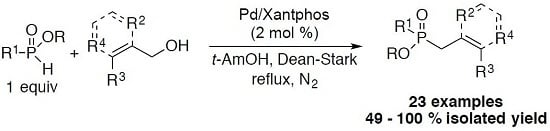
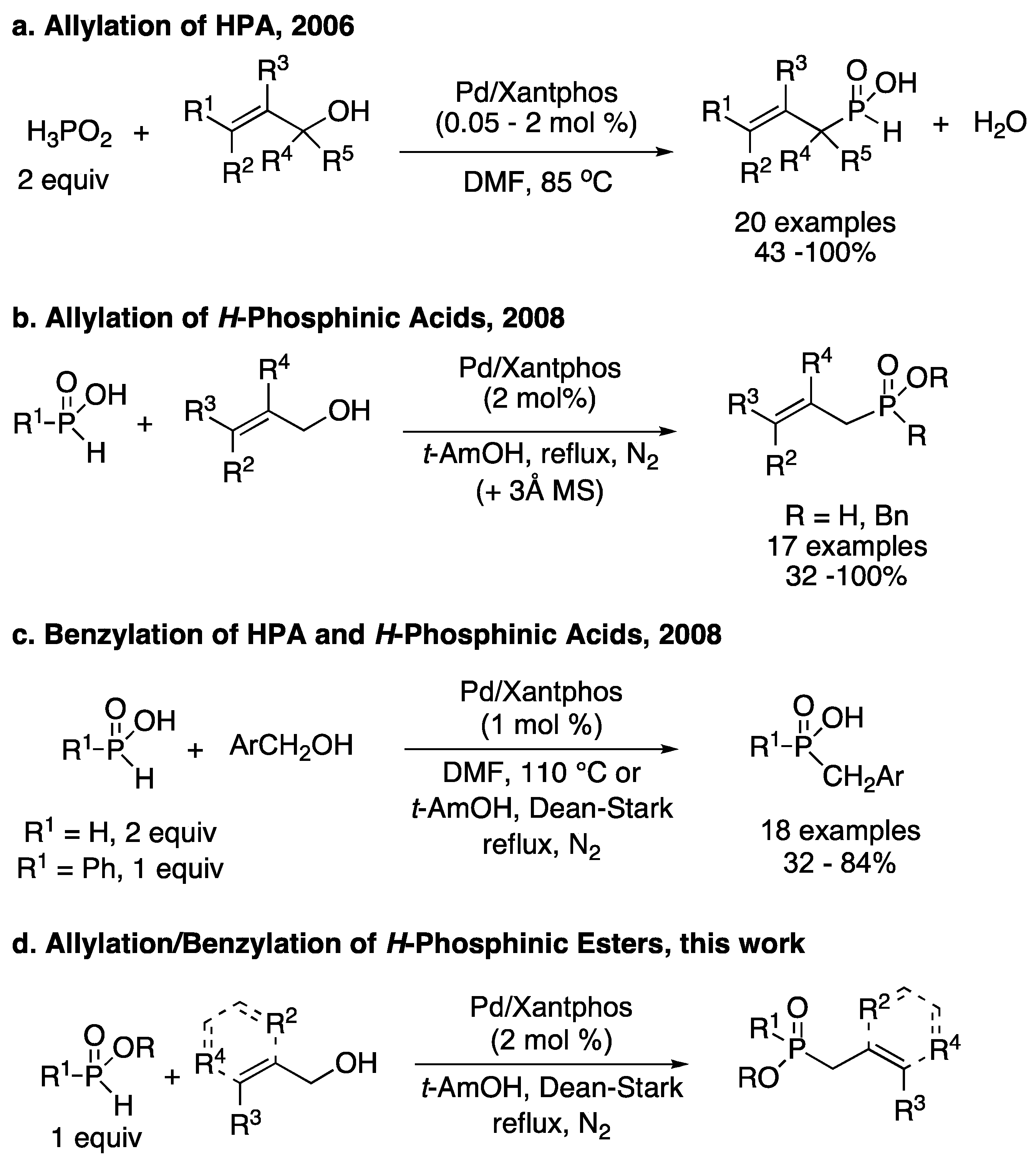
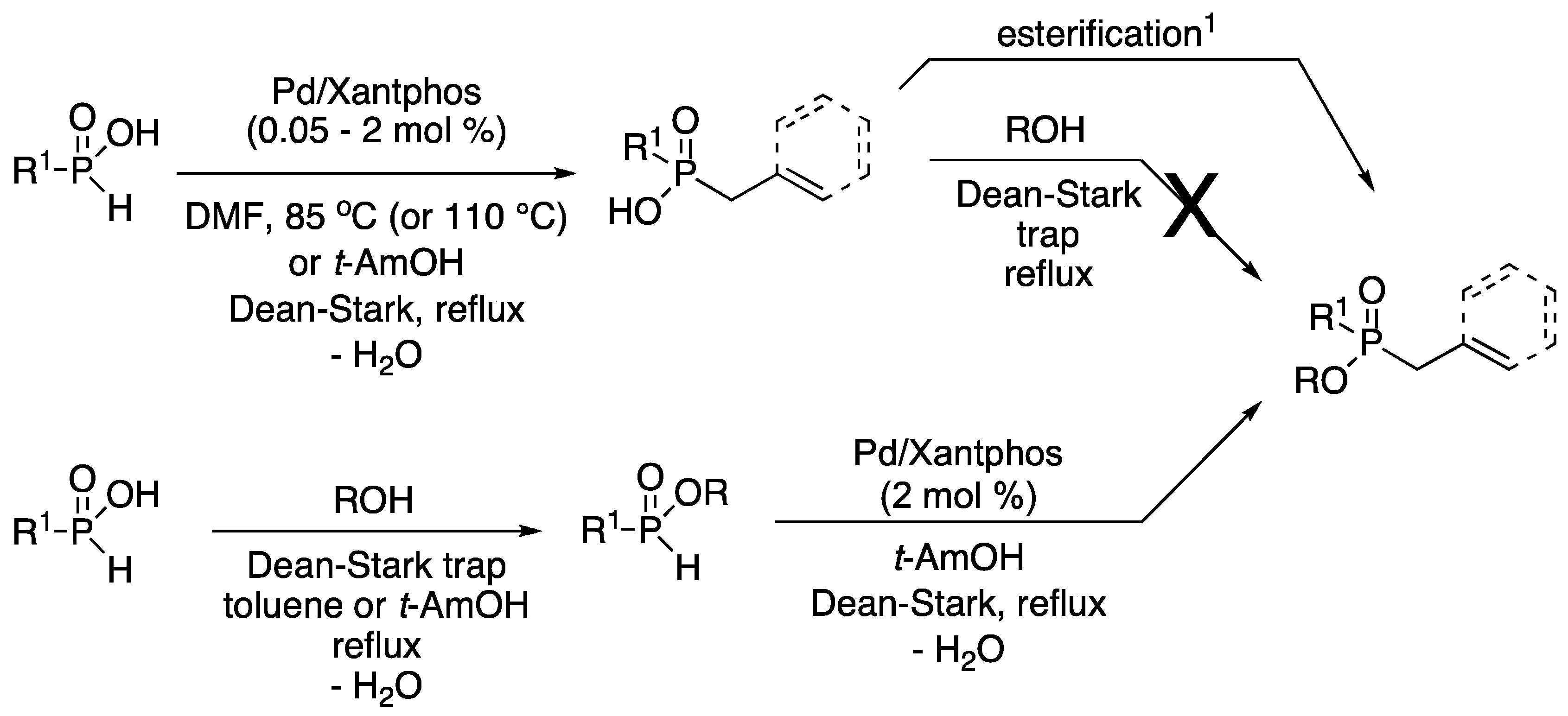
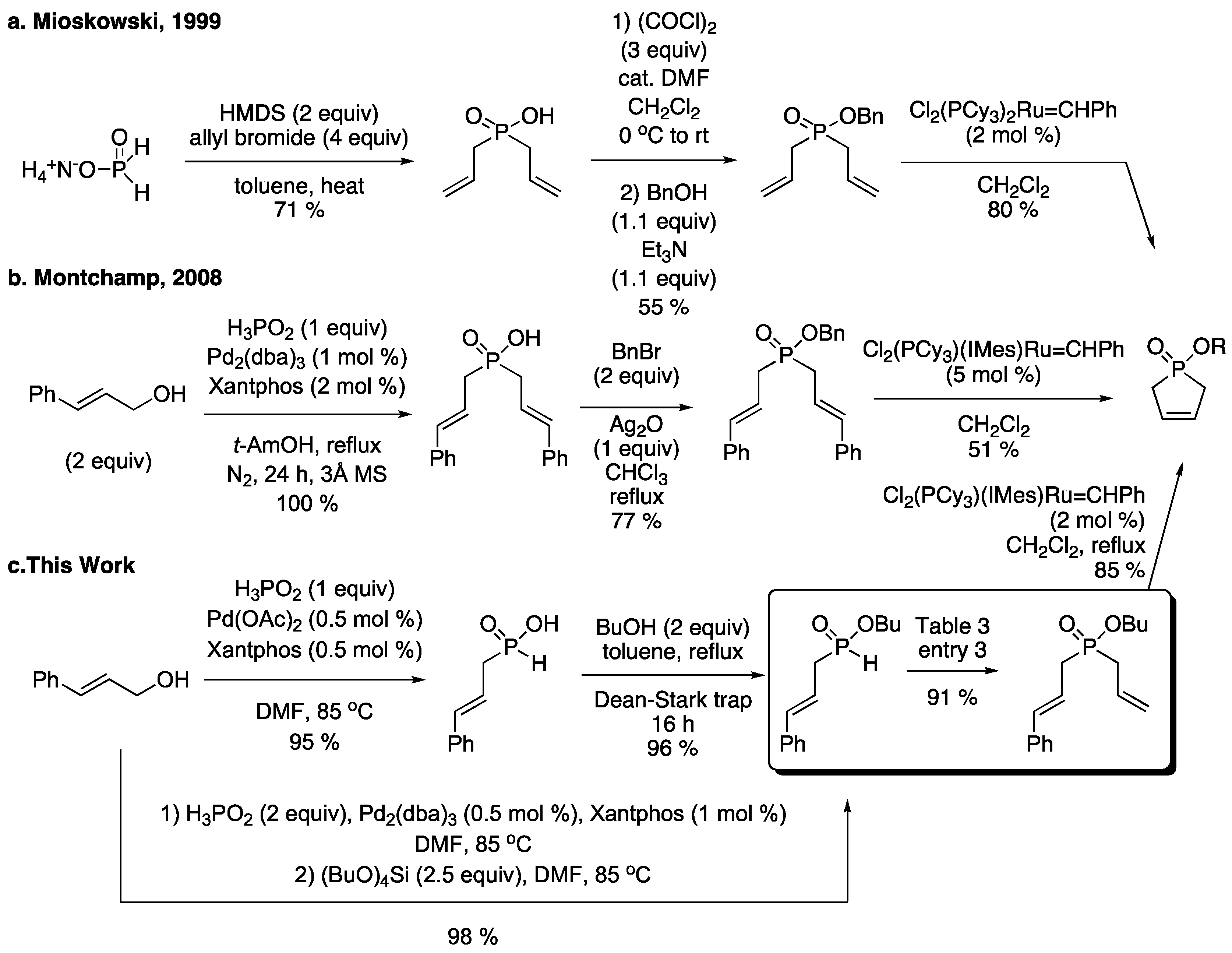
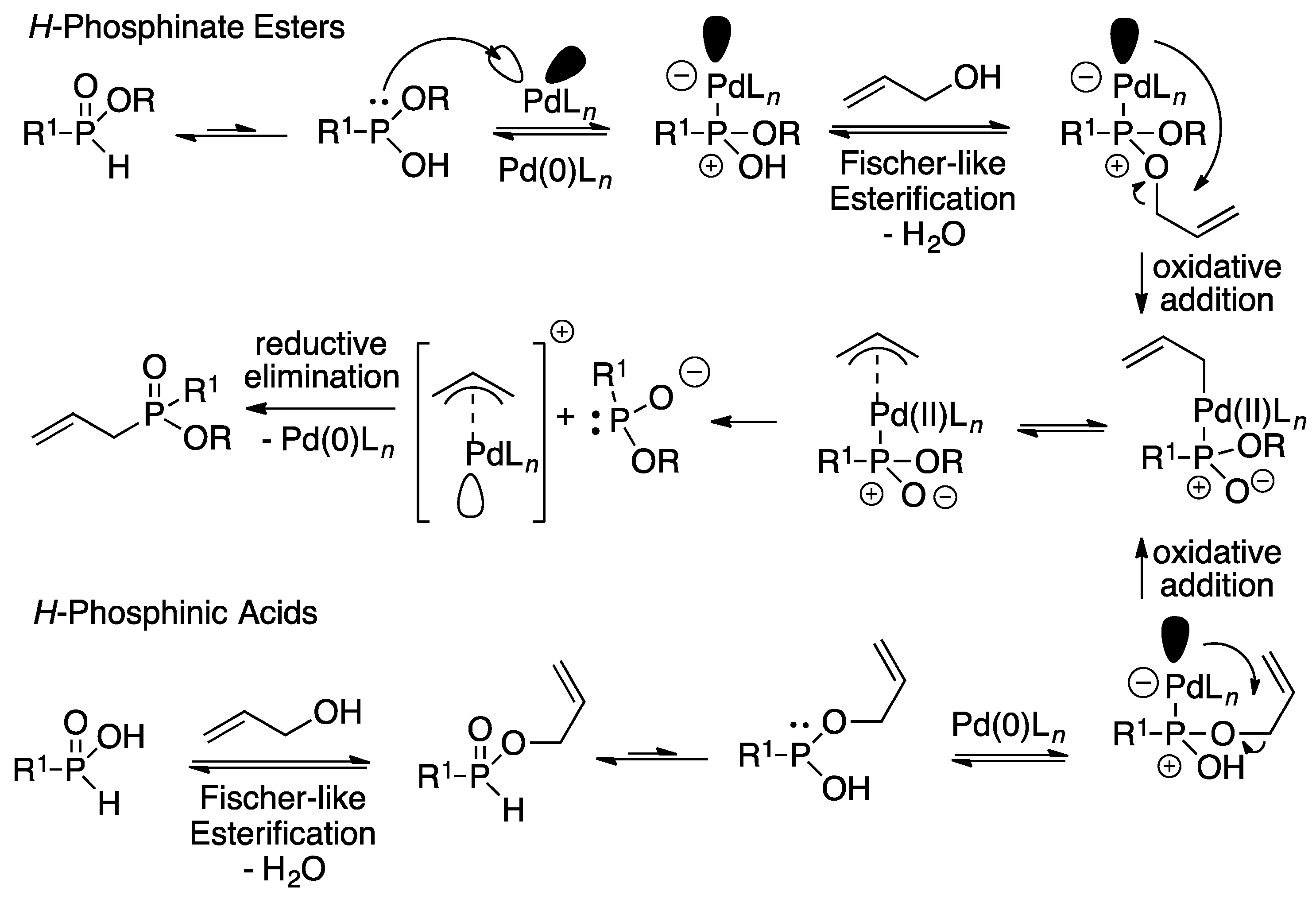
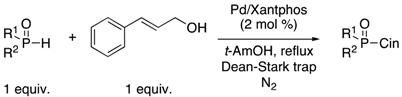
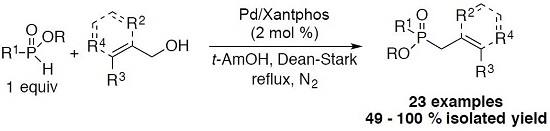
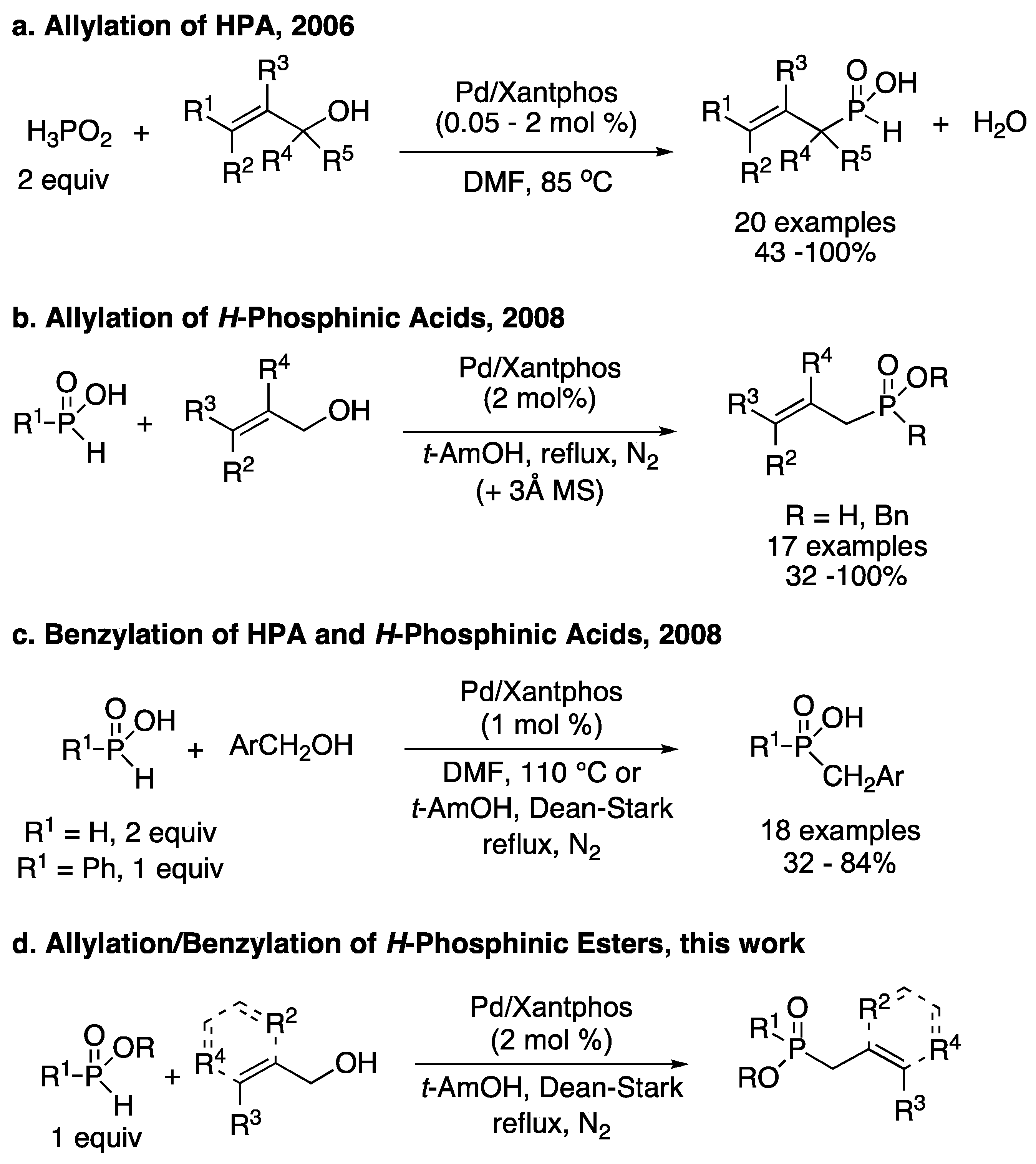
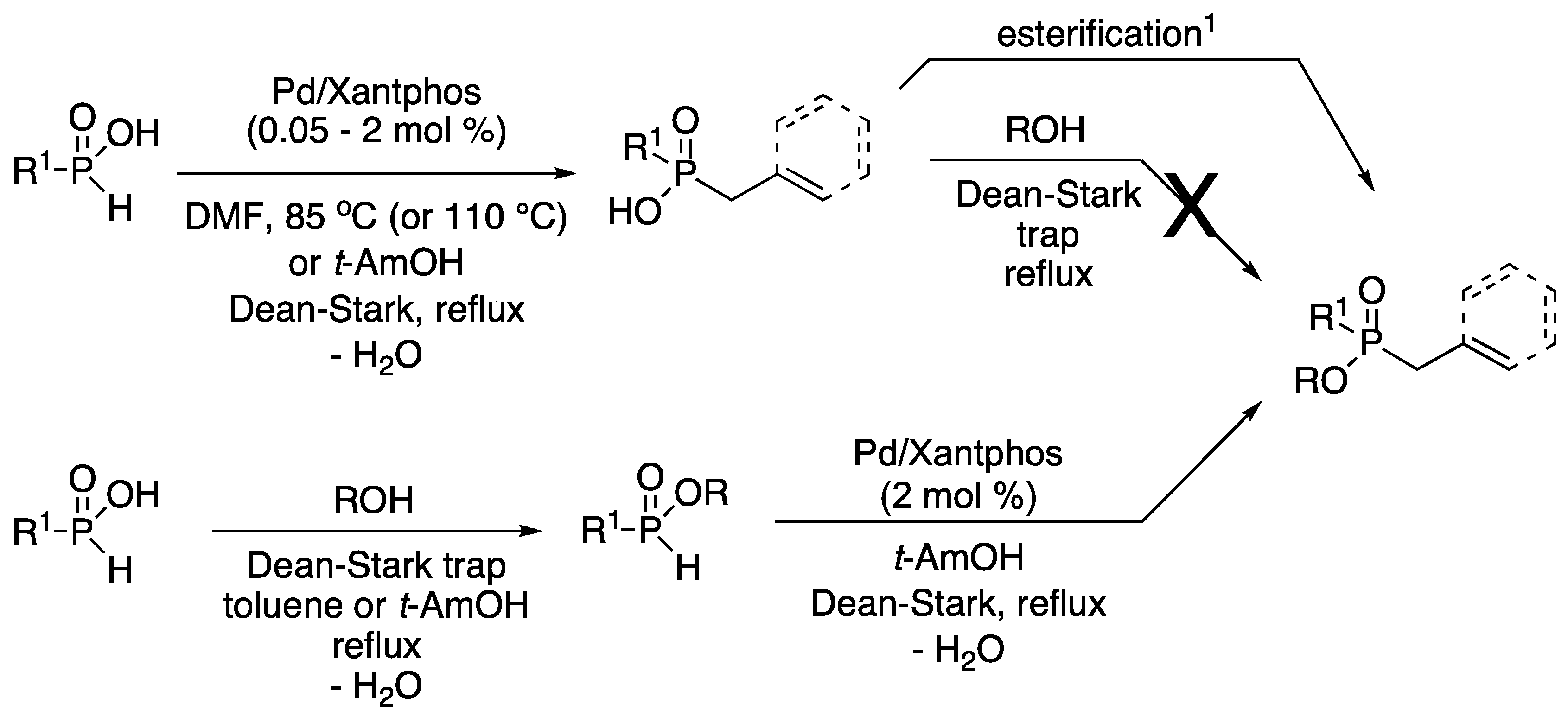
3.1. General Procedure for the Allylation/Benzylation of H-Phosphinates and Related Compounds
To a solution of the appropriate
H-phosphinate ester (1 equiv.) in
t-amyl alcohol (10 mL), tris(dibenzylideneacetone)dipalladium(0) Pd
2(dba)
3 (1 mol %), Xantphos (2 mol %), and the corresponding alcohol (1 equiv.) were added. The reaction mixture was stirred at reflux for 24 h under N
2 in a flask equipped with a Dean-Stark trap. After cooling down the reaction to room temperature (rt), the solvent was removed under vacuum and the residue obtained was purified by column chromatography on silica gel using a mixture of hexane/ethyl acetate to afford the different products. The NMR spectra of the products can be found in the
Supplementary Materials.
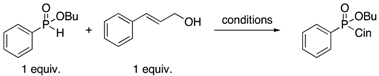
Butyl cinnamyl phenylphosphinate (Table 2, Entry 1) [
13]. General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and butyl phenyl-
H-phosphinate (198 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as a yellow oil (310 mg, 99%).
31P-NMR (CDCl
3, 162 MHz) δ = 39.1 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.73–7.80 (m, 2H), 7.47–7.54 (m, 1H), 7.40–7.46 (m, 2H), 7.21–7.27 (m, 4H), 7.14–7.20 (m, 1H), 6.33 (dd,
J = 5.0 and 15.8 Hz, 1H), 6.04–6.15 (m, 1H), 3.93 (dm,
J = 95 Hz, 2H), 2.89 (dd,
J = 7.6 and 18.5 Hz, 2H), 1.63 (quint.,
J = 6.8 Hz, 2H), 1.37 (dsextuplet,
J = 1.7 and 7.5 Hz, 2H), 0.87 (t,
J = 7.4 Hz, 3H).
Cyclohexyl cinnamyl phenylphosphinate (Table 2, Entry 2). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and cyclohexyl phenyl-
H-phosphinate (224 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as a yellow oil (311 mg, 91%).
31P-NMR (CDCl
3, 162 MHz) δ = 37.7 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.76–7.84 (m, 2H), 7.49–7.55 (m, 1H), 7.41–7.47 (m, 2H), 7.23–7.30 (m, 4H), 7.16–7.22 (m, 1H), 6.35 (dd,
J = 5.0 and 15.8 Hz, 1H), 6.06–6.17 (m, 1H), 4.30–4.41 (m, 1H), 2.89 (ddd,
J = 1.0, 7.6 and 18.4 Hz, 2H), 1.97–2.07 (m, 1H), 1.57–1.79 (m, 4H), 1.39–1.52 (m, 2H), 1.15–1.36 (m, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 137.0 (d,
JPCCCC = 3.3 Hz), 134.9 (d,
JPCC = 13.2 Hz), 132.2 (d,
JPCCCC = 2.7 Hz), 131.7 (d,
JPCCC = 9.6 Hz, 2C), 131.5 (d,
JPC = 128 Hz), 128.5 (2C), 128.4 (d,
JPCC = 12.5 Hz, 2C), 127.4, 126.1, 126.1, 118.9 (d,
JPCCC = 10.3 Hz), 74.7 (d,
JPOC = 6.9 Hz), 35.9 (d,
JPC = 97.3 Hz), 34.2 (d,
JPOCC = 2.9 Hz), 33.7 (d,
JPOC = 4.1 Hz), 25.1, 23.6, 23.6; HRMS (EI+)
m/
z calcd for C
21H
26O
2P ([M + H]
+) 341.1665, found 341.1675.
Benzyl cinnamyl phenylphosphinate (Table 2, Entry 3). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and benzyl phenyl-
H-phosphinate (232 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (248 mg, 71%).
31P-NMR (CDCl
3, 162 MHz) δ = 41.4 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.80–7.87 (m, 2H), 7.54–7.60 (m, 1H), 7.45–7.52 (m, 2H), 7.31–7.40 (m, 5H), 7.26–7.31 (m, 4H), 7.20–7.25 (m, 1H), 6.38 (dd,
J = 5.2 and 15.9 Hz, 1H), 6.08–6.19 (m, 1H), 5.18 (dd,
J = 7.3 and 11.8 Hz, 1H), 4.88 (dd,
J = 7.2 and 11.8 Hz, 1H), 2.99 (dd,
J = 7.6 and 18.7 Hz, 2H);
13C-NMR (101 MHz, CDCl
3): δ = 136.9 (d,
JPCCCC = 3.5 Hz), 136.4 (d,
JPOCC = 6.8 Hz), 135.3 (d,
JPCC = 13.3 Hz), 132.6 (d,
JPCCCC = 2.5 Hz), 131.9 (d,
JPCCC = 9.7 Hz, 2C), 130.1 (d,
JPC = 125 Hz), 128.7 (d,
JPCC = 12.3 Hz, 2C), 128.6 (2C), 128.5 (2C), 128.4, 127.9 (2C), 127.6, 126.3, 126.2, 118.4 (d,
JPCCC = 10.4 Hz), 66.3 (d,
JPOC = 6.4 Hz), 35.3 (d,
JPC = 96.3 Hz); HRMS (EI+)
m/
z calcd for C
22H
22O
2P ([M + H]
+) 349.1357, found 349.1379.
Cyclohexyl cinnamyl octylphosphinate (Table 2, Entry 4). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and cyclohexyl octyl-
H-phosphinate (260 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 50:50 to 0:100) to afford the product as an orange oil (328 mg, 87%).
31P-NMR (CDCl
3, 162 MHz) δ = 51.6 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.24–7.35 (m, 4H), 7.16–7.22 (m, 1H), 6.47 (dd,
J = 4.5 and 15.8 Hz, 1H), 6.10–6.22 (m, 1H), 4.34–4.46 (m, 1H), 2.71 (dd,
J = 7.7 and 17.3 Hz, 2H), 1.83–1.94 (m, 2H), 1.42–1.75 (m, 8H), 1.15–1.37 (m, 14 H), 0.84 (t,
J = 6.8 Hz, 3 H);
13C-NMR (101 MHz, CDCl
3): δ = 136.8 (d,
JPCCCC = 3.0 Hz), 134.4 (d,
JPCC = 12.6 Hz), 128.5 (2C), 127.5, 126.1, 126.1, 119.7 (d,
JPCCC = 9.4 Hz), 73.7 (d,
JPOC = 7.0 Hz), 34.6 (d,
JPC = 82.2 Hz), 34.2 (d,
JPOCC = 5.6 Hz), 34.1, 31.8, 30.8 (d,
JPCC = 15.1 Hz), 29.0, 29.0, 28.3 (d,
JPC = 93.1 Hz), 25.2, 23.7 (2C), 22.6, 21.6 (d,
JPCCC = 4.2 Hz), 14.1; HRMS (EI+)
m/
z calcd for C
23H
38O
2P ([M + H]
+) 377.2609, found 377.2531.
Butyl bis cinnamylphosphinate (Table 2, Entry 5). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and butyl cinnamyl-
H-phosphinate (238 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 50:50 to 0:100) to afford the product as an orange oil (354 mg, 100%).
31P-NMR (CDCl
3, 162 MHz) δ = 49.9 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.21–7.33 (m, 8H), 7.14–7.20 (m, 2H), 6.45 (dd,
J = 4.6 and 15.8 Hz, 2H), 6.11–6.22 (m, 2H), 4.02 (dt,
J = 6.8 and 7.0 Hz, 2H), 2.73 (dd,
J = 7.6 and 17.5 Hz, 4H), 1.61 (quint.,
J = 7.2 Hz, 2H), 1.36 (sextuplet,
J = 7.5 Hz, 2H), 0.88 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 136.7 (d,
JPCCCC = 3.0 Hz, 2C), 135.0 (d,
JPCC = 13.1 Hz, 2C), 128.6 (4C), 127.7 (2C), 126.2 (2C), 126.2 (2C), 118.9 (d,
JPCCC = 9.6 Hz, 2C), 64.6 (d,
JPOC = 7.2 Hz), 33.3 (d,
JPC = 88.7 Hz, 2C), 32.8 (d,
JPOCC = 5.6 Hz), 18.8, 13.7; HRMS (EI+)
m/
z calcd for C
22H
28O
2P ([M + H]
+) 355.1827, found 355.1831.
Butyl 2-[(4-methoxyphenyl)ethyl] cinnamylphosphinate (Table 2, Entry 6). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and butyl 2-[4-methoxyphenyl)ethyl]-
H-phosphinate (256 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (261 mg, 70%).
31P-NMR (CDCl
3, 162 MHz) δ = 52.3 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.29–7.38 (m, 4H), 7.22–7.28 (m, 1H), 7.10–7.15 (m, 2H), 6.81–6.87 (m, 2H), 6.45 (dd,
J = 4.6 and 15.8 Hz, 1H), 6.11–6.22 (m, 1H), 4.06 (dt,
J = 6.6 and 6.7 Hz, 2H), 3.78 (s, 3H), 2.83–2.99 (m, 2H), 2.73 (ddd,
J = 0.7, 7.2 and 16.8 Hz, 2H), 2.01–2.12 (m, 2H), 1.68 (quint.,
J = 7.5 Hz, 2H), 1.43 (sextuplet,
J = 7.5 Hz, 2H), 0.95 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 158.2, 136.7 (d,
JPCCCC = 3.2 Hz), 134.8 (d,
JPCCC = 12.9 Hz), 133.0 (d,
JPCC = 14.6 Hz), 129.1 (2C), 128.6 (2C), 127.7, 126.2, 126.2, 119.1 (d,
JPCCC = 9.4 Hz, 2C), 114.0 (2C), 64.4 (d,
JPOC = 7.0 Hz), 55.3, 34.0 (d,
JPC = 86.0 Hz), 32.8 (d,
JPOCC = 5.7 Hz), 29.7 (d,
JPC = 90.4 Hz), 27.0 (d,
JPCC = 3.5 Hz), 18.9, 13.7; HRMS (EI+)
m/
z calcd for C
22H
29O
3P ([M + H]
+) 373.1933, found 373.1855.
Butyl (2-ethylphthalimide) cinnamylphosphinate (Table 2, Entry 7). General procedure was used with cinnamyl alcohol (1.7 mL, 12.94 mmol, 1 equiv.) and butyl (2-ethylphthalimide)-
H-phosphinate (3.3 g, 12.94 mmol, 1 equiv.). The crude obtained was purified by column chromatography (dichloromethane/acetone 100:0 to 90:10) to afford the product as an orange oil (4.87 g, 100%).
31P-NMR (CDCl
3, 162 MHz) δ = 49.1 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.82–7.90 (m, 2H), 7.68–7.75 (m, 2H), 7.20–7.44 (m, 5H), 6.62 (dd,
J = 4.7 and 15.8 Hz, 1H), 6.16–6.27 (m, 1H), 3.95–4.12 (m, 4H), 2.88 (dd,
J = 7.5 and 17.5 Hz, 2H), 2.17–2.34 (m, 2H), 1.58 (quint.,
J = 7.1 Hz, 2H), 1.35 (sextuplet,
J = 7.4 Hz, 2H), 0.89 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 167.8 (2C), 136.6 (d,
JPCCCC = 3.2 Hz), 135.3 (d,
JPCC = 13.1 Hz), 134.1 (2C), 132.0 (2C), 128.6 (2C), 127.7, 126.3, 126.3, 123.4 (2C), 118.5 (d,
JPCCC = 9.6 Hz), 64.6 (d,
JPOC = 7.0 Hz), 33.7 (d,
JPC = 88.4 Hz), 32.6 (d,
JPCC = 5.9 Hz), 31.7, 26.5 (d,
JPC = 89.7 Hz), 18.8, 13.6; HRMS (EI+)
m/
z calcd for C
23H
27NO
4P ([M + H]
+) 412.1678, found 412.1676.
Butyl (acetoxymethyl) cinnamylphosphinate (Table 2, Entry 8). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and butyl (acetoxymethyl)-
H-phosphinate (194 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (239 mg, 77%).
31P-NMR (CDCl
3, 162 MHz) δ = 43.6 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.27–7.32 (m, 2H), 7.21–7.27 (m, 2H), 7.14–7.20 (m, 1H), 6.50 (dd,
J = 5.0 and 15.9 Hz, 1H), 6.06–6.17 (m, 1H), 4.43 (dd,
J = 7.8 and 14.5 Hz, 1H), 4.27 (dd,
J = 4.2 and 14.5 Hz, 1H), 4.03 (dm,
J = 39.6 Hz, 2H), 2.79 (ddd,
J = 1.0, 7.7 and 18.5 Hz, 2H), 2.03 (s, 3H), 1.60 (quint.,
J = 7.5 Hz, 2H), 1.34 (sextuplet,
J = 7.5 Hz, 2H), 0.86 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 170.0 (d,
JPCOC = 6.8 Hz), 136.5 (d,
JPCCCC = 3.5 Hz), 135.5 (d,
JPCC = 13.3 Hz), 128.6 (2C), 127.8, 126.3, 126.2, 117.5 (d,
JPCCC = 10.2 Hz), 65.2 (d,
JPOC = 6.9 Hz), 57.9 (d,
JPC = 108 Hz), 32.6 (d,
JPOCC = 5.8 Hz), 32.5 (d,
JPC = 93.1 Hz), 20.5 (d,
JPOCCC = 8.8 Hz), 18.7, 13.6; HRMS (EI+)
m/
z calcd for C
16H
24O
4P ([M + H]
+) 311.1407, found 311.1401.
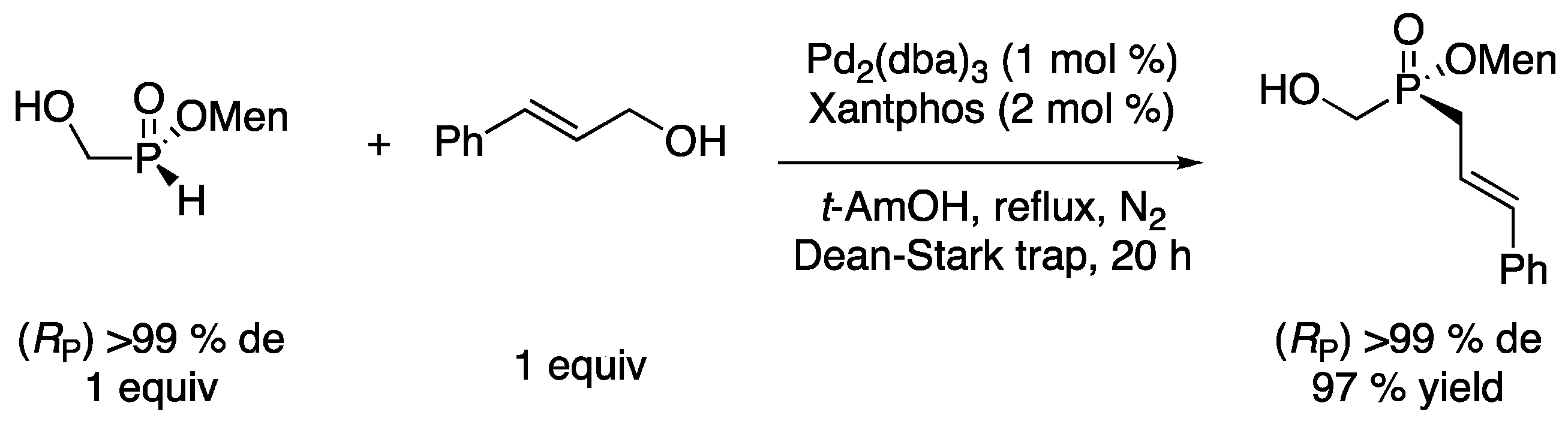
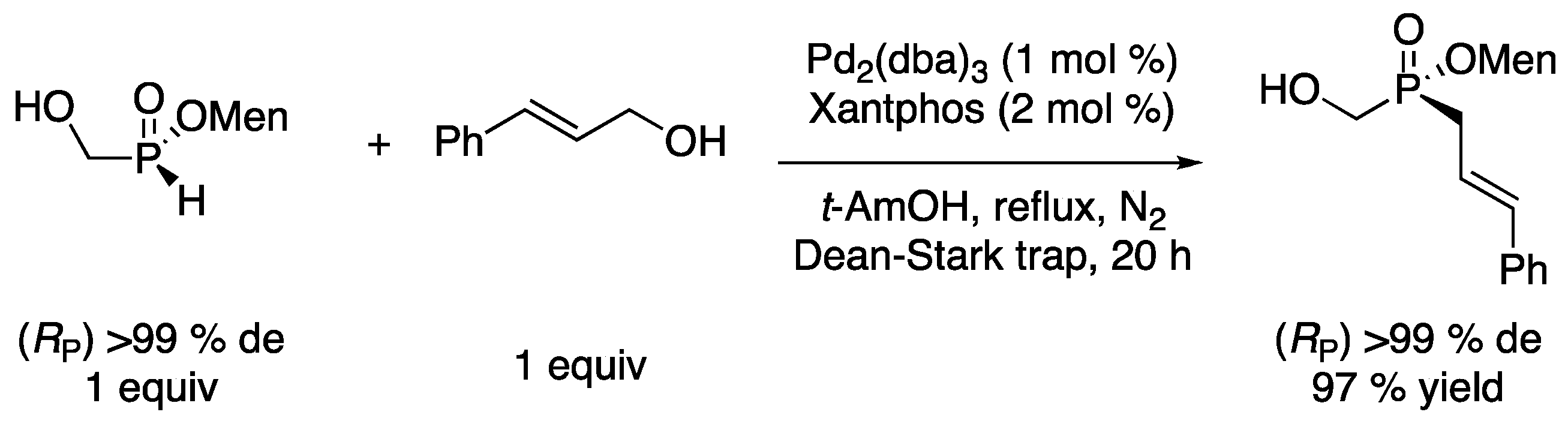
(Rp)/(Sp)-Menthyl cinnamyl(hydroxymethyl)phosphinate (Table 2, Entry 9) [
14]. To a solution of
(Rp)/(Sp)-menthyl (hydroxymethyl)-
H-phosphinate (2.34 g, 10 mmol, 1 equiv, 56:44 diastereoisomeric ratio) in
t-amyl alcohol (30 mL), Pd
2(dba)
3 (92 mg, 0.1 mmol, 1 mol %), Xantphos (116 mg, 0.2 mmol, 2 mol %), and cinnamyl alcohol (1.3 mL, 10 mmol, 1 equiv.) were added. The reaction mixture was stirred at reflux for 20 h under N
2 in a flask equipped with a Dean-Stark trap. After cooling down the reaction to rt, the solvent was removed under vacuum and the residue obtained was purified by column chromatography (dichloromethane/acetone 100:0 to 90:10) to afford the product as a white solid (3.19 g, 91%, 56:44 diastereoisomeric ratio).
31P-NMR (162 MHz, CDCl
3): δ = 48.4 (s, 56%) and 48.1 (s, 44%).
6-(Cinnamyl)-6H-dibenzo[c,e][1,2λ5]oxaphosphinine-6-oxide (Table 2, Entry 11). General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and DOPO (216 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 50:50 to 0:100) to afford the product as a yellow solid (318 mg, 96%).
31P-NMR (CDCl
3, 162 MHz) δ = 27.4 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.89–7.99 (m, 2H), 7.68–7.74 (m, 1H), 7.49–7.55 (m, 1H), 7.36–7.42 (m, 1H), 7.22–7.30 (m, 6H), 7.16–7.21 (m, 2H), 6.35 (dd,
J = 5.3 and 15.8 Hz, 1H), 6.02–6.13 (m, 1H), 3.08 (ddt,
J = 1.3, 7.6 and 18.2 Hz, 2H);
13C-NMR (101 MHz, CDCl
3): δ = 149.6 (d,
JPOC = 8.2 Hz), 136.5 (d,
JPCCCC = 3.6 Hz), 135.9 (d,
JPCCC = 6.1 Hz), 135.8 (d,
JPCC = 13.8 Hz), 133.4 (d,
JPCCCC = 2.3 Hz), 130.7, 130.5 (d,
JPCCC = 10.2 Hz), 128.5 (2C), 128.4 (d,
JPCC = 13.0 Hz), 127.7 (d,
JPCCCC = 2.2 Hz), 126.2, 126.2, 125.2, 124.6, 124.2 (d,
JPC = 120 Hz), 123.8 (d,
JPCCC = 9.6 Hz), 122.3 (d,
JPCCC = 10.5 Hz), 120.4 (d,
JPCCC = 6.3 Hz), 117.2 (d,
JPCC = 11.0 Hz), 34.5 (d,
JPC = 93.8 Hz); HRMS (EI+)
m/
z calcd for C
21H
18O
2P ([M + H]
+) 333.1039, found 333.1048.
Diethyl cinnamylphosphonate (Table 2, Entry 12) [
15]. General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and diethylphosphite (138 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 50:50 to 0:100) to afford the product as an orange oil (240 mg, 94%).
31P-NMR (CDCl
3, 162 MHz) δ = 34.8 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.34–7.40 (m, 2H), 7.28–7.34 (m, 2H), 7.21–7.26 (m, 1H), 6.53 (dd,
J = 5.1 and 15.8 Hz, 1H), 6.12–6.23 (m, 1H), 4.08–4.18 (m, 4H), 2.76 (dd,
J = 7.6 and 22.2 Hz, 2H), 1.32 (t,
J = 6.1 Hz, 6H).
Cinnamyl diphenylphosphine oxide (Table 2, Entry 13) [
16]. General procedure was used with cinnamyl alcohol (0.13 mL, 1 mmol, 1 equiv.) and diphenyl phosphine oxide (202 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as a yellow oil (292 mg, 92%).
31P-NMR (CDCl
3, 162 MHz) δ = 28.9 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.75–7.82 (m, 4H), 7.46–7.58 (m, 6H), 7.18–7.29 (m, 5H), 6.44 (dd,
J = 4.5 and 15.8 Hz, 1H), 6.14–6.25 (m, 1H), 3.31 (ddd,
J = 1.2, 7.5 and 15.0 Hz, 2H).
Butyl allyl phenylphosphinate (Table 3, Entry 1) [
17]. General procedure was used with allyl alcohol (0.13 mL, 2 mmol, 2 equiv.) and butyl phenyl-
H-phosphinate (198 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as a yellow oil (234 mg, 98%).
31P-NMR (CDCl
3, 162 MHz) δ = 35.1 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.66–7.75 (m, 2H), 7.45–7.51 (m, 1H), 7.36–7.43 (m, 2H), 5.61–5.75 (m, 1H), 4.95–5.09 (m, 1H), 3.93–4.03 (m, 1H), 3.69–3.80 (m, 1H), 3.31 (dd,
J = 7.0 and 18.5 Hz, 2H), 1.58 (quint.,
J = 7.1 Hz, 2H), 1.32 (sextuplet,
J = 1.9 and 7.4 Hz, 2H), 0.83 (t,
J = 7.4 Hz, 3H).
Cyclohexyl allyl phenylphosphinate (Table 3, Entry 2). General procedure was used with allyl alcohol (0.13 mL, 2 mmol, 2 equiv.) and cyclohexyl phenyl-
H-phosphinate (224 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (247 mg, 94%).
31P-NMR (CDCl
3, 162 MHz) δ = 37.7 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.70–7.79 (m, 2H), 7.46–7.52 (m, 1H), 7.38–7.45 (m, 2H), 5.64–5.78 (m, 1H), 4.97–5.11 (m, 2H), 4.22–4.33 (m, 1H), 2.65–2.76 (m, 2H), 1.92–2.02 (m, 1H), 1.51–1.75 (m, 4H), 1.34–1.47 (m, 2H), 1.12–1.32 (m, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 132.1 (d,
JPCCCC = 2.7 Hz), 131.7 (d,
JPCCC = 9.6 Hz, 2C), 131.4 (d,
JPC = 126 Hz), 128.3 (d,
JPCC = 12.5 Hz, 2C), 127.4 (d,
JPCCC = 9.2 Hz), 120.1 (d,
JPCC = 13.0 Hz), 74.6 (d,
JPOC = 6.9 Hz), 36.6 (d,
JPC = 97.5 Hz), 34.2 (d,
JPOCC = 3.1 Hz), 33.7 (d,
JPOCC = 4.3 Hz), 25.1, 23.6, 23.6; HRMS (EI+)
m/
z calcd for C
15H
22O
2P ([M + H]
+) 265.1352, found 265.1359.
Butyl allyl cinnamylphosphinate (Table 3, Entry 3) [
6]. General procedure was used with allyl alcohol (0.15 mL, 2 mmol, 2 equiv.) and butyl cinnamyl-
H-phosphinate (238 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (254 mg, 91%).
31P-NMR (CDCl
3, 162 MHz) δ = 48.0 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.23–7.34 (m, 4H), 7.15–7.21 (m, 1H), 6.46 (dd,
J = 4.7 and 15.8 Hz, 1H), 6.08–6.19 (m, 1H), 5.72–5.86 (m, 1H), 5.14–5.23 (m, 2H), 4.00 (dt,
J = 6.8 and 6.9 Hz, 2H), 2.72 (ddd,
J = 0.7, 7.6 and 17.6 Hz, 2H), 2.60 (dd,
J = 7.5 and 17.2 Hz, 2H), 1.60 (quint.,
J = 7.1 Hz, 2H), 1.35 (sextuplet,
J = 7.5 Hz, 2H), 0.88 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 136.7 (d,
JPCCCC = 3.3 Hz), 135.0 (d,
JPCC = 12.9 Hz), 128.6 (2C), 127.7 (d,
JPCCC = 8.0 Hz), 127.6, 126.2, 126.1, 120.3 (d,
JPCC = 12.6 Hz), 118.8 (d,
JPCCC = 9.6 Hz), 64.5 (d,
JPOC = 7.2 Hz), 33.8 (d,
JPC = 88.7 Hz), 32.9 (d,
JPC = 89.0 Hz), 32.7 (d,
JPOCC = 5.7 Hz), 18.8, 13.6.
Cyclohexyl 3-[(2-methyl-2-propene)phenyl] phenylphosphinate (Table 3, Entry 4). General procedure was used with
trans-2-methyl-3-phenyl-2-propen-1-ol (0.15 mL, 1 mmol, 1 equiv.) and cyclohexyl phenyl-
H-phosphinate (224 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (330 mg, 93%).
31P-NMR (CDCl
3, 162 MHz) δ = 39.5 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.77–7.85 (m, 2H), 7.48–7.54 (m, 1H), 7.40–7.47 (m, 2H), 7.22–7.29 (m, 2H), 7.11–7.17 (m, 1H), 7.03–7.09 (m, 2H), 6.09 (d,
J = 5.5 Hz, 1H), 4.29–4.40 (m, 1H), 2.76–2.91 (m, 2H), 1.98–2.07 (m, 1H), 1.92 (dd,
J = 1.3 and 3.5 Hz, 3H), 1.59–1.79 (m, 4H), 1.39–1.49 (m, 2H), 1.17–1.36 (m, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 137.8 (d,
JPCCCC = 3.7 Hz), 132.1 (d,
JPCCCC = 2.7 Hz), 131.9 (d,
JPCCC = 9.5 Hz, 2C), 131.6 (d,
JPC = 125 Hz), 130.0 (d,
JPCC = 11.5 Hz), 129.3 (d,
JPCCC = 10.3 Hz), 128.7 (d,
JPCCCCC = 2.9 Hz, 2C), 128.3 (d,
JPCC = 12.4 Hz, 2C), 128.0 (2C), 126.3, 74.7 (d,
JPOC = 7.0 Hz), 42.8 (d,
JPC = 95.2 Hz), 34.3 (d,
JPOCC = 2.9 Hz), 33.7 (d,
JPOCC = 4.3 Hz), 25.2, 23.6 (2C), 19.5 (d,
JCCC = 2.1 Hz); HRMS (EI+)
m/
z calcd for C
22H
28O
2P ([M + H]
+) 355.1827, found 355.1849.
Butyl 3-[(2-methyl-2-propene)phenyl] cinnamylphosphinate (Table 3, Entry 5). General procedure was used with
trans-2-methyl-3-phenyl-2-propen-1-ol (0.15 mL, 1 mmol, 1 equiv.) and butyl cinnamyl-
H-phosphinate (238 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 50:50 to 0:100) to afford the product as an orange oil (283 mg, 77%).
31P-NMR (CDCl
3, 162 MHz) δ = 48.2 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.19–7.38 (m, 10H), 6.54 (dd,
J = 4.4 and 15.8 Hz, 1H), 6.43 (d,
J = 4.8 Hz, 1H), 6.19–6.30 (m, 1H), 4.10 (dt,
J = 6.7 and 6.8 Hz, 2H), 2.84 (dd,
J = 7.6 and 17.1 Hz, 2H), 2.77 (d,
J = 17.3 Hz, 2H), 2.07 (d,
J = 3.1 Hz, 3H), 1.68 (quint.,
J = 7.4 Hz, 2H), 1.42 (sextuplet,
J = 7.5 Hz, 2H), 0.93 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 137.5 (d,
JPCCCC = 3.5 Hz), 136.8 (d,
JPCCCC = 3.1 Hz), 135.0 (d,
JPCC = 12.7 Hz), 129.9 (d,
JPCCC = 11.7 Hz), 129.8 (d,
JPCC = 10.0 Hz), 128.9, 128.8, 128.6 (2C), 128.2 (2C), 127.7, 126.6, 126.2, 126.2, 119.2 (d,
JPCCC = 9.9 Hz), 64.6 (d,
JPOC = 7.2 Hz), 40.6 (d,
JPC = 86.7 Hz), 33.5 (d,
JPC = 88.0 Hz), 32.9 (d,
JPOCC = 5.7 Hz), 19.6 (d,
JPCCC = 1.9 Hz), 18.9, 13.7; HRMS (EI+)
m/
z calcd for C
23H
30O
2P ([M + H]
+) 369.1983, found 369.1906.
Butyl (2-methylprop-2-ene) cinnamylphosphinate (Table 3, Entry 6). General procedure was used with methylallyl alcohol (0.154 mL, 1 mmol, 1 equiv.) and butyl cinnamyl-
H-phosphinate (238 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (148 mg, 51%).
31P-NMR (CDCl
3, 162 MHz) δ = 47.4 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.30–7.39 (m, 4H), 7.22–7.28 (m, 1H), 6.52 (dd,
J = 4.6 and 15.8 Hz, 1H), 6.14–6.25 (m, 1H), 4.97–5.01 (m, 1H), 4.95 (d,
J = 4.5 Hz, 1H), 4.06 (dt,
J = 6.8 Hz, 2H), 2.81 (dd,
J = 7.6 and 17.3 Hz, 2H), 2.62 (d,
J = 17.4 Hz, 2H), 1.92 (t,
J = 1.2 Hz, 3H), 1.66 (quint.,
J = 7.5 Hz, 2H), 1.41 (sextuplet,
J = 7.6 Hz, 2H), 0.93 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 136.8 (d,
JPCCCC = 3.3 Hz), 136.7 (d,
JPCCC = 9.0 Hz), 134.9 (d,
JPCC = 13.1 Hz), 128.6 (2C), 127.6, 126.2, 126.2, 119.2 (d,
JPCCC = 9.7 Hz), 115.7 (d,
JPCC = 10.9 Hz), 64.6 (d,
JPOC = 7.2 Hz), 37.6 (d,
JPC = 87.4 Hz), 33.1 (d,
JPC = 88.3 Hz), 32.8 (d,
JPOCC = 5.8 Hz), 24.1 (d,
JPCCC = 2.2 Hz), 18.8, 13.7; HRMS (EI+)
m/
z calcd for C
17H
26O
2P ([M + H]
+) 293.1670, found 293.1693.
Butyl myrtenyl phenylphosphinate (Table 3, Entry 7). General procedure was used with myrtenol (0.17 mL, 1 mmol, 1 equiv.) and butyl phenyl-
H-phosphinate (198 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (246 mg, 74%).
31P-NMR (CDCl
3, 162 MHz) δ = 40.3 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.64–7.72 (m, 2H), 7.41–7.47 (m, 1H), 7.33–7.40 (m, 2H), 5.15–5.21 (m, 1H), 3.88–3.97 (m, 1H), 3.61–3.71 (m, 1H), 2.52–2.76 (m, 2H), 2.17–2.26 (m, 1H), 2.00–2.12 (m, 3H), 1.87–1.96 (m, 1H), 1.53 (quint.,
J = 7.2 Hz, 2H), 1.30 (sextuplet,
J = 7.5 Hz, 2H), 1.14 (d,
J = 10.4 Hz, 3H), 0.99 (dd,
J = 8.7 and 20.1 Hz, 1H), 0.81 (t,
J = 7.6 Hz, 3H), 0.72 (d,
J = 4.0 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 137.8 (d,
JPCC = 10.4 Hz), 132.0 (d,
JPCCCC = 2.8 Hz, 0.5C), 132.0 (d,
JPCCCC = 2.8 Hz, 0.5C), 131.8 (d,
JPCCC = 9.7 Hz), 131.8 (d,
JPCCC = 9.8 Hz), 131.0 (d,
JPC = 123 Hz), 131.0 (d,
JPC = 123 Hz), 128.2 (d,
JPCC = 12.3 Hz, 2C), 122.2 (d,
JPCCC = 12.6 Hz, 0.5C), 122.2 (d,
JPCCC = 12.6 Hz, 0.5C), 64.1 (d,
JPOC = 7.0 Hz, 0.5C), 64.0 (d,
JPOC = 7.0 Hz, 0.5C), 46.9 (d,
JPOCC = 2.8 Hz, 0.5C), 46.7 (d,
JPOCC = 2.4 Hz, 0.5C), 40.1 (0.5C), 40.1 (0.5C), 38.7 (d,
JPC = 97.6 Hz), 37.9 (d,
JPCCCC = 2.2 Hz, 0.5C), 37.9 (d,
JPOCC = 2.2 Hz, 0.5C), 32.5 (0.5C), 32.5 (0.5C), 31.6 (d,
JPCCCC = 2.4 Hz, 0.5C), 31.5 (d,
JPCCCC = 2.0 Hz, 0.5C), 26.1 (0.5C), 26.1 (0.5C), 21.0 (0.5C), 20.9 (0.5C), 18.7, 13.6; HRMS (EI+)
m/
z calcd for C
20H
30O
2P ([M + H]
+) 333.1983, found 333.1906.
Cyclohexyl benzyl octylphosphinate (Table 3, Entry 8) [
1]. General procedure was used with benzyl alcohol (0.11 mL, 1 mmol, 1 equiv.) and cyclohexyl octyl-
H-phosphinate (260 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 50:50 to 0:100) to afford the product as an orange oil (226 mg, 65%).
31P-NMR (CDCl
3, 162 MHz) δ = 52.1 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.22–7.37 (m, 5H), 4.30–4.42 (m, 1H), 3.13 (d,
J = 16.7 Hz, 2H), 1.86–1.95 (m, 2H), 1.40–1.84 (m, 10H), 1.17–1.37 (m, 12H), 0.89 (t,
J = 6.9 Hz, 3H).
Butyl benzyl cinnamylphosphinate (Table 3, Entry 9). General procedure was used with benzyl alcohol (0.11 mL, 1 mmol, 1 equiv.) and butyl cinnamyl-
H-phosphinate (238 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (187 mg, 57%).
31P-NMR (CDCl
3, 162 MHz) δ = 48.5 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.21–7.39 (m, 10H), 6.45 (dd,
J = 4.2 and 15.8 Hz, 1H), 6.08–6.20 (m, 1H), 3.93–4.07 (m, 2H), 3.20 (d,
J = 16.8 Hz, 2H), 2.70 (dd,
J = 7.6 and 17.2 Hz, 2H), 2.01–2.12 (m, 2H), 1.62 (quint.,
J = 7.3 Hz, 2H), 1.37 (sextuplet,
J = 7.5 Hz, 2H), 0.91 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 136.8 (d,
JPCCCC = 3.1 Hz), 135.1 (d,
JPCCC = 13.0 Hz), 131.6 (d,
JPCC = 7.3 Hz), 129.9 (d,
JPCCC = 5.8 Hz, 2C), 128.7 (d,
JPCCCC = 2.6 Hz, 2C), 128.6 (2C), 127.7, 127.0 (d,
JPCCCCC = 3.0 Hz), 126.2, 126.2, 118.9 (d,
JPCCC = 9.8 Hz), 64.8 (d,
JPOC = 7.0 Hz), 36.0 (d,
JPC = 87.1 Hz), 33.2 (d,
JPC = 88.0 Hz), 32.8 (d,
JPOCC = 5.9 Hz), 27.0 (d,
JPCC = 3.5 Hz), 18.8, 13.7; HRMS (EI+)
m/
z calcd for C
20H
26O
2P ([M + H]
+) 329.1665, found 329.1671.
Cyclohexyl 1-(naphthylmethyl) phenylphosphinate (Table 3, Entry 10). General procedure was used with 1-naphthalenemethanol (158 mg, 1 mmol, 1 equiv.) and cyclohexyl phenyl-
H-phosphinate (224 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (266 mg, 73%).
31P-NMR (CDCl
3, 162 MHz) δ = 35.5 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 7.95–8.02 (m, 1H), 7.77–7.84 (m, 1H), 7.69–7.75 (m, 1H), 7.56–7.65 (m, 2H), 7.39–7.48 (m, 3H), 7.29–7.37 (m, 3H), 7.22–7.28 (m, 1H), 4.22–4.33 (m, 1H), 3.76 (d,
J = 17.8 Hz, 2H), 1.77–1.86 (m, 1H), 1.45–1.67 (m, 4H), 1.09–1.44 (m, 5H);
13C-NMR (101 MHz, CDCl
3): δ = 133.8 (d,
JPCCCC = 2.1 Hz), 132.2 (d,
JPCCCC = 4.3 Hz), 132.1 (d,
JPCCCC = 2.2 Hz), 132.0 (d,
JPCCC = 9.5 Hz, 2C), 131.4 (d,
JPC = 124 Hz), 128.6 (d,
JPCCC = 6.7 Hz), 128.4, 128.4 (d,
JPCC = 8.0 Hz), 128.2 (d,
JPCC = 12.4 Hz, 2C), 127.5 (d,
JPCCCC = 3.8 Hz), 125.7, 125.5, 125.2 (d,
JPCCCC = 3.6 Hz), 124.6, 74.9 (d,
JPOC = 6.8 Hz), 35.9 (d,
JPC = 96.1 Hz), 34.0 (d,
JPOCC = 2.6 Hz), 33.5 (d,
JPOCC = 4.0 Hz), 25.1, 23.4 (2C); HRMS (EI+)
m/
z calcd for C
22H
28O
2P ([M + H]
+) 355.1827, found 355.1849.
Butyl (1-methylnaphthalene) cinnamylphosphinate (Table 3, Entry 11). General procedure was used with 1-naphthalenemethanol (316 mg, 2 mmol, 2 equiv.) and butyl cinnamyl-
H-phosphinate (238 mg, 1 mmol, 1 equiv.). The crude obtained was purified by column chromatography (hexane/ethyl acetate 100:0 to 0:100) to afford the product as an orange oil (186 mg, 49%).
31P-NMR (CDCl
3, 162 MHz) δ = 47.5 (s);
1H-NMR (CDCl
3, 400 MHz) δ = 8.09–8.14 (m, 1H), 7.85–7.90 (m, 1H), 7.76–7.82 (m, 1H), 7.49–7.58 (m, 2H), 7.42–7.48 (m, 1H), 7.29–7.36 (m, 5H), 7.23–7.29 (m, 1H), 6.40 (dd,
J = 4.5 and 15.8 Hz, 1H), 6.09–6.20 (m, 1H), 3.83–4.01 (m, 2H), 3.62–3.78 (m, 2H), 2.75 (dd,
J = 7.6 and 16.8 Hz, 2H), 1.51 (quint.,
J = 7.4 Hz, 2H), 1.27 (sextuplet,
J = 7.6 Hz, 2H), 0.85 (t,
J = 7.4 Hz, 3H);
13C-NMR (101 MHz, CDCl
3): δ = 136.7 (d,
JPCCCC = 3.1 Hz), 135.0 (d,
JPCC = 12.8 Hz), 134.0 (d,
JPCCCC = 2.4 Hz), 132.1 (d,
JPCCC = 4.6 Hz), 128.8, 128.6 (2C), 128.5 (d,
JPCCCC = 6.7 Hz), 128.1 (d,
JPCC = 8.2 Hz), 127.9 (d,
JPCCCC = 3.8 Hz), 127.7, 126.2 (3C), 125.9, 125.4 (d,
JPCCCC = 3.6 Hz), 124.4 (d,
JPCCCCC = 1.2 Hz), 118.9 (d,
JPCCC = 9.9 Hz), 64.9 (d,
JPOC = 7.3 Hz), 33.7 (d,
JPC = 88.2 Hz), 33.2 (d,
JPC = 87.7 Hz), 32.7 (d,
JPOCC = 5.9 Hz), 18.7, 13.6; HRMS (EI+)
m/
z calcd for C
24H
28O
2P ([M + H]
+) 379.1827, found 379.1750.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}