
Reduced Reactivity of Amines against Nucleophilic Substitution via Reversible Reaction with Carbon Dioxide

Abstract
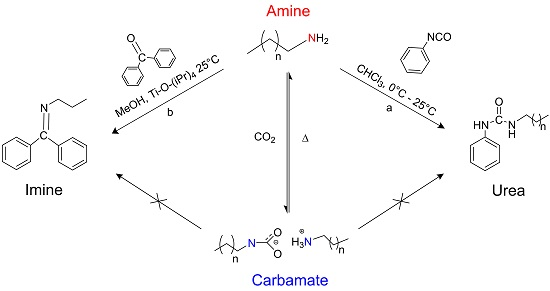
: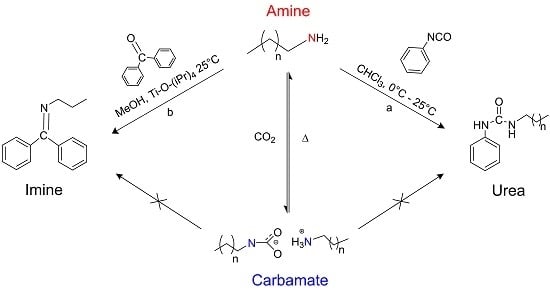
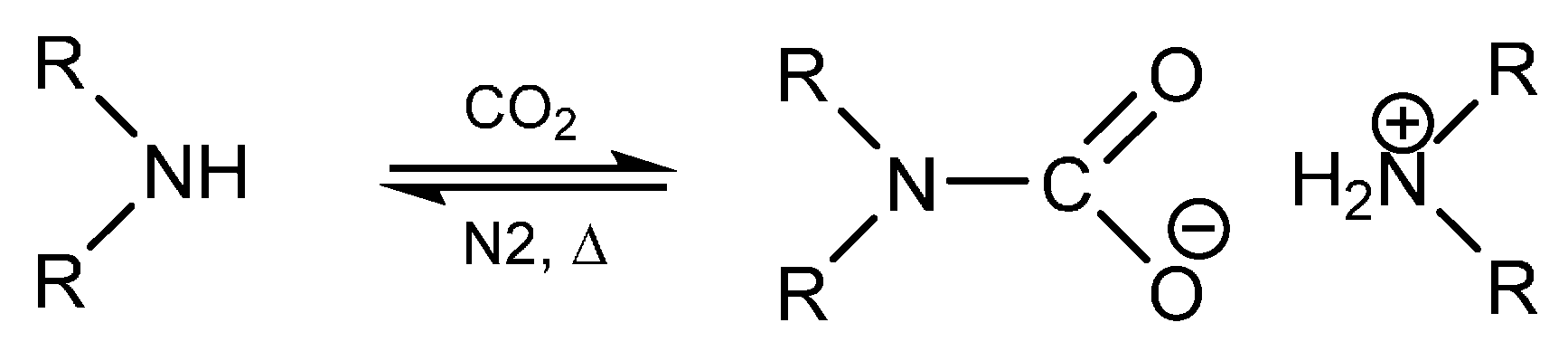
1. Introduction
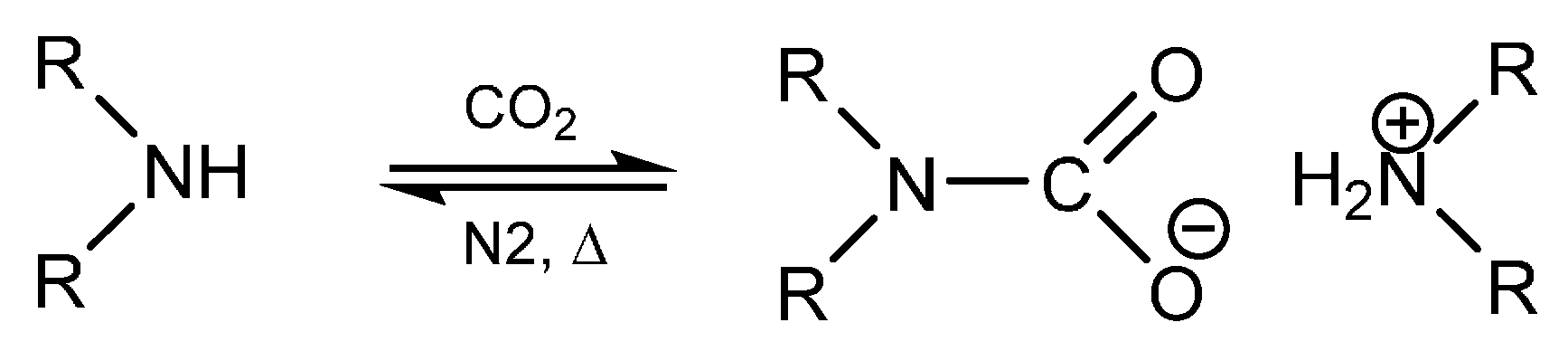
2. Results and Discussion
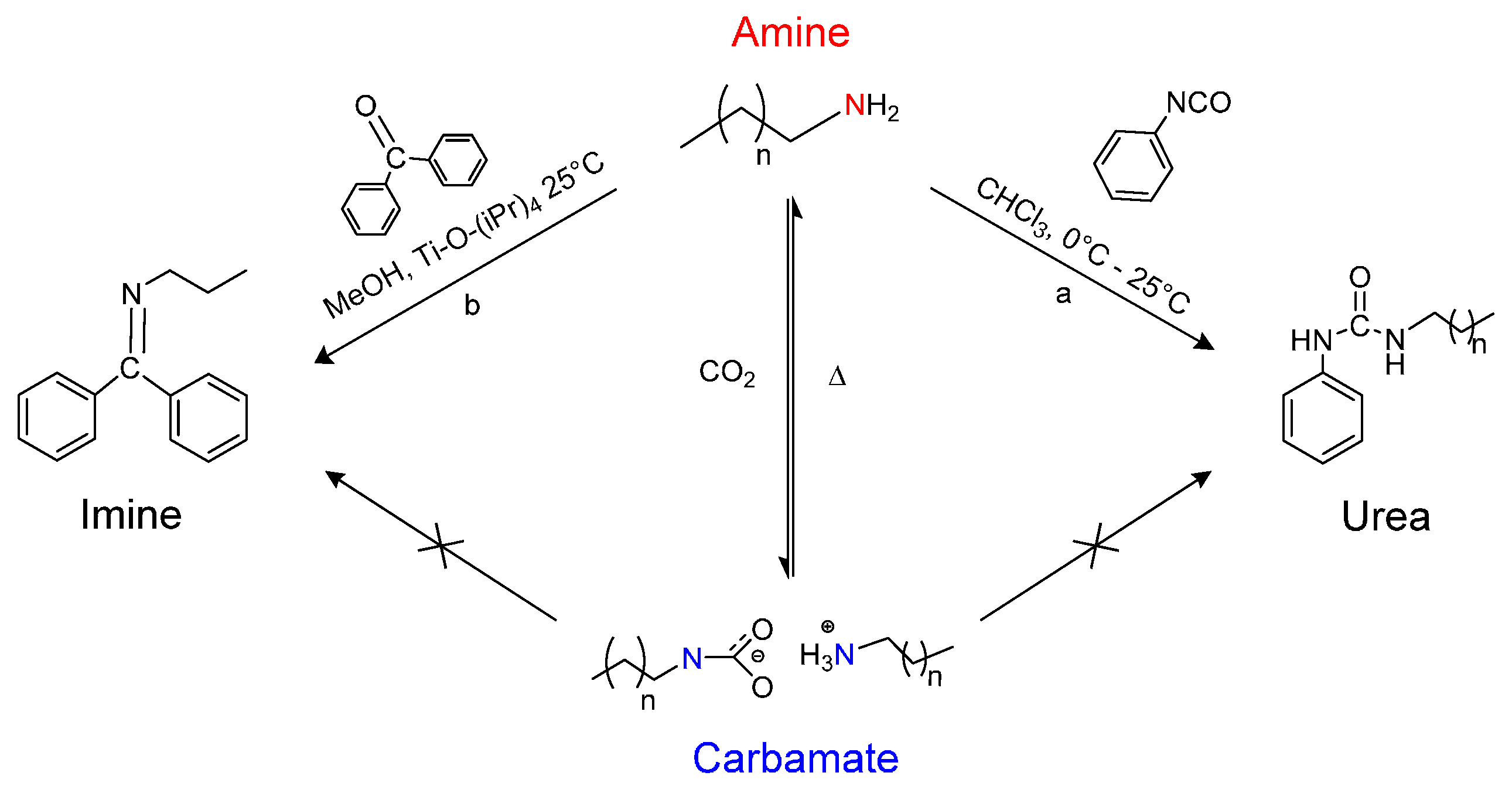
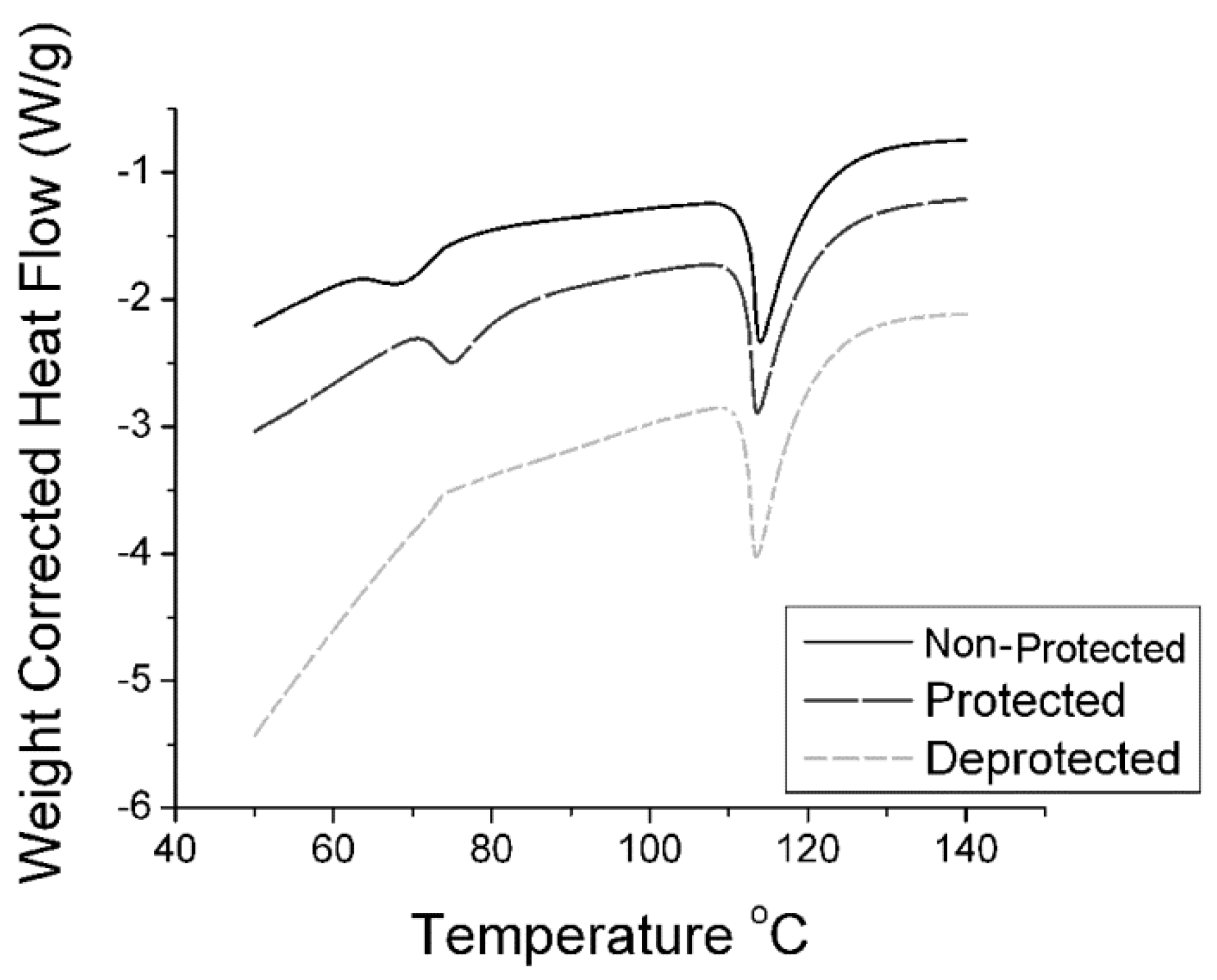
2.1. Synthesis of n-Alkyl, n-Phenyl Urea

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amine | n | Percentage Yield (%) | ||
|---|---|---|---|---|
| Non-Protected | Protected | De-Protected | ||
| Propylamine | 1 | 95 | 28 | 85 |
| Hexylamine | 4 | 85 | 20 | 80 |
| Decylamine | 8 | 91 | 38 | 93 |
| Octadecylamine | 16 | 90 | 69 | 72 |
| Entry | MP (°C) | Recrystalization Temp. (°C) |
|---|---|---|
| n-phenyl, n-propyl urea | 112–114 | 89 |
| n-phenyl, n-hexyl urea | 68–70 | 47 |
| n-phenyl, n-decyl urea | 81–83 | 61 |
| n-phenyl, n-stearyl urea | 95–97 | 84 |
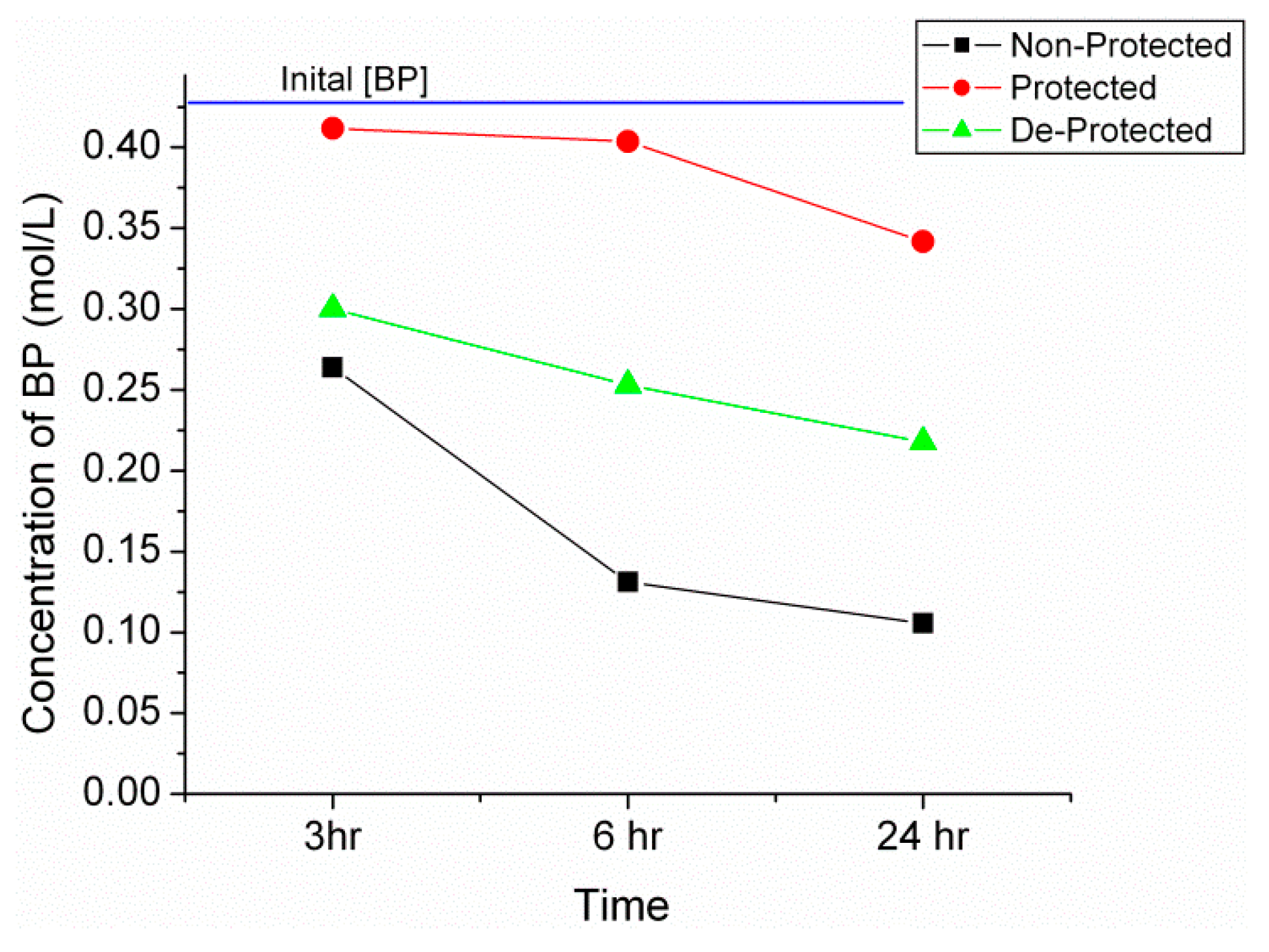
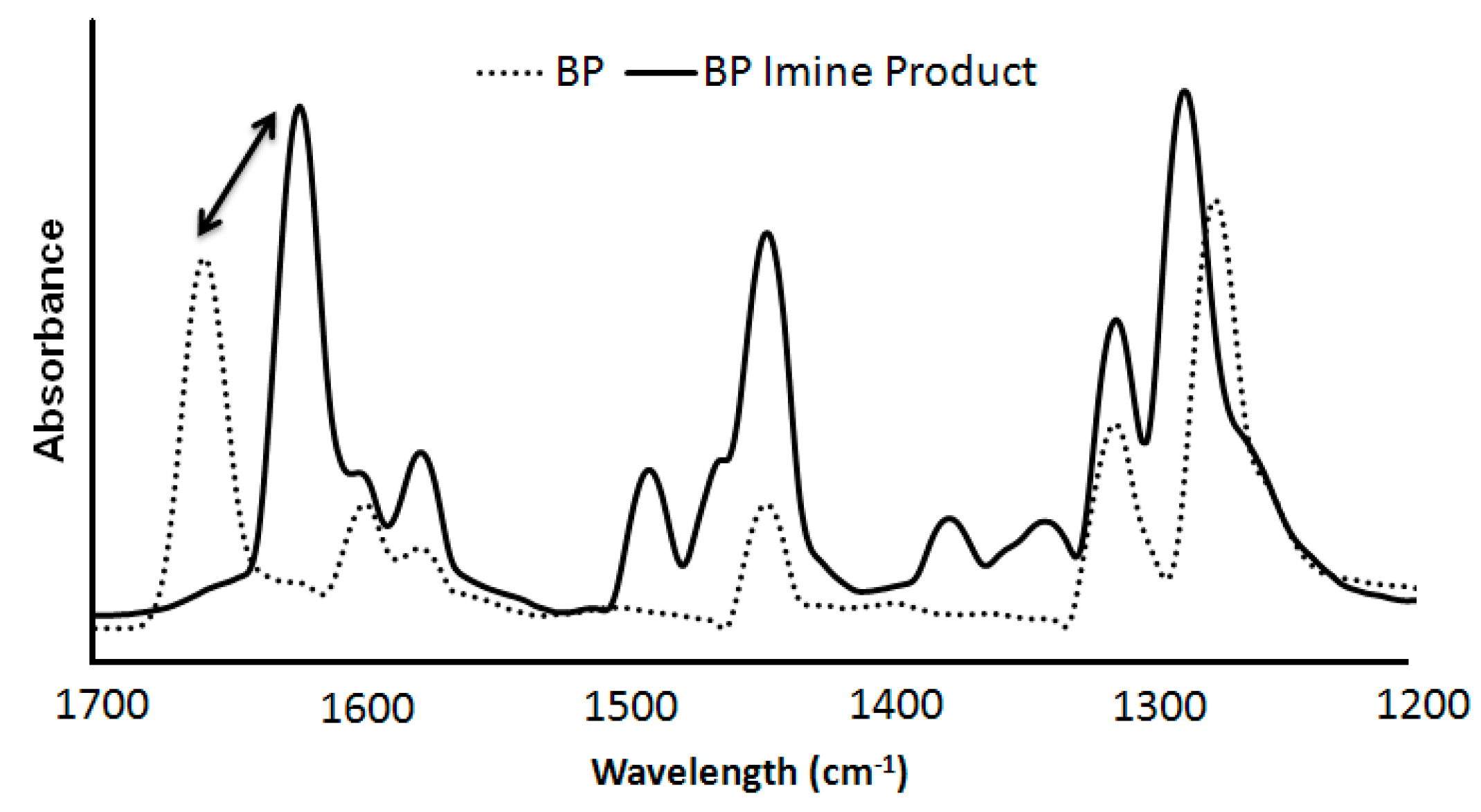
2.2. Synthesis of n-Propyl, Benzophenone(BP) Imine
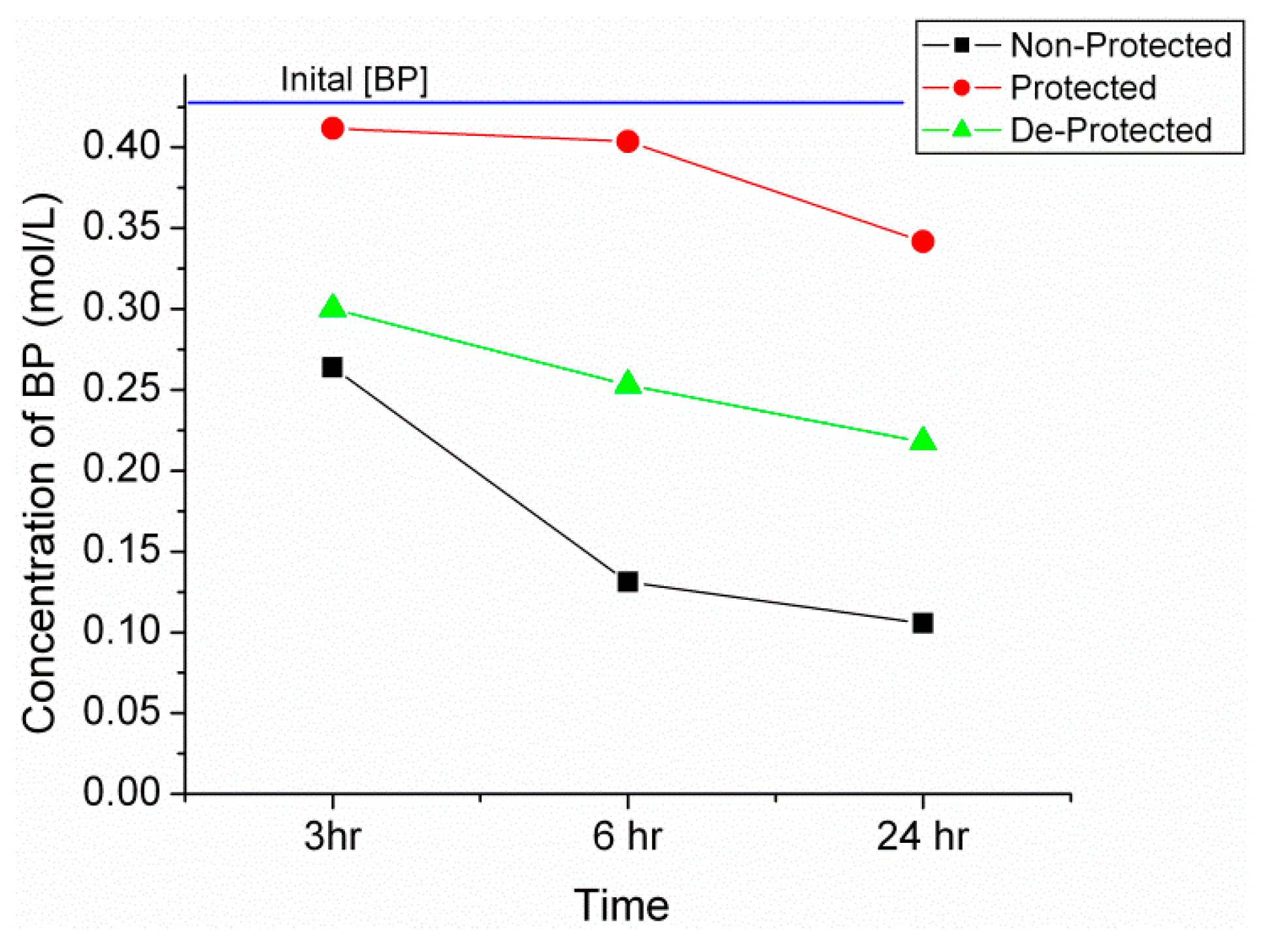

| Sample | Time (h) | Calculated IR Conversion (%) | Conversion from GC (%) |
|---|---|---|---|
| Non-protected | 3 | 35 | 33 |
| Non-protected | 12 | 55 | 65 |
| Protected | 3 | 16 | 18 |
| Protected | 12 | 26 | 20 * |
3. Materials and Methods
3.1. General Amine Protection
3.2. General Amine De-Protection
3.3. General Characterization
3.4. Synthesis of n-Alkyl, n-Phenyl Urea
3.5. Synthesis of n-Propyl, Benzophenone(BP) Imine
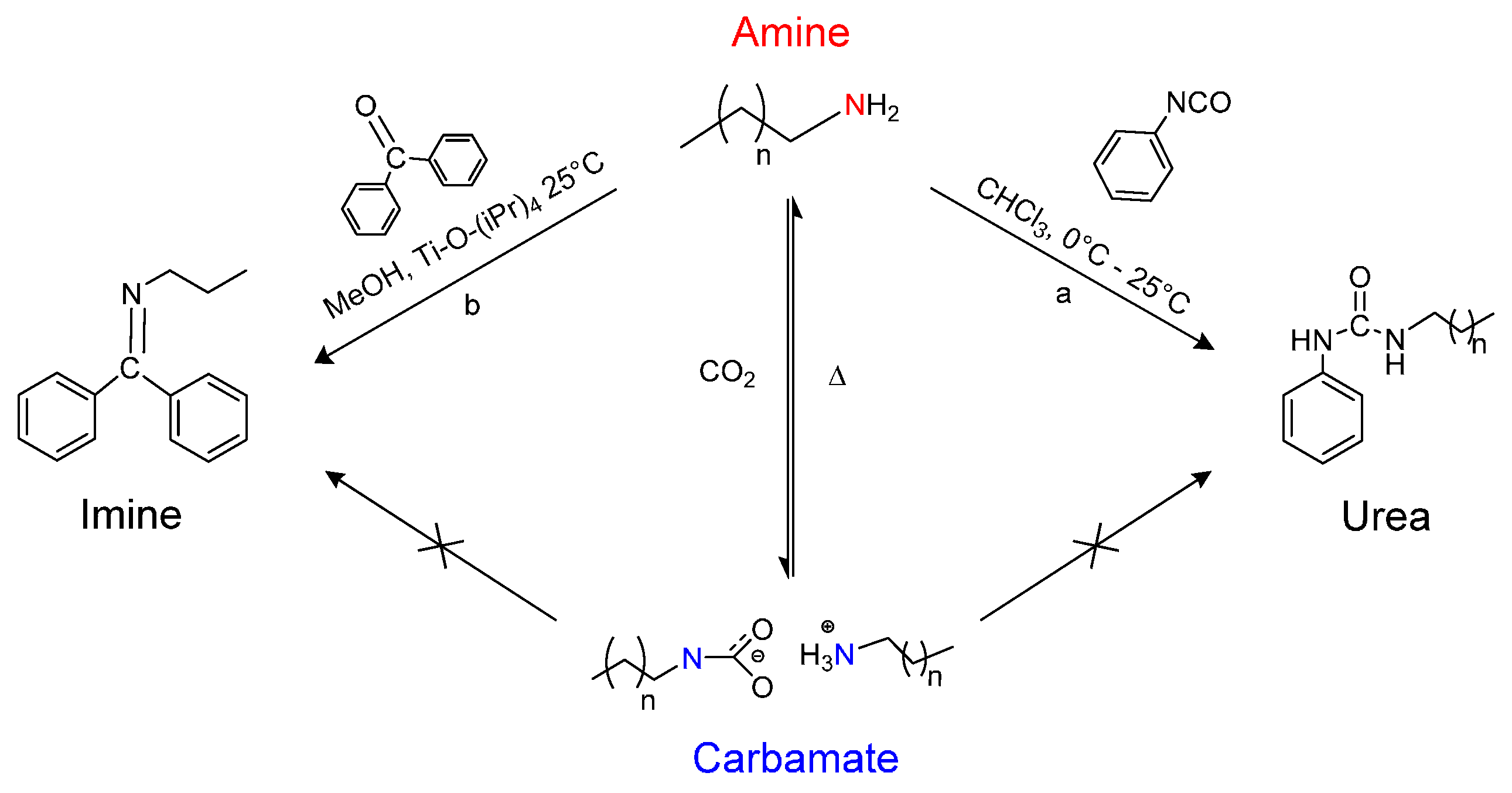
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jensen, A.; Faurholt, C. Studies on carbamates. V. The carbamates of alpha-alanine and beta-alanine. Acta Chem. Scand. 1952, 6, 385–394. [Google Scholar] [CrossRef]
- Jensen, A.; Jensen, B.J.; Faurholt, C. Studies on carbamates. Vi. The carbamate of glycine. Acta Chem. Scand. 1952, 6, 395–397. [Google Scholar] [CrossRef]
- Olsen, J.; Vejlby, K.; Faurholt, C. Studies on carbamates. Vii. The carbamates of n-propylamine and iso-propylamine. Acta Chem. Scand. 1952, 6, 398–403. [Google Scholar] [CrossRef]
- Werner, E.A. Cxvii.-the constitution of carbamides. Part xi. The mechanism of the synthesis of urea from ammonium carbamate. The preparation of certain mixed tri-substituted carbamates and dithiocarbamates. J. Chem. Soc. Trans. 1920, 117, 1046–1053. [Google Scholar]
- Fichter, F.; Becker, B. Über die bildung ysmmetrisch dialkylierter harnstoffe durch erhitzen der entsprechenden carbaminate. Ber. Dtsch. Chem. Ges. 1911, 44, 3481–3485. [Google Scholar] [CrossRef]
- Murphy, L.J.; Robertson, K.N.; Kemp, R.A.; Tuononen, H.M.; Clyburne, J.A.C. Structurally simple complexes of CO2. Chem. Commun. 2015, 51, 3942–3956. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.Y.; Yang, L.P.; Hu, W.H. Organic reactions with carbon dioxide. Prog. Chem. 2009, 21, 1217–1228. [Google Scholar]
- Jessop, P.G.; Heldebrant, D.J.; Li, X.; Eckertt, C.A.; Liotta, C.L. Green chemistry: Reversible nonpolar-to-polar solvent. Nature 2005, 436, 1102–1102. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Weiss, R.G. Chemically reversible organogels via “latent” gelators. Aliphatic amines with carbon dioxide and their ammonium carbamates. Langmuir 2002, 18, 7124–7135. [Google Scholar] [CrossRef]
- Xu, H.; Rudkevich, D.M. CO2 in supramolecular chemistry: Preparation of switchable supramolecular polymers. Chem. Eur. J. 2004, 10, 5432–5442. [Google Scholar] [CrossRef] [PubMed]
- George, M.; Weiss, R.G. Primary alkyl amines as latent gelators and their organogel adducts with neutral triatomic molecules. Langmuir 2003, 19, 1017–1025. [Google Scholar] [CrossRef]
- Mohammed, F.S.; Wuttigul, S.; Kitchens, C.L. Dynamic surface properties of amino-terminated self-assembled monolayers incorporating reversible CO2 chemistry. Ind. Eng. Chem. Res. 2011, 50, 8034–8041. [Google Scholar] [CrossRef]
- Blasucci, V.M.; Hart, R.; Pollet, P.; Liotta, C.L.; Eckert, C.A. Reversible ionic liquids designed for facile separations. Fluid Phase Equilib. 2010, 294, 1–6. [Google Scholar] [CrossRef]
- Pollet, P.; Eckert, C.A.; Liotta, C.L. Switchable solvents. Chem. Sci. 2011, 2, 609–614. [Google Scholar] [CrossRef]
- Jessop, P.G.; Mercer, S.M.; Heldebrant, D.J. CO2-triggered switchable solvents, surfactants, and other materials. Energy Environ. Sci. 2012, 5, 7240–7253. [Google Scholar] [CrossRef]
- Kerton, F. Tunable and switchable solvent systems. In Alternative Solvents for Green Chemistry, 2nd ed.; RSC Publishing: London, UK, 2013; pp. 262–284. [Google Scholar]
- Switzer, J.R.; Ethier, A.L.; Flack, K.M.; Biddinger, E.J.; Gelbaum, L.; Pollet, P.; Eckert, C.A.; Liotta, C.L. Reversible ionic liquid stabilized carbamic acids: A pathway toward enhanced CO2 capture. Ind. Eng. Chem. Res. 2013, 52, 13159–13163. [Google Scholar] [CrossRef]
- Pollet, P.; Davey, E.A.; Urena-Benavides, E.E.; Eckert, C.A.; Liotta, C.L. Solvents for sustainable chemical processes. Green Chem. 2014, 16, 1034–1055. [Google Scholar] [CrossRef]
- Jessop, P.G. Switchable solvents as media for synthesis and separations. Aldrichimica Acta 2015, 48, 18–21. [Google Scholar]
- Blasucci, V.; Hart, R.; Mestre, V.L.; Hahne, D.J.; Burlager, M.; Huttenhower, H.; Thio, B.J.R.; Pollet, P.; Liotta, C.L.; Eckert, C.A. Single component, reversible ionic liquids for energy applications. Fuel 2010, 89, 1315–1319. [Google Scholar] [CrossRef]
- Shannon, M.S.; Bara, J.E. Reactive and reversible ionic liquids for CO2 capture and acid gas removal. Sep. Sci. Technol. 2012, 47, 178–188. [Google Scholar] [CrossRef]
- Phan, L.; Chiu, D.; Heldebrant, D.J.; Huttenhower, H.; John, E.; Li, X.W.; Pollet, P.; Wang, R.Y.; Eckert, C.A.; Liotta, C.L.; et al. Switchable solvents consisting of amidine/alcohol or guanidine/alcohol mixtures. Ind. Eng. Chem. Res. 2008, 47, 539–545. [Google Scholar] [CrossRef]
- Yang, Y.L.; Xie, H.B.; Liu, E.H. Acylation of cellulose in reversible ionic liquids. Green Chem. 2014, 16, 3018–3023. [Google Scholar] [CrossRef]
- Liu, Y.X.; Jessop, P.G.; Cunningham, M.; Eckert, C.A.; Liotta, C.L. Switchable surfactants. Science 2006, 313, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Alauzun, J.; Besson, E.; Mehdi, A.; Reye, C.; Corriu, R.J.P. Reversible covalent chemistry of CO2: An opportunity for nano-structured hybrid organic-inorganic materials. Chem. Mater. 2008, 20, 503–513. [Google Scholar] [CrossRef]
- Wittmann, K.; Wisniewski, W.; Mynott, R.; Leitner, W.; Kranemann, C.L.; Rische, T.; Eilbracht, P.; Kluwer, S.; Ernsting, J.M.; Elsevier, C.L. Supercritical carbon dioxide as solvent and temporary protecting group for rhodium-catalyzed hydroaminomethylation. Chem. Eur. J. 2001, 7, 4584–4589. [Google Scholar] [CrossRef]
- Xie, X.; Liotta, C.L.; Eckert, C.A. CO2-protected amine formation from nitrile and imine hydrogenation in gas-expanded liquids. Ind. Eng. Chem. Res. 2004, 43, 7907–7911. [Google Scholar] [CrossRef]
- Tom, N.J.; Simon, W.M.; Frost, H.N.; Ewing, M. Deprotection of a primary boc group under basic conditions. Tetrahedron Lett. 2004, 45, 905–906. [Google Scholar] [CrossRef]
- Lutz, C.; Lutz, V.; Knochel, P. Enantioselective synthesis of 1,2-, 1,3- and 1,4- aminoalcohols by the addition of dialkylzincs to 1,2-, 1,3- and 1,4- aminoaldehydes. Tetrahedron 1998, 54, 6385–6402. [Google Scholar] [CrossRef]
- Ribiere, P.; Declerck, V.; Martinez, J.; Lamaty, F. 2-(trimethylsilyl)ethanesulfonyl (or ses) group in amine protection and activation. Chem. Rev. 2006, 106, 2249–2269. [Google Scholar] [CrossRef] [PubMed]
- Zajac, M.A. An application of borane as a protecting group for pyridine. J. Org. Chem. 2008, 73, 6899–6901. [Google Scholar] [CrossRef] [PubMed]
- Chankeshwara, S.V.; Chakraborti, A.K. Catalyst-free chemoselective n-tert-butyloxycarbonylation of amines in water. Org. Lett. 2006, 8, 3259–3262. [Google Scholar] [CrossRef] [PubMed]
- Agami, C.; Couty, F. The reactivity of the n-boc protecting group: An underrated feature. Tetrahedron 2002, 58, 2701–2724. [Google Scholar] [CrossRef]
- Heydari, A.; Khaksar, S.; Tajbakhsh, M. 1,1,1,3,3,3-hexafluoroisopropanol: A recyclable organocatalyst for n-boc protection of amines. Synthesis 2008, 2008, 3126–3130. [Google Scholar] [CrossRef]
- Perron, V.R.; Abbott, S.; Moreau, N.; Lee, D.; Penney, C.; Zacharie, B. A method for the selective protection of aromatic amines in the presence of aliphatic amines. Synthesis 2009, 2009, 283–289. [Google Scholar]
- Peeters, A.; Ameloot, R.; de Vos, D.E. Carbon dioxide as a reversible amine-protecting agent in selective michael additions and acylations. Green Chem. 2013, 15, 1550–1557. [Google Scholar] [CrossRef]
- Ethier, A.; Switzer, J.; Rumple, A.; Medina-Ramos, W.; Li, Z.; Fisk, J.; Holden, B.; Gelbaum, L.; Pollet, P.; Eckert, C.; et al. The effects of solvent and added bases on the protection of benzylamines with carbon dioxide. Processes 2015, 3, 497–513. [Google Scholar] [CrossRef]
- Bathini, T.; Rawat, V.S.; Bojja, S. In situ protection and deprotection of amines for iron catalyzed oxidative amidation of aldehydes. Tetrahedron Lett. 2015, 56, 5656–5660. [Google Scholar] [CrossRef]
- Perveen, S.; Khan, K.M.; Lodhi, M.A.; Choudhary, M.I.; Voelter, W. Urease and alpha-chymotrypsin inhibitory effects of selected urea derivatives. Lett. Drug Des. Discov. 2008, 5, 401–405. [Google Scholar]
- Zhang, W.; Sita, L.R. Investigation of dynamic intra- and intermolecular processes within a tether-length dependent series of group 4 bimetallic initiators for stereomodulated degenerative transfer living ziegler-natta propene polymerization. Adv. Synth. Catal. 2008, 350, 439–447. [Google Scholar] [CrossRef]
- lzdebski, J.; Pawlak, D. A new convenient method for the synthesis of symmetrical and unsymmetrical n,n’-disubstituted ureas. Synthesis 2002, 1989, 423–425. [Google Scholar] [CrossRef]
- Abdel-Magid, A.; Carson, K.G.; Harris, B.D.; Maryanoff, C.A.; Shah, R.D. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. Studies on direct and indirect reductive amination procedures 1. J. Org. Chem. 1996, 61, 3849–3862. [Google Scholar] [CrossRef] [PubMed]
- Kumpaty, H.J.; Bhattacharyya, S.; Rehr, E.W.; Gonzalez, A.M. Selective access to secondary amines by a highly controlled reductive mono-n-alkylation of primary amines. Synthesis 2003, 2206–2210. [Google Scholar] [CrossRef]
- Salmi, C.; Letourneux, Y.; Brunel, J.M. Efficient diastereoselective titanium(iv) reductive amination of ketones. Lett. Org. Chem. 2006, 3, 384–389. [Google Scholar] [CrossRef]
- Salmi, C.; Letourneux, Y.; Brunel, J.M. Efficient synthesis of various secondary amines through a titanium(iv) isopropoxide-mediated reductive amination of ketones. Lett. Org. Chem. 2006, 3, 396–401. [Google Scholar] [CrossRef]
- Salmi, C.; Loncle, C.; Letourneux, Y.; Brunel, J.M. Efficient preparation of secondary aminoalcohols through a Ti(iv) reductive amination procedure. Application to the synthesis and antibacterial evaluation of new 3 beta-n-[hydroxyalkyl]aminosteroid derivatives. Tetrahedron 2008, 64, 4453–4459. [Google Scholar] [CrossRef]
- Moretti, I.; Torre, G. A convenient method for the preparation of n-alkyl benzophenone imines. Synthesis 1970, 1970, 141. [Google Scholar] [CrossRef]
- Valli, V.L.K.; Alper, H. A simple, convenient, and efficient method for the synthesis of isocyanates from urethanes. J. Org. Chem. 1995, 60, 257–258. [Google Scholar] [CrossRef]
- Balaraman, E.; Gunanathan, C.; Zhang, J.; Shimon, L.J.W.; Milstein, D. Efficient hydrogenation of organic carbonates, carbamates and formates indicates alternative routes to methanol based on CO2 and CO. Nat. Chem. 2011, 3, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, F.S.; Kitchens, C.L. Reduced Reactivity of Amines against Nucleophilic Substitution via Reversible Reaction with Carbon Dioxide. Molecules 2016, 21, 24. https://doi.org/10.3390/molecules21010024
Mohammed FS, Kitchens CL. Reduced Reactivity of Amines against Nucleophilic Substitution via Reversible Reaction with Carbon Dioxide. Molecules. 2016; 21(1):24. https://doi.org/10.3390/molecules21010024
Chicago/Turabian StyleMohammed, Fiaz S., and Christopher L. Kitchens. 2016. "Reduced Reactivity of Amines against Nucleophilic Substitution via Reversible Reaction with Carbon Dioxide" Molecules 21, no. 1: 24. https://doi.org/10.3390/molecules21010024
APA StyleMohammed, F. S., & Kitchens, C. L. (2016). Reduced Reactivity of Amines against Nucleophilic Substitution via Reversible Reaction with Carbon Dioxide. Molecules, 21(1), 24. https://doi.org/10.3390/molecules21010024