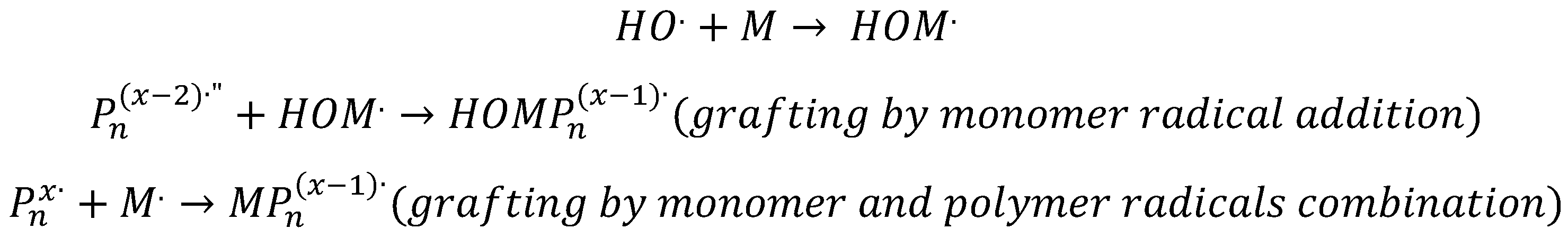{kind=link}