
Advances in Organic and Organic-Inorganic Hybrid Polymeric Supports for Catalytic Applications



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
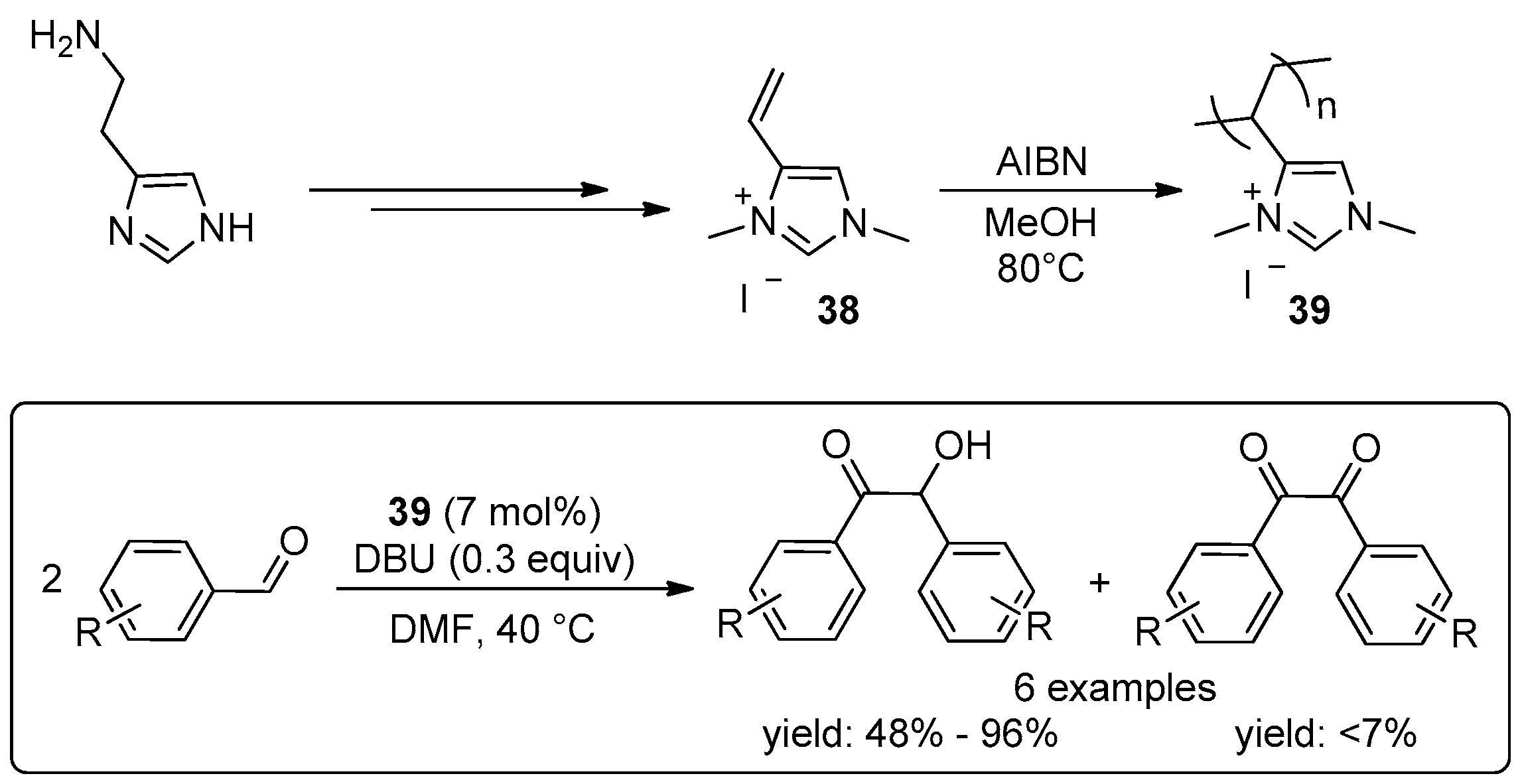{kind=link}
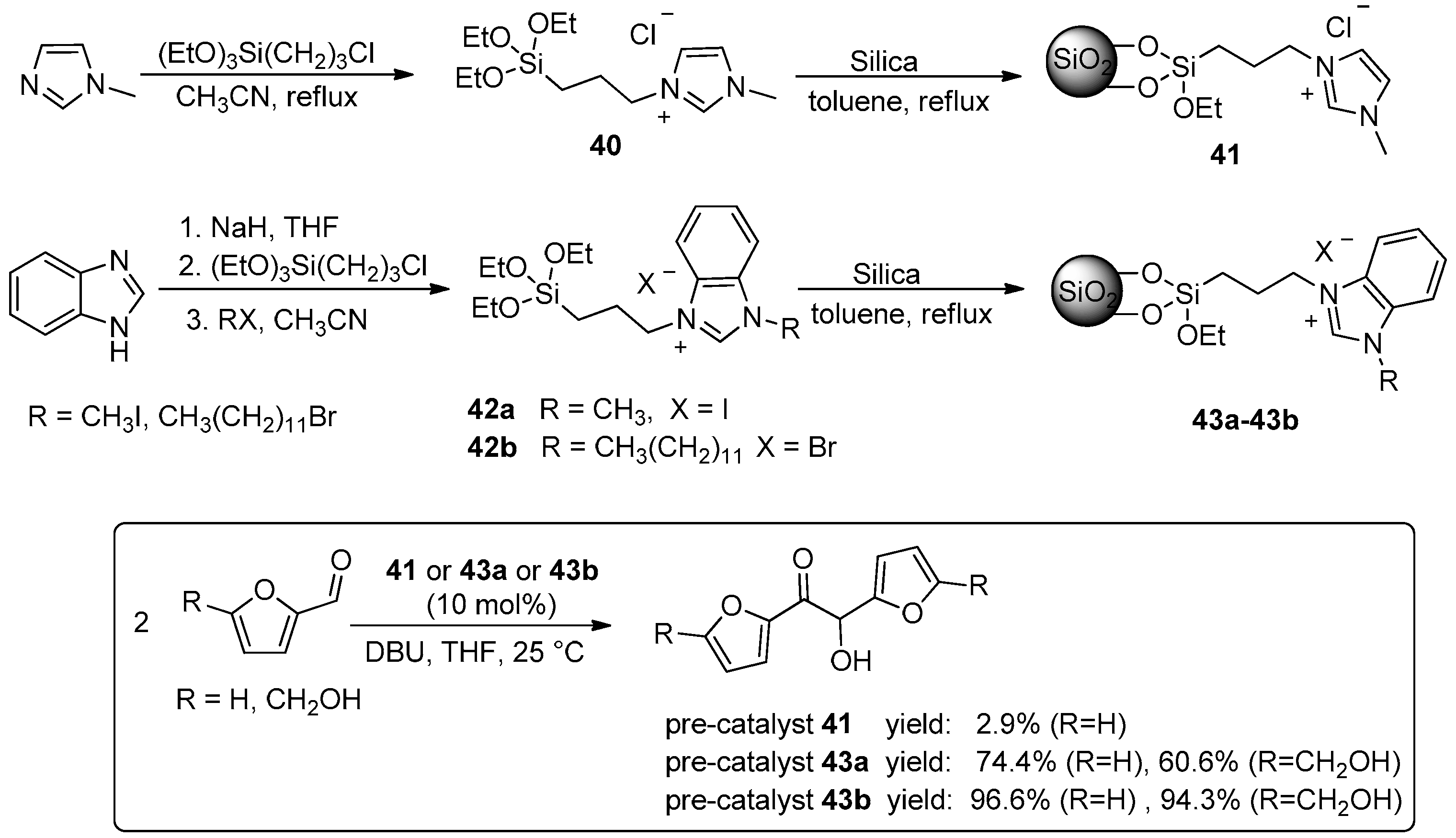{kind=link}
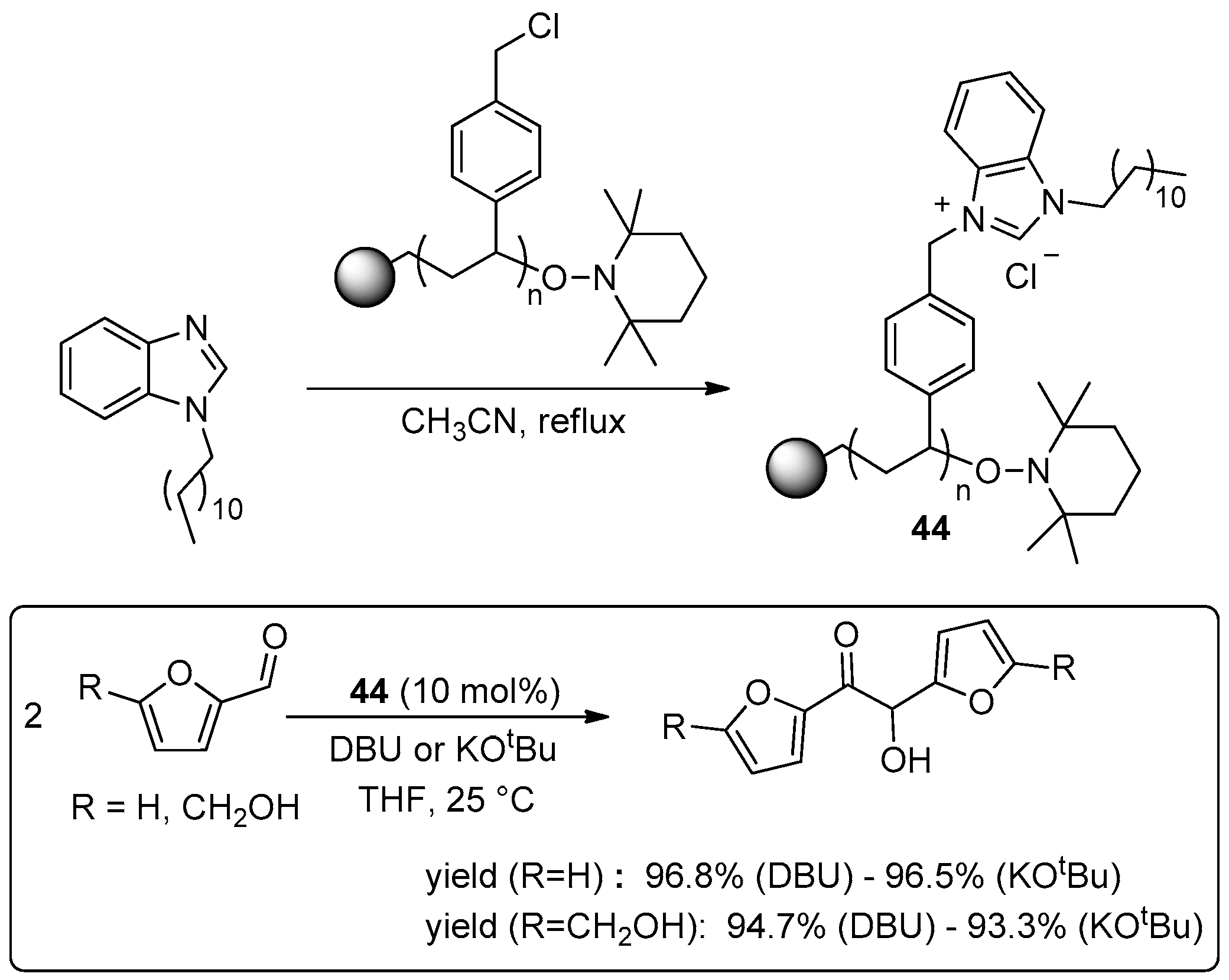{kind=link}
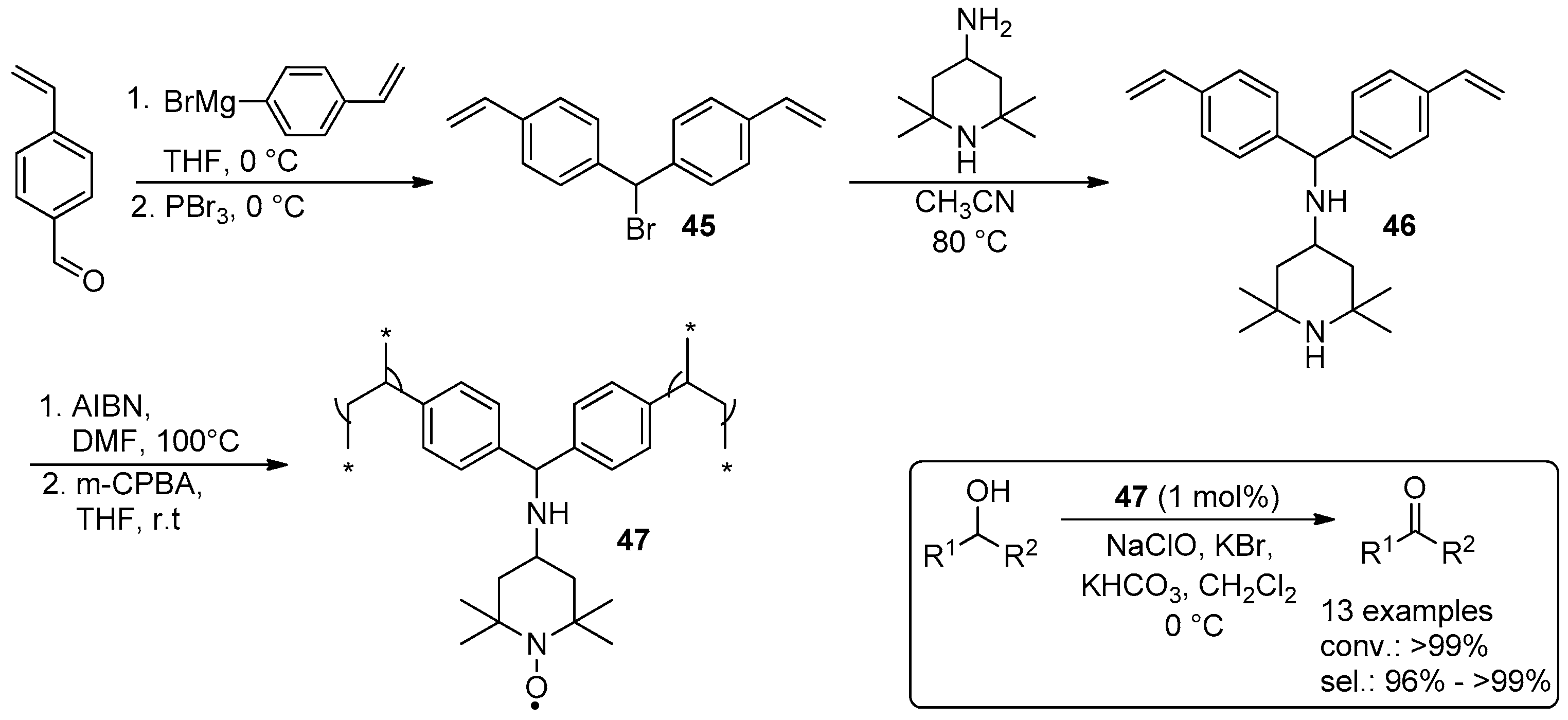{kind=link}
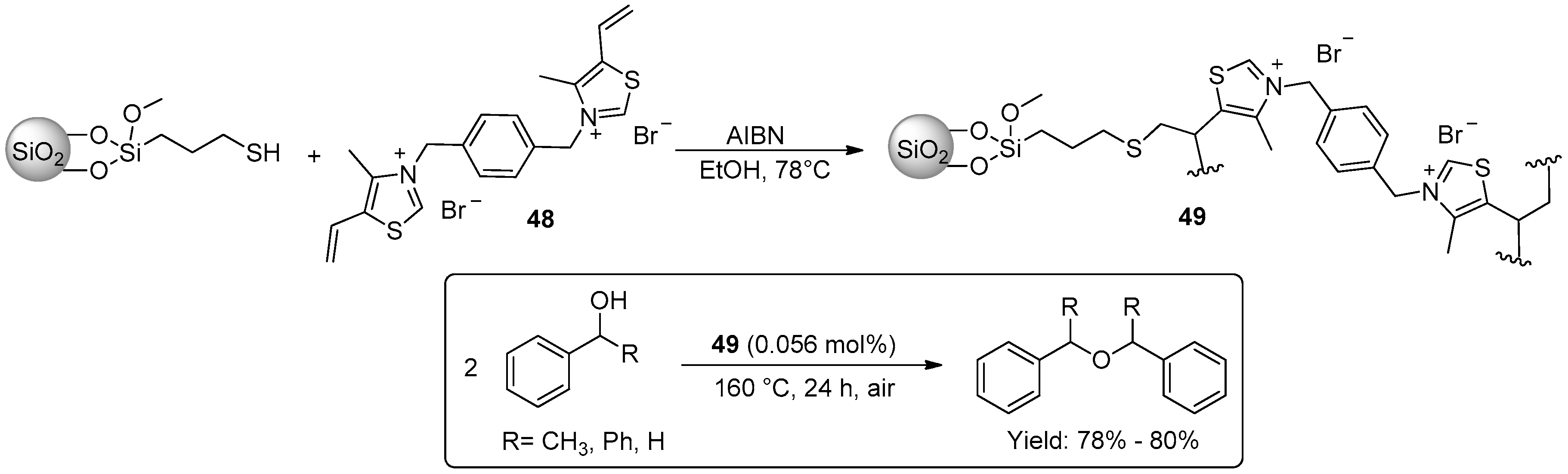{kind=link}
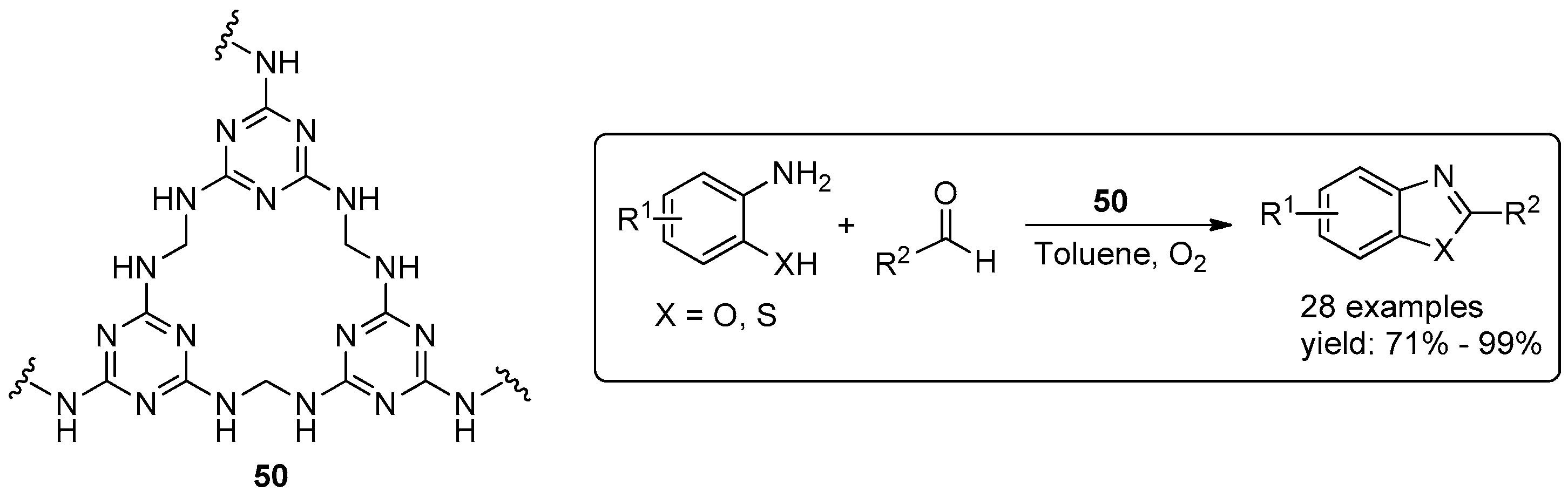{kind=link}
{kind=link}
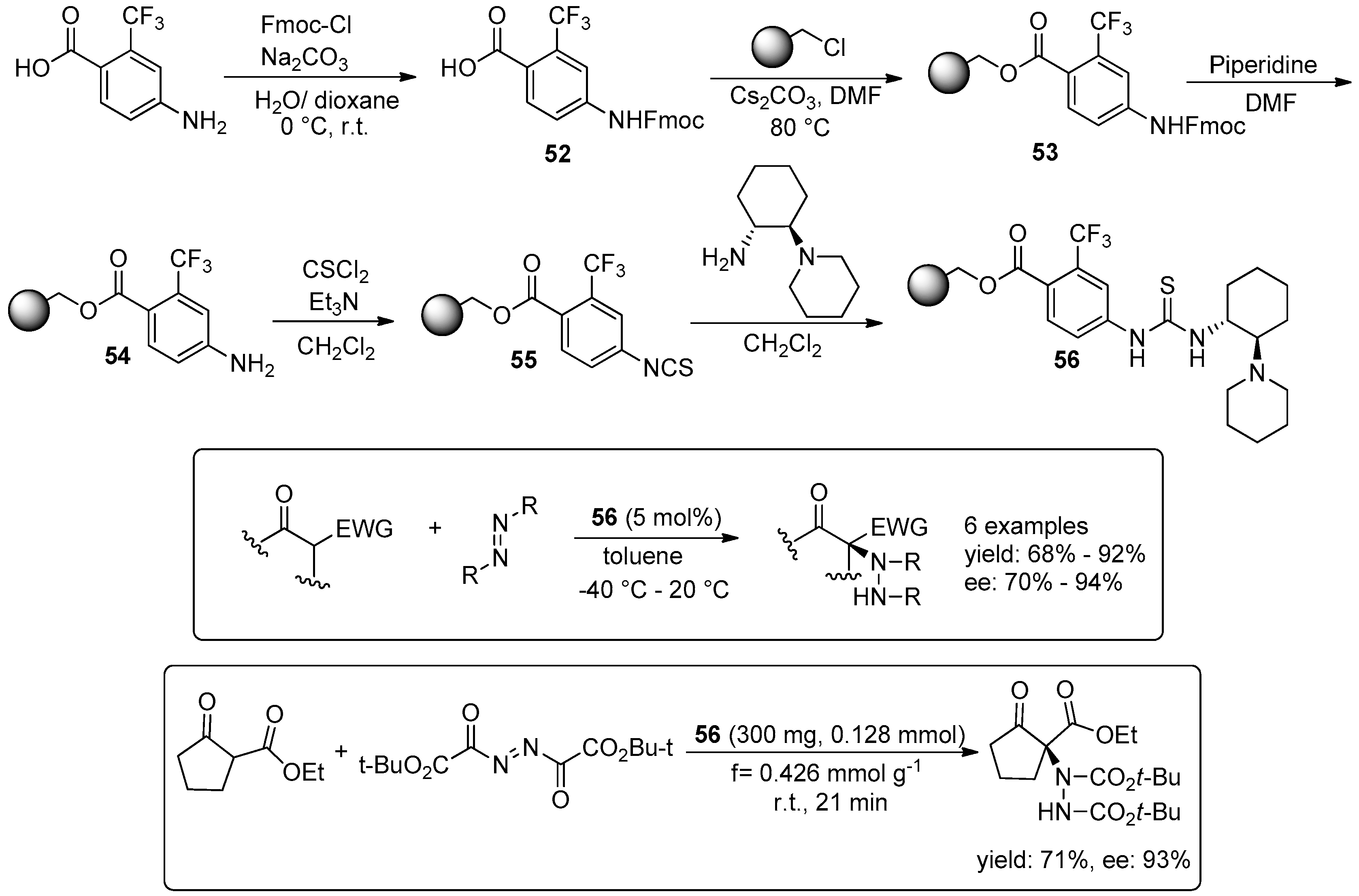{kind=link}
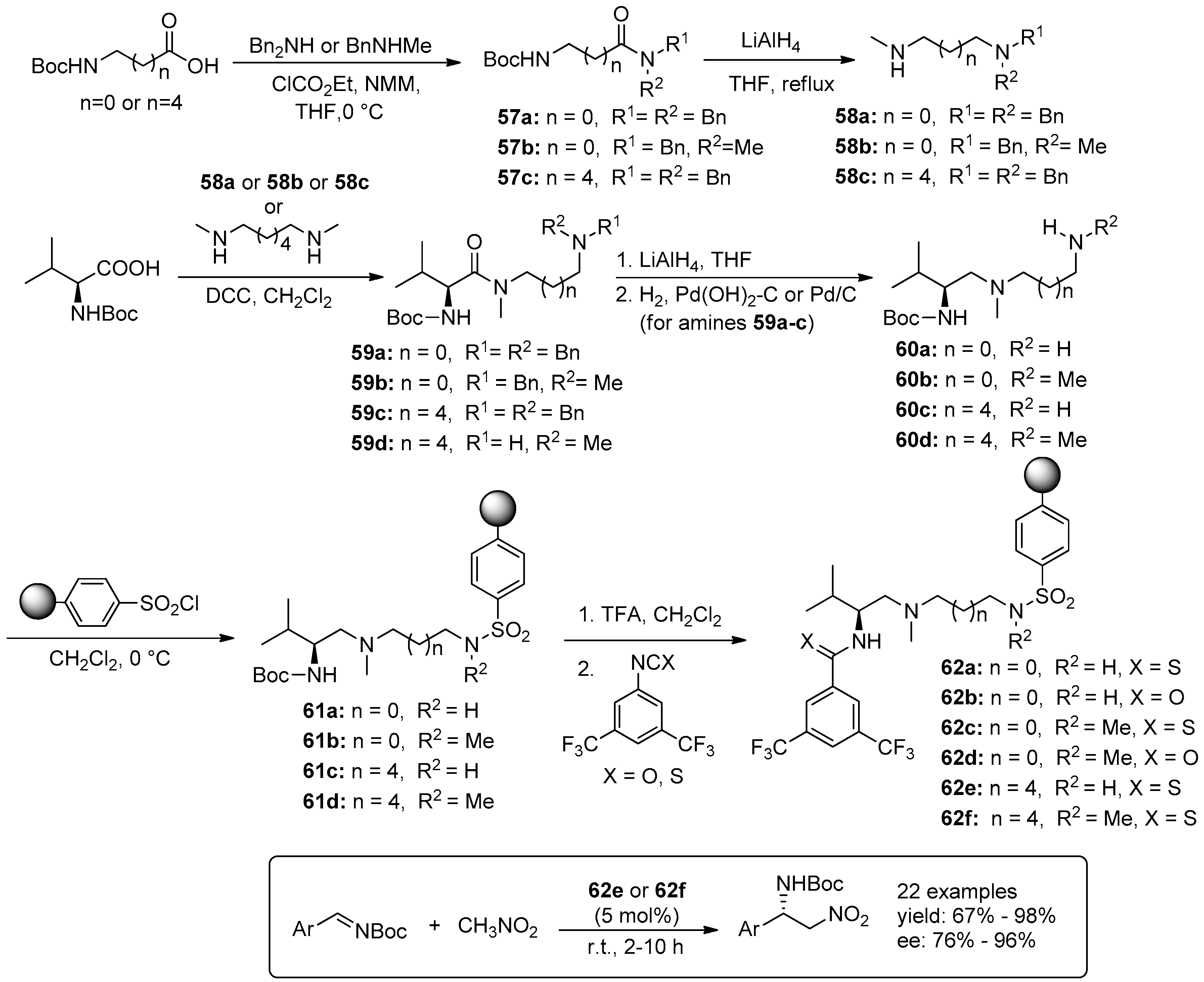{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
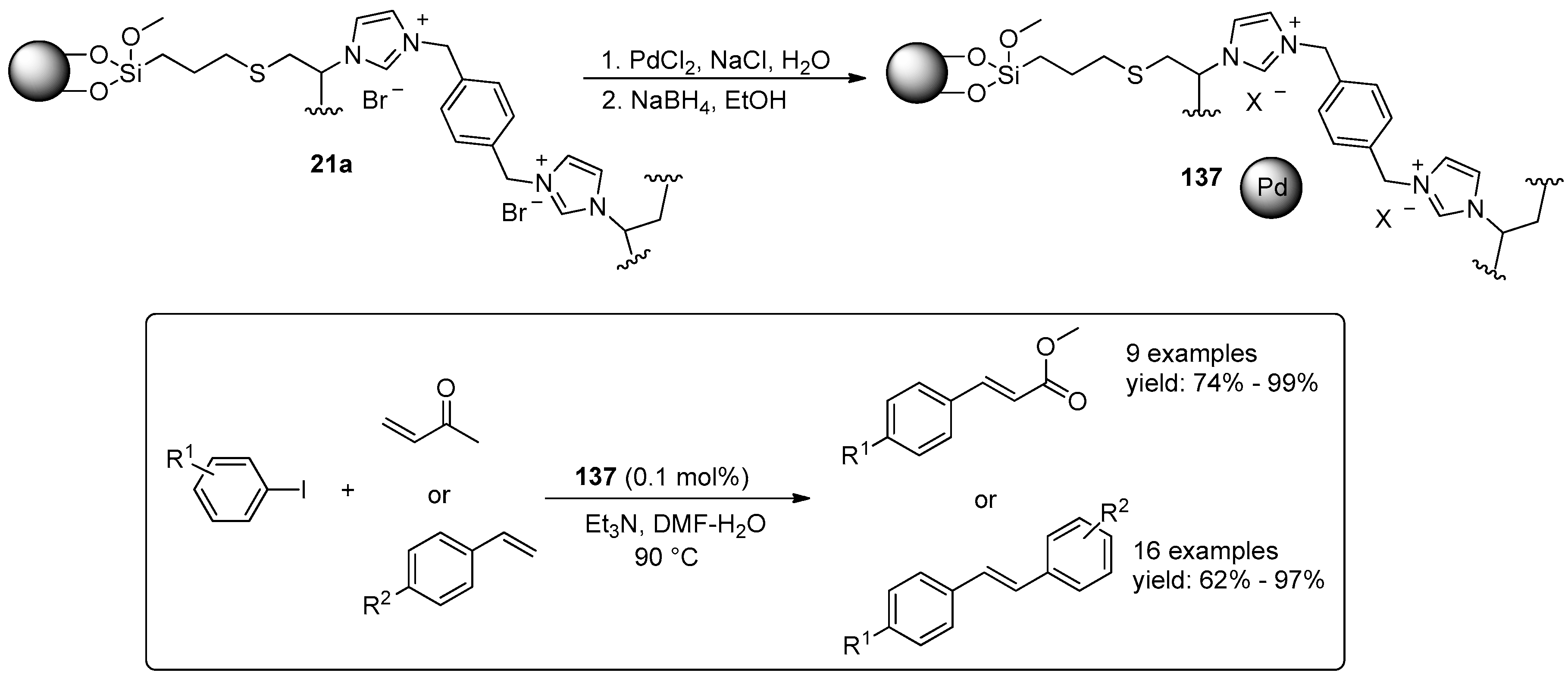{kind=link}
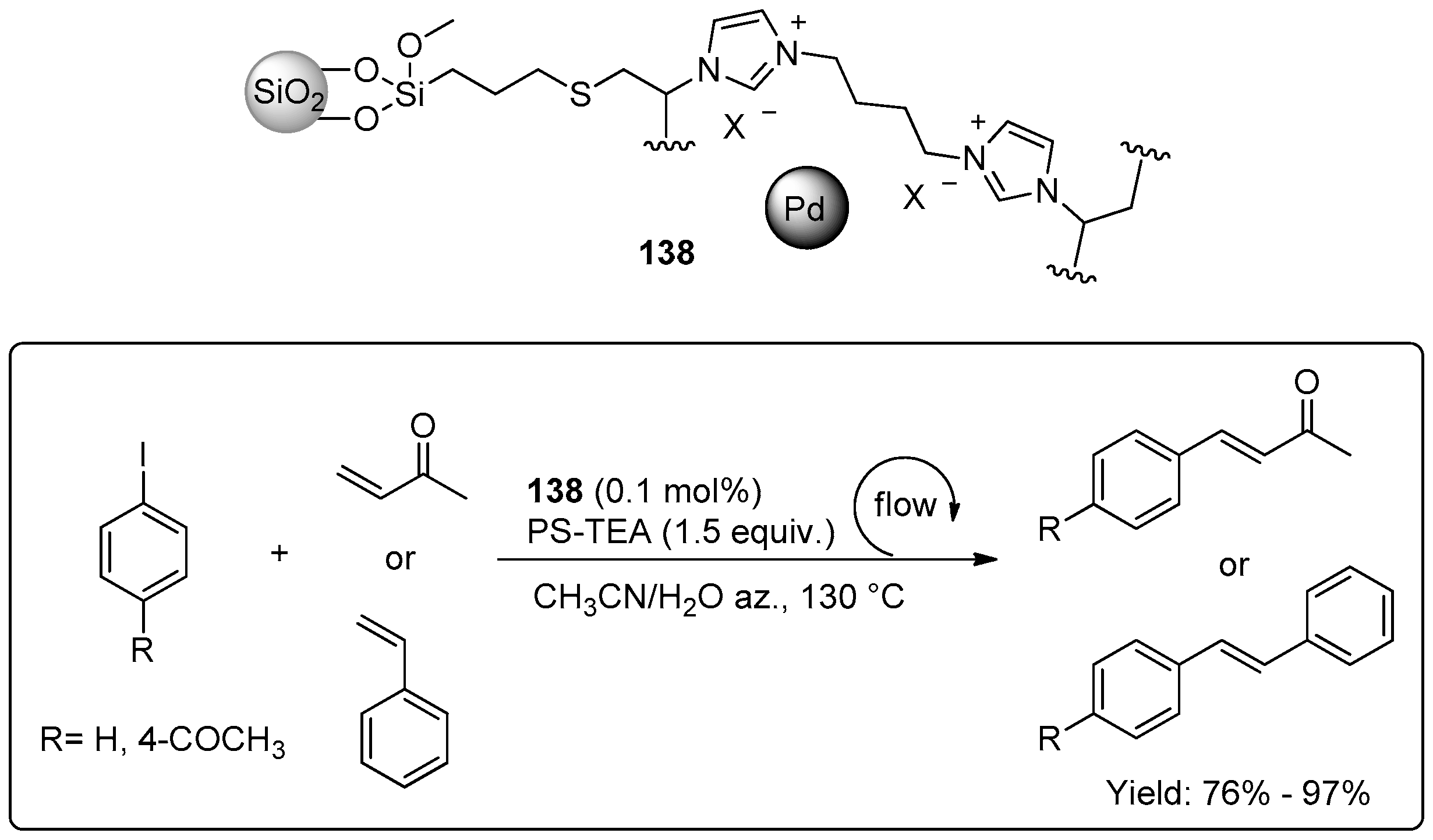{kind=link}
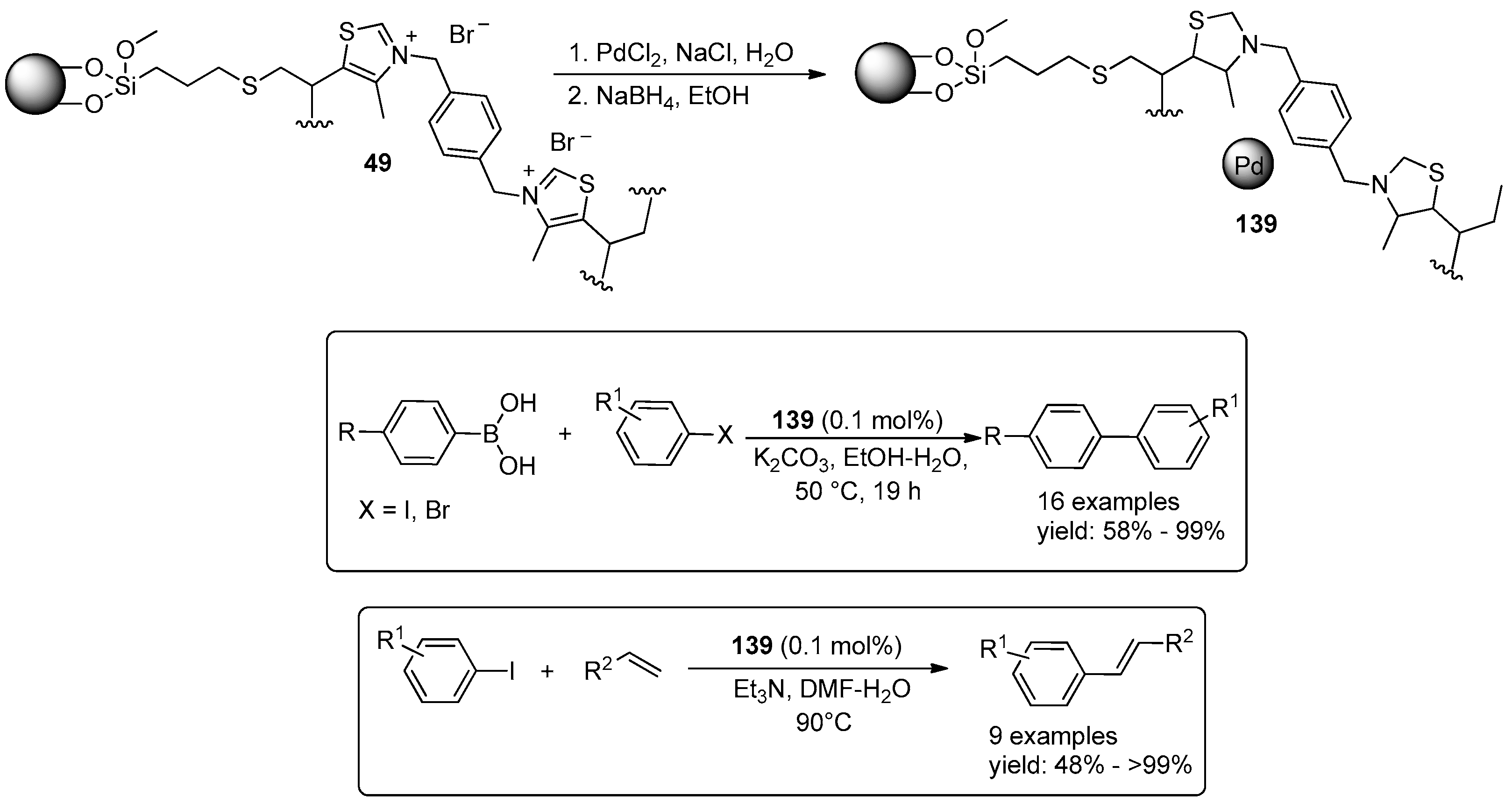{kind=link}
{kind=link}
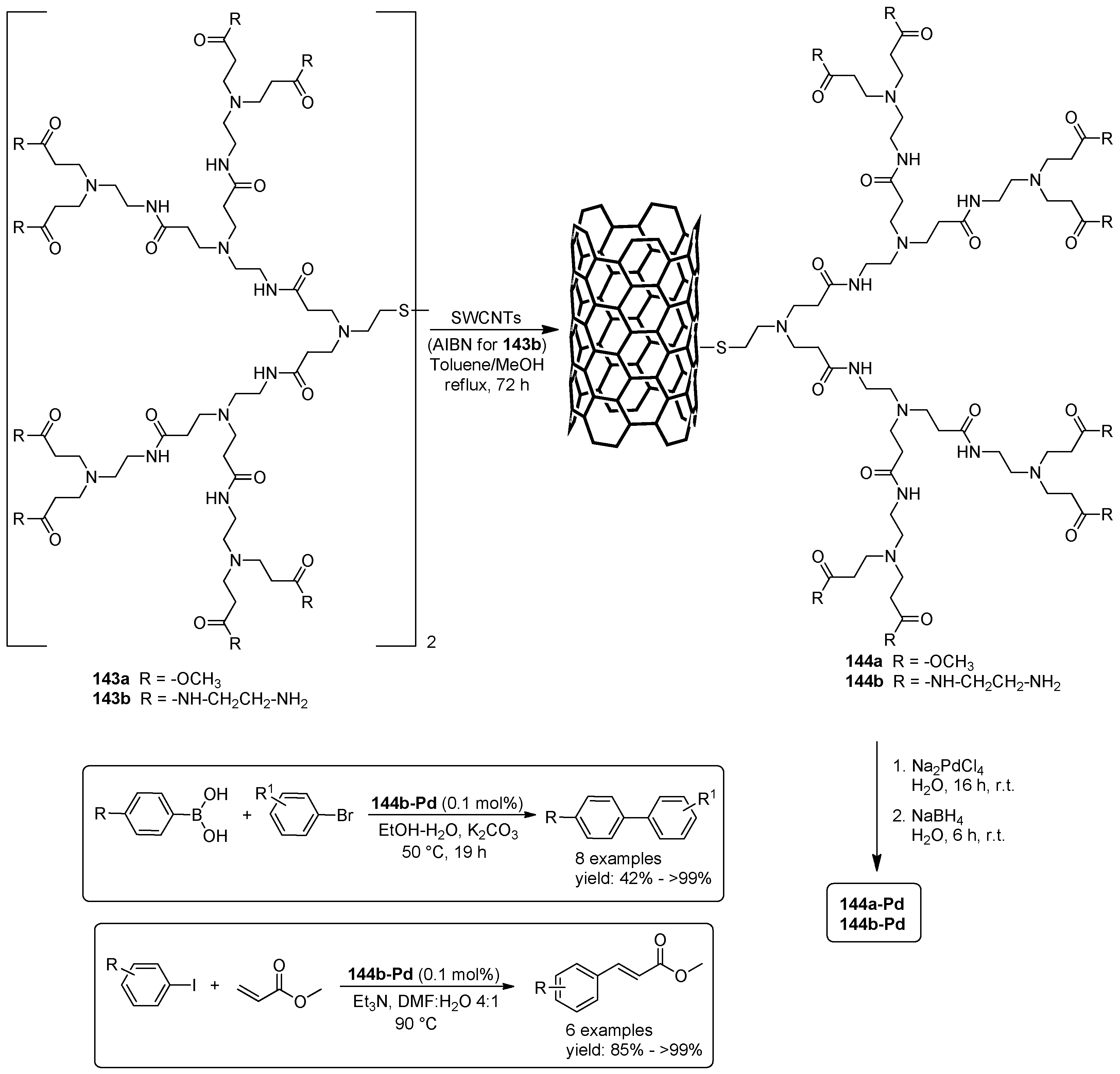{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Organocatalysed Non-Asymmetric Reactions
2.1. Brønsted Acid Catalysed Reactions
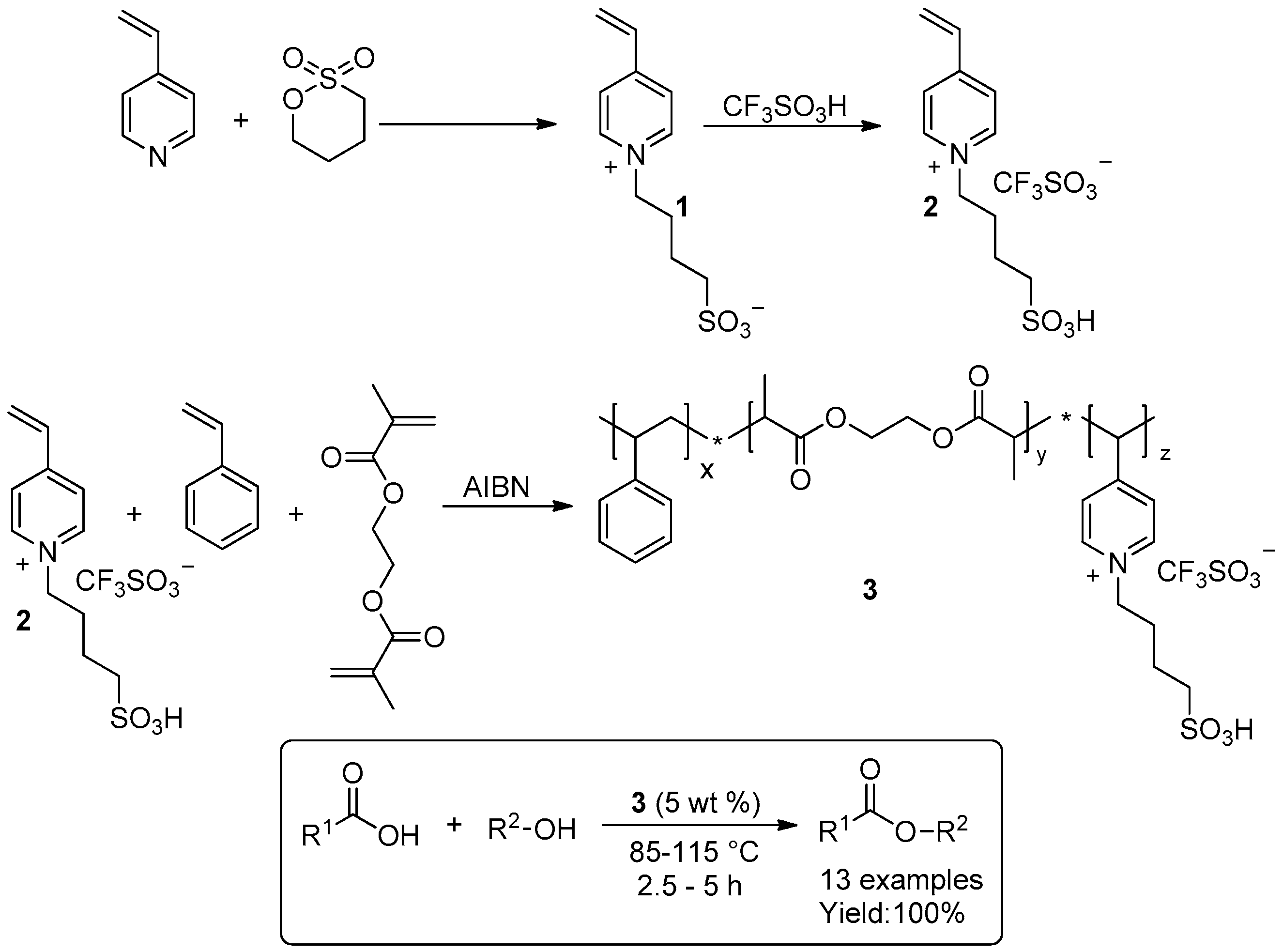
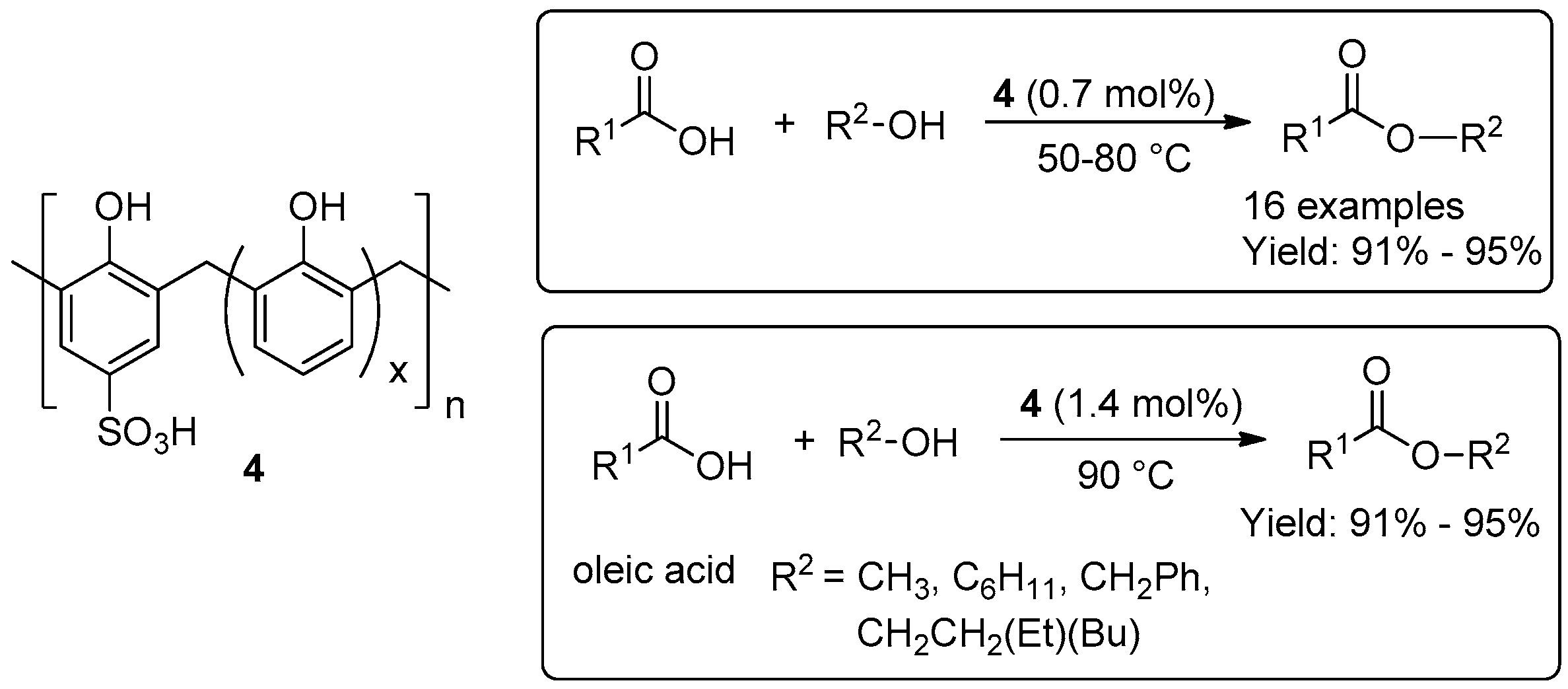
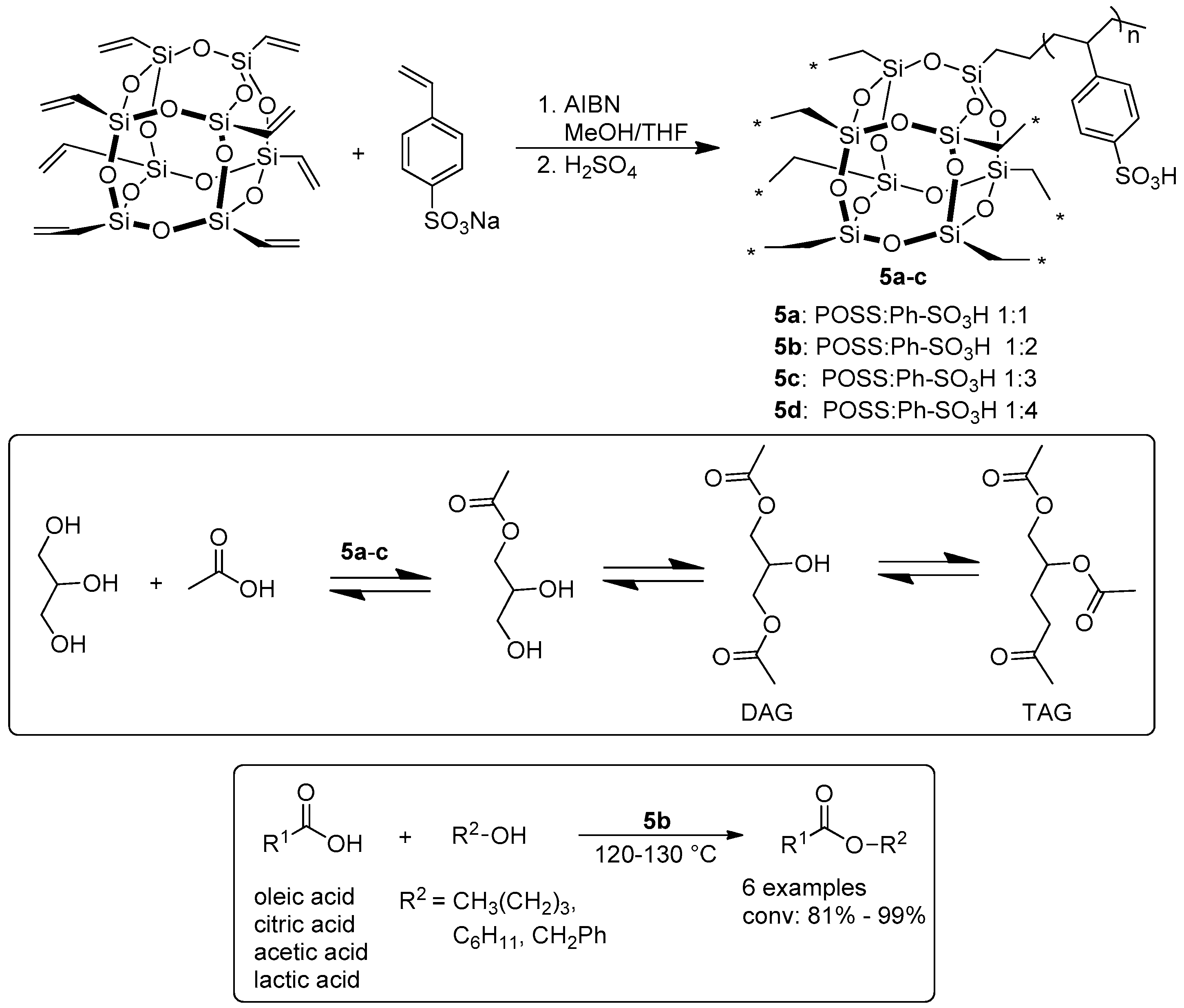
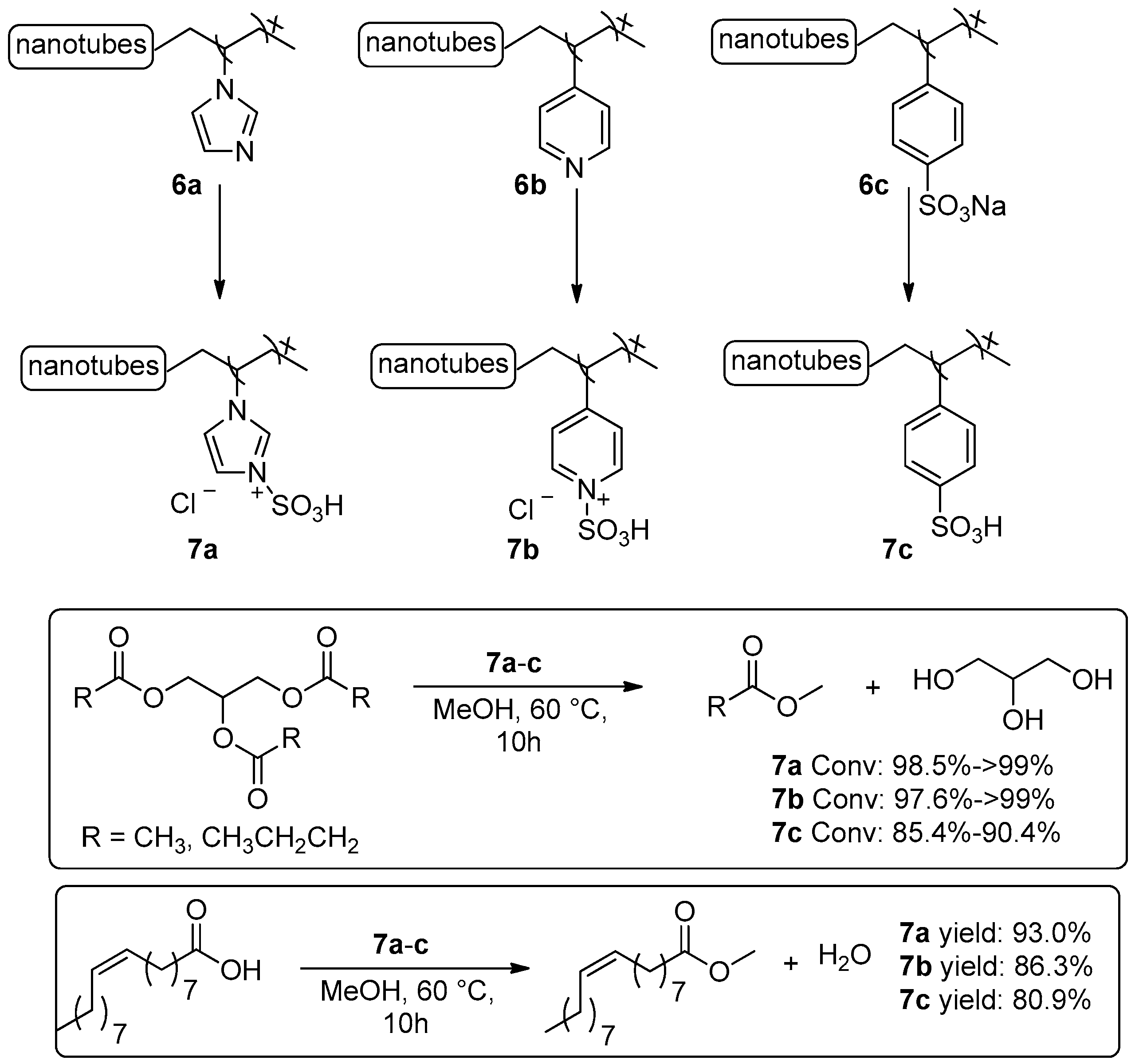
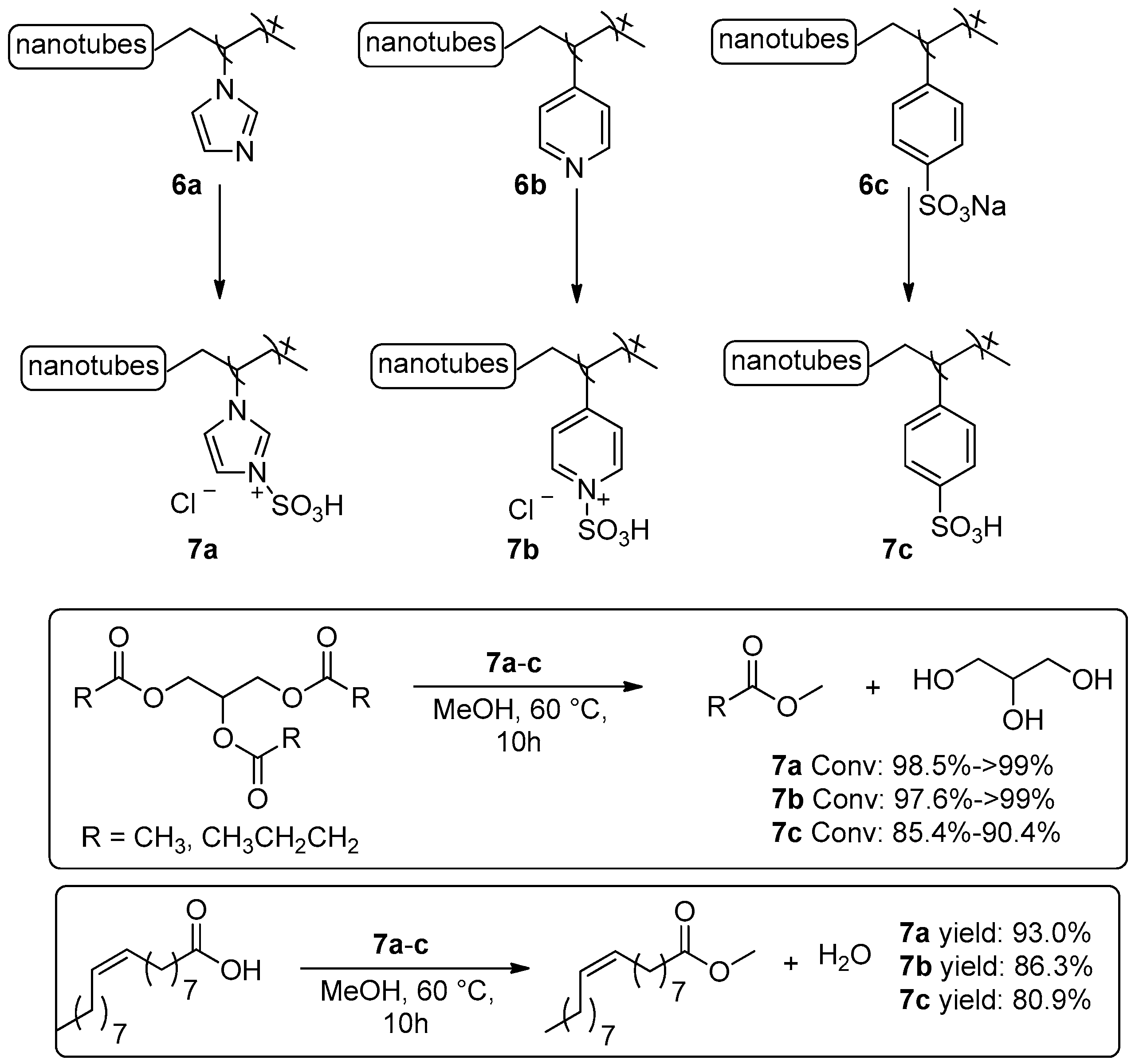
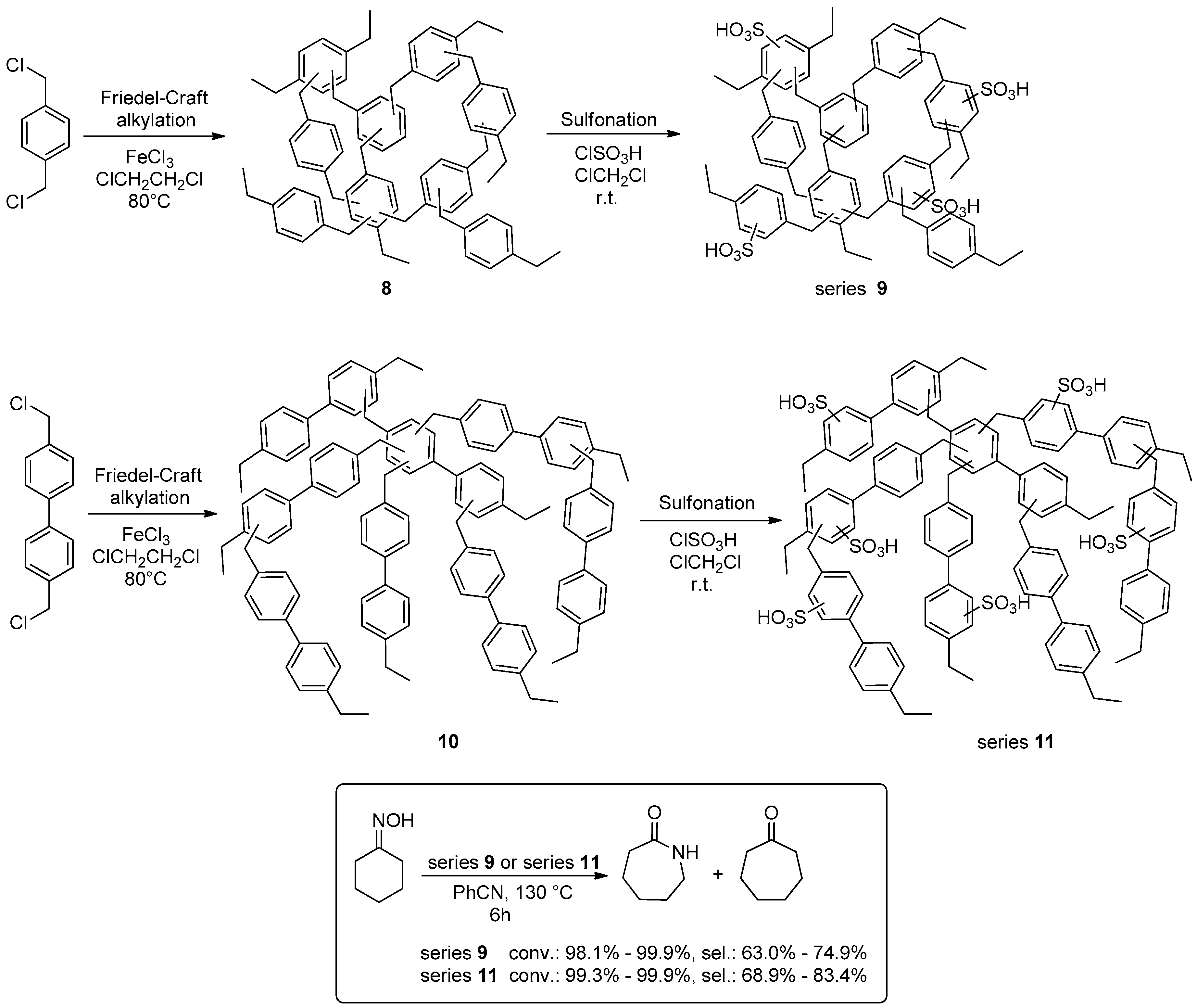
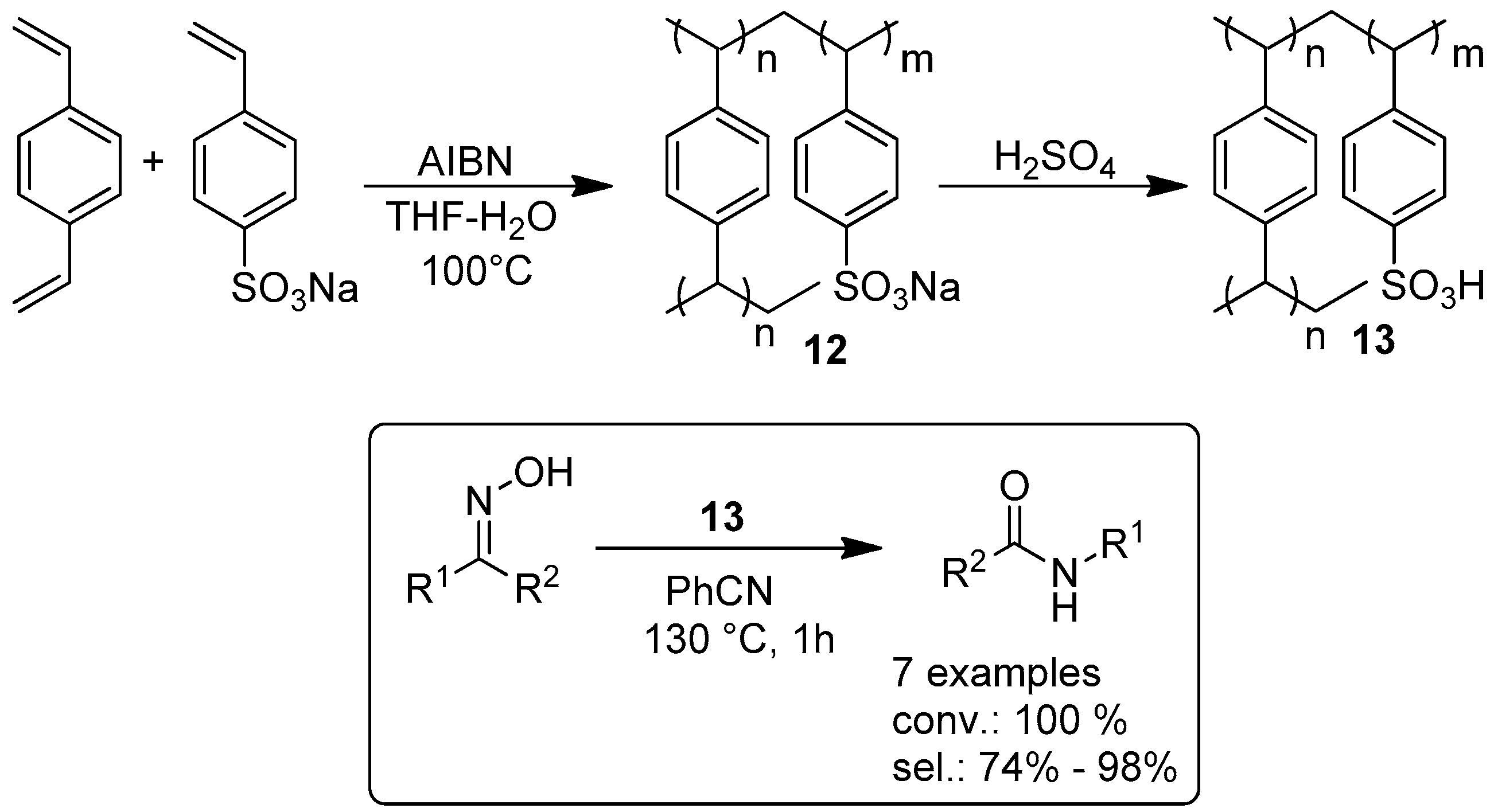
2.1.1. Esterification Reactions
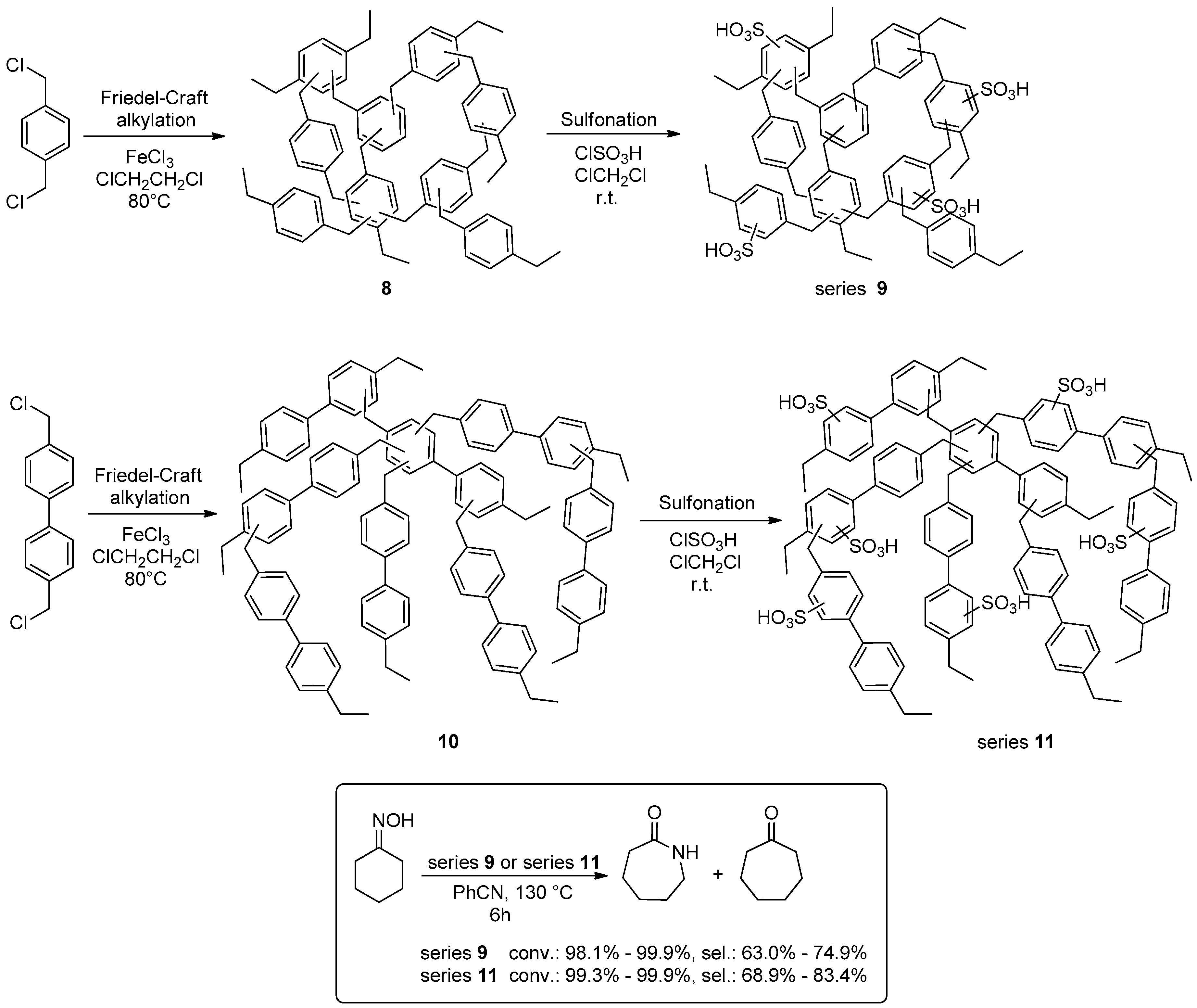
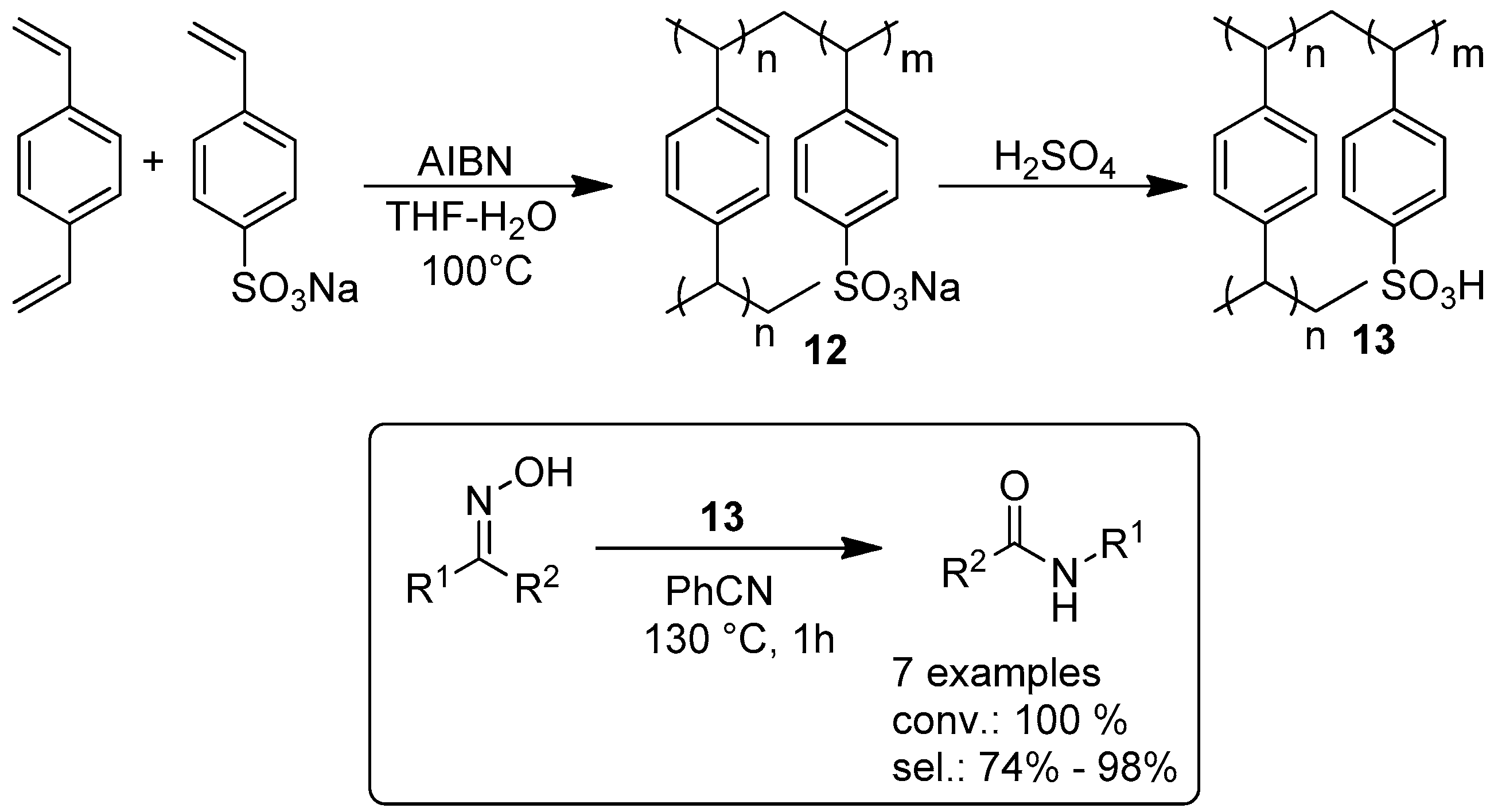
2.1.2. Beckmann Reactions
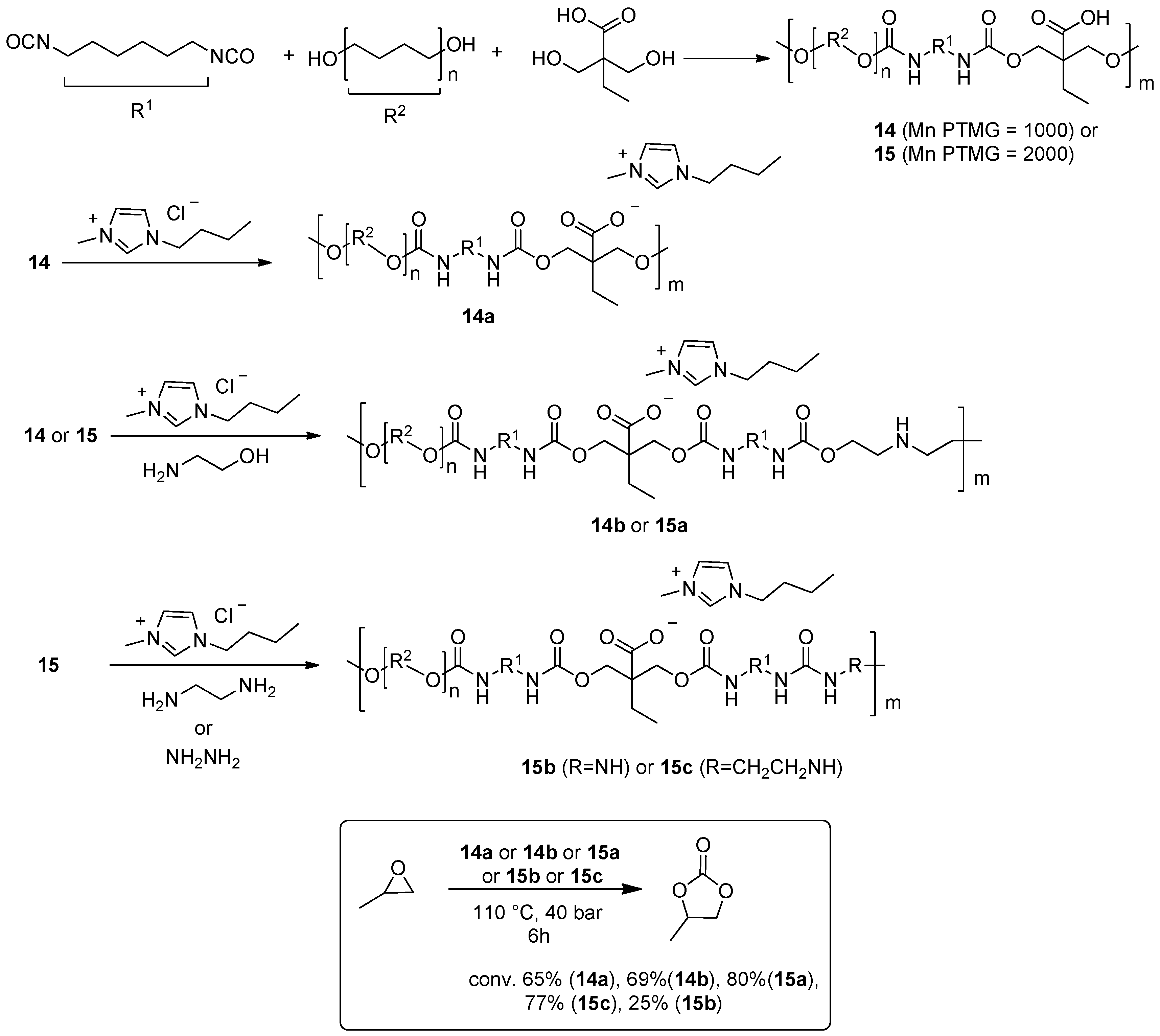
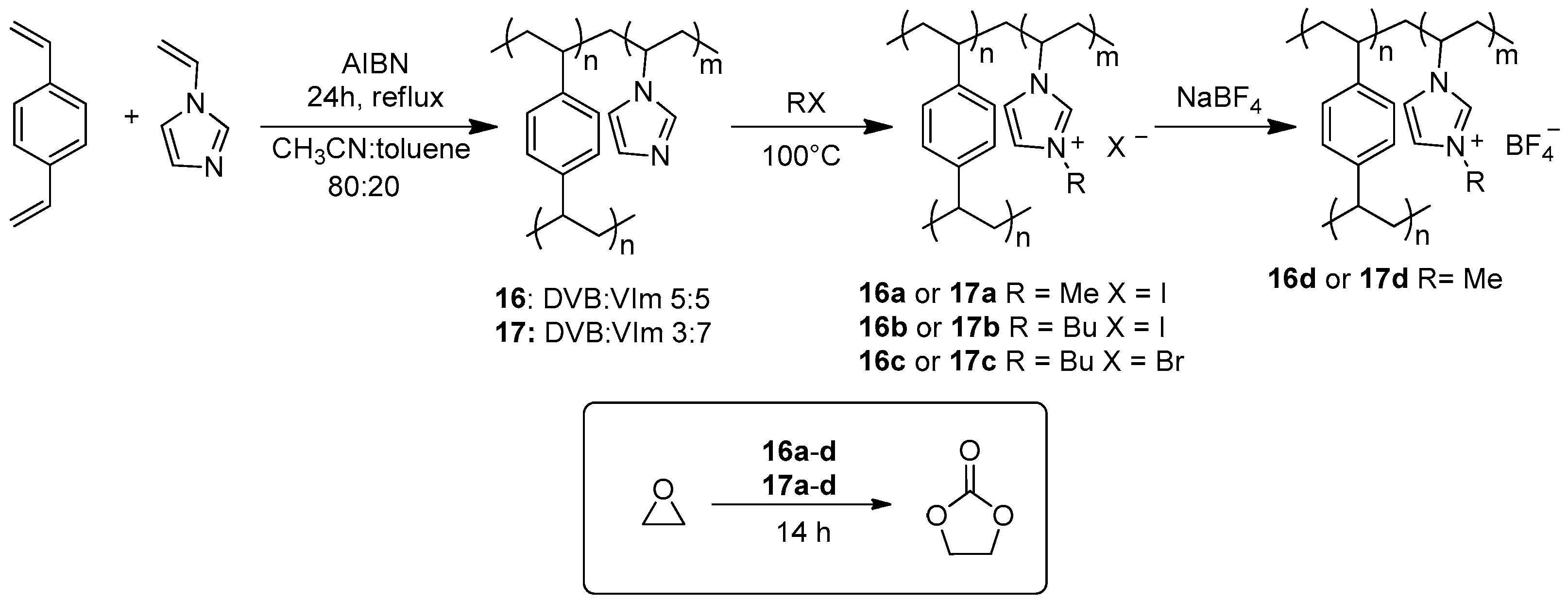
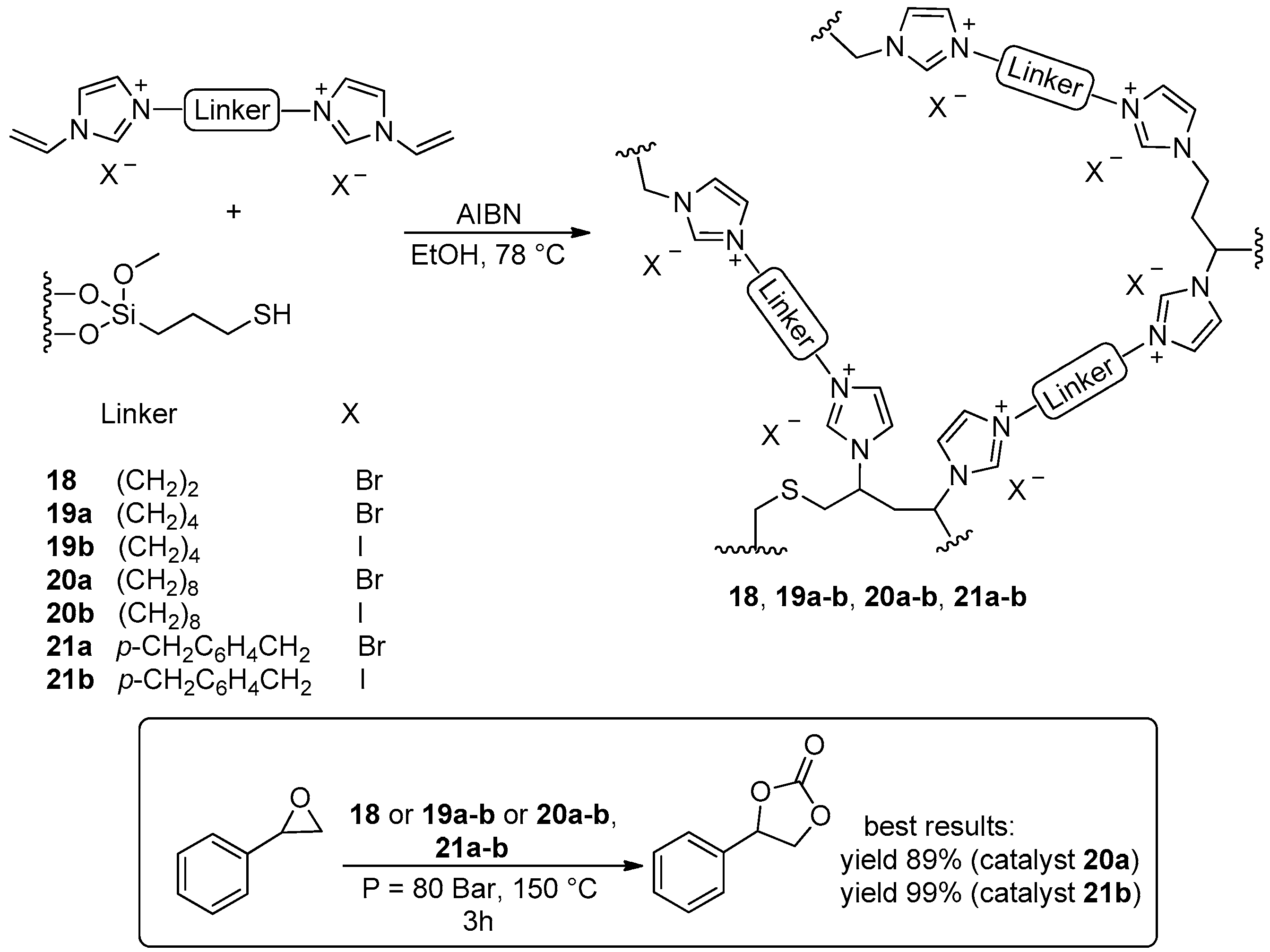
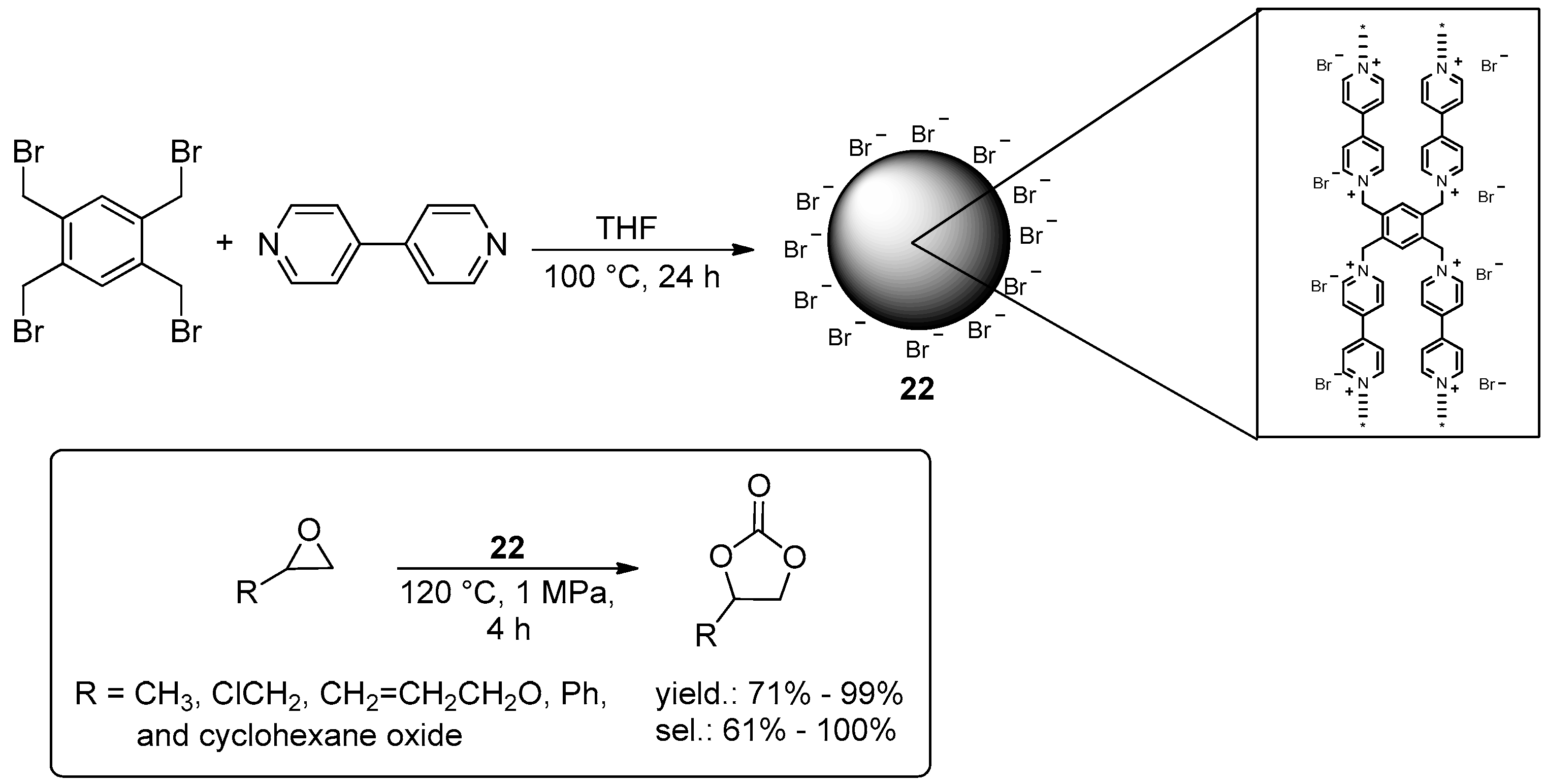
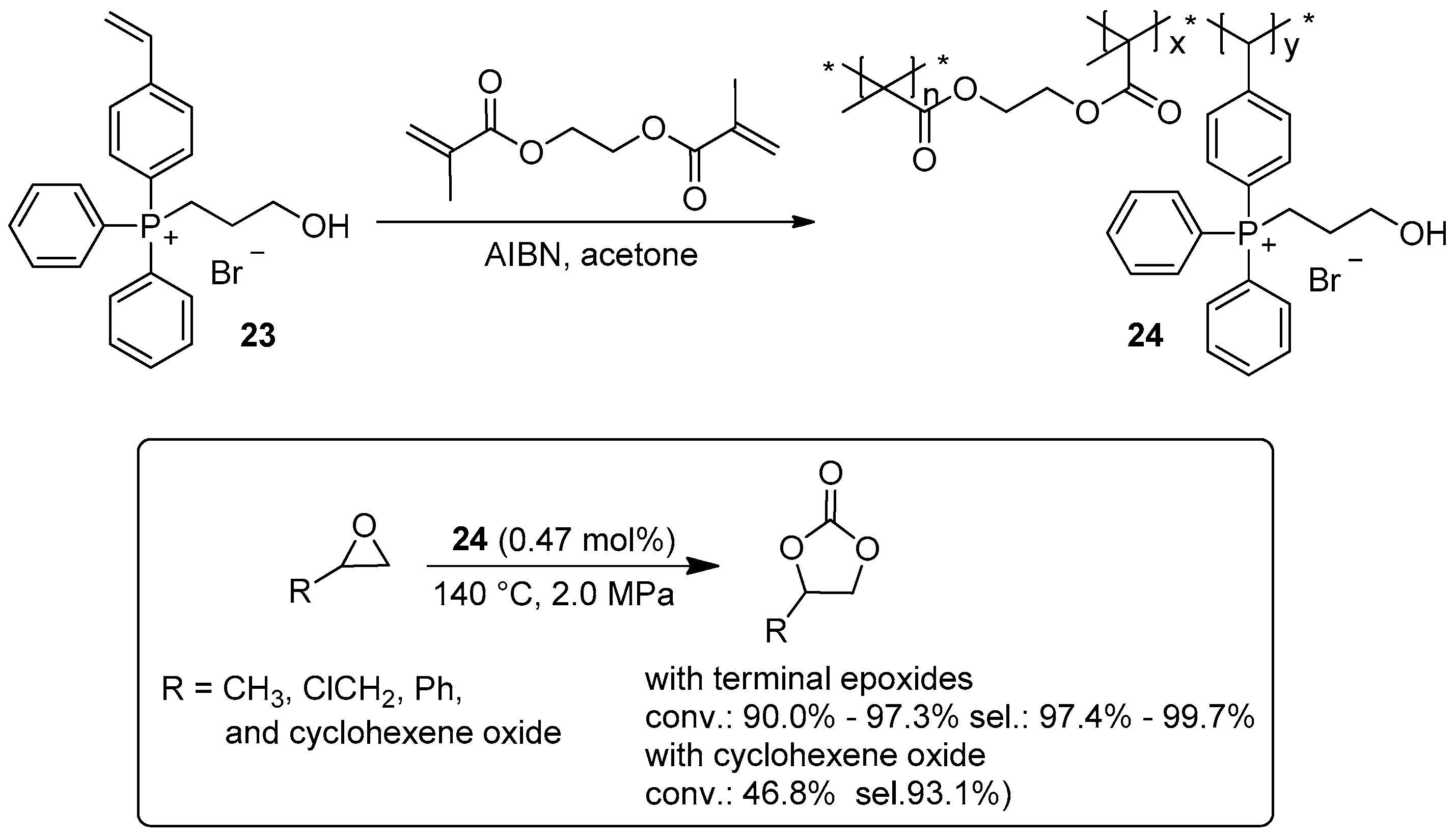
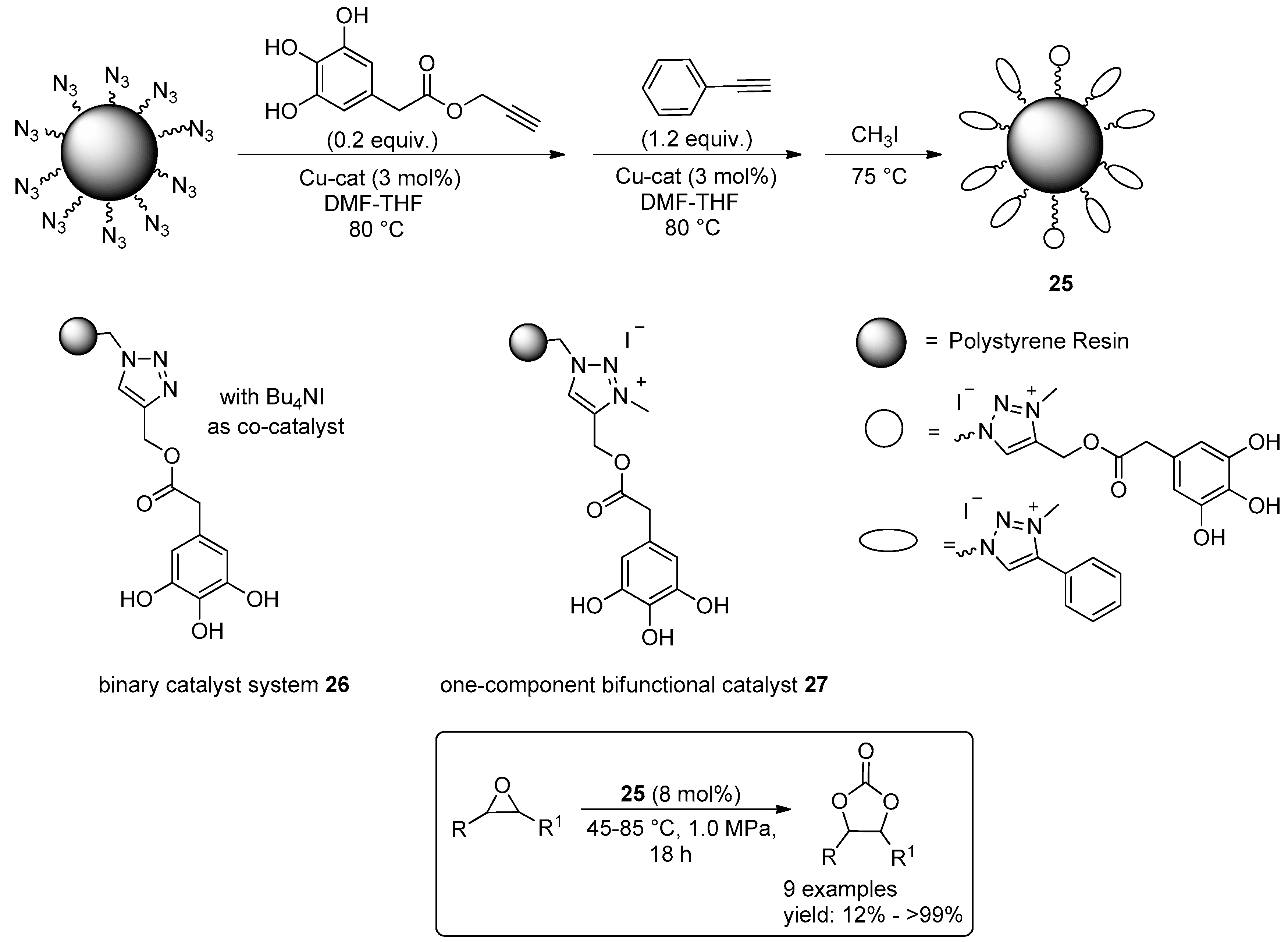
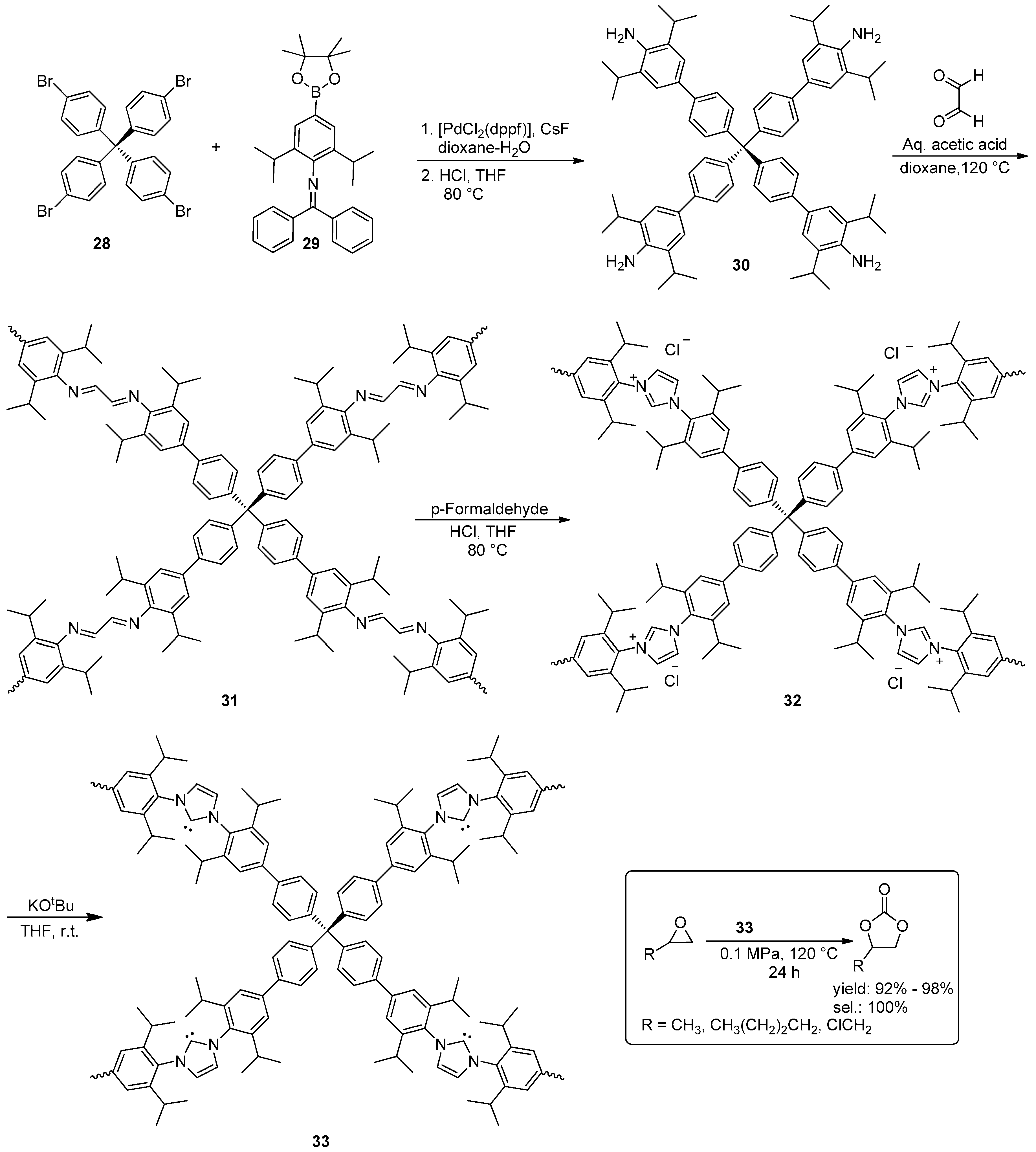
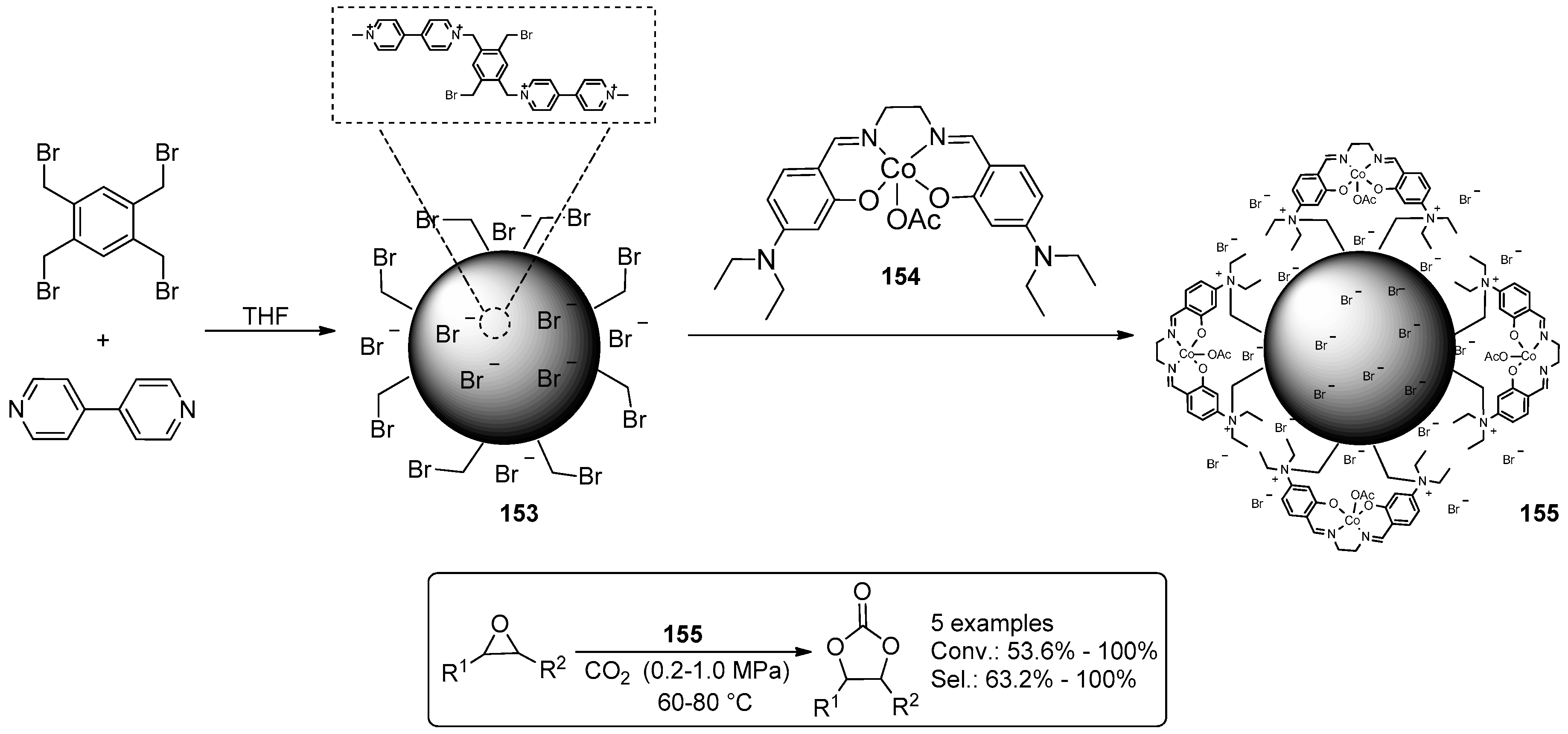
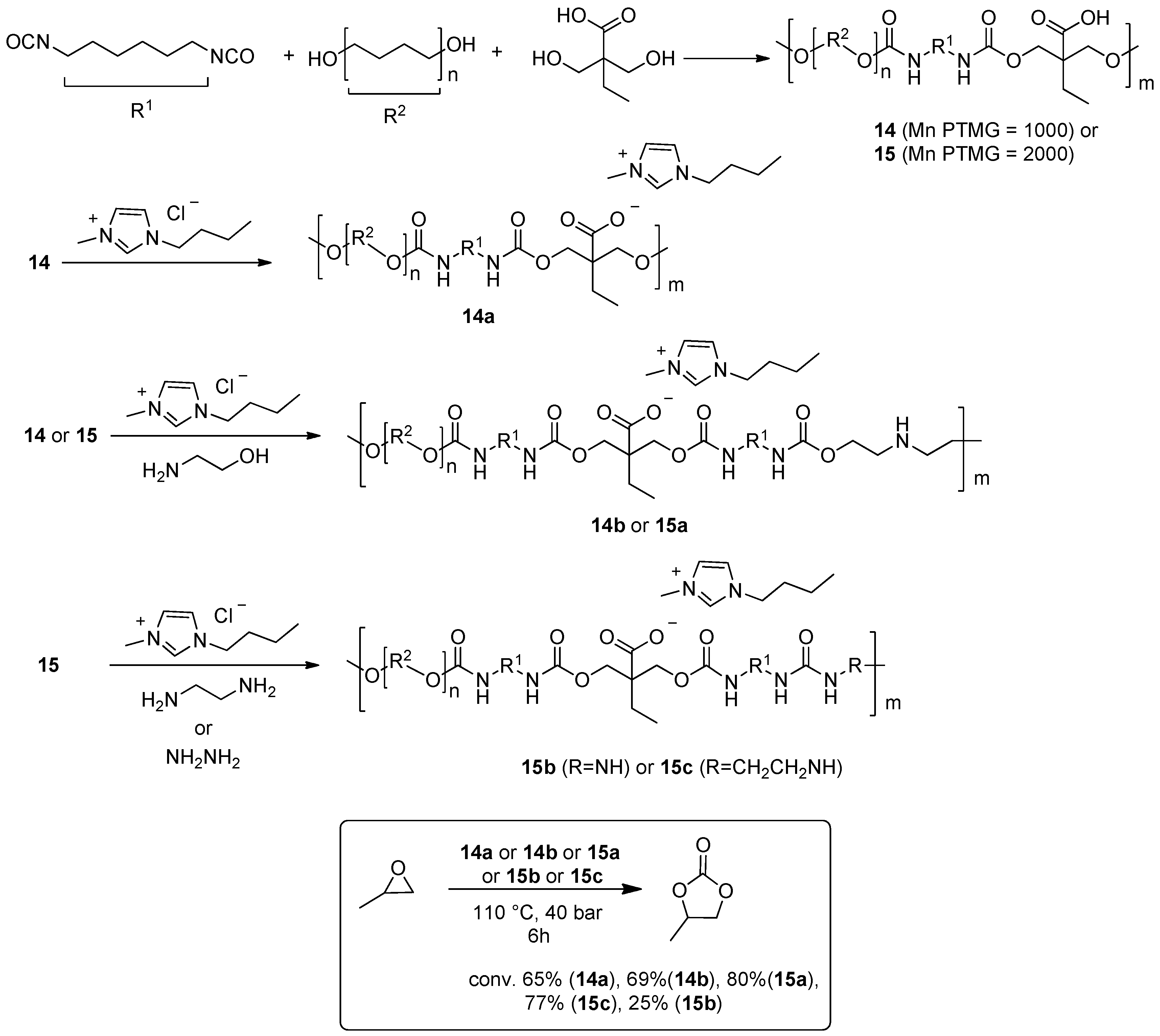
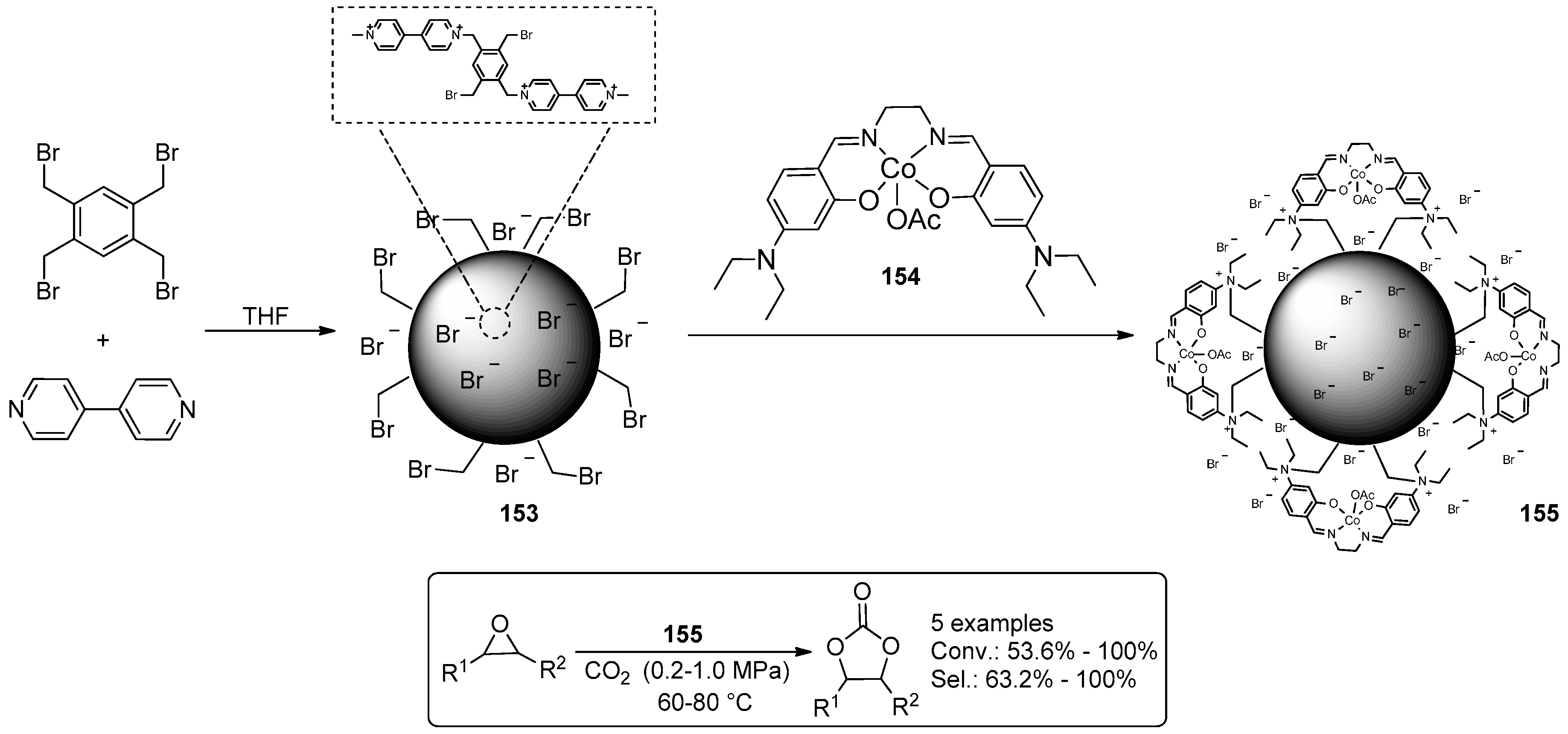
2.2. Synthesis of Cyclic Carbonates
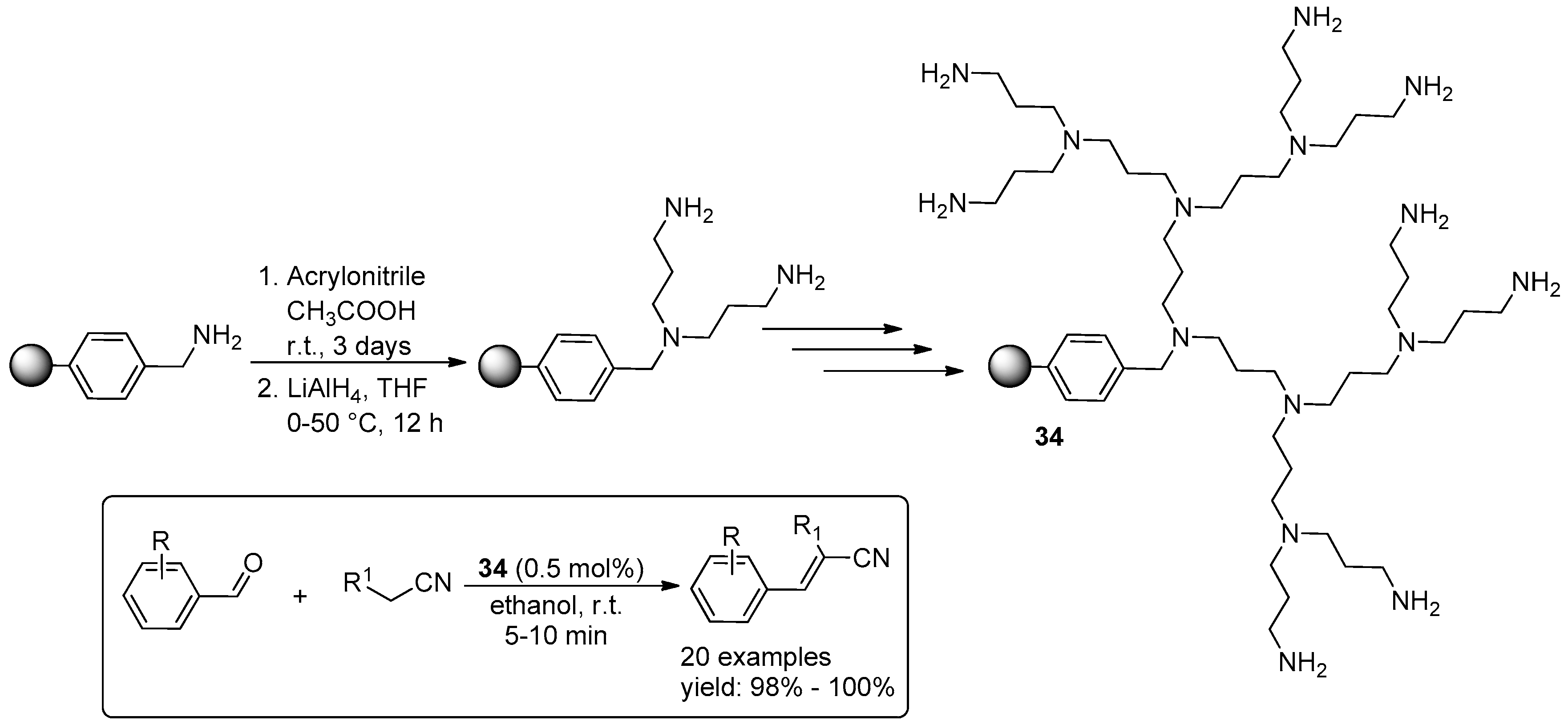
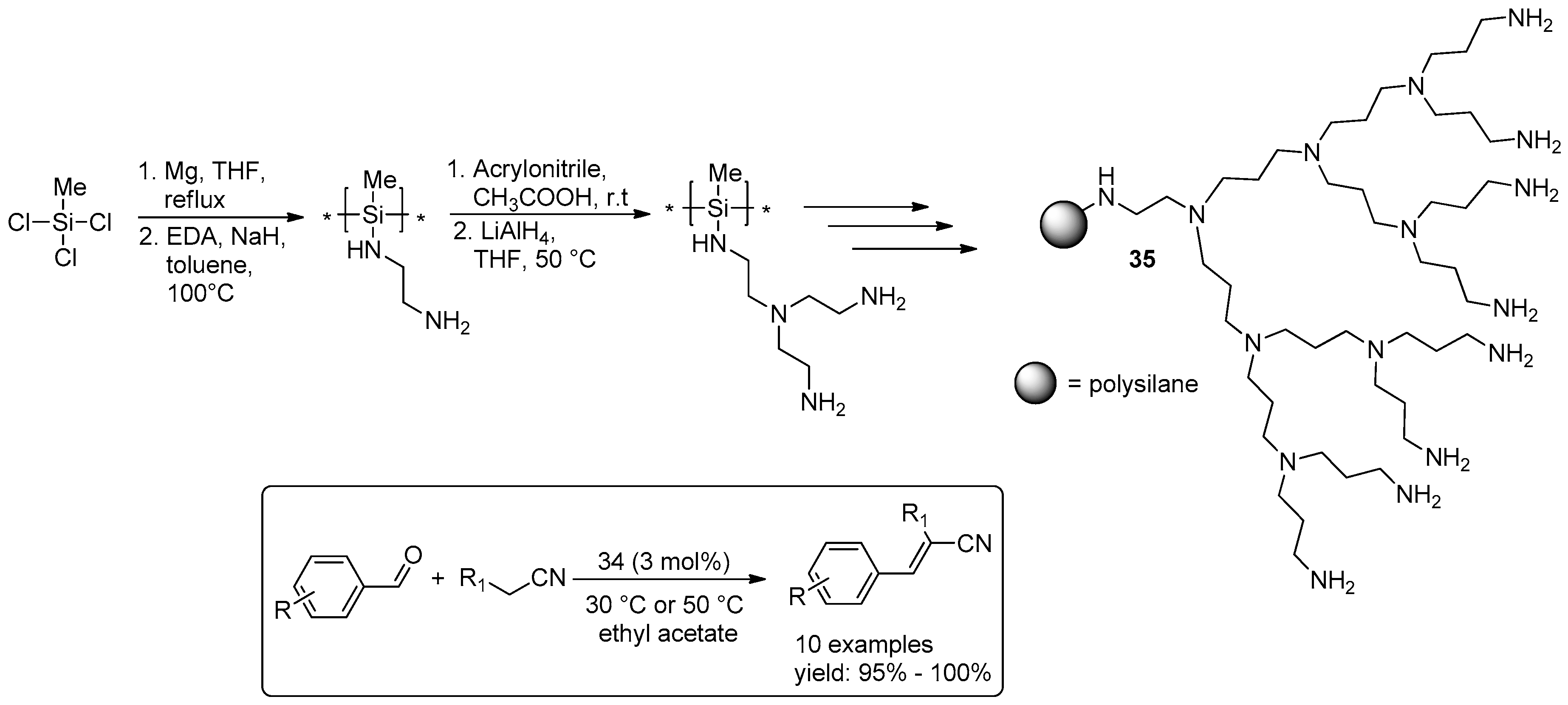
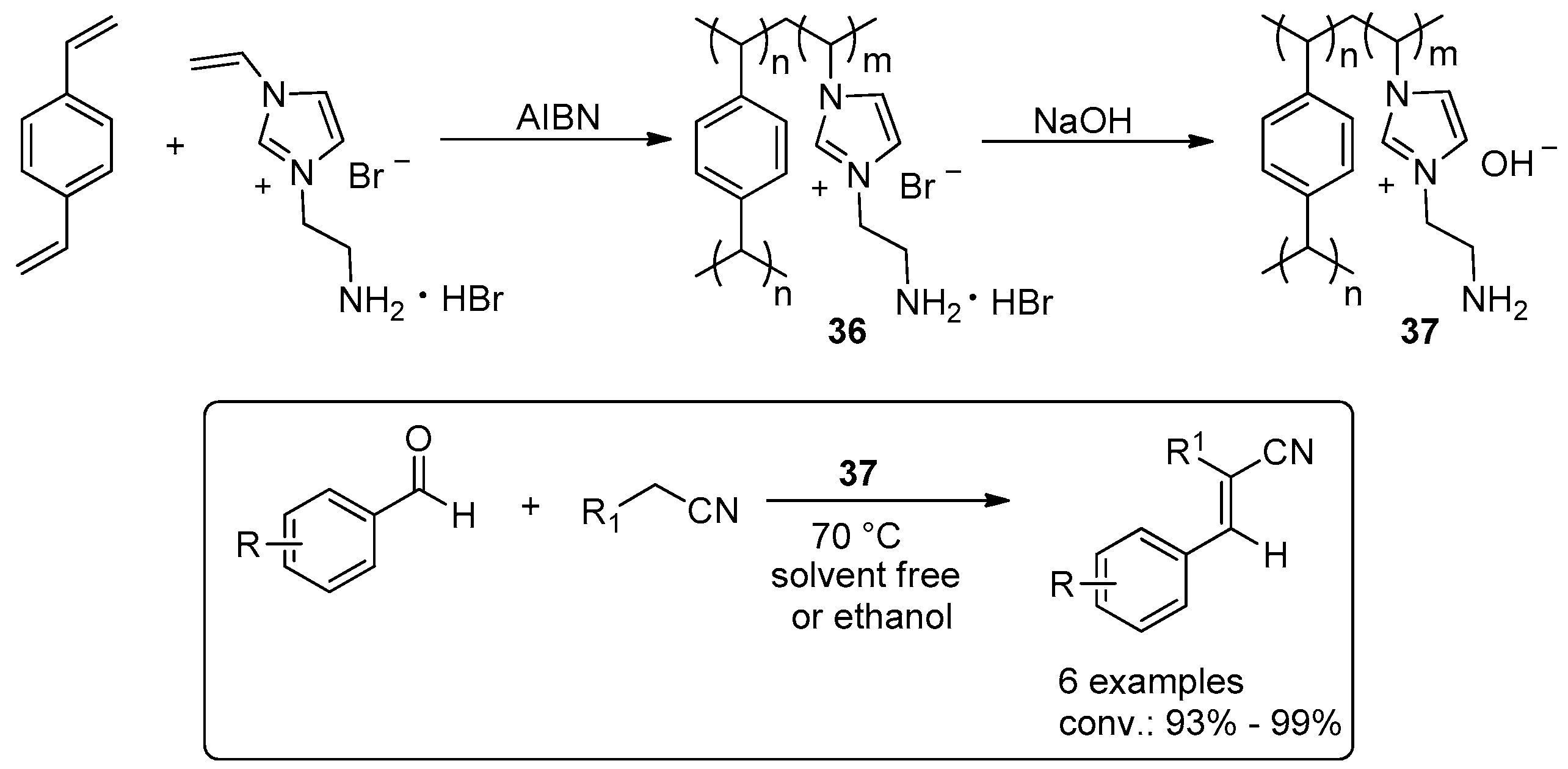
2.3. C–C Bond Forming Reactions
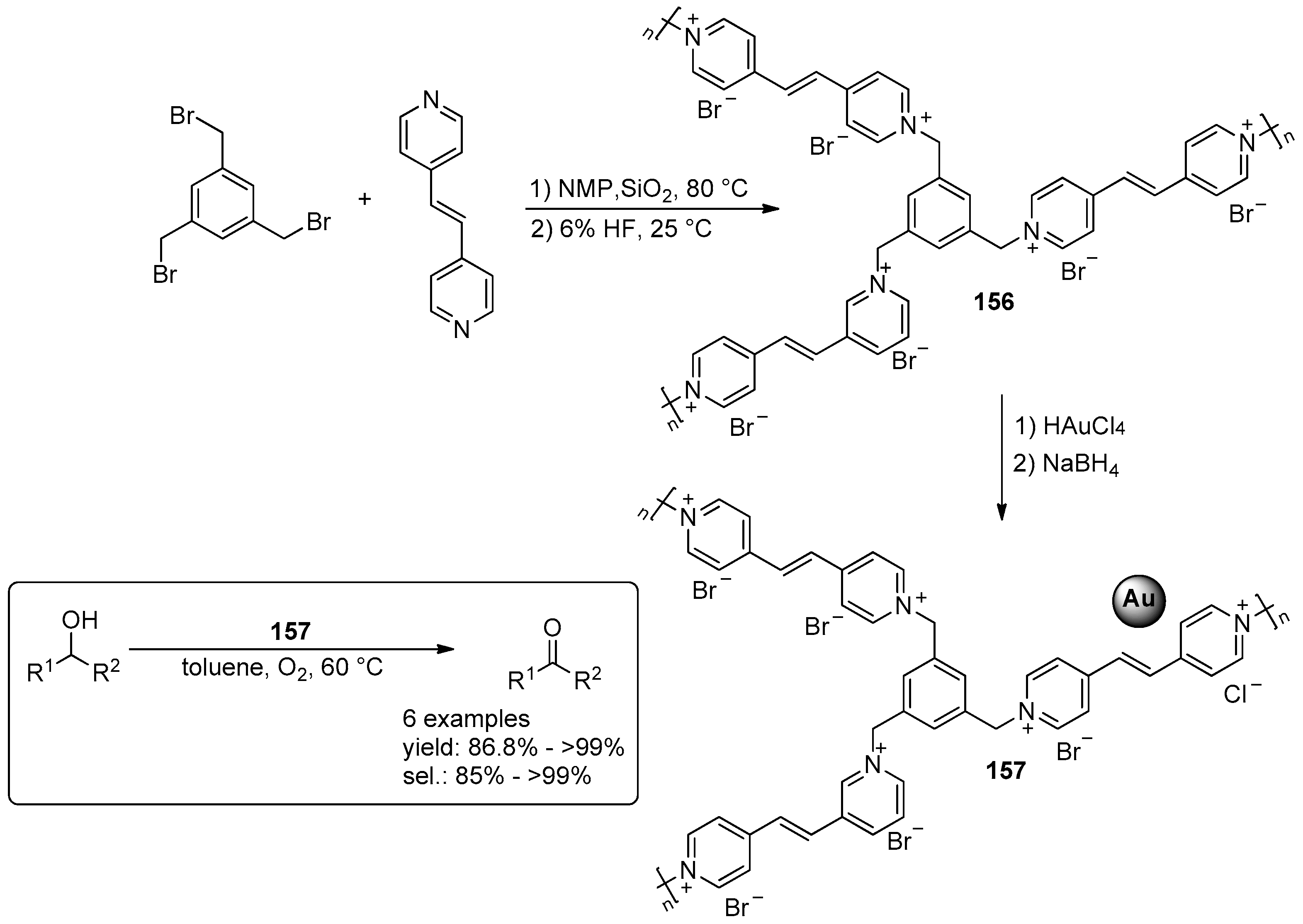
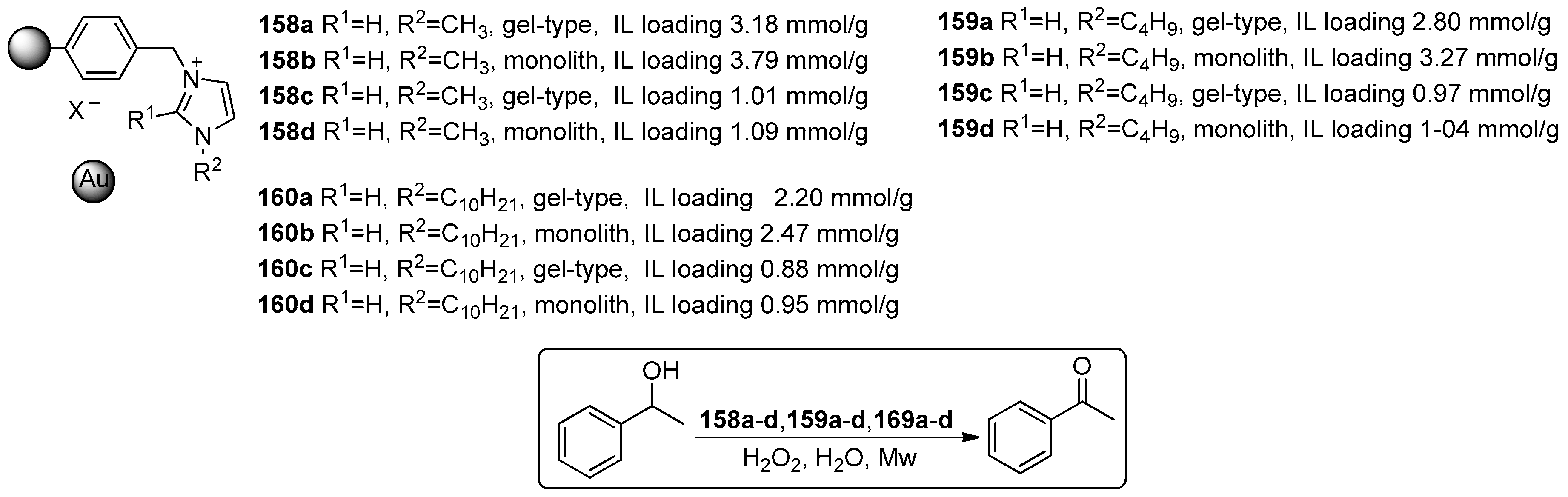
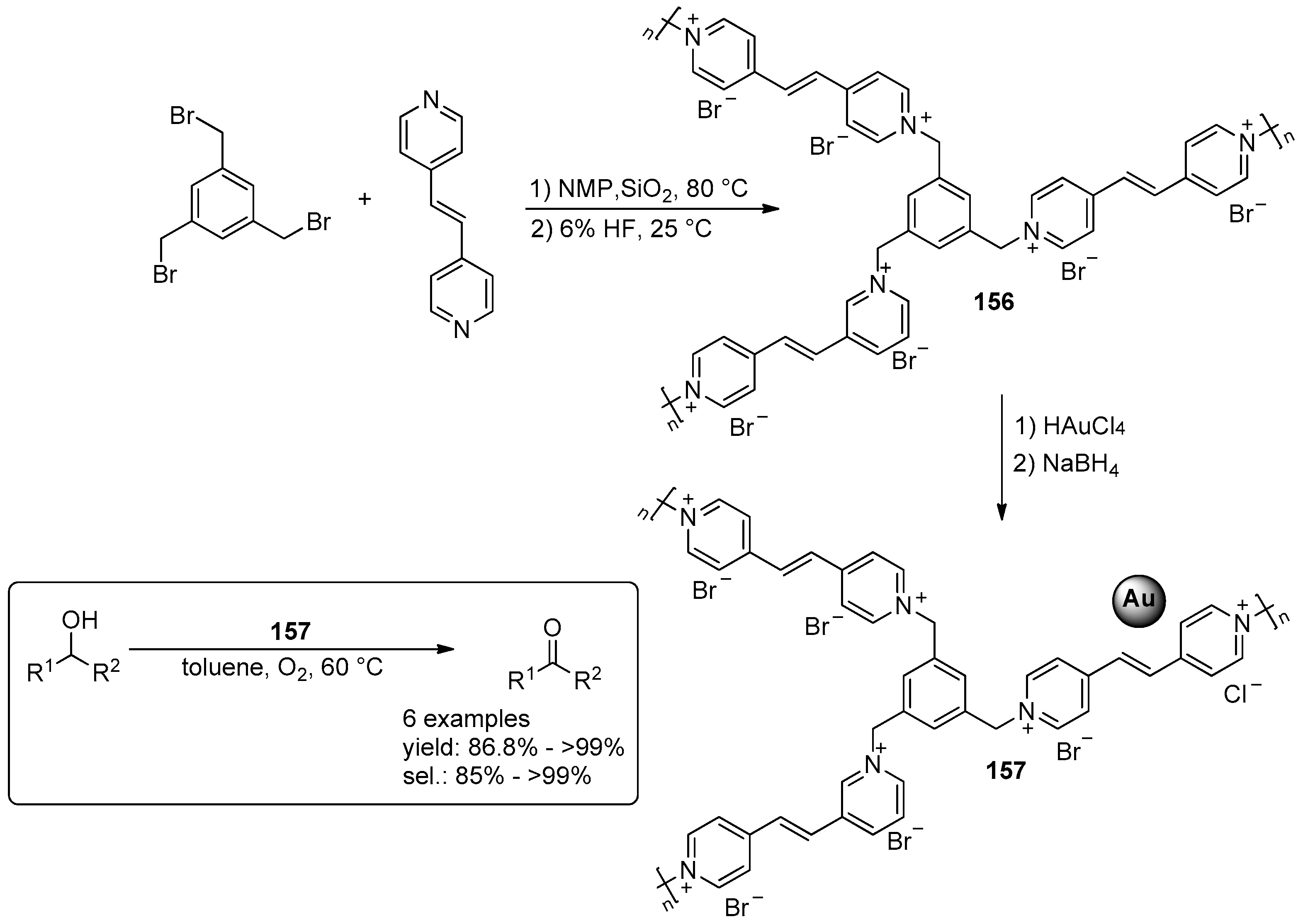
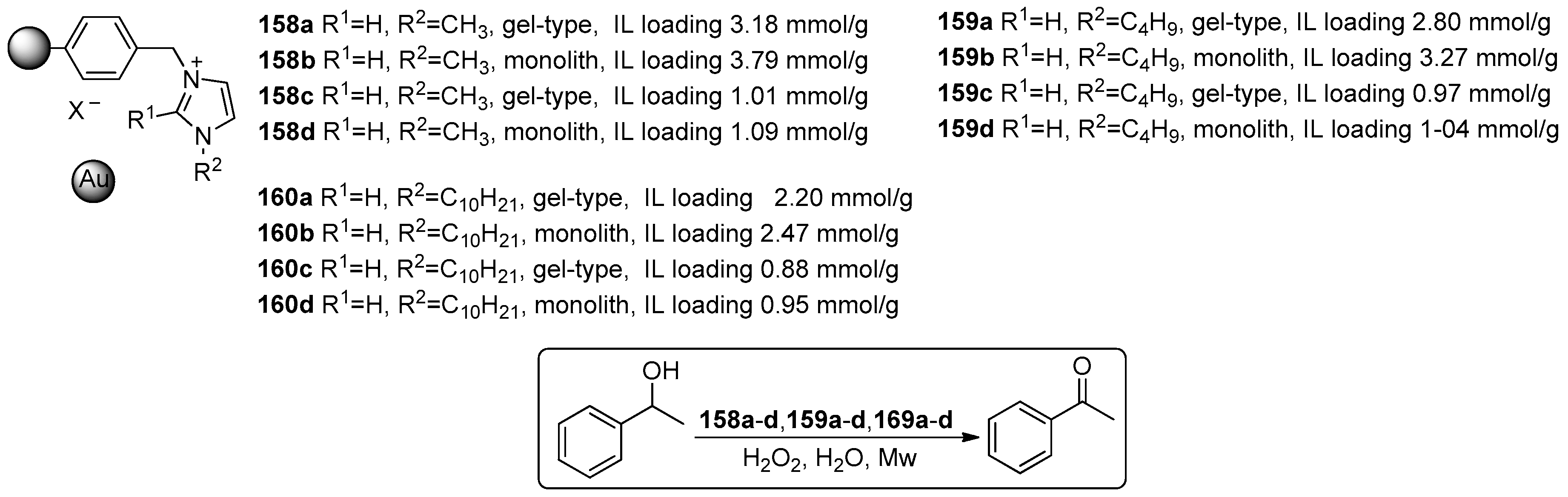
2.4. Miscellaneous Examples
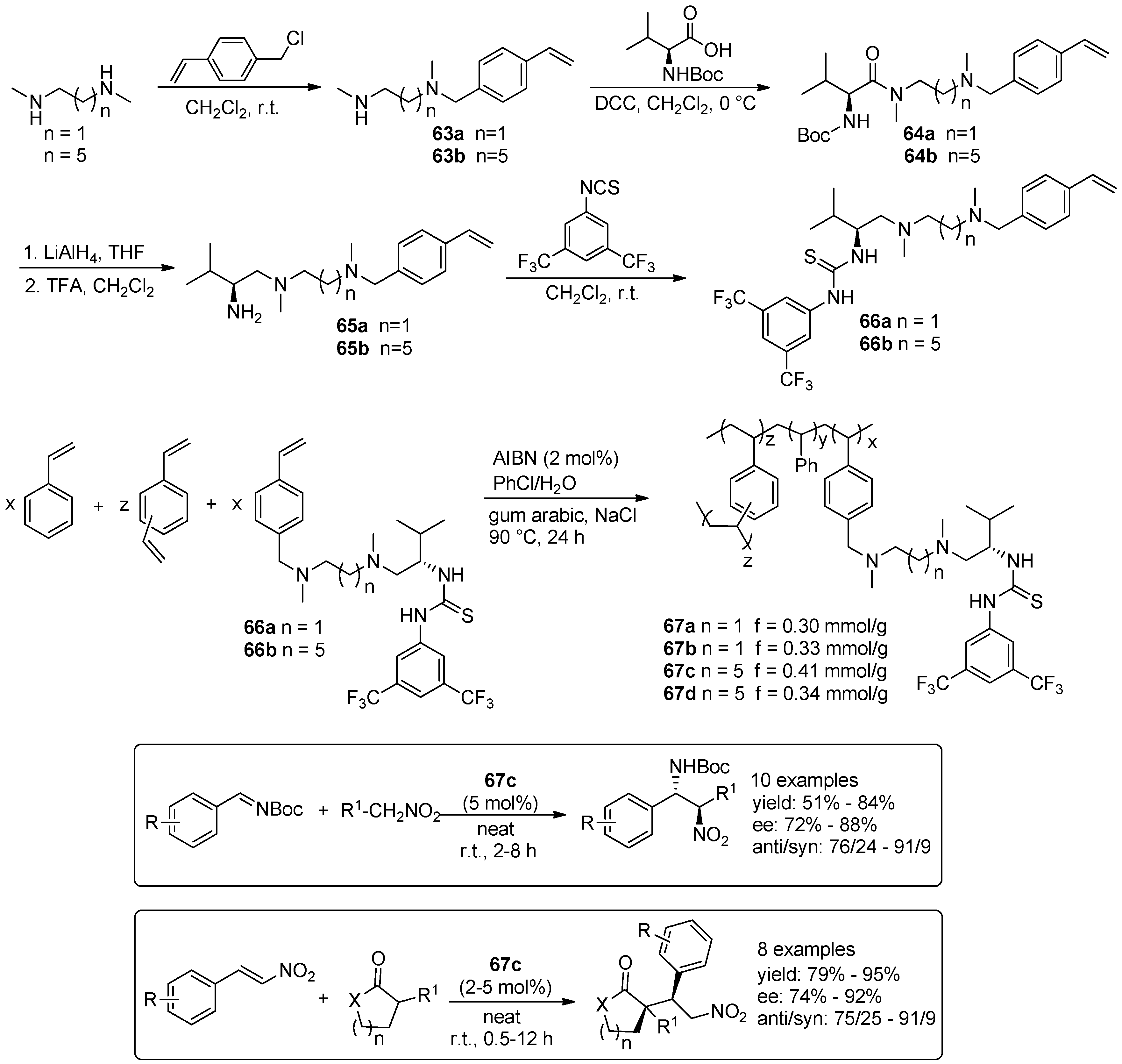
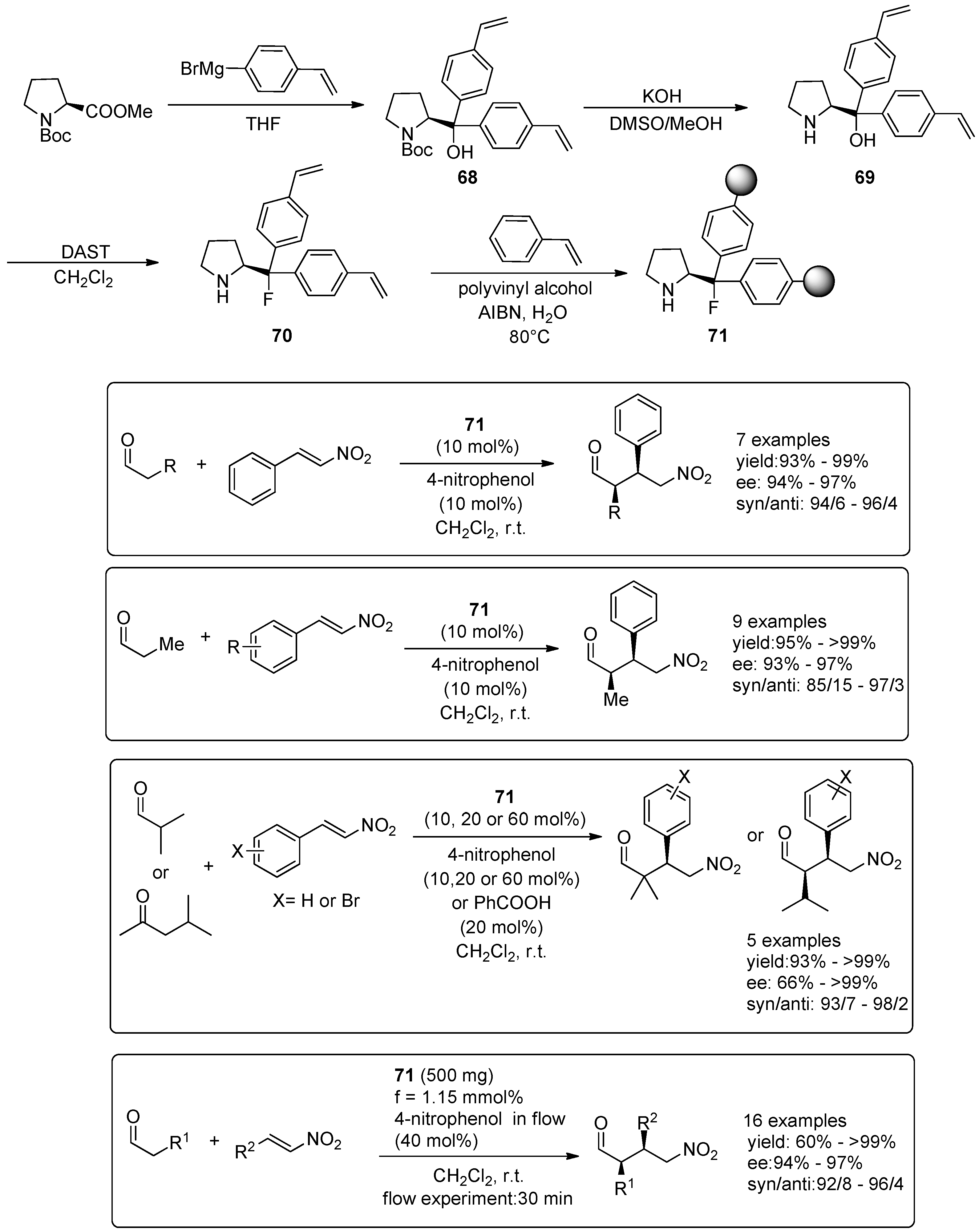
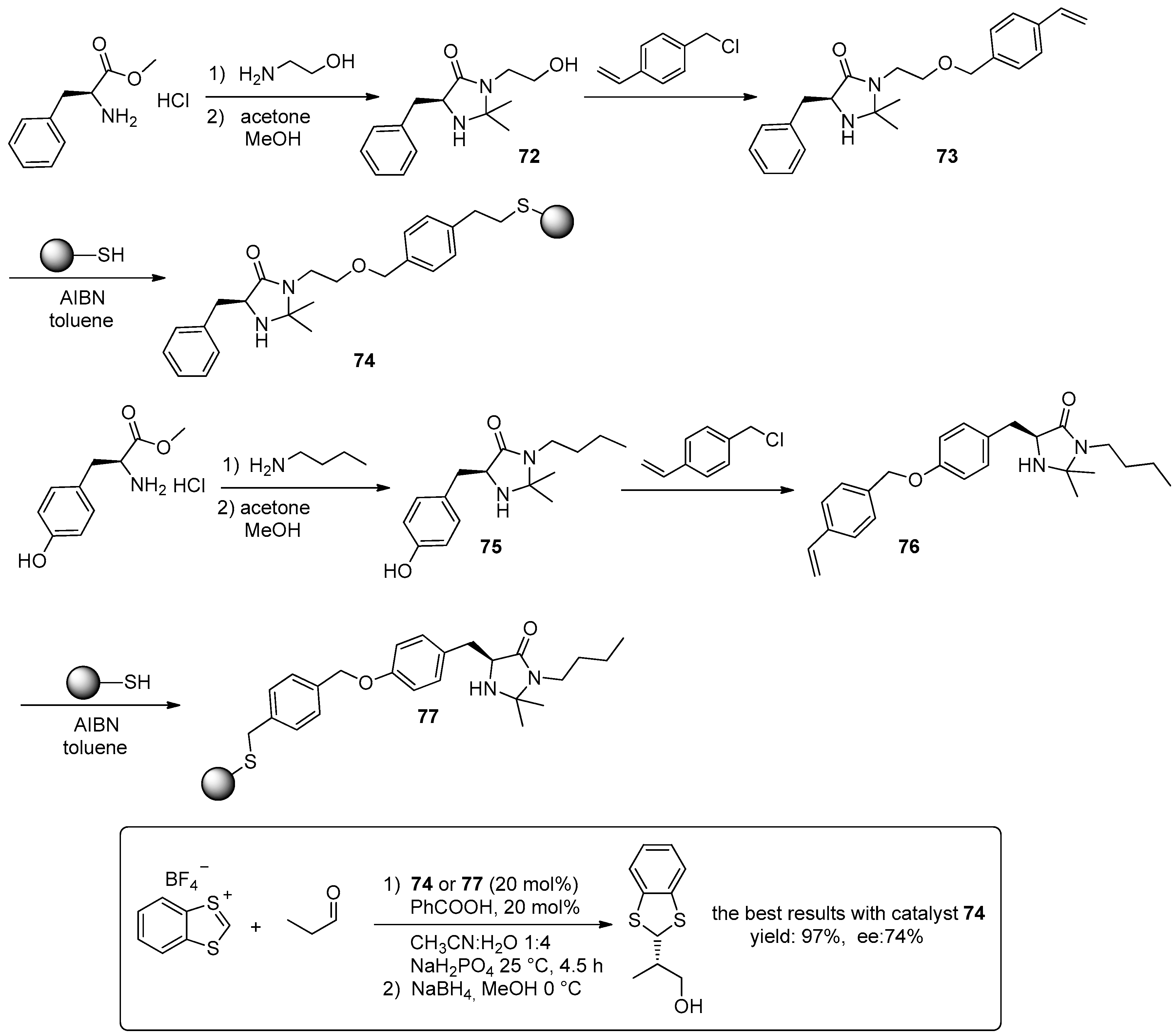
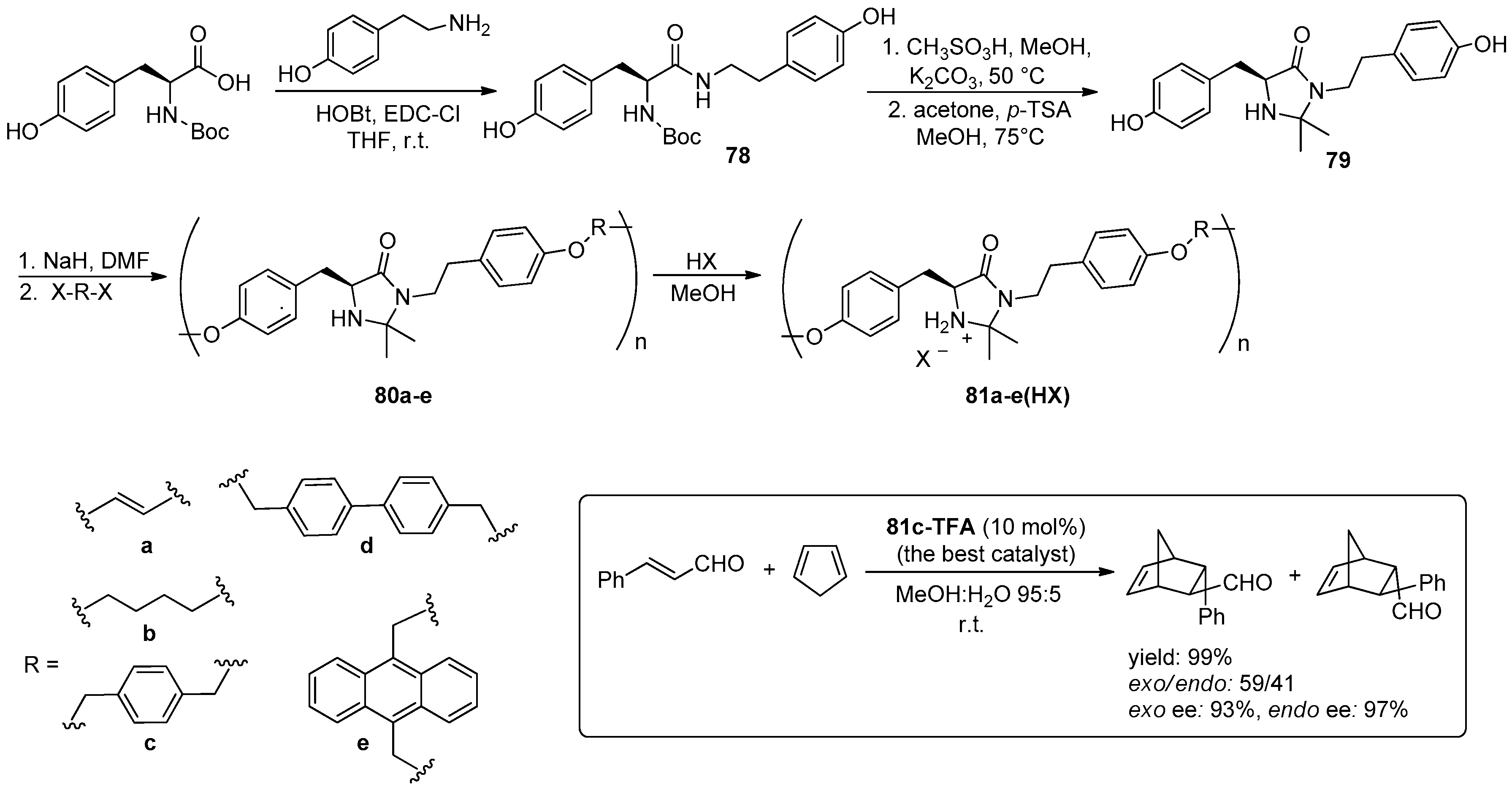
3. Organocatalysed Asymmetric Reactions
4. Metal-Catalysed Non-Asymmetric Reactions
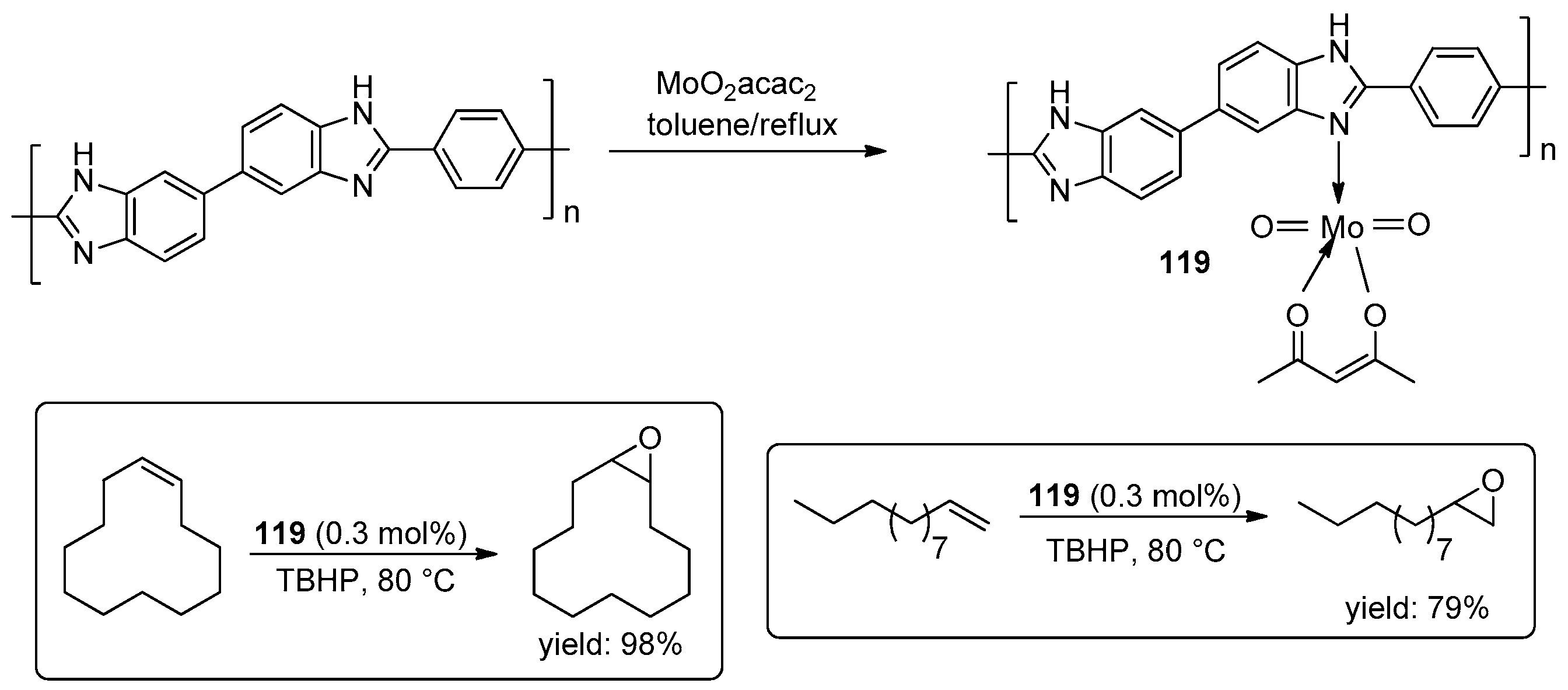
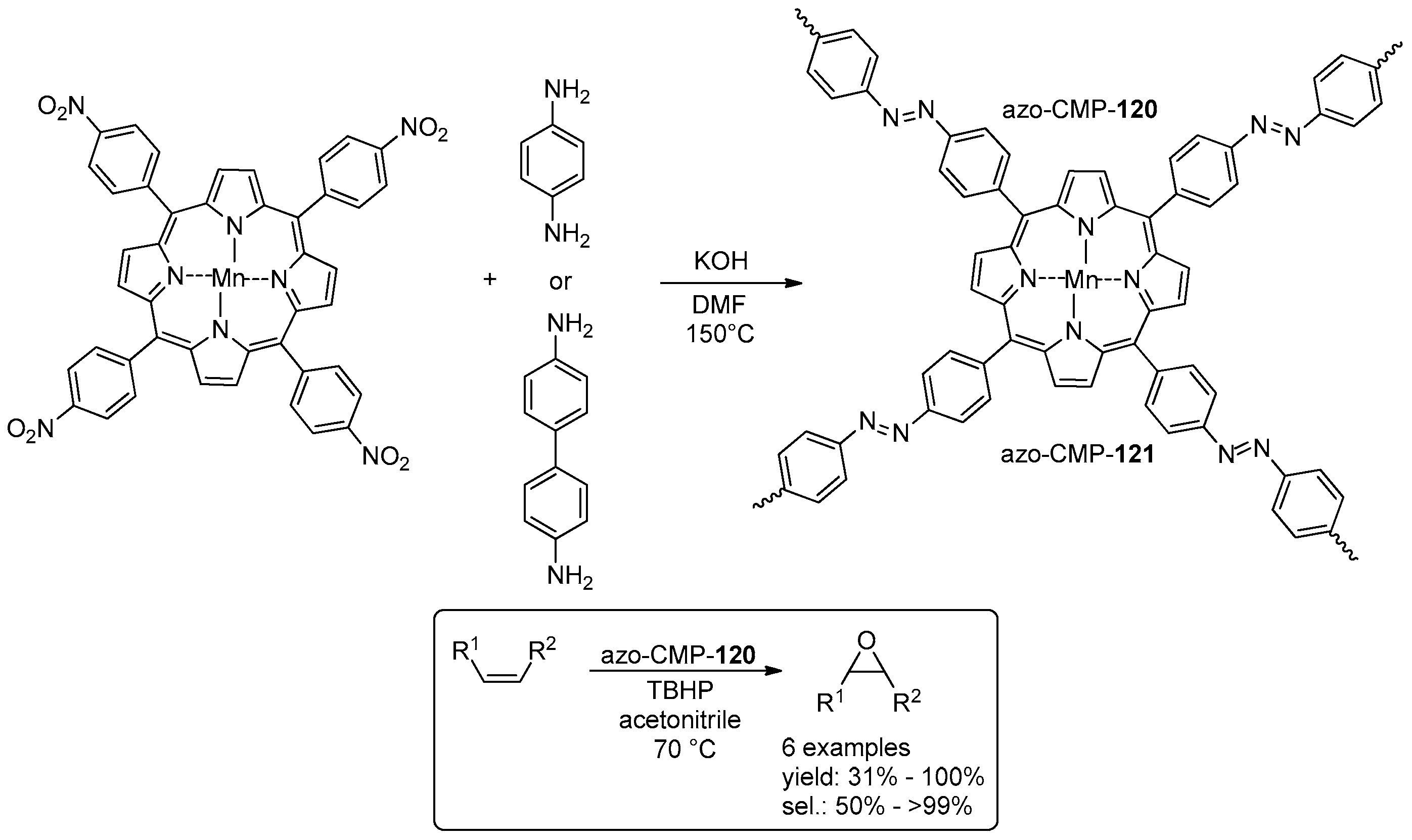
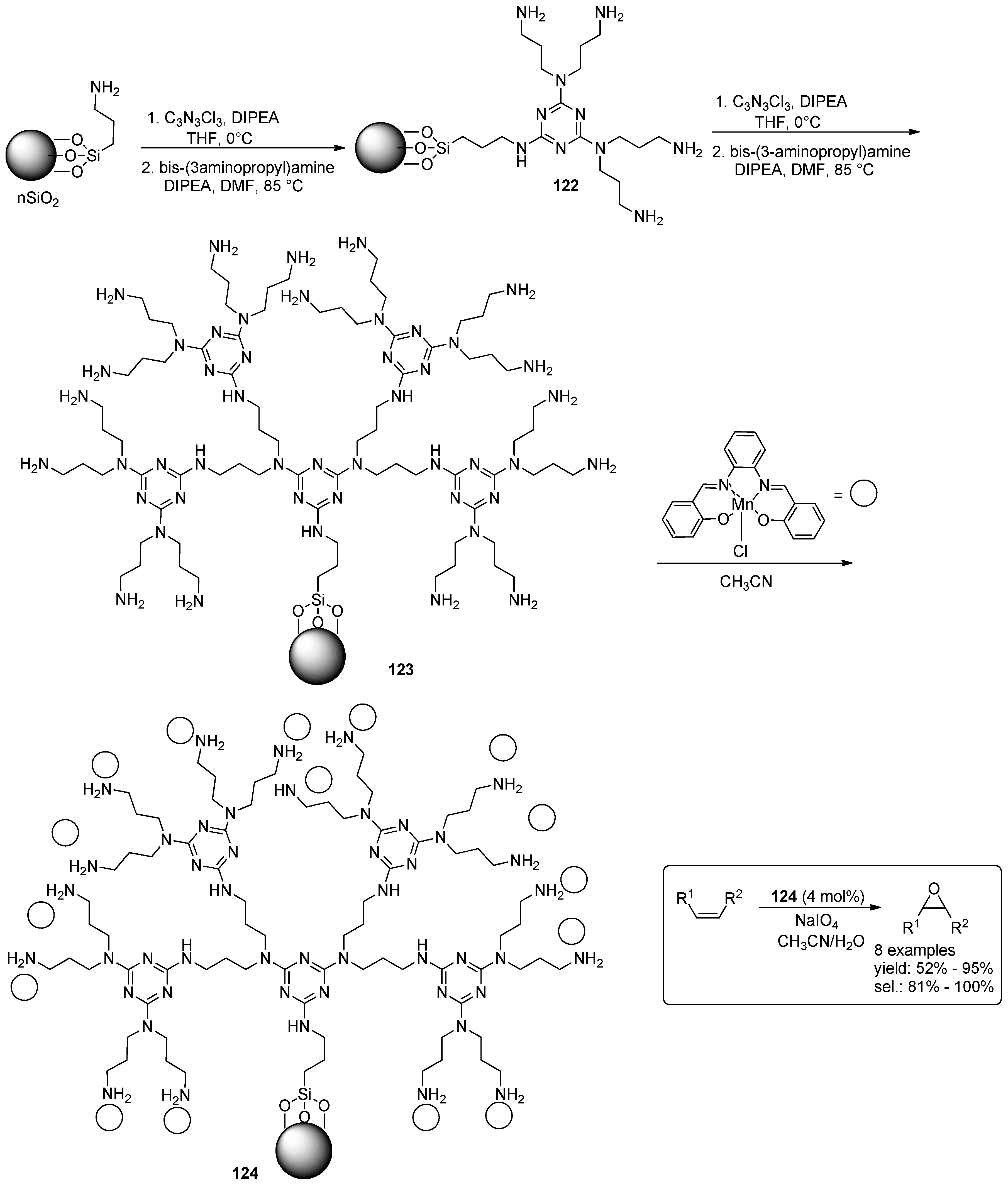
4.1. Epoxidation of Alkenes
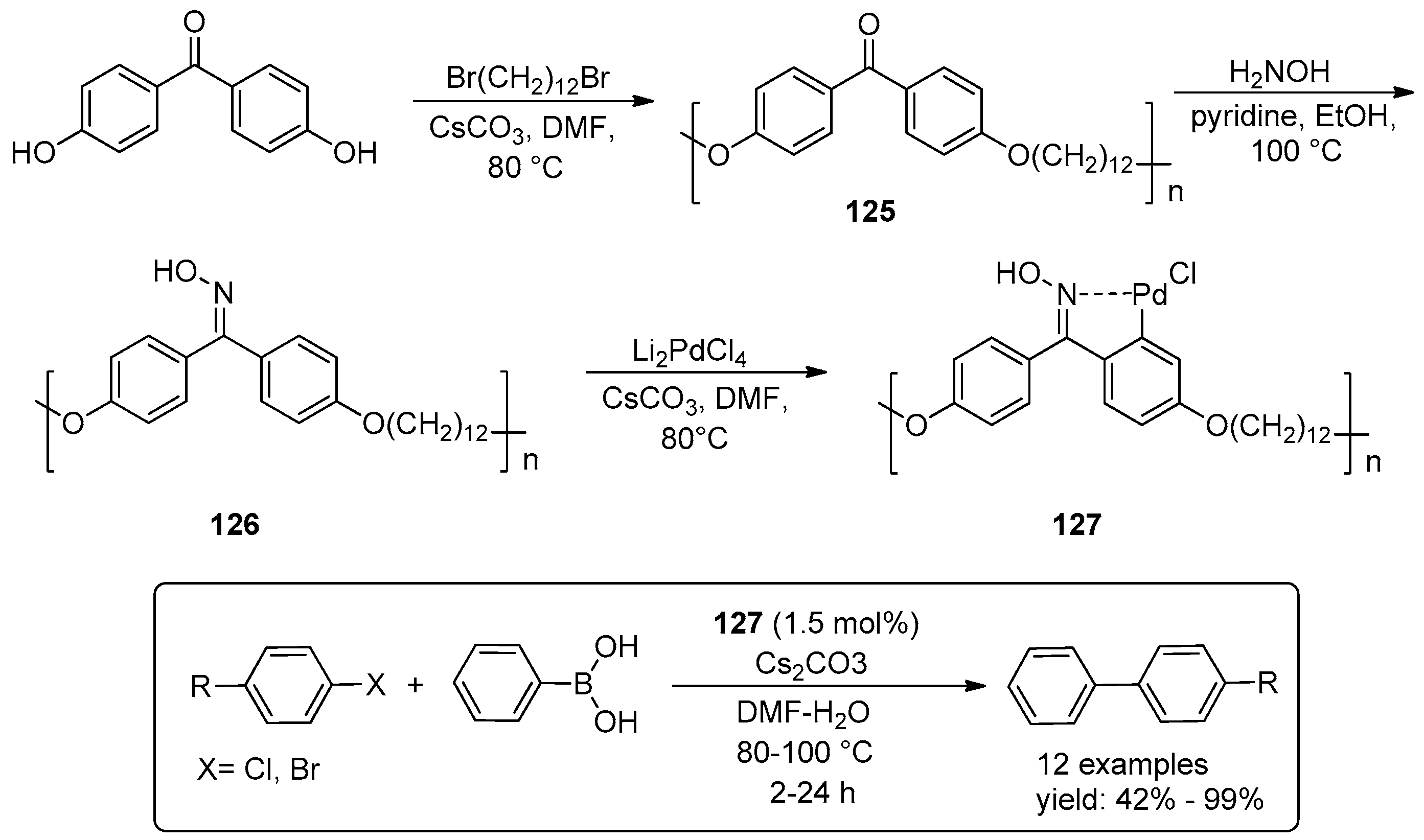
4.2. C–C Coupling Reactions
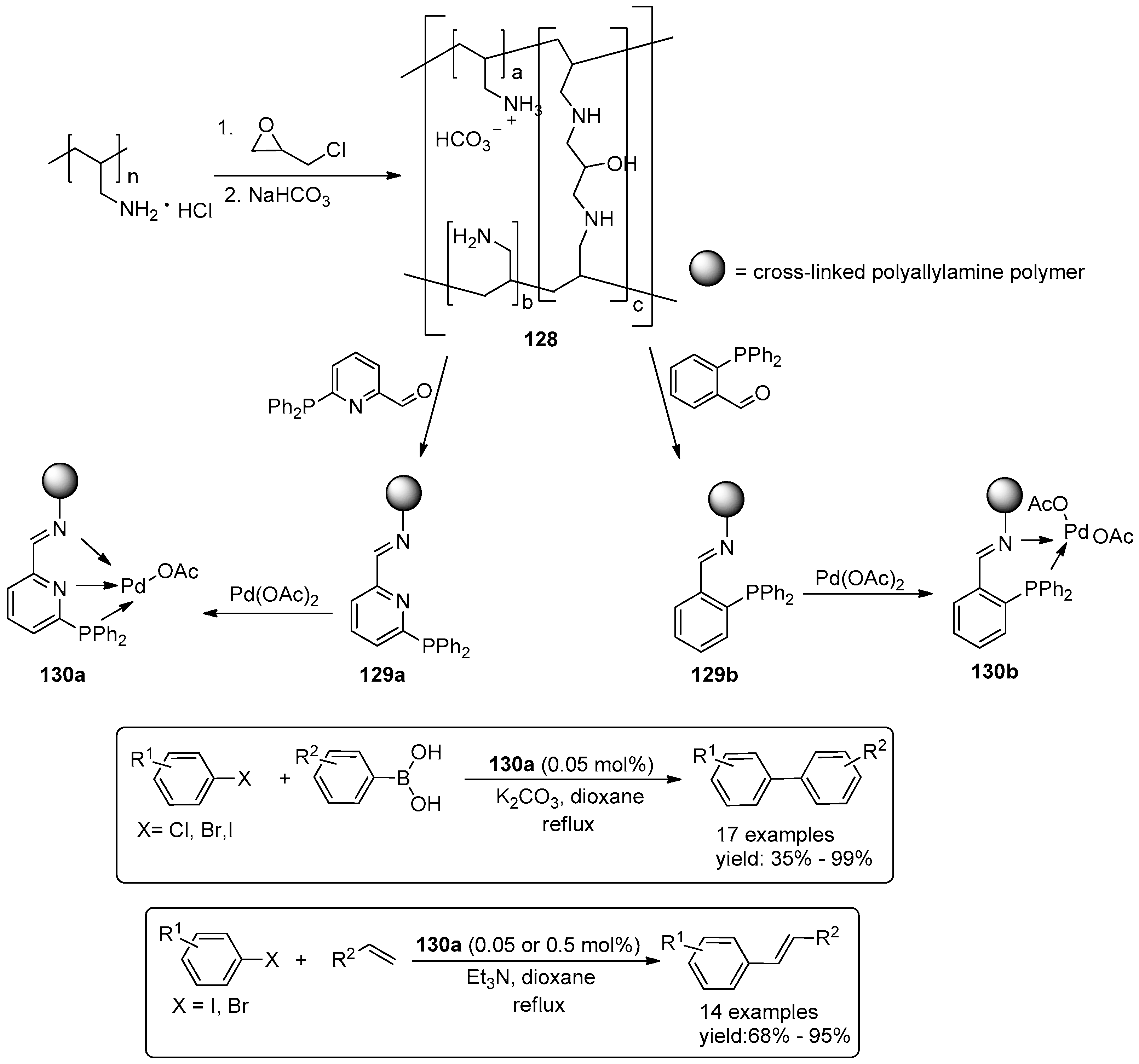
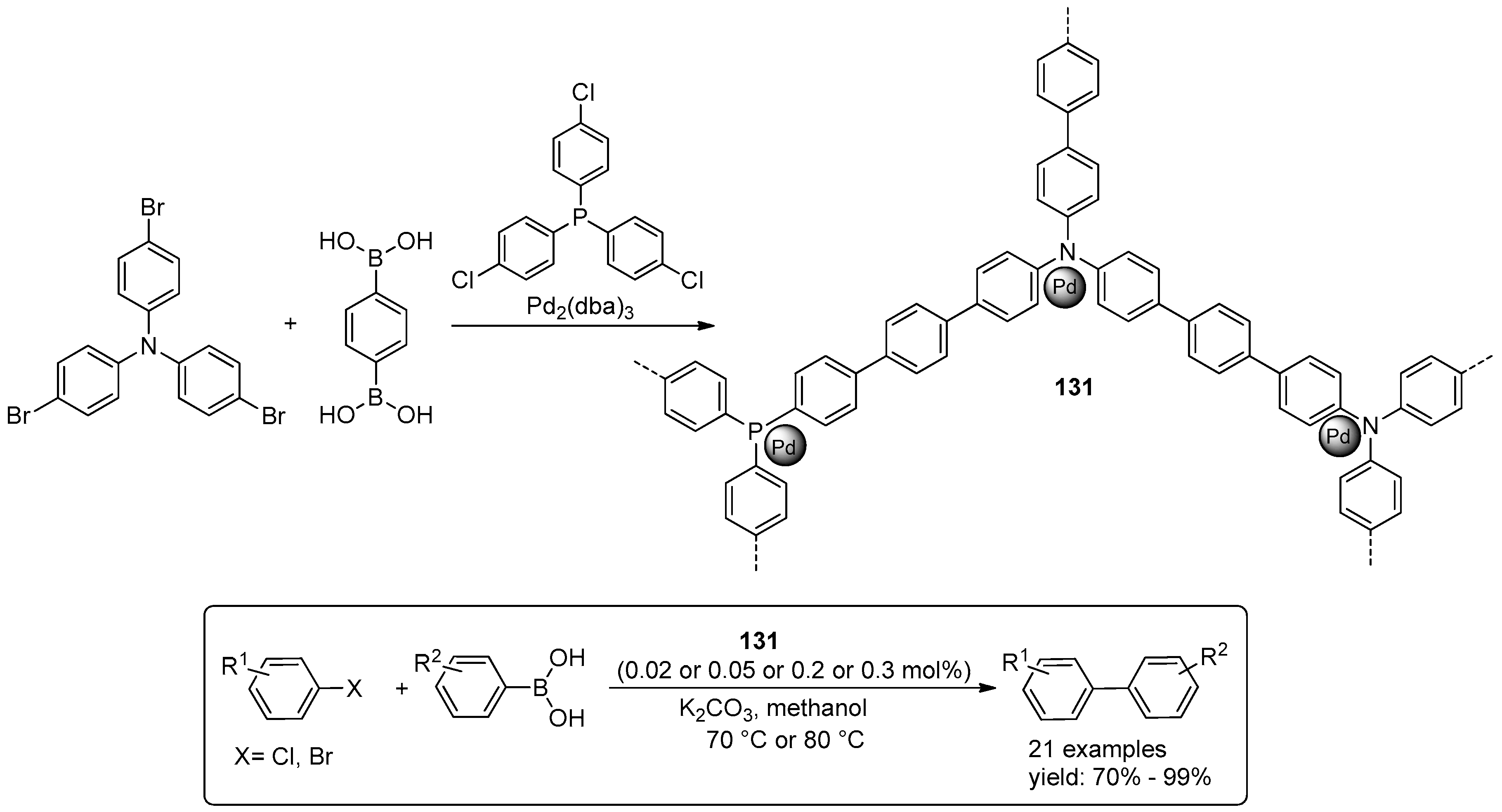
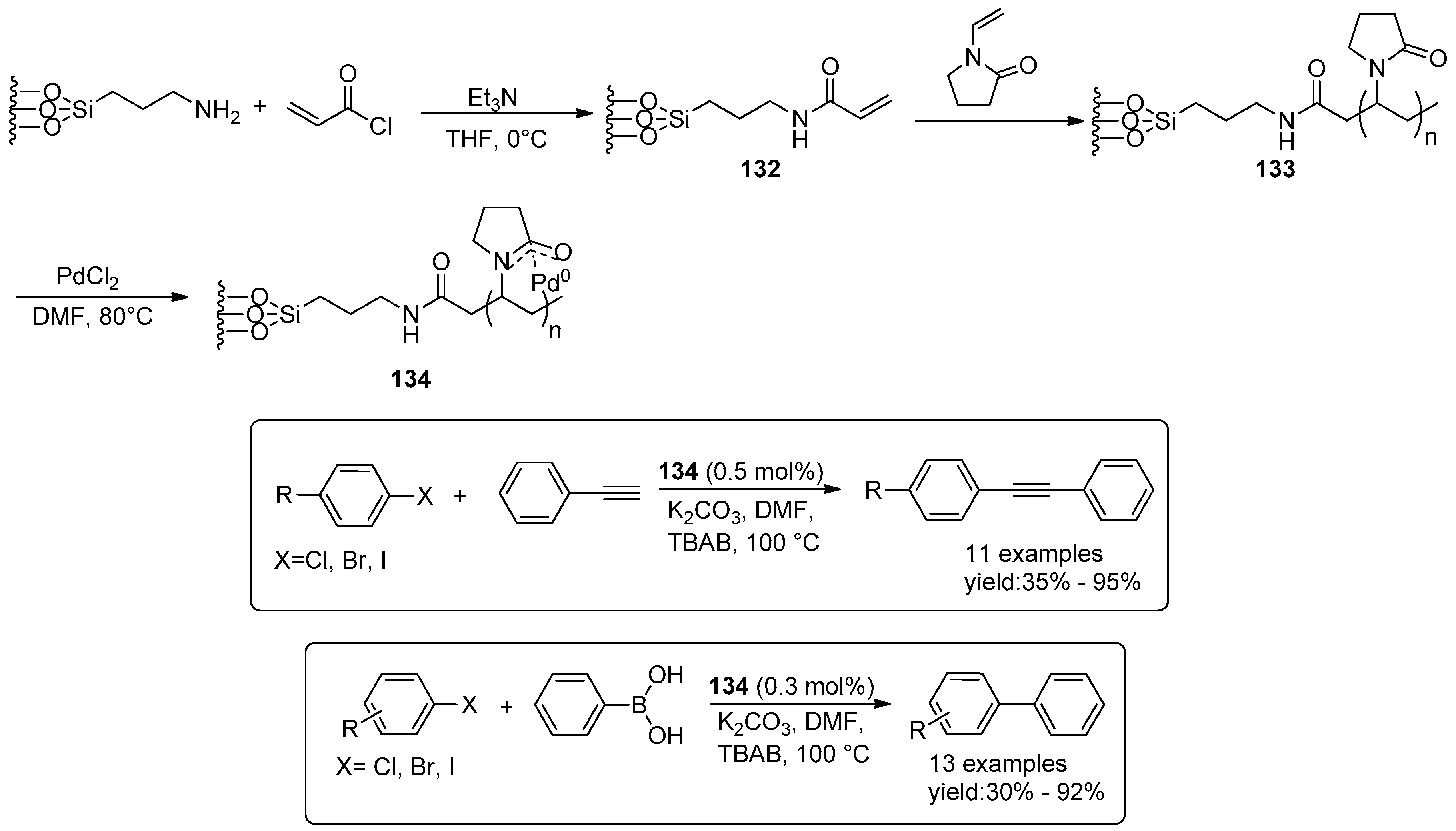
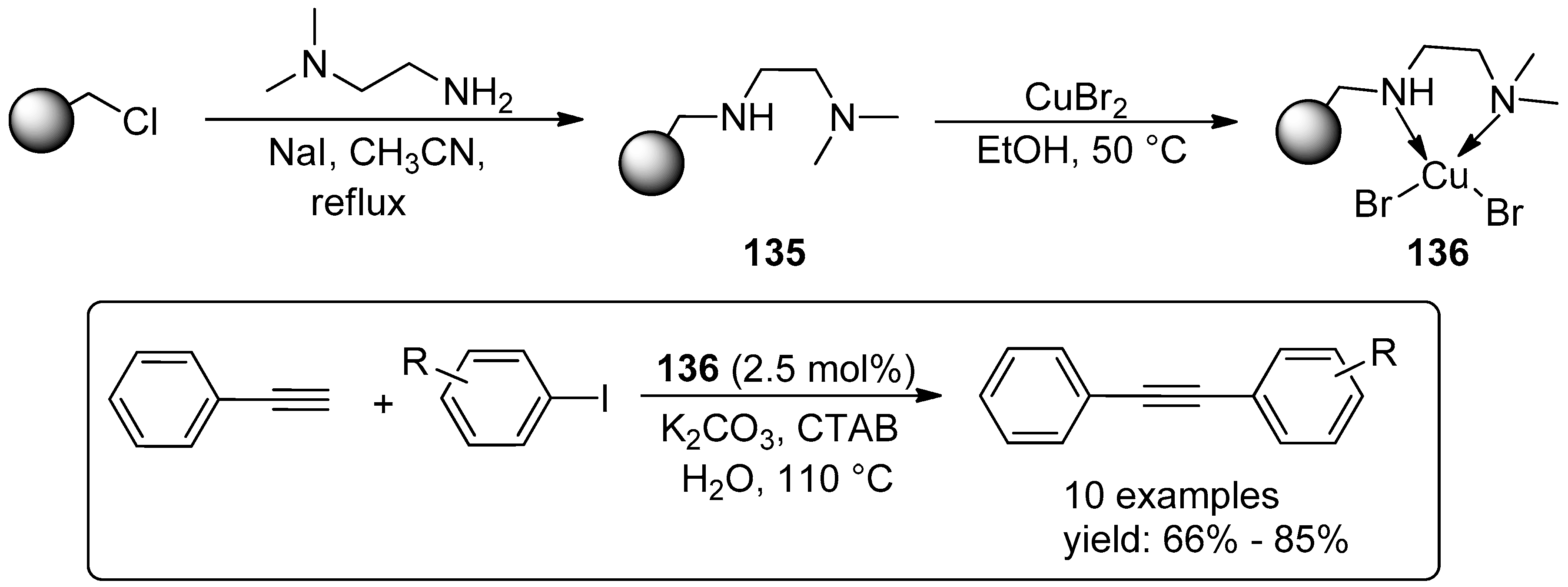
4.3. Miscellaneous Examples
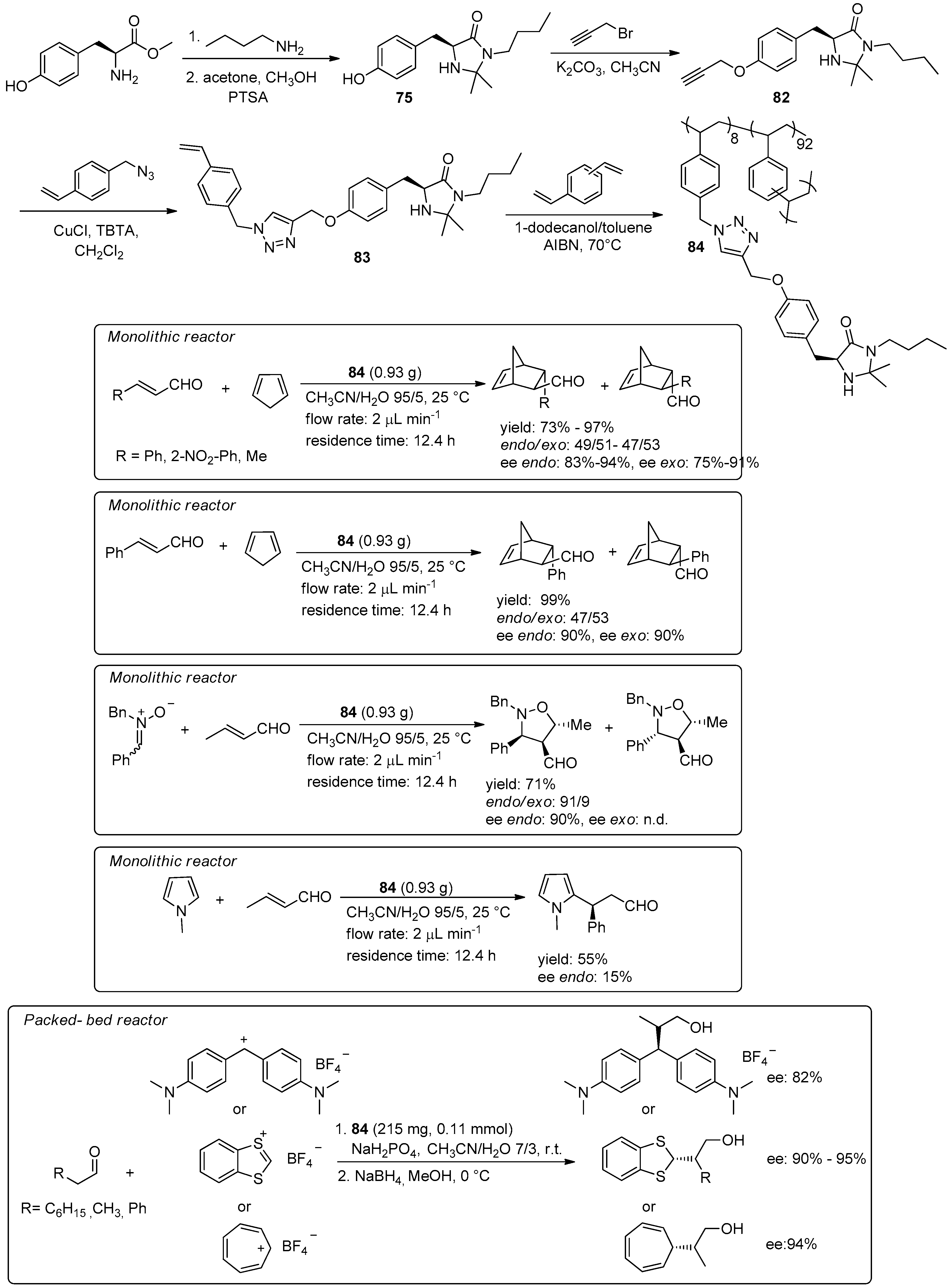
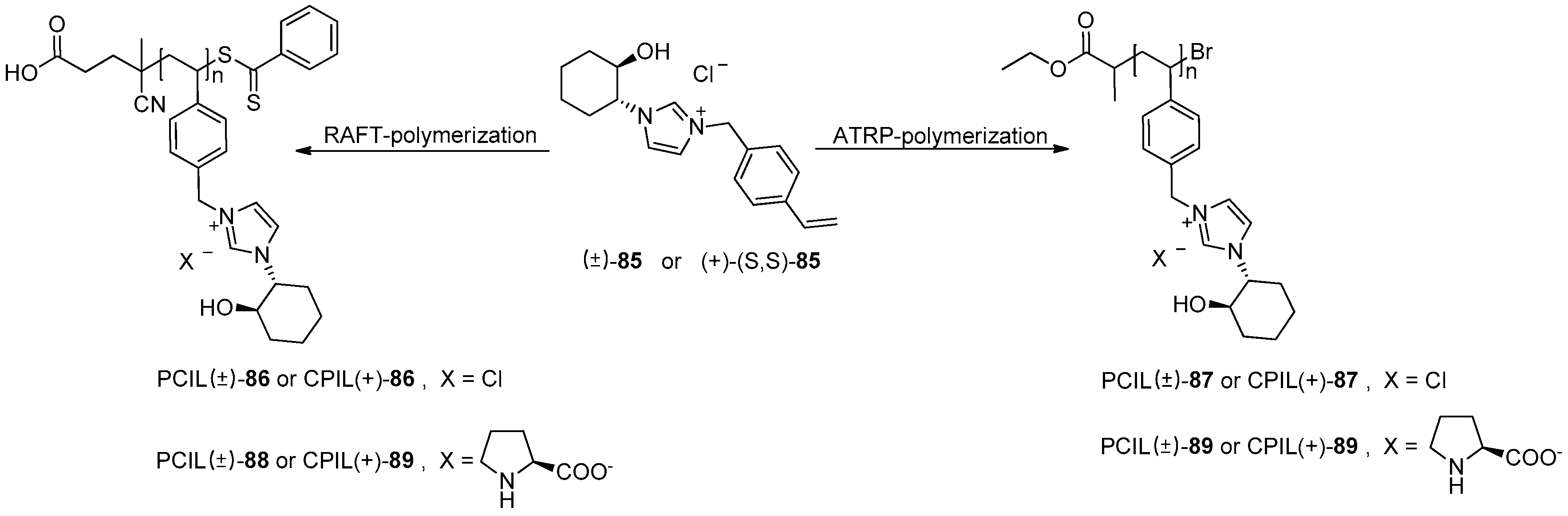
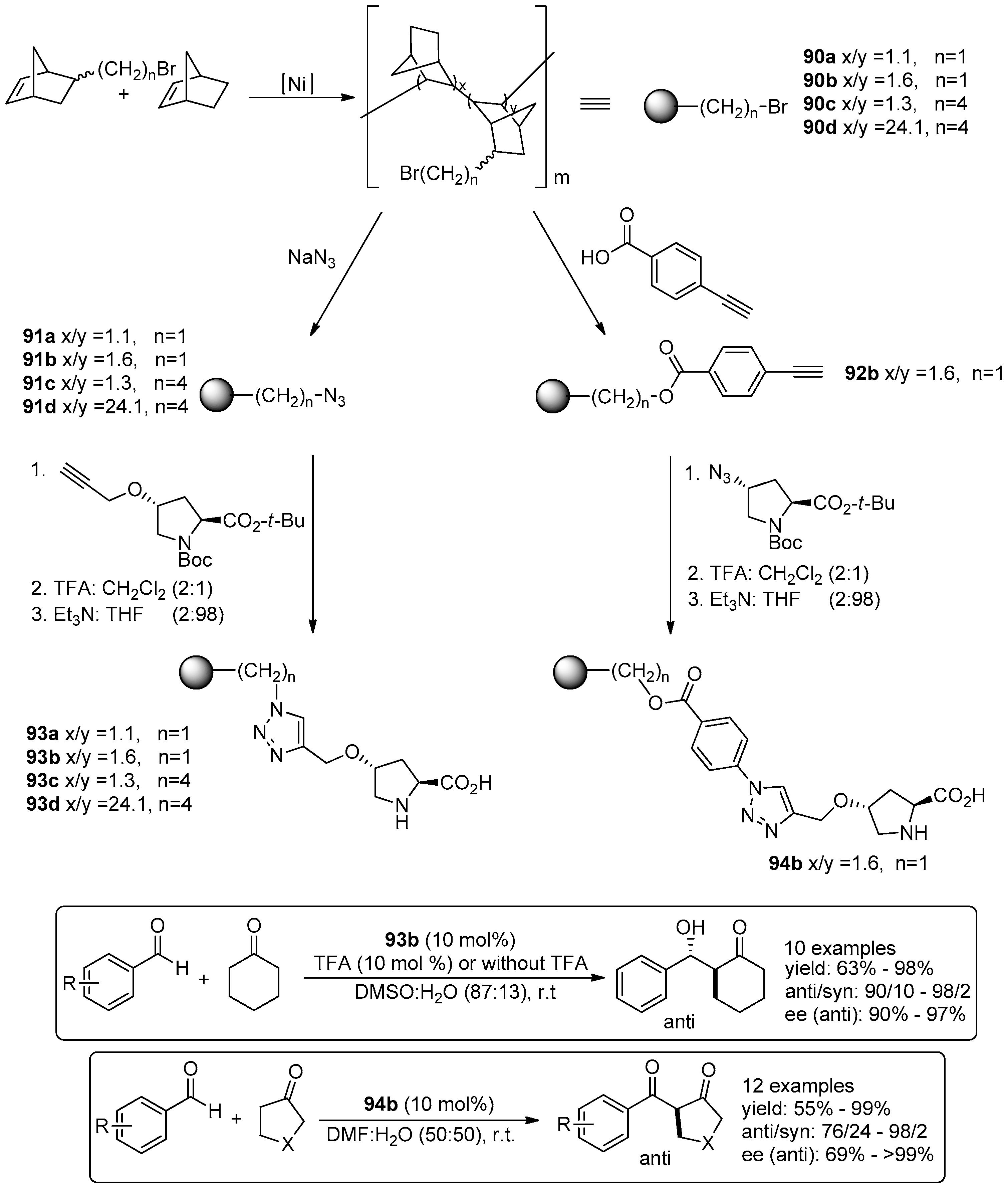
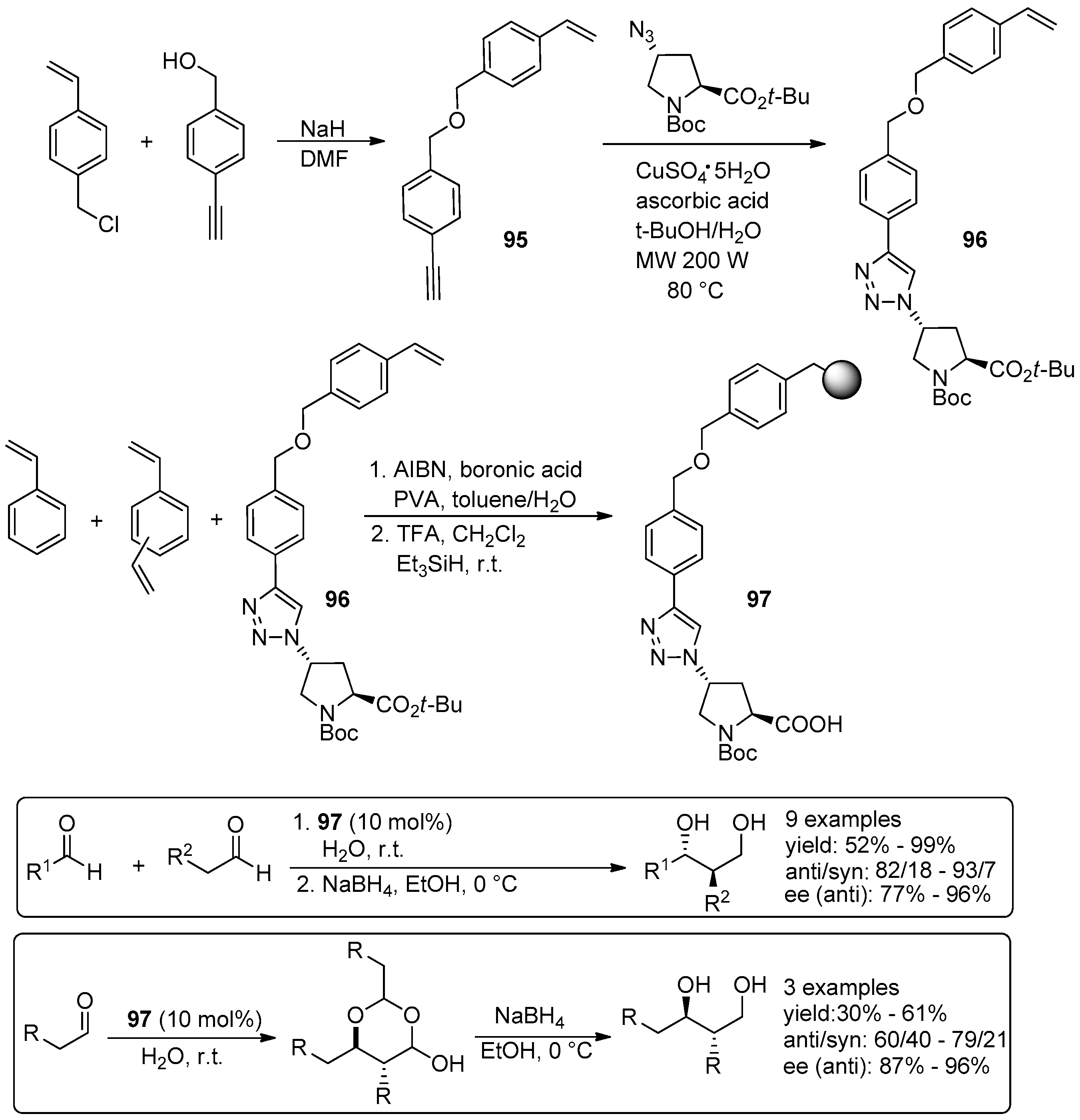
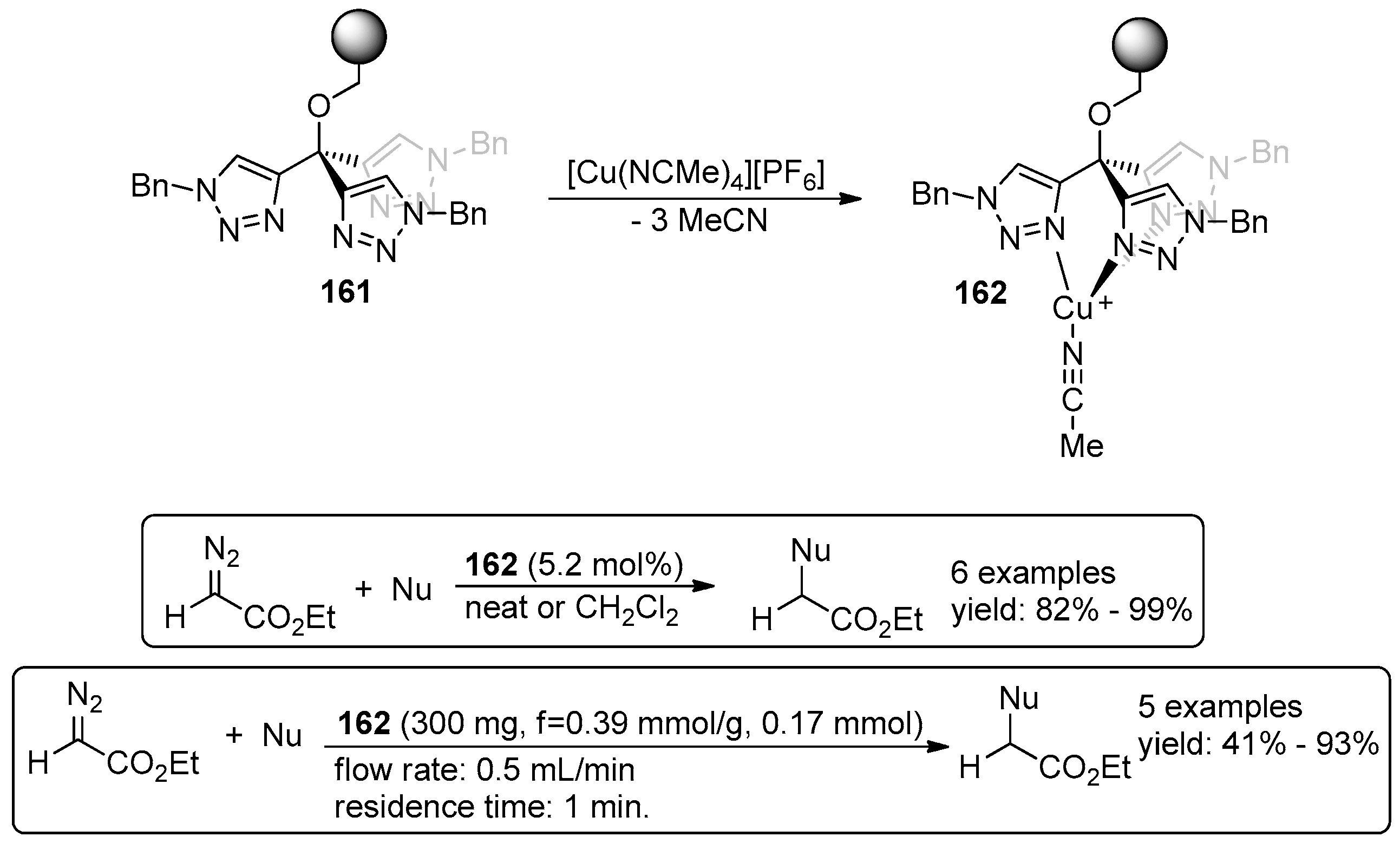
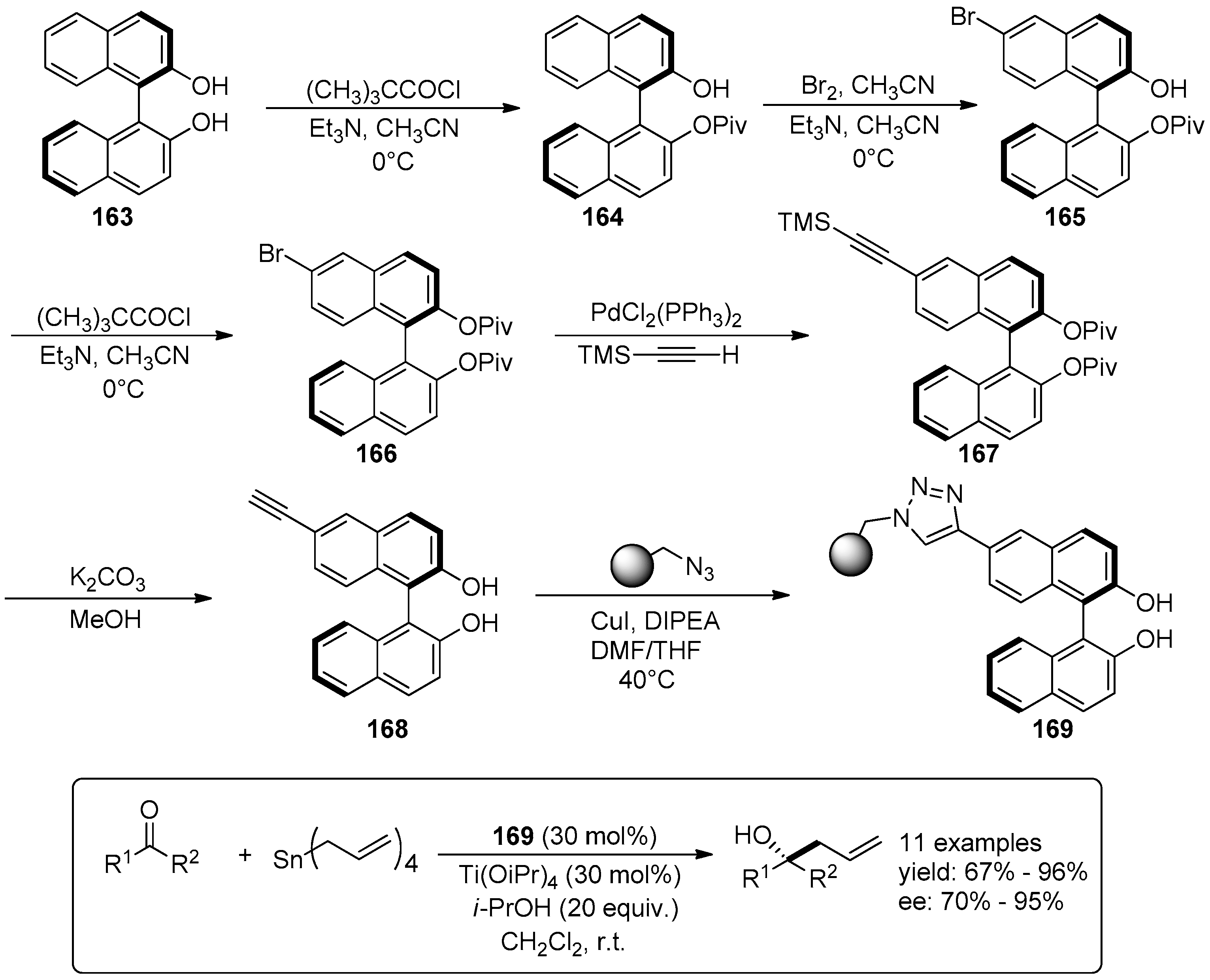
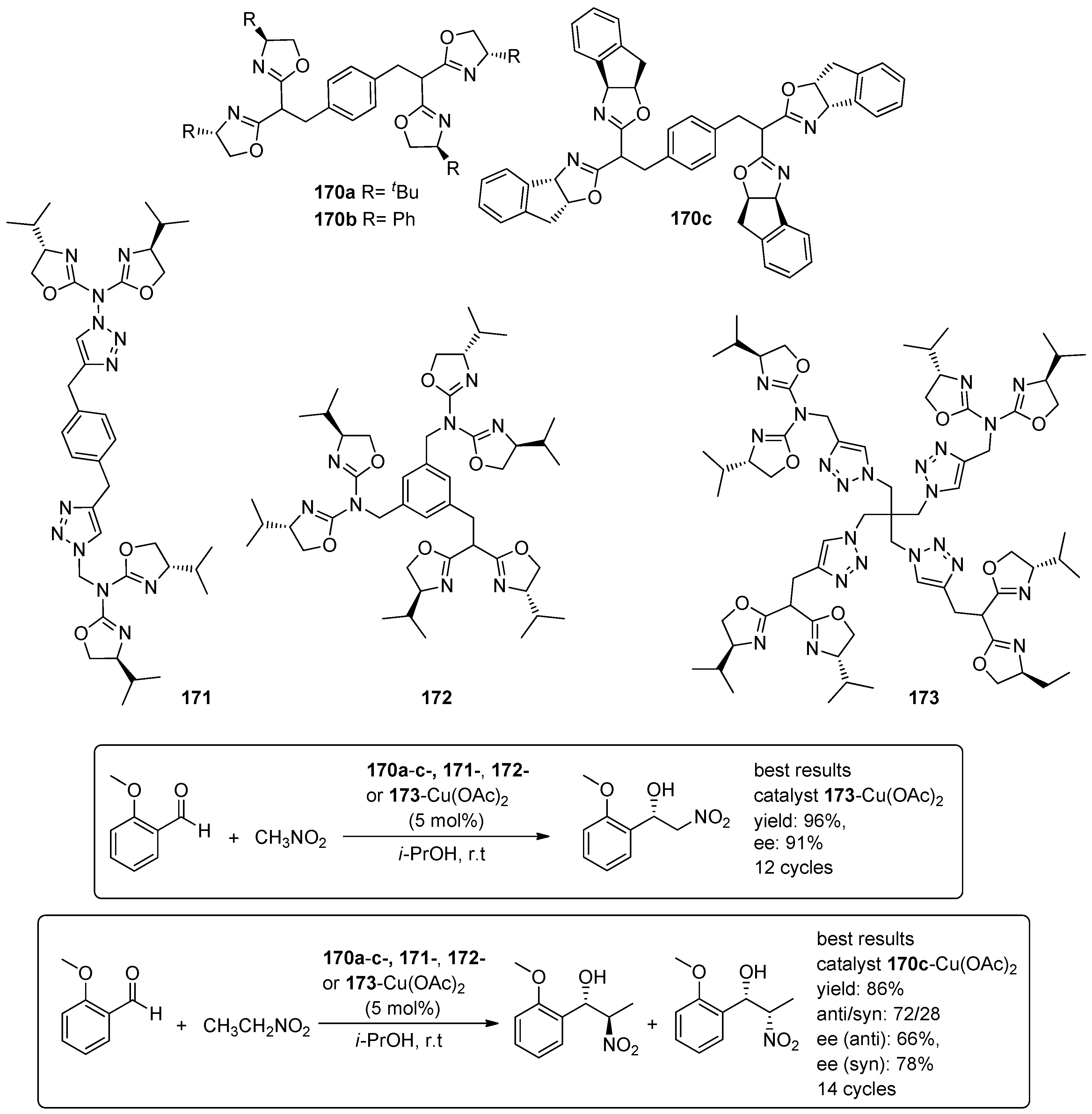
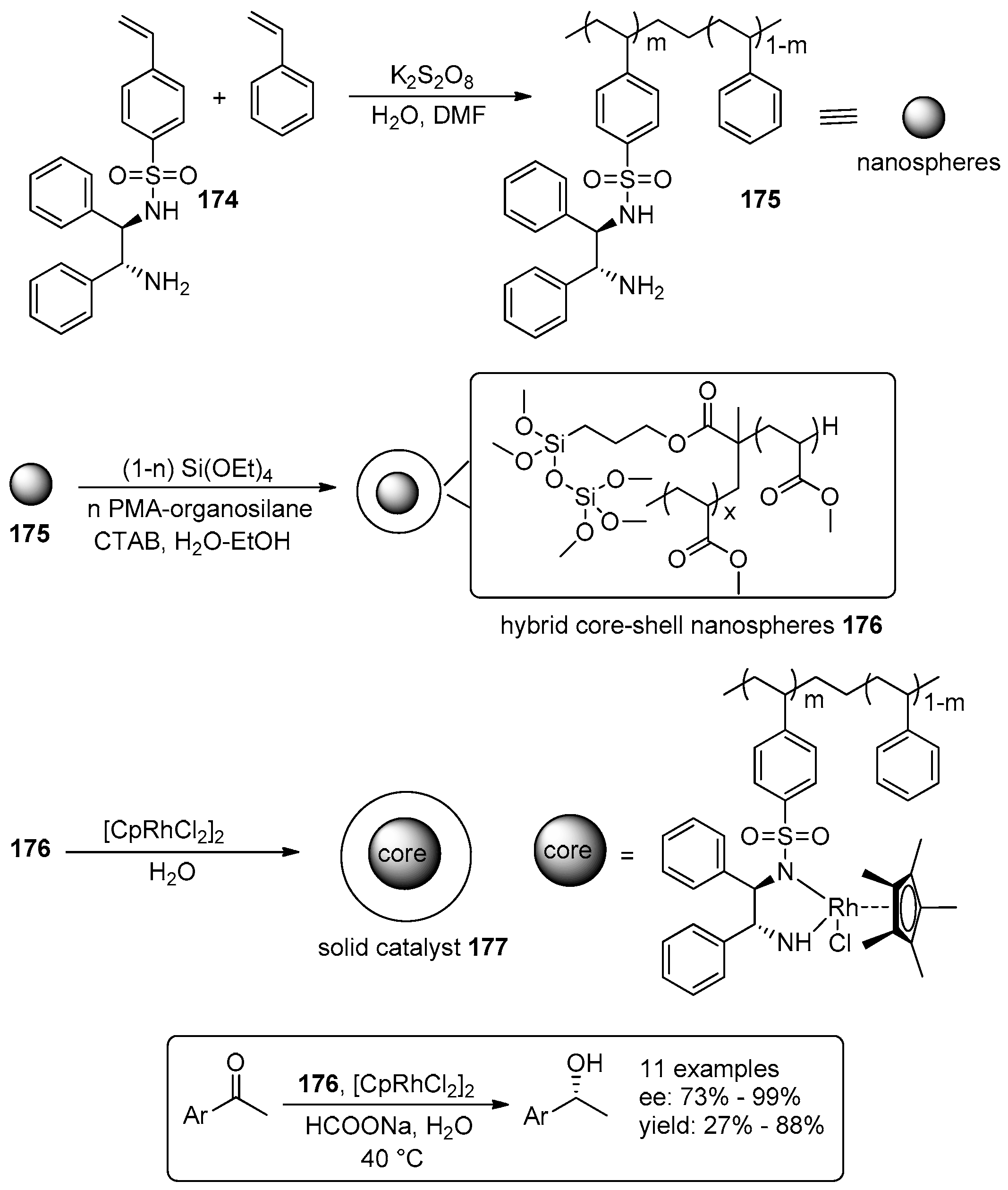
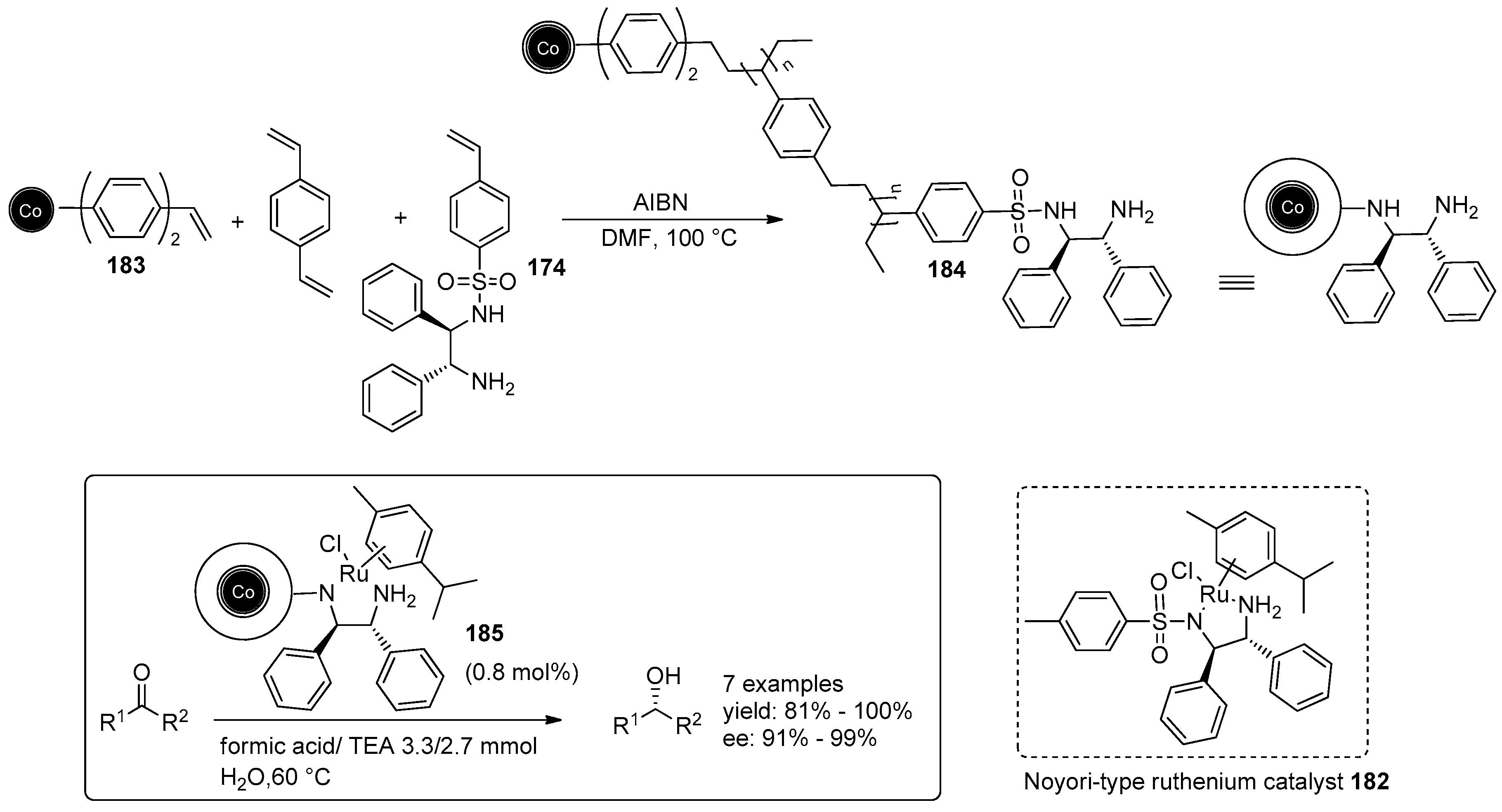
5. Metal-Catalysed Asymmetric Reactions
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hübner, S.; de Vries, J.G.; Farina, V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts? Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar] [CrossRef]
- Toy, P.H. Reengineering Classic Organic Reactions Using Polymeric Tools. Pure Appl. Chem. 2014, 86, 1651–1661. [Google Scholar] [CrossRef]
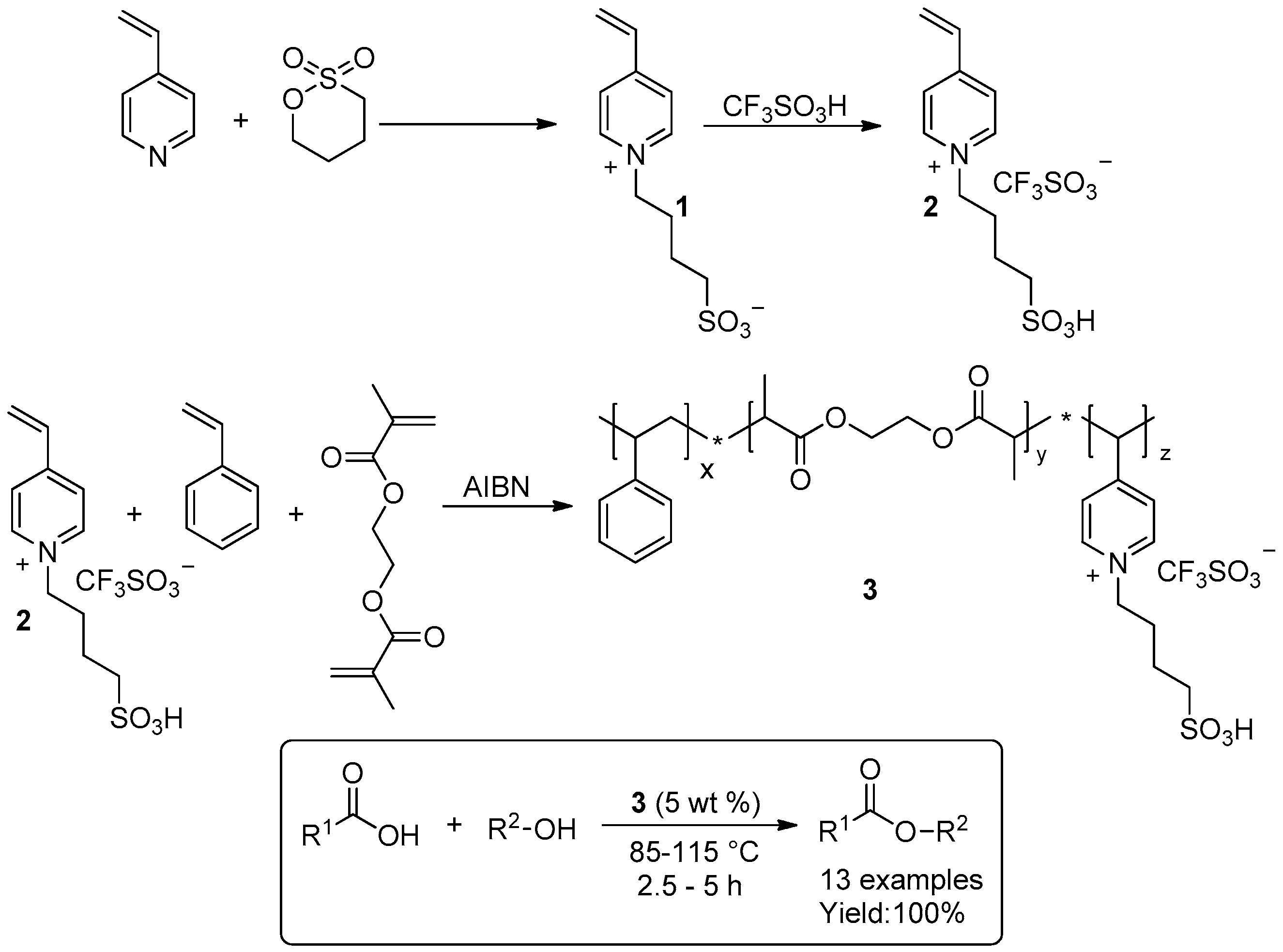
- Zhang, J.; Zhang, S.; Han, J.; Hu, Y.; Yan, R. Uniform acid poly ionic liquid-based large particle and its catalytic application in esterification reaction. Chem. Eng. J. 2015, 271, 269–275. [Google Scholar] [CrossRef]
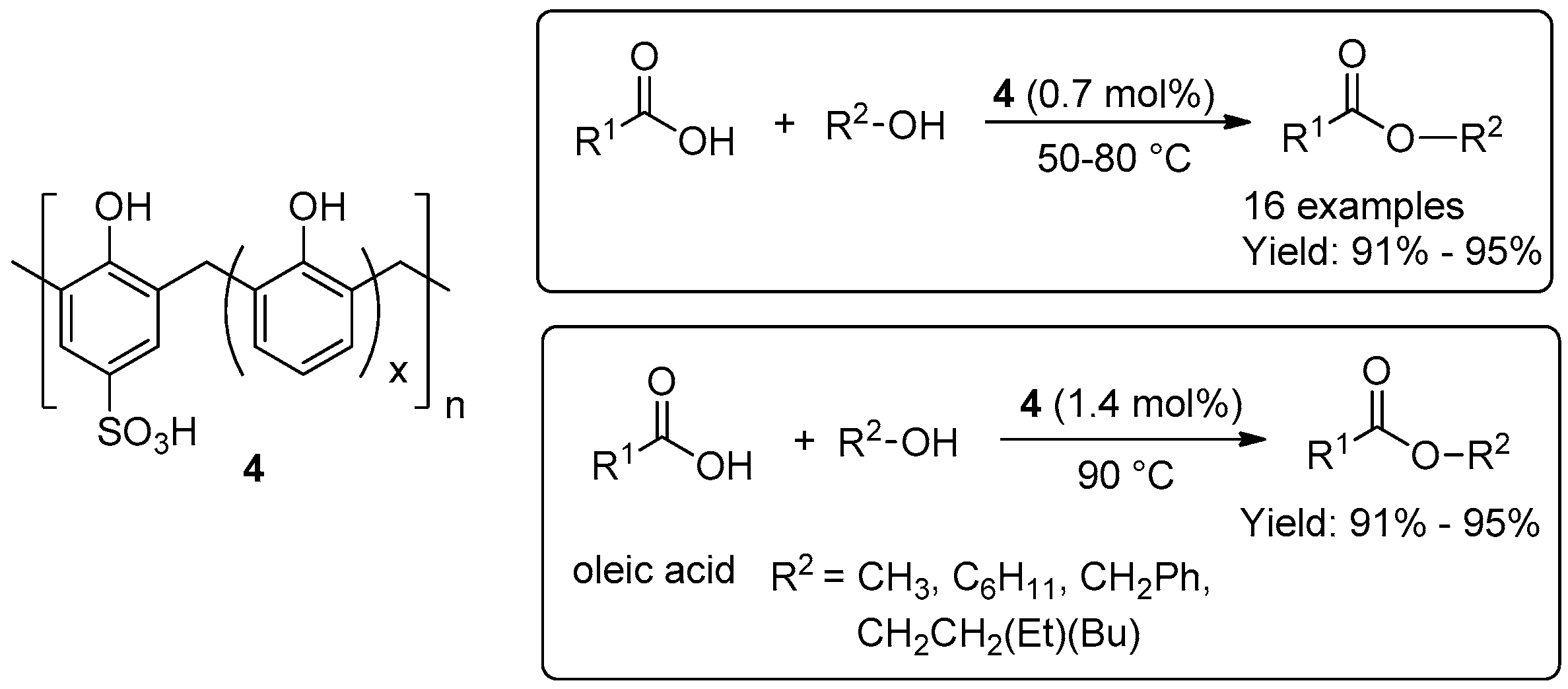
- Minakawa, M.; Baek, H.; Yamada, Y.M.A.; Han, J.W.; Uozumi, Y. Direct Dehydrative Esterification of Alcohols and Carboxylic Acids with a Macroporous Polymeric Acid Catalyst. Org. Lett. 2013, 15, 5798–5801. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Han, J.; Jung, B.Y.; Baek, H.; Yamada, Y.M.A.; Uozumi, Y.; Lee, Y.S. Production of Valuable Esters from Oleic Acid with a Porous Polymeric Acid Catalyst without Water Removal. Synlett 2016, 27, 29–32. [Google Scholar]
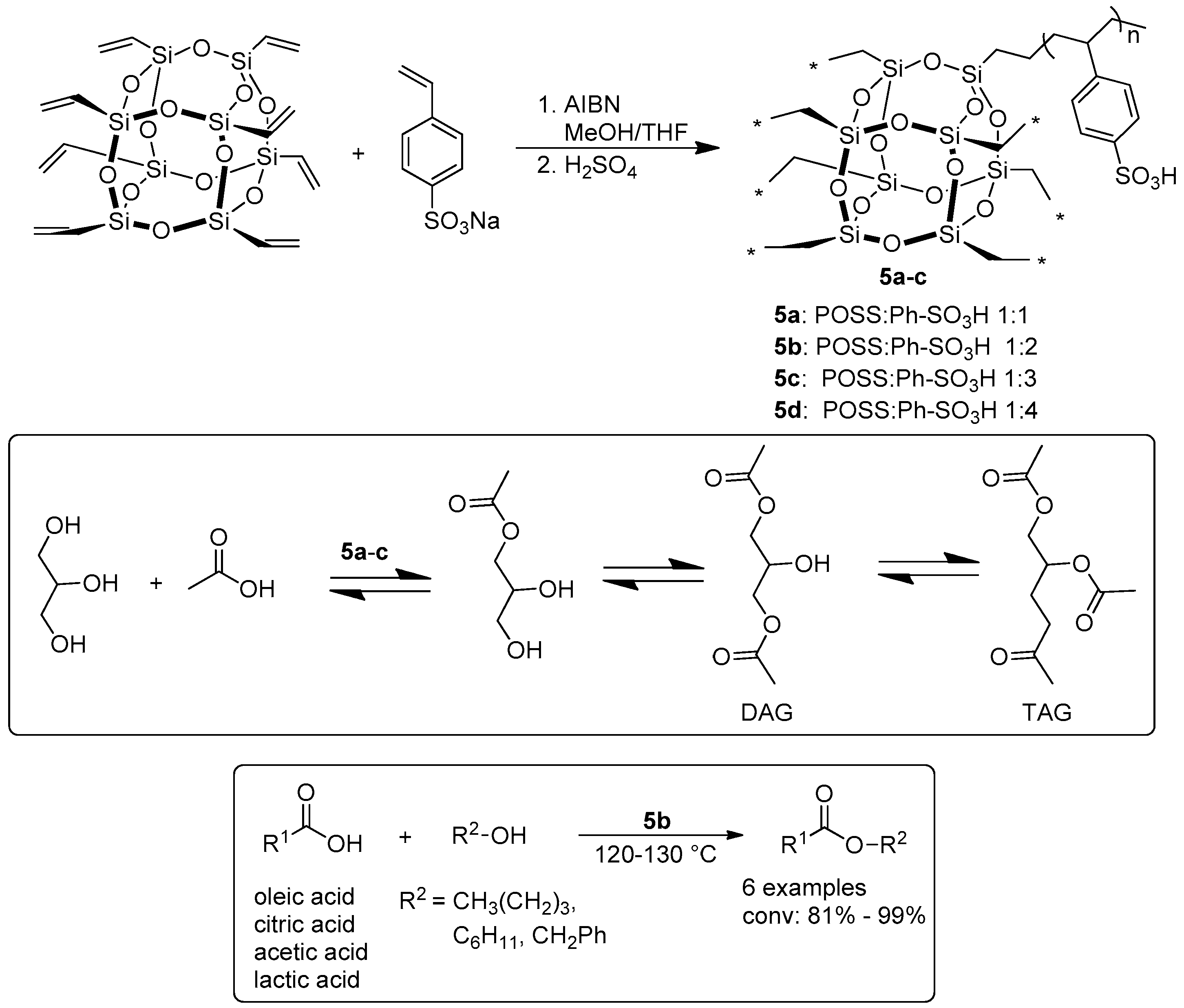
- Leng, Y.; Zhao, J.; Jiang, P.; Lu, D. POSS-derived solid acid catalysts with excellent hydrophobicity for highly efficient transformations of glycerol. Catal. Sci. Technol. 2016, 6, 875–881. [Google Scholar] [CrossRef]
- Yang, B.; Li, J.; Wang, J.; Xu, H.; Guang, S.; Li, C. Poly(vinyl pyrrolidone-co-octavinyl polyhedral oligomeric silsesquioxane) Hybrid Nanocomposites: Preparation, Thermal Properties, and Tg Improvement Mechanism. J. Appl. Polym. Sci. 2009, 111, 2963–2969. [Google Scholar] [CrossRef]
- Wang, W.; Jie, X.; Fei, M.; Jiang, H. Synthesis of core-shell particles by batch emulsion polymerization of styrene and octavinyl polyhedral oligomeric silsesquioxane. J. Polym. Res. 2011, 18, 13–17. [Google Scholar] [CrossRef]
- Wang, B.; Shen, Y.; Sun, J.; Xu, F.; Sun, R. Conversion of platform chemical glycerol to cyclic acetals promoted by acidic ionic liquids. RSC Adv. 2014, 4, 18917–18923. [Google Scholar] [CrossRef]
- Singh, S.; Patel, A. Selective Green Esterification and Oxidation of Glycerol over 12-Tungstophosphoric Acid Anchored to MCM-48. Ind. Eng. Chem. Res. 2014, 53, 14592–14600. [Google Scholar] [CrossRef]
- Liu, H.; Chen, J.; Chen, L.; Xu, Y.; Guo, X.; Fang, D. Carbon Nanotube-Based Solid Sulfonic Acids as Catalysts for Production of Fatty Acid Methyl Ester via Transesterification and Esterification. ACS Sustain. Chem. Eng. 2016, 4, 3140–3150. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Chen, G.; Li, D.; Zhou, Y.; Yang, X.; Wang, J. Hypercrosslinked organic polymer based carbonaceous catalytic materials: Sulfonic acid functionality and nano-confinement effect. Appl. Catal. B Environ. 2015, 176–177, 718–730. [Google Scholar] [CrossRef]
- Li, D.; Mao, D.; Li, J.; Zhou, Y.; Wang, J. In situ functionalized sulfonic copolymer toward recyclable heterogeneous catalyst for efficient Beckmann rearrangement of cyclohexanone oxime. Appl. Catal. A Gen. 2016, 510, 125–133. [Google Scholar] [CrossRef]
- Liu, F.; Kong, W.; Qi, C.; Zhu, L.; Xiao, F.-S. Design and Synthesis of Mesoporous Polymer-Based Solid Acid Catalysts with Excellent Hydrophobicity and Extraordinary Catalytic Activity. ACS Catal. 2012, 2, 565–572. [Google Scholar] [CrossRef]
- Rojas, M.F.; Bernard, F.L.; Aquino, A.; Borges, J.; Vecchia, F.D.; Menezes, S.; Ligabue, R.; Einloft, S. Poly(ionic liquid)s as efficient catalyst in transformation of CO2 to cyclic carbonate. J. Mol. Catal. A Chem. 2014, 392, 83–88. [Google Scholar] [CrossRef]
- Dani, A.; Groppo, E.; Barolo, C.; Vitillo, J.G.; Bordiga, S. Design of high surface area poly(ionic liquid)s to convert carbon dioxide into ethylene carbonate. J. Mater. Chem. A 2015, 3, 8508–8518. [Google Scholar] [CrossRef]
- Agrigento, P.; Al-Amsyar, S.M.; Soree, B.; Taherimehr, M.; Gruttadauria, M.; Aprile, C.; Pescarmona, P.P. Synthesis and high-throughput testing of multilayered supported ionic liquid catalysts for the conversion of CO2 and epoxides into cyclic carbonates. Catal. Sci. Technol. 2014, 4, 1598–1607. [Google Scholar] [CrossRef]
- Giacalone, F.; Gruttadauria, M. Covalently Supported Ionic Liquid Phases: An Advanced Class of Recyclable Catalytic Systems. ChemCatChem 2016, 8, 664–684. [Google Scholar] [CrossRef]
- Xin, B.; Hao, J. Imidazolium-based ionic liquids grafted on solid surfaces. Chem. Soc. Rev. 2014, 43, 7171–7187. [Google Scholar] [CrossRef] [PubMed]
- Aprile, C.; Giacalone, F.; Agrigento, P.; Liotta, L.F.; Martens, J.A.; Pescarmona, P.P.; Gruttadauria, M. Multilayered Supported Ionic Liquids as Catalysts for Chemical Fixation of Carbon Dioxide: A High-Throughput Study in Supercritical Conditions. ChemSusChem 2011, 4, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Lu, D.; Jiang, P.; Zhang, C.; Zhao, J.; Zhang, W. Highly cross-linked cationic polymer microspheres as an efficient catalyst for facile CO2 fixation. Catal. Commun. 2016, 74, 99–103. [Google Scholar] [CrossRef]
- Dai, W.-L.; Jin, B.; Luo, S.-L.; Luo, X.-B.; Tu, X.-M.; Au, C.-T. Functionalized phosphonium-based ionic liquids as efficient catalysts for the synthesis of cyclic carbonate from expoxides and carbon dioxide. Appl. Catal. A Gen. 2014, 470, 183–188. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, Y.; Tan, Y.; Luo, X.; Tu, X. Reusable and efficient polymer nanoparticles grafted with hydroxyl-functionalized phosphonium-based ionic liquid catalyst for cycloaddition of CO2 with epoxides. Appl. Catal. A Gen. 2016, 514, 43–50. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Henseler, A.H.; Ayats, C.; Kleij, A.W.; Pericas, M.A. Conversion of oxiranes and CO2 to organic cyclic carbonates using a recyclable, bifunctional polystyrene-supported organocatalyst. Green Chem. 2014, 16, 1552–1559. [Google Scholar] [CrossRef]
- Talapaneni, S.N.; Buyukcakir, O.; Je, S.H.; Srinivasan, S.; Seo, Y.; Polychronopoulou, K.; Coskun, A. Nanoporous Polymers Incorporating Sterically Confined N-Heterocyclic Carbenes for Simultaneous CO2 Capture and Conversion at Ambient Pressure. Chem. Mater. 2015, 27, 6818–6826. [Google Scholar] [CrossRef]
- Gopalakrishna Panicker, R.K.; Krishnapillai, S. Synthesis of on resin poly(propylene imine) dendrimer and its use as organocatalyst. Tetrahedron Lett. 2014, 55, 2352–2354. [Google Scholar] [CrossRef]
- Krishnan, G.R.; Sreekumar, K. First Example of Organocatalysis by Polystyrene-Supported PAMAM Dendrimers: Highly Efficient and Reusable Catalyst for Knoevenagel Condensations. Eur. J. Org. Chem. 2008, 2008, 4763–4768. [Google Scholar] [CrossRef]
- Mangala, K.; Sreekumar, K. Dendrimer functionalized polysilane: An efficient and recyclable organocatalyst. J. Appl. Polym. Sci. 2015, 132, 41593–41599. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Chen, G.; Guo, Z.; Zhou, Y.; Wang, J. Hydrophobic Mesoporous Poly(ionic liquid)s towards Highly Efficient and Contamination-Resistant Solid-Base Catalysts. ChemCatChem 2015, 7, 993–1003. [Google Scholar] [CrossRef]
- Seo, U.R.; Chung, Y.K. Poly(4-vinylimidazolium) iodides: A highly recyclable organocatalyst precursor for benzoin condensation reaction. RSC Adv. 2014, 4, 32371–32374. [Google Scholar] [CrossRef]
- Seo, U.R.; Chung, Y.K. Poly(4-vinylimidazolium)s/Diazabicyclo[5.4.0]undec-7-ene/Zinc(II) Bromide-Catalyzed Cycloaddition of Carbon Dioxide to Epoxides. Adv. Synth. Catal. 2014, 356, 1955–1961. [Google Scholar] [CrossRef]
- Pinaud, J.; Vignolle, J.; Gnanou, Y.; Taton, D. Poly(N-heterocyclic-carbene)s and their CO2 Adducts as Recyclable Polymer-Supported Organocatalysts for Benzoin Condensation and Transesterification Reactions. Macromolecules 2011, 44, 1900–1908. [Google Scholar] [CrossRef]
- Coupillaud, P.; Pinaud, J.; Guidolin, N.; Vignolle, J.; Fèvre, M.; Veaudecrenne, E.; Mecerreyes, D.; Taton, D. Poly(ionic liquid)s based on imidazolium hydrogen carbonate monomer units as recyclable polymer-supported N-heterocyclic carbenes: Use in organocatalysis. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 4530–4540. [Google Scholar]
- Wang, L.; Chen, E.Y.X. Recyclable Supported Carbene Catalysts for High-Yielding Self-Condensation of Furaldehydes into C10 and C12 Furoins. ACS Catal. 2015, 5, 6907–6917. [Google Scholar] [CrossRef]
- Iwamoto, K.-I.; Kimura, H.; Oike, M.; Sato, M. Methylene-bridged bis(benzimidazolium) salt as a highly efficient catalyst for the benzoin reaction in aqueous media. Org. Biomol. Chem. 2008, 6, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, Y.; Sun, Q.; Pan, S.; Meng, X.; Xiao, F.-S. Porous Polymerized Organocatalysts Rationally Synthesized from the Corresponding Vinyl-Functionalized Monomers as Efficient Heterogeneous Catalysts. ACS Catal. 2015, 5, 1556–1559. [Google Scholar] [CrossRef]
- Brunel, D.; Fajula, F.; Nagy, J.B.; Deroide, B.; Verhoef, M.J.; Veum, L.; Peters, J.A.; van Bekkum, H. Comparison of two MCM-41 grafted TEMPO catalysts in selective alcohol oxidation. Appl. Catal. A Gen. 2001, 213, 73–82. [Google Scholar] [CrossRef]
- Bivona, L.A.; Quertinmont, F.; Beejapur, H.A.; Giacalone, F.; Buaki-Sogo, M.; Gruttadauria, M.; Aprile, C. Thiazolium-Based Catalysts for the Etherification of Benzylic Alcohols under Solvent-Free Conditions. Adv. Synth. Catal. 2015, 357, 800–810. [Google Scholar] [CrossRef]
- Yang, D.; Liu, P.; Zhang, N.; Wei, W.; Yue, M.; You, J.; Wang, H. Mesoporous Poly(melamine–formaldehyde): A Green and Recyclable Heterogeneous Organocatalyst for the Synthesis of Benzoxazoles and Benzothiazoles Using Dioxygen as Oxidant. ChemCatChem 2014, 6, 3434–3439. [Google Scholar] [CrossRef]
- Tan, M.X.; Sum, Y.N.; Ying, J.Y.; Zhang, Y. A mesoporous poly-melamine-formaldehyde polymer as a solid sorbent for toxic metal removal. Energy Environ. Sci. 2013, 6, 3254–3259. [Google Scholar] [CrossRef]
- Tan, M.X.; Gu, L.; Li, N.; Ying, J.Y.; Zhang, Y. Mesoporous poly-melamine-formaldehyde (mPMF)—A highly efficient catalyst for chemoselective acetalization of aldehydes. Green Chem. 2013, 15, 1127–1132. [Google Scholar] [CrossRef]
- Tan, M.X.; Zhang, Y.; Ying, J.Y. Mesoporous Poly(Melamine–Formaldehyde) Solid Sorbent for Carbon Dioxide Capture. ChemSusChem 2013, 6, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Strappaveccia, G.; Bianchi, L.; Ziarelli, S.; Santoro, S.; Lanari, D.; Pizzo, F.; Vaccaro, L. PS-BEMP as a basic catalyst for the phospha-Michael addition to electron-poor alkenes. Org. Biomol. Chem. 2016, 14, 3521–3525. [Google Scholar] [CrossRef] [PubMed]
- Bonollo, S.; Lanari, D.; Longo, J.M.; Vaccaro, L. E-factor minimized protocols for the polystyryl-BEMP catalyzed conjugate additions of various nucleophiles to a,b-unsaturated carbonyl compounds. Green Chem. 2012, 14, 164–169. [Google Scholar] [CrossRef]
- Zvagulis, A.; Bonollo, S.; Lanari, D.; Pizzo, F.; Vaccaro, L. 2-tert-Butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine Supported on Polystyrene (PS-BEMP) as an Efficient Recoverable and Reusable Catalyst for the Phenolysis of Epoxides under Solvent-Free Conditions. Adv. Synth. Catal. 2010, 352, 2489–2496. [Google Scholar] [CrossRef]
- Itsuno, S.; Hassan, M.M. Polymer-immobilized chiral catalysts. RSC Adv. 2014, 4, 52023–52043. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F. Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques, and Applications; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Kasaplar, P.; Ozkal, E.; Rodriguez-Escrich, C.; Pericas, M.A. Enantioselective a-amination of 1,3-dicarbonyl compounds in batch and flow with immobilized thiourea organocatalysts. Green Chem. 2015, 17, 3122–3129. [Google Scholar] [CrossRef]
- Pedrosa, R.; Andres, J.M.; Avila, D.P.; Ceballos, M.; Pindado, R. Chiral ureas and thioureas supported on polystyrene for enantioselective aza-Henry reactions under solvent-free conditions. Green Chem. 2015, 17, 2217–2225. [Google Scholar] [CrossRef]
- Andrés, J.M.; de La Cruz, N.; Valle, M.; Pedrosa, R. Bottom-Up Synthesis of Supported Thioureas and Their Use in Enantioselective Solvent-Free Aza-Henry and Michael Additions. ChemPlusChem 2016, 81, 86–92. [Google Scholar] [CrossRef]
- Sagamanova, I.; Rodríguez-Escrich, C.; Molnár, I.G.; Sayalero, S.; Gilmour, R.; Pericàs, M.A. Translating the Enantioselective Michael Reaction to a Continuous Flow Paradigm with an Immobilized, Fluorinated Organocatalyst. ACS Catal. 2015, 5, 6241–6248. [Google Scholar] [CrossRef]
- Salvo, A.M.P.; Giacalone, F.; Noto, R.; Gruttadauria, M. Recyclable Heterogeneous and Low-Loading Homogeneous Chiral Imidazolidinone Catalysts for α-Alkylation of Aldehydes. ChemPlusChem 2014, 79, 857–862. [Google Scholar] [CrossRef]
- Itsuno, S.; Oonami, T.; Takenaka, N.; Haraguchi, N. Synthesis of Chiral Polyethers Containing Imidazolidinone Repeating Units and Application as Catalyst in Asymmetric Diels–Alder Reaction. Adv. Synth. Catal. 2015, 357, 3995–4002. [Google Scholar] [CrossRef]
- Chiroli, V.; Benaglia, M.; Puglisi, A.; Porta, R.; Jumde, R.P.; Mandoli, A. A chiral organocatalytic polymer-based monolithic reactor. Green Chem. 2014, 16, 2798–2806. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Puglisi, A.; Mandoli, A.; Gualandi, A.; Cozzi, P.G. A Catalytic Reactor for the Organocatalyzed Enantioselective Continuous Flow Alkylation of Aldehydes. ChemSusChem 2014, 7, 3534–3540. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, E.; Izquierdo, D.F.; Marti-Centelles, V.; Luis, S.V.; Tenhu, H.; Garcia-Verdugo, E. An enzymatic biomimetic system: Enhancement of catalytic efficiency with new polymeric chiral ionic liquids synthesised by controlled radical polymerisation. Polym. Chem. 2014, 5, 1437–1446. [Google Scholar] [CrossRef]
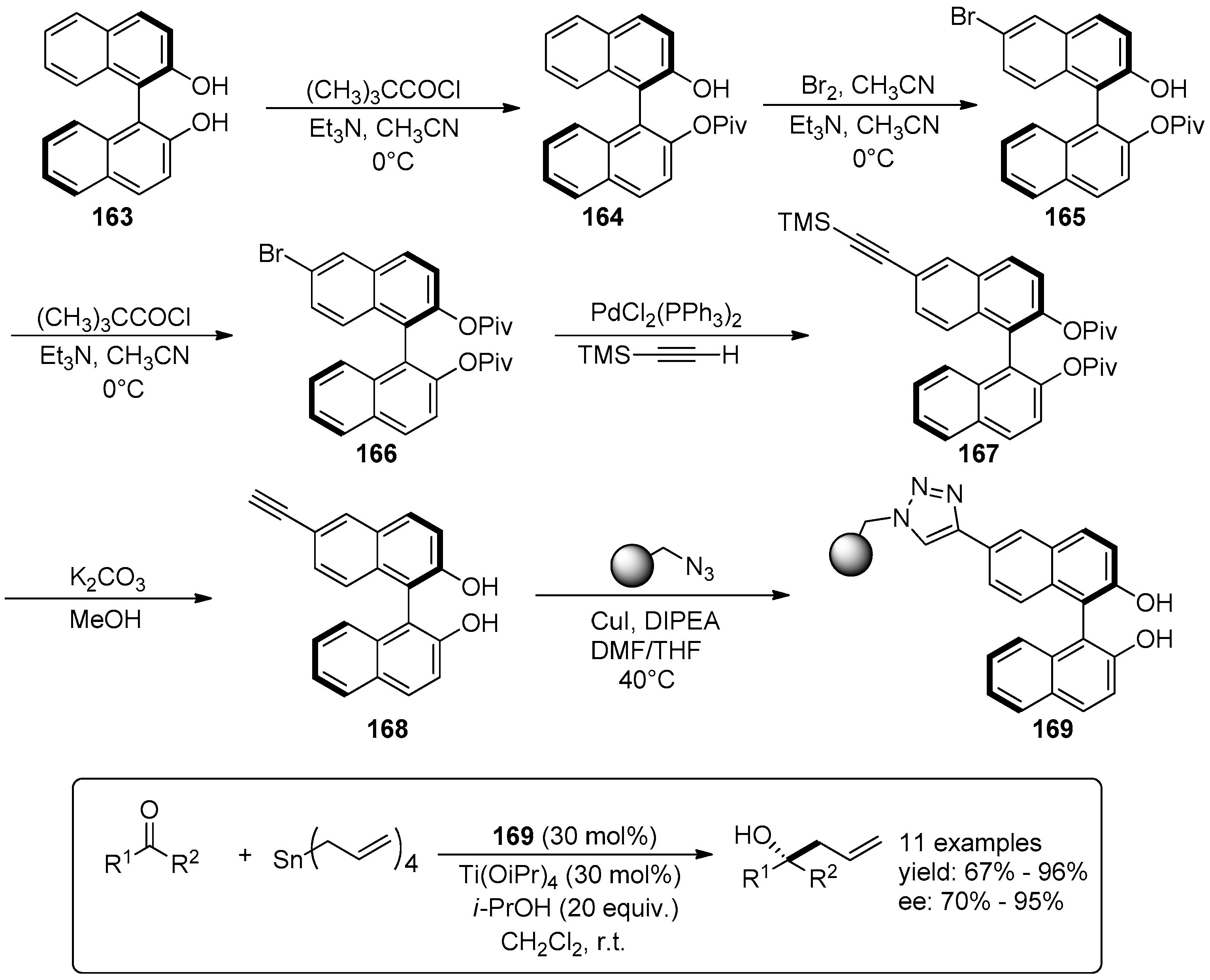
- Sagamanova, I.K.; Sayalero, S.; Martinez-Arranz, S.; Albeniz, A.C.; Pericas, M.A. Asymmetric organocatalysts supported on vinyl addition polynorbornenes for work in aqueous media. Catal. Sci. Technol. 2015, 5, 754–764. [Google Scholar] [CrossRef]
- Martínez-Arranz, S.; Albéniz, A.C.; Espinet, P. Versatile Route to Functionalized Vinylic Addition Polynorbornenes. Macromolecules 2010, 43, 7482–7487. [Google Scholar] [CrossRef]
- Llanes, P.; Sayalero, S.; Rodriguez-Escrich, C.; Pericas, M.A. Asymmetric cross- and self-aldol reactions of aldehydes in water with a polystyrene-supported triazolylproline organocatalyst. Green Chem. 2016, 18, 3507–3512. [Google Scholar] [CrossRef]
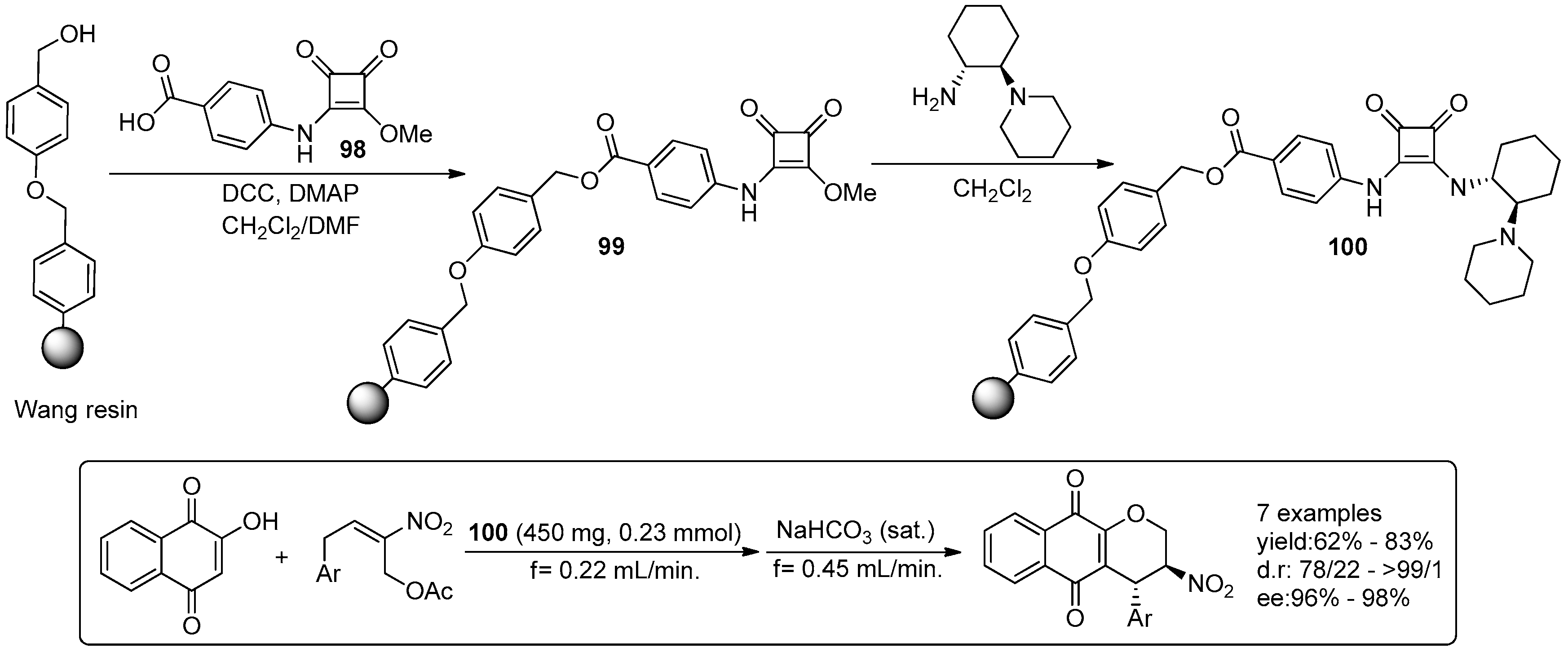
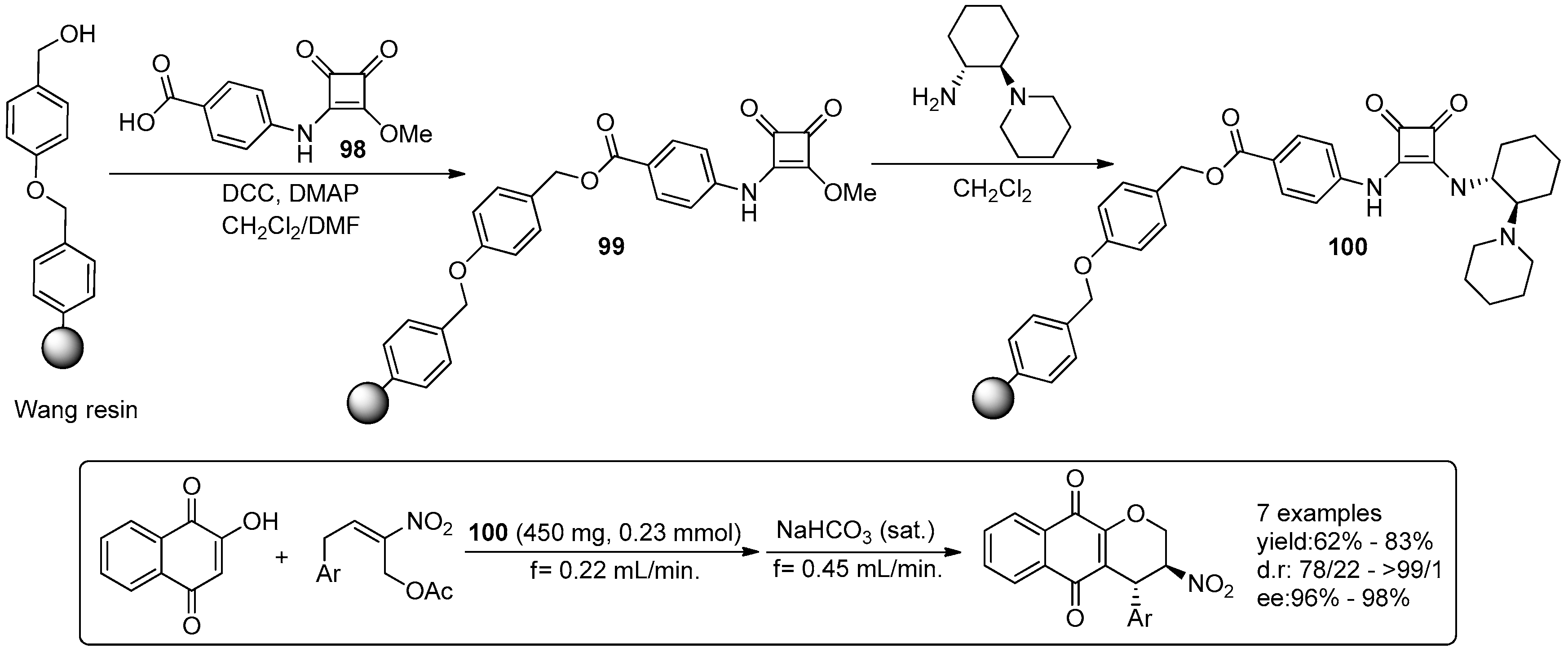
- Osorio-Planes, L.; Rodriguez-Escrich, C.; Pericas, M.A. Removing the superfluous: A supported squaramide catalyst with a minimalistic linker applied to the enantioselective flow synthesis of pyranonaphthoquinones. Catal. Sci. Technol. 2016, 6, 4686–4689. [Google Scholar] [CrossRef]
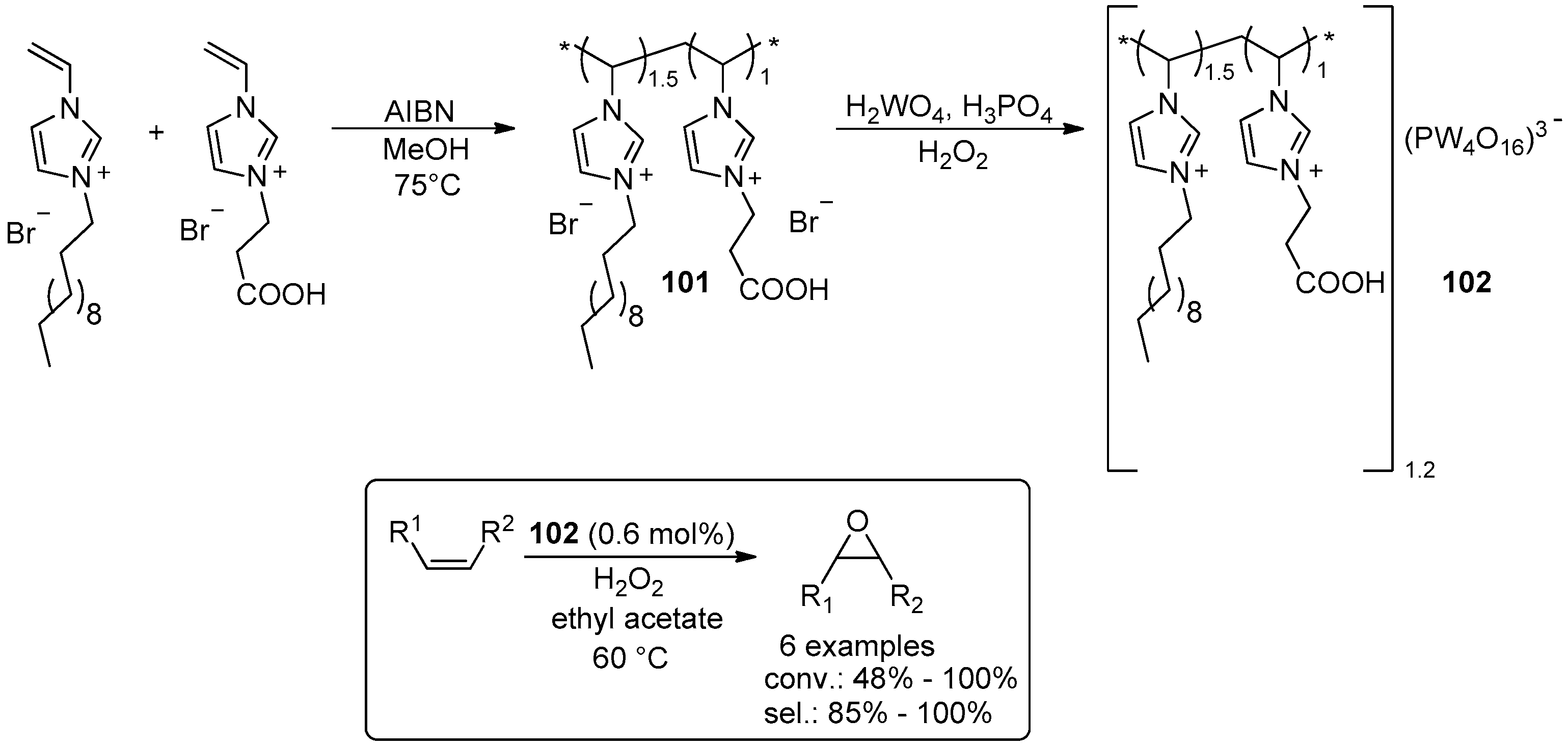
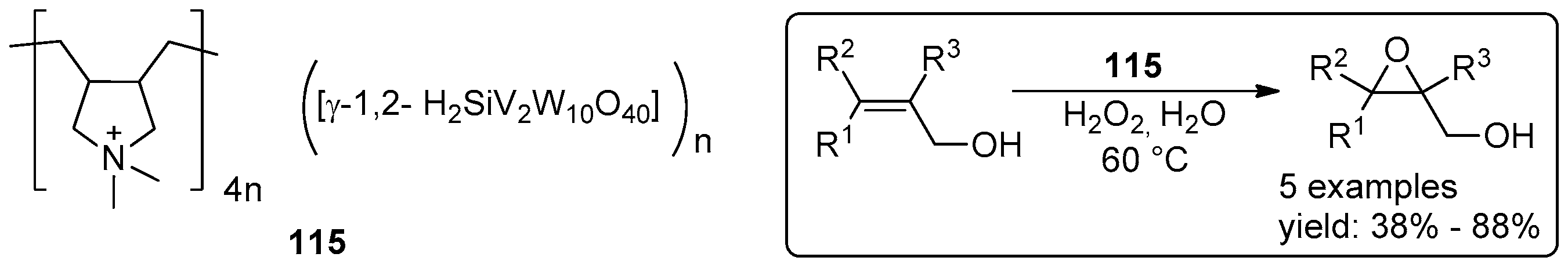
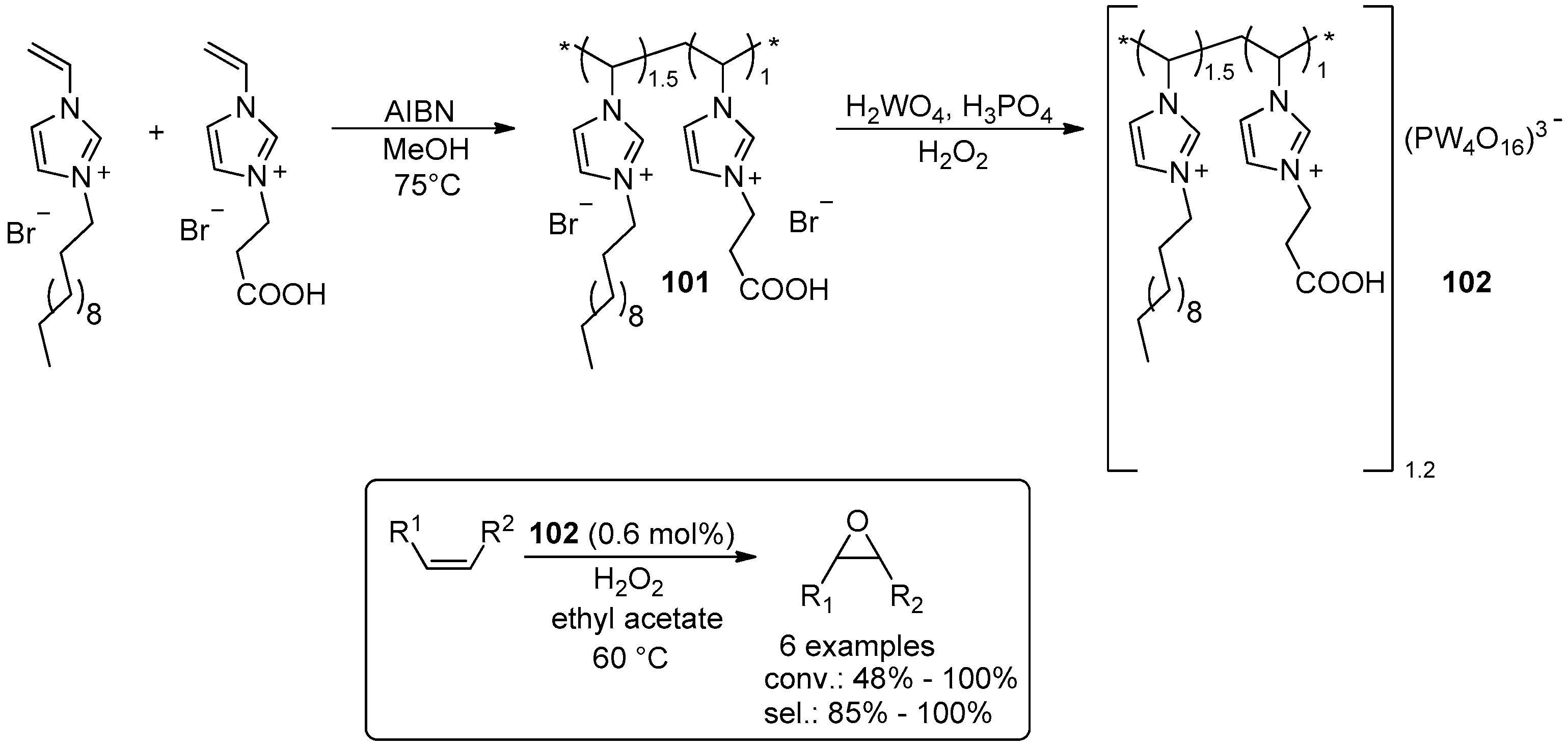
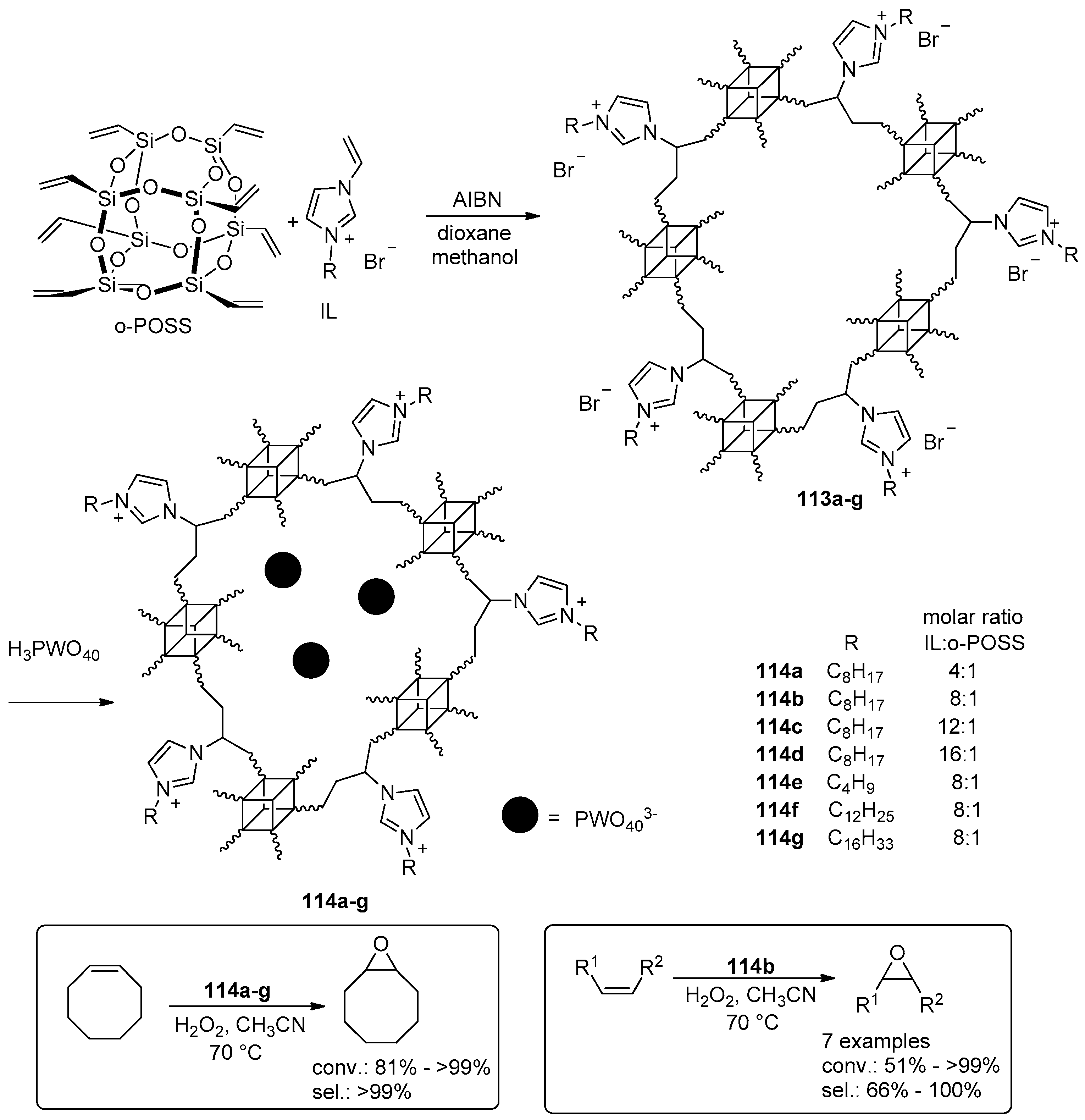
- Leng, Y.; Wu, J.; Jiang, P.; Wang, J. Amphiphilic phosphotungstate-paired ionic copolymer as a highly efficient catalyst for triphase epoxidation of alkenes with H2O2. Catal. Sci. Technol. 2014, 4, 1293–1300. [Google Scholar] [CrossRef]
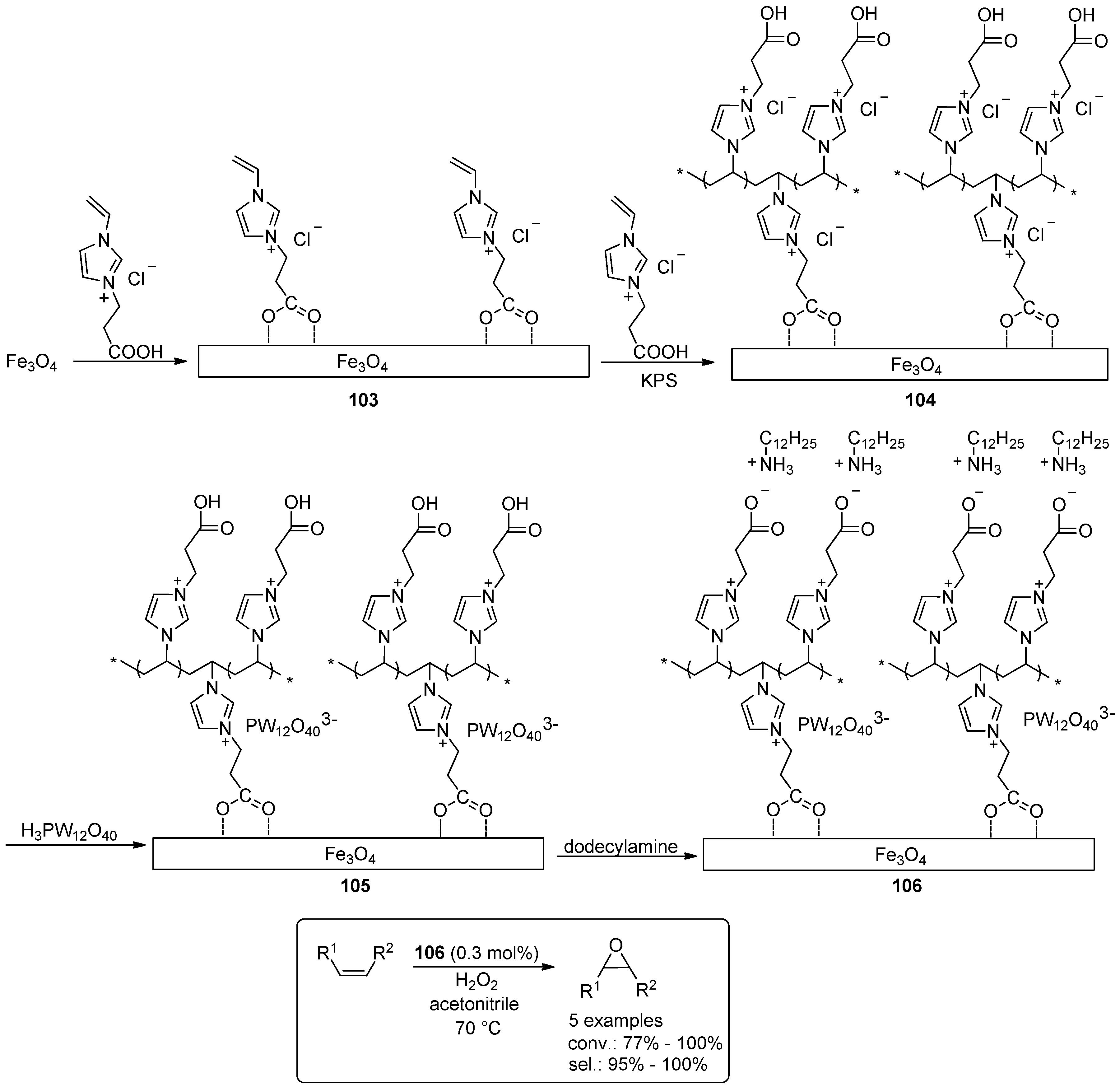
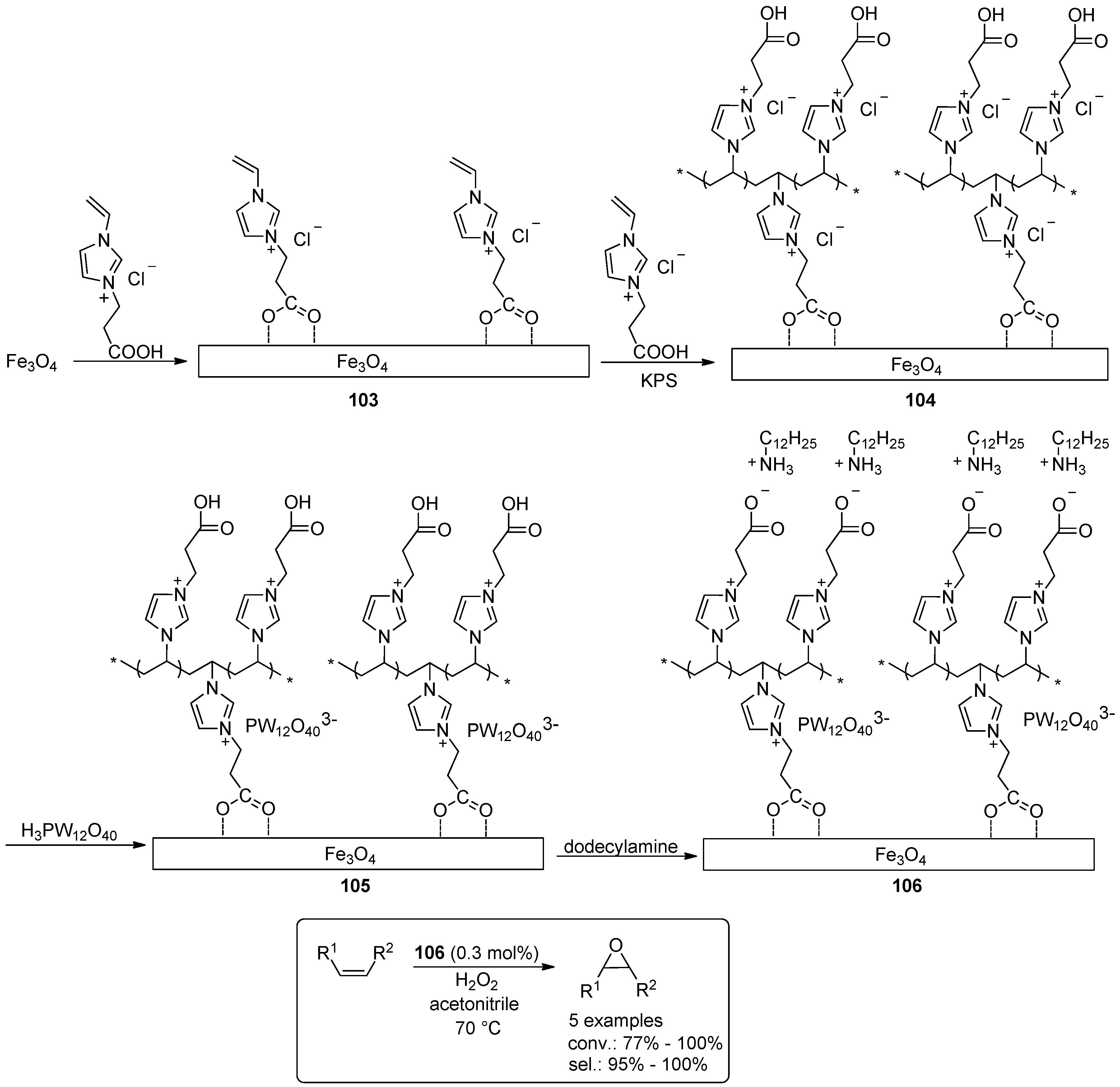
- Leng, Y.; Zhao, J.; Jiang, P.; Wang, J. Amphiphilic Polyoxometalate-Paired Polymer Coated Fe3O4: Magnetically Recyclable Catalyst for Epoxidation of Bio-Derived Olefins with H2O2. ACS Appl. Mater. Interfaces 2014, 6, 5947–5954. [Google Scholar] [CrossRef] [PubMed]
- Seino, M.; Wang, W.; Lofgreen, J.E.; Puzzo, D.P.; Manabe, T.; Ozin, G.A. Low-k Periodic Mesoporous Organosilica with Air Walls: POSS-PMO. J. Am. Chem. Soc. 2011, 133, 18082–18085. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Ou, J.; Zhang, Z.; Dong, J.; Zou, H. Ring-opening polymerization reaction of polyhedral oligomeric silsesquioxanes (POSSs) for preparation of well-controlled 3D skeletal hybrid monoliths. Chem. Commun. 2013, 49, 231–233. [Google Scholar] [CrossRef] [PubMed]
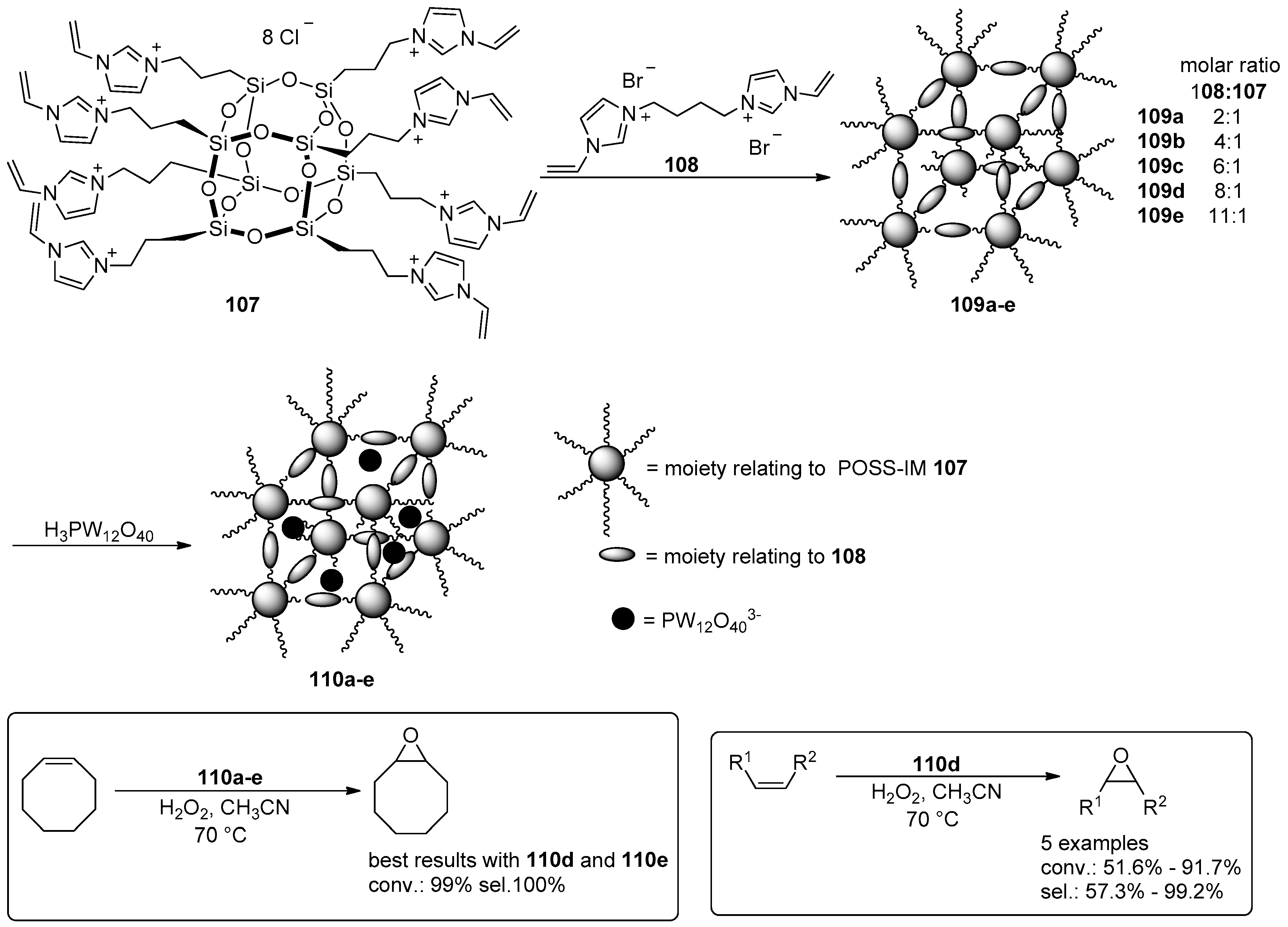
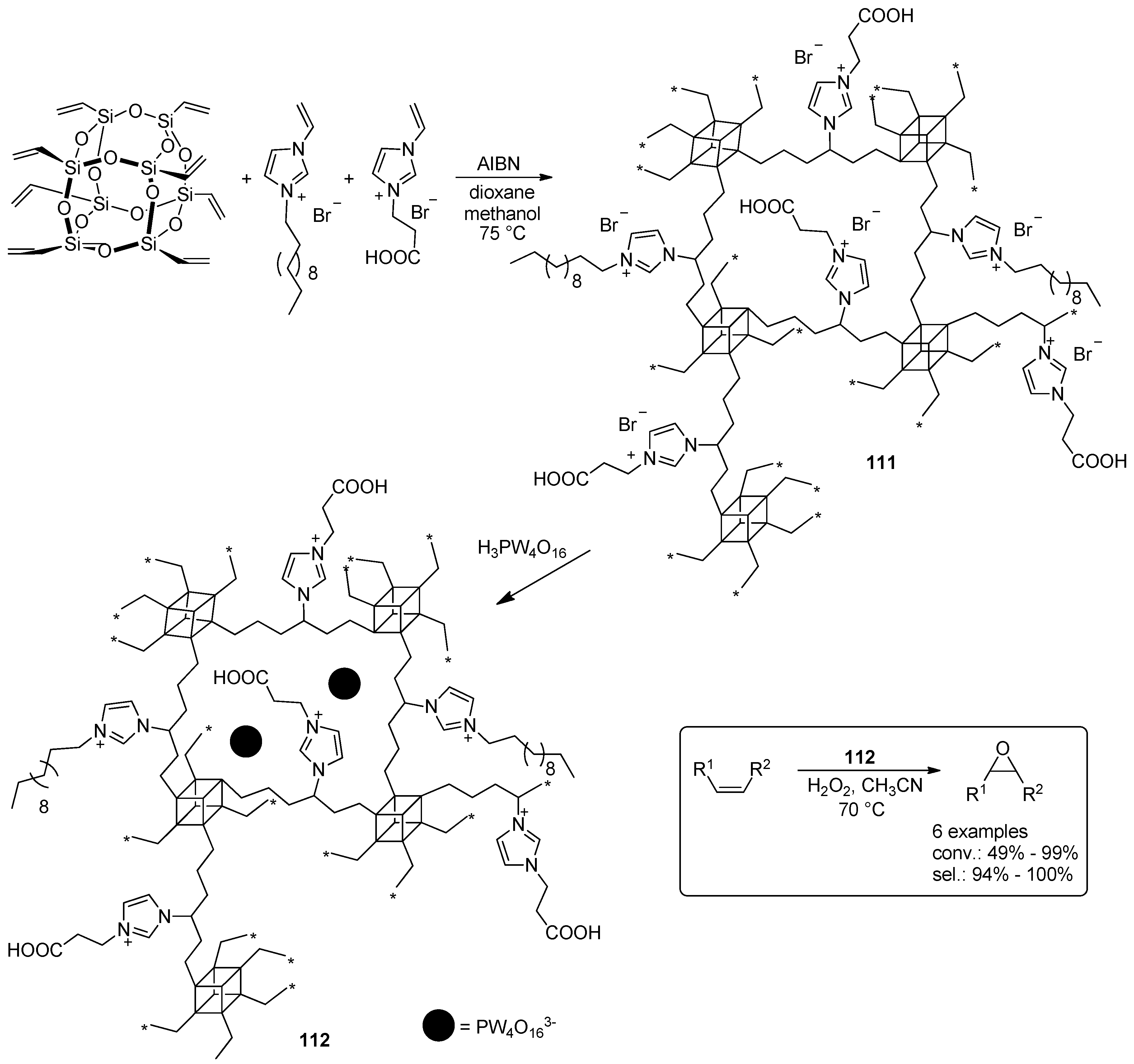
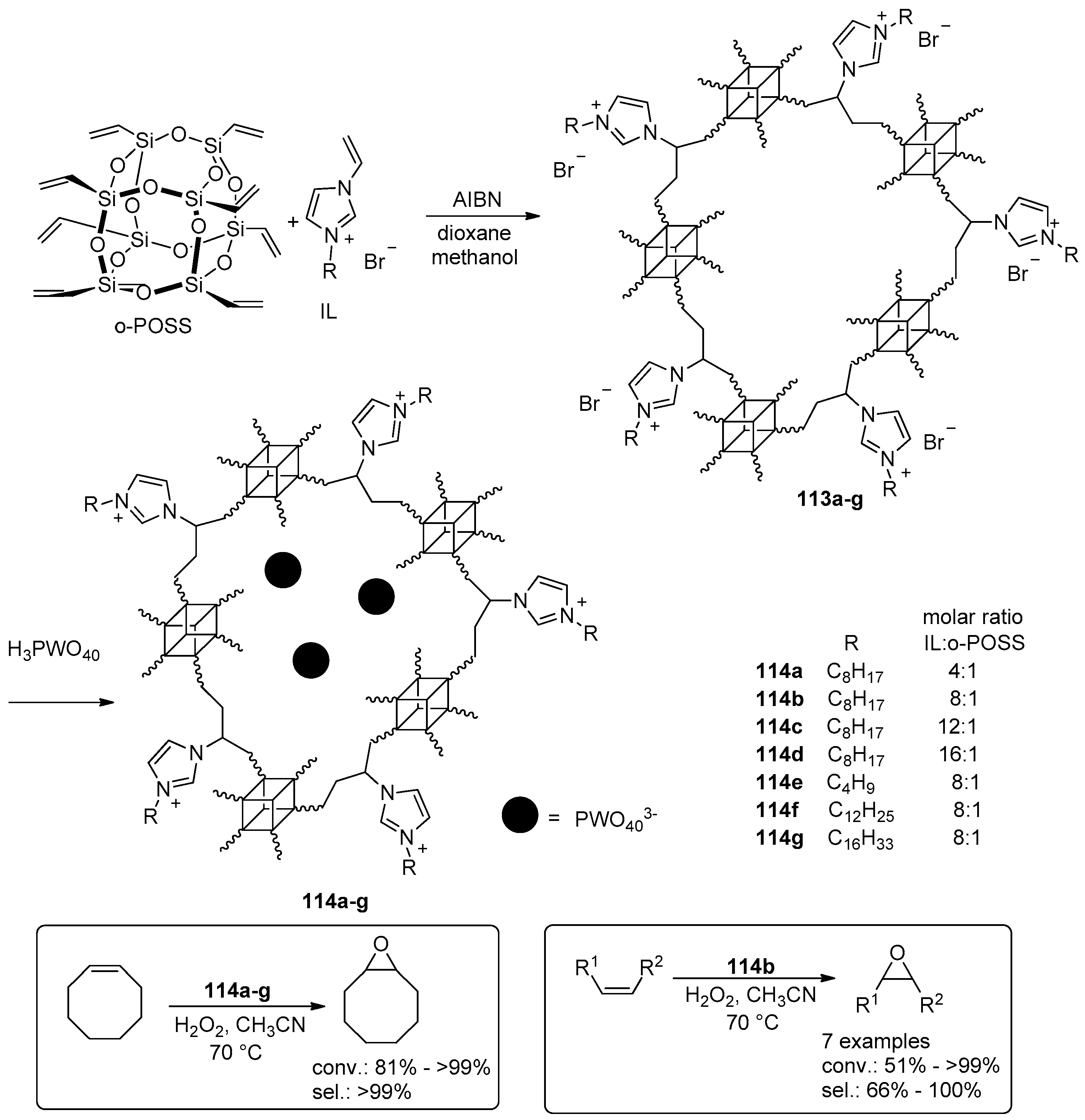
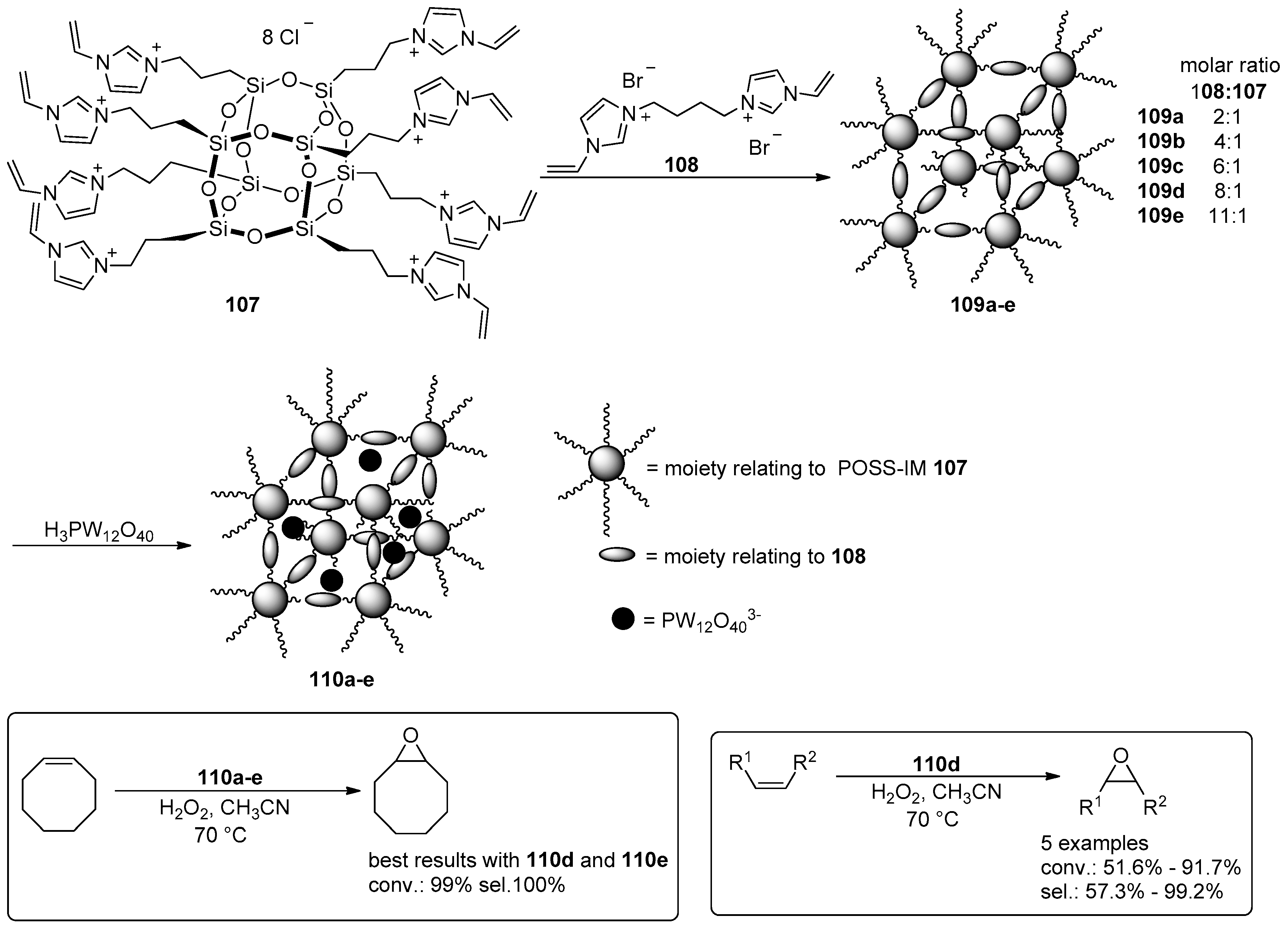
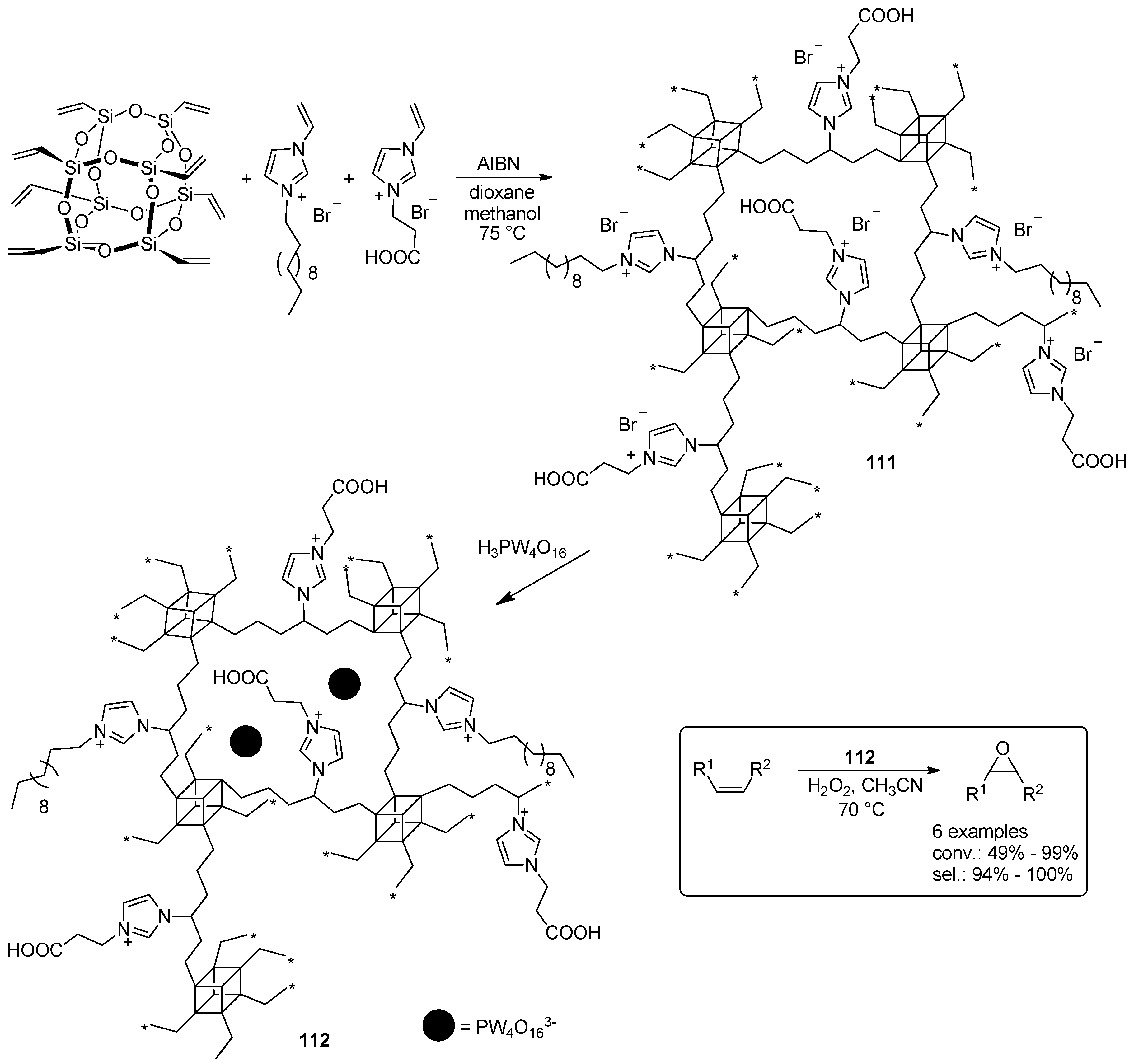
- Leng, Y.; Liu, J.; Jiang, P.; Wang, J. POSS-Derived Mesostructured Amphiphilic Polyoxometalate-based Ionic Hybrids as Highly Efficient Epoxidation Catalysts. ACS Sustain. Chem. Eng. 2015, 3, 170–176. [Google Scholar] [CrossRef]
- Leng, Y.; Zhao, J.; Jiang, P.; Wang, J. Amphiphilic porous polyhedral oligomeric silsesquioxanes (POSS) incorporated polyoxometalate-paired polymeric hybrids: Interfacial catalysts for epoxidation reactions. RSC Adv. 2015, 5, 17709–17715. [Google Scholar] [CrossRef]
- Zhao, J.; Leng, Y.; Jiang, P.; Wang, J.; Zhang, C. POSS-derived mesoporous ionic copolymer-polyoxometalate catalysts with a surfactant function for epoxidation reactions. New J. Chem. 2016, 40, 1022–1028. [Google Scholar] [CrossRef]
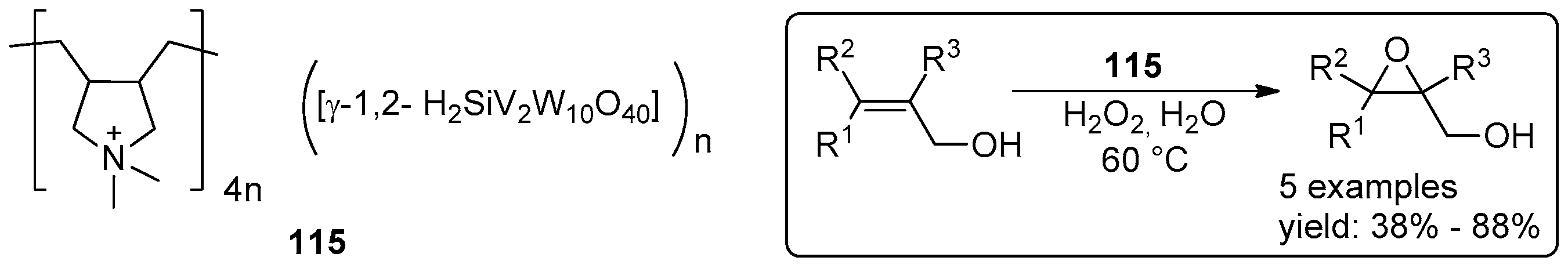
- Zhao, W.; Yang, C.; Cheng, Z.; Zhang, Z. A reusable catalytic system for sulfide oxidation and epoxidation of allylic alcohols in water catalyzed by poly(dimethyl diallyl) ammonium/polyoxometalate. Green Chem. 2016, 18, 995–998. [Google Scholar] [CrossRef]
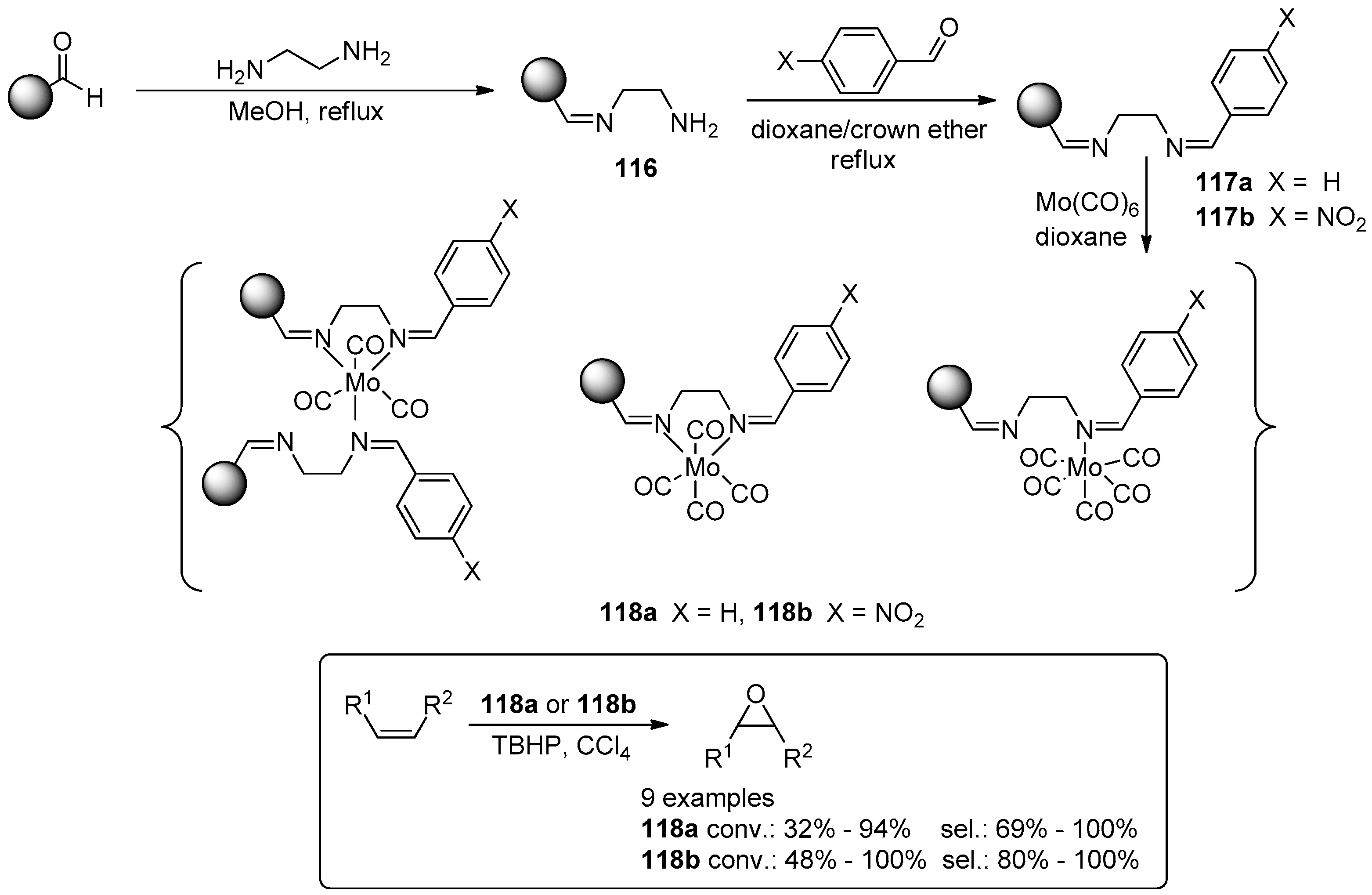
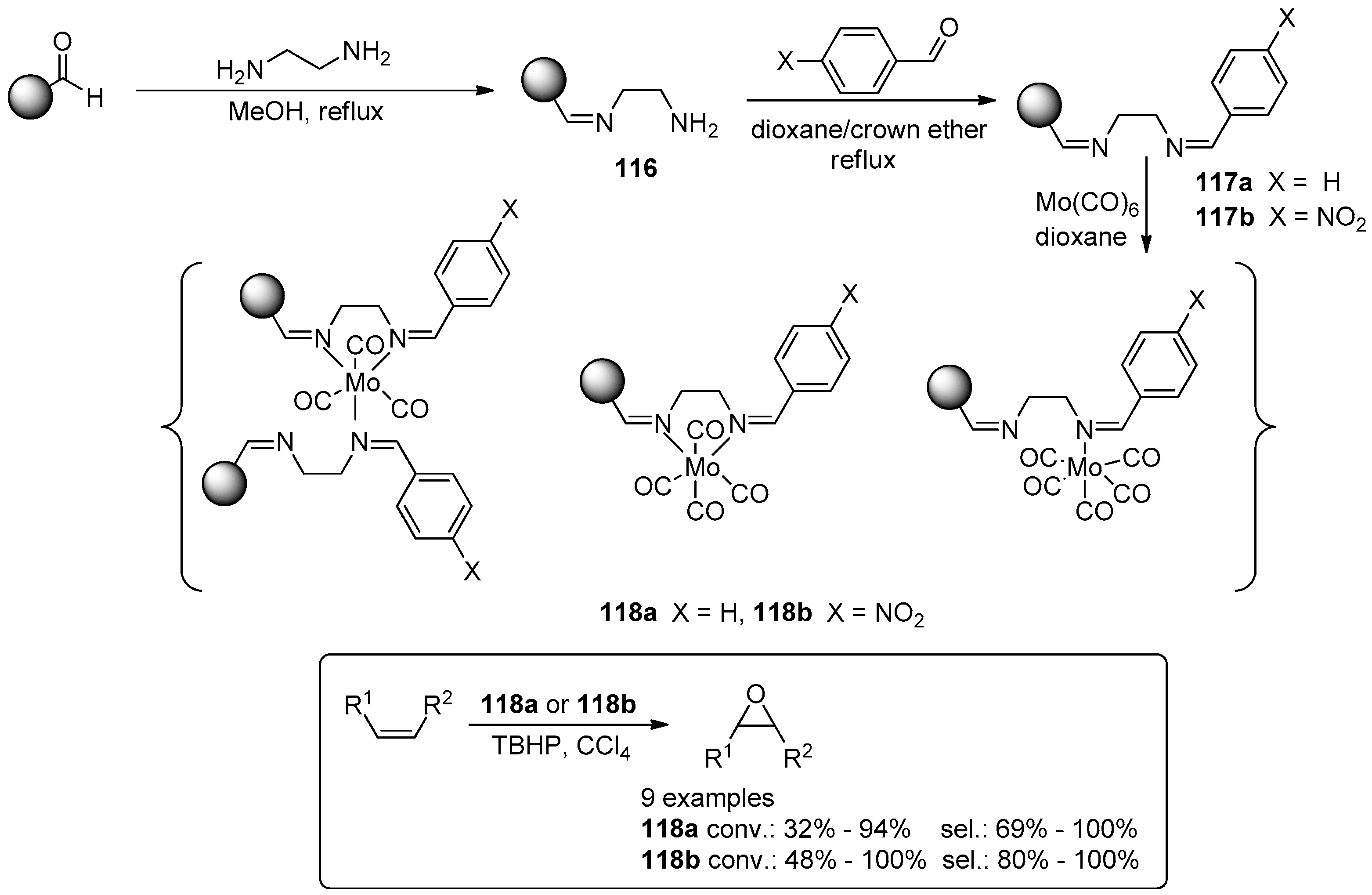
- Vaz, P.D.; Nunes, C.D. The Versatility of Immobilized Mo Complexes in Organic Transformations—Epoxidation and Metathesis Reactions. Curr. Org. Chem. 2012, 16, 89–114. [Google Scholar] [CrossRef]
- Grivani, G.; Halili, A. Polymer-supported diimine molybdenum carbonyl complexes as highly reusable and efficient pre-catalysts in epoxidation of alkenes. J. Iran. Chem. Soc. 2014, 11, 163–168. [Google Scholar] [CrossRef]
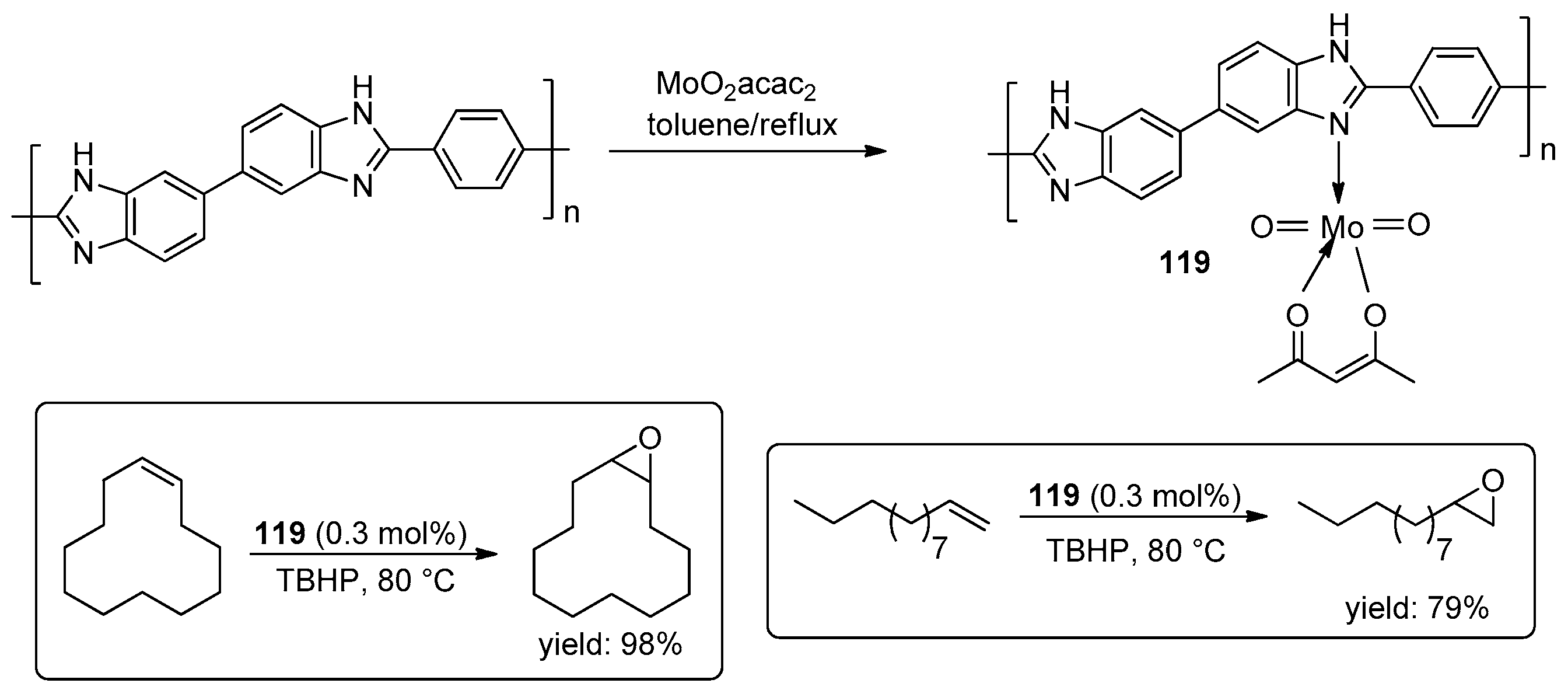
- Mbeleck, R.; Mohammed, M.L.; Ambroziak, K.; Sherrington, D.C.; Saha, B. Efficient epoxidation of cyclododecene and dodecene catalysed by polybenzimidazole supported Mo(VI) complex. Catal. Today 2015, 256, 287–293. [Google Scholar] [CrossRef]
- Zhang, W.; Jiang, P.; Wang, Y.; Zhang, J.; Zhang, P. Directing two azo-bridged covalent metalloporphyrinic polymers as highly efficient catalysts for selective oxidation. Appl. Catal. A Gen. 2015, 489, 117–122. [Google Scholar] [CrossRef]
- Fardjahromi, M.A.; Moghadam, M.; Tangestaninejad, S.; Mirkhani, V.; Mohammadpoor-Baltork, I. Manganese(iii)salophen supported on a silica containing triazine dendrimer: An efficient catalyst for epoxidation of alkenes with sodium periodate. RSC Adv. 2016, 6, 20128–20134. [Google Scholar] [CrossRef]
- Isfahani, A.L.; Mohammadpoor-Baltork, I.; Mirkhani, V.; Khosropour, A.R.; Moghadam, M.; Tangestaninejad, S.; Kia, R. Palladium Nanoparticles Immobilized on Nano-Silica Triazine Dendritic Polymer (Pdnp-nSTDP): An Efficient and Reusable Catalyst for Suzuki–Miyaura Cross-Coupling and Heck Reactions. Adv. Synth. Catal. 2013, 355, 957–972. [Google Scholar] [CrossRef]
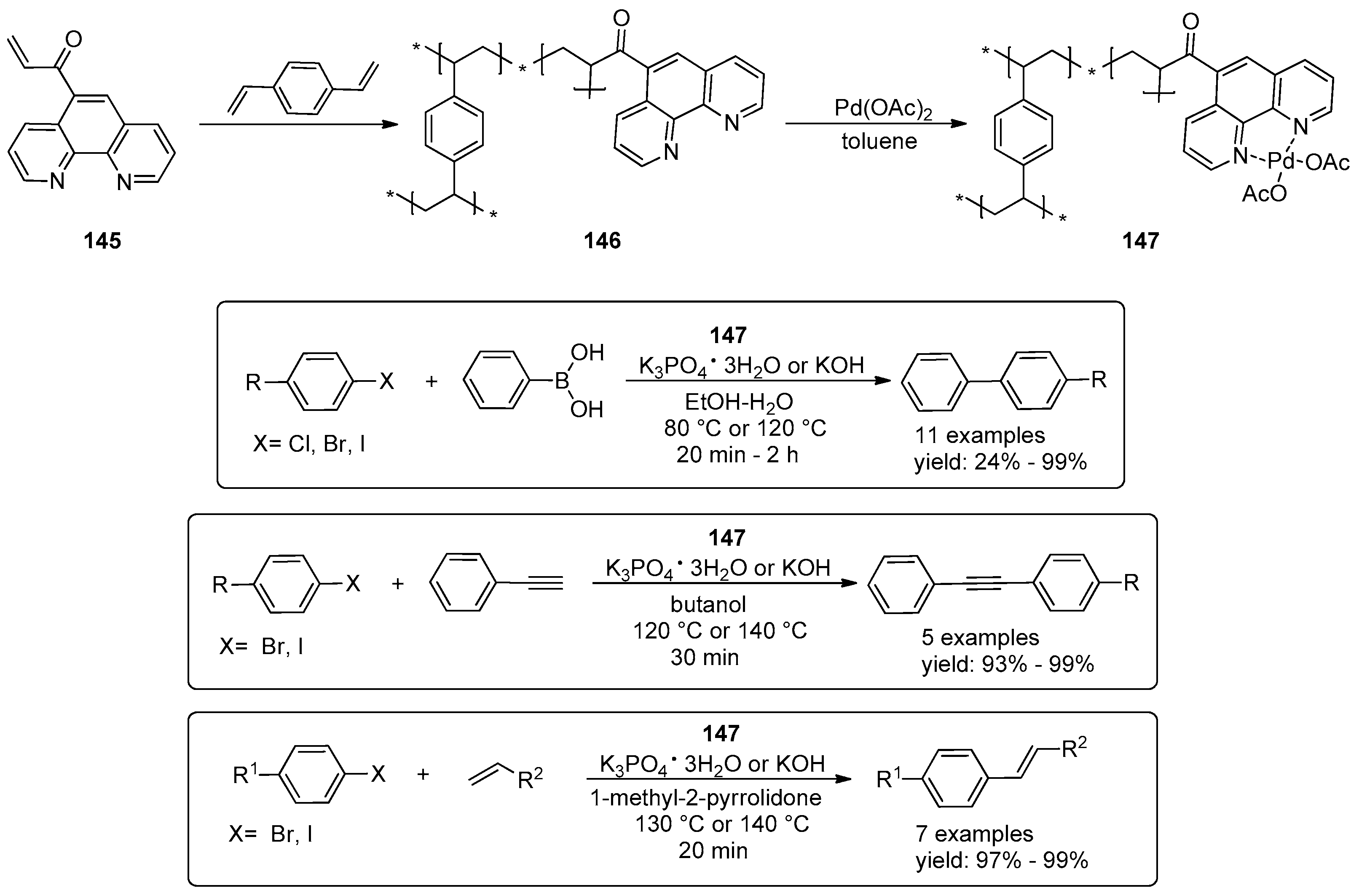
- Yang, Y.-C.; Toy, P.H. Self-Supported Ligands as a Platform for Catalysis: Use of a Polymeric Oxime in a Recyclable Palladacycle Precatalyst for Suzuki–Miyaura Reactions. Synlett 2014, 25, 1319–1324. [Google Scholar]
- Liu, X.; Zhao, X.; Lu, M. Novel polymer supported iminopyridylphosphine palladium(II) complexes: An efficient catalyst for Suzuki–Miyaura and Heck cross-coupling reactions. J. Organomet. Chem. 2014, 768, 23–27. [Google Scholar] [CrossRef]
- Chen, T.; Mao, F.; Qi, Z.; Li, Y.; Chen, R.; Wang, Y.; Huang, J. Immobilized palladium nanoparticles within polymers as active catalysts for Suzuki–Miyaura reaction. RSC Adv. 2016, 6, 16899–16903. [Google Scholar] [CrossRef]
- Tamami, B.; Farjadian, F.; Ghasemi, S.; Allahyari, H.; Mirzadeh, M. Palladium Nanoparticles Supported on Poly(N-vinylpyrrolidone)-Grafted Silica as an Efficient Catalyst for Copper-Free Sonogashira and Suzuki Cross-Coupling Reactions. J. Braz. Chem. Soc. 2015, 26, 1591–1598. [Google Scholar] [CrossRef]
- Kodicherla, B.; Perumgani, C.P.; Mandapati, M.R. A reusable polystyrene-supported copper (II) catalytic system for N-arylation of indoles and Sonogashira coupling reactions in water. Appl. Catal. A Gen. 2014, 483, 110–115. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Liotta, L.F.; Salvo, A.M.P.; Giacalone, F.; La Parola, V.; Aprile, C.; Noto, R. Multi-Layered, Covalently Supported Ionic Liquid Phase (mlc-SILP) as Highly Cross-Linked Support for Recyclable Palladium Catalysts for the Suzuki Reaction in Aqueous Medium. Adv. Synth. Catal. 2011, 353, 2119–2130. [Google Scholar] [CrossRef]
- Pavia, C.; Giacalone, F.; Bivona, L.A.; Salvo, A.M.P.; Petrucci, C.; Strappaveccia, G.; Vaccaro, L.; Aprile, C.; Gruttadauria, M. Evidences of release and catch mechanism in the Heck reaction catalyzed by palladium immobilized on highly cross-linked-supported imidazolium salts. J. Mol. Catal. A Chem. 2014, 387, 57–62. [Google Scholar] [CrossRef]
- Buscemi, R.; Giacalone, F.; Orecchio, S.; Gruttadauria, M. Cross-Linked Imidazolium Salts as Scavengers for Palladium. ChemPlusChem 2014, 79, 421–426. [Google Scholar] [CrossRef]
- Petrucci, C.; Strappaveccia, G.; Giacalone, F.; Gruttadauria, M.; Pizzo, F.; Vaccaro, L. An E-Factor Minimized Protocol for a Sustainable and Efficient Heck Reaction in Flow. ACS Sustain. Chem. Eng. 2014, 2, 2813–2819. [Google Scholar] [CrossRef]
- Bivona, L.A.; Giacalone, F.; Vaccaro, L.; Aprile, C.; Gruttadauria, M. Cross-Linked Thiazolidine Network as Support for Palladium: A New Catalyst for Suzuki and Heck Reactions. ChemCatChem 2015, 7, 2526–2533. [Google Scholar] [CrossRef]
- Bukowska, A.; Bukowski, W.; Bester, K.; Flaga, S. Linkage of the PAMAM type dendrimer with the gel type resin based on glycidyl methacrylate terpolymer as a method of preparation of the polymer support for the recyclable palladium catalyst for Suzuki–Miyaura cross-coupling reactions. RSC Adv. 2015, 5, 49036–49044. [Google Scholar] [CrossRef]
- Tang, M.X.; Redemann, C.T.; Szoka, F.C. In Vitro Gene Delivery by Degraded Polyamidoamine Dendrimers. Bioconjug. Chem. 1996, 7, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Giacalone, F.; Campisciano, V.; Calabrese, C.; La Parola, V.; Syrgiannis, Z.; Prato, M.; Gruttadauria, M. Single-Walled Carbon Nanotube–Polyamidoamine Dendrimer Hybrids for Heterogeneous Catalysis. ACS Nano 2016, 10, 4627–4636. [Google Scholar] [CrossRef] [PubMed]
- Gruttadauria, M.; Giacalone, F.; Noto, R. "Release and catch" catalytic systems. Green Chem. 2013, 15, 2608–2618. [Google Scholar] [CrossRef]
- Dai, Z.; Chen, F.; Sun, Q.; Ji, Y.; Wang, L.; Meng, X.; Xiao, F.-S. A Pd-metalated porous organic polymer as a highly efficient heterogeneous catalyst for C–C couplings. Chin. J. Catal. 2016, 37, 54–60. [Google Scholar] [CrossRef]
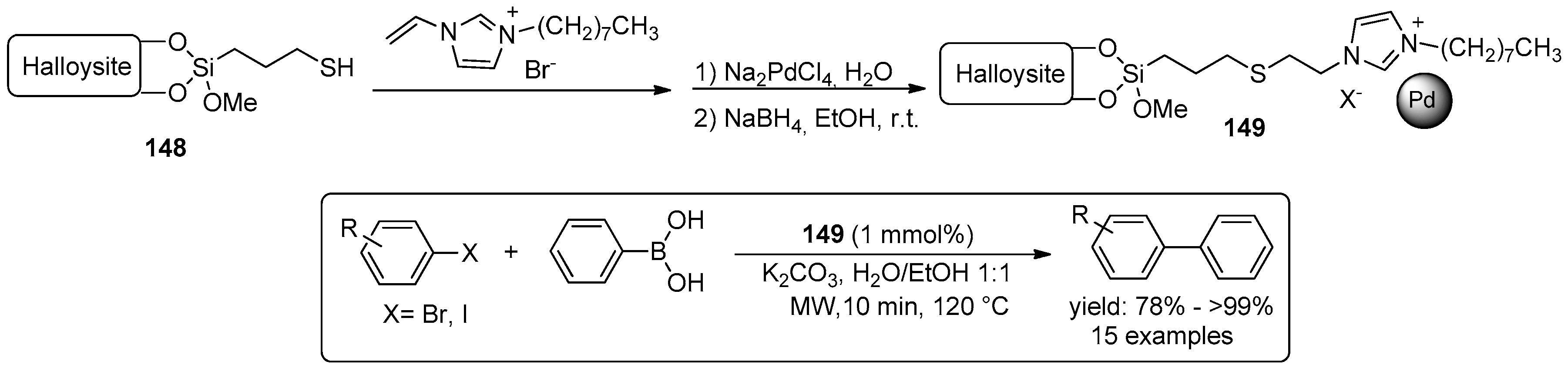
- Massaro, M.; Riela, S.; Lazzara, G.; Gruttadauria, M.; Milioto, S.; Noto, R. Green conditions for the Suzuki reaction using microwave irradiation and a new HNT-supported ionic liquid-like phase (HNT-SILLP) catalyst. Appl. Organomet. Chem. 2014, 28, 234–238. [Google Scholar] [CrossRef]
- Massaro, M.; Riela, S.; Cavallaro, G.; Gruttadauria, M.; Milioto, S.; Noto, R.; Lazzara, G. Eco-friendly functionalization of natural halloysite clay nanotube with ionic liquids by microwave irradiation for Suzuki coupling reaction. J. Organomet. Chem. 2014, 749, 410–415. [Google Scholar] [CrossRef]
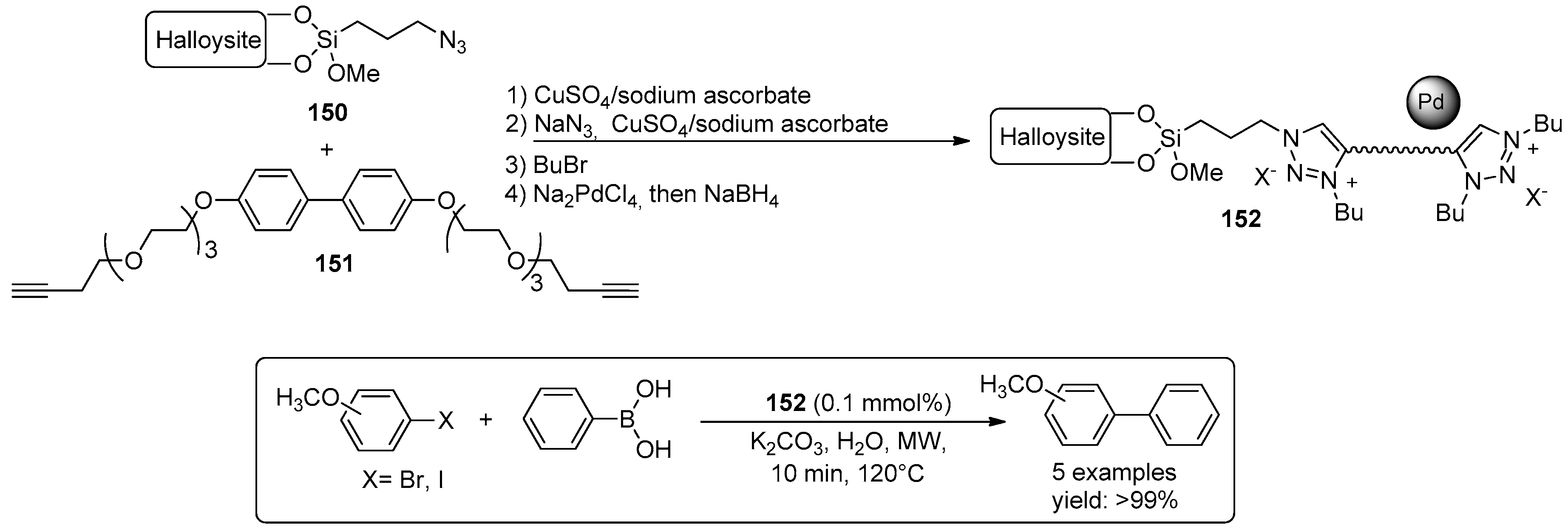
- Massaro, M.; Riela, S.; Cavallaro, G.; Colletti, C.G.; Milioto, S.; Noto, R.; Parisi, F.; Lazzara, G. Palladium supported on Halloysite-triazolium salts as catalyst for ligand free Suzuki cross-coupling in water under microwave irradiation. J. Mol. Catal. A Chem. 2015, 408, 12–19. [Google Scholar] [CrossRef]
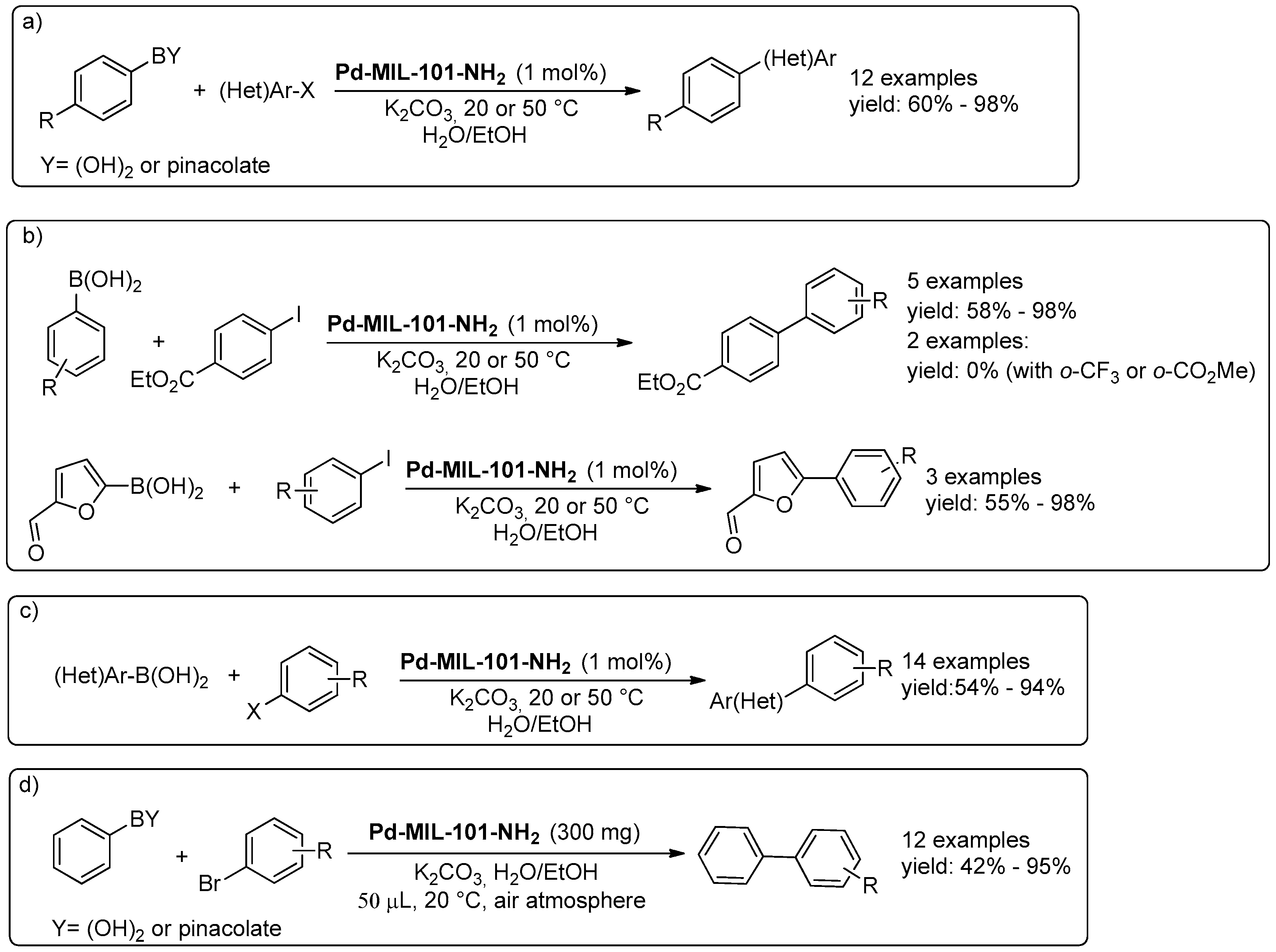
- Pascanu, V.; Hansen, P.R.; Bermejo Gómez, A.; Ayats, C.; Platero-Prats, A.E.; Johansson, M.J.; Pericàs, M.À.; Martín-Matute, B. Highly Functionalized Biaryls via Suzuki–Miyaura Cross-Coupling Catalyzed by Pd@MOF under Batch and Continuous Flow Regimes. ChemSusChem 2015, 8, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Férey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surblé, S.; Margiolaki, I. A Chromium Terephthalate-Based Solid with Unusually Large Pore Volumes and Surface Area. Science 2005, 309, 2040–2042. [Google Scholar] [CrossRef] [PubMed]
- Bernt, S.; Guillerm, V.; Serre, C.; Stock, N. Direct covalent post-synthetic chemical modification of Cr-MIL-101 using nitrating acid. Chem. Commun. 2011, 47, 2838–2840. [Google Scholar] [CrossRef] [PubMed]
- Pascanu, V.; Yao, Q.; Bermejo Gómez, A.; Gustafsson, M.; Yun, Y.; Wan, W.; Samain, L.; Zou, X.; Martín-Matute, B. Sustainable Catalysis: Rational Pd Loading on MIL-101Cr-NH2 for More Efficient and Recyclable Suzuki–Miyaura Reactions. Chem. Eur. J. 2013, 19, 17483–17493. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Lu, D.; Zhang, C.; Jiang, P.; Zhang, W.; Wang, J. Ionic Polymer Microspheres Bearing a CoIII–Salen Moiety as a Bifunctional Heterogeneous Catalyst for the Efficient Cycloaddition of CO2 and Epoxides. Chem. Eur. J. 2016, 22, 8368–8375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Qiao, Z.-A.; Jiang, X.; Veith, G.M.; Dai, S. Nanoporous Ionic Organic Networks: Stabilizing and Supporting Gold Nanoparticles for Catalysis. Nano Lett. 2015, 15, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, J.; Porcar, R.; Lozano, P.; Burguete, M.I.; García-Verdugo, E.; Luis, S.V. Microwave-Assisted Selective Oxidation of 1-Phenyl Ethanol in Water Catalyzed by Metal Nanoparticles Immobilized onto Supported Ionic Liquidlike Phases. ACS Catal. 2015, 5, 4743–4750. [Google Scholar] [CrossRef]
- Isabel Burguete, M.; Garcia-Verdugo, E.; Luis, S.V.; Restrepo, J.A. Preparation of polymer-supported gold nanoparticles based on resins containing ionic liquid-like fragments: Easy control of size and stability. Phys. Chem. Chem. Phys. 2011, 13, 14831–14838. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, J.; Lozano, P.; Burguete, M.I.; García-Verdugo, E.; Luis, S.V. Gold nanoparticles immobilized onto supported ionic liquid-like phases for microwave phenylethanol oxidation in water. Catal. Today 2015, 255, 97–101. [Google Scholar] [CrossRef]
- Roy, R. A Primer on the Taguchi Method; Society of Manufacturing Engineers: Dearborn, MI, USA, 1990. [Google Scholar]
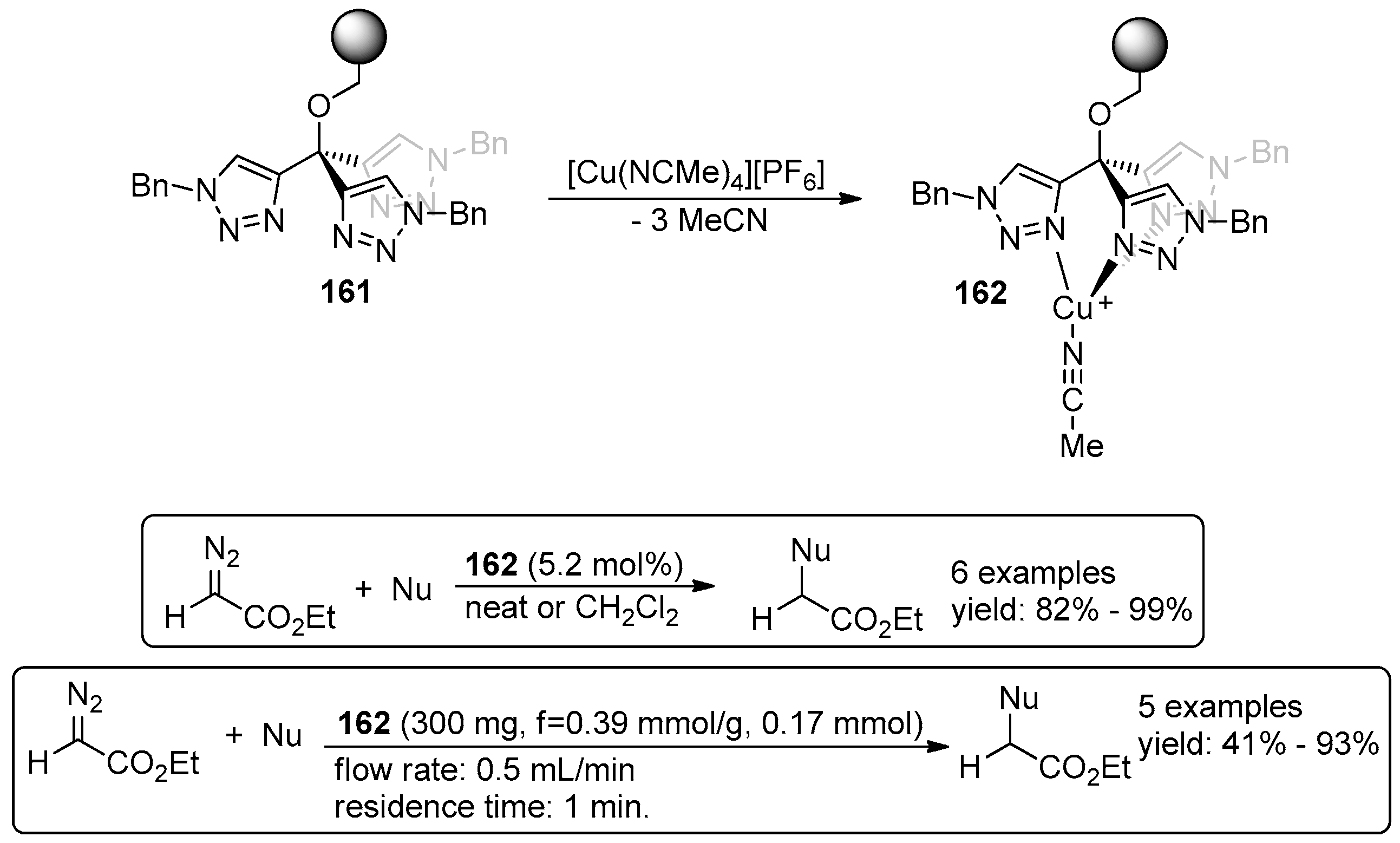
- Maestre, L.; Ozkal, E.; Ayats, C.; Beltran, A.; Diaz-Requejo, M.M.; Perez, P.J.; Pericas, M.A. A fully recyclable heterogenized Cu catalyst for the general carbene transfer reaction in batch and flow. Chem. Sci. 2015, 6, 1510–1515. [Google Scholar] [CrossRef]
- Yadav, J.; Stanton, G.R.; Fan, X.; Robinson, J.R.; Schelter, E.J.; Walsh, P.J.; Pericas, M.A. Asymmetric Allylation of Ketones and Subsequent Tandem Reactions Catalyzed by a Novel Polymer-Supported Titanium–BINOLate Complex. Chem. Eur. J. 2014, 20, 7122–7127. [Google Scholar] [CrossRef] [PubMed]
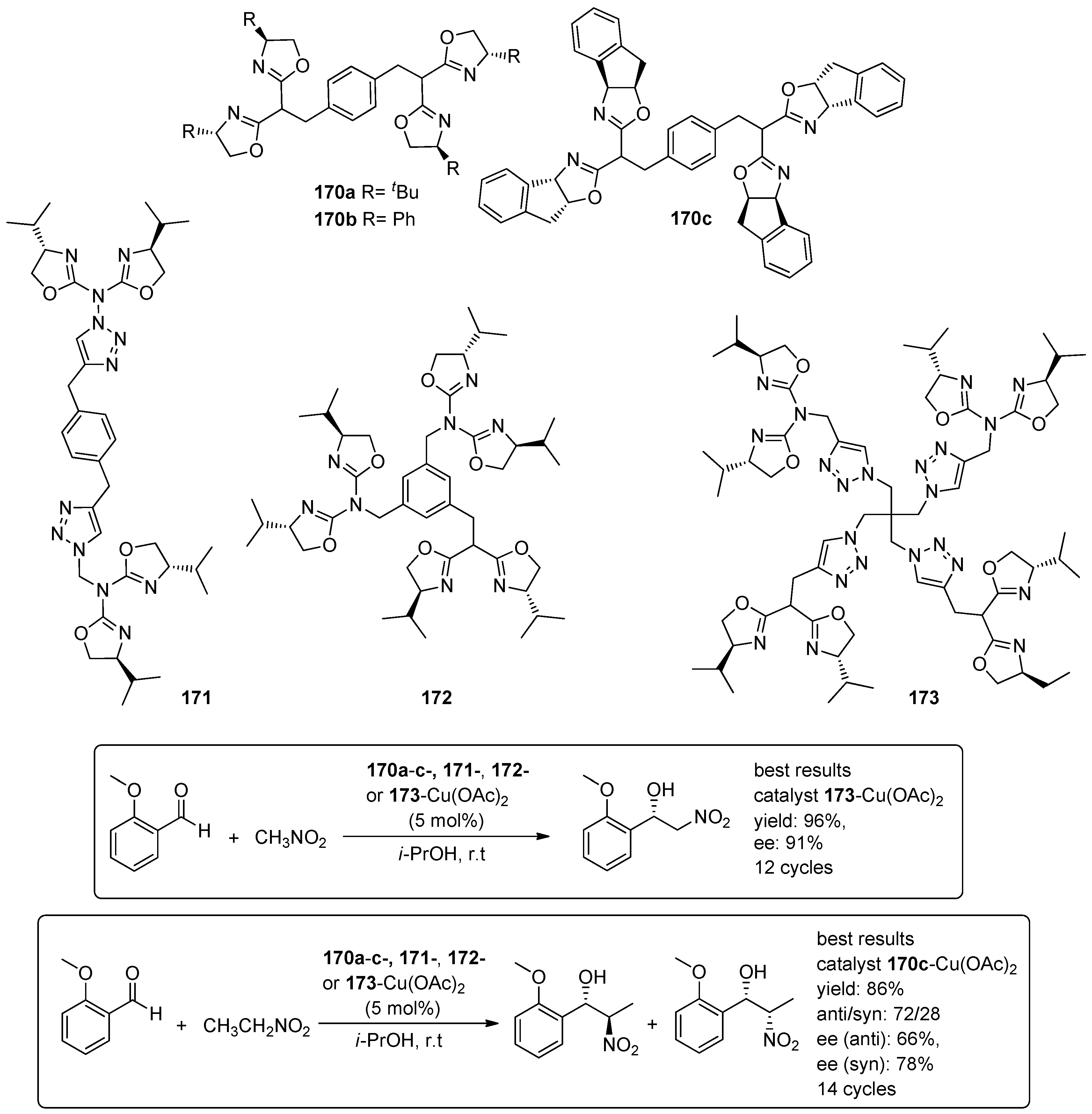
- Angulo, B.; Garcia, J.I.; Herrerias, C.I.; Mayoral, J.A.; Minana, A.C. Polytopic bis(oxazoline)-based ligands for recoverable catalytic systems applied to the enantioselective Henry reaction. Org. Biomol. Chem. 2015, 13, 9314–9322. [Google Scholar] [CrossRef] [PubMed]
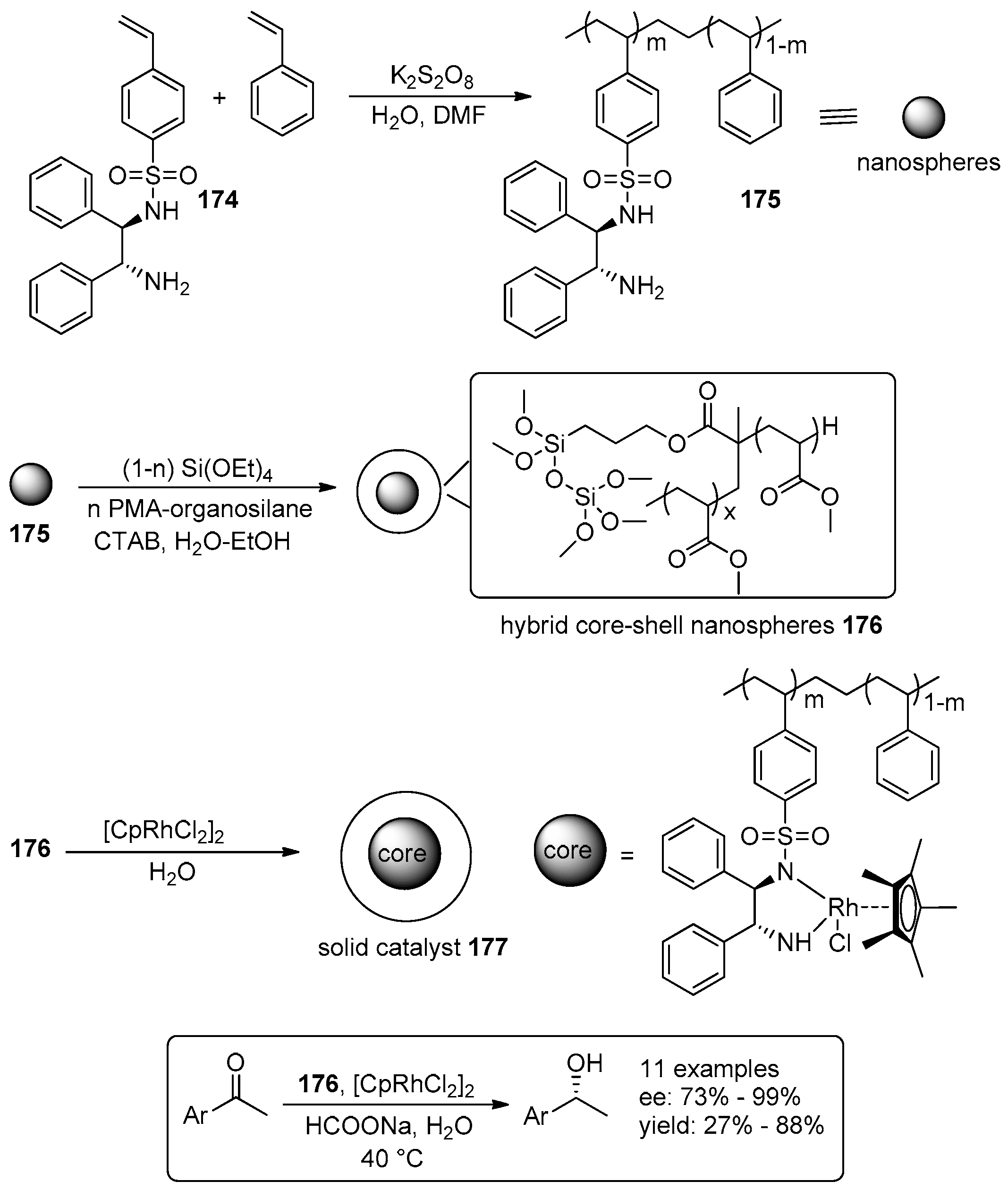
- Wei, J.; Zhang, X.; Zhang, X.; Zhao, Y.; Li, R.; Yang, Q. Facile Synthesis of Hybrid Core–Shell Nanospheres for the Asymmetric Transfer Hydrogenation of Aromatic Ketones. ChemCatChem 2014, 6, 1368–1374. [Google Scholar]
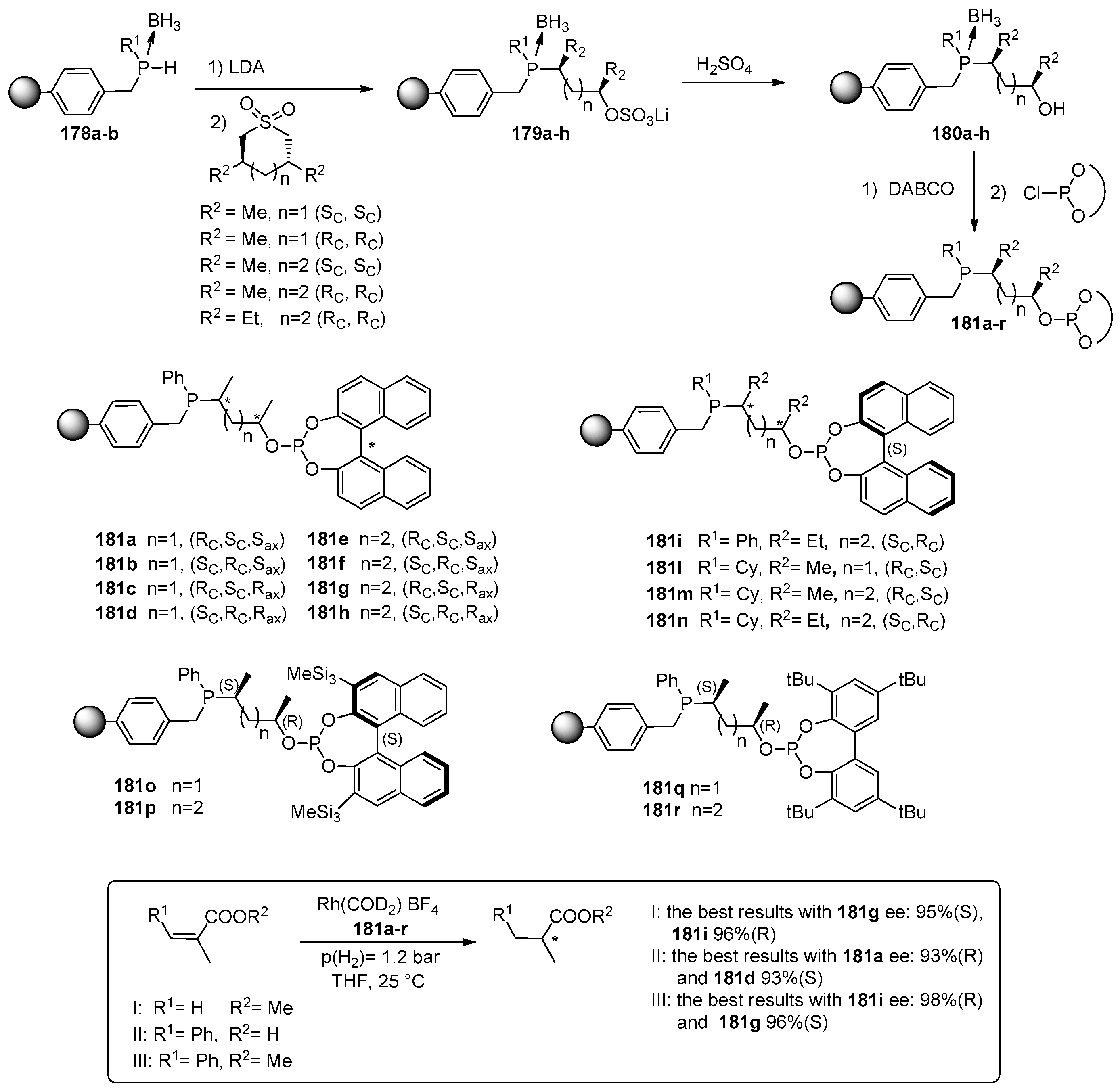
- Heutz, F.J.L.; Kamer, P.C.J. Modular solid-phase synthesis, catalytic application and efficient recycling of supported phosphine–phosphite ligand libraries. Dalton Trans. 2016, 45, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
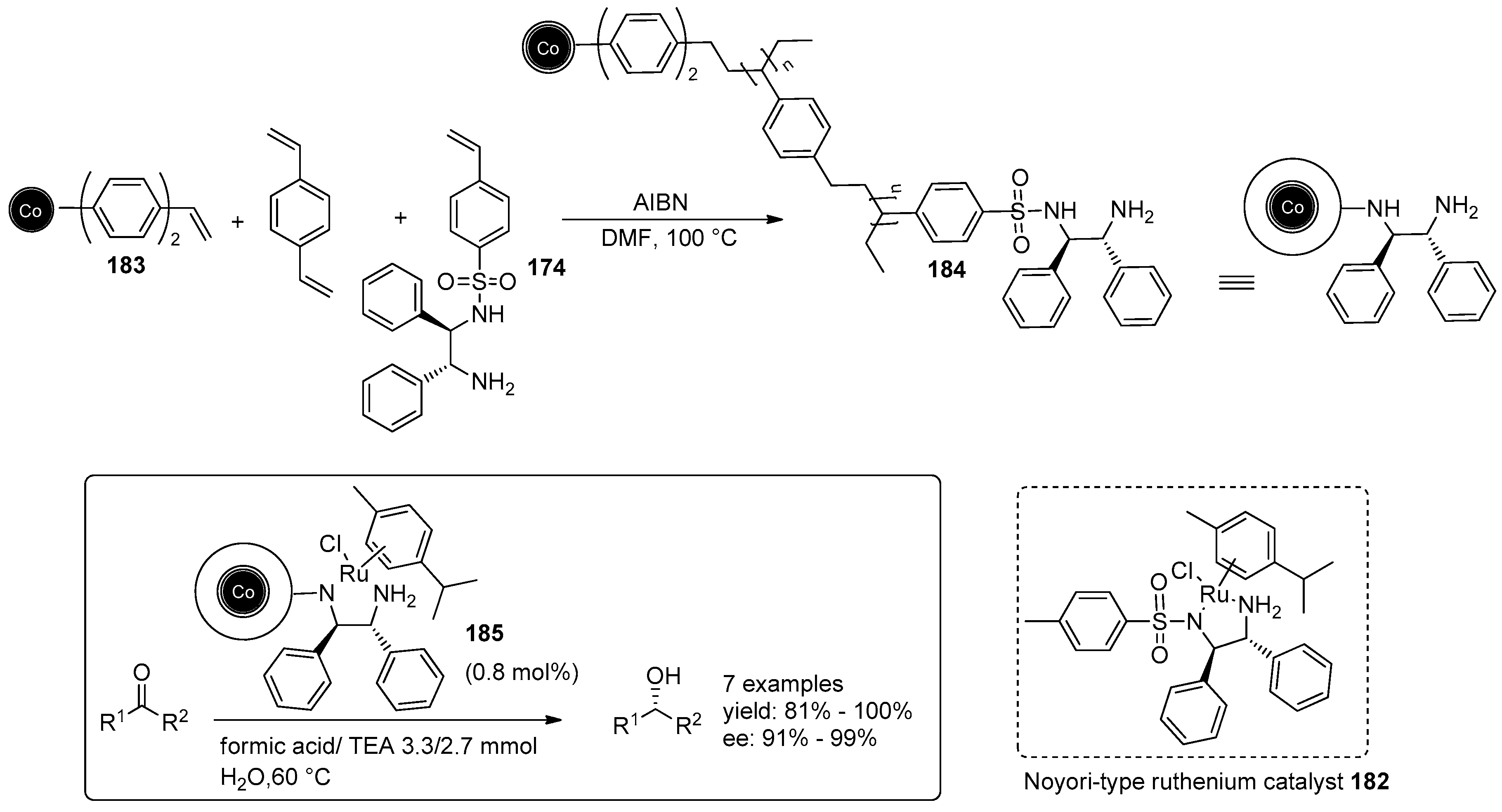
- Eichenseer, C.M.; Kastl, B.; Pericàs, M.A.; Hanson, P.R.; Reiser, O. Synthesis and Application of Magnetic Noyori-Type Ruthenium Catalysts for Asymmetric Transfer Hydrogenation Reactions in Water. ACS Sustain. Chem. Eng. 2016, 4, 2698–2705. [Google Scholar] [CrossRef]

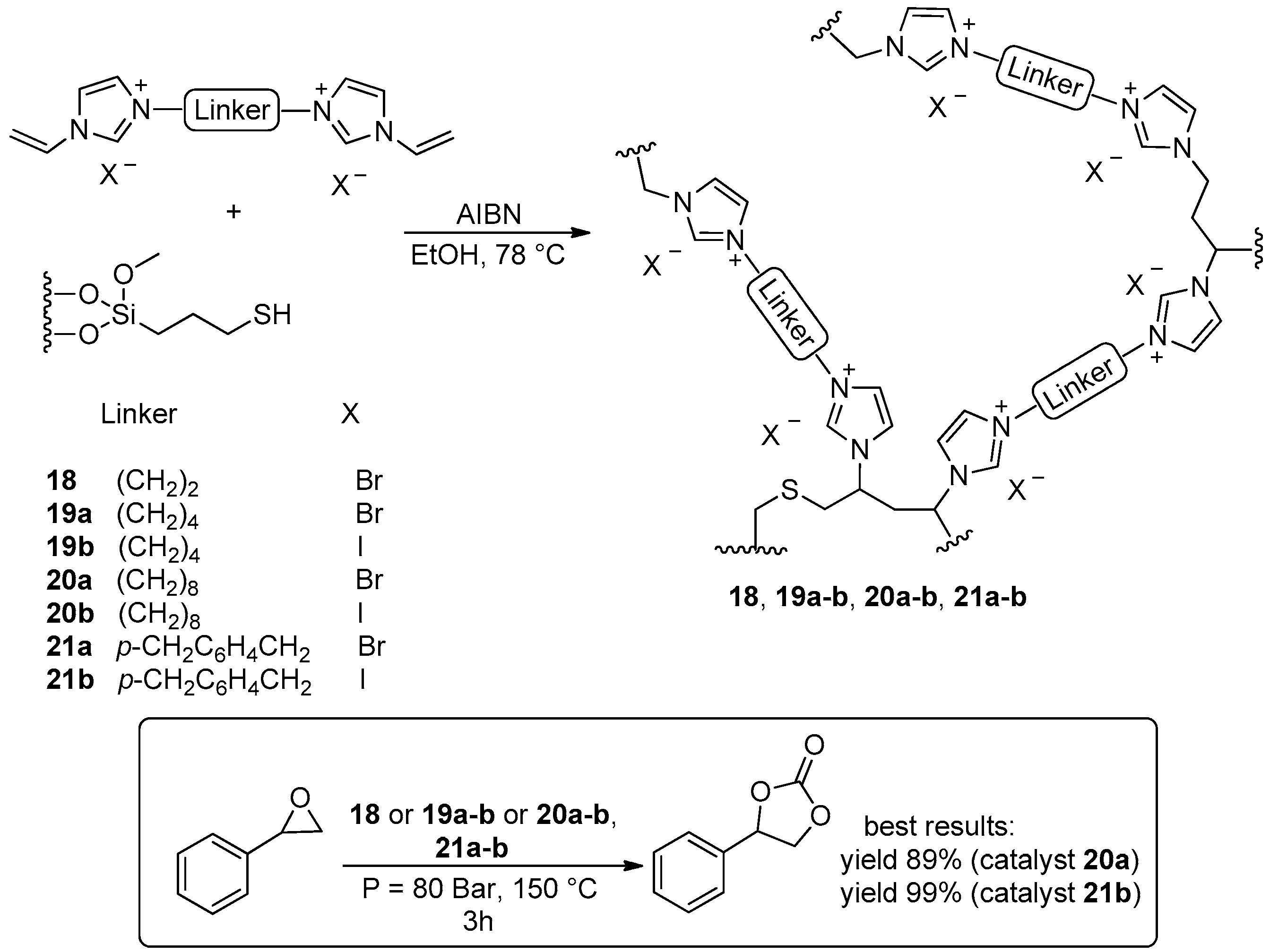


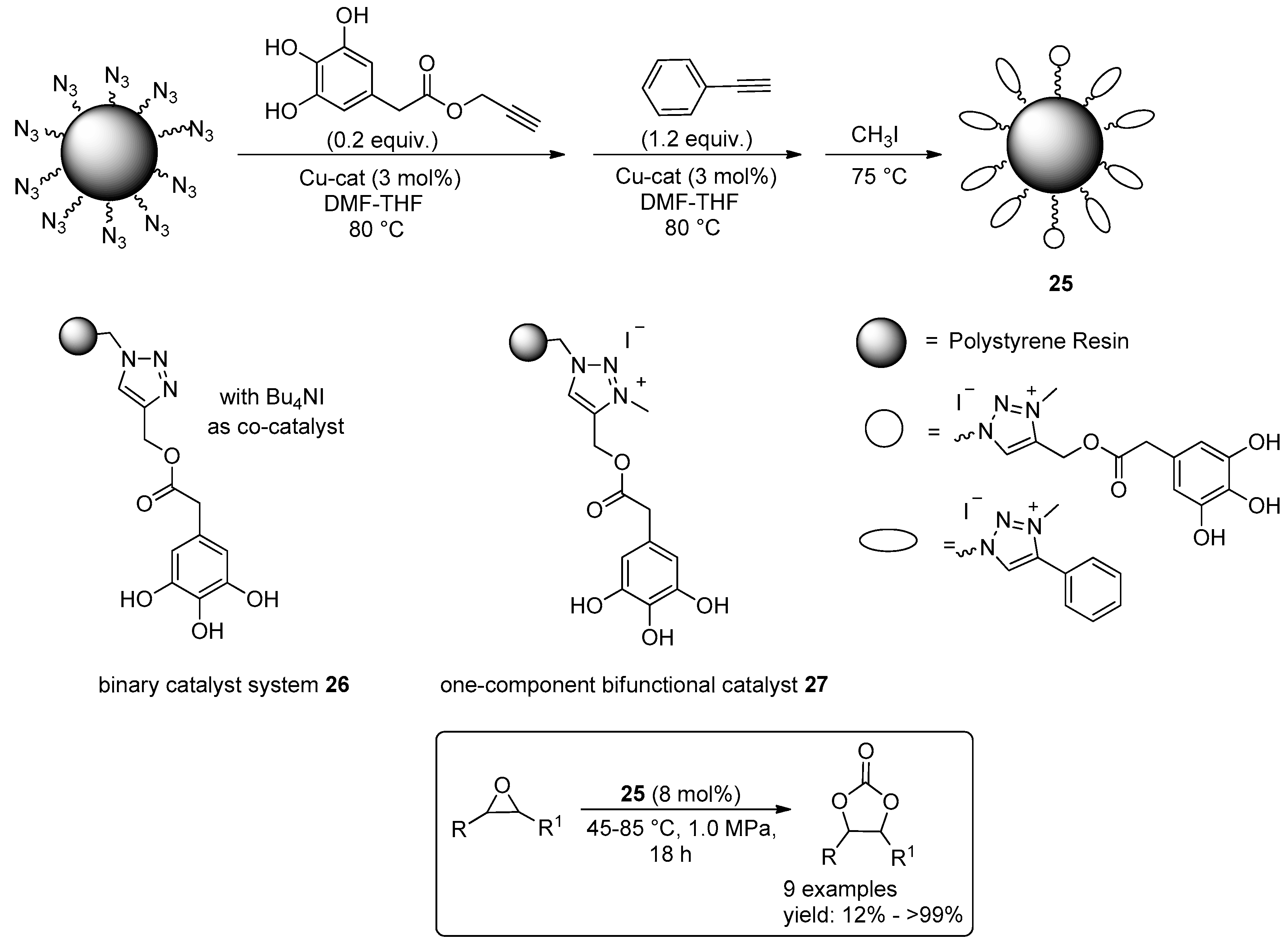
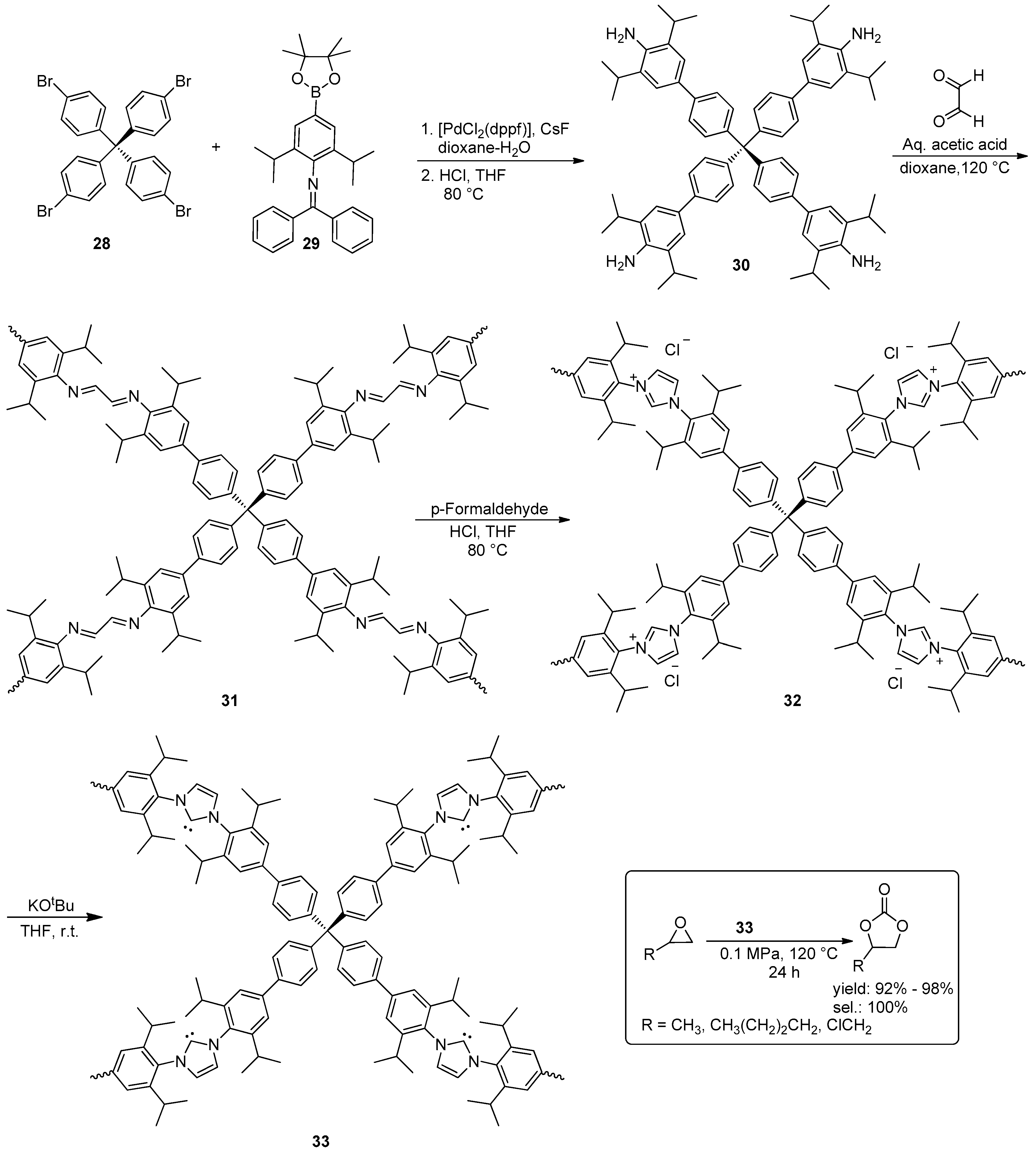
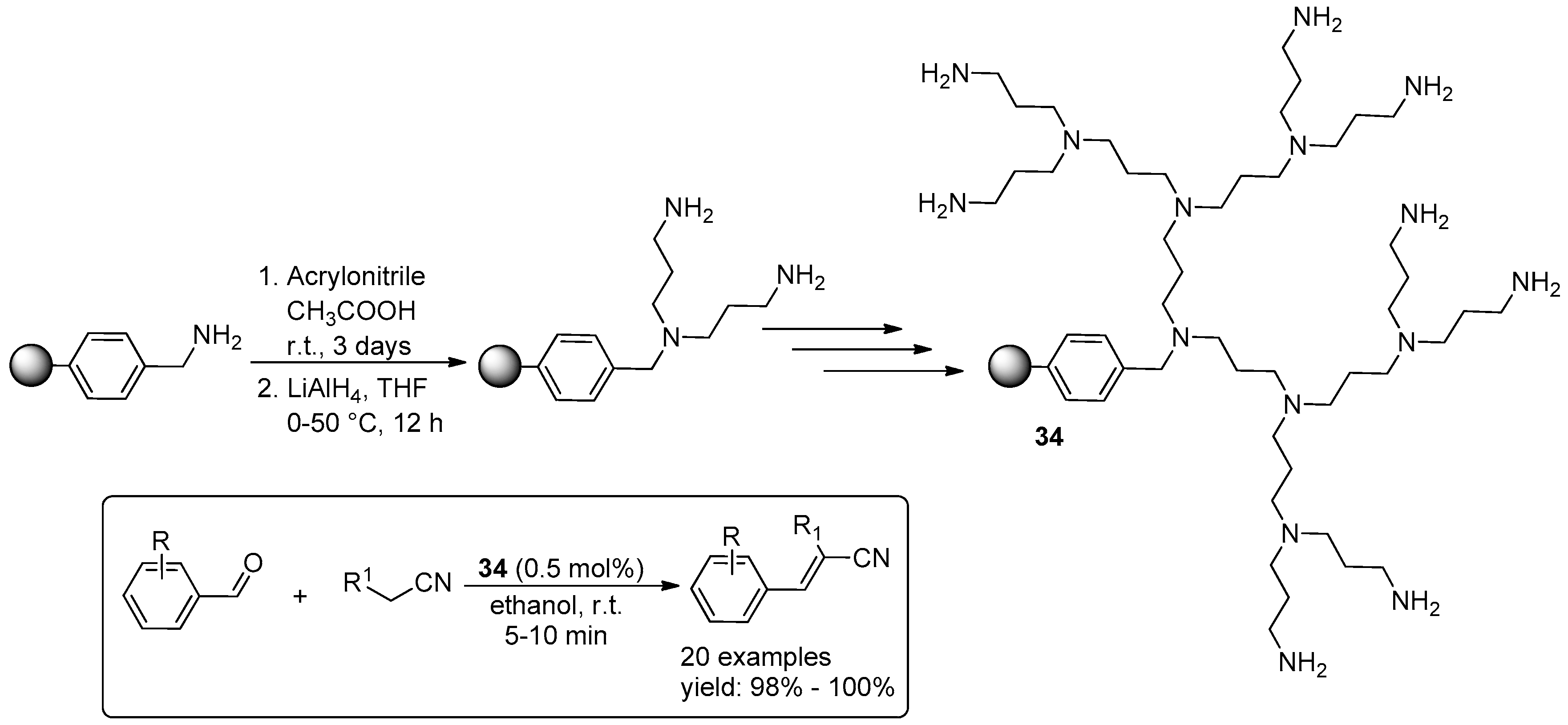
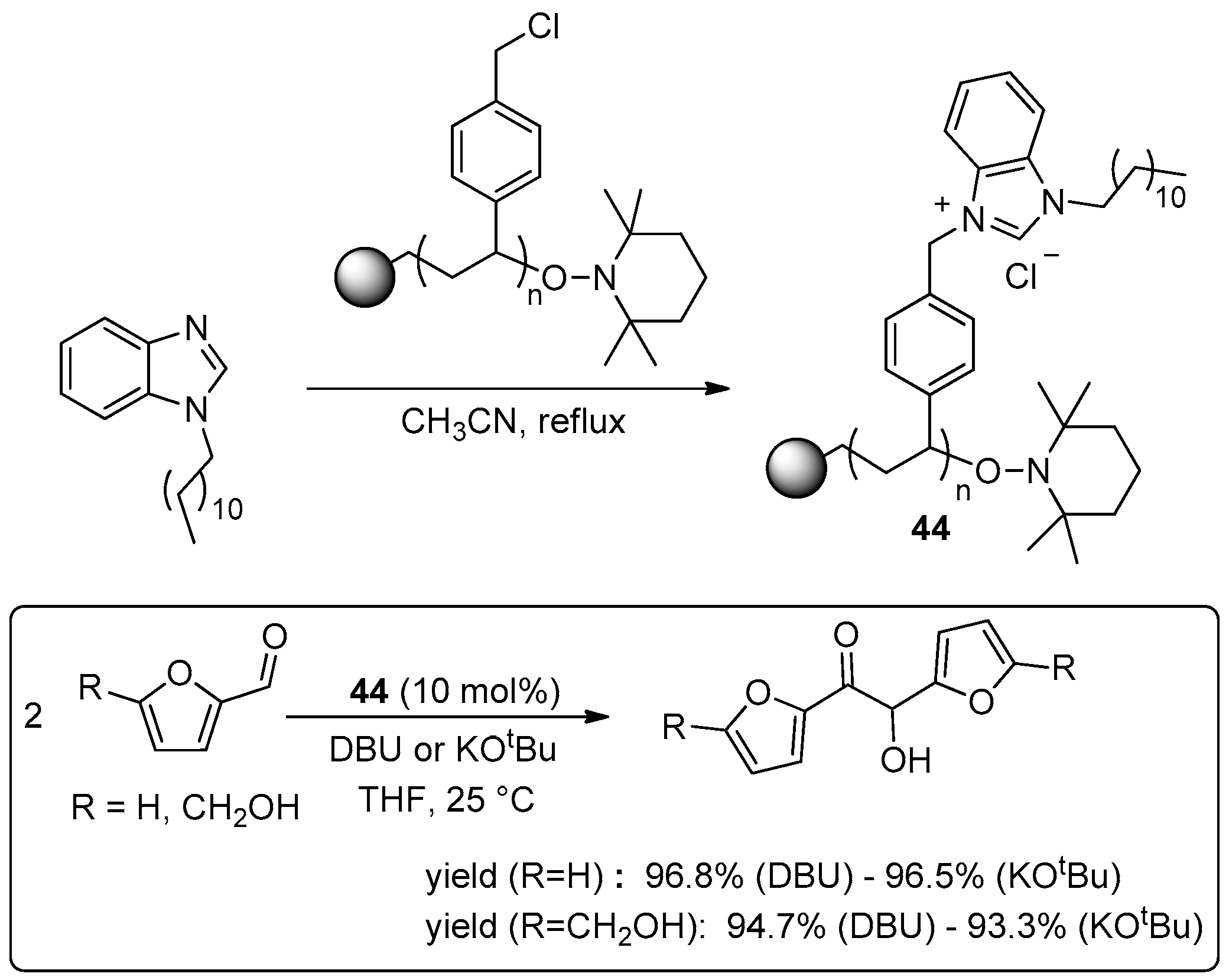
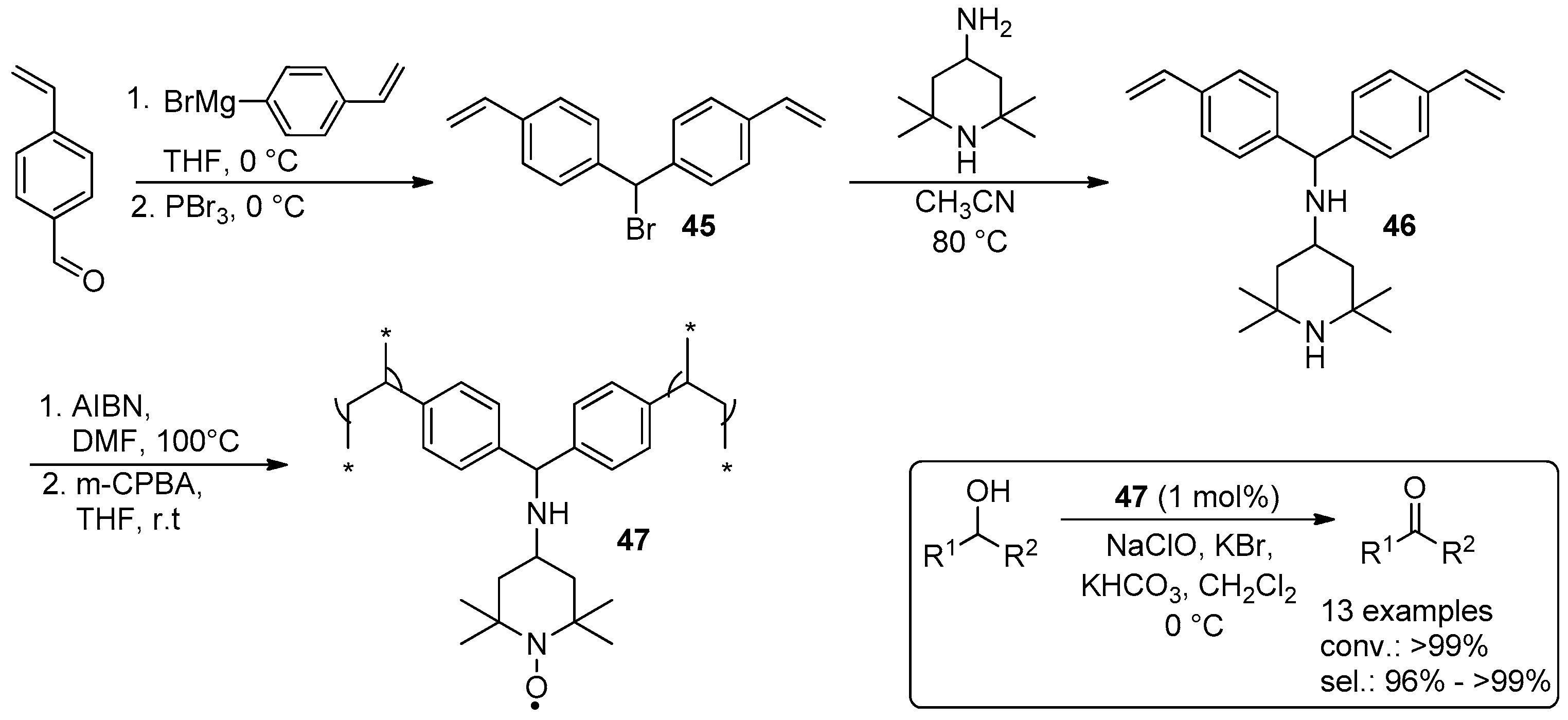
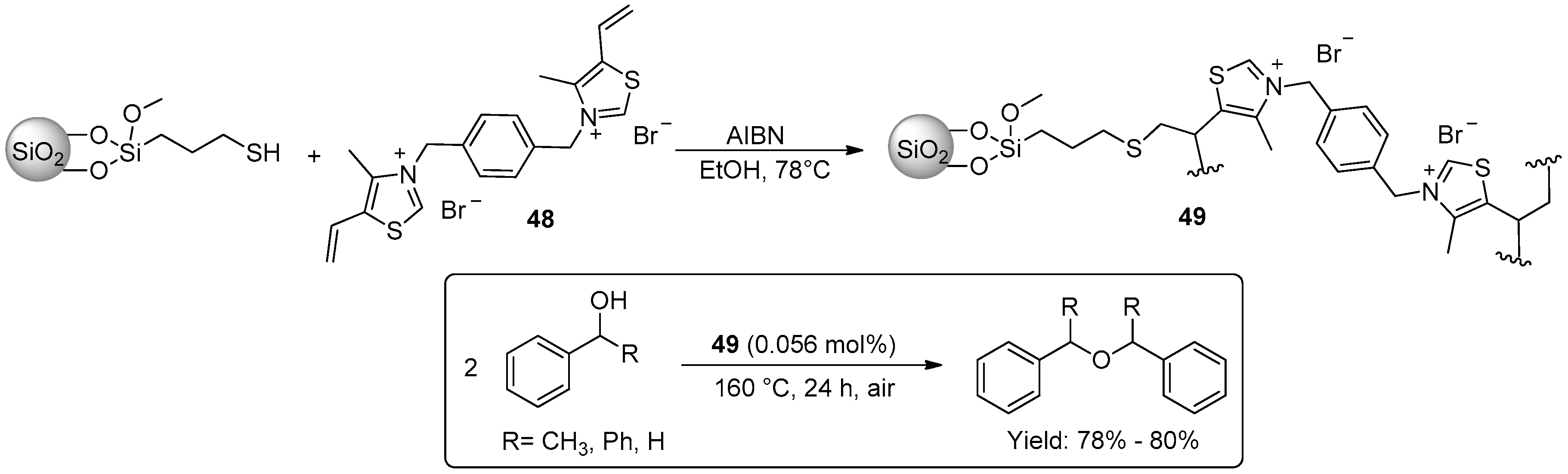
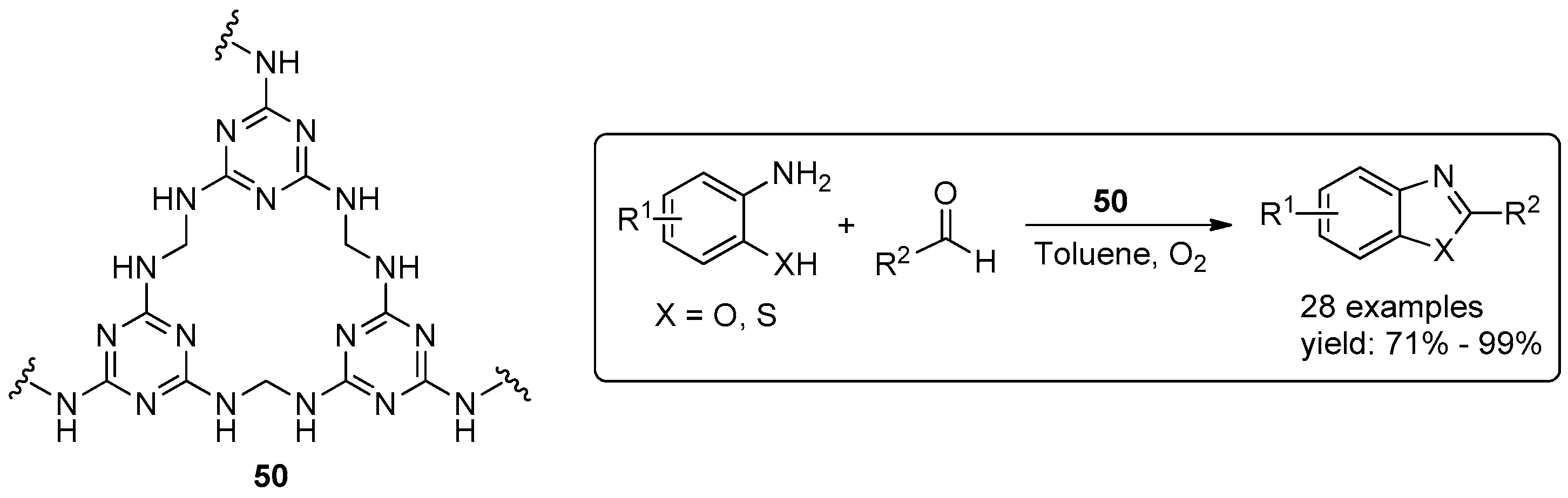
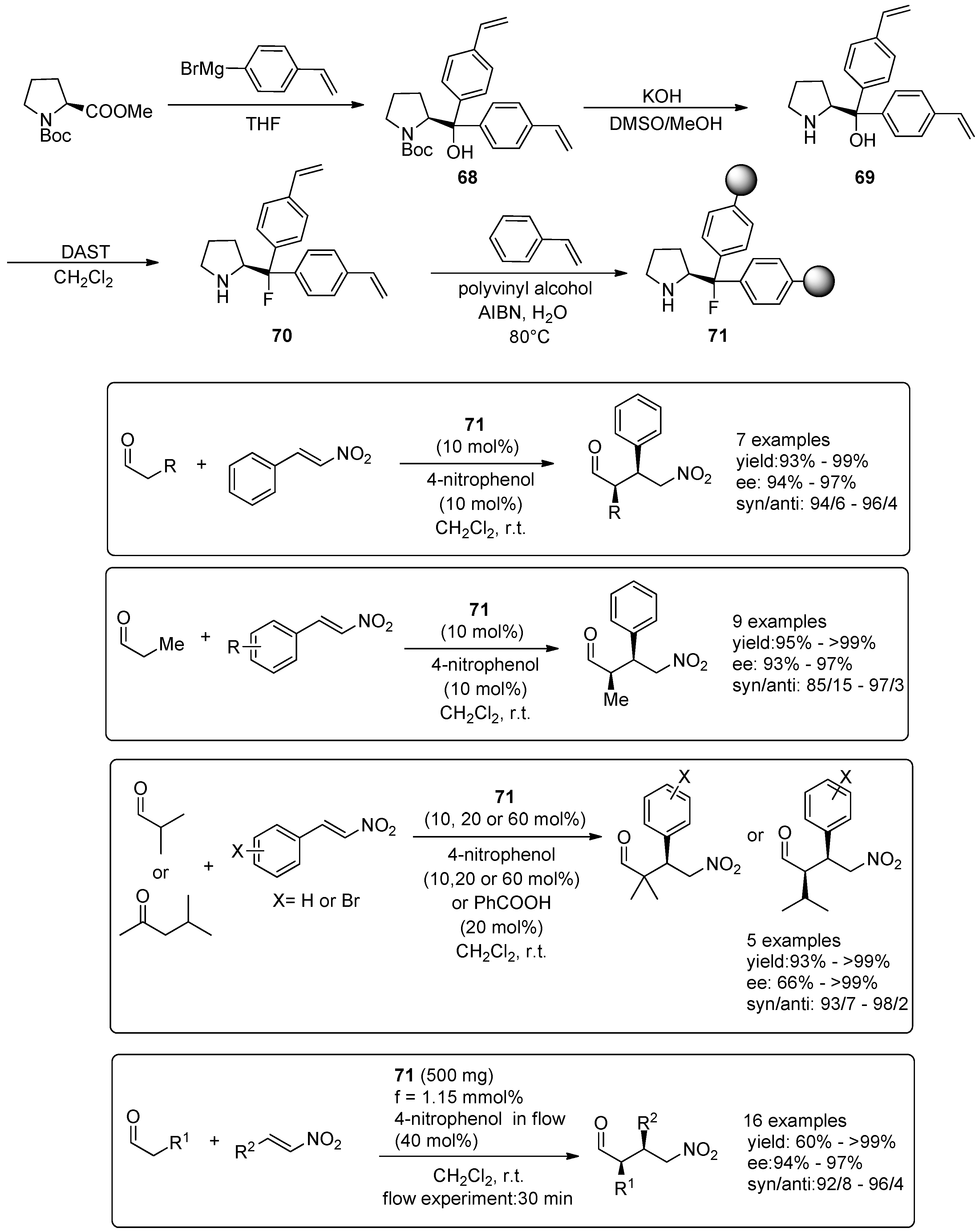
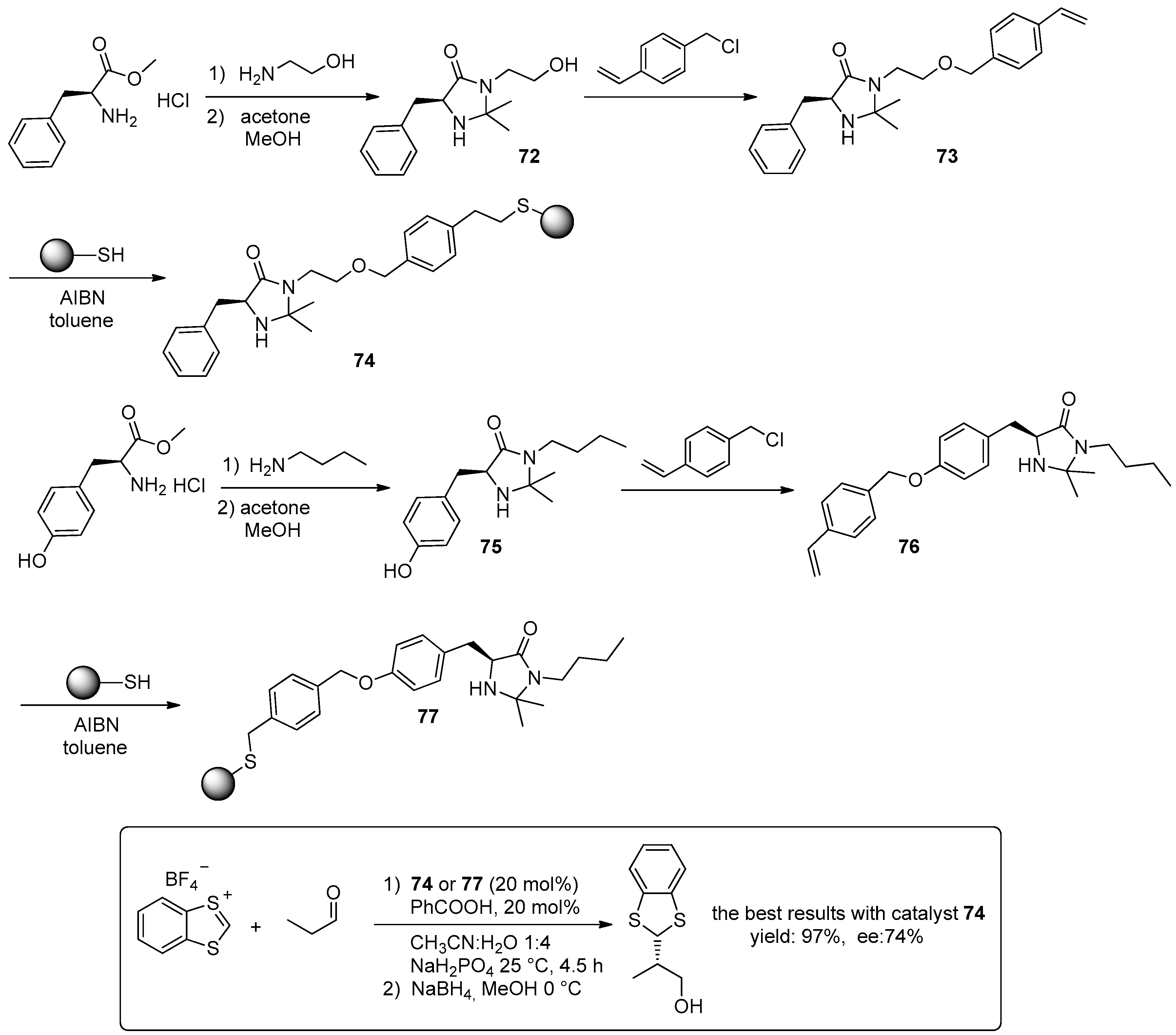
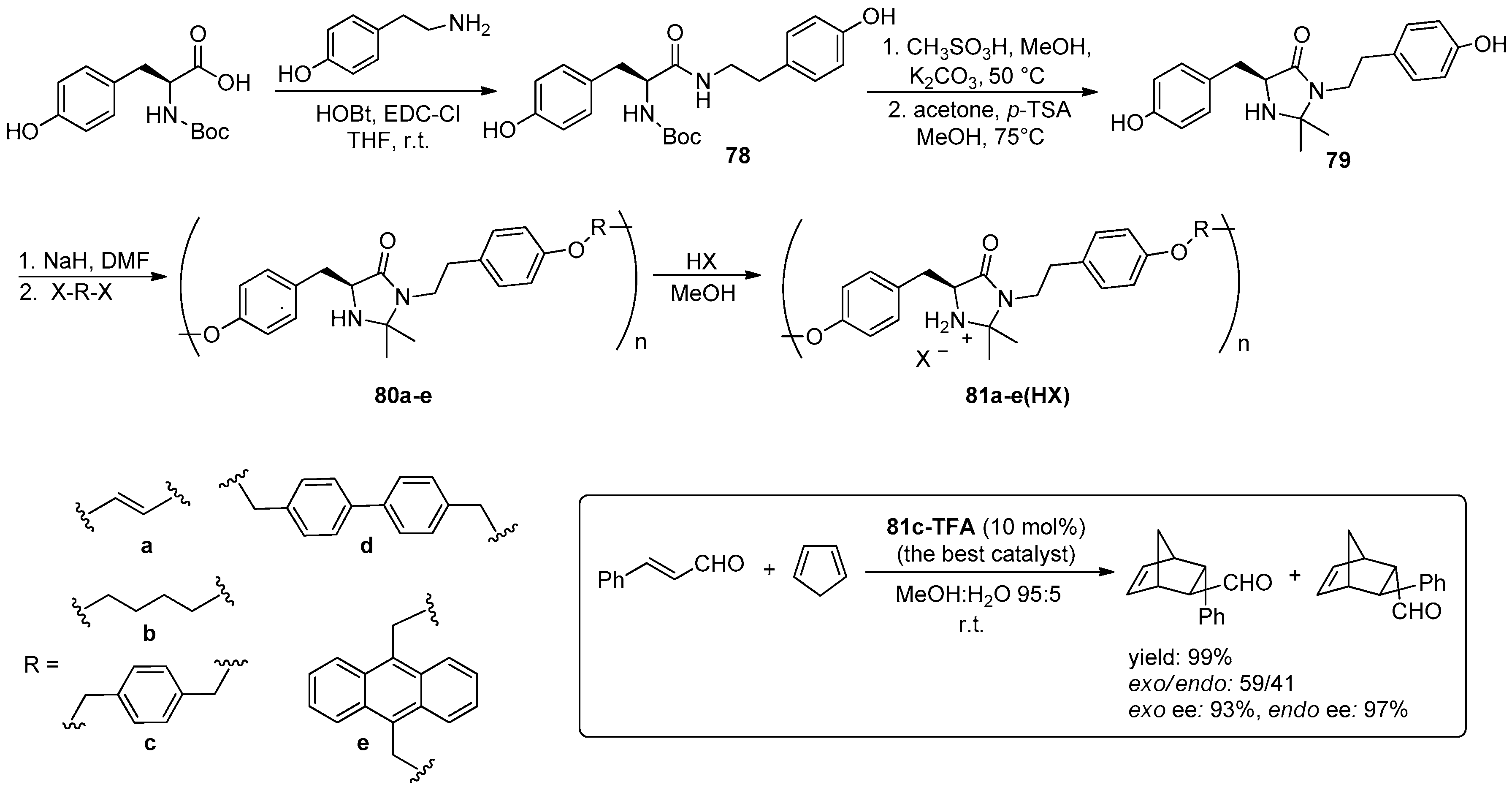
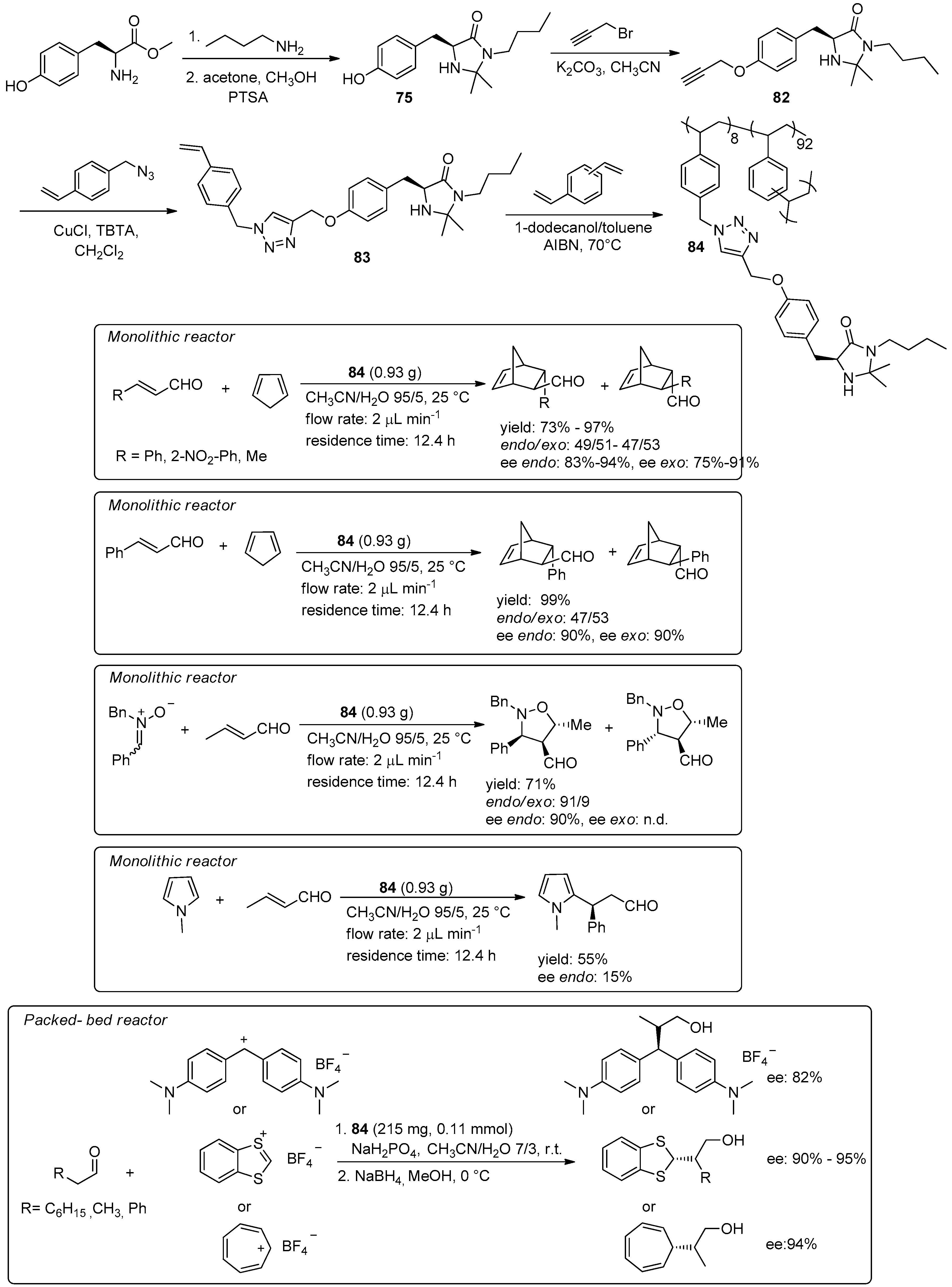
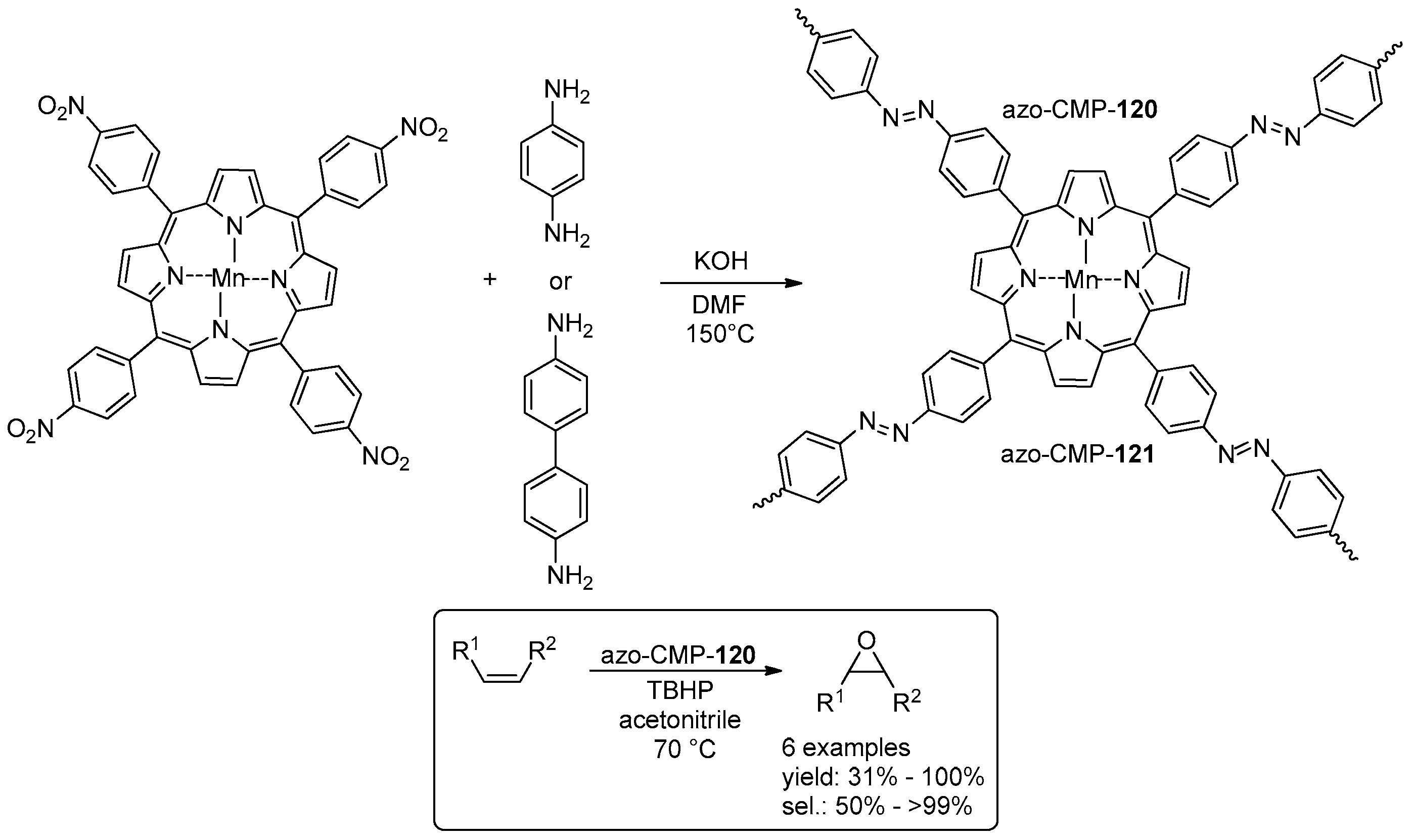
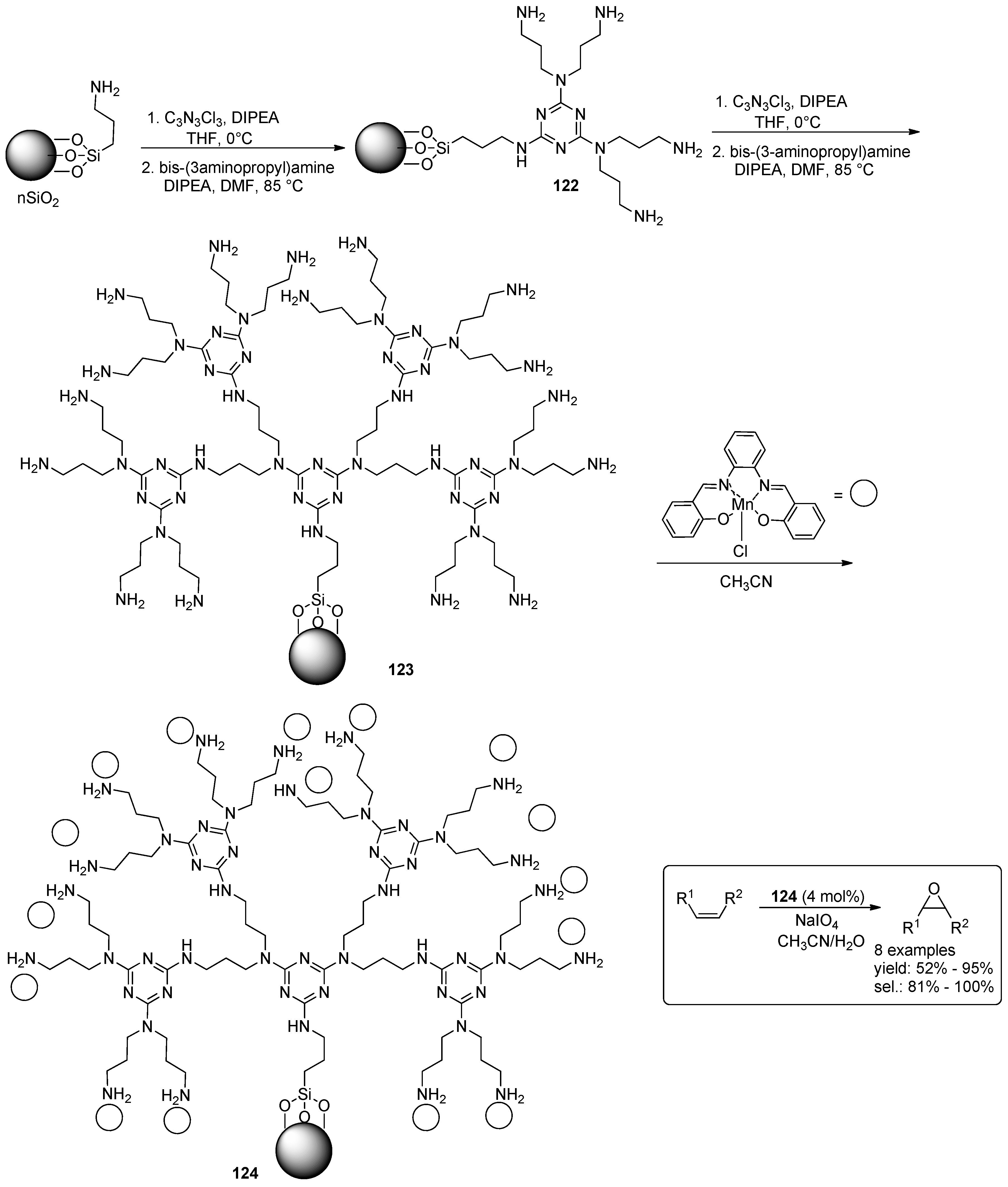
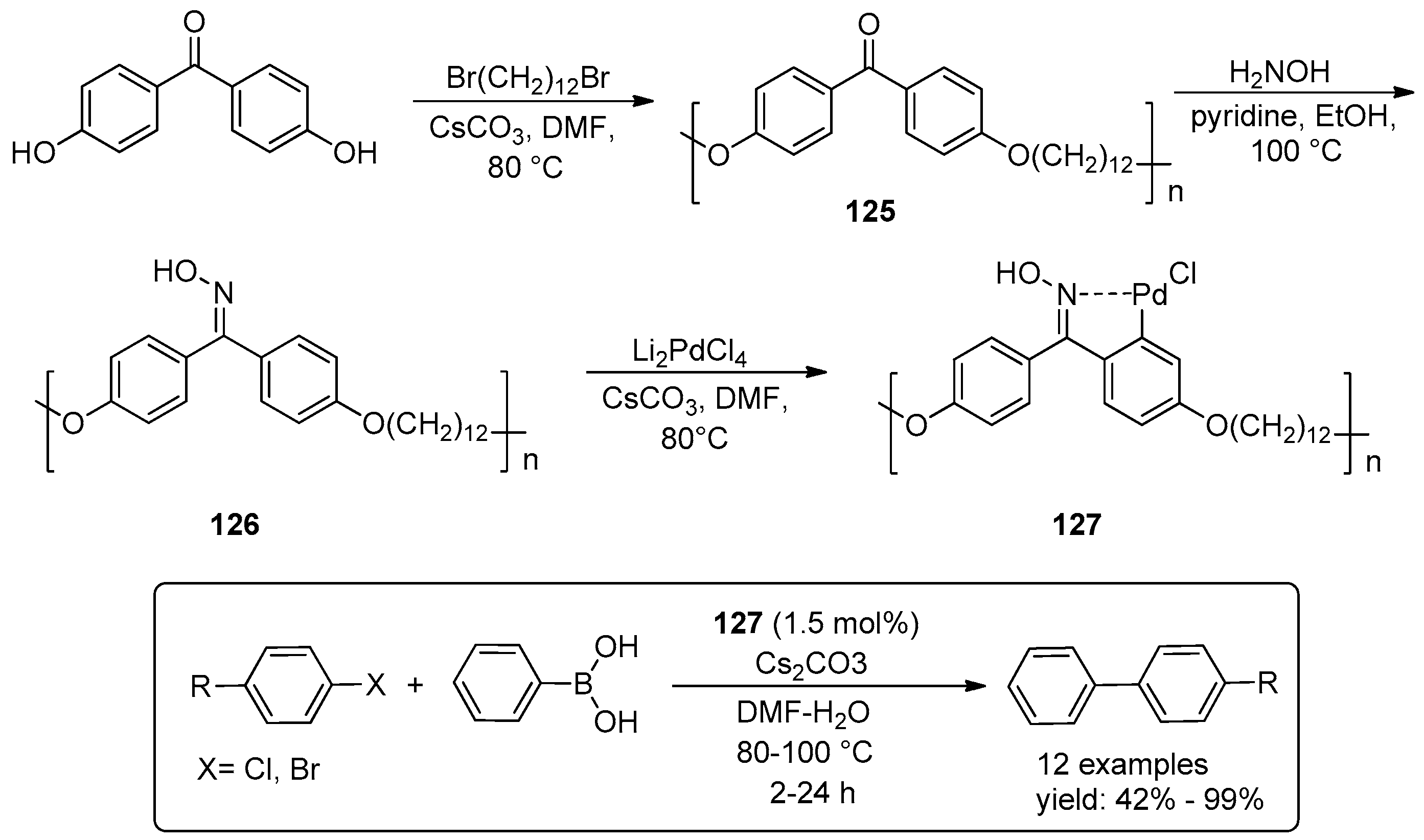
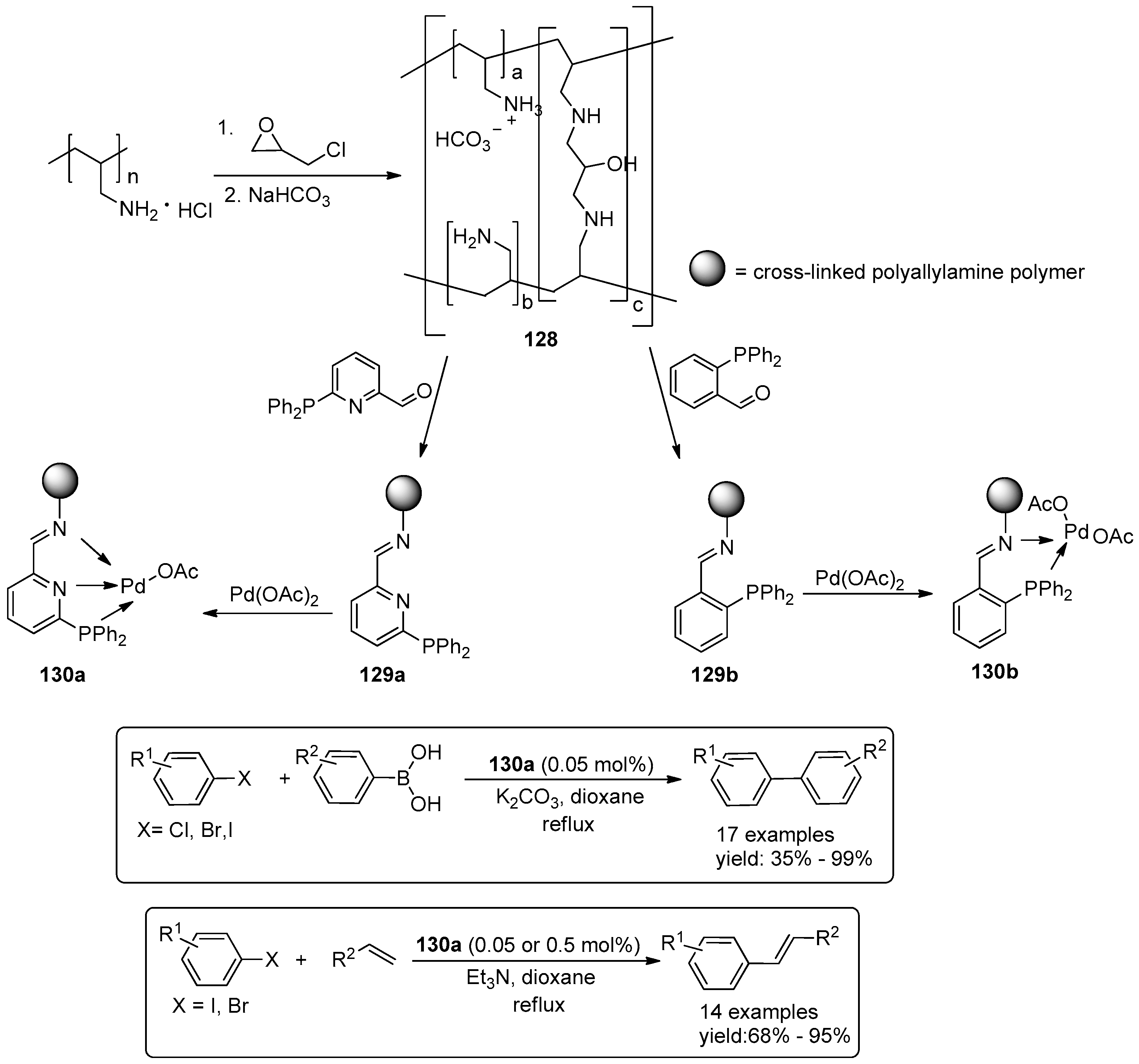
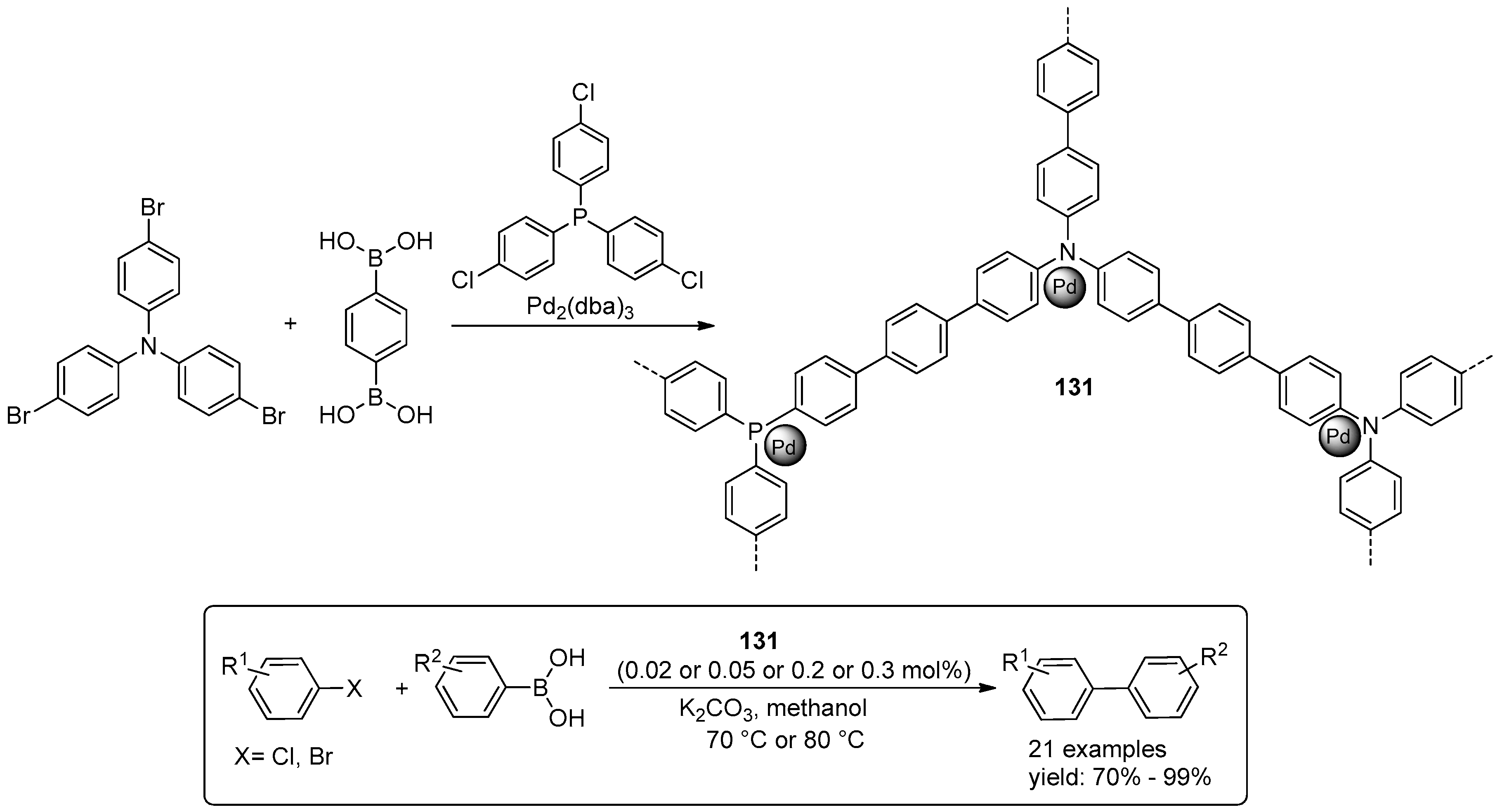
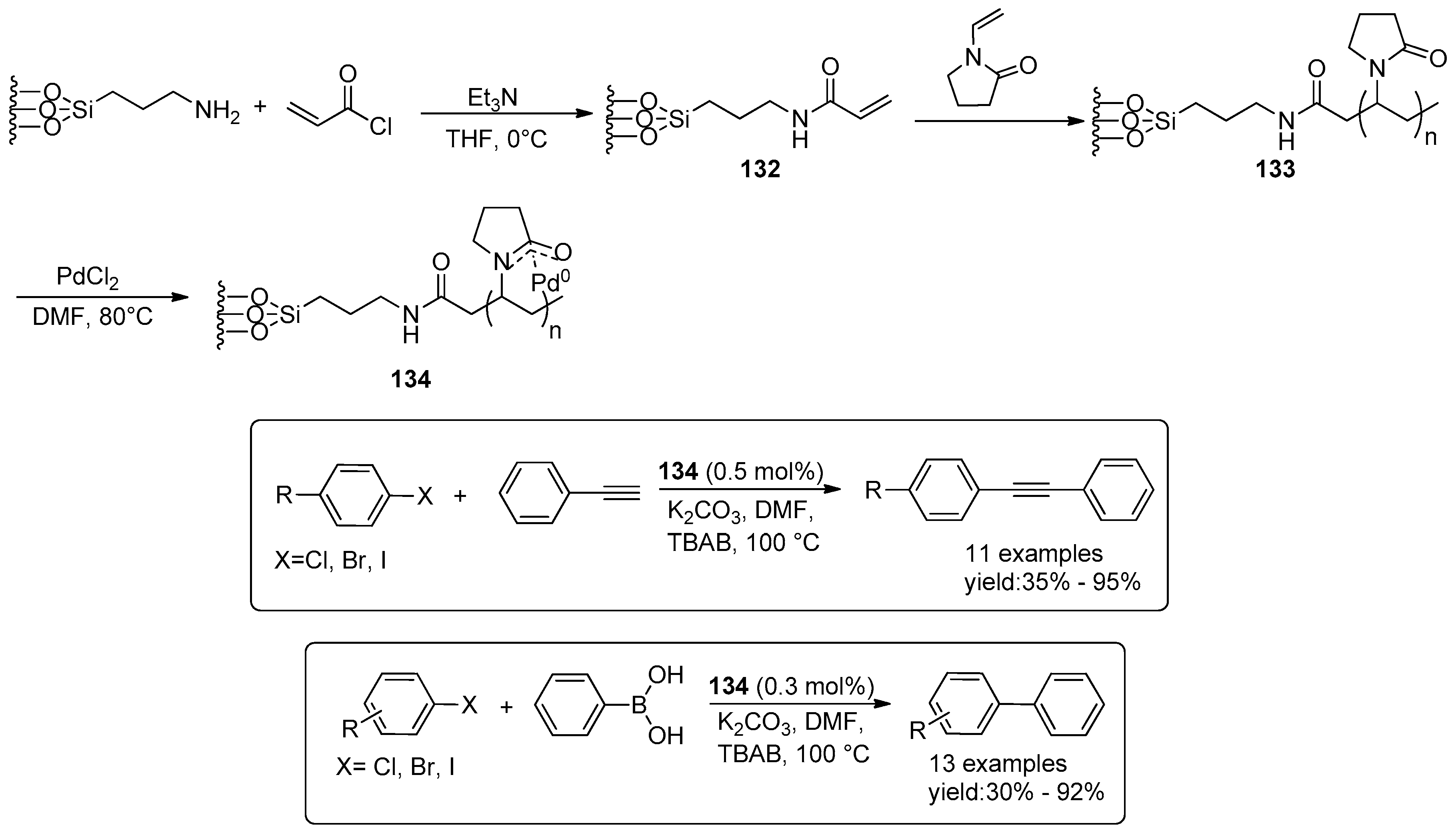
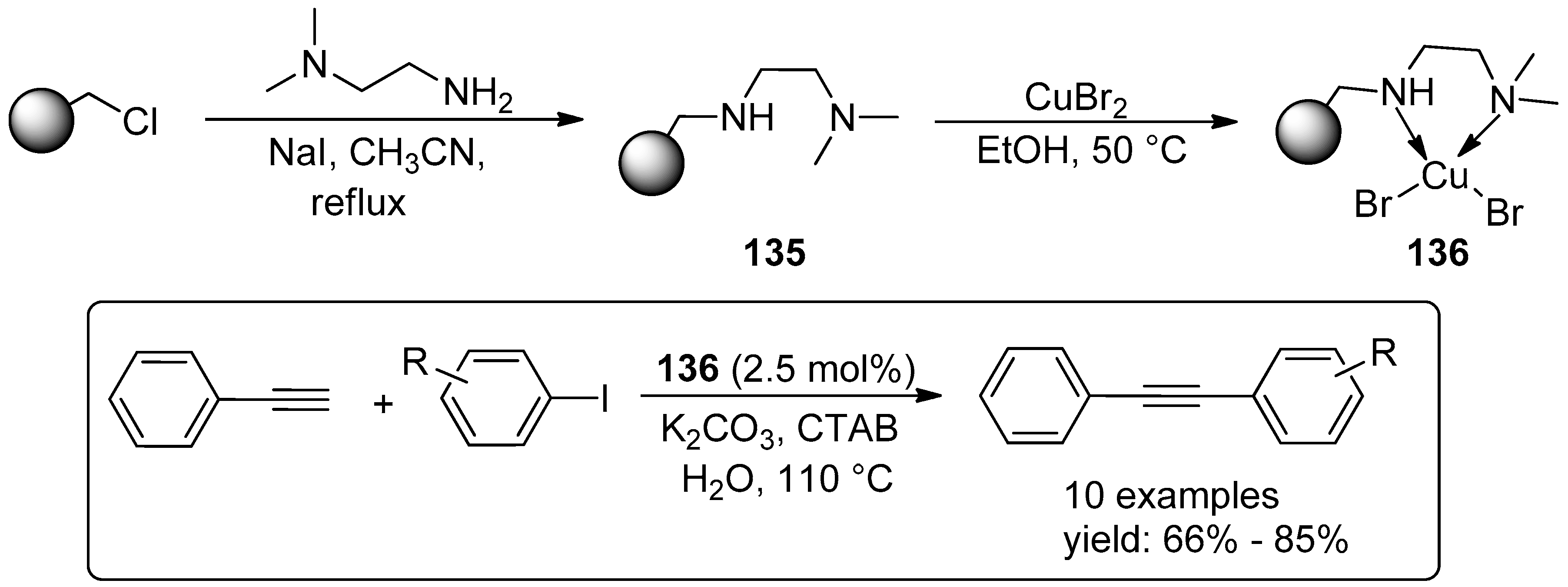
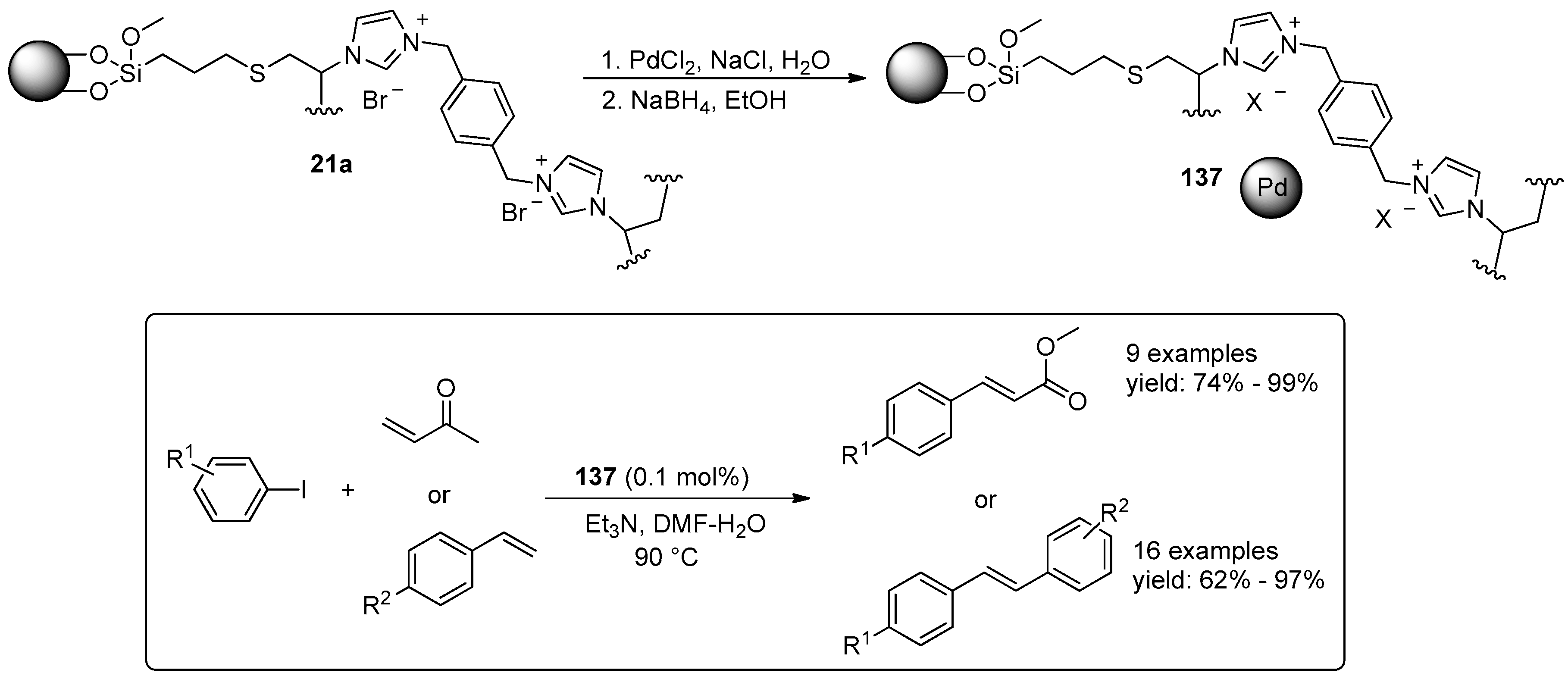
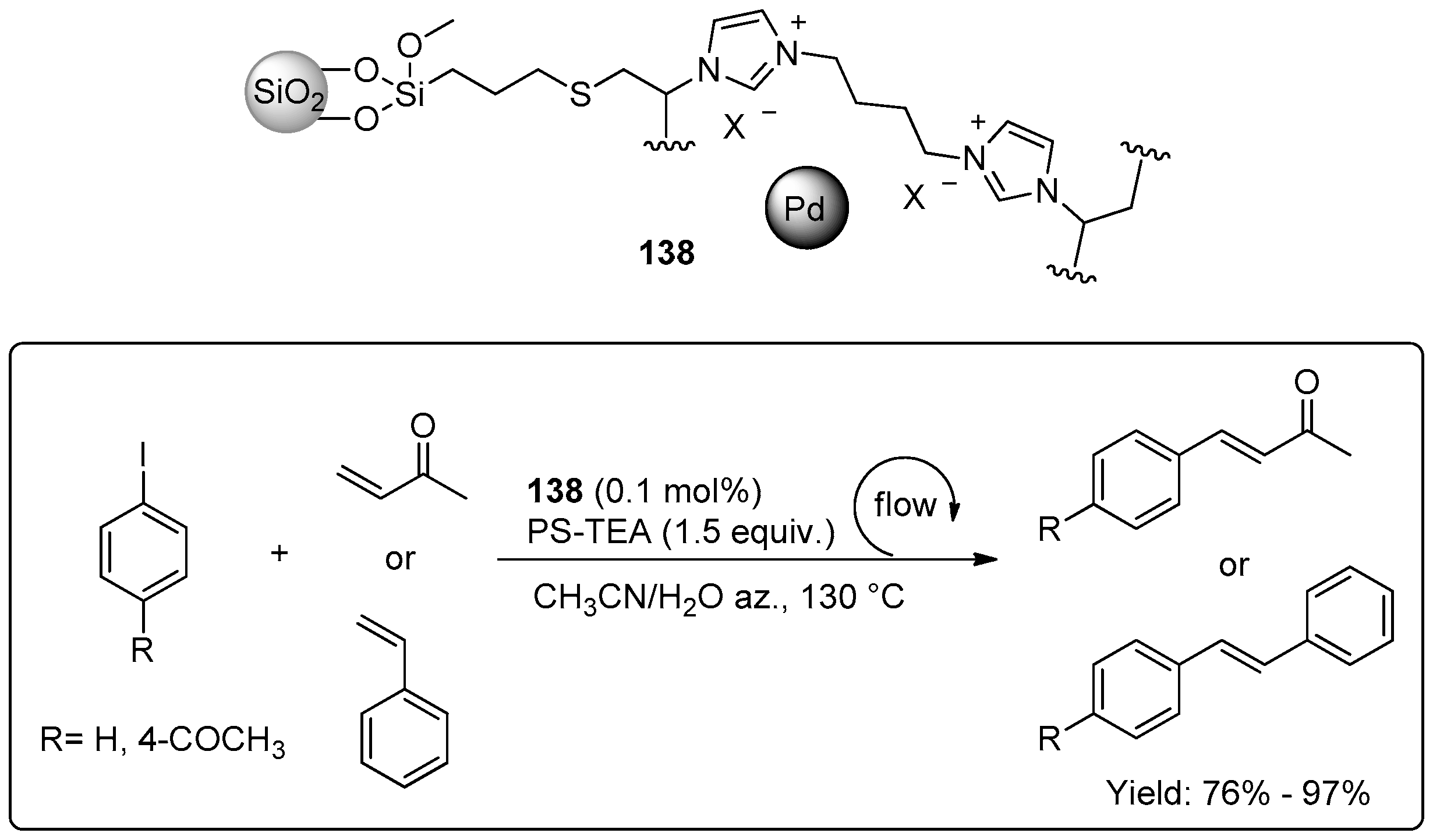
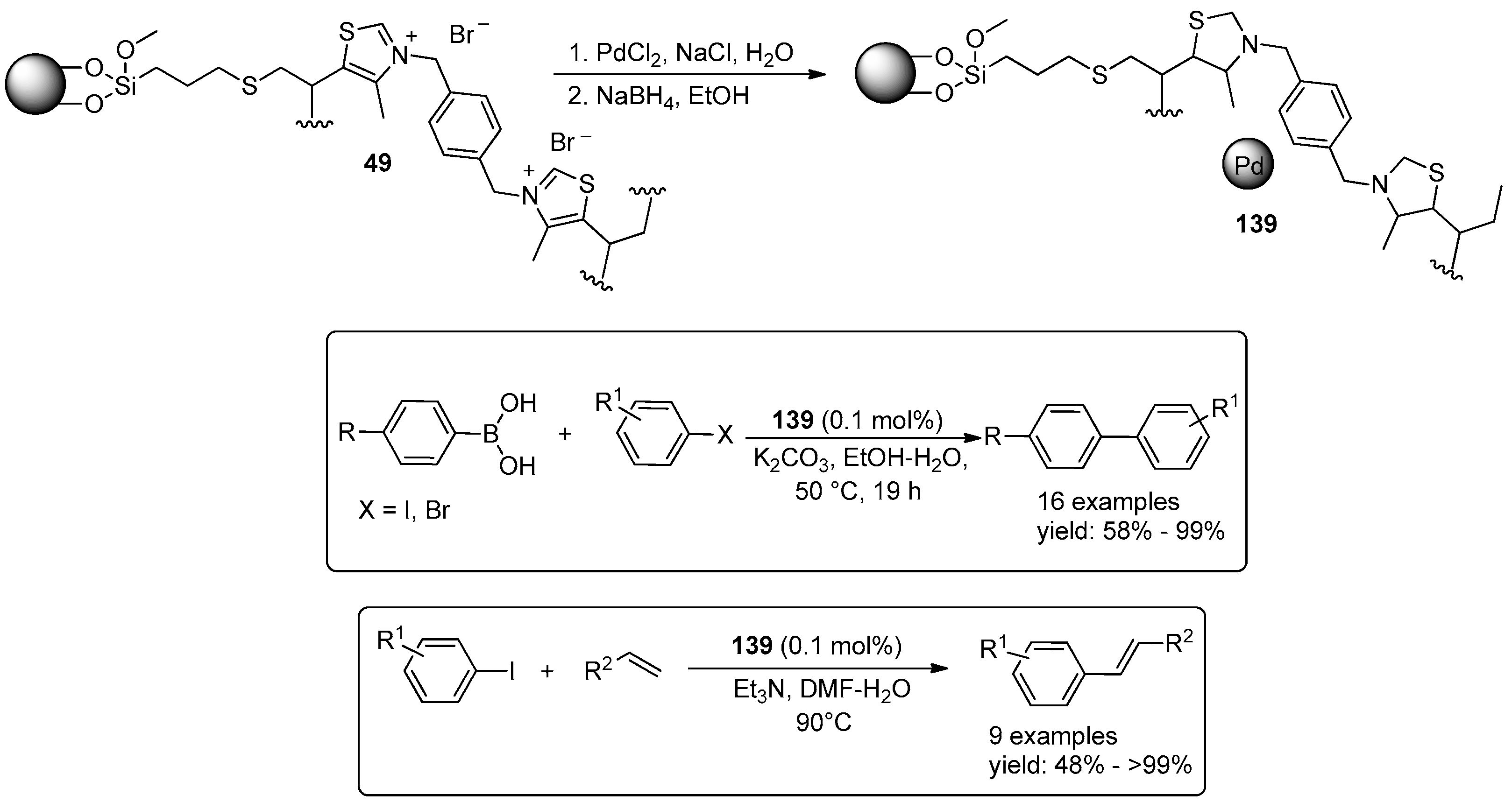
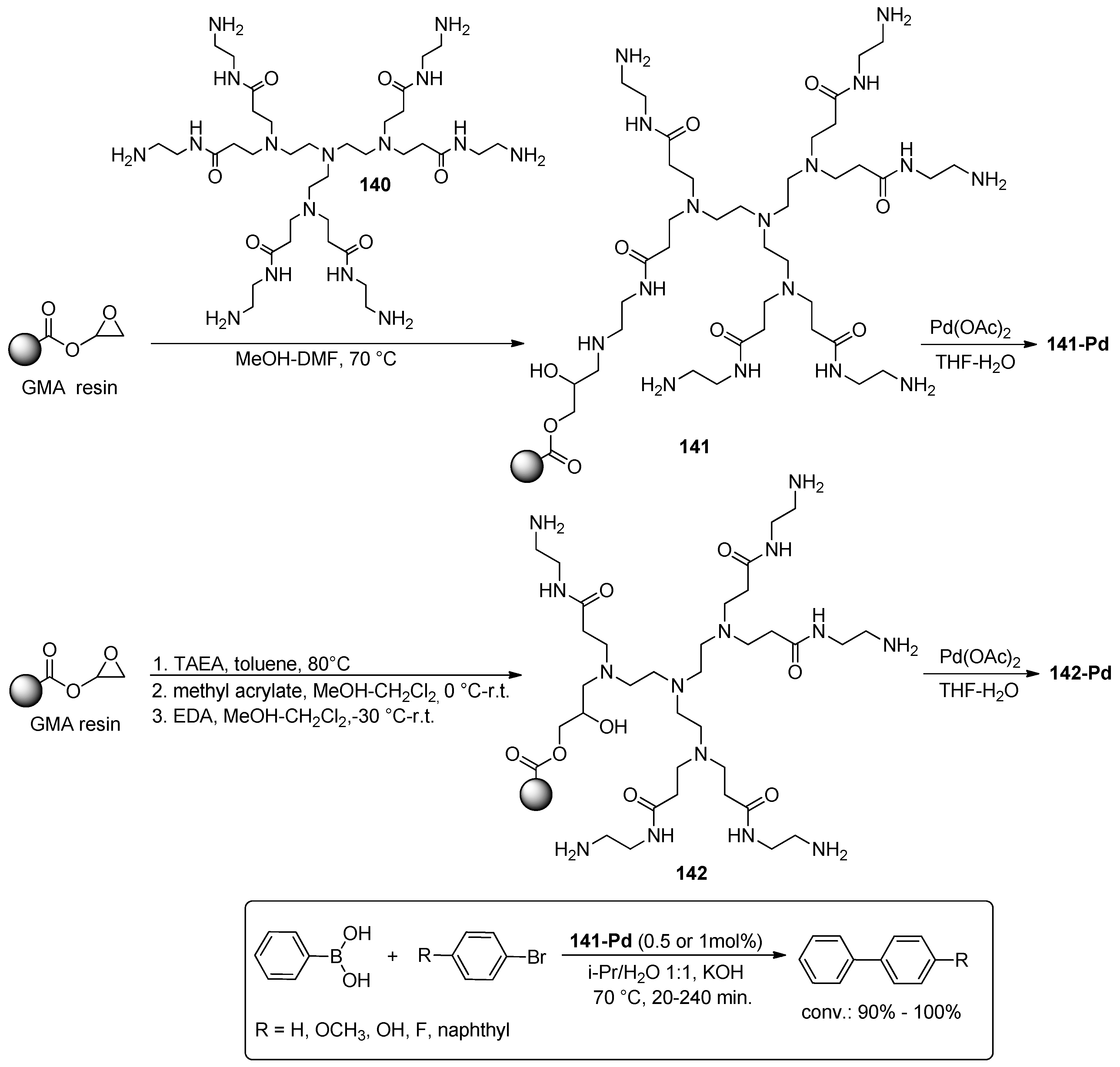
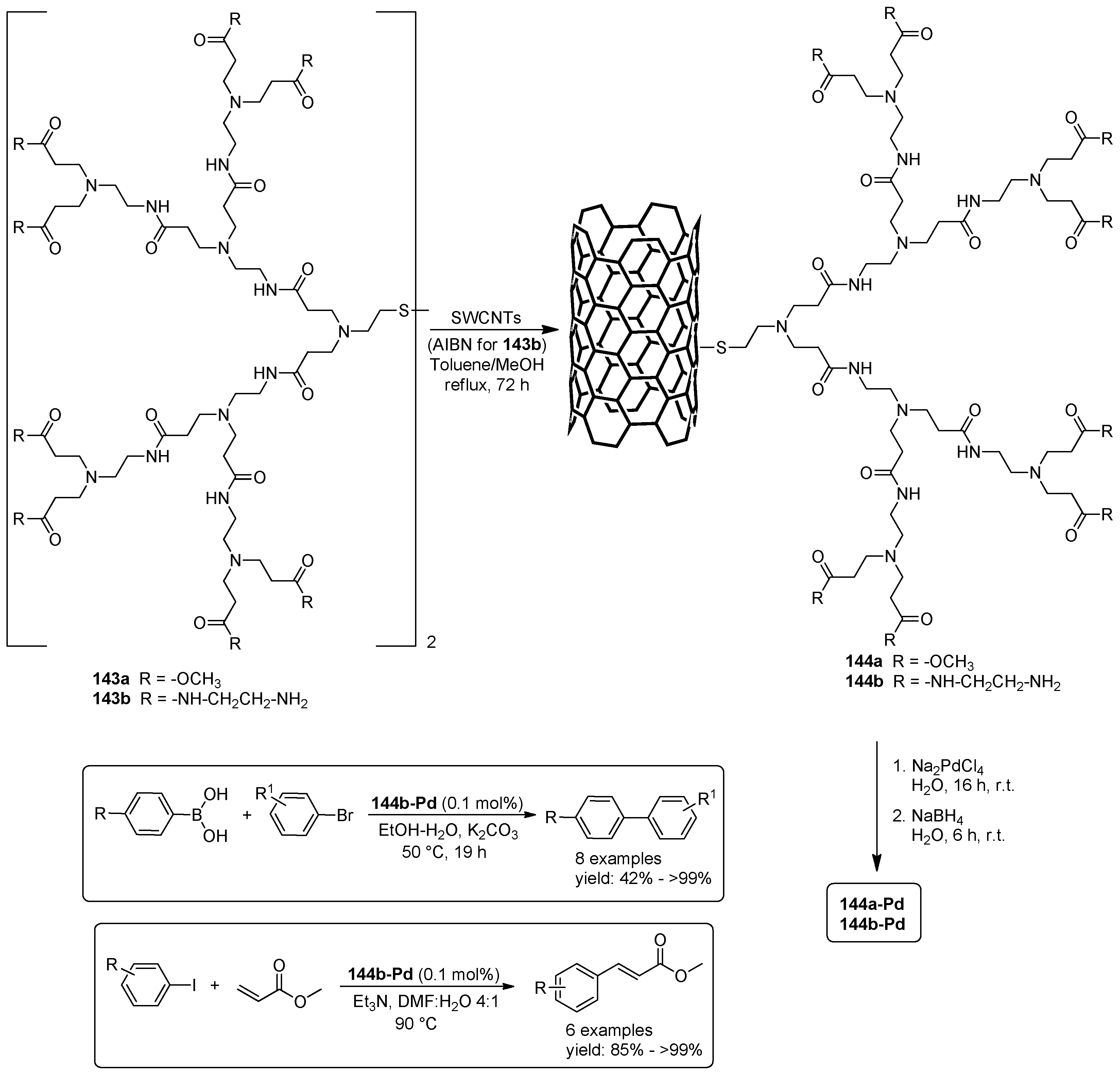
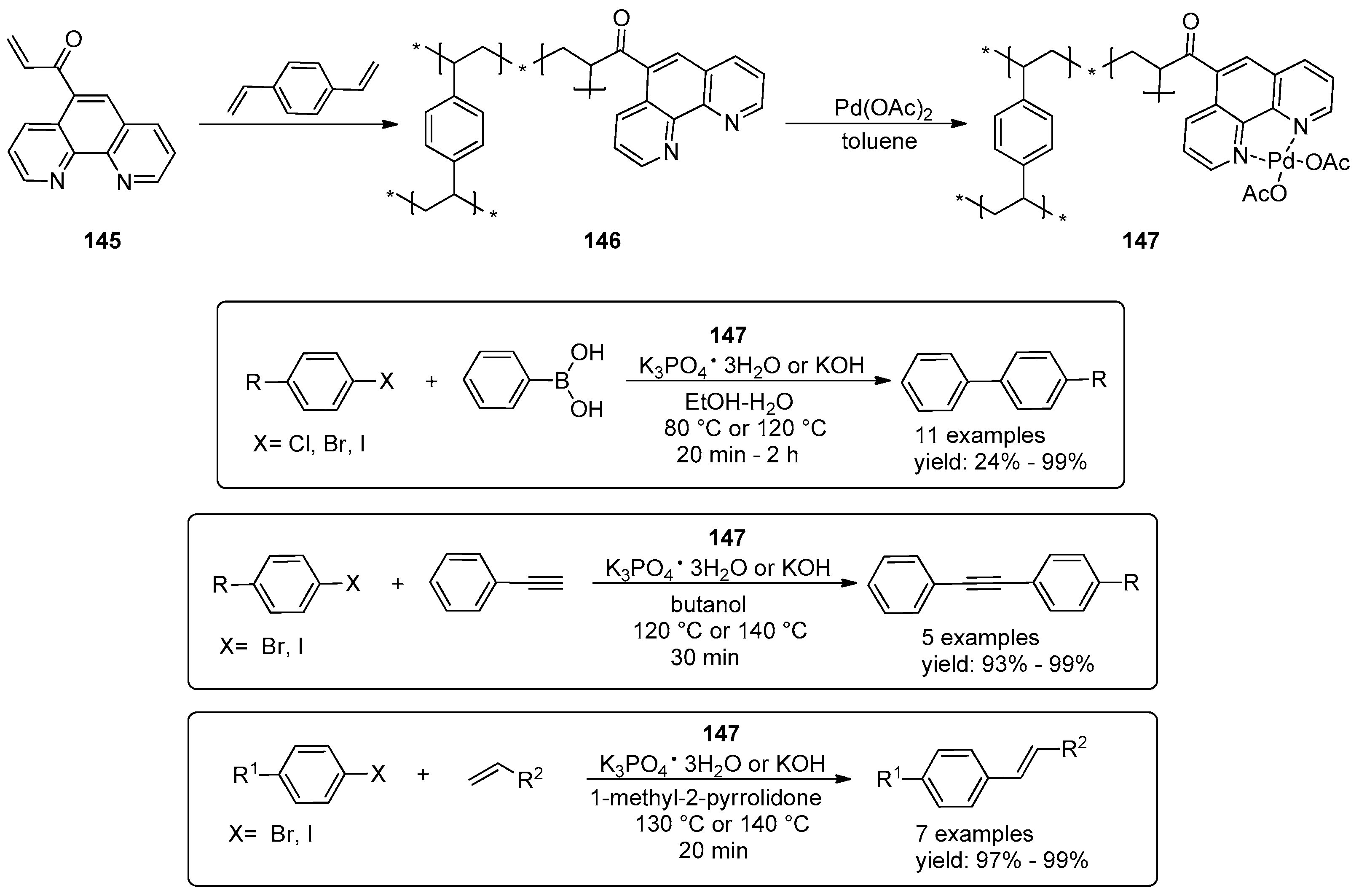
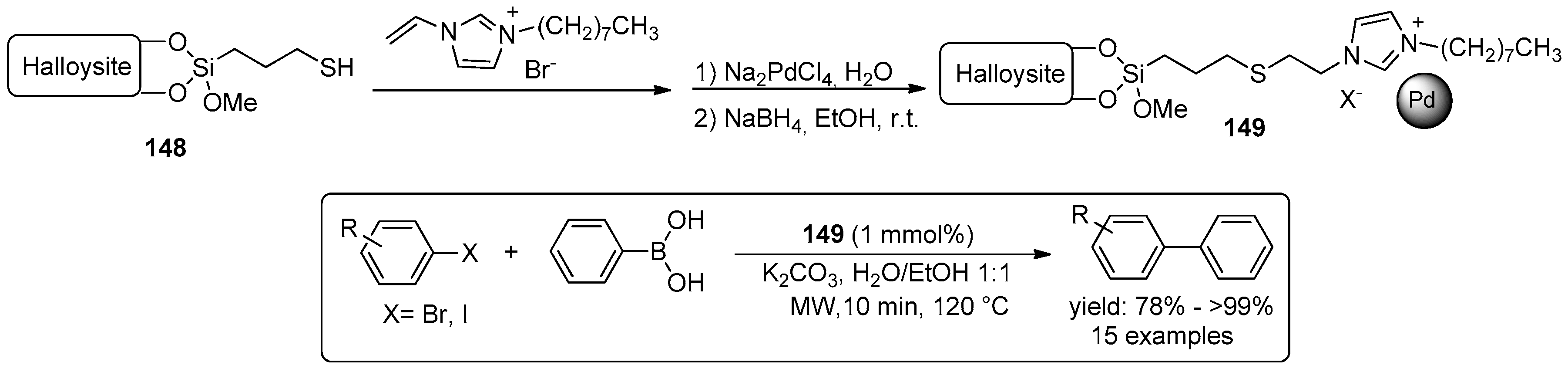
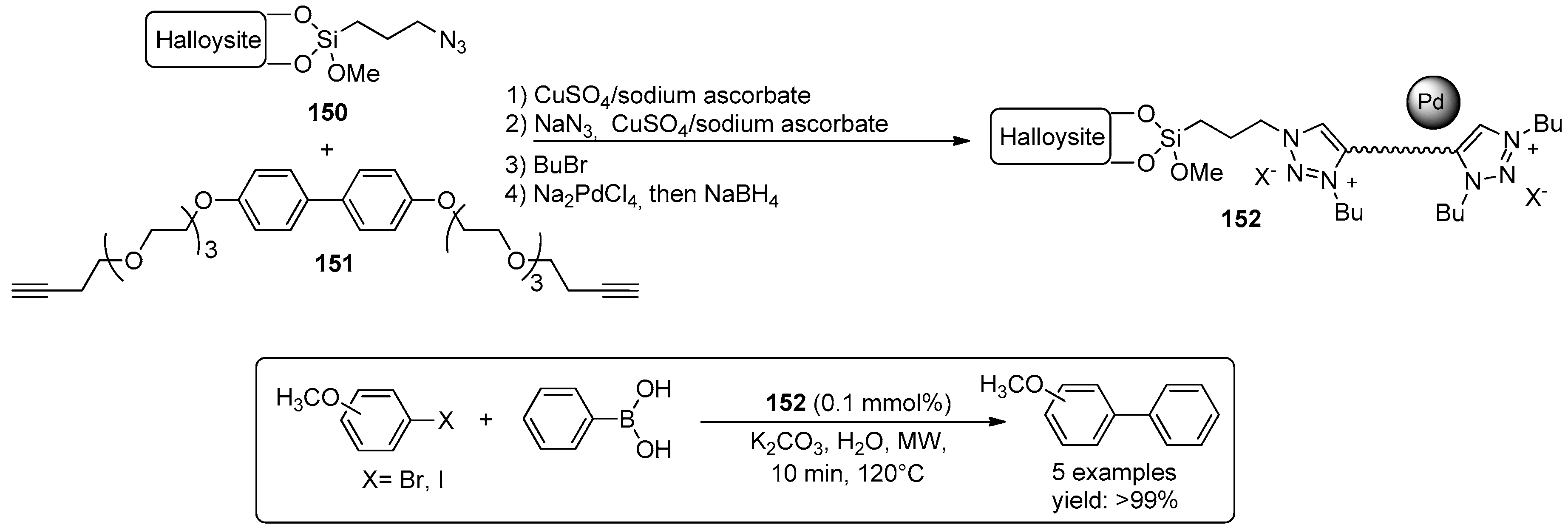
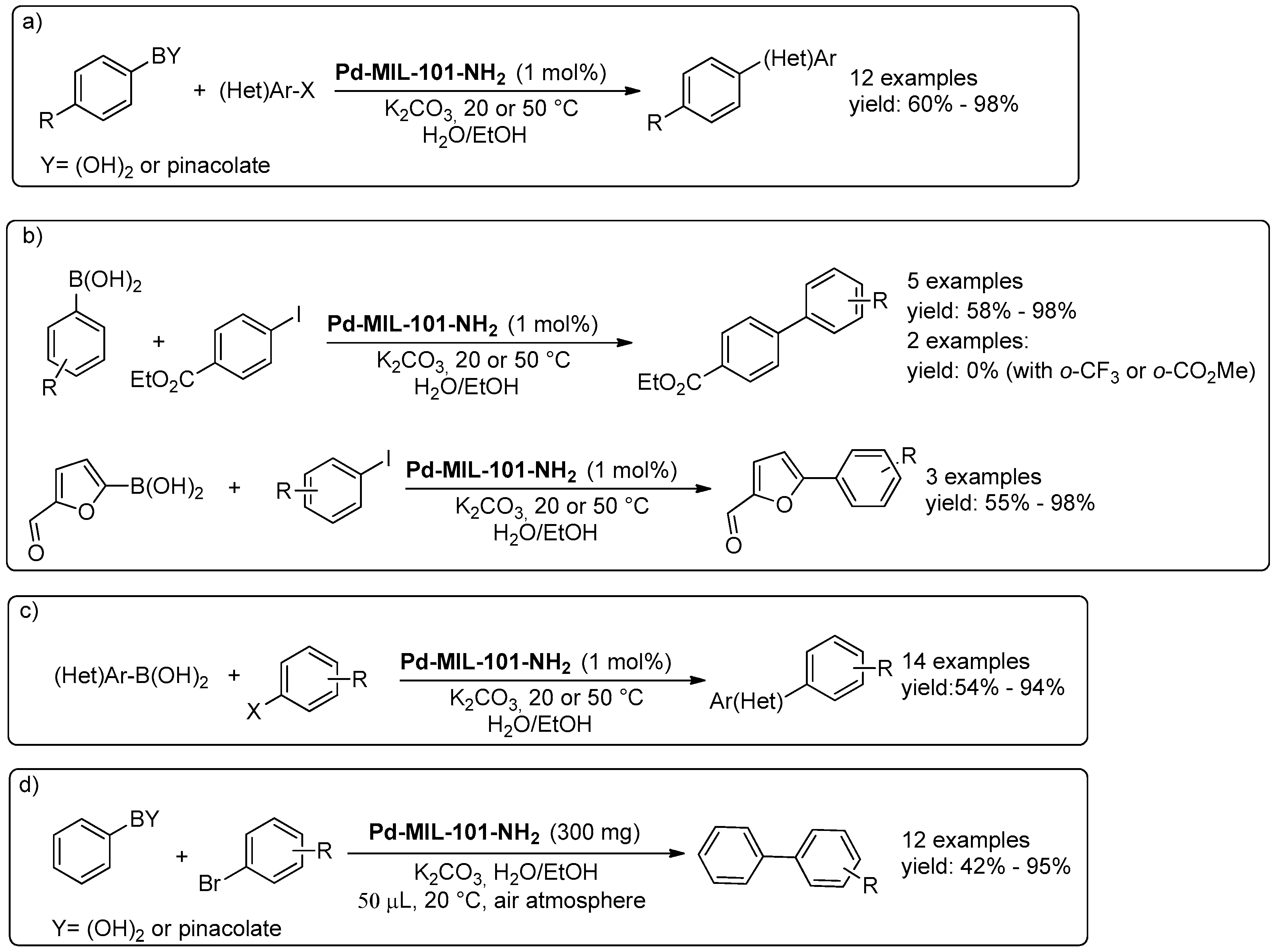

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvo, A.M.P.; Giacalone, F.; Gruttadauria, M. Advances in Organic and Organic-Inorganic Hybrid Polymeric Supports for Catalytic Applications. Molecules 2016, 21, 1288. https://doi.org/10.3390/molecules21101288
Salvo AMP, Giacalone F, Gruttadauria M. Advances in Organic and Organic-Inorganic Hybrid Polymeric Supports for Catalytic Applications. Molecules. 2016; 21(10):1288. https://doi.org/10.3390/molecules21101288
Chicago/Turabian StyleSalvo, Anna Maria Pia, Francesco Giacalone, and Michelangelo Gruttadauria. 2016. "Advances in Organic and Organic-Inorganic Hybrid Polymeric Supports for Catalytic Applications" Molecules 21, no. 10: 1288. https://doi.org/10.3390/molecules21101288
APA StyleSalvo, A. M. P., Giacalone, F., & Gruttadauria, M. (2016). Advances in Organic and Organic-Inorganic Hybrid Polymeric Supports for Catalytic Applications. Molecules, 21(10), 1288. https://doi.org/10.3390/molecules21101288