
Extended Functional Groups (EFG): An Efficient Set for Chemical Characterization and Structure-Activity Relationship Studies of Chemical Compounds




Abstract
:
1. Introduction
2. Results and Discussion
2.1. Functional Groups as Descriptors

{kind=link}
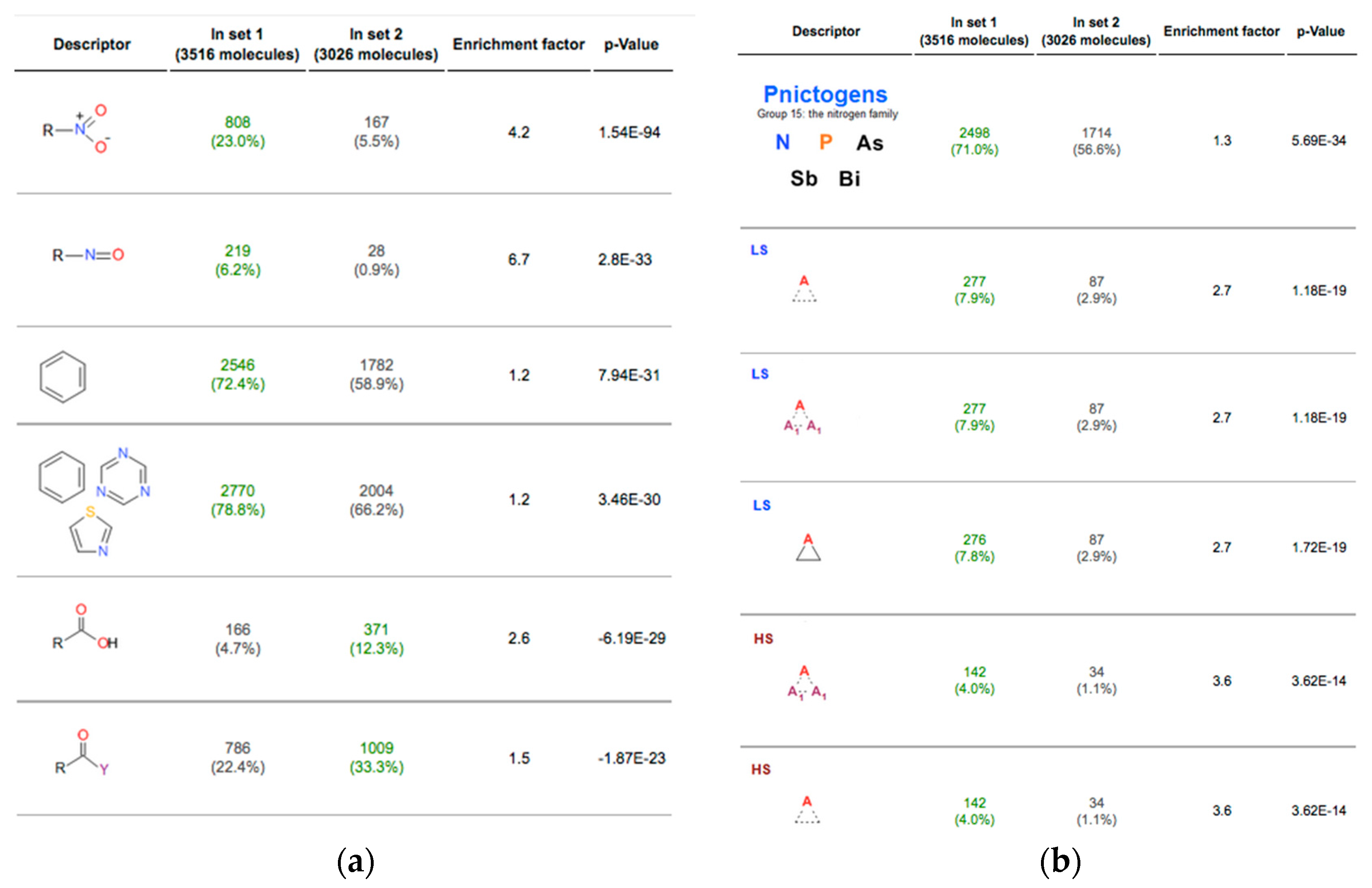{kind=link}
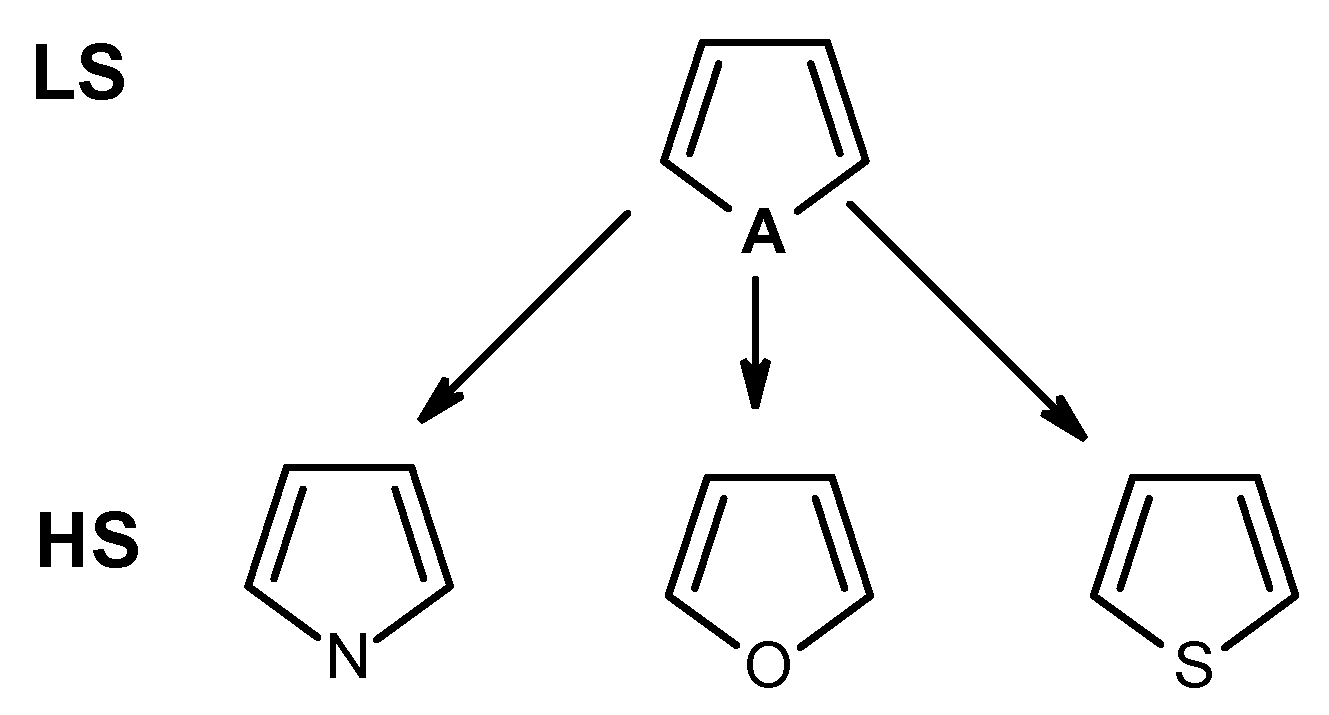{kind=link}
| Property | Original Models | Based on CheckMol-FG | New EFG Descriptors | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Best descriptors a | N | Method | RMSE | R2 | RMSE | R2 | RMSE | R2 | |
| Environmental toxicity against T. Pyriformis [17] | Estate | 644 | ASNN | 0.44 ± 0.02 | 0.83 ± 0.02 | 0.8 ± 0.03 | 0.44 ± 0.04 | 0.48 ± 0.03 | 0.8 ± 0.02 |
| logP for Pt(II/IV) complexes [18] | Fragmentor (out of 12) | 233 | ASNN | 0.43± 0.03 | 0.92± 0.02 | 1.42 ± 0.07 | 0.16 ± 0.05 | 0.45± 0.03 | 0.91± 0.02 |
| HIV inhibition [19] | Dragon (out of 10) | 286 | ASNN | 0.48± 0.03 | 0.87± 0.02 | 0.68 ± 0.03 | 0.75 ± 0.03 | 0.55 ± 0.03 | 0.83 ± 0.02 |
| Melting point [12] | CDK (out of 10) | 47427 | ASNN | 39.1 ± 0.2 | 0.76 ± 0.01 | 50.6 ± 0.2 | 0.59 ± 0.01 | 45.1 ± 0.2 | 0.67 ± 0.01 |
| Melting point [13] | Fragmentor (out of 12) | 275133 | LibSVM | 35.4 ± 0.1 | 0.69 ± 0.01 | 47.5 ± 0.1 | 0.46 ± 0.01 | 40.5 ± 0.1 | 0.61 ± 0.01 |
| Lowest Effect Level (LEL) toxicity prediction challenge [14,20] | Adriana (out of 10) | 483 | ASNN | 0.93 ± 0.03 | 0.22 ± 0.04 | 0.98 ± 0.05 | 0.16 ± 0.04 | 0.97 ± 0.05 | 0.17 ± 0.04 |
| Solubility in water [11] | Estate | 1311 | ASNN | 0.62 ± 0.2 | 0.91 ± 0.01 | 1.25 ± 0.04 | 0.65 ± 0.02 | 0.66 ± 0.02 | 0.90 ± 0.01 |
| Pyrolysis point [13] | Estate | 13769 | LibSVM | 35.6 ± 0.2 | 0.55 ± 0.01 | 42.1 ± 0.3 | 0.38 ± 0.01 | 38.7 ± 0.2 | 0.47 ± 0.01 |
| PTB1B inhibition [21] | Dragon | 2237 | ASNN | 0.77 ± 0.02 | 0.71 ± 0.02 | 0.96 ± 0.02 | 0.55 ± 0.02 | 0.81 ± 0.02 | 0.68 ± 0.02 |
| Estrogen Receptor binding [15] | ALOGPS + Estate | 1677 | ASNN | 0.062 ± 0.004 | 0.58 ± 0.06 | 0.084 ± 0.006 | 0.33 ± 0.04 | 0.079 ± 0.006 | 0.34 ± 0.04 |
| Property | Original Models | CheckMol-FG Descriptors | EFG Descriptors | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Best descriptors a | N | Method | BA | MCC | BA | MCC | BA | MCC | |
| AMES test [24] | Estate | 4361 | ASNN | 77.5% ± 0.6% | 0.55 ± 0.01 | 74.4% ± 0.6% | 0.49 ± 0.01 | 76.2% ± 0.6% | 0.53 ± 0.01 |
| Ready biodegradability [25] | CDK (out of 7) | 1884 | ASNN | 86.7% ± 0.8% | 0.72 ± 0.02 | 75% ± 1% | 0.49 ± 0.02 | 83.2%± 0.9% | 0.65 ± 0.02 |
| Solubility in DMSO [26] | ALOGPS + Estate (out of 9) | 50620 | ASNN | 73.8% ± 0.4% | 0.24 ± 0.01 | 57.1% ± 0.4% | 0.07 ± 0.01 | 71.5% ± 0.5% | 0.22 ± 0.01 |
| CYP450 inhibition [27] | Dragon (out of 10) | 3737 | J48 | 82.1% ± 0.6% | 0.64 ± 0.01 | 78.9% ± 0.7% | 0.59 ± 0.01 | 79.5% ± 0.7% | 0.59 ± 0.01 |
| Pyrolysis/ Melting point classification [13] | Estate (out of 10) | 241699 | LibSVM | 78.2%± 0.2% | 0.33 ± 0.01 | 53.1% ± 0.2% | 0.04 ± 0.01 | 74.2%± 0.2% | 0.27 ± 0.01 |
| Androgen receptor binding [28] | Dragon | 744 | ASNN | 77% ± 2% | 0.54 ± 0.03 | 70% ± 2% | 0.41 ± 0.04 | 77% ± 2% | 0.54 ± 0.03 |
| Ransthyretin receptor binding [28] | Dragon | 162 | ASNN | 89% ± 3.0% | 0.79 ± 0.05 | 83% ± 3% | 0.67 ± 0.06 | 86% ± 3% | 0.72 ± 0.06 |
| Estrogen Receptor binding [15] | Dragon (out of 11) | 1677 | ASNN | 74% ± 2.0% | 0.39 ± 0.03 | 62% ± 2% | 0.33 ± 0.04 | 72% ± 2% | 0.34 ± 0.03 |
| Azeotropes classification [16] | Adriana | 465 | RF | 78% ± 2% | 0.55 ± 0.04 | 77% ± 2% | 0.55 ± 0.04 | 75% ± 2% | 0.49 ± 0.04 |
| ATAD5 genotoxicity [29] | Dragon | 9363 | ASNN | 78% ± 1% | 0.28 ± 0.01 | 58% ± 1% | 0.07 ± 0.01 | 74% ± 1% | 0.22 ± 0.01 |
2.2. Analysis of Datasets
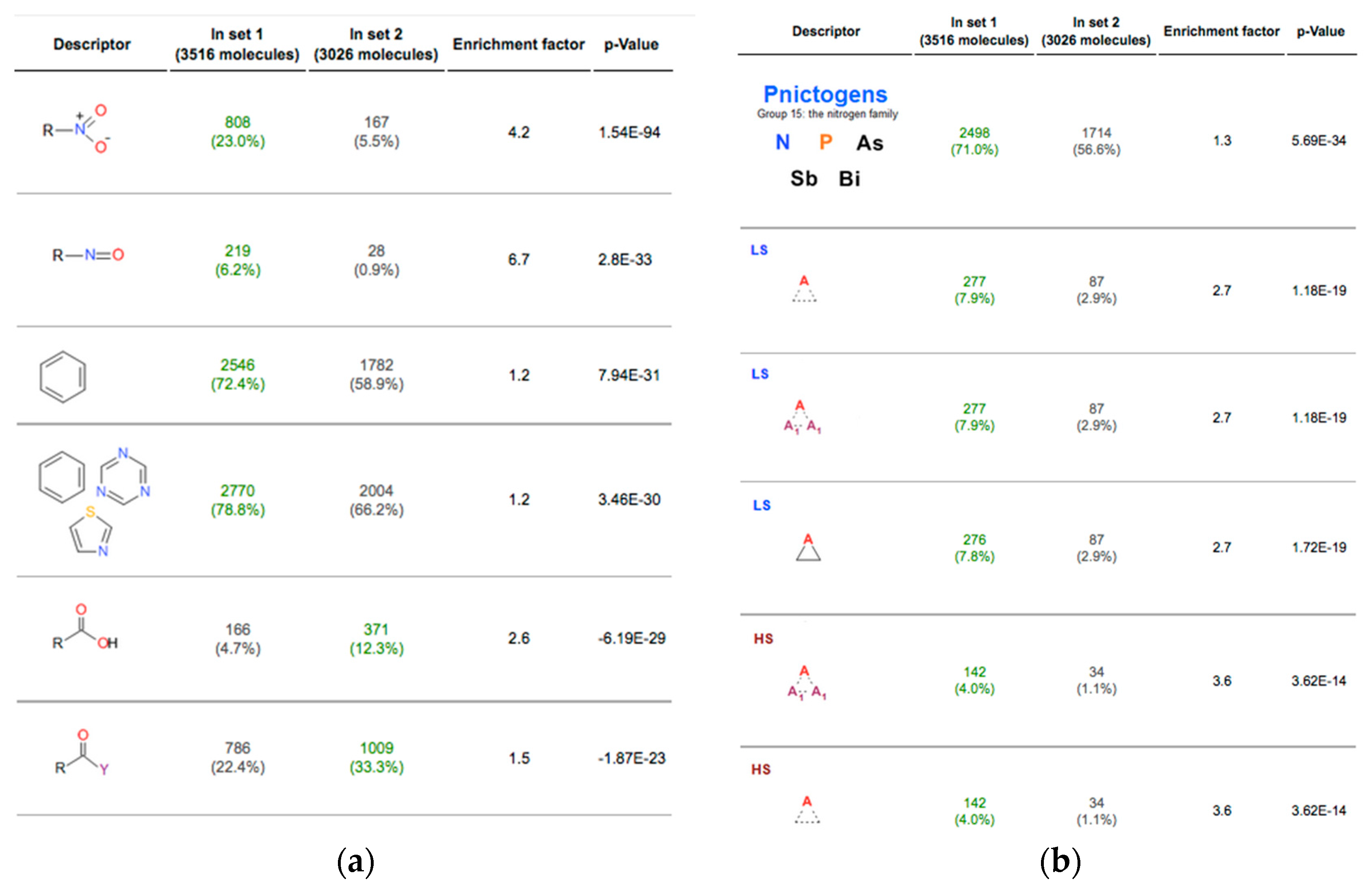
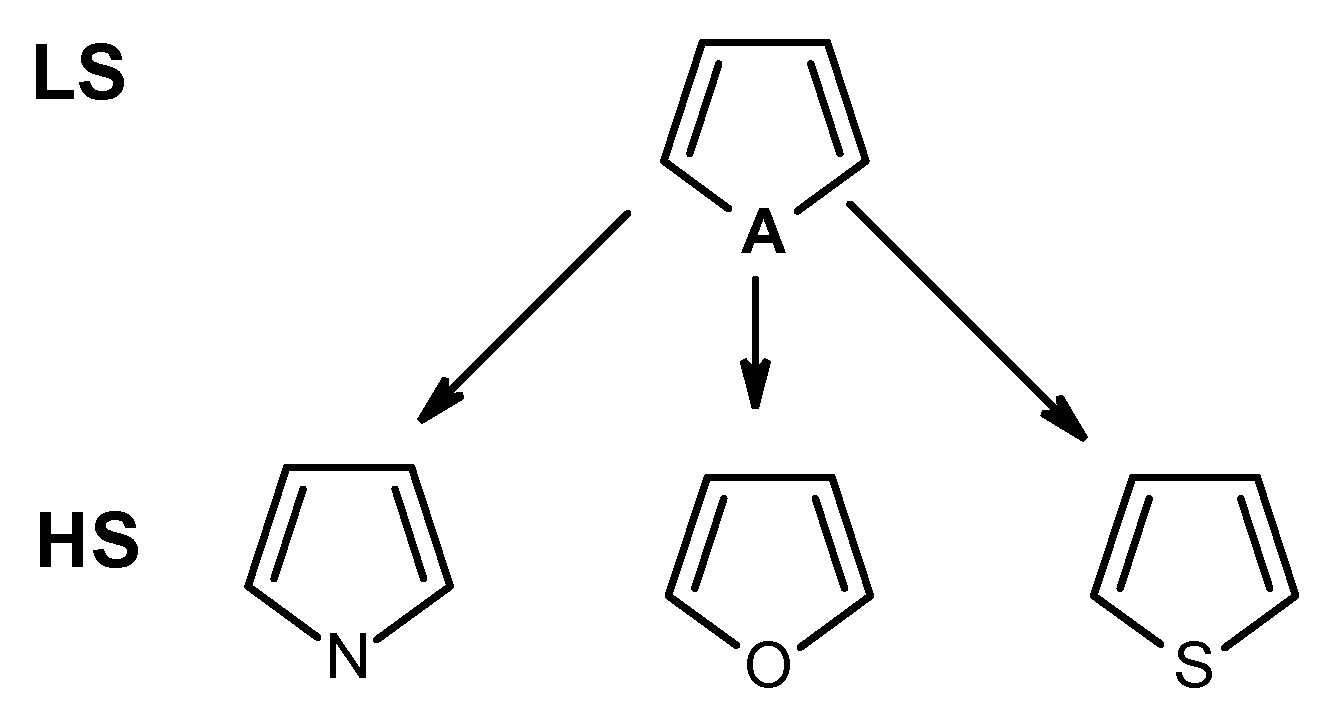
3. Methods
3.1. Extension of Functional Groups (FG) Recognized by the CheckMol
3.2. Technical Implementation
4. Conclusions
Supplementary Materials
Acknowledgements
Author Contributions
Conflicts of Interest
References
- International Union of Pure and Applied Chemistry (IUPAC). Functional Group. Available online: http://goldbook.iupac.org/F02555.html (accessed on 18 December 2015).
- Fredenslund, A.; Jones, R.L.; Prausnitz, J.M. Group-contribution estimation of activity coefficients in nonideal liquid mixtures. AIChE J. 1975, 21, 1086–1099. [Google Scholar] [CrossRef]
- Varnek, A.; Fourches, D.; Horvath, D.; Klimchuk, O.; Gaudin, C.; Vayer, P.; Solov’ev, V.; Hoonakker, F.; Tetko, I.V.; Marcou, G. ISIDA—Platform for virtual screening based on fragment and pharmacophoric descriptors. Curr. Comput. Aided Drug Des. 2008, 4, 191–198. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Glen, R.C.; Reiling, S. Similarity searching of chemical databases using atom environment descriptors (MOLPRINT 2D): Evaluation of performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- The Checkmol/Matchmol Homepage. Available online: http://merian.pch.univie.ac.at/~nhaider/cheminf/cmmm.html (accessed on 15 October 2015).
- Yang, C.; Tarkhov, A.; Marusczyk, J.; Bienfait, B.; Gasteiger, J.; Kleinoeder, T.; Magdziarz, T.; Sacher, O.; Schwab, C.H.; Schwoebel, J.; et al. New publicly available chemical query language, CSRML, to support chemotype representations for application to data mining and modeling. J. Chem. Inf. Model. 2015, 55, 510–528. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.J.; Dumontier, M.; Ling, S.; Haider, N.; Hogue, C.W. CO: A chemical ontology for identification of functional groups and semantic comparison of small molecules. FEBS Lett. 2005, 579, 4685–4691. [Google Scholar] [CrossRef] [PubMed]
- Poongavanam, V.; Haider, N.; Ecker, G.F. Fingerprint-based in silico models for the prediction of P-glycoprotein substrates and inhibitors. Bioorg. Med. Chem. 2012, 20, 5388–5395. [Google Scholar] [CrossRef]
- Haider, N. Functionality pattern matching as an efficient complementary structure/reaction search tool: An open-source approach. Molecules 2010, 15, 5079–5092. [Google Scholar] [CrossRef]
- Tetko, I.V.; Tanchuk, V.Y.; Kasheva, T.N.; Villa, A.E.P. Estimation of aqueous solubility of chemical compounds using E-state indices. J. Chem. Inf. Comput. Sci. 2001, 41, 1488–1493. [Google Scholar] [CrossRef]
- Tetko, I.V.; Sushko, Y.; Novotarskyi, S.; Patiny, L.; Kondratov, I.; Petrenko, A.E.; Charochkina, L.; Asiri, A.M. How accurately can we predict the melting points of drug-like compounds? J. Chem. Inf. Model. 2014, 54, 3320–3329. [Google Scholar] [CrossRef]
- Tetko, I.V.; Lowe, D.; Williams, A. The development of models to predict melting and pyrolysis point data associated with several hundred thousand compounds mined from patents. J. Cheminform. 2015, in press. [Google Scholar]
- Novoratskyi, S.; Sushko, Y.; Abdelaziz, A.; Korner, R.; Vogt, J.; Tetko, I.V. Why Rank-I submission of the ToxCast EPA in vitro to in vivo challenge to predict lowest effect level (LEL) does not use in vitro measurements? Chem. Res. Toxicol. 2015. submitted. [Google Scholar]
- CERAPP—Collaborative Estrogen Receptor Activity Prediction Project. Available online: http://www.epa.gov/chemical-research/cerapp-collaborative-estrogen-receptor-activity-prediction-project-0 (accessed on 18 December 2015).
- Oprisiu, I.; Novotarskyi, S.; Tetko, I.V. Modeling of non-additive mixture properties using the Online CHEmical database and Modeling environment (OCHEM). J. Cheminform. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Sushko, I.; Pandey, A.K.; Zhu, H.; Tropsha, A.; Papa, E.; Oberg, T.; Todeschini, R.; Fourches, D.; Varnek, A. Critical assessment of QSAR models of environmental toxicity against Tetrahymena pyriformis: Focusing on applicability domain and overfitting by variable selection. J. Chem. Inf. Model. 2008, 48, 1733–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetko, I.V.; Varbanov, H.; Galanski, M.; Platts, J.A.; Gabano, E. Prediction of LogP for Pt(II) and Pt(IV) complexes: Comparison of statistical and quantum-chemistry based approaches. J. Inorg. Biochem. 2016. [Google Scholar] [CrossRef]
- Nizami, B.; Tetko, I.V.; Koorbanally, N.A.; Honarparvar, B. QSAR models and scaffold-based analysis of non-nucleoside HIV RT inhibitors. Chemom. Intell. Lab. 2015, 148, 134–144. [Google Scholar] [CrossRef]
- TopCoder. Available online: http://www.topcoder.com/epa/toxcast/ (accessed on 15 October 2015).
- Yu, V.T.; Tanin, O.V.; Vovk, A.I. QSAR models for predicting protein tyrosine phosphatase 1B inhibition by structurally diverse inhibitors. J. Org. Pharm. Chem. 2013, 11, 51–56. [Google Scholar]
- Tetko, I.V. Associative neural network. Meth. Mol. Biol. 2008, 458, 185–202. [Google Scholar]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 21–27. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi, S.; Korner, R.; Pandey, A.K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Muller, K.R.; et al. Applicability domains for classification problems: Benchmarking of distance to models for Ames mutagenicity set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef] [PubMed]
- Vorberg, S.; Tetko, I.V. Modeling the biodegradability of chemical compounds using the online chemical modeling environment (OCHEM). Mol. Inf. 2014, 33, 73–85. [Google Scholar] [CrossRef]
- Tetko, I.V.; Novotarskyi, S.; Sushko, I.; Ivanov, V.; Petrenko, A.E.; Dieden, R.; Lebon, F.; Mathieu, B. Development of dimethyl sulfoxide solubility models using 163,000 molecules: Using a domain applicability metric to select more reliable predictions. J. Chem. Inf. Model. 2013, 53, 1990–2000. [Google Scholar] [CrossRef]
- Novotarskyi, S.; Sushko, I.; Korner, R.; Pandey, A.K.; Tetko, I.V. A comparison of different QSAR approaches to modeling CYP450 1A2 inhibition. J. Chem. Inf. Model. 2011, 51, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Rybacka, A.; Ruden, C.; Tetko, I.V.; Andersson, P.L. Identifying potential endocrine disruptors among industrial chemicals and their metabolites—Development and evaluation of in silico tools. Chemosphere 2015, 139, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Abdelaziz, A.; Spahn-Langguth, H.; Schramm, K.W.; Tetko, I.V. Consensus approach for modeling HTS assays using in silico descriptors. Front. Environ. Sci. 2015. submitted. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Frank, E.; Hall, M.; Trigg, L.; Holmes, G.; Witten, I.H. Data mining in bioinformatics using WEKA. Bioinformatics 2004, 20, 2479–2481. [Google Scholar] [CrossRef]
- Kazius, J.; McGuire, R.; Bursi, R. Derivation and validation of toxicophores for mutagenicity prediction. J. Med. Chem. 2005, 48, 312–320. [Google Scholar] [CrossRef]
- Sushko, I.; Salmina, E.; Potemkin, V.A.; Poda, G.; Tetko, I.V. Toxalerts: A web server of structural alerts for toxic chemicals and compounds with potential adverse reactions. J. Chem. Inf. Model. 2012, 52, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Huynh, M.H.; Hiskey, M.A.; Chavez, D.E.; Naud, D.L.; Gilardi, R.D. Synthesis, characterization, and energetic properties of diazido heteroaromatic high-nitrogen C-N compound. J. Am. Chem. Soc. 2005, 127, 12537–12543. [Google Scholar] [CrossRef]
- Kaim, W. The coordination chemistry of 1,2,4,5-tetrazines. Coord. Chem. Rev. 2002, 230, 127–139. [Google Scholar] [CrossRef]
- Tetko, I.V. The perspectives of computational chemistry modeling. J. Comput. Aided Mol. Des. 2012, 26, 135–136. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salmina, E.S.; Haider, N.; Tetko, I.V. Extended Functional Groups (EFG): An Efficient Set for Chemical Characterization and Structure-Activity Relationship Studies of Chemical Compounds. Molecules 2016, 21, 1. https://doi.org/10.3390/molecules21010001
Salmina ES, Haider N, Tetko IV. Extended Functional Groups (EFG): An Efficient Set for Chemical Characterization and Structure-Activity Relationship Studies of Chemical Compounds. Molecules. 2016; 21(1):1. https://doi.org/10.3390/molecules21010001
Chicago/Turabian StyleSalmina, Elena S., Norbert Haider, and Igor V. Tetko. 2016. "Extended Functional Groups (EFG): An Efficient Set for Chemical Characterization and Structure-Activity Relationship Studies of Chemical Compounds" Molecules 21, no. 1: 1. https://doi.org/10.3390/molecules21010001
APA StyleSalmina, E. S., Haider, N., & Tetko, I. V. (2016). Extended Functional Groups (EFG): An Efficient Set for Chemical Characterization and Structure-Activity Relationship Studies of Chemical Compounds. Molecules, 21(1), 1. https://doi.org/10.3390/molecules21010001