
Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases

 and
and
Abstract
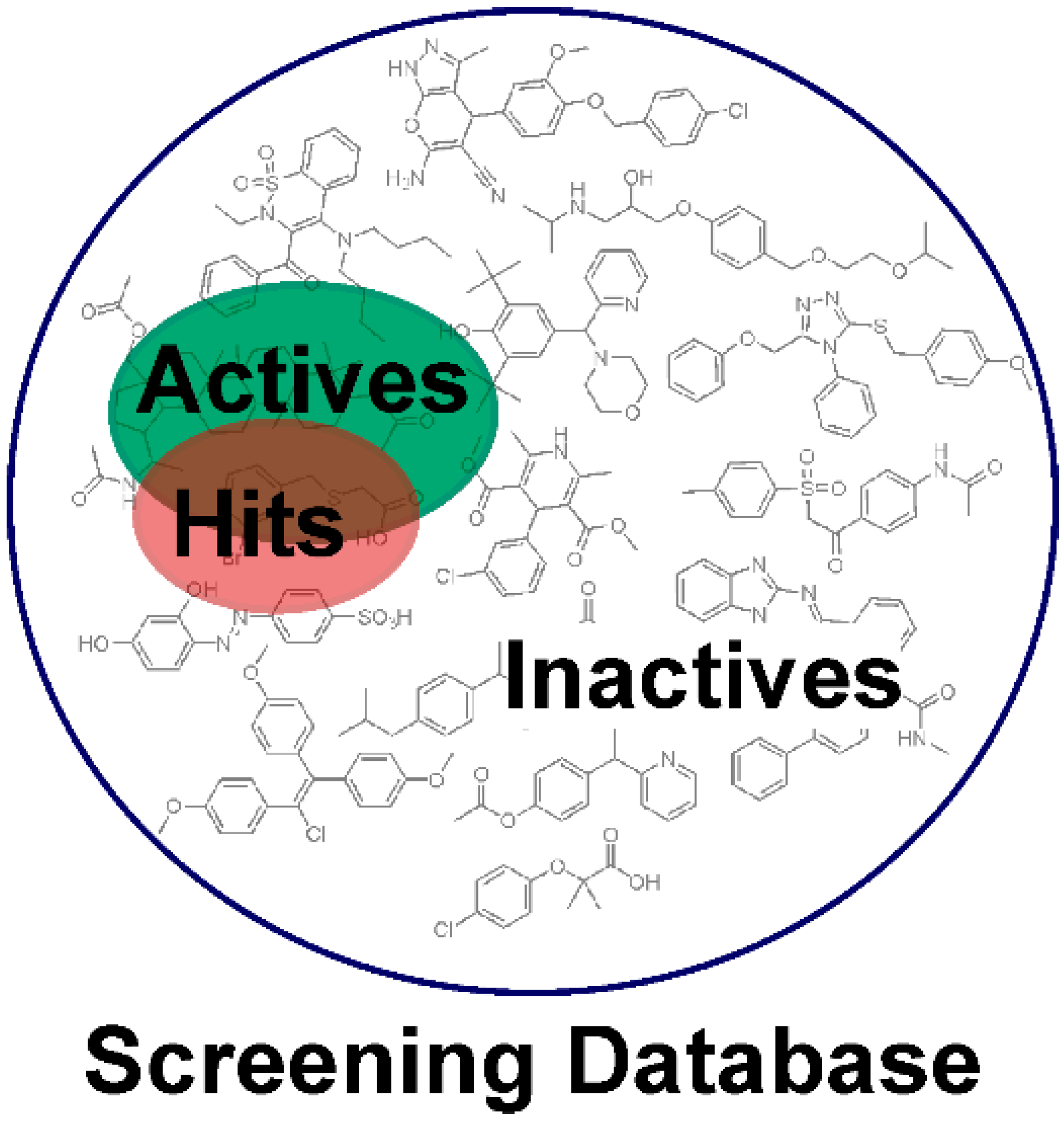
1. Introduction
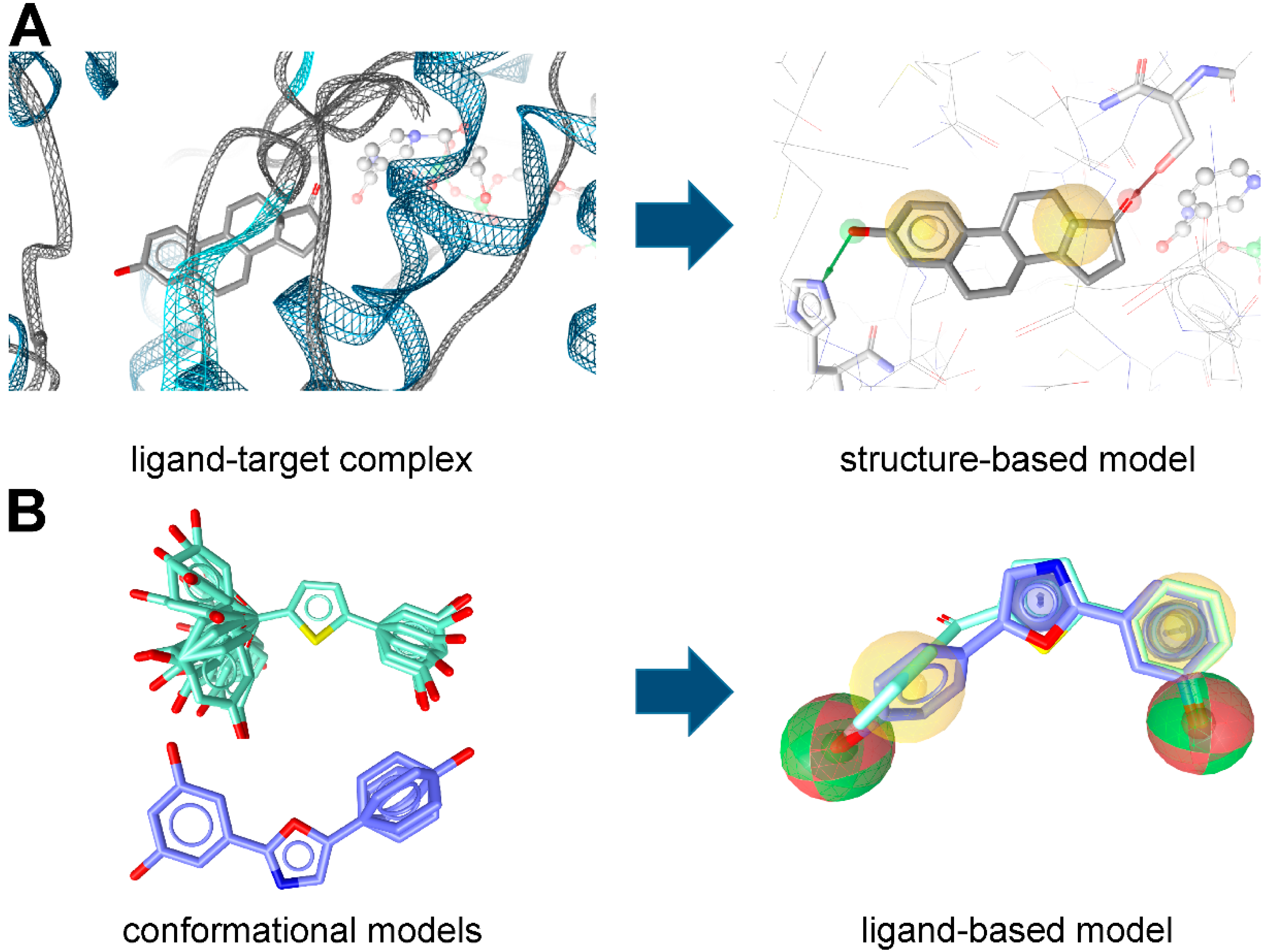
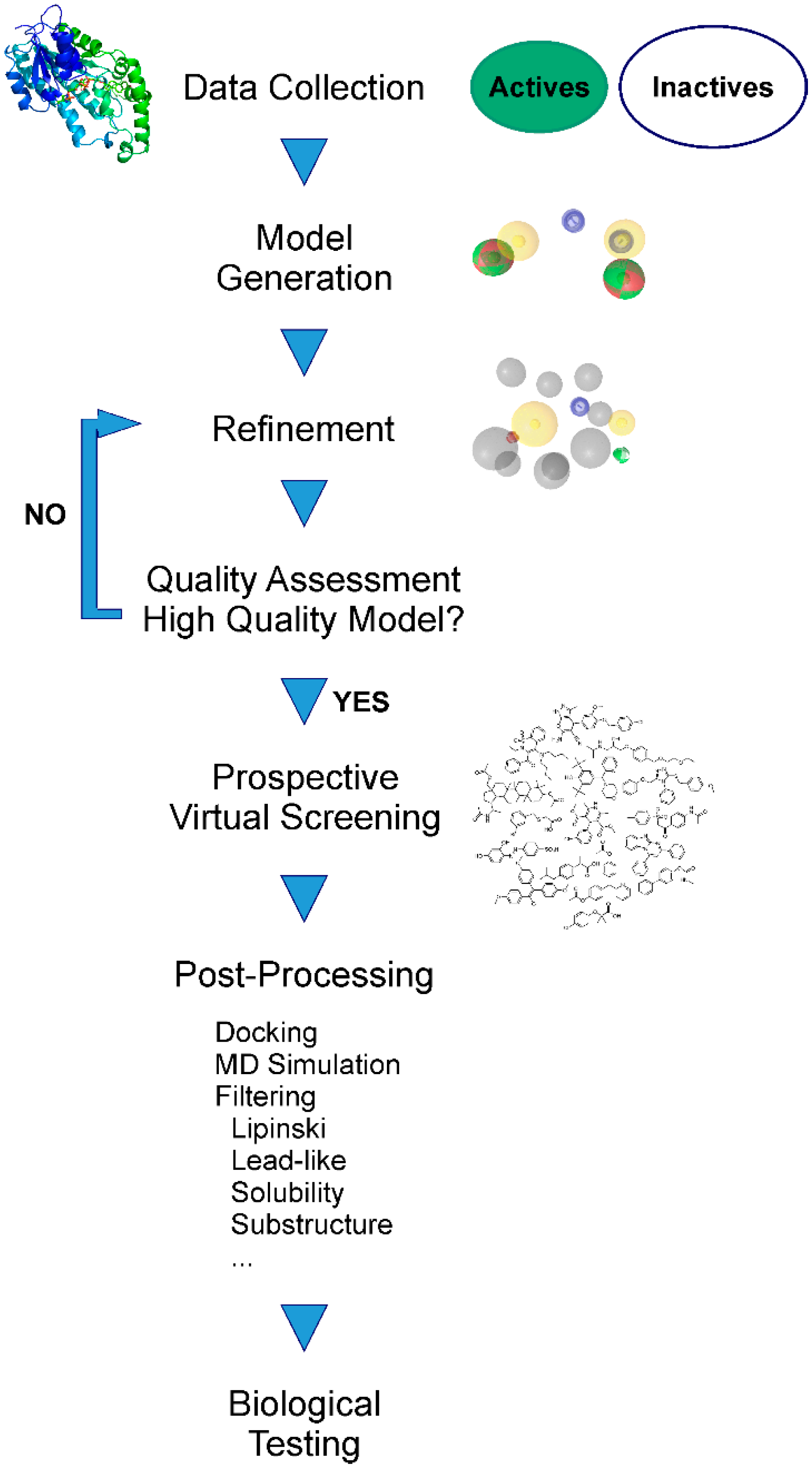
Pharmacophore Modeling
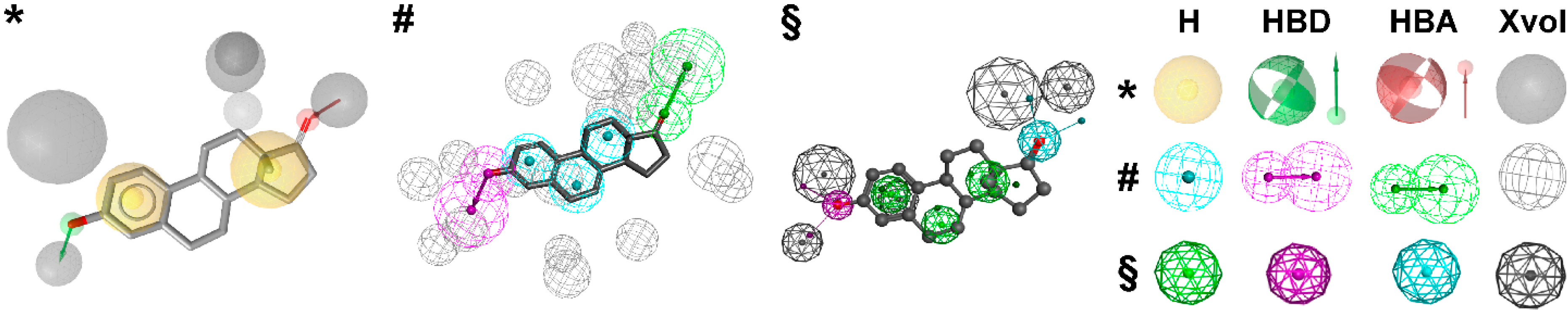
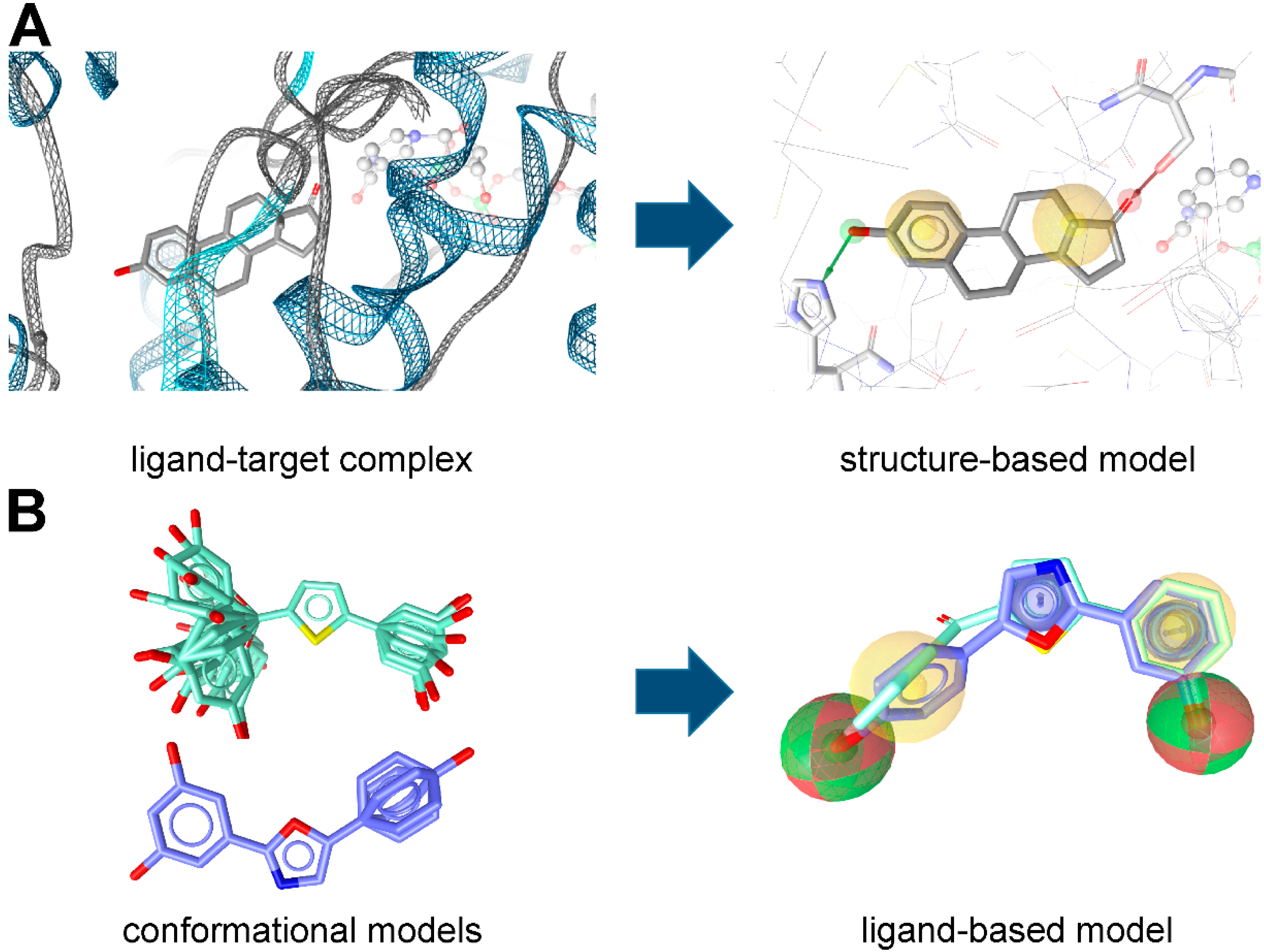
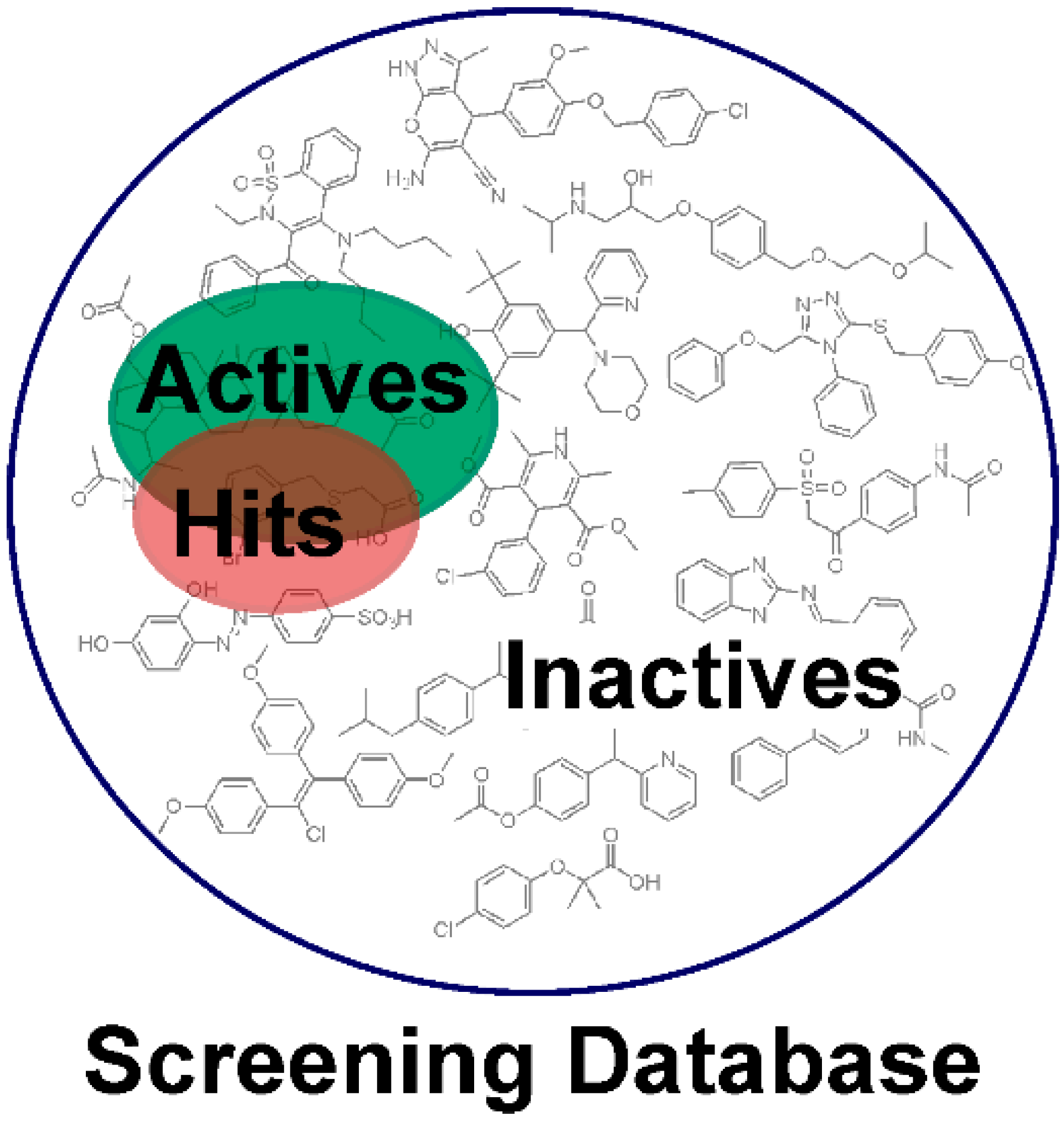

2. Applications of Pharmacophore-Based VS
2.1. Drug Discovery
2.1.1. Lead Identification
2.1.2. Structure-Activity Relationships
2.1.3. Scaffold Hopping
2.1.4. Selectivity Profiling
2.1.5. Combination with Other Techniques
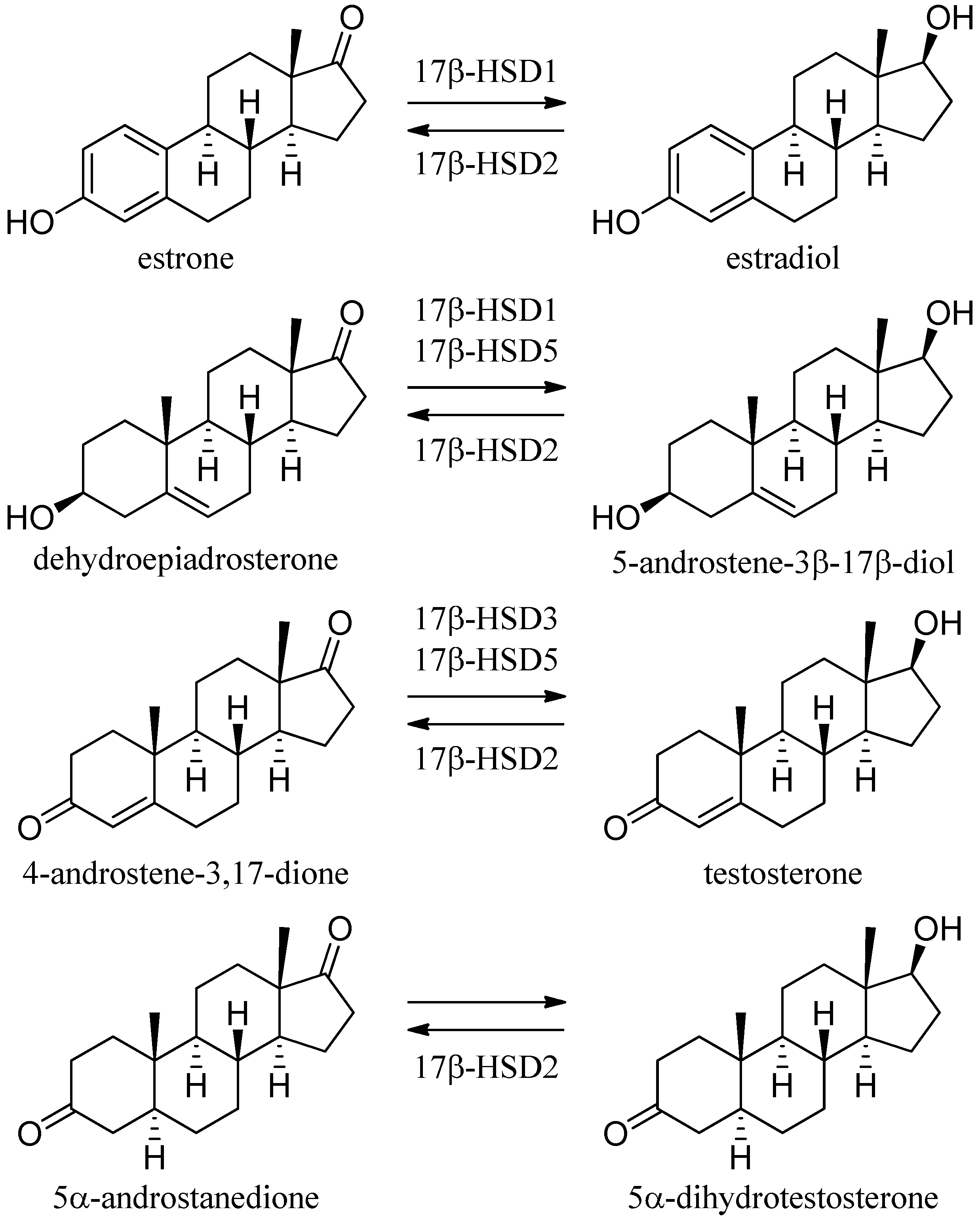
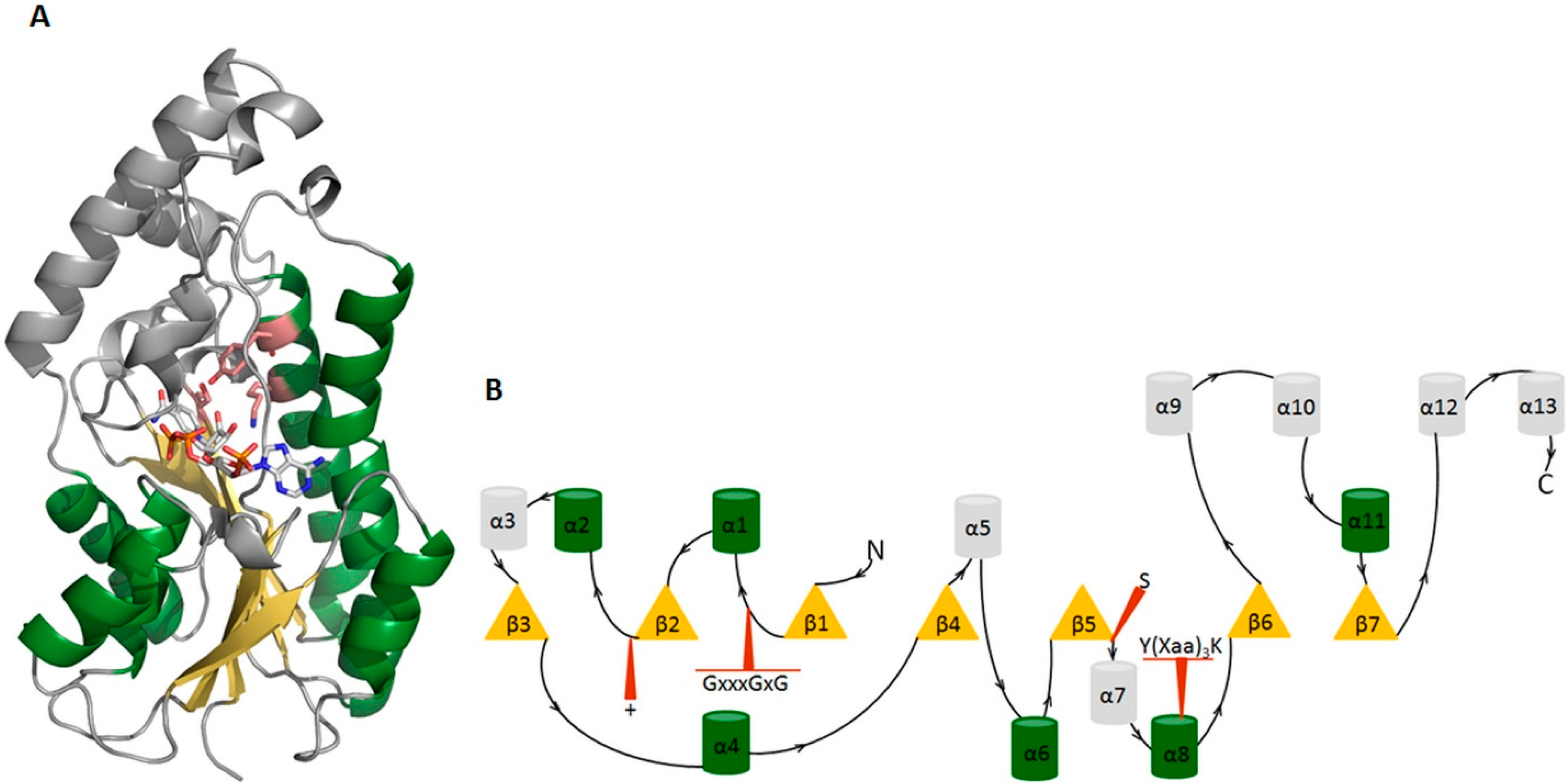
2.2. The Short-Chain Dehydrogenase/Reductase Superfamily
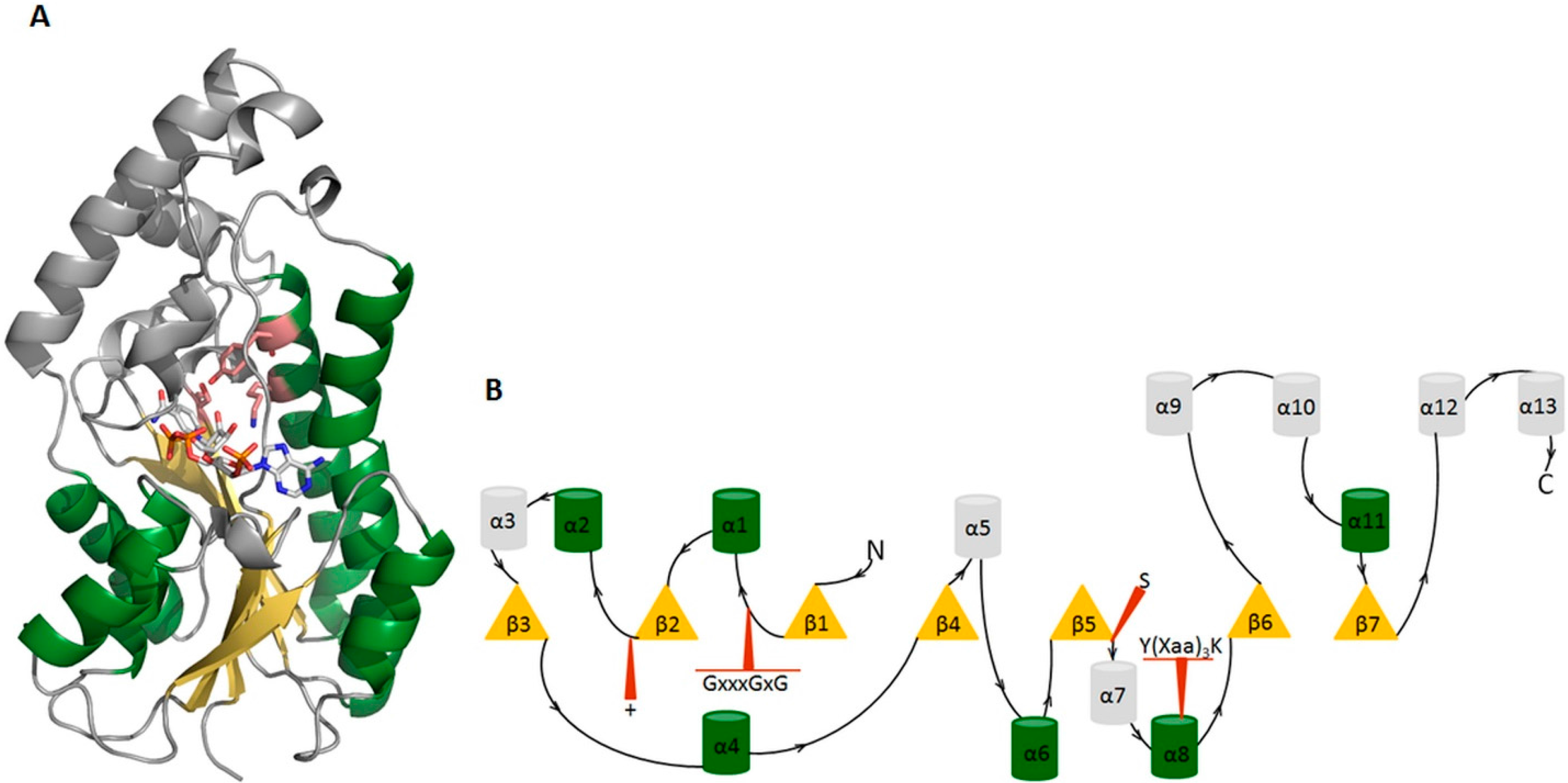
3. Examples from the SDR Family
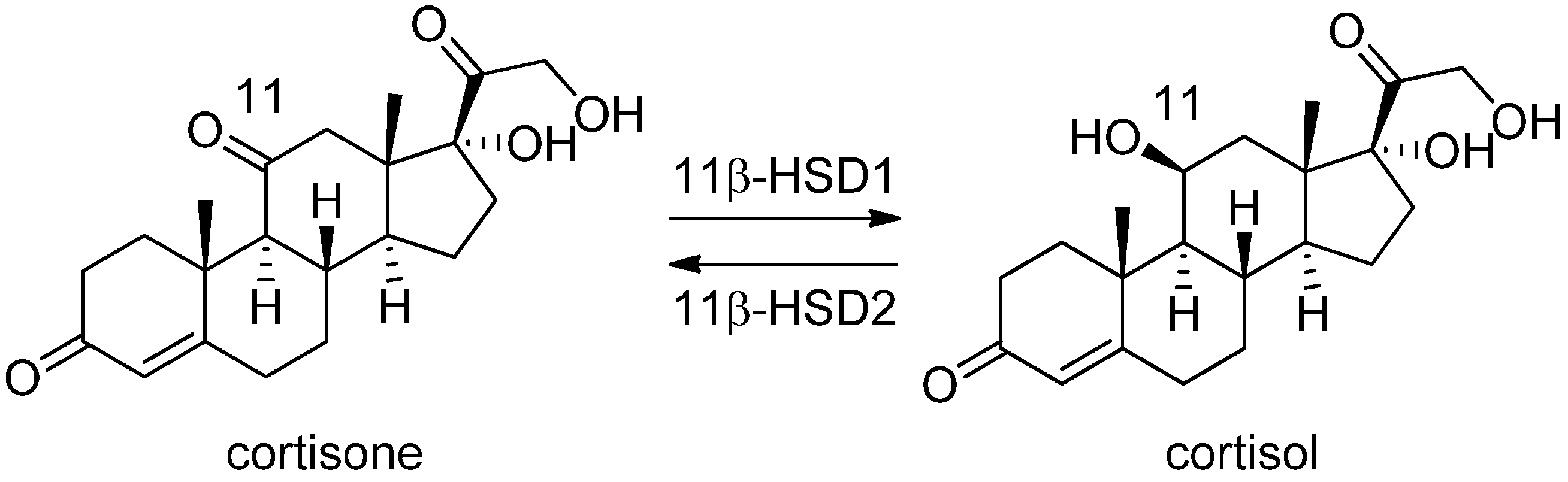
3.1. 11β-Hydroxysteroid Dehydrogenase Type 1
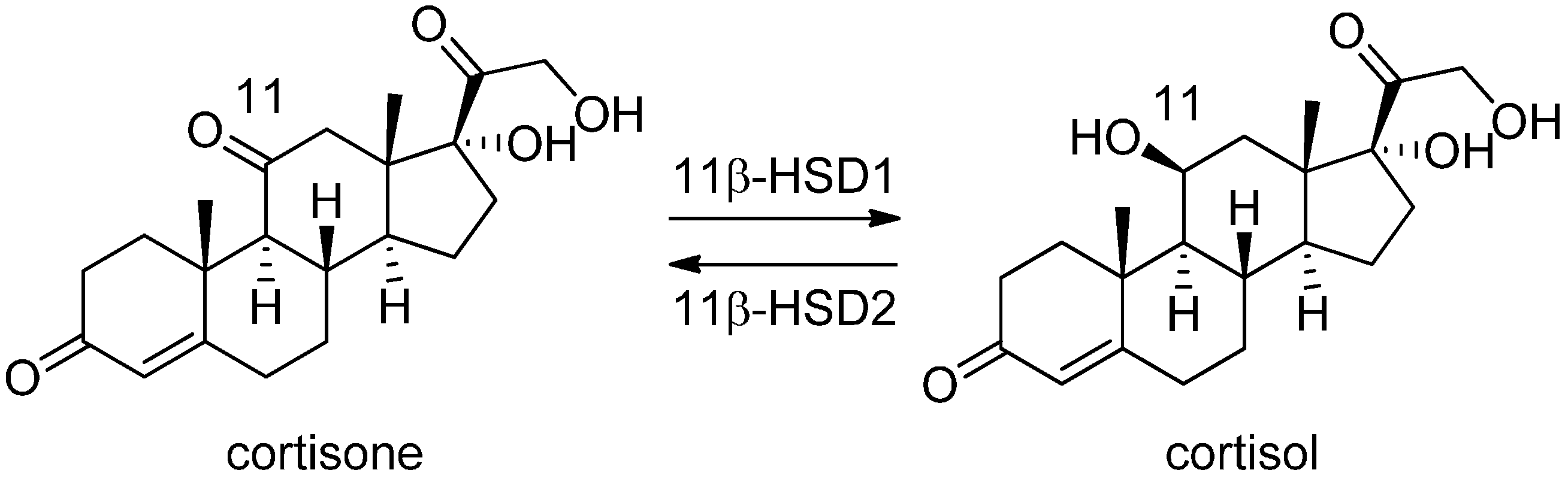
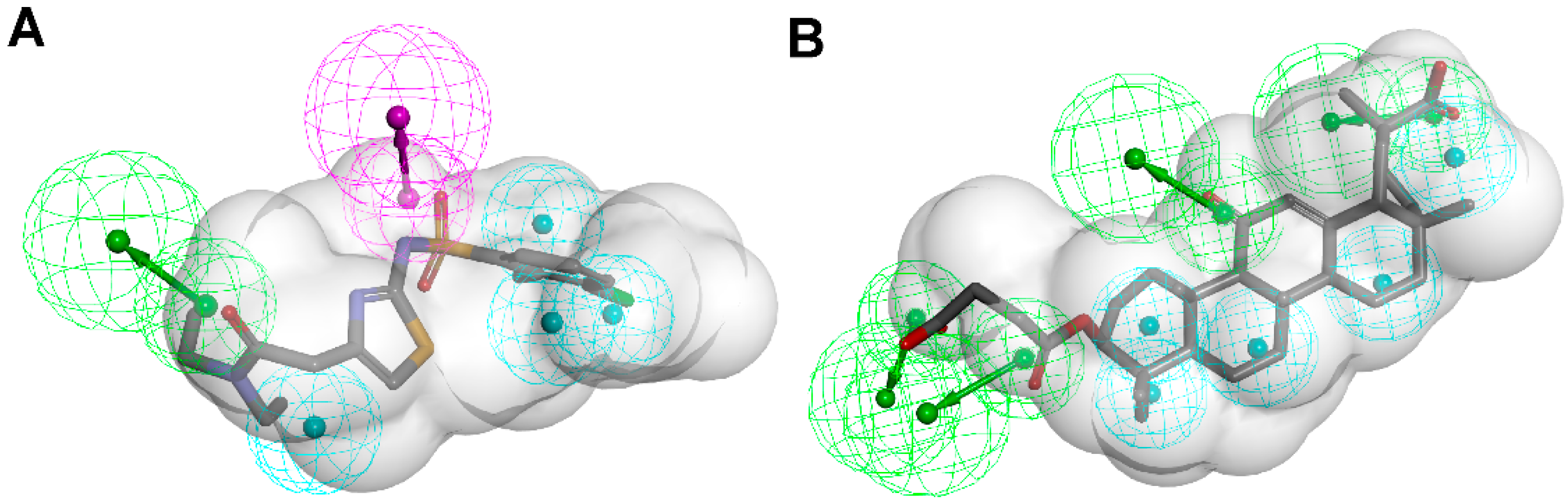
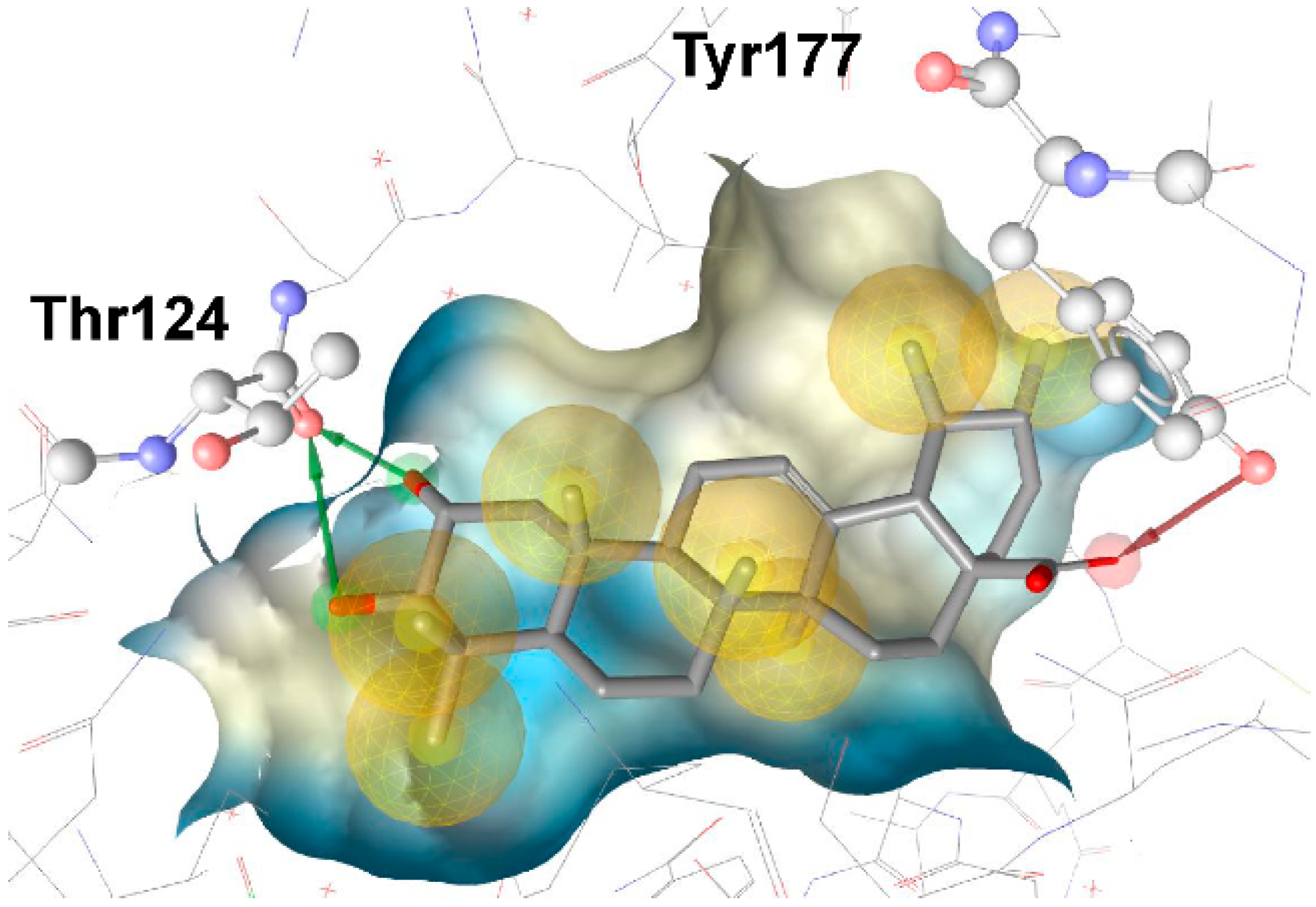
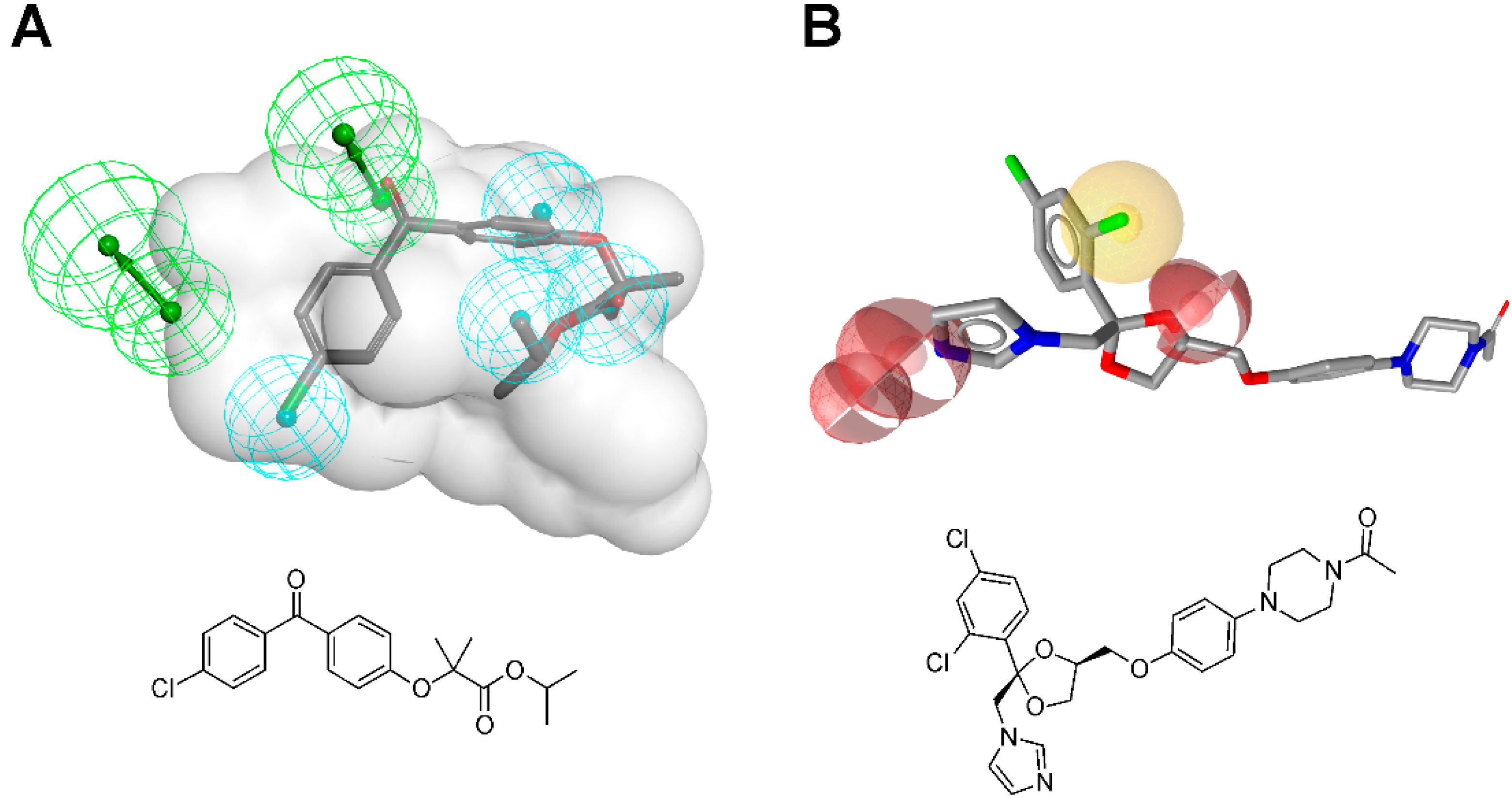
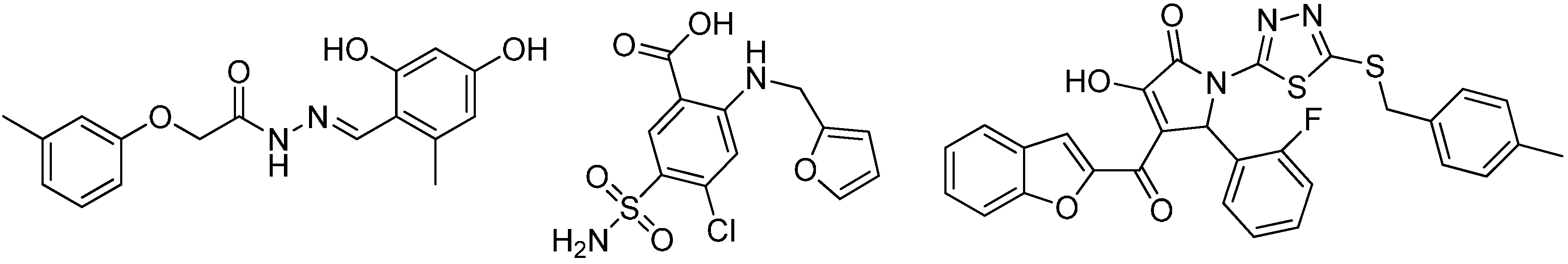
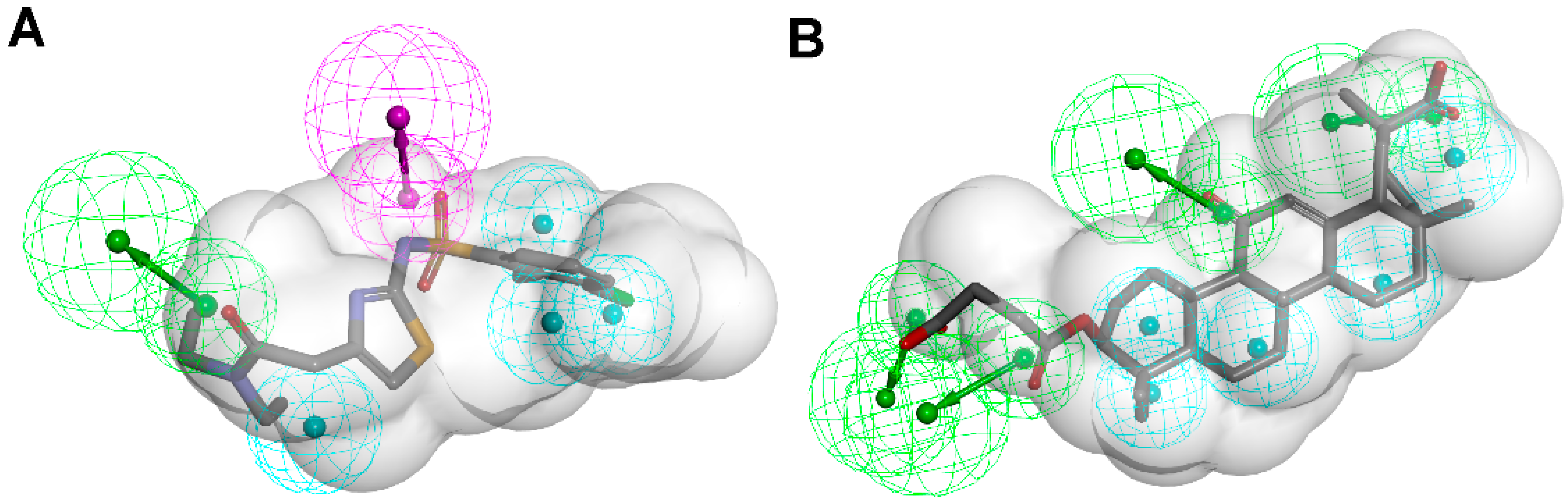
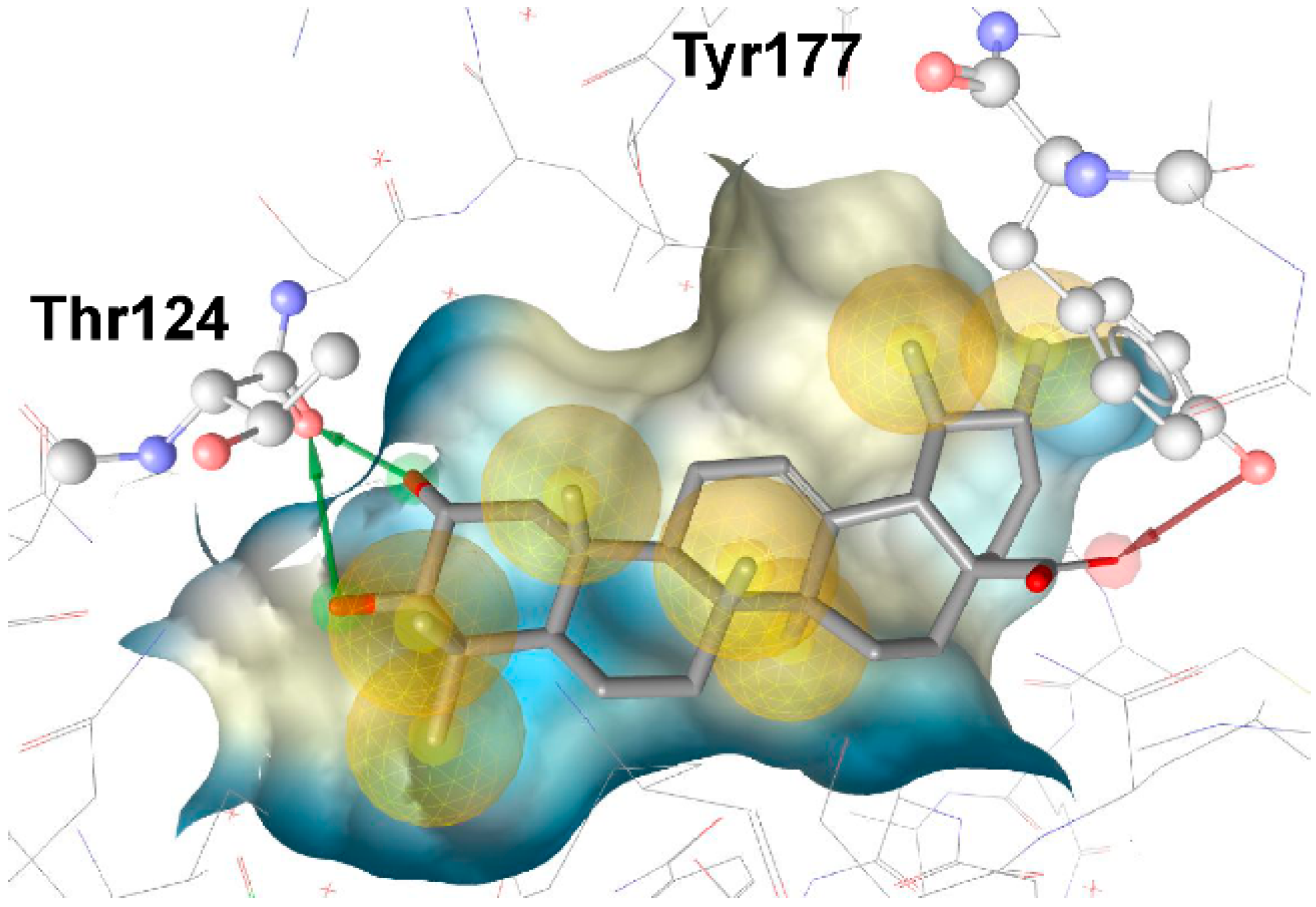
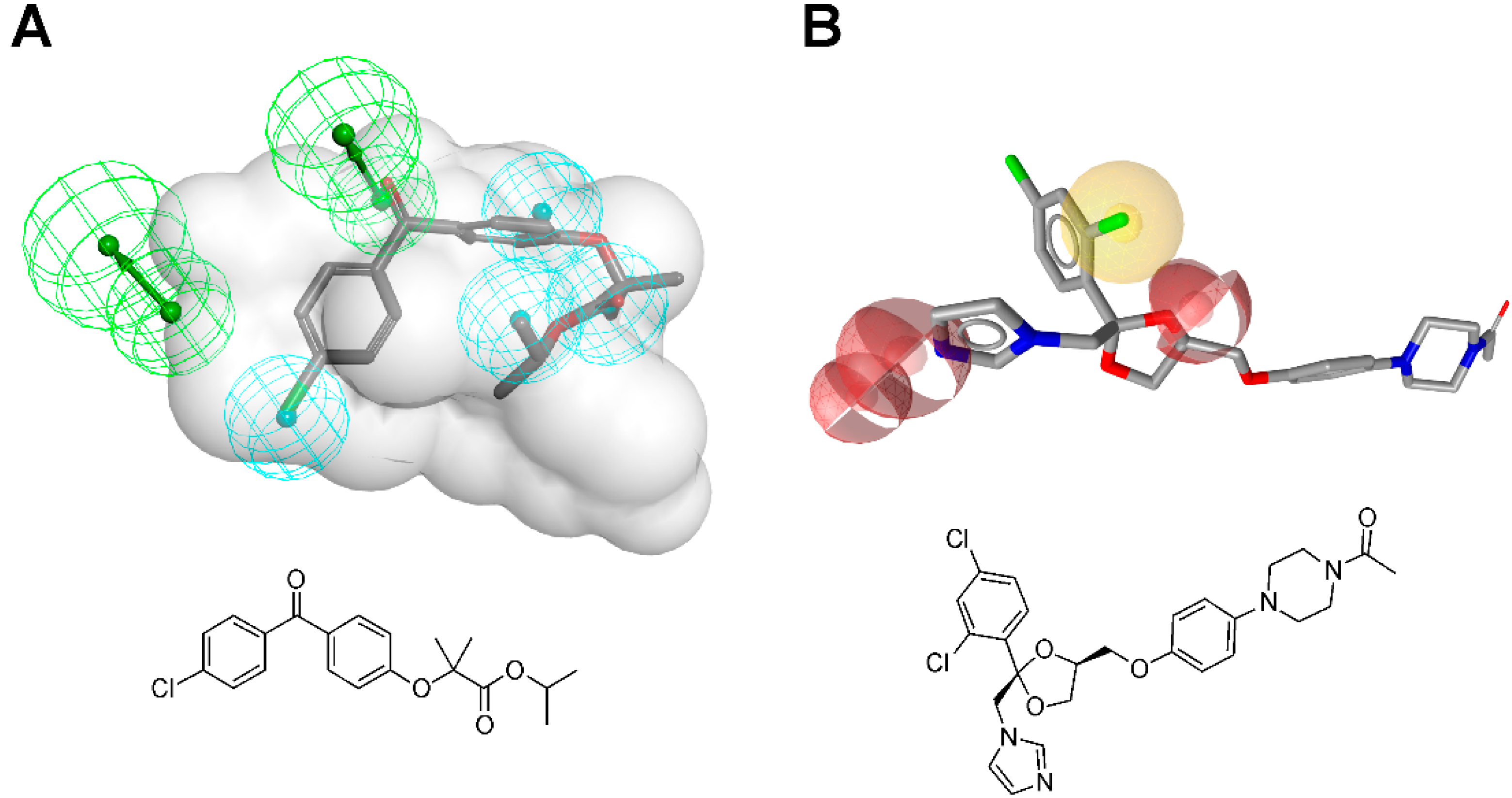
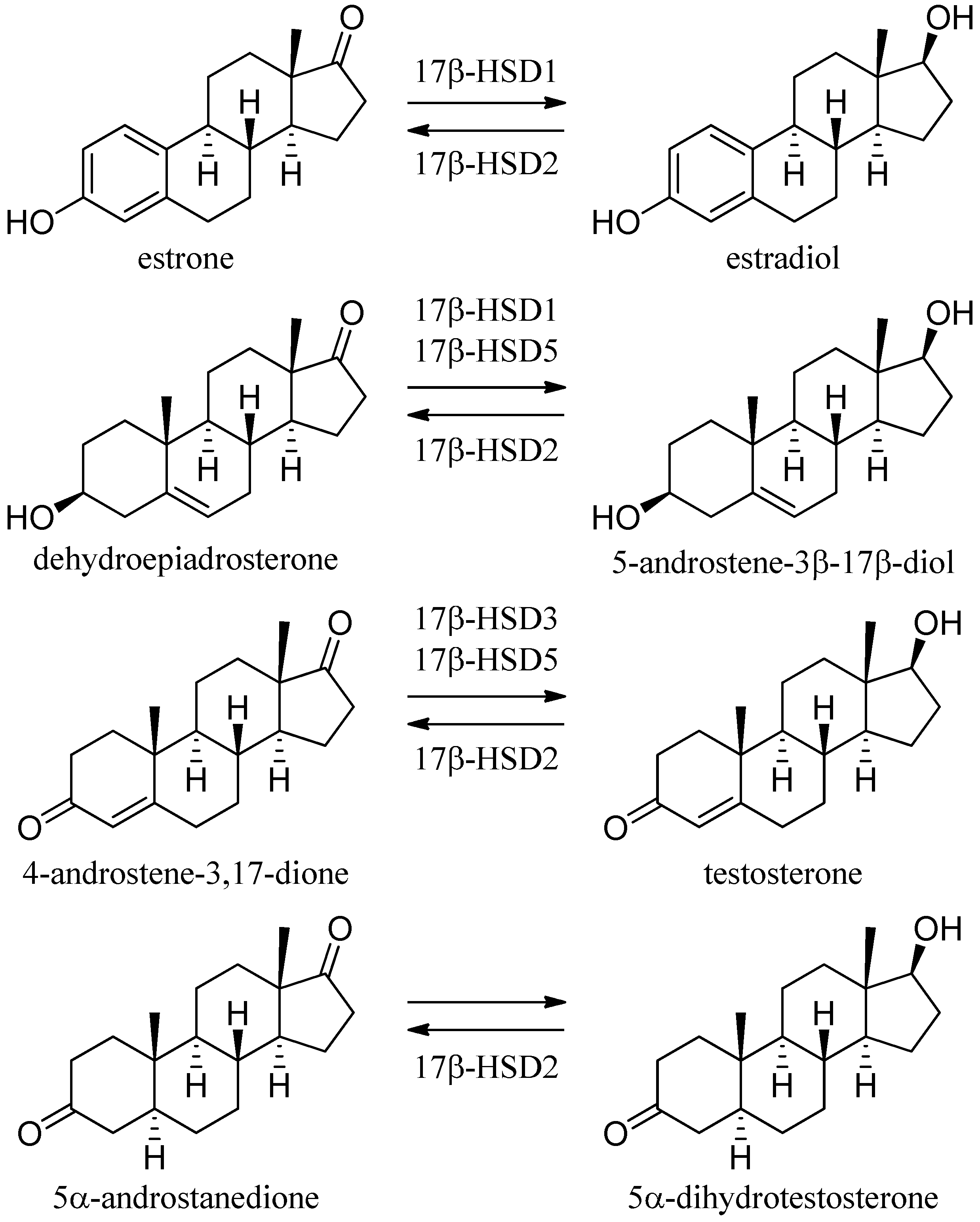
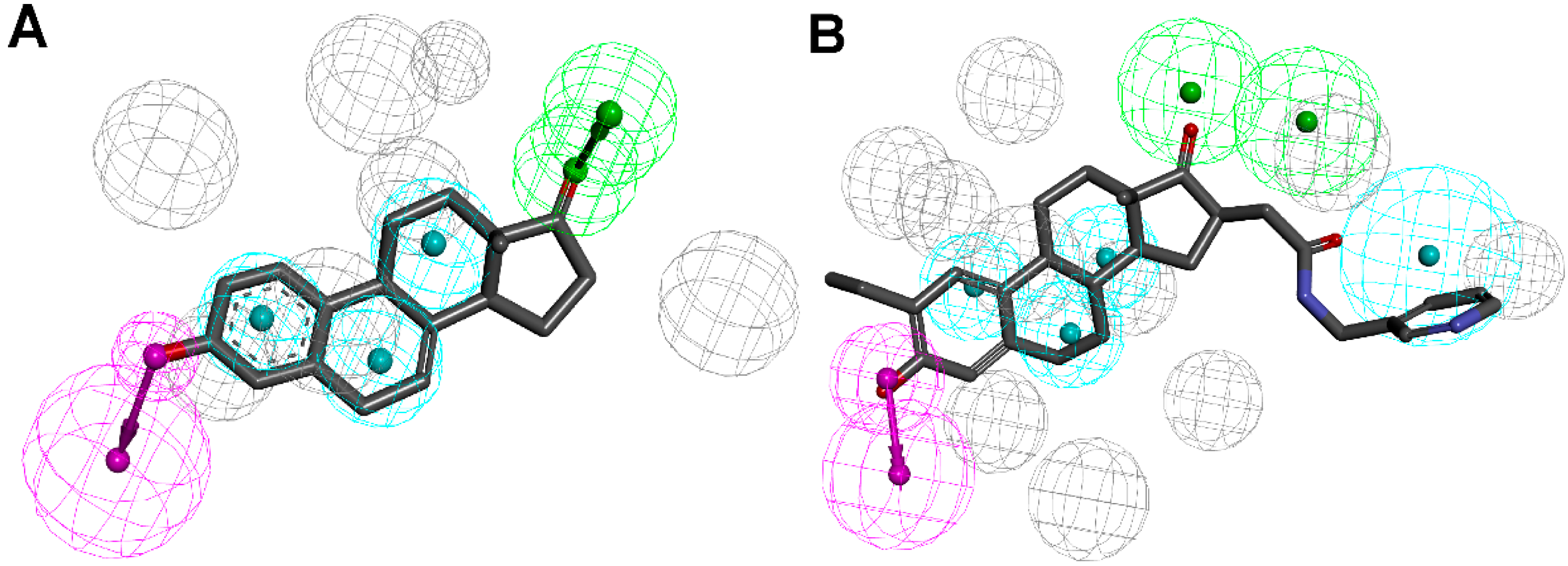
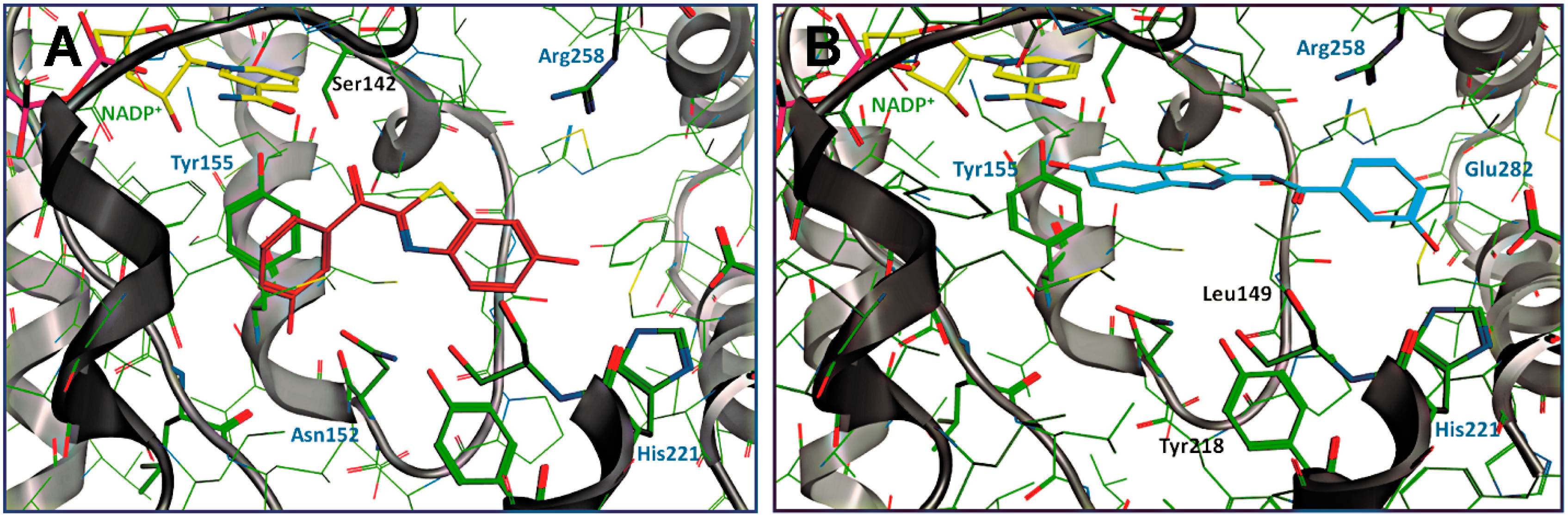
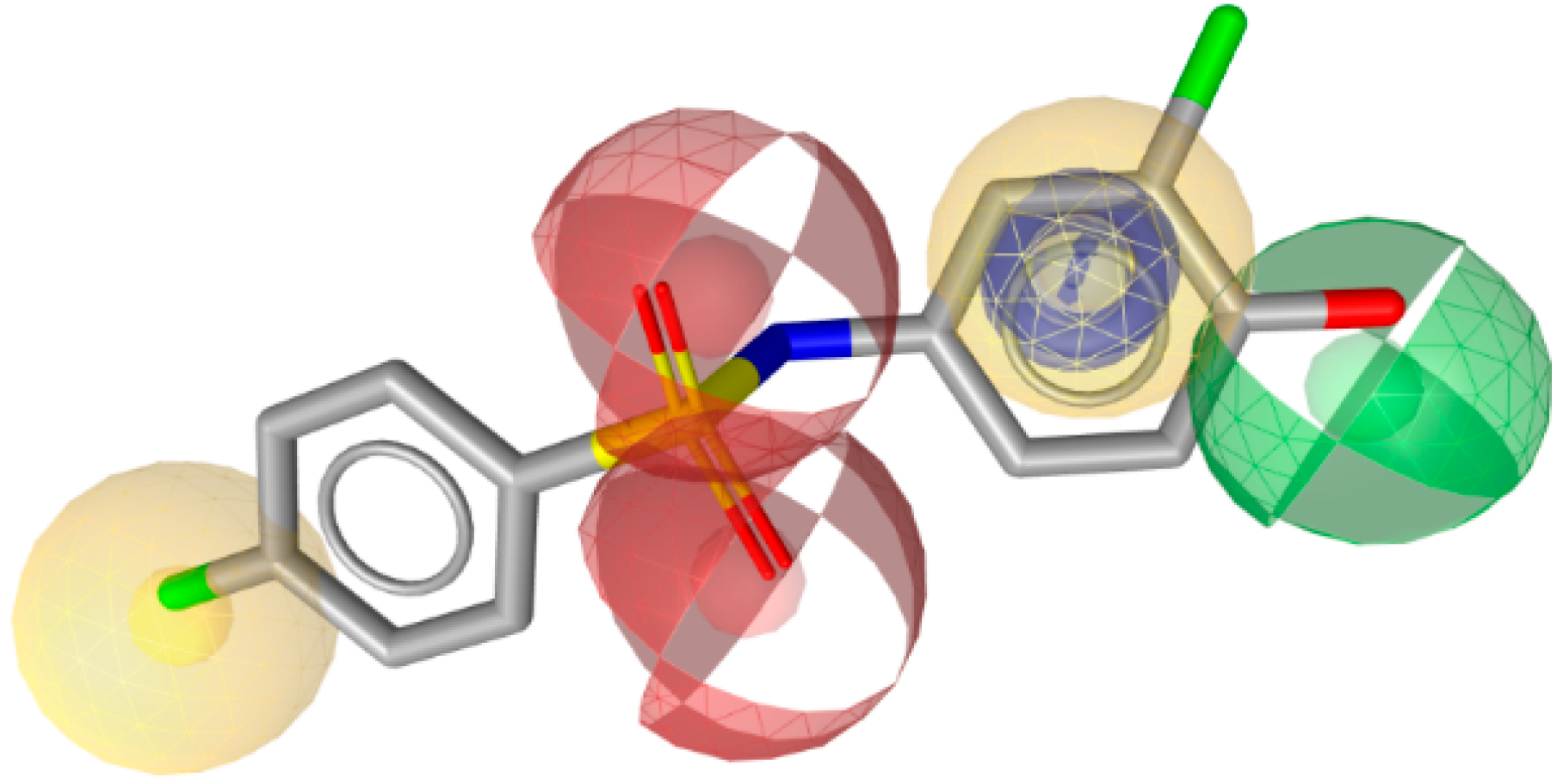
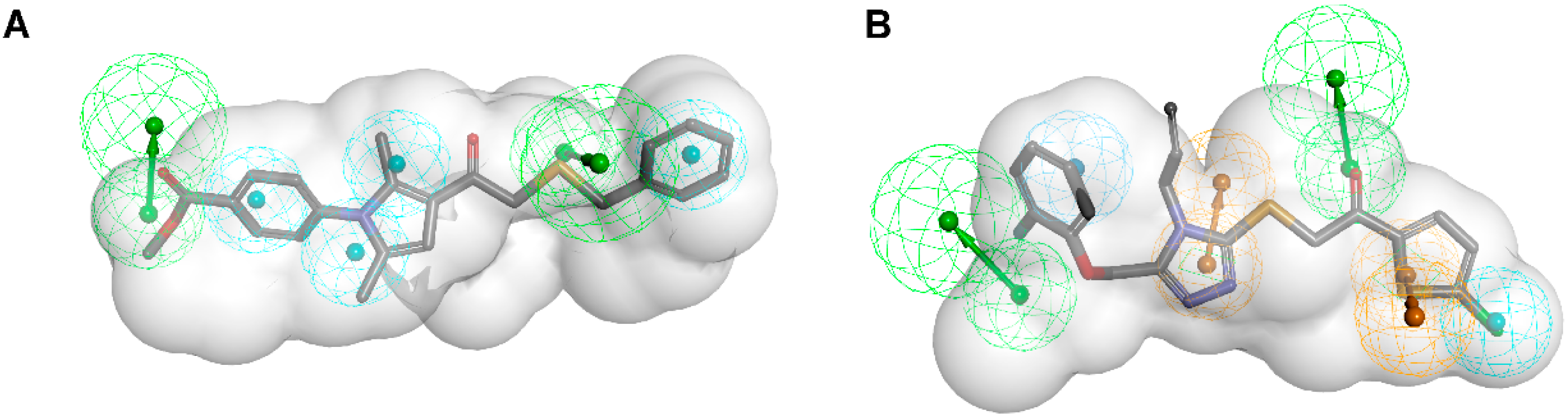
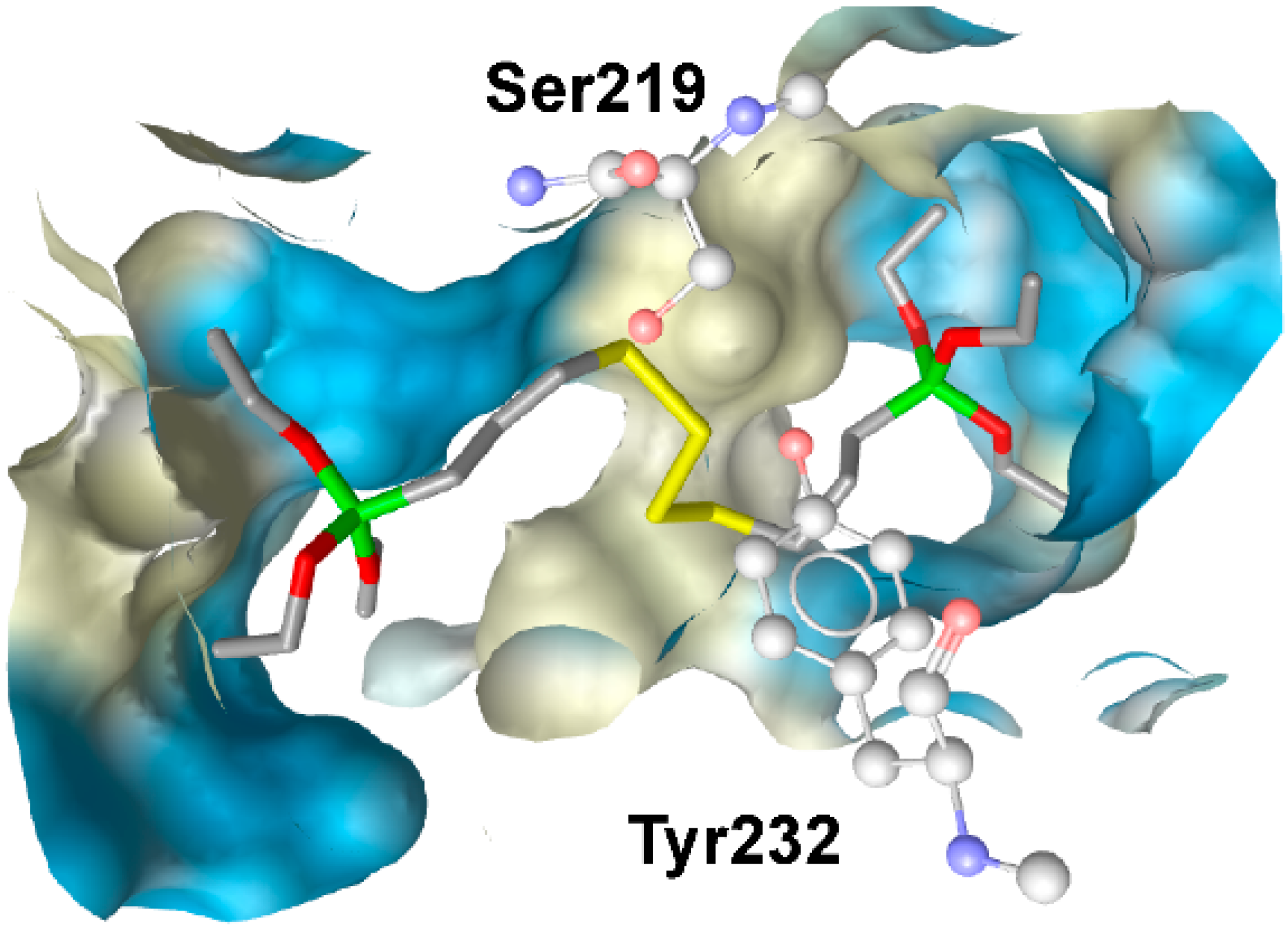
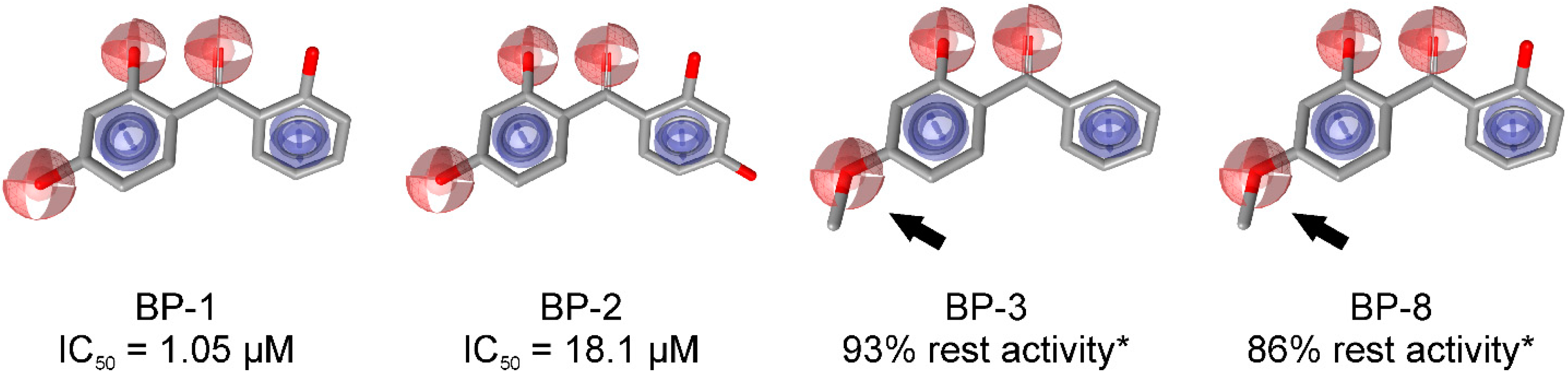
3.1.1. 17β-Hydroxysteroid Dehydrogenase Type 1

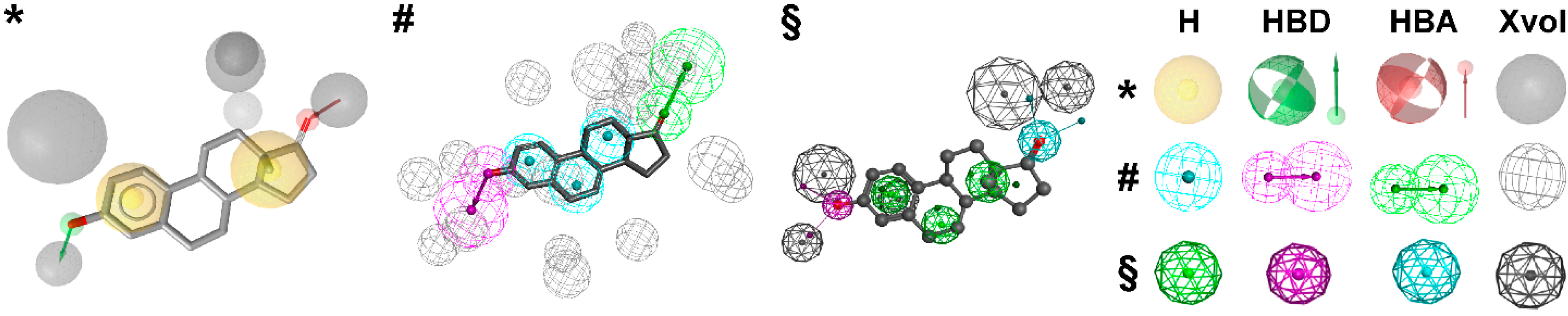{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference Study Aim | Pharmacophore Model | Database Used for VS | Hits | Biological Testing | |||||
|---|---|---|---|---|---|---|---|---|---|
| Most Active Hit | Number of Virtual Hits | Tested in Vitro | Actives | Assay | IC50 | Selectivity | |||
| Schuster and Maurer et al. [106] 11β-HSD1 inhibitors | Ligand-based using Catalyst | Asinex Gold and Platinum, Bionet 2003, ChemBridge DBS, Clab and IDC, Enamine 03, Interbioscreen 03 nat and syn, Maybridge 2003, NCI, Specs 09 03 |  | 16/20304 | 15 | 2 | Lysate | 2.03 and 7.59 μM | Against 11β-HSD2, 17β-HSD1, and 17β-HSD2 |
| 11β-HSD1 selective (4 H, 1 HBA, 1 HBD, and shape restriction) | |||||||||
| 11β-HSD unselective (5 H, 4 HBA and shape restriction) |  | 107/1776579 | 15 | 5 | Lysate | 11β-HSD1 0.144–2.81 μM 11β-HSD2 0.06–3.95 μM | Most of them against 17β-HSD1 and 17β-HSD2 | ||
| Hofer et al. [107] Lead optimization | 11β-HSD1 selective from Schuster and Maurer et al. [106] | In-house database | 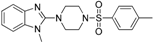 | - | - | - | Lysate | 0.7 μM | Against 11β-HSD2 |
| Rollinger et al. [108] Natural compounds inhibiting 11β-HSD1 | 11β-HSD1 selective from Schuster and Maurer et al. [106] | DIOS (Natural products in-house database) | 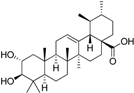 corosolic acid | 172 | 1 | 1 | Lysate | 0.81 μM | Against 11β-HSD2 |
| Vuorinen et al. [29] Refinement study | Refined models from Schuster and Maurer et al. [106] Using Discovery Studio | In-house database, DIOS | 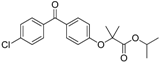 fenofibrate | 463 | 9 | 3 | Lysate | Considered as active if remaining enzyme activity ≤55% at test substance concentration of 20 μM or ≤65% at test substance concentration of 10 μM 5%–40% | Two preferentially inhibited 11β-HSD2, one was unselective |
| 11β-HSD1 selective | |||||||||
| 11β-HSD2 selective | In-house database, Specs, Maybridge | 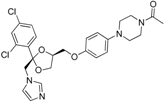 ketoconazole | 444 | 25 | 2 | 11%–61% Enzyme rest activity | One preferentially inhibited 11β-HSD1 and one was unselective | ||
| 11β-HSD unselective | EDC, In-house database | 38 | 4 | 36%–49% Enzyme rest activity | Two preferentially inhibited 11β-HSD1 one preferentially inhibited 11β-HSD2 | ||||
| Vuorinen et al. [112] Mode of action study | Refined 11β-HSD1 model from Vuorinen et al. [29] | DIOS | 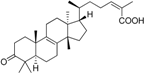 | 305/6702 | 2 | 2 | Lysate | 1.94 μM and 2.15 μM | Against 11β-HSD2 |
| Yang et al. [113] 11β-HSD1 inhibitors | Ligand-based Using Catalyst (4 H, 1 HBA, 1 AR) | SPECS | 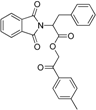 Active against human 11β-HSD1 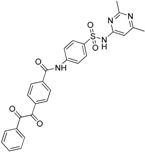 Active against human and mouse 11β-HSD1 | 3000 Selected by docking (these 3000 were fitted in the pharmacophore model) | 121 (39 out of docking and 82 from pharmacophore modeling) | 11 | Scintillation proximity assay | Human 11β-HSD1 0.26–14.6 μM Nine compounds Mouse 11β-HSD1 0.48–12.49 μM | Only tested against mouse 11β-HSD2 not tested toward the human 11β-HSD2 |
| Yang et al. [119] 11β-HSD1 inhibitors | Two structure-based models using LigandScout (PDB code 2IRW) (3 H, 1HBD, 1 HBA) | SPECS | 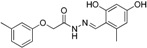 | 1000 Selected for each model | 56 | Nine human and six mouse | Scintillation proximity assay | Human 11β-HSD1 0.85–7.98 μM Mouse 11β-HSD1 0.44–8.48 μM | Against 11β-HSD2 |

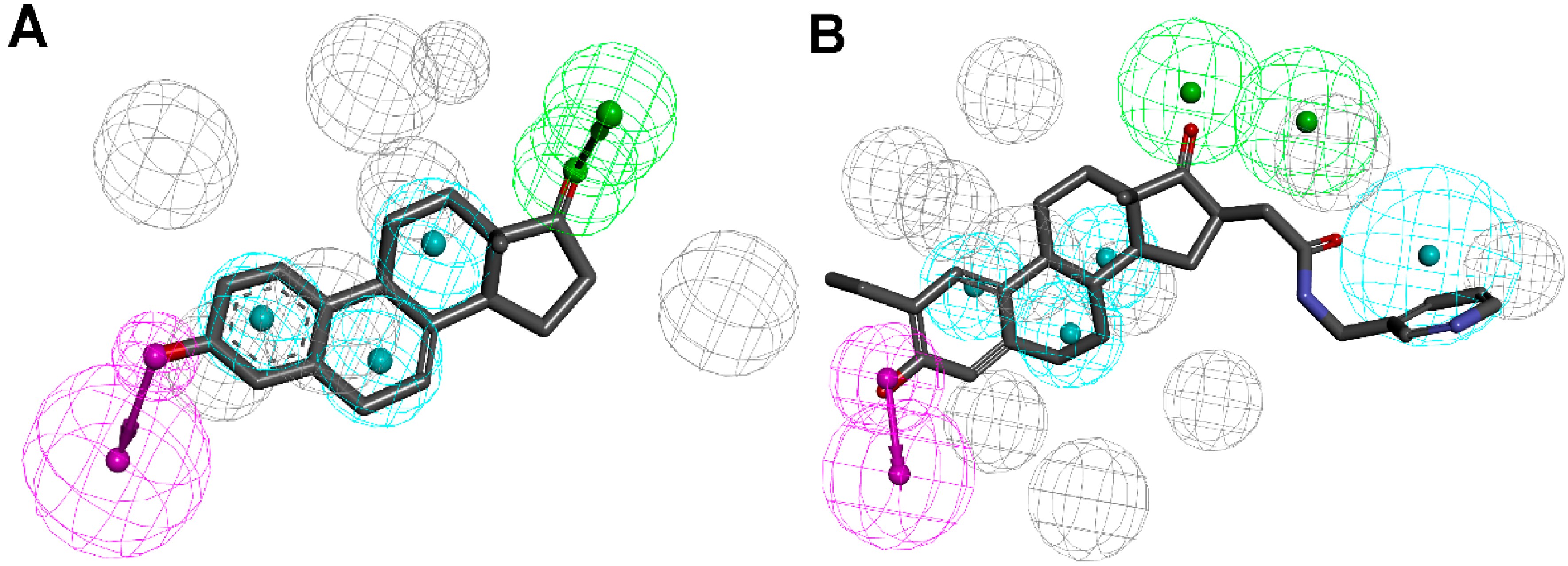

3.1.2. 17β-Hydroxysteroid Dehydrogenase Type 2
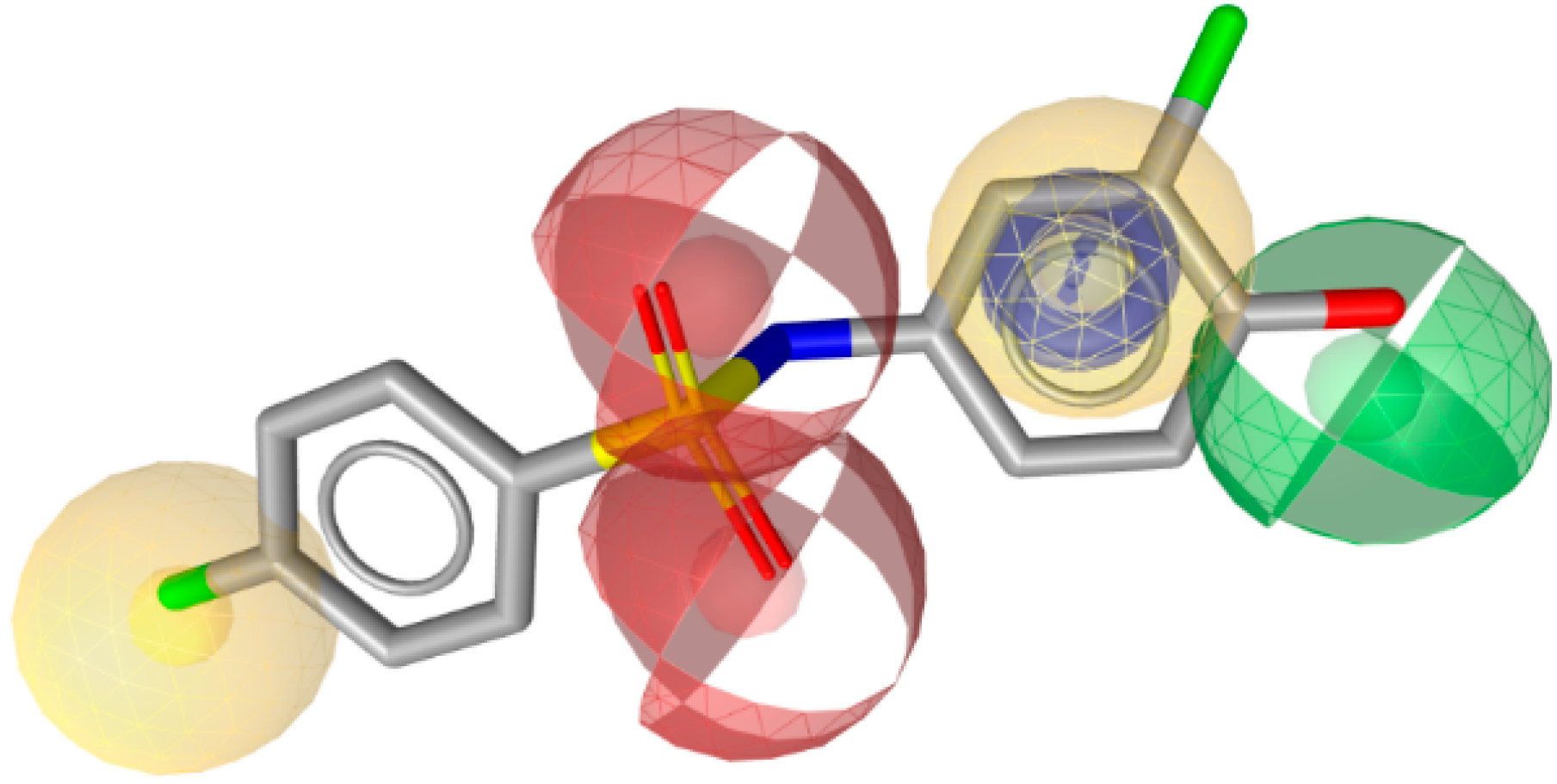
3.1.3. 17β-Hydroxysteroid Dehydrogenase Type 3
| Reference Study Aim | Pharmacophore Model | Database Used for VS | Hits | Biological Testing | |||||
|---|---|---|---|---|---|---|---|---|---|
| Most Active Hit | Number of Virtual Hits | Tested in Vitro | Actives | Assay | IC50 | Selectivity | |||
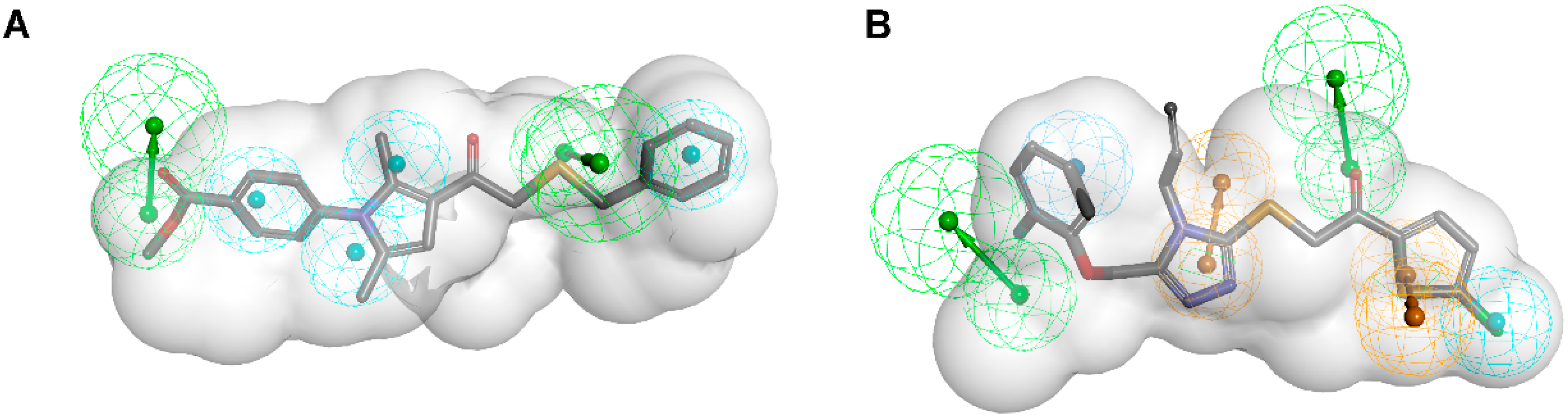
| Schuster and Nashev et al. [133] 17β-HSD1 inhibitors | Structure-based Using LigandScout and Catalyst 1I5R model (4 H, 2HBA, 2 HBD) Based on a hybrid inhibitor | NCI, SPECS |  | 1559/340042 | 14 | 4, IC50 < 50 μM | Lysates | 5.7–47 μM | Selective over 17β-HSD2, 17β-HSD3, 17β-HSD5 and 11β-HSD1, except one compound, which was not selective towards 17β-HSD5 and 11β-HSD1 However, one compound inhibited 17β-HSD3 and 11β-HSD1 but not 17β-HSD1 and another compound inhibited 11β-HSD1 only |
| Sparado et al. [134] 17β-HSD1 inhibitors and lead optimization | Ligand-based By superimposing co-crystallized ligands using MOE (5 H, 3 HBA, 1 HBD, 1 AR) | In-house database |  | -/37 | - | 1 | Cell-free | 34% Enzyme inhibition with 10 μM test compounds | Selectivity of optimized compounds tested against 17β-HSD2 and ERα and ERβ |
| Reference Study Aim | Pharmacophore Model | Database Used for VS | Hits | Biological Testing | |||||
|---|---|---|---|---|---|---|---|---|---|
| Most Active Hit | Number of Hits after Filtering | Tested in Vitro | Actives | Assay | Enzyme Inhibition | Selectivity | |||
| Schuster et al. [146] 17β-HSD3 inhibitors | Ligand-based Using Catalyst | Asinex Gold and Platinum, ChemBridge, Enamine, IF-Labs, Maybridge, Specs, Vitas-M |  | 3921/1712102 | 15 | 2 | Lysates | Inhibition >40% with 2 μM test compounds as threshold 41.3% and 50.8% | Selective over 17β-HSD2, 17β-HSD4, 17β-HSD7, 11β-HSD1, and 11β-HSD2, acceptable selectivity over 17β-HSD1 and 17β-HSD5. However, several hits inhibited 17β-HSD5 more potently than 17β-HSD3 |
| Model 1: steroidal training compounds (four H, two HBA) | |||||||||
| Model 2: non-steroidal training compounds (one H, two HBA, two AR, one H-AR) |  | 8190/1712102 | 16 | 2 | 55.6% and 57.5% | Selective over 17β-HSD2, 17β-HSD4, 17β-HSD7, and 11β-HSD2, acceptable selectivity over 17β-HSD1 One hit was not selective over 17β-HSD5 and the other not over 11β-HSD1. However, several hits inhibited 17β-HSD5 more potently than 17β-HSD3 | |||
3.2. Applications in Toxicology
3.2.1. Anti-Target Screening
3.2.2. Parallel Screening
3.2.3. Examples
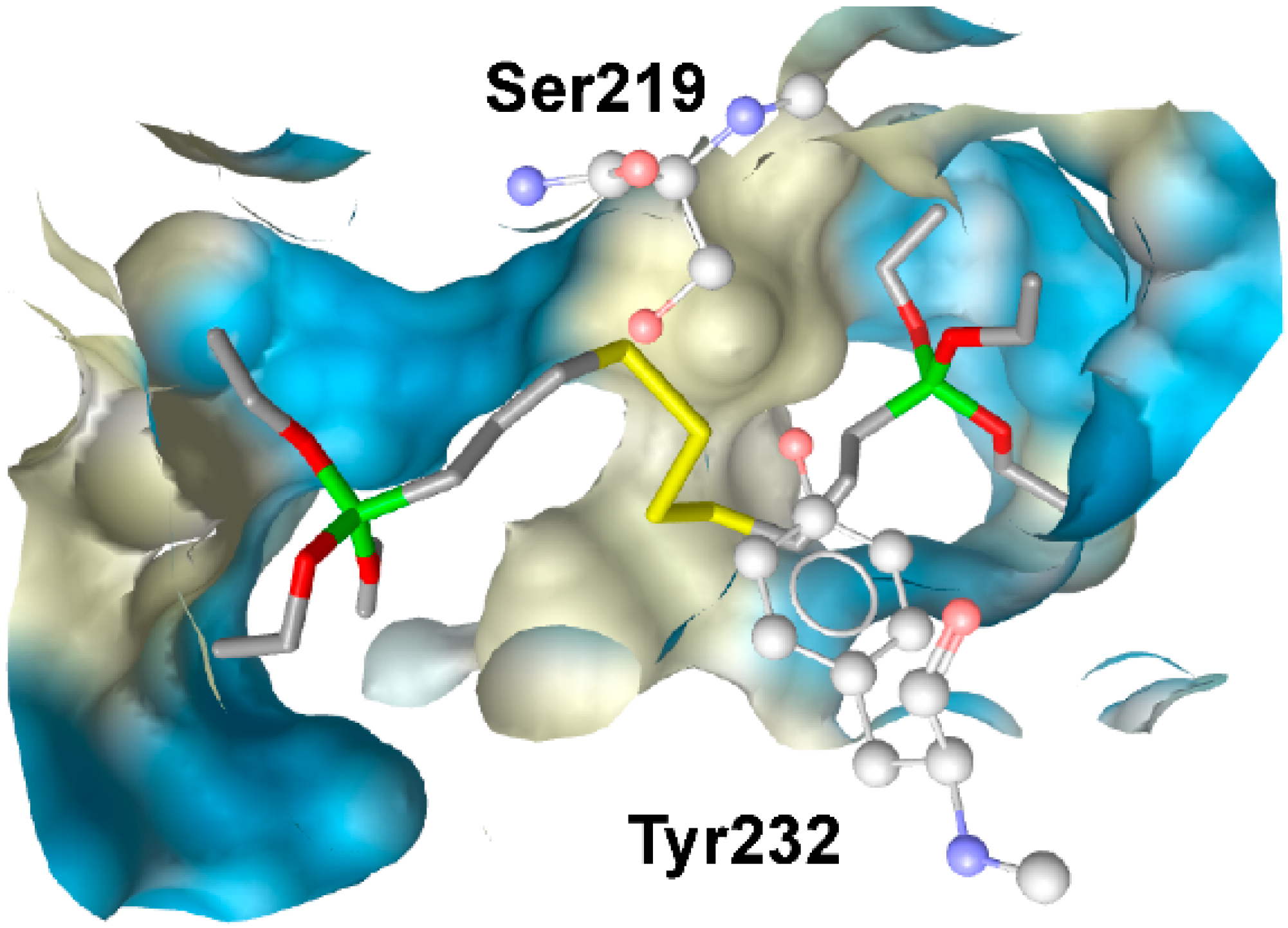
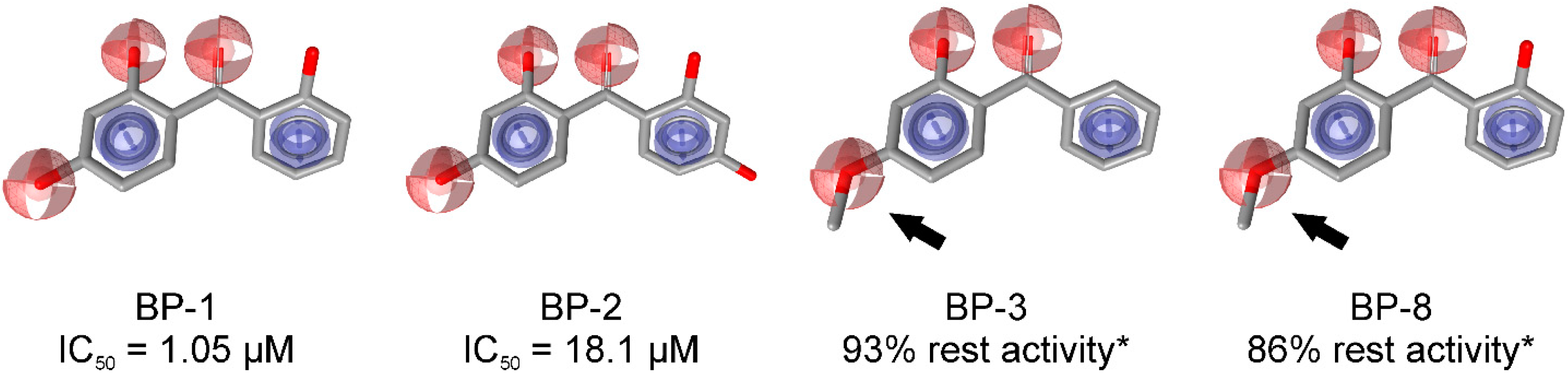
4. Limitations
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3-BC | 3-benzylidene camphor |
| 4-MBC | 4-methylbenzylidene camphor |
| AKR | aldo-keto reductase |
| AME | apparent mineralocorticoid excess |
| AR | aromatic features |
| BP | benzophenone |
| CETP | cholesterylester transfer protein |
| CYP | cytochrome P450 |
| DHEA | dehydroepiandrosterone |
| DHT | 5α-dihydrotestosterone |
| DUD-E | Directory of Useful Decoys, Enhanced |
| EDCs | endocrine disrupting chemicals |
| ER | estrogen receptor |
| GA | glycyrrhetinic acid |
| H | hydrophobic feature |
| HBA | hydrogen bond acceptor |
| HBD | hydrogen bond donor |
| hERG | human ether-a-go-go related gene |
| HSD | hydroxysteroid dehydrogenase |
| HTS | high-throughput screening |
| IUPAC | International Union of Pure and Applied Chemistry |
| MD | molecular dynamics |
| MOE | Molecular Operating Environment |
| MR | mineralocorticoid receptor |
| NADP | nicotinamide adenine dinucleotide phosphate |
| PAINS | Pan-Assay Interference Compounds |
| PDB | Protein Data Bank |
| PPAR γ | peroxisome proliferator-activated receptor γ |
| ROC-AUC | area under the receiver operating characteristics curve |
| SAR | structure-activity relationship |
| SDR | short-chain dehydrogenase/reductase |
| VS | virtual screening |
| XVols | exclusion volumes |
References
- Ehrlich, P. Über die constitution des diphtheriegiftes. Deutsch. Med. Wochschr. 1898, 24, 597–600. [Google Scholar] [CrossRef]
- Güner, O.F.; Bowen, J.P. Setting the record straight: The origin of the pharmacophore concept. J. Chem. Inf. Model. 2014, 54, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Schueler, F.W. Chemobiodynamics and Drug Design; McGraw-Hill: New York, NY, USA, 1960. [Google Scholar]
- Wermuth, G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (iupac recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Sawicki, M.W.; Erman, M.; Puranen, T.; Vihko, P.; Ghosh, D. Structure of the ternary complex of human 17β-hydroxysteroid dehydrogenase type 1 with 3-hydroxyestra-1,3,5,7-tetraen-17-one (equilin) and NADP+. Proc. Natl. Acad. Sci. USA 1999, 96, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. Ligandscout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment; Dassault Systèmes: San Diego, CA, USA, 2015. [Google Scholar]
- Molecular Operating Environment (MOE); Chemical Computing Group Inc.: Montreal, QC, Canada, 2015.
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sutter, J.; Li, J.; Maynard, A.J.; Goupil, A.; Luu, T.; Nadassy, K. New features that improve the pharmacophore tools from accelrys. Curr. Comput.-Aided Drug Des. 2011, 7, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, W.; Fang, H.; Perkins, R.; Tong, W.; Hong, H. Homology modeling, molecular docking, and molecular dynamics simulations elucidated alpha-fetoprotein binding modes. BMC Bioinform. 2013, 14 (Suppl. 14), S6. [Google Scholar]
- Bey, E.; Marchais-Oberwinkler, S.; Kruchten, P.; Frotscher, M.; Werth, R.; Oster, A.; Algül, O.; Neugebauer, A.; Hartmann, R.W. Design, synthesis and biological evaluation of bis(hydroxyphenyl) azoles as potent and selective non-steroidal inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) for the treatment of estrogen-dependent diseases. Bioorg. Med. Chem. 2008, 16, 6423–6435. [Google Scholar] [CrossRef] [PubMed]
- Oster, A.; Hinsberger, S.; Werth, R.; Marchais-Oberwinkler, S.; Frotscher, M.; Hartmann, R.W. Bicyclic substituted hydroxyphenylmethanones as novel inhibitors of 17β-hydroxysteroid dehydrogenase type 1 (17β-hsd1) for the treatment of estrogen-dependent diseases. J. Med. Chem. 2010, 53, 8176–8186. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Kaserer, T.; Schuster, D. Pharmacophore modeling and screening. In In silico Drug Discovery and Design: Theory, Methods, Challenges and Applications; Cavasotto, C., Ed.; CRC Press: Boca Raton, FL, USA, 2015; pp. 123–153. [Google Scholar]
- Vuorinen, A.; Schuster, D. Methods for generating and applying pharmacophore models as virtual screening filters and for bioactivity profiling. Methods 2015, 71, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Zagury, J.-F.; Montes, M. Benchmarking data sets for the evaluation of virtual ligand screening methods: Review and perspectives. J. Chem. Inf. Model. 2015, 55, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Heikamp, K.; Bajorath, J. Comparison of confirmed inactive and randomly selected compounds as negative training examples in support vector machine-based virtual screening. J. Chem. Inf. Model. 2013, 53, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. Chembl: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. Drugbank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.J.; Harland, L.; Groth, P.; Pettifer, S.; Chichester, C.; Willighagen, E.L.; Evelo, C.T.; Blomberg, N.; Ecker, G.; Goble, C.; et al. Open phacts: Semantic interoperability for drug discovery. Drug Discov. Today 2012, 17, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Dix, D.J.; Houck, K.A.; Martin, M.T.; Richard, A.M.; Setzer, R.W.; Kavlock, R.J. The toxcast program for prioritizing toxicity testing of environmental chemicals. Toxicol. Sci. 2007, 95, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Kavlock, R.J.; Austin, C.P.; Tice, R.R. Toxicity testing in the 21st century: Implications for human health risk assessment. Risk Anal. 2009, 29, 485–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. Pubchem’s bioassay database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.M.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual screening using protein−ligand docking: Avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3d virtual screening protocols: Rmsd comparisons, enrichment assessments, and decoy selection—What can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Nashev, L.G.; Odermatt, A.; Rollinger, J.M.; Schuster, D. Pharmacophore model refinement for 11β-xydroxysteroid dehydrogenase inhibitors: Search for modulators of intracellular glucocorticoid concentrations. Mol. Inf. 2014, 33, 15–25. [Google Scholar] [CrossRef]
- Güner, F.; Henry, R. Metric for analyzing hit-lists and pharmacophores. In Pharmacophore Perception, Development, and Use in Drug Design; Güner, O.F., Ed.; International University Line: La Jolla, CA, USA, 2000; pp. 193–212. [Google Scholar]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.C.; Andrade, C.H. Assessing the performance of 3d pharmacophore models in virtual screening: How good are they? Curr. Top. Med. Chem. 2013, 13, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Tanrikulu, Y.; Krüger, B.; Proschak, E. The holistic integration of virtual screening in drug discovery. Drug Discov. Today 2013, 18, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Spetea, M.; Music, M.; Rief, S.; Fink, M.; Kirchmair, J.; Schütz, J.; Wolber, G.; Langer, T.; Stuppner, H.; et al. Morphinans and isoquinolines: Acetylcholinesterase inhibition, pharmacophore modeling, and interaction with opioid receptors. Bioorganic Med. Chem. 2010, 18, 5071–5080. [Google Scholar] [CrossRef] [PubMed]
- Polgár, T.; Baki, A.; Szendrei, G.I.; Keserűu, G.M. Comparative virtual and experimental high-throughput screening for glycogen synthase kinase-3β inhibitors. J. Med. Chem. 2005, 48, 7946–7959. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1b. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Gao, J.; Wang, M. Development of a complex scintillation proximity assay for high throughput screening of ppar[gamma] modulators. Acta Pharmacol. Sin. 2005, 26, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Murgueitio, M.S.; Henneke, P.; Glossmann, H.; Santos-Sierra, S.; Wolber, G. Prospective virtual screening in a sparse data scenario: Design of small-molecule tlr2 antagonists. ChemMedChem 2014, 9, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Krautscheid, Y.; Senning, C.J.Å.; Sartori, S.B.; Singewald, N.; Schuster, D.; Stuppner, H. Pharmacophore modeling, virtual screening, and in vitro testing reveal haloperidol, eprazinone, and fenbutrazate as neurokinin receptors ligands. J. Chem. Inf. Model. 2014, 54, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.Y.; Lee, H.Y.; Park, J.; Lee, J.-Y.; Chang, B.H.; No, K.T.; Nam, K.-Y.; Hwang, J.S. Identification of novel rab27a/melanophilin blockers by pharmacophore-based virtual screening. Appl. Biochem. Biotechnol. 2014, 172, 1882–1897. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Wang, Y.; Ouyang, P.K.; She, J.; He, M. 3d-qsar based pharmacophore modeling and virtual screening for identification of novel g protein-coupled receptor 40 agonists. Curr. Comput.-Aided Drug Des. 2015, 11, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tiwari, S.; Srivastava, K.K.; Siddiqi, M.I. Identification of novel inhibitors of mycobacterium tuberculosis pkng using pharmacophore based virtual screening, docking, molecular dynamics simulation, and their biological evaluation. J. Chem. Inf. Model. 2015, 55, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Temml, V.; Voss, C.V.; Dirsch, V.M.; Schuster, D. Discovery of new liver x receptor agonists by pharmacophore modeling and shape-based virtual screening. J. Chem. Inf. Model. 2014, 54, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Debnath, B.; Odde, S.; Bensman, T.; Ho, H.; Beringer, P.M.; Neamati, N. Discovery of novel cxcr2 inhibitors using ligand-based pharmacophore models. J. Chem. Inf. Model. 2015, 55, 1720–1738. [Google Scholar] [CrossRef] [PubMed]
- Lepailleur, A.; Freret, T.; Lemaître, S.; Boulouard, M.; Dauphin, F.; Hinschberger, A.; Dulin, F.; Lesnard, A.; Bureau, R.; Rault, S. Dual histamine h3r/serotonin 5-ht4r ligands with antiamnesic properties: Pharmacophore-based virtual screening and polypharmacology. J. Chem. Inf. Model. 2014, 54, 1773–1784. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.J.; dos Santos, D.J.V.A.; Ferreira, M.-J.U.; Guedes, R.C. Toward a better pharmacophore description of p-glycoprotein modulators, based on macrocyclic diterpenes from euphorbia species. J. Chem. Inf. Model. 2011, 51, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Flohr, S.; Kurz, M.; Kostenis, E.; Brkovich, A.; Fournier, A.; Klabunde, T. Identification of nonpeptidic urotensin ii receptor antagonists by virtual screening based on a pharmacophore model derived from structure−activity relationships and nuclear magnetic resonance studies on urotensin ii. J. Med. Chem. 2002, 45, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Hessler, G.; Baringhaus, K.-H. The scaffold hopping potential of pharmacophores. Drug Discov. Today Technol. 2010, 7, e263–e269. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, D.; Pakfeifer, P.; Hering, S.; Ecker, G.F. Novel scaffolds for modulation of trpv1 identified with pharmacophore modeling and virtual screening. Future Med. Chem. 2015, 7, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Ayan, D.; Maltais, R.; Roy, J.; Poirier, D. A new nonestrogenic steroidal inhibitor of 17beta-hydroxysteroid dehydrogenase type i blocks the estrogen-dependent breast cancer tumor growth induced by estrone. Mol. Cancer Ther. 2012, 11, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Delvoux, B.; D’Hooghe, T.; Kyama, C.; Koskimies, P.; Hermans, R.J.; Dunselman, G.A.; Romano, A. Inhibition of type 1 17beta-hydroxysteroid dehydrogenase impairs the synthesis of 17beta-estradiol in endometriosis lesions. J. Clin. Endocr. Metab. 2014, 99, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Marchais-Oberwinkler, S.; Henn, C.; Moller, G.; Klein, T.; Negri, M.; Oster, A.; Spadaro, A.; Werth, R.; Wetzel, M.; Xu, K.; et al. 17beta-hydroxysteroid dehydrogenases (17beta-hsds) as therapeutic targets: Protein structures, functions, and recent progress in inhibitor development. J. Steroid Biochem. Mol. Biol. 2011, 125, 66–82. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Wittenberg, L.O.; Basu, S.; Jeyaraj, D.A.; Gourzoulidou, E.; Reinecke, K.; Odermatt, A.; Waldmann, H. Compound library development guided by protein structure similarity clustering and natural product structure. Proc. Natl. Acad. Sci. USA 2004, 101, 16721–16726. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.A.; Schuffenhauer, A.; Scheck, M.; Wetzel, S.; Casaulta, M.; Odermatt, A.; Ertl, P.; Waldmann, H. Charting biologically relevant chemical space: A structural classification of natural products (sconp). Proc. Natl. Acad. Sci. USA 2005, 102, 17272–17277. [Google Scholar] [CrossRef] [PubMed]
- Guasch, L.; Sala, E.; Castell-Auví, A.; Cedó, L.; Liedl, K.R.; Wolber, G.; Muehlbacher, M.; Mulero, M.; Pinent, M.; Ardévol, A.; et al. Identification of ppargamma partial agonists of natural origin (i): Development of a virtual screening procedure and in vitro validation. PLoS ONE 2012, 7, e50816. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of three” for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J. Chem. Med. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Noha, S.M.; Fischer, K.; Koeberle, A.; Garscha, U.; Werz, O.; Schuster, D. Discovery of novel, non-acidic mPGES-1 inhibitors by virtual screening with a multistep protocol. Bioorganic Med. Chem. 2015, 23, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.L.; Jornvall, H.; Persson, B.; Oppermann, U. Medium- and short-chain dehydrogenase/reductase gene and protein families: The SDR superfamily: Functional and structural diversity within a family of metabolic and regulatory enzymes. Cell. Mol. Life Sci. 2008, 65, 3895–3906. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; He, X.Y.; Isaacs, C.; Dobkin, C.; Miller, D.; Philipp, M. Roles of 17β-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. J. Steroid Biochem. Mol. Biol. 2014, 143, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Gathercole, L.L.; Lavery, G.G.; Morgan, S.A.; Cooper, M.S.; Sinclair, A.J.; Tomlinson, J.W.; Stewart, P.M. 11β-hydroxysteroid dehydrogenase 1: Translational and therapeutic aspects. Endocr. Rev. 2013, 34, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Luu-The, V. Assessment of steroidogenesis and steroidogenic enzyme functions. J. Steroid Biochem. Mol. Biol. 2013, 137, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Vuorinen, A.; Odermatt, A.; Schuster, D. In silico methods in the discovery of endocrine disrupting chemicals. J. Steroid Biochem. Mol. Biol. 2013, 137, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Vitku, J.; Starka, L.; Bicikova, M.; Hill, M.; Heracek, J.; Sosvorova, L.; Hampl, R. Endocrine disruptors and other inhibitors of 11β-hydroxysteroid dehydrogenase 1 and 2: Tissue-specific consequences of enzyme inhibition. J. Steroid Biochem. Mol. Biol. 2016, 155, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Nashev, L.G.; Vuorinen, A.; Praxmarer, L.; Chantong, B.; Cereghetti, D.; Winiger, R.; Schuster, D.; Odermatt, A. Virtual screening as a strategy for the identification of xenobiotics disrupting corticosteroid action. PLoS ONE 2012, 7, e46958. [Google Scholar] [CrossRef] [PubMed]
- Odermatt, A.; Nashev, L.G. The glucocorticoid-activating enzyme 11β-hydroxysteroid dehydrogenase type 1 has broad substrate specificity: Physiological and toxicological considerations. J. Steroid Biochem. Mol. Biol. 2010, 119, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maser, E.; Oppermann, U.C. Role of type-1 11β-hydroxysteroid dehydrogenase in detoxification processes. Eur. J. Biochem. 1997, 249, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Maser, E. Xenobiotic carbonyl reduction and physiological steroid oxidoreduction. The pluripotency of several hydroxysteroid dehydrogenases. Biochem. Pharmacol. 1995, 49, 421–440. [Google Scholar] [CrossRef]
- Nashev, L.G.; Schuster, D.; Laggner, C.; Sodha, S.; Langer, T.; Wolber, G.; Odermatt, A. The uv-filter benzophenone-1 inhibits 17β-hydroxysteroid dehydrogenase type 3: Virtual screening as a strategy to identify potential endocrine disrupting chemicals. Biochem. Pharmacol. 2010, 79, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Zhao, B.; Li, X.W.; Hu, G.X.; Su, Y.; Chu, Y.; Akingbemi, B.T.; Lian, Q.Q.; Ge, R.S. Effects of phthalates on 3β-hydroxysteroid dehydrogenase and 17beta-hydroxysteroid dehydrogenase 3 activities in human and rat testes. Chem.-Biol. Interact. 2012, 195, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chu, Y.; Hardy, D.O.; Li, X.K.; Ge, R.S. Inhibition of 3β- and 17β-hydroxysteroid dehydrogenase activities in rat leydig cells by perfluorooctane acid. J. Steroid Biochem. Mol. Biol. 2009, 118, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.; Holmes, M.; Seckl, J. 11beta-hydroxysteroid dehydrogenases: Intracellular gate-keepers of tissue glucocorticoid action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef] [PubMed]
- Kratschmar, D.V.; Vuorinen, A.; Da Cunha, T.; Wolber, G.; Classen-Houben, D.; Doblhoff, O.; Schuster, D.; Odermatt, A. Characterization of activity and binding mode of glycyrrhetinic acid derivatives inhibiting 11beta-hydroxysteroid dehydrogenase type 2. J. Steroid Biochem. Mol. Biol. 2011, 125, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Kannisto, K.; Pietilainen, K.H.; Ehrenborg, E.; Rissanen, A.; Kaprio, J.; Hamsten, A.; Yki-Jarvinen, H. Overexpression of 11β-hydroxysteroid dehydrogenase-1 in adipose tissue is associated with acquired obesity and features of insulin resistance: Studies in young adult monozygotic twins. J. Clin. Endocrinol. Metab. 2004, 89, 4414–4421. [Google Scholar] [CrossRef] [PubMed]
- Kotelevtsev, Y.; Holmes, M.C.; Burchell, A.; Houston, P.M.; Schmoll, D.; Jamieson, P.; Best, R.; Brown, R.; Edwards, C.R.; Seckl, J.R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc. Natl. Acad. Sci. USA 1997, 94, 14924–14929. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, R.S.; Wake, D.J.; Nair, S.; Bunt, J.; Livingstone, D.E.; Permana, P.A.; Tataranni, P.A.; Walker, B.R. Subcutaneous adipose 11β-hydroxysteroid dehydrogenase type 1 activity and messenger ribonucleic acid levels are associated with adiposity and insulinemia in pima indians and caucasians. J. Clin. Endocrinol. Metab. 2003, 88, 2738–2744. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Paterson, J.; Shinyama, H.; Morton, N.M.; Mullins, J.J.; Seckl, J.R.; Flier, J.S. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001, 294, 2166–2170. [Google Scholar] [CrossRef] [PubMed]
- Masuzaki, H.; Yamamoto, H.; Kenyon, C.J.; Elmquist, J.K.; Morton, N.M.; Paterson, J.M.; Shinyama, H.; Sharp, M.G.; Fleming, S.; Mullins, J.J.; et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J. Clin. Investig. 2003, 112, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Paterson, J.M.; Morton, N.M.; Fievet, C.; Kenyon, C.J.; Holmes, M.C.; Staels, B.; Seckl, J.R.; Mullins, J.J. Metabolic syndrome without obesity: Hepatic overexpression of 11β-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc. Natl. Acad. Sci. USA 2004, 101, 7088–7093. [Google Scholar] [CrossRef] [PubMed]
- Paulmyer-Lacroix, O.; Boullu, S.; Oliver, C.; Alessi, M.C.; Grino, M. Expression of the mRNA coding for 11β-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: An in situ hybridization study. J. Clin. Endocrinol. Metab. 2002, 87, 2701–2705. [Google Scholar] [CrossRef] [PubMed]
- Rask, E.; Walker, B.R.; Soderberg, S.; Livingstone, D.E.; Eliasson, M.; Johnson, O.; Andrew, R.; Olsson, T. Tissue-specific changes in peripheral cortisol metabolism in obese women: Increased adipose 11β-hydroxysteroid dehydrogenase type 1 activity. J. Clin. Endocrinol. Metab. 2002, 87, 3330–3336. [Google Scholar] [CrossRef] [PubMed]
- Valsamakis, G.; Anwar, A.; Tomlinson, J.W.; Shackleton, C.H.; McTernan, P.G.; Chetty, R.; Wood, P.J.; Banerjee, A.K.; Holder, G.; Barnett, A.H.; et al. 11β-hydroxysteroid dehydrogenase type 1 activity in lean and obese males with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2004, 89, 4755–4761. [Google Scholar] [CrossRef] [PubMed]
- Kipari, T.; Hadoke, P.W.; Iqbal, J.; Man, T.Y.; Miller, E.; Coutinho, A.E.; Zhang, Z.; Sullivan, K.M.; Mitic, T.; Livingstone, D.E.; et al. 11β-hydroxysteroid dehydrogenase type 1 deficiency in bone marrow-derived cells reduces atherosclerosis. FASEB J. 2013, 27, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Hermanowski-Vosatka, A.; Balkovec, J.M.; Cheng, K.; Chen, H.Y.; Hernandez, M.; Koo, G.C.; Le Grand, C.B.; Li, Z.; Metzger, J.M.; Mundt, S.S.; et al. 11β-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J. Exp. Med. 2005, 202, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.A.; Search, D.J.; Lupisella, J.A.; Ostrowski, J.; Guan, B.; Chen, J.; Yang, W.P.; Truong, A.; He, A.; Zhang, R.; et al. 11β-hydroxysteroid dehydrogenase type 1 gene knockout attenuates atherosclerosis and in vivo foam cell formation in hyperlipidemic apoe−/− mice. PLoS ONE 2013, 8, e53192. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.J.; Thieringer, R.; Springer, M.S.; Wright, S.D.; Hermanowski-Vosatka, A.; Plump, A.; Balkovec, J.M.; Cheng, K.; Ding, G.J.; Kawka, D.W.; et al. 11β-HSD1 inhibition reduces atherosclerosis in mice by altering proinflammatory gene expression in the vasculature. Physiol. Genom. 2013, 45, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Qi, H.; Zhong, Y.; Lv, S.; Yu, J.; Liu, J.; Wang, L.; Bi, J.; Kong, X.; Di, W.; et al. 11β-hydroxysteroid dehydrogenase type 1 selective inhibitor bvt.2733 protects osteoblasts against endogenous glucocorticoid induced dysfunction. Endocr. J. 2013, 60, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Rauz, S.; Cheung, C.M.; Wood, P.J.; Coca-Prados, M.; Walker, E.A.; Murray, P.I.; Stewart, P.M. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 lowers intraocular pressure in patients with ocular hypertension. QJM 2003, 96, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Rauz, S.; Walker, E.A.; Shackleton, C.H.; Hewison, M.; Murray, P.I.; Stewart, P.M. Expression and putative role of 11β-hydroxysteroid dehydrogenase isozymes within the human eye. Investig. Ophthalmol. Visual Sci. 2001, 42, 2037–2042. [Google Scholar]
- Anderson, S.; Carreiro, S.; Quenzer, T.; Gale, D.; Xiang, C.; Gukasyan, H.; Lafontaine, J.; Cheng, H.; Krauss, A.; Prasanna, G. In vivo evaluation of 11β-hydroxysteroid dehydrogenase activity in the rabbit eye. J. Ocul. Pharmacol. Ther. 2009, 25, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Sooy, K.; Webster, S.P.; Noble, J.; Binnie, M.; Walker, B.R.; Seckl, J.R.; Yau, J.L. Partial deficiency or short-term inhibition of 11β-hydroxysteroid dehydrogenase type 1 improves cognitive function in aging mice. J. Neurosci. 2010, 30, 13867–13872. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.L.; McNair, K.M.; Noble, J.; Brownstein, D.; Hibberd, C.; Morton, N.; Mullins, J.J.; Morris, R.G.; Cobb, S.; Seckl, J.R. Enhanced hippocampal long-term potentiation and spatial learning in aged 11β-hydroxysteroid dehydrogenase type 1 knock-out mice. J. Neurosci. 2007, 27, 10487–10496. [Google Scholar] [CrossRef] [PubMed]
- Yau, J.L.; Noble, J.; Seckl, J.R. 11β-hydroxysteroid dehydrogenase type 1 deficiency prevents memory deficits with aging by switching from glucocorticoid receptor to mineralocorticoid receptor-mediated cognitive control. J. Neurosci. 2011, 31, 4188–4193. [Google Scholar] [CrossRef] [PubMed]
- Sooy, K.; Noble, J.; McBride, A.; Binnie, M.; Yau, J.L.; Seckl, J.R.; Walker, B.R.; Webster, S.P. Cognitive and disease-modifying effects of 11ss-hydroxysteroid dehydrogenase type 1 inhibition in male tg2576 mice, a model of Alzheimer’s disease. Endocrinology 2015, 156, 4592–4603. [Google Scholar] [CrossRef] [PubMed]
- Mohler, E.G.; Browman, K.E.; Roderwald, V.A.; Cronin, E.A.; Markosyan, S.; Scott Bitner, R.; Strakhova, M.I.; Drescher, K.U.; Hornberger, W.; Rohde, J.J.; et al. Acute inhibition of 11β-hydroxysteroid dehydrogenase type-1 improves memory in rodent models of cognition. J. Neurosci. 2011, 31, 5406–5413. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Tahrani, A.A.; Morgan, S.A.; Otranto, M.; Desmouliere, A.; Abrahams, L.; Hassan-Smith, Z.; Walker, E.A.; Rabbitt, E.H.; Cooper, M.S.; et al. 11β-hydroxysteroid dehydrogenase blockade prevents age-induced skin structure and function defects. J. Clin. Investig. 2013, 123, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Tiganescu, A.; Hupe, M.; Uchida, Y.; Mauro, T.; Elias, P.M.; Holleran, W.M. Increased glucocorticoid activation during mouse skin wound healing. J. Endocrinol. 2014, 221, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Youm, J.K.; Park, K.; Uchida, Y.; Chan, A.; Mauro, T.M.; Holleran, W.M.; Elias, P.M. Local blockade of glucocorticoid activation reverses stress- and glucocorticoid-induced delays in cutaneous wound healing. Wound Repair Regen. 2013, 21, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.S.; Goldberg, F.W.; Turnbull, A.V. Medicinal chemistry of inhibitors of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1). J. Med. Chem. 2014, 57, 4466–4486. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Potter, B.V. Crystal structures of 11beta-hydroxysteroid dehydrogenase type 1 and their use in drug discovery. Future Med. Chem. 2011, 3, 367–390. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Maurer, E.M.; Laggner, C.; Nashev, L.G.; Wilckens, T.; Langer, T.; Odermatt, A. The discovery of new 11β-hydroxysteroid dehydrogenase type 1 inhibitors by common feature pharmacophore modeling and virtual screening. J. Med. Chem. 2006, 49, 3454–3466. [Google Scholar] [CrossRef] [PubMed]
- Hofer, S.; Kratschmar, D.V.; Schernthanner, B.; Vuorinen, A.; Schuster, D.; Odermatt, A.; Easmon, J. Synthesis and biological analysis of benzazol-2-yl piperazine sulfonamides as 11β-hydroxysteroid dehydrogenase 1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5397–5400. [Google Scholar] [CrossRef] [PubMed]
- Rollinger, J.M.; Kratschmar, D.V.; Schuster, D.; Pfisterer, P.H.; Gumy, C.; Aubry, E.M.; Brandstötter, S.; Stuppner, H.; Wolber, G.; Odermatt, A. 11β-hydroxysteroid dehydrogenase 1 inhibiting constituents from eriobotrya japonica revealed by bioactivity-guided isolation and computational approaches. Bioorganic Med. Chem. 2010, 18, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Gumy, C.; Thurnbichler, C.; Aubry, E.M.; Balazs, Z.; Pfisterer, P.; Baumgartner, L.; Stuppner, H.; Odermatt, A.; Rollinger, J.M. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 by plant extracts used as traditional antidiabetic medicines. Fitoterapia 2009. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Kavanagh, K.; Svensson, S.; Elleby, B.; Hult, M.; Von Delft, F.; Marsden, B.; Jornvall, H.; Abrahmsen, L.; Oppermann, U. Structure of human 11β-hydroxysteroid dehydrogenase in complex with nadp and carbenoxolone. PDB Entry 2BEL 2004. [Google Scholar] [CrossRef]
- Vuorinen, A.; Seibert, J.; Papageorgiou, V.P.; Rollinger, J.M.; Odermatt, A.; Schuster, D.; Assimopoulou, A.N. Pistacia lentiscus oleoresin: Virtual screening and identification of masticadienonic and isomasticadienonic acids as inhibitors of 11β-hydroxysteroid dehydrogenase 1. Planta Med. 2015, 81, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Dou, W.; Lou, J.; Leng, Y.; Shen, J. Discovery of novel inhibitors of 11β-hydroxysteroid dehydrogenase type 1 by docking and pharmacophore modeling. Bioorg. Med. Chem. Lett. 2008, 18, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- Hosfield, D.J.; Wu, Y.; Skene, R.J.; Hilgers, M.; Jennings, A.; Snell, G.P.; Aertgeerts, K. Conformational flexibility in crystal structures of human 11β-hydroxysteroid dehydrogenase type i provide insights into glucocorticoid interconversion and enzyme regulation. J. Biol. Chem. 2005, 280, 4639–4648. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.A.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput.-Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Catalyst Version 4.10; Accelrys Software Inc.: San Diego, CA, USA, 2005.
- Arampatzis, S.; Kadereit, B.; Schuster, D.; Balazs, Z.; Schweizer, R.A.; Frey, F.J.; Langer, T.; Odermatt, A. Comparative enzymology of 11β-hydroxysteroid dehydrogenase type 1 from six species. J. Mol. Endocrinol. 2005, 35, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Barf, T.; Vallgarda, J.; Emond, R.; Haggstrom, C.; Kurz, G.; Nygren, A.; Larwood, V.; Mosialou, E.; Axelsson, K.; Olsson, R.; et al. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11beta-hydroxysteroid dehydrogenase type 1. J. Med. Chem. 2002, 45, 3813–3815. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shen, Y.; Chen, J.; Jiang, Q.; Leng, Y.; Shen, J. Structure-based virtual screening for identification of novel 11β-HSD1 inhibitors. Eur. J. Med. Chem. 2009, 44, 1167–1171. [Google Scholar] [CrossRef] [PubMed]
- Moeller, G.; Adamski, J. Integrated view on 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2009, 301, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Poirier, D. Inhibitors of 17β-hydroxysteroid dehydrogenases. Curr. Med. Chem. 2003, 10, 453–477. [Google Scholar] [CrossRef] [PubMed]
- Lukacik, P.; Kavanagh, K.L.; Oppermann, U. Structure and function of human 17β-hydroxysteroid dehydrogenases. Mol. Cell. Endocrinol. 2006, 248, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jansson, A. 17β-hydroxysteroid dehydrogenase enzymes and breast cancer. J. Steroid Biochem. Mol. Biol. 2009, 114, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Oduwole, O.O.; Li, Y.; Isomaa, V.V.; Mantyniemi, A.; Pulkka, A.E.; Soini, Y.; Vihko, P.T. 17β-hydroxysteroid dehydrogenase type 1 is an independent prognostic marker in breast cancer. Cancer Res. 2004, 64, 7604–7609. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Ando, A.; Shiba, E.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Involvement of up-regulation of 17β-hydroxysteroid dehydrogenase type 1 in maintenance of intratumoral high estradiol levels in postmenopausal breast cancers. Int. J. Cancer 2001, 94, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Smuc, T.; Pucelj, M.R.; Sinkovec, J.; Husen, B.; Thole, H.; Rizner, T.L. Expression analysis of the genes involved in estradiol and progesterone action in human ovarian endometriosis. Gynecol. Endocrinol. 2007, 23, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Cornel, K.M.; Kruitwagen, R.F.; Delvoux, B.; Visconti, L.; van de Vijver, K.K.; Day, J.M.; van Gorp, T.; Hermans, R.J.; Dunselman, G.A.; Romano, A. Overexpression of 17β-hydroxysteroid dehydrogenase type 1 increases the exposure of endometrial cancer to 17β-estradiol. J. Clin. Endocrinol. Metab. 2012, 97, E591–E601. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Shozu, M.; Murakami, K.; Segawa, T.; Shinohara, K.; Nomura, K.; Inoue, M. Increased expression of type i 17β-hydroxysteroid dehydrogenase enhances in situ production of estradiol in uterine leiomyoma. J. Clin. Endocrinol. Metab. 2004, 89, 5661–5668. [Google Scholar] [CrossRef] [PubMed]
- Hoffren, A.M.; Murray, C.M.; Hoffmann, R.D. Structure-based focusing using pharmacophores derived from the active site of 17β-hydroxysteroid dehydrogenase. Curr. Pharm. Des. 2001, 7, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Krazeisen, A.; Breitling, R.; Moller, G.; Adamski, J. Phytoestrogens inhibit human 17β-hydroxysteroid dehydrogenase type 5. Mol. Cell. Endocrinol. 2001, 171, 151–162. [Google Scholar] [CrossRef]
- Berube, M.; Poirier, D. Synthesis of simplified hybrid inhibitors of type 1 17β-hydroxysteroid dehydrogenase via cross-metathesis and sonogashira coupling reactions. Org. Lett. 2004, 6, 3127–3130. [Google Scholar] [CrossRef] [PubMed]
- Fournier, D.; Poirier, D.; Mazumdar, M.; Lin, S.X. Design and synthesis of bisubstrate inhibitors of type 1 17β-hydroxysteroid dehydrogenase: Overview and perspectives. Eur. J. Med. Chem. 2008, 43, 2298–2306. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Nashev, L.G.; Kirchmair, J.; Laggner, C.; Wolber, G.; Langer, T.; Odermatt, A. Discovery of nonsteroidal 17β-hydroxysteroid dehydrogenase 1 inhibitors by pharmacophore-based screening of virtual compound libraries. J. Med. Chem. 2008, 51, 4188–4199. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, A.; Negri, M.; Marchais-Oberwinkler, S.; Bey, E.; Frotscher, M. Hydroxybenzothiazoles as new nonsteroidal inhibitors of 17beta-hydroxysteroid dehydrogenase type 1 (17β-HSD1). PLoS ONE 2012, 7, e29252. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, A.; Frotscher, M.; Hartmann, R.W. Optimization of hydroxybenzothiazoles as novel potent and selective inhibitors of 17β-HSD1. J. Med. Chem. 2012, 55, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Karkola, S.; Alho-Richmond, S.; Wahala, K. Pharmacophore modelling of 17β-HSD1 enzyme based on active inhibitors and enzyme structure. Mol. Cell. Endocrinol. 2009, 301, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Einstein, M.; Geissler, W.M.; Chan, H.K.; Elliston, K.O.; Andersson, S. Expression cloning and characterization of human 17β-hydroxysteroid dehydrogenase type 2, a microsomal enzyme possessing 20 α-hydroxysteroid dehydrogenase activity. J. Biol. Chem. 1993, 268, 12964–12969. [Google Scholar] [PubMed]
- Puranen, T.J.; Kurkela, R.M.; Lakkakorpi, J.T.; Poutanen, M.H.; Itaranta, P.V.; Melis, J.P.; Ghosh, D.; Vihko, R.K.; Vihko, P.T. Characterization of molecular and catalytic properties of intact and truncated human 17β-hydroxysteroid dehydrogenase type 2 enzymes: Intracellular localization of the wild-type enzyme in the endoplasmic reticulum. Endocrinology 1999, 140, 3334–3341. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Qiu, Q.Q.; Debear, J.; Lathrop, W.F.; Bertolini, D.R.; Tamburini, P.P. 17β-hydroxysteroid dehydrogenases in human bone cells. J. Bone Miner. Res. 1998, 13, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Vihko, P.; Isomaa, V.; Ghosh, D. Structure and function of 17β-hydroxysteroid dehydrogenase type 1 and type 2. Mol. Cell. Endocrinol. 2001, 171, 71–76. [Google Scholar] [CrossRef]
- Vuorinen, A.; Engeli, R.; Meyer, A.; Bachmann, F.; Griesser, U.J.; Schuster, D.; Odermatt, A. Ligand-based pharmacophore modeling and virtual screening for the discovery of novel 17β-hydroxysteroid dehydrogenase 2 inhibitors. J. Med. Chem. 2014, 57, 5995–6007. [Google Scholar] [CrossRef] [PubMed]
- Geissler, W.M.; Davis, D.L.; Wu, L.; Bradshaw, K.D.; Patel, S.; Mendonca, B.B.; Elliston, K.O.; Wilson, J.D.; Russell, D.W.; Andersson, S. Male pseudohermaphroditism caused by mutations of testicular 17β-hydroxysteroid dehydrogenase 3. Nat. Genet. 1994, 7, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Koh, E.; Noda, T.; Kanaya, J.; Namiki, M. Differential expression of 17β-hydroxysteroid dehydrogenase isozyme genes in prostate cancer and noncancer tissues. Prostate 2002, 53, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Legeza, B.; Balazs, Z.; Nashev, L.G.; Odermatt, A. The microsomal enzyme 17β-hydroxysteroid dehydrogenase 3 faces the cytoplasm and uses NADPH generated by glucose-6-phosphate dehydrogenase. Endocrinology 2013, 154, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Tsachaki, M.; Birk, J.; Egert, A.; Odermatt, A. Determination of the topology of endoplasmic reticulum membrane proteins using redox-sensitive green-fluorescence protein fusions. Biochim. Biophys. Acta 2015, 1853, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Schuster, D.; Kowalik, D.; Kirchmair, J.; Laggner, C.; Markt, P.; Aebischer-Gumy, C.; Strohle, F.; Moller, G.; Wolber, G.; Wilckens, T.; et al. Identification of chemically diverse, novel inhibitors of 17β-hydroxysteroid dehydrogenase type 3 and 5 by pharmacophore-based virtual screening. J. Steroid Biochem. Mol. Biol. 2011, 125, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Vicker, N.; Sharland, C.M.; Heaton, W.B.; Gonzalez, A.M.; Bailey, H.V.; Smith, A.; Springall, J.S.; Day, J.M.; Tutill, H.J.; Reed, M.J.; et al. The design of novel 17β-hydroxysteroid dehydrogenase type 3 inhibitors. Mol. Cell. Endocrinol. 2009, 301, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.M.; Schuster, D.; Edtbauer, M.; Saxena, P.; Mair, C.E.; Kirchebner, J.; Matuszczak, B.; Baburin, I.; Hering, S.; Rollinger, J.M. Experimentally validated herg pharmacophore models as cardiotoxicity prediction tools. J. Chem. Inf. Model. 2014, 54, 2887–2901. [Google Scholar] [CrossRef] [PubMed]
- Duwensee, K.; Schwaiger, S.; Tancevski, I.; Eller, K.; van Eck, M.; Markt, P.; Linder, T.; Stanzl, U.; Ritsch, A.; Patsch, J.R.; et al. Leoligin, the major lignan from edelweiss, activates cholesteryl ester transfer protein. Atherosclerosis 2011, 219, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kaserer, T.; Höferl, M.; Müller, K.; Elmer, S.; Ganzera, M.; Jäger, W.; Schuster, D. In silico predictions of drug-drug interactions caused by cyp1a2, 2c9, and 3a4 inhibition—A comparative study of virtual screening performance. Mol. Inf. 2015, 34, 431–457. [Google Scholar] [CrossRef]
- Blumberg, B.; Iguchi, T.; Odermatt, A. Endocrine disrupting chemicals. J. Steroid Biochem. Mol. Biol. 2011, 127, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hampl, R.; Kubatova, J.; Starka, L. Steroids and endocrine disruptors-history, recent state of art and open questions. J. Steroid Biochem. Mol. Biol. 2016, 155, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Mune, T.; Rogerson, F.M.; Nikkila, H.; Agarwal, A.K.; White, P.C. Human hypertension caused by mutations in the kidney isozyme of 11β-hydroxysteroid dehydrogenase. Nat. Genet. 1995, 10, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Harbison, M.D.; Krozowski, Z.S.; Funder, J.W.; Shackleton, C.H.L.; Hanauskeabel, H.M.; Wei, J.Q.; Hertecant, J.; Moran, A.; Neiberger, R.E.; et al. Several homozygous mutations in the gene for 11β-hydroxysteroid dehydrogenase type-2 in patients with apparent mineralocorticoid excess. J. Clin. Endocrinol. Metab. 1995, 80, 3145–3150. [Google Scholar] [PubMed]
- Lindsay, R.S.; Lindsay, R.M.; Edwards, C.R.; Seckl, J.R. Inhibition of 11β-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension 1996, 27, 1200–1204. [Google Scholar] [CrossRef] [PubMed]
- Nyirenda, M.J.; Lindsay, R.S.; Kenyon, C.J.; Burchell, A.; Seckl, J.R. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J. Clin. Investig. 1998, 101, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Boehmer, A.L.; Brinkmann, A.O.; Sandkuijl, L.A.; Halley, D.J.; Niermeijer, M.F.; Andersson, S.; de Jong, F.H.; Kayserili, H.; de Vroede, M.A.; Otten, B.J.; et al. 17β-hydroxysteroid dehydrogenase-3 deficiency: Diagnosis, phenotypic variability, population genetics, and worldwide distribution of ancient and de novo mutations. J. Clin. Endocrinol. Metab. 1999, 84, 4713–4721. [Google Scholar] [CrossRef] [PubMed]
- Phelan, N.; Williams, E.L.; Cardamone, S.; Lee, M.; Creighton, S.M.; Rumsby, G.; Conway, G.S. Screening for mutations in 17β-hydroxysteroid dehydrogenase and androgen receptor in women presenting with partially virilised 46,xy disorders of sex development. Eur. J. Endocrinol. 2015, 172, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kannan, K. Characteristic profiles of benzonphenone-3 and its derivatives in urine of children and adults from the United States and China. Environ. Sci. Technol. 2013, 47, 12532–12538. [Google Scholar] [CrossRef] [PubMed]
- Temml, V.; Kaserer, T.; Kutil, Z.; Landa, P.; Vanek, T.; Schuster, D. Pharmacophore modelling for cyclooxygenase-1 and 2 inhibitors with ligandscout in comparison to discovery studio. Future Med. Chem. 2014, 6, 1869–1881. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.; Smondyrev, A.; Knoll, E.; Rao, S.; Shaw, D.; Friesner, R. Phase: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput.-Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen interactions in protein-ligand complexes: Implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust, but verify: On the importance of chemical structure curation in cheminformatics and qsar modeling research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaserer, T.; Beck, K.R.; Akram, M.; Odermatt, A.; Schuster, D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules 2015, 20, 22799-22832. https://doi.org/10.3390/molecules201219880
Kaserer T, Beck KR, Akram M, Odermatt A, Schuster D. Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules. 2015; 20(12):22799-22832. https://doi.org/10.3390/molecules201219880
Chicago/Turabian StyleKaserer, Teresa, Katharina R. Beck, Muhammad Akram, Alex Odermatt, and Daniela Schuster. 2015. "Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases" Molecules 20, no. 12: 22799-22832. https://doi.org/10.3390/molecules201219880
APA StyleKaserer, T., Beck, K. R., Akram, M., Odermatt, A., & Schuster, D. (2015). Pharmacophore Models and Pharmacophore-Based Virtual Screening: Concepts and Applications Exemplified on Hydroxysteroid Dehydrogenases. Molecules, 20(12), 22799-22832. https://doi.org/10.3390/molecules201219880