
Nucleophilic Substitution on 2-Monosubstituted Quinoxalines Giving 2,3-Disubstituted Quinoxalines: Investigating the Effect of the 2-Substituent


Abstract
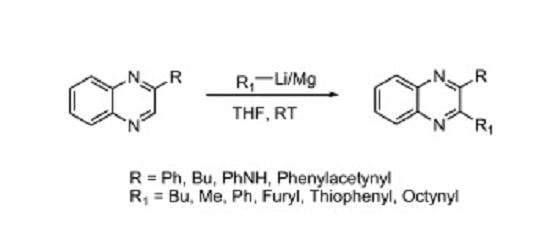
:
1. Introduction
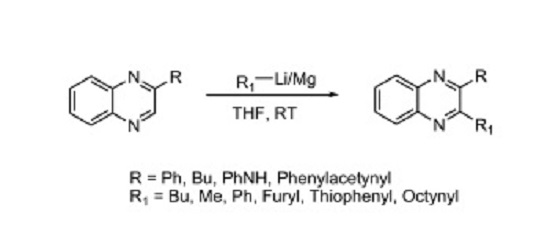
2. Results
3. Experimental Section
3.1. General Information
3.2. Synthetic Procedure
3.2.1. 2-Benzensulfonyloxyquinoxaline (1)
3.2.2. 2-Phenylquinoxaline (2)
General Methods for Nucleophilic Substitution on 2-Phenylquinoxaline
3.2.3. 3-Butyl-2-phenylquinoxaline (2a)
3.2.4. 3-Isopropyl-2-phenylquinoxaline (2b)
3.2.5. 2,3-Diphenylquinoxaline (2c)
3.2.6. 2-Phenyl-3-(2-phenylethynyl)quinoxaline (2d)
3.2.7. 3-(Oct-1-ynyl)-2-phenylquinoxaline (2e)
3.2.8. 2-Phenyl-3-(thiophen-2-yl)quinoxaline (2f)
3.2.9. 2-(Furan-2-yl)-3-phenylquinoxaline (2g)
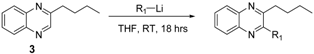
3.2.10. 2-Butylquinoxaline (3)
3.2.11. 2,3-Dibutylquinoxaline (3a)
3.2.12. 2-Butyl-3-(furan-2-yl)quinoxaline (3b)
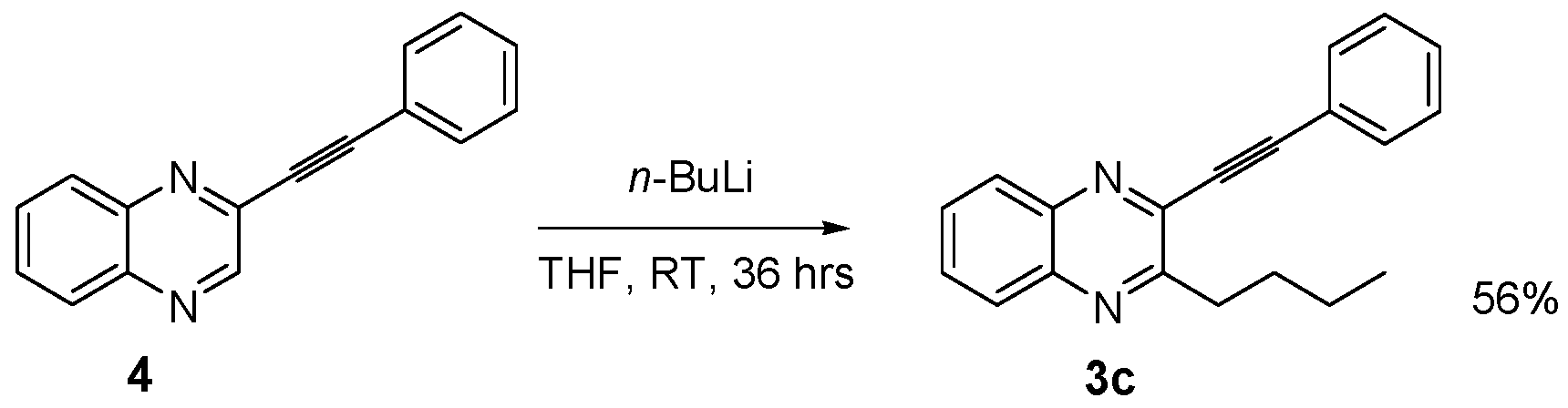
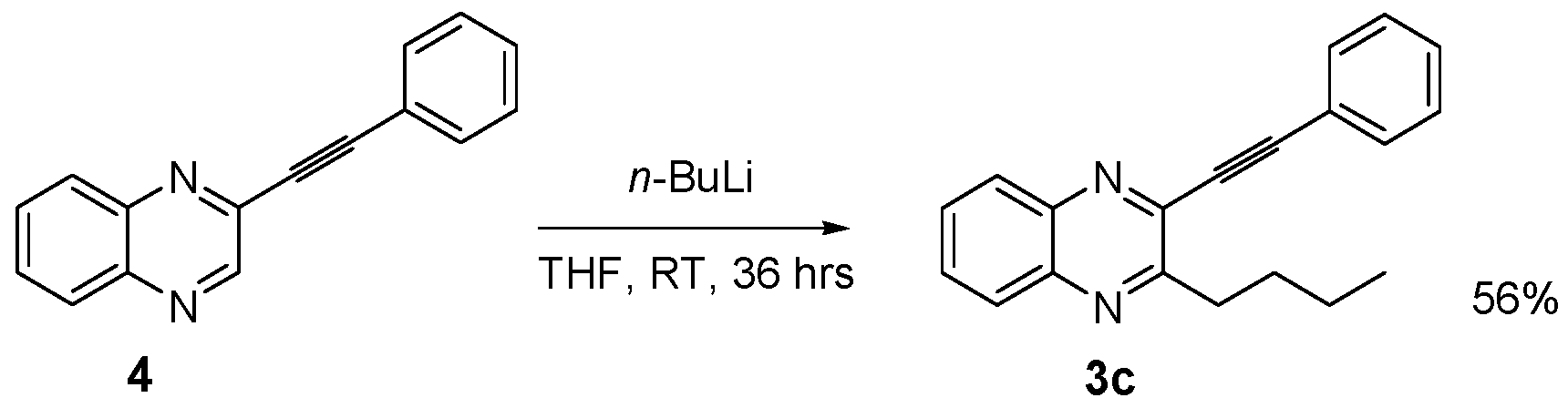
3.2.13. 3-Butyl-2-(2-phenylethynyl)quinoxaline (3c)
3.2.14. 2-(2-Phenylethynyl)quinoxaline (4)
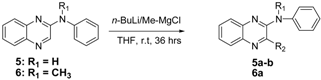
3.2.15. N-Phenylquinoxalin-2-amine (5)
3.2.16. 3-Butyl-N-phenylquinoxalin-2-amine (5a)
3.2.17. 3-Methyl-N-phenylquinoxalin-2-amine (5b)
3.2.18. N-Methyl-N-phenylquinoxalin-2-amine (6)
3.2.19. 3-Butyl-N-methyl-N-phenylquinoxalin-2-amine (7a)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, D.P.; Deivedi, S.K.; Hashim, S.R.; Singhal, R.G. Synthesis and antimicrobial activity of some new quinoxaline derivatives. Pharmaceuticals 2010, 3, 2416–2425. [Google Scholar] [CrossRef]
- Selvam, P.; Chandramohan, M.; Pannecouque, C.; de Clercq, E. Studies on antiviral activity of 2,3-diphenylquinoxaline. Int. J. Pharm Ind. Res. 2011, 1, 138–140. [Google Scholar]
- Ramalingam, P.; Ganapaty, S.; Rao, C.B. In vitro antitubercular and antimicrobial activities of 1-substituted quinoxaline-2,3(1H,4H)-diones. Biorg. Med. Chem. Lett. 2010, 20, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.; Bergman, J.; Graslund, A. 1H-NMR studies of the interaction between a self-complementary deoxyoligonucleotide duplex and indolo[2,3-b]quinoxaline derivatives active against herpes virus. Eur. J. Biochem. 1991, 197, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Sasikumar, P.; Velmurugan, D. In-vitro evaluation and molecular docking calculation of tricyclic phthalimide quinoxaline analogues as novel inhibitors of hiv-1 integrase using GLIDE and GOLD. Int. J. Pharm. Sci. Drug Res. 2014, 6, 60–66. [Google Scholar]
- Potey, L.C.; Kosalge, S.B.; Hadke, M.A. Synthesis and antimicrobial activity of quinoxaline sulfonamide. Int. J. Adv. Sci. Technol. 2013, 2, 126–134. [Google Scholar]
- Agboke, A.; Attama, A.; Qkoye, C.; Jackson, C. Evaluation of effectiveness of various combinations of penicillin groups commonly used in Nigeria clinics on selected microorganisms. Innov. J. Med. Health. Sci. 2014, 4, 93–98. [Google Scholar]
- Le Douaron, G.; Ferrié, L.; Sepulveda-Diaz, J.E.; Amar, M.; Harfouche, A.; Séon-Méniel, B.; Raisman-Vozari, R.; Michel, P.P.; Figadère, B. New 6-aminoquinoxaline derivatives with neuroprotective effect on dopaminergic neurons in cellular and animal Parkinson disease models. J. Med. Chem. 2016, 59, 6169–6186. [Google Scholar] [CrossRef] [PubMed]
- Le Douaron, G.; Schmidt, F.; Amar, M.; Kadar, H.; Debortoli, L.; Latini, A.; Séon-Méniel, B.; Ferrié, L.; Michel, P.P.; Touboul, D.; et al. Neuroprotective effects of a brain permeant 6-aminoquinoxaline derivative in cell culture conditions that model the loss of dopaminergic neurons in Parkinson disease. Eur. J. Med. Chem. 2015, 89, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, B.C.G.; Wallace, J.M.; Tamariz, J. A novel palladium-catalyzed synthesis of 1,2-dihydroquinoxalines and 3,4-dihydroquinoxalinones. Org. Lett. 2002, 4, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Issidorides, C.H.; Haddadin, M.J. Benzofurazan oxide. II. reactions with enolate anions. J. Org. Chem. 1966, 31, 4067–4068. [Google Scholar] [CrossRef]
- Abu-Hashem, A.A. Synthesis, reactions and biological activity of quinoxaline derivatives. Am. J. Org. Chem. 2015, 5, 14–56. [Google Scholar]
- Noorulla, S.M.P.; Sreenivasulu, N.; Khan, A.; Sayeed, A. Antibacterial activity of novel substituted quinoxaline heterocycles. Pharmanest 2011, 2, 229–238. [Google Scholar]
- Cho, C.S.; Ren, W.X. A recyclable copper catalysis in quinoxaline synthesis from α-hydroxyketones and o-phenylenediamines. J. Organomet. Chem. 2009, 694, 3215–3217. [Google Scholar] [CrossRef]
- Khaksar, S.; Rostamnezhad, F. A novel one-pot synthesis of quinoxaline derivatives in fluorinated alcohols. Korean Chem. Soc. 2012, 33, 2581–2584. [Google Scholar] [CrossRef]
- Soleymani, R.; Niakan, N.; Tayeb, S.; Hakimi, S. Synthesis of novel aryl quinoxaline derivatives by new catalytic methods. Oriental J. Chem. 2012, 28, 687–701. [Google Scholar] [CrossRef]
- Makosza, M. Nucleophilic substitution of hydrogen in electron-deficient arenes, a general process of great practical value. Chem. Soc. Rev. 2010, 39, 2855–2868. [Google Scholar] [CrossRef] [PubMed]
- Makosza, M. Vicarious nucleophilic substitution of hydrogen. Russ. Chem. Rev. 1989, 58, 1298–1317. [Google Scholar] [CrossRef]
- Kovalev, I.S.; Kopchuk, D.S.; Zyryanov, G.V.; Rusinov, V.L.; Chupakhin, O.N.; Charushin, V.N. Organolithium compounds in the nucleophilic substitution of hydrogen in arenes and hetarenes. Russ. Chem. Rev. 2015, 84, 1191–1225. [Google Scholar] [CrossRef]
- Hui, X.; Schmidt, F.; Fakhfakh, M.F.; Franck, X.; Figadère, B. Novel highly regioselective syntheses of unsymmetrical 2,3-disubstituted quinoxalines. Heterocycles 2007, 72, 353–361. [Google Scholar]
- Azev, Y.A.; Oprina, E.D.; Golomolzin, B.V.; Ermakova, O.S.; Bakulev, V.S. A simple means of preparing quinoxaline derivatives: Direct introduction of C-nucleophiles into the quinoxaline nucleus by substituting a hydrogen atom. Pharm. Chem. J. 2013, 47, 172–175. [Google Scholar] [CrossRef]
- Badr, M.Z.A.; El-Naggar, G.M.; El-Sherief, H.A.H.; Abdel-Rahman, A.E.; Aly, M.F. Reaction of quinoxaline derivatives with nucleophilic reagents. Bull. Chem. Soc. Jpn. 1983, 56, 326–330. [Google Scholar] [CrossRef]
- Zhuo, F.; Xie, W.; Yang, Y.; Zhang, L.; Wang, P.; Yuan, R.; Da, C. TMEDA-assisted effective direct ortho arylation of electron-deficient N-heteroarenes with aromatic Grignard reagents. J. Org. Chem. 2013, 78, 3243–3249. [Google Scholar] [CrossRef] [PubMed]
- Prokhorov, A.M.; Makosza, M.; Chupakhin, O.N. Direct introduction of acetylene moieties into azines by SNH methodology. Tetrahedron Lett. 2009, 50, 1444–1446. [Google Scholar] [CrossRef]
- Epifani, E.; Florio, S.; Ingrosso, G.; Sgarra, R.; Stasi, F. Reaction of quimoxalines with β,γ-unsaturated grignard reagents: Synthesis of allyl-, allenyl-, propargyl-quinoxaline derivatives. Tetrahedron 1987, 43, 2767–2778. [Google Scholar] [CrossRef]
- Wienhöfer, E.; Kauffmann, T. Lineare verknüpfung von 5 und 6 verschiedenen arenen (1). Tetrahedron Lett. 1974, 15, 2347–2350. [Google Scholar] [CrossRef]
- Nxumalo, W.; Dinsmore, A. Preparation of 6-ethynylpteridine derivatives by Sonogashira coupling. Heterocycles 2013, 87, 78–79. [Google Scholar] [CrossRef]
- Nxumalo, W.; Dinsmore, A.S. Negishi coupling of pteridine-O-sulfonates. S. Afr. J. Chem. 2013, 66, 42–46. [Google Scholar]
- Negishi, E.; King, A.O.; Okukado, N. Selective carbon-carbon bond formation via transition metal catalysis. 3. A highly selective synthesis of unsymmetrical biaryls and diarylmethanes by the nickel- or palladium-catalyzed reaction of aryl- and benzylzinc derivatives with aryl halides. J. Org. Chem. 1977, 42, 1821–1823. [Google Scholar] [CrossRef]
- Sonogashira, K. Development of Pd-Cu catalyzed cross-coupling of terminal acetylenes with sp2-carbon halides. J. Organomet. Chem. 2002, 653, 46–49. [Google Scholar] [CrossRef]
- Guram, A.S.; Buchwald, S.L. Palladium-catalyzed aromatic aminations with in situ generated aminostannanes. J. Am. Chem. Soc. 1994, 116, 7901–7902. [Google Scholar] [CrossRef]
- Darabantu, M.; Boully, L.; Turck, A.; Ple, N. Synthesis of new polyaza heterocycles. Part 42: Diazines. Tetrahedron 2005, 61, 2897–2905. [Google Scholar] [CrossRef]
- Cho, C.S.; Ren, W.X.; Shim, S.C. Ketones as a new synthon for quinoxaline synthesis. Tetrahedron Lett. 2007, 48, 4665–4667. [Google Scholar] [CrossRef]
- Rekha, M.; Kathyayini, H.; Nagaraju, N. Catalytic activity of manganese oxide supported on alumina in the synthesis of quinoxalines. Front. Chem. Sci. Eng. 2013, 7, 415–421. [Google Scholar] [CrossRef]
- Wang, W.; Shen, Y.; Meng, X.; Zhao, M.; Chen, Y.; Chen, B. Copper-catalyzed synthesis of quinoxalines with o-phenylenediamine and terminal alkyne in the presence of bases. Org. Lett. 2011, 13, 4514–4517. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Georg, B.E. Thiophene series. XLVII. 2-Thienyl phenyl diketone and its oxime. Eur. J. Org. Chem. 1939, 540, 14–24. [Google Scholar]
- Gomez-Sanchez, A.; Antinolo, M.Y.; Gonzalez, F.G. The influence of the aromatic nucleus on the dehydration of polyhydroxy chains. V. The structure of Maurer’s glucazidone. Span. Roya. Soc. Chem. 1954, 50B, 431–440. [Google Scholar]
- Das, B.; Venkateswarlu, K.; Suneel, K.; Majhi, A. An efficient and convenient protocol for the synthesis of quinoxalines and dihydropyrazines via cyclization-oxidation processes using HClO4/SiO2 as a heterogeneous recyclable catalyst. Tetrahedron Lett. 2007, 48, 5371–5374. [Google Scholar] [CrossRef]
- Fontana, F.; Minisci, F.; Barbosa, M.C.N.; Vismara, E. Homolytic alkylation of heteroaromatic bases: the problem of monoalkylation. Tetrahedron 1990, 46, 2525–2538. [Google Scholar] [CrossRef]
- Supabphol, A.; Muangman, V.; Chavasiri, W.; Supabphol, R.; Gritsanapan, W. N-acetylcysteine inhibits proliferation, adhesion, migration and invasion of human bladder cancer cells. J. Med. Ass. Thail. 2009, 92, 1171–1177. [Google Scholar]
- Saari, R.; Törmä, J.; Nevalainen, T. Microwave-assisted synthesis of quinoline, isoquinoline, quinoxaline and quinazoline derivatives as CB2 receptor agonists. Bioorg. Med. Chem. 2011, 19, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
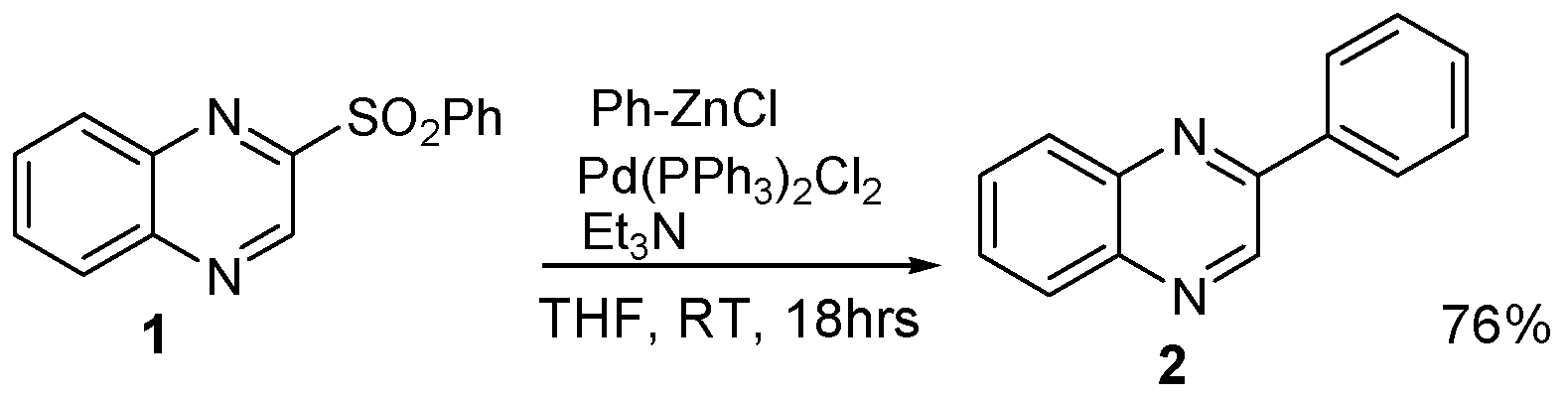

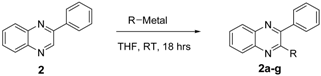
| Entry | Product | R-Metal | R | % Yield |
|---|---|---|---|---|
| 1 | 2a | n-BuLi | -Butyl | 66 |
| 2 | 2b | Isopropyl-MgCl | -Isopropyl | 54 |
| 3 | 2c | Phenyl-MgBr | -Phenyl | 22 |
| 4 | 2d | 2-Phenylethynyl-Li | -2-Phenylethynyl | 47 |
| 5 | 2e | Oct-1-ynyl-Li | -Oct-1-ynyl | 14 |
| 6 | 2f | Thiopen-2-yl-Li | -Thiophen-2-yl | 22 |
| 7 | 2g | Furan-2-yl-Li | -furan-2-yl | 46 |
| Entry | Product | R1 | % Yield |
|---|---|---|---|
| 1 | 3a | -Butyl | 97 |
| 2 | 3b | -Furan-2-yl | 91 |
| 3 | 2a | -Phenyl | 95 |
| 4 | 3c | -Phenylacetylene | 65 |
| Entry | Product | R1 | R2 | % Yield |
|---|---|---|---|---|
| 1 | 5a | -H | -Butyl | 32 |
| 2 | 5b | -H | -Methyl | 42 |
| 3 | 6a | -CH3 | -Butyl | 62 |
{kind=link}
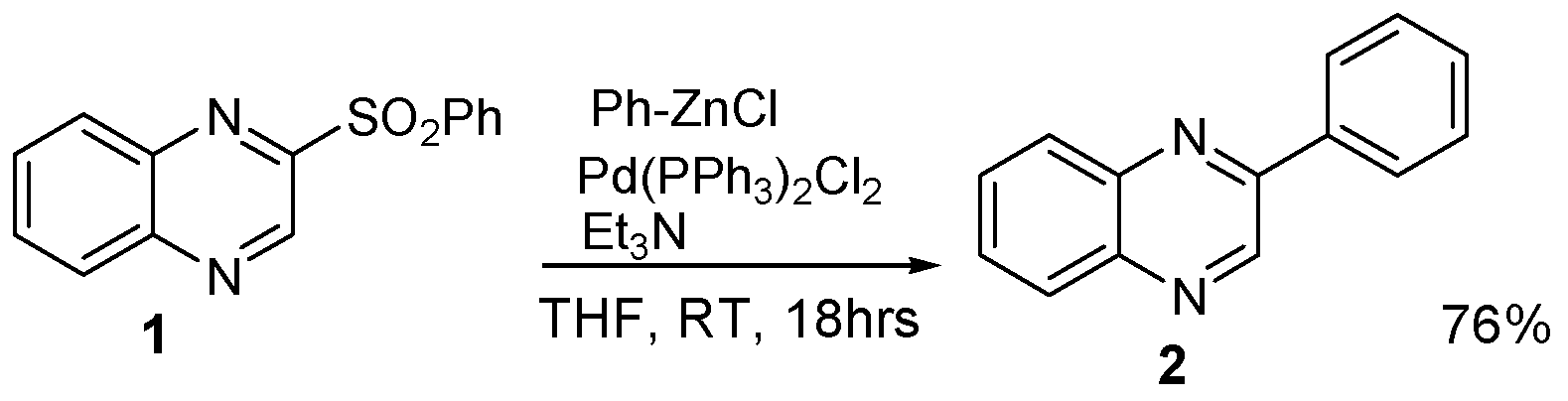{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndlovu, N.T.; Nxumalo, W. Nucleophilic Substitution on 2-Monosubstituted Quinoxalines Giving 2,3-Disubstituted Quinoxalines: Investigating the Effect of the 2-Substituent. Molecules 2016, 21, 1304. https://doi.org/10.3390/molecules21101304
Ndlovu NT, Nxumalo W. Nucleophilic Substitution on 2-Monosubstituted Quinoxalines Giving 2,3-Disubstituted Quinoxalines: Investigating the Effect of the 2-Substituent. Molecules. 2016; 21(10):1304. https://doi.org/10.3390/molecules21101304
Chicago/Turabian StyleNdlovu, Ndumiso Thamsanqa, and Winston Nxumalo. 2016. "Nucleophilic Substitution on 2-Monosubstituted Quinoxalines Giving 2,3-Disubstituted Quinoxalines: Investigating the Effect of the 2-Substituent" Molecules 21, no. 10: 1304. https://doi.org/10.3390/molecules21101304
APA StyleNdlovu, N. T., & Nxumalo, W. (2016). Nucleophilic Substitution on 2-Monosubstituted Quinoxalines Giving 2,3-Disubstituted Quinoxalines: Investigating the Effect of the 2-Substituent. Molecules, 21(10), 1304. https://doi.org/10.3390/molecules21101304