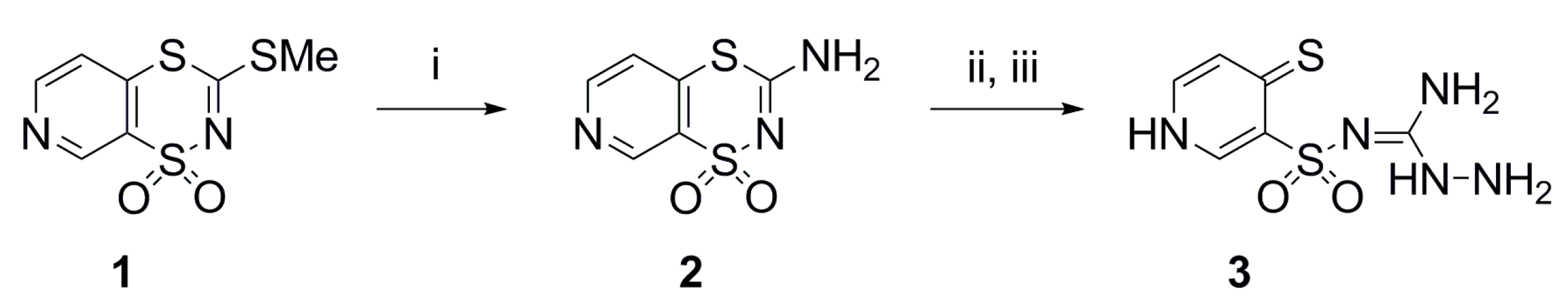
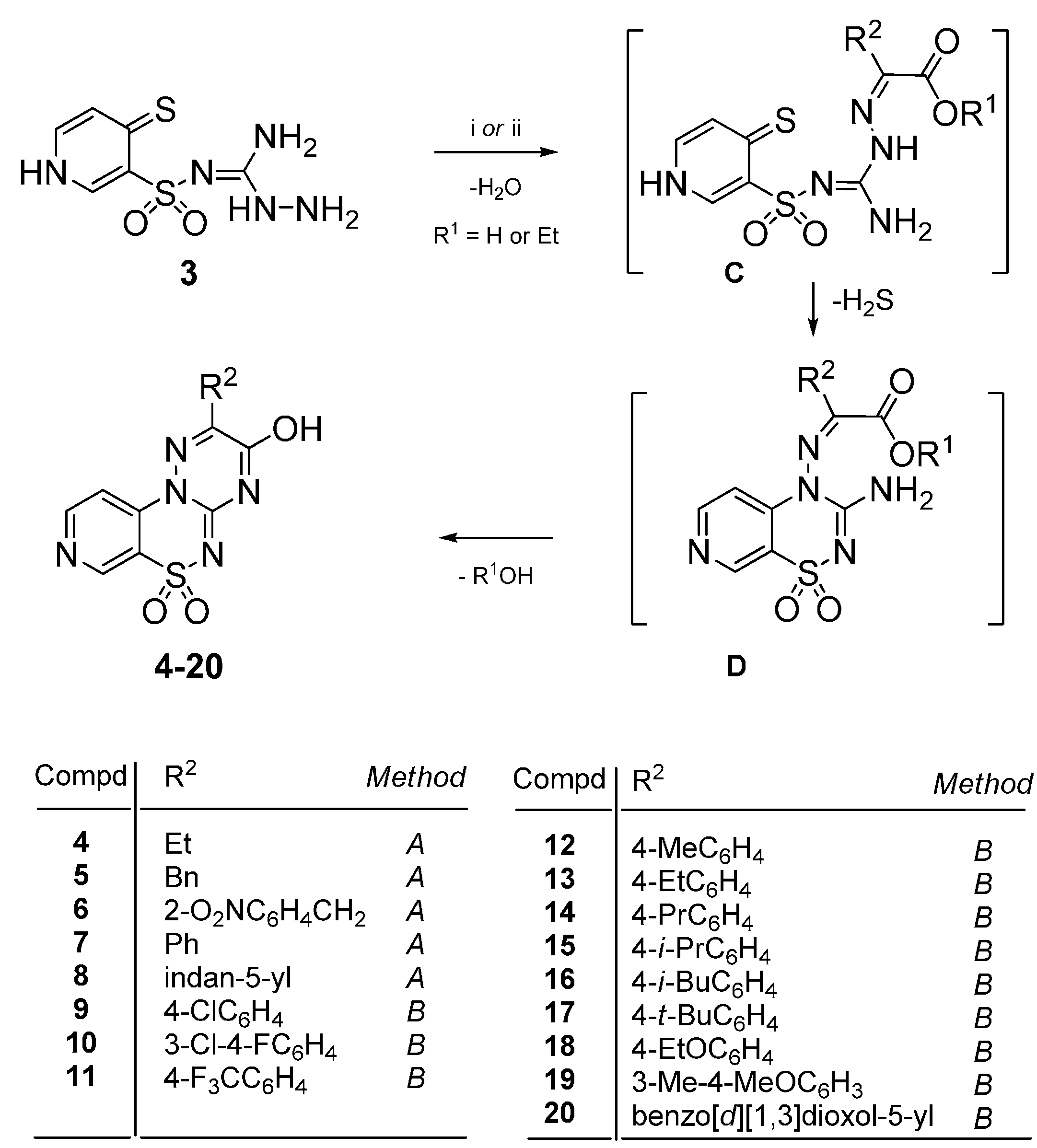
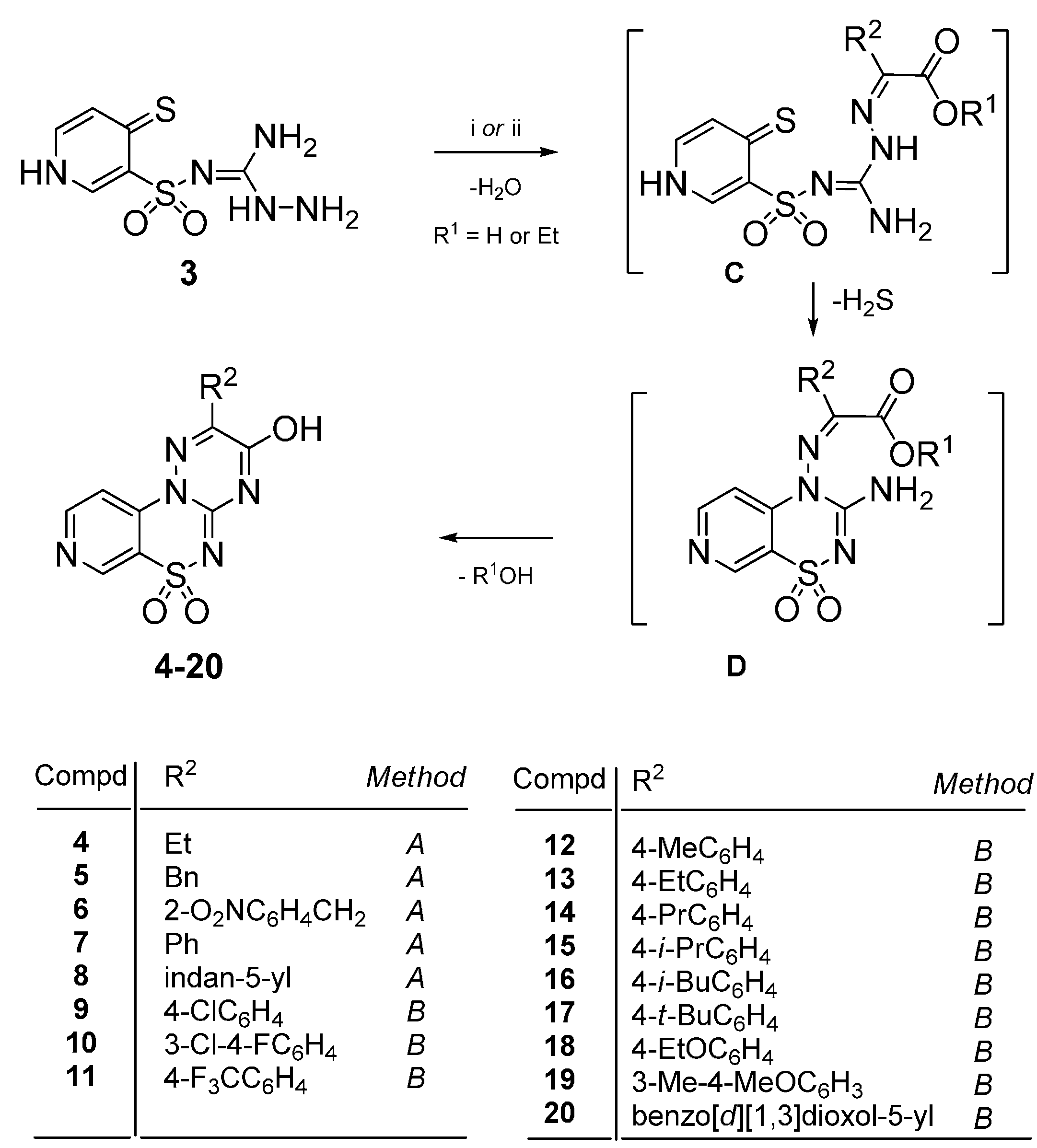
4.2. Synthesis
4.2.1. General Procedure for the Preparation of 2-Substituted-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxides 4–8
Method A: The mixture of 3 (1 mmol) and the appropriate 2-oxoalkanoic acid (1 mmol) in glacial acetic acid (3 mL) was stirred at reflux for 50–90 h. After standing overnight at room the precipitate was filtered off and purified by extraction of contaminations with boiling ethanol. In this manner the following compounds 4–8 were obtained.
2-Ethyl-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (4). The mixture of 3 and 2-oxobutanoic acid (0.102 g), gave compound 4 as a white solid powder (0.085 g, 30%) after 50 h of heating at reflux, m.p. 302–303 °C; IR (KBr) νmax 3525 (OH), 1619 (C=C), 1586 (C=N), 1312, 1165 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 1.22 (t, J = 7.32 Hz, 3H), 2.71 (q, J = 7.00 Hz, 2H), 7.98 (d, J = 5.86 Hz, 1H), 8.89 (d, J = 5,86 Hz, 1H), 9.08 (s, 1H) ppm; 13C-NMR (DMSO-d6): δ 10.11, 23.84, 111.23, 120.53, 142.33, 144.69, 146.52, 154.14, 154.23, 154.31 ppm; anal. C 42.69, H 3.07, N 24.70% calcd. for C10H9N5O3S, C 43.01, H 3.25, N 25.08%.
2-Benyzl-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (5). The mixture of 3 and 2-oxo-3-phenylpropanoic acid (0.164 g), gave compound 5 as a white solid powder (0.076 g, 22%) after 90 h of heating at reflux, m.p. 303–305 °C; IR (KBr) νmax 3425 (OH), 1638 (C=C), 1587 (C=N), 1321, 1178 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 4.04 (br. s, 2H), 7.27 (m, 2H), 7.36 (m, 2H), 7.79 (br. s, 1H), 8.86 (br. s, 1H), 9.08 (br. s, 1H) ppm; 13C-NMR (DMSO-d6) δ 36.24, 111.02, 120.55, 127.53, 129.05, 130.04, 135.89, 142.29, 144.66, 146.59, 151.79, 154.15, 154.24 ppm; anal. C 52.45, H 3.07, N 20.12% calcd. for C15H11N5O3S, C 52.78, H 3.25, N 20.52%.
3-Hydroxy-2-(2-nitrobenzyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (6). The mixture of 3 and 3-(2-nitrophenyl)-2-oxopropanoic acid (0.209 g) gave compound 6 as a yellow solid powder (0.119 g, 31%) after 90 h of heating at reflux, m.p. 277–279 °C; IR (KBr) νmax 3442 (OH), 1636 (C=C), 1588 (C=N), 1311, 1177 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 4.42 (s, 2H), 7.36 (d, J = 5.86 Hz, 1H), 7.64 (m, 1H), 7.75 (t, J = 7.32 Hz, 1H), 8.11 (d, J = 7.81 Hz, 1H), 8.80 (d, J = 5.86 Hz, 1H) 9.06 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 33.53, 110.43, 120.49, 125.57, 129.59, 130.39, 133.87, 134.42, 142.11, 144.54, 146.63, 149.64, 150.97, 154.11 ppm; anal. C 47.02, H 2.46, N 21.50% calcd. for C15H10N6O5S, C 46.63, H 2.61, N 21.75%.
3-Hydroxy-2-phenylpyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (7). The mixture of 3 and 2-phenyl-2-oxoacetic acid (0.150 g), gave compound 7 as a white solid powder (0.150 g, 45%) after 60 h of heating at reflux, m.p. 349–350 °C; IR (KBr) νmax 3424 (OH), 1629 (C=C), 1311, 1173 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 7.55 (t, 1H), 7.59 (t, 2H), 8.10 (t, 2H), 8.11 (d, J = 5.86 Hz, 1H), 8.91 (d, J = 5.86 Hz, 1H), 9.13 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 111.56, 120.78, 129.14, 129.85, 131.23, 132.00, 142.29, 144.39, 146.60, 147.54, 154.26, 154.36 ppm; LC-MS IT-TOF m/z [M + H]+ calcd. for C14H9N5O3S: 327.32, found: 328.04, tR = 22 min. Anal. C 50.99, H 3.07, N 21.05% calcd. for C14H9N5O3S, C 51.37, H 2.77, N 21.40%.
2-(2,3-Dihydro-1H-inden-5-yl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-di-oxide (8). The mixture of 3 and 2-(2,3-dihydro-1H-inden-5-yl)-2-oxoacetic acid (0.190 g), gave compound 8 as a white solid powder (0.098 g, 27%) after 50 h of heating at reflux, m.p. 349–350 °C; IR (KBr) νmax 3425 (OH), 1637 (C=C), 1587 (C=N), 1320, 1186 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 2.05 (br. s, 2H), 2.81–3.04 (m, 4H), 7.38 (d, J = 6.35 Hz, 1H), 7.89 (d, J = 6.35 Hz, 1H), 7.94 (br. s, 1H), 8.10 (br. s, 1H), 8.92 (br. s, 1H), 9.11 (br. s, 1H) ppm; 13C-NMR (DMSO-d6) δ 25.72, 32.93, 33.09, 111.57, 120.76, 124.91, 125.50, 128.14, 129.19, 142.30, 144.36, 144.74, 146.56, 147.70, 148.41, 154.34 ppm; anal. C 55.70, H 3.26, N 18.84% calcd. for C17H13N5O3S, C 55.58, H 3.57, N 19.06%.
4.2.2. General Procedure for the Preparation of 2-Aryl-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxides 9–20
Method B: The mixture of 3 (1 mmol) and the appropriate ethyl phenylglyoxylate (1 mmol) in glacial acetic acid (3 mL) was stirred at reflux for 22–26 h. After standing overnight at room temperature the precipitate was filtered off and purified by extraction of the impurities with boiling ethanol. In this manner the following compounds 9–20 were obtained.
2-(4-Chlorophenyl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (9). The mixture of 3 and ethyl 2-(4-chlorophenyl)-2-oxoacetate (0.213 g) gave compound 9 as a white solid powder (0.088 g, 24%), m.p. 366–368 °C; IR (KBr) νmax 3431 (OH), 1634 (C=C), 1588 (C=N), 1323, 1180 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 7.63 (d, J = 8.30 Hz, 2H), 8.13 (m, 3H), 8.92 (d, J = 5.86 Hz, 1H), 9.13 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 111.62, 120.78, 129.26, 130.03, 131.64, 136.69, 142.29, 144.45, 146.46, 146.68, 154.32 ppm; anal. C 46.08, H 2.05, N 19.02% calcd. for C14H8ClN5O3S, C 46.48, H 2.23, N 19.36%.
2-(3-Chloro-4-fluorophenyl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (10). The mixture of 3 and ethyl 2-(3-chloro-4-fluorophenyl)-2-oxoacetate (0.230 g) gave compound 10 as a white solid powder (0.104 g, 27%), m.p. 389–392 °C; IR (KBr) νmax 3422 (OH), 1642 C=C), 1591 (C=N), 1318, 1182 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 7.62 (t, J = 8.79 Hz, 1H), 8.11–8.22 (m, 2H), 8.23–8.33 (m, 1H), 8.92 (d, J = 5.86 Hz, 1H), 9.14 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 111.76, 117.84, 118.02, 120.45, 120.60, 120.77, 128.55, 130.99, 131.06, 131.91, 142.14, 144.30, 145.56, 146.57, 154.19, 154.45, 158.60, 160.61 ppm; anal. C 44.06, H 1.86, N 18.21% calcd. for C14H7ClFN5O3S, C 44.28, H 1.86, N 18.44%.
3-Hydroxy-2-(4-trifluormethylphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (11). The mixture of 3 and ethyl 2-oxo-2-(4-trifluoromethylphenyl)acetate (0.246 g) gave compound 11 as a white solid powder (0.084 g, 21%), m.p. 378–380 °C; IR (KBr) νmax 3431 (OH), 1642 (C=C), 1589 (C=N), 1382, 1182 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 7.92 (m, 2H), 8.13 (br. s, 1H), 8.29 (d, J = 6.84 Hz, 2H), 8.92 (m, 1H), 9.14 (br. s, 1H) ppm; 13C-NMR (DMSO-d6) δ 111.54, 120.80, 126.04, 130.71, 131.51, 131.77, 135.07, 142.18, 144.35, 146.60, 146.66, 154.17, 154.43 ppm; anal. C 45.25,H 1.81, N 17.42% calcd. for C15H8F3N5O3S, C 45.57, H 2.04, N 17.72%.
3-Hydroxy-2-(p-tolyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (12). The mixture of 3 and ethyl 2-oxo-2-p-tolylacetate (0.192 g) gave compound 12 as a white solid powder (0.123 g, 36%), m.p. 359–360 °C; IR (KBr) νmax 3430 (OH), 1633 (C=C), 1587 (C=N), 1321, 1176 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 2.39 (s, 3H), 7.36 (m, J = 7.81 Hz, 2H), 8.03 (m, J = 7.81 Hz, 2H), 8.11 (d, J = 5.86 Hz, 1H), 8.91 (d, J = 5.86 Hz, 1H), 9.12 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 21.80, 111.56, 120.77, 128.48, 129.74, 129.76, 142.12, 142.30, 144.37, 146.58, 147.27, 154.33 ppm; anal. C 52.9, H 3.07, N 20.36% calcd. for C15H11N5O3S, C 52.78, H 3.25, N 20.52%.
2-(4-Ethylphenyl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (13). The mixture of 3 and ethyl 2-(4-ethylphenyl)-2-oxoacetate (0.206 g) gave compound 13 as a white solid powder (0.084, 23%), m.p. 375–379 °C; IR (KBr) νmax 3441 (OH), 1633 (C=C), 1587 (C=N), 1321, 1176 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 1.21 (t, J = 7.57 Hz, 3H), 2.69 (q, J = 7.32 Hz, 2H), 7.39 (d, J = 8.30 Hz, 2H), 8.03 (d, J = 8.30 Hz, 2H), 8.11 (d, J = 5.86 Hz, 1H), 8.91 (d, J = 5.86 Hz, 1H), 9.12 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 15.76, 28.77, 111.56, 120.95, 128.50, 128.68, 129.87, 142.41, 144.44, 146.34, 147.66, 148.36, 154.15, 154.23 ppm; anal. C 53.81, H 3.53, N 19.41% calcd. for C16H13N5O3S, C 54.08, H 3.69, N 19.71%.
3-Hydroxy-2-(4-propylphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (14). The mixture of 3 and ethyl 2-oxo-2-(4-propylphenyl)acetate (0.220 g) gave compound 14 as a white solid powder (0.092, 25%), m.p. 343–345 °C; IR (KBr) νmax 3417 (OH), 1627 (C=C), 1583 (C=N), 1318, 1167 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 0.91 (t, J = 7.32 Hz, 3H), 1.56–1.58 (m, 2H), 2.63 (t, J = 7.57 Hz, 2H), 7.37 (d, J = 8.30 Hz, 2H), 8.03 (d, J = 8.30 Hz, 2H), 8.11 (d, J = 5.86 Hz, 1H), 8.91 (d, J = 5.86 Hz, 1H), 9.12 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 14.30, 24.61, 37.79, 111.57, 120.77, 128.76, 129.15, 129.81, 142.31, 144.37, 146.58, 146.65, 147.38, 154.33 ppm; anal. C 55.27, H 4.09, N 18.86% calcd. for C17H15N5O3S, C 55.02, H 3.96, N 18.54%.
3-Hydroxy-2-(4-isopropylphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (15). The mixture of 3 and ethyl 2-(4-isopropylphenyl)-2-oxoacetate (0.220 g) gave compound 15 as a white solid powder (0.092 g, 25%), m.p. 361–362 °C; IR (KBr) νmax 3430 (OH), 1638 (C=C), 1588 (C=N), 1322, 1182 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 1.24 (d, J = 6.84 Hz, 6H), 2.89–3.03 (m, 1H), 7.42 (d, J = 7.81 Hz, 2H), 8.02 (d, J = 8.30 Hz, 2H), 8.10 (d, J = 5.86 Hz, 1H), 8.91 (d, J = 5.86 Hz, 1H), 9.12 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 24.19, 34.07, 111.57, 120.91, 127.06, 128.82, 129.95, 142.42, 144.49, 146.37, 147.70, 152.86, 154.18, 154.29 ppm; anal. C 54.93, H 3.95, N 18.75% calcd. for C17H15N5O3S, C 55.27, H 4.09, N 18.96%.
3-Hydroxy 2-(4-isobutylphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (16). The mixture of 3 and ethyl 2-(4-isobutylphenyl)-2-oxoacetate (0.234 g) gave compound 16 as a white solid powder (0.085 g, 22%), m.p. 359–361 °C; IR (KBr) νmax 3428 (OH), 1634 (C=C), 1589 (C=N), 1322, 1177 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 0.88 (d, J = 6.35 Hz, 6H), 1.89 (m, 1H), 2.53 (d, J = 6.84 Hz, 2H), 7.34 (d, J = 7.81 Hz, 2H), 8.03 (d, J = 7.81 Hz, 2H), 8.12 (d, J = 5.86 Hz, 1H), 8.91 (d, J = 5.37 Hz, 1H), 9.12 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 22.79, 30.23, 45.06, 111.62, 120.76, 128.74, 129.69, 142.37, 144.48, 145.72, 146.42, 147.51, 154.26 ppm; anal. C 55.98, H 4.36, N 18.05% calcd. for C18H17N5O3S, C 56.38, H 4.47, N 18.27%.
2-(4-tert-Butylpheny)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (17). The mixture of 3 and ethyl 2-(4-tert-butylphenyl)-2-oxoacetate (0.234 g) gave compound 17 as a white solid powder (0.234 g, 37%), m.p. 385–387 °C; IR (KBr) νmax 3444 (OH), 1638 (C=C), 1589 (C=N), 1324, 1183 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 1.32 (br. s., 9H), 7.56 (d, J = 7.32 Hz, 2H), 8.01 (d, J = 7.32 Hz, 2H), 8.10 (d, J = 5.37 Hz, 1H), 8.91 (d, J = 5.37 Hz, 1H), 9.12 (br. s., 1H) ppm; 13C-NMR (DMSO-d6) δ 31.58, 35.40, 111.53, 120.76, 125.97, 128.51, 129.69, 142.31, 144.41, 146.58, 147.47, 154.30, 154.90 ppm; anal. C 56.10, H 4.46, N 18.14% calcd. for C18H17N5O3S, C 56.38, H 4.47, N 18.27%.
2-(4-Ethoxyphenyl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (18). The mixture of 3 and ethyl 2-(4-ethoxyphenyl)-2-oxoacetate (0.222 g) gave compound 18 as a white solid powder (0.081 g, 21%), m.p. 321–322 °C; IR (KBr) νmax 3543 (OH), 1626 (C=C), 1581 (C=N), 1316, 1167 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 1.36 (t, J = 7.08 Hz, 3H), 4.12 (q, J = 7.16 Hz, 2H), 7.08 (d, J = 8.79 Hz, 2H), 8.12 (m, 1H), 8.91 (d, J = 5.86 Hz, 1H), 9.11 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 15.11, 64.25, 111.59, 115.18, 120.95, 123.45, 131.61, 142.44, 144.46, 146.32, 147.03, 154.12, 154.41, 161.87 ppm; anal. C 51.55, H 3.40, N 18.49% calcd. for C16H13N5O4S, C 51.75, H 3.53, N 18.86%.
2-(4-Methoxy-3-methylphenyl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (19). The mixture of 3 and ethyl 2-(4-methoxy-3-methylphenyl)-2-oxoacetate (0.236 g) gave compound 19 as a white solid powder (0.091 g, 24%), m.p. 318–320 °C; IR (KBr) νmax 3430 (OH), 1632 (C=C), 1591 (C=N), 1327, 1170 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 2.24 (s, 3H), 3.87 (s, 3H), 7.09 (d, J = 8.79 Hz, 1H), 7.93 (s, 1H), 8.06 (dd, J = 8.79 Hz, 1H), 8.14 (d, J = 5.86 Hz, 1H), 8.92 (d, J = 5.86 Hz, 1H), 9.11 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 16.83, 56.28, 110.69, 111.64, 120.74, 123.08, 126.53, 129.77, 131.42, 142.33, 144.34, 146.53, 146.85, 154.31, 154.43, 160.67 ppm; anal. C 51.88, H 3.45, N 18.50% calcd. for C16H13N5O4S, C 51.75, H 3.53, N 18.86%.
2-(Benzo[d][1,3]dioxol-5-yl)-3-hydroxypyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (20). The mixture of 3 and ethyl 2-(benzo[d][1,3]dioxol-5-yl)-2-oxoacetate (0.222 g) gave compound 20 as a white solid powder (0.084 g, 23%), m.p. 319–320 °C; IR (KBr) νmax 3424 (OH), 1632 (C=C), 1588 (C=N), 1321, 1174 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 6.14 (s, 2H), 7.09 (d, J = 8.30 Hz, 1H), 7.64 (s, 1H), 7.76–7.84 (m, 1H), 8.17 (d, J = 5.86 Hz, 1H), 8.90 (d, J = 6.35 Hz, 1H), 9.11 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 102.50, 108.92, 109.27, 111.74, 120.66, 124.88, 125.43, 142.26, 144.29, 146.39, 146.59, 148.11, 150.58, 154.30 ppm; anal. C 48.41, H 2.36, N 18.51% calcd. for C15H9N5O3S, C 48.52, H 2.44, N 18.86%.
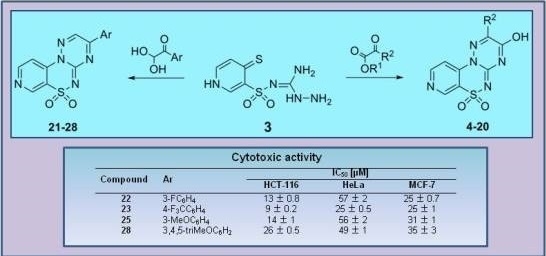
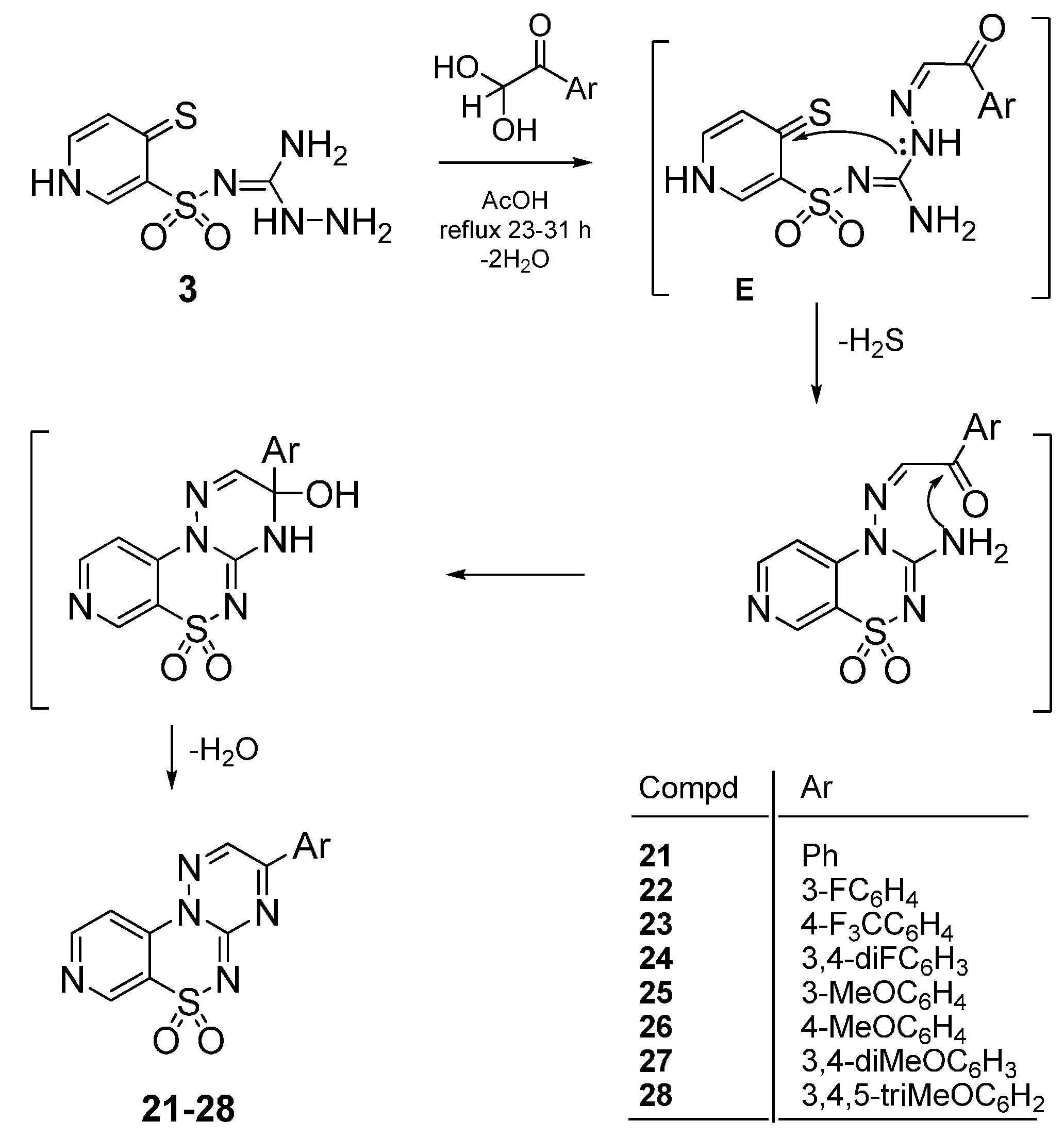
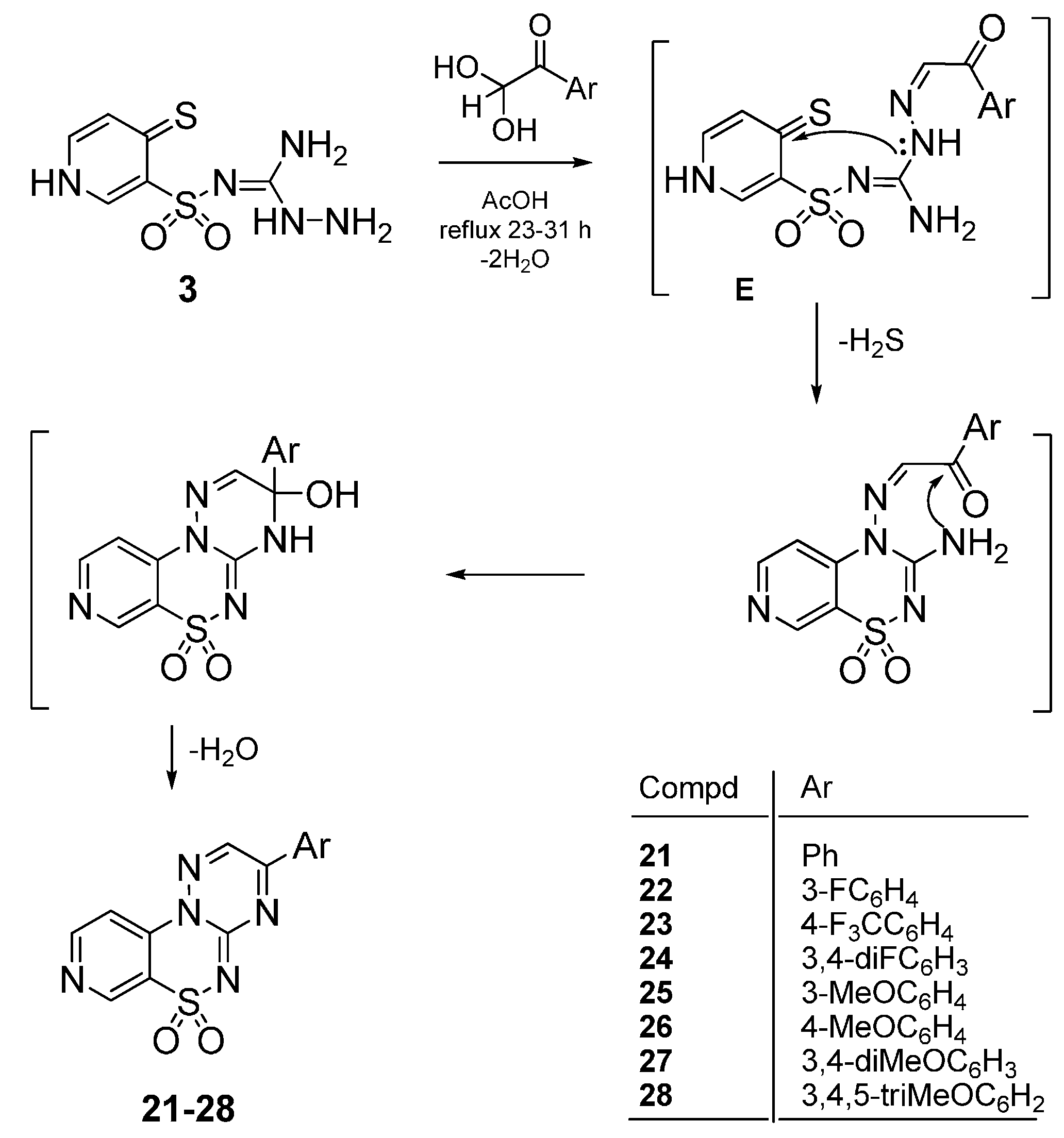
4.2.3. General Procedure for the Preparation of 3-Phenylpyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]-thiadiazine 6,6-dioxides 21–28
The mixture of 3 (2.5 mmol) and the appropriate phenylglyoxal hydrate (2.5 mmol) in glacial acetic acid (6 mL) was stirred at reflux for 23–31 h. After standing at room temperature, the precipi-tate was filtered off and dried. A final purification was described below.
3-Phenylpyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (21). The mixture of 3 and 1-phenyl-2,2-dihydroxyethanone (0.380 g) gave compound 21 as a dark yellow solid powder. The product was purified by extraction of contaminants with boiling ethanol (0.437 g, 56%), m.p. 259–261 °C; IR (KBr) νmax 1575 (C=N), 1315, 1123 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 7.69 (m, 2H); 7.80 (m, 1H); 8.15 (d, J = 4.88 Hz, 1H); 8.43 (d, J = 7.32 Hz, 2H); 9.00 (d, J = 5.37 Hz, 1H); 9.24 (br. s, 1H); 9.51 (br. s, 1H) ppm; 13C-NMR (DMSO-d6) δ 111.95, 120.76, 129.91, 130.20, 132.24, 135.58, 137.37, 142.24, 146.70, 146.87, 154.19, 162.63 ppm; anal. C 53.78, H 2.94, N 22.25% calcd. for C14H9N5O3S, C 54.01, H 2.91, N 22.50%.
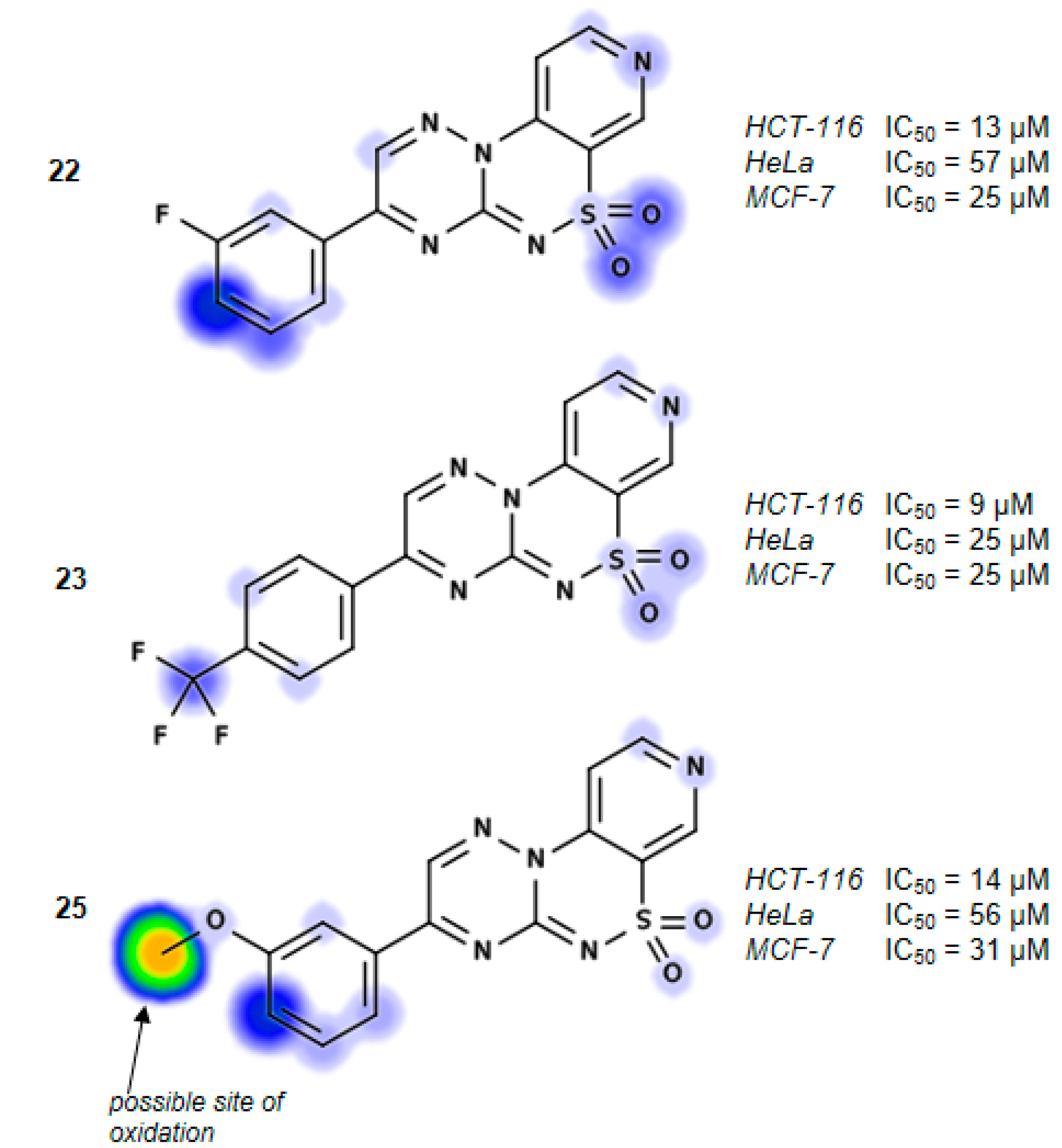
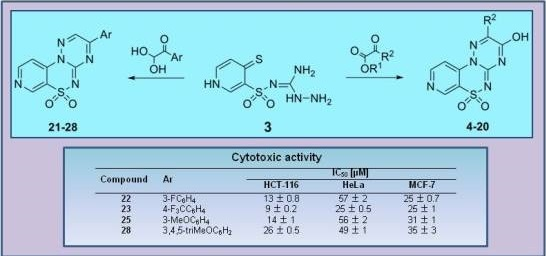
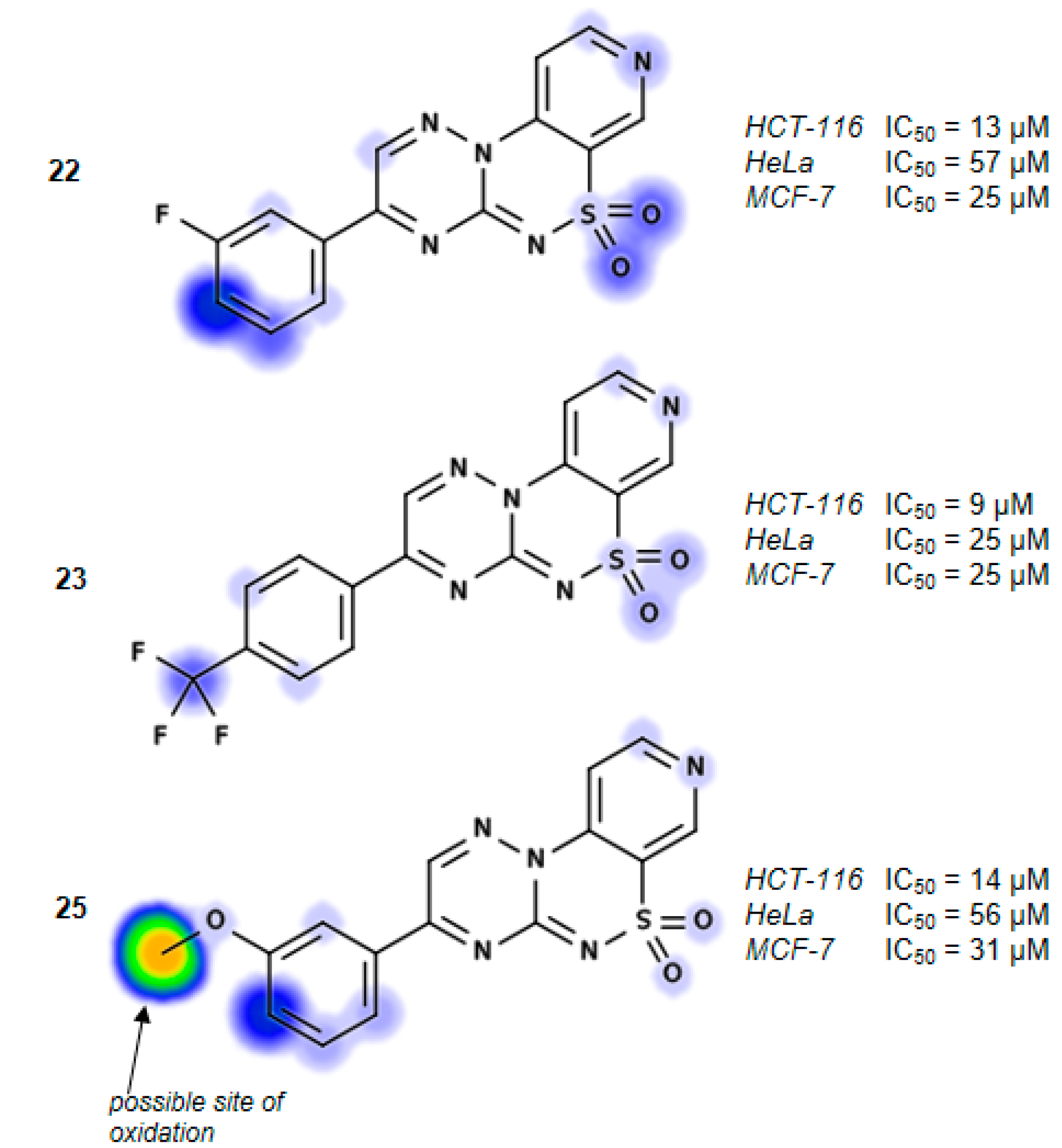
3-(3-Fluorophenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (22). The mixture of 3 and 1-(3-fluorophenyl)-2,2-dihydroxyethanone (0.425 g) gave compound 22 as a white solid powder. The product was purified by extraction of contaminants with boiling benzene and crystallization from acetonitrile (0.172 g, 20%), m.p. 264–266 °C; IR (KBr) νmax 1580 (C=N), 1306, 1119 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 7.68 (m, 1H), 7.75 (m, 1H), 8.18 (d, 1H), 8.28 (m, 2H), 9.02 (d, 1H), 9.24 (s, 1H), 9.51 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 112.17, 116.38, 116.57, 120.89, 122.46, 122.63, 126.26, 126.28, 132.57, 132.64, 134.68, 134.74, 137.50, 142.36, 146.71, 147.09, 154.45, 161.91, 162.25, 164.21 ppm; anal. C 50.83, H 2.49, N 20.92% calcd. for C14H8FN5O2S, C 51.06, H 2.45, N 21.27%.
3-[4-(Trifluoromethyl)phenyl]pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (23). The mixture of 3 and 2,2-dihydroxy-1-[4-(trifluoromethyl)phenyl]ethanone (0.550 g) gave compound 23 as a white solid powder. The product was purified by extraction of contaminants with boiling benzene and crystallization from acetonitrile (0.151 g, 15%), m.p. 292–295 °C; IR (KBr) νmax 1594 (C=N), 1324, 1117 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 8.07–8.18 (m, 3H), 8.60–8.62 (m, 2H), 9.02–9.03 (m, 1H), 9.25 (s, 1H), 9.56 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 112.20, 120.89, 127.13, 127.15, 130.80, 136.16, 137.63, 142.33, 146.73, 147.12, 154.50, 162.03 ppm; anal. C 47.65, H 2.13,N 18.24% calcd. for C15H8F3N5O2S, C 47.50, H 2.13, N 18.46%.
3-(3,4-Difluorophenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (24). The mixture of 3 and 1-(3,4-difluorophenyl)-2,2-dihydroxyethanone (0.470 g) gave compound 24 as a white solid powder. The product was purified by extraction of contaminants with boiling ethanol (0.197 g, 24%), m.p. 309–311 °C; IR (KBr) νmax 1582 (C=N), 1313, 1120 (SO2) cm−1; 1H-NMR (DMSO-d6)δ 7.70 (t, J = 7.57 Hz, 1H), 8.18 (d, J = 5.86 Hz, 1H), 8.30 (d, J = 8.30 Hz, 1H), 8.44 (d, J = 10.74 Hz, 1H), 9.02 (d, J = 5.86 Hz, 1H), 9.25 (s, 1H), 9.50 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 110.98, 113.24, 115.50, 122.52, 126.23, 131.58, 131.72, 131.98, 132.04, 133.14, 136.99, 142.11, 144.45, 146.69, 147.73, 160.26 ppm; anal. C 48.12, H 2.13, N 19.88% calcd. for C14H7F2N5O2S, C 48.42, H 2.03, N 20.17%.
3-(3-Methoxyphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (25). The mixture of 3 and 2,2-dihydroxy-1-(3-methoxyphenyl)ethanone (0.455 g) gave compound 25 as a yellow solid powder. The product was purified by extraction of contaminants with boiling ethanol (0.400 g, 47%), m.p. 270–272 °C; IR (KBr) νmax 1586 (C=N), 1299, 1164 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 3.90 (s, 3H), 7.39 (m, 1H), 7.62 (t, 1H), 7.91 (s, 1H), 8.03 (m, 1H), 8,16 (m, 1H), 9.02 (m, 1H), 9.23 (s, 1H), 9.52 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 55.65, 111.50, 113.53, 120.25, 121.34, 122.00, 131.00, 133.08, 137.00, 141.74, 146.14, 146.80, 153.75, 160.01, 161.90 ppm; anal. C 52.44, H 3.07, N 20.29% calcd. for C15H11N5O3S, C 52.78, H 3.25, N 20.52%.
3-(4-Methoxyphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (26). The mixture of 3 and 2,2-dihydroxy-1-(4-methoxyphenyl)ethanone (0.455 g) gave compound 26 as a yellow solid powder. The product was purified by crystallization from acetonitrile (0.169 g, 25%), m.p. 276–278 °C; IR (KBr) νmax 1579 (C=N), 1301, 1164 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 3.93 (s, 3H), 7.25 (d, 2H), 8.12–8.13 (m, 2H), 8.44–8.46 (m, 2H), 8.98–8.99 (m, 1H), 9.20 (s, 1H), 9.46 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 56.72, 112.00, 116.10, 121.03, 124.72, 132.75, 137.45, 142.50, 146.94, 154.25, 161.56, 165.93 ppm; anal. C 52.65, H 3.03, N 20.34% calcd. for C15H11N5O3S, C 52.78, H 3.25, N 20.52%.
3-(3,4-Dimethoxyphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (27). The mixture of 3 and 1-(3,4-dimethoxyphenyl)-2,2-dihydroxyethanone (0.530 g) gave compound 27 as an orange solid powder. The product was purified by extraction of contaminants with boiling ethanol (0.490 g, 53%), m.p. 264–265 °C; IR (KBr) νmax 1566 (C=N), 1302, 1111(SO2) cm−1; 1H-NMR (DMSO-d6) δ 3.91 (s, 3H), 3.94 (s, 3H), 7.27–7.28 (d, 1H), 7.86–7.90 (m, 1H), 8.12–8.13 (s, 1H), 8.17–8.21 (m, 1H), 8.98–8.99 (m,1H), 9.20 (s, 1H), 9.51 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 55.37, 55.69, 111.24, 111.54, 116.03, 122.62, 123.62, 127.69, 128.21, 137.73, 141.83, 144.37, 147.40, 147.95, 149.98, 158.12 ppm; anal. C 51.37H 3.18, N 18.52% calcd. for C16H13N5O4S, C 51.75, H 3.53, N 18.86%.
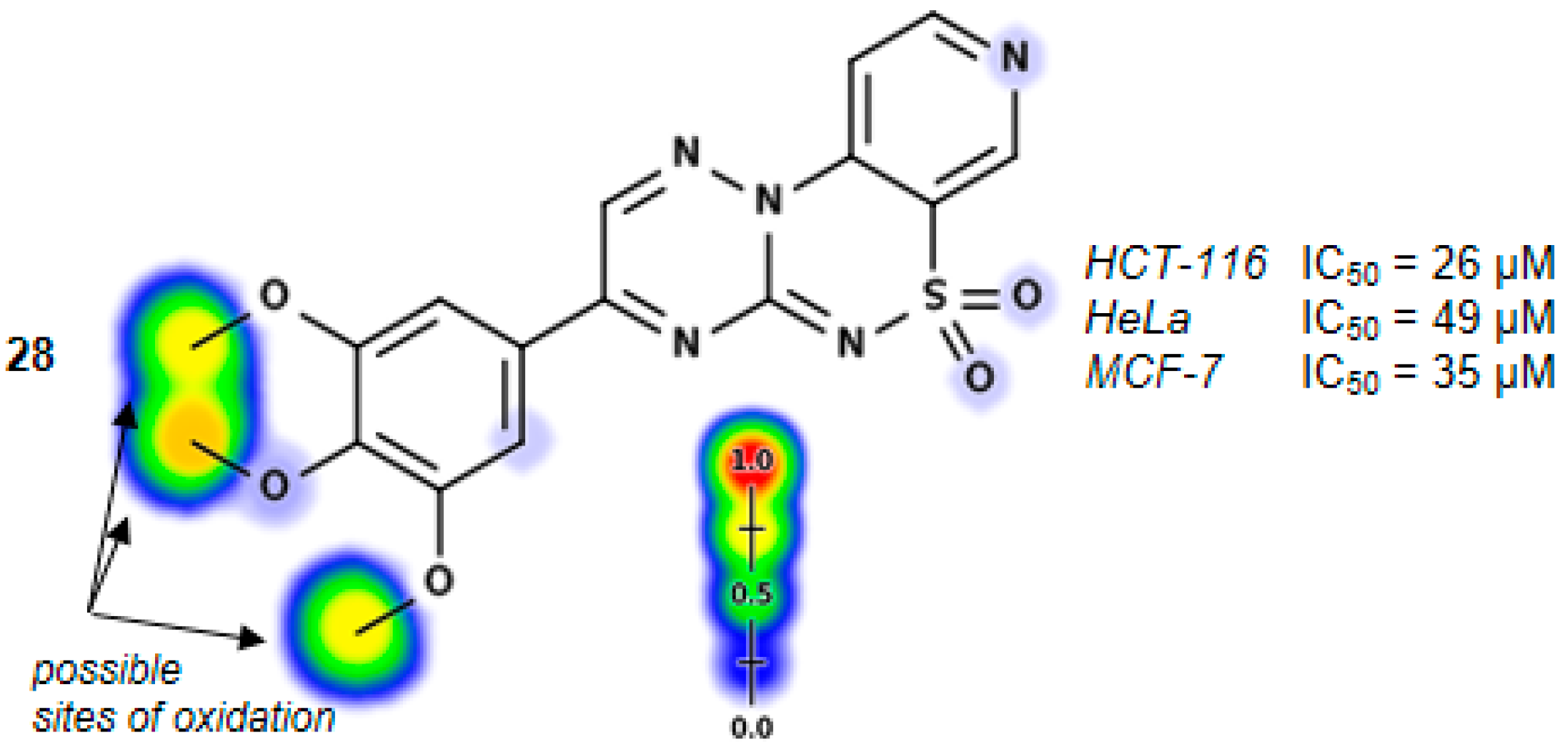
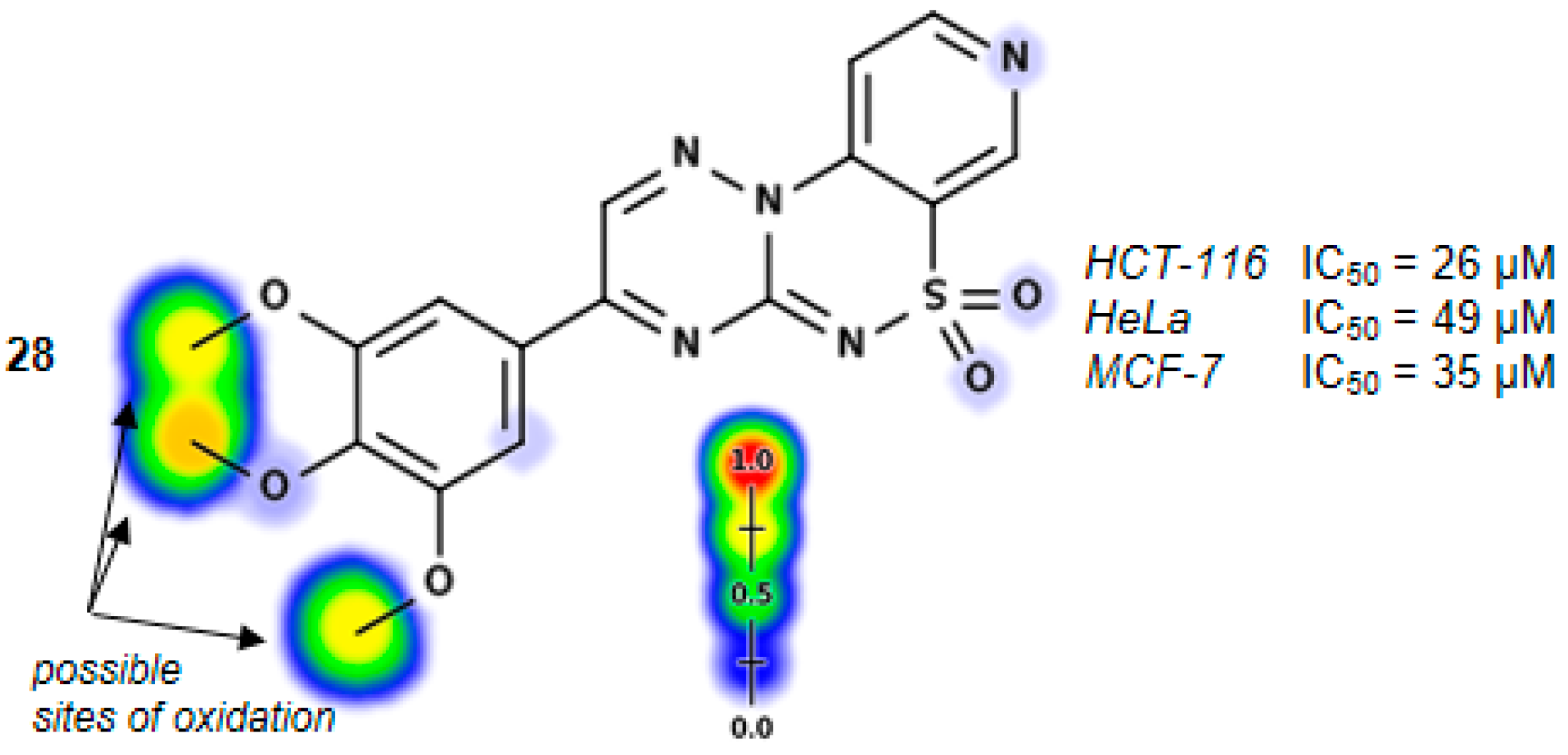
3-(3,4,5-Trimethoxyphenyl)pyrido[4,3-e][1,2,4]triazino[3,2-c][1,2,4]thiadiazine 6,6-dioxide (28). The mixture of 3 and 2,2-dihydroxy-1-(3,4,5-trimethoxyphenyl)ethanone (0.605 g) gave compound 28 as a red solid powder. The product was purified by extraction of contaminants with boiling acetonitrile (0.283 g, 28%), m.p. 257–259 °C; IR (KBr) νmax 1572 (C=N), 1316, 1114 (SO2) cm−1; 1H-NMR (DMSO-d6) δ 3.84 (s, 3H), 3.94 (s, 6H), 7.73 (s, 2H), 8.15–8.16 (m, 1H), 9.00–9.01 (m, 1H), 9.22 (s, 1H), 9.59 (s, 1H) ppm; 13C-NMR (DMSO-d6) δ 55.74, 61.12, 108.28, 116.12, 122.61, 125.65, 137.64,141.92, 144.52, 146.15, 147.12, 147.81, 153.22 ppm; anal. C 50.63, H 3.65, N 17.30% calcd. for C17H15N5O5S, C 50.87, H 3.77, N 17.45%.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}