
Generation of Aptamers with an Expanded Chemical Repertoire


Abstract
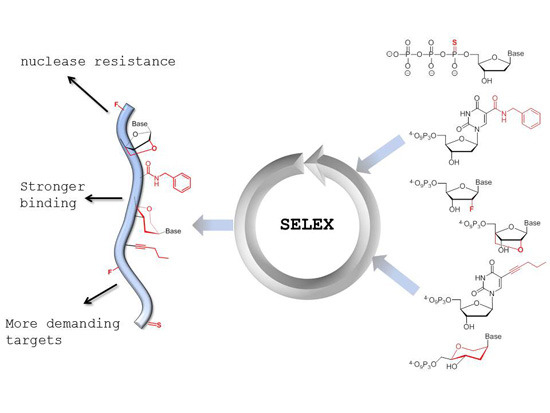
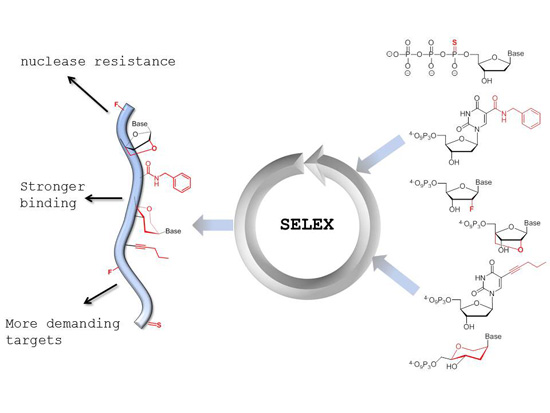
:
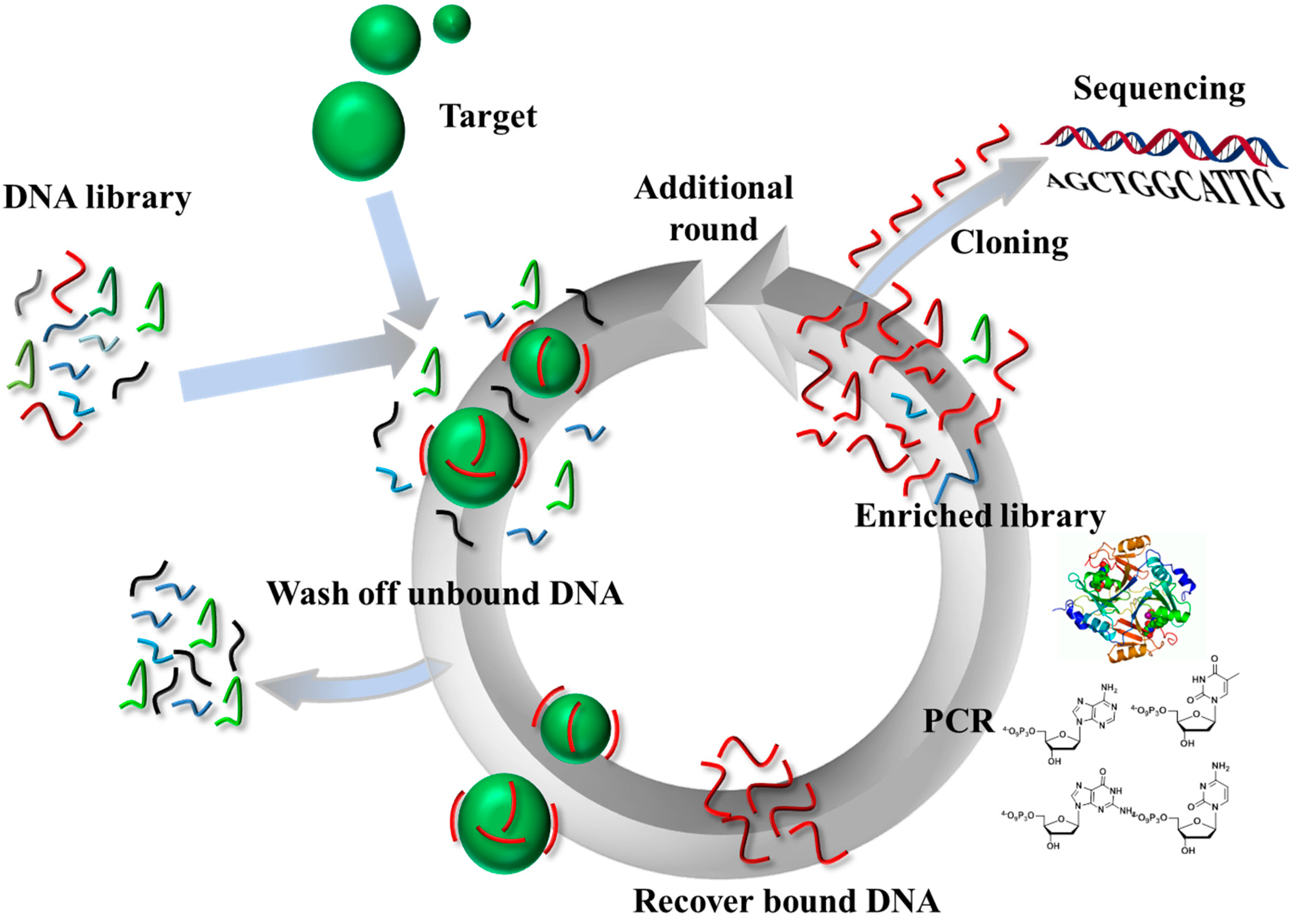
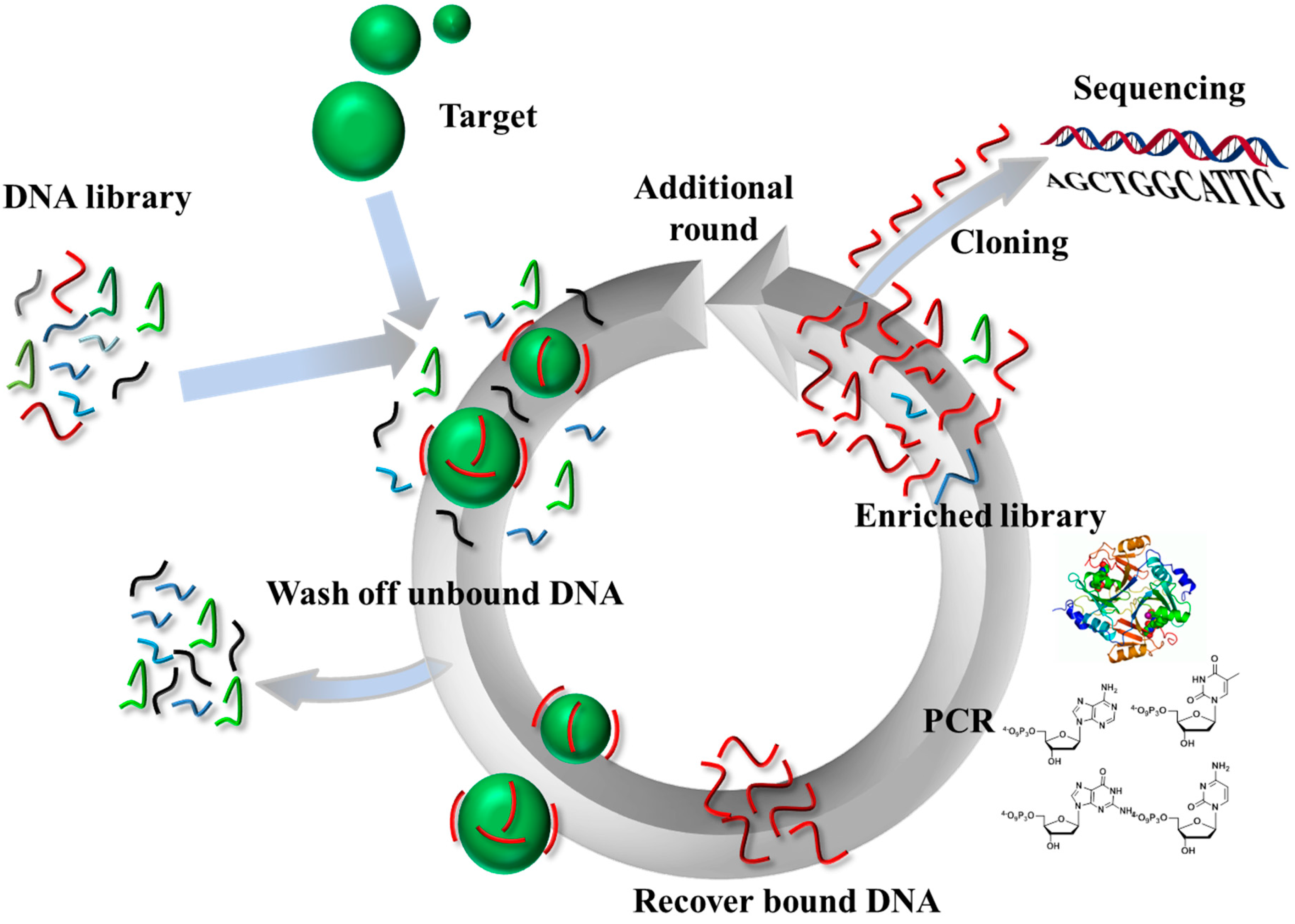
1. Introduction
2. Modified Aptamers for Therapeutic and Diagnostic Applications
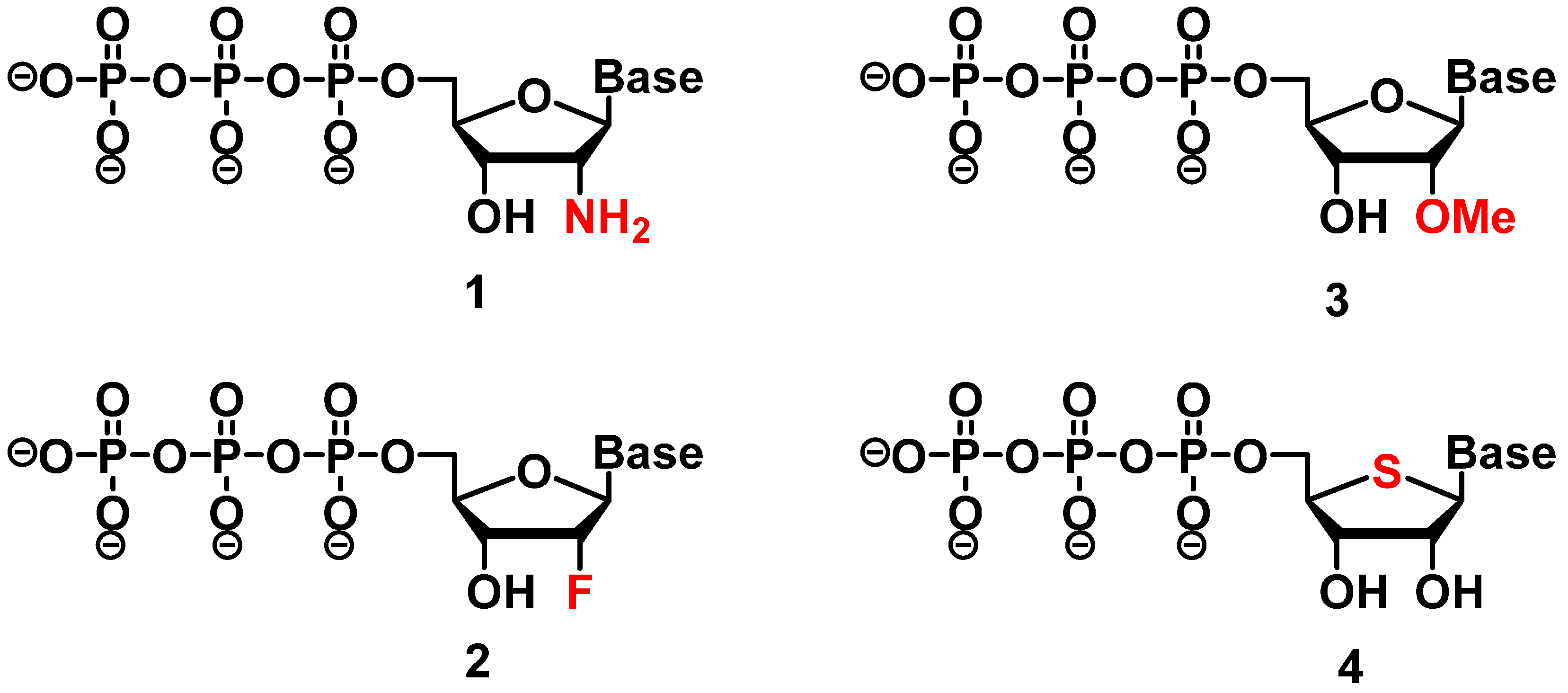
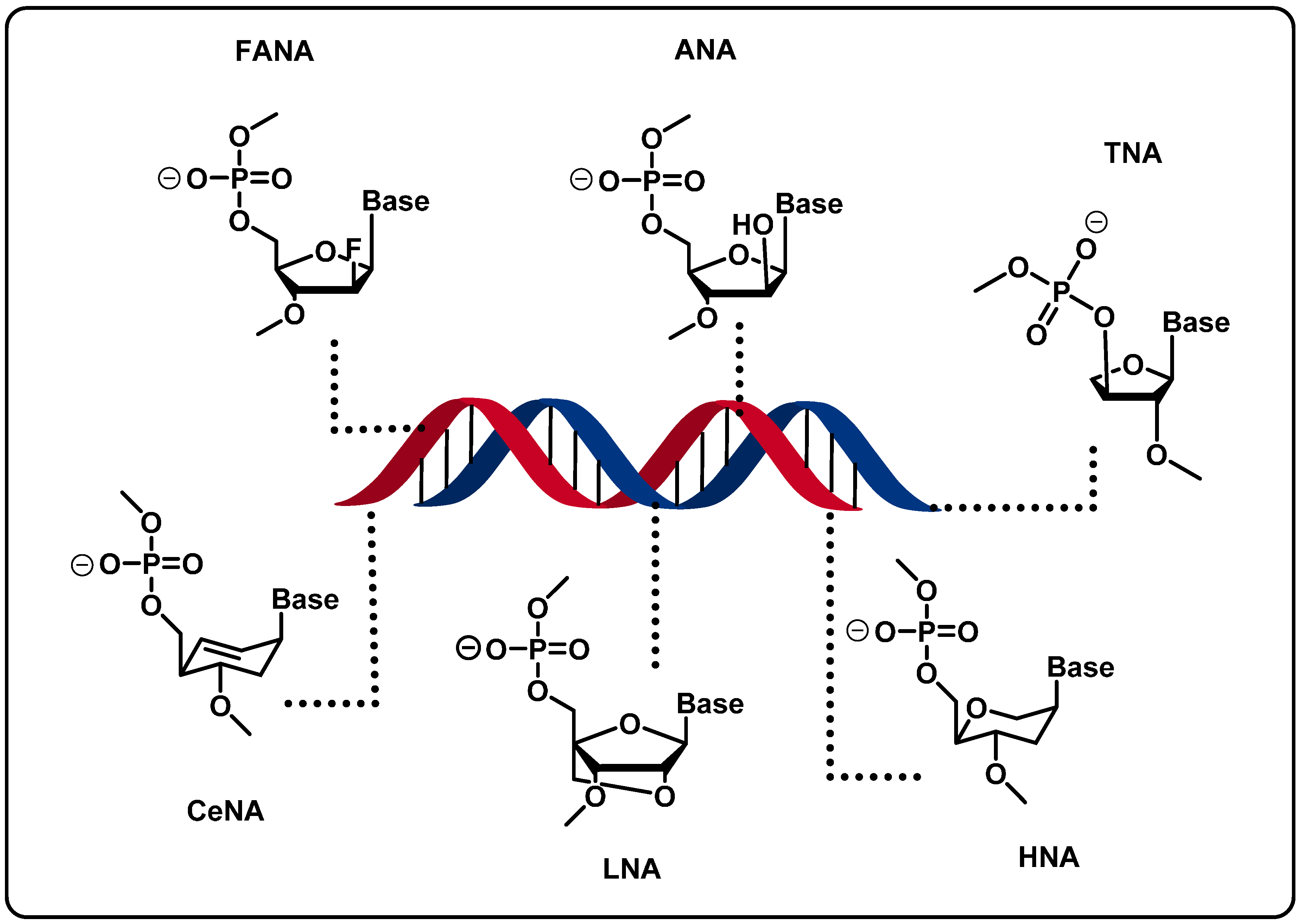
2.1. Sugar Modifications
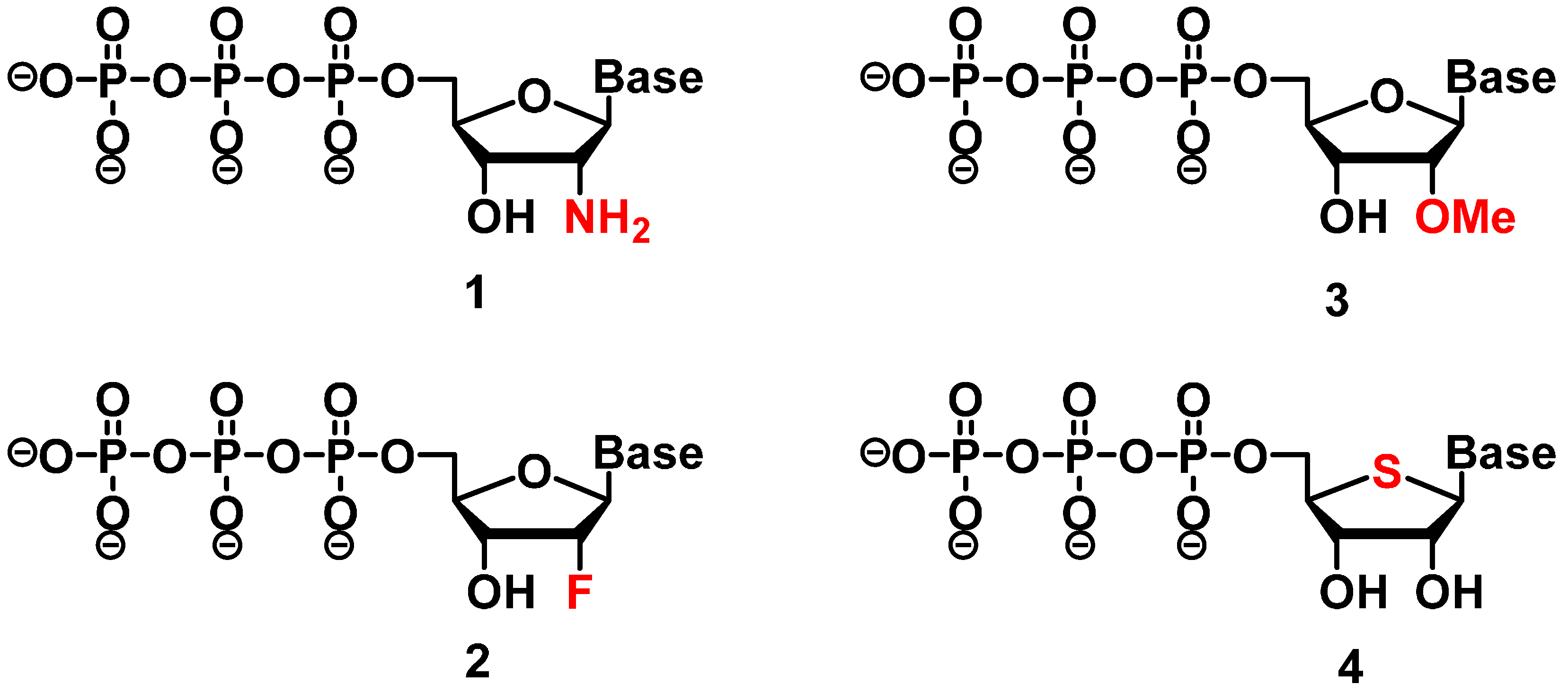

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aptamer Name | Aptamer Target | Kd Value (nM) | Reference |
|---|---|---|---|
| E07 | Epidermal growth factor receptor (EGFR) | 2.4 | [60] |
| CL4 | Epidermal growth factor receptor (EGFR) | 10 | [61] |
| S2 | Prostate-specific antigen (PSA) | 630 | [63] |
| A15 | Brain penetrating aptamer | - | [66] |
| R-F t2 | NS5B replicase, essential for the replication of hepatitis C virus (HCV) | 2.6 | [67] |
| Gint4.T | Platelet-derived growth factor receptor β (PDGFRβ) | 9.6 | [68] |
| GL21.T | Transmembrane tyrosine kinase receptor (RTK) Axl | 12 | [69] |
| G-3 | C-C chemokine receptor type 5 (CCR5) | 110 | [70] |
| C26-50 | N-methyl-d-aspartate (NMDA) receptor ion channel | 120 | [71] |
| Apt1 | CD44, a cell-surface glycoprotein that serves as a cancer stem cell marker | 81.3 | [72] |
| B-68 | HIV-1Ba-L glycoprotein 120 | 52 | [73] |
| GL44 | Human U87MG glioma cells | 38 | [74] |
| RNA 14-16 | p68 RNA helicase, which is involved in colorectal cancer | 13,8 | [65] |
| FAIR-6 | Interleukin-6 receptor (IL-6R) | 40.9 | [75] |
| CD28Apt2, CD28Apt7 | CD28 costimulatory receptor for the activation of T lymphocytes | 40, 60 | [76] |
| 9C7 | OX40 costimulatory receptor | 1.7 | [77] |
| αV-1, β3-1 | αV and β3 subunits of integrin αVβ3 | 2.7, 6.5 | [78] |
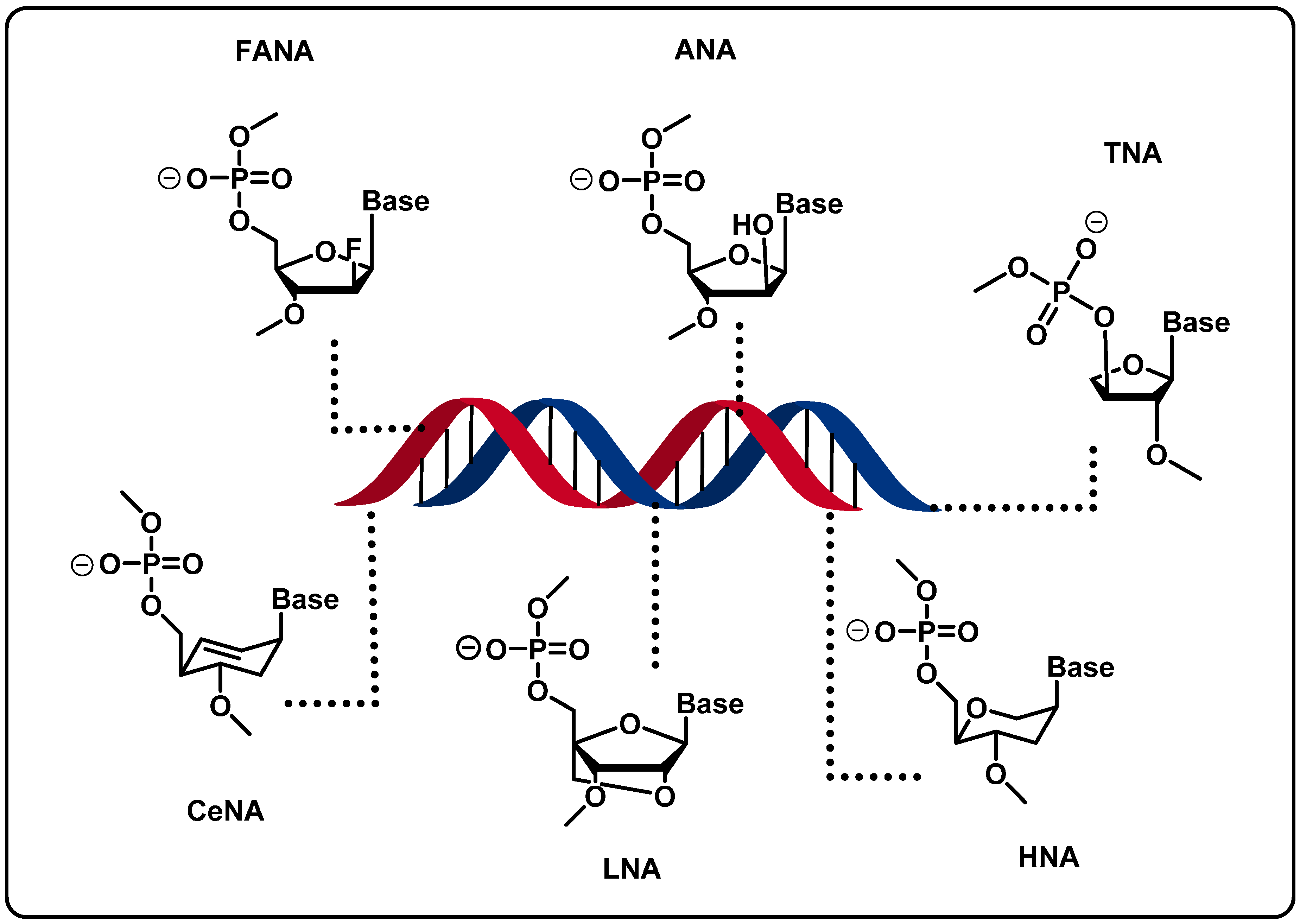
2.2. Backbone Modifications
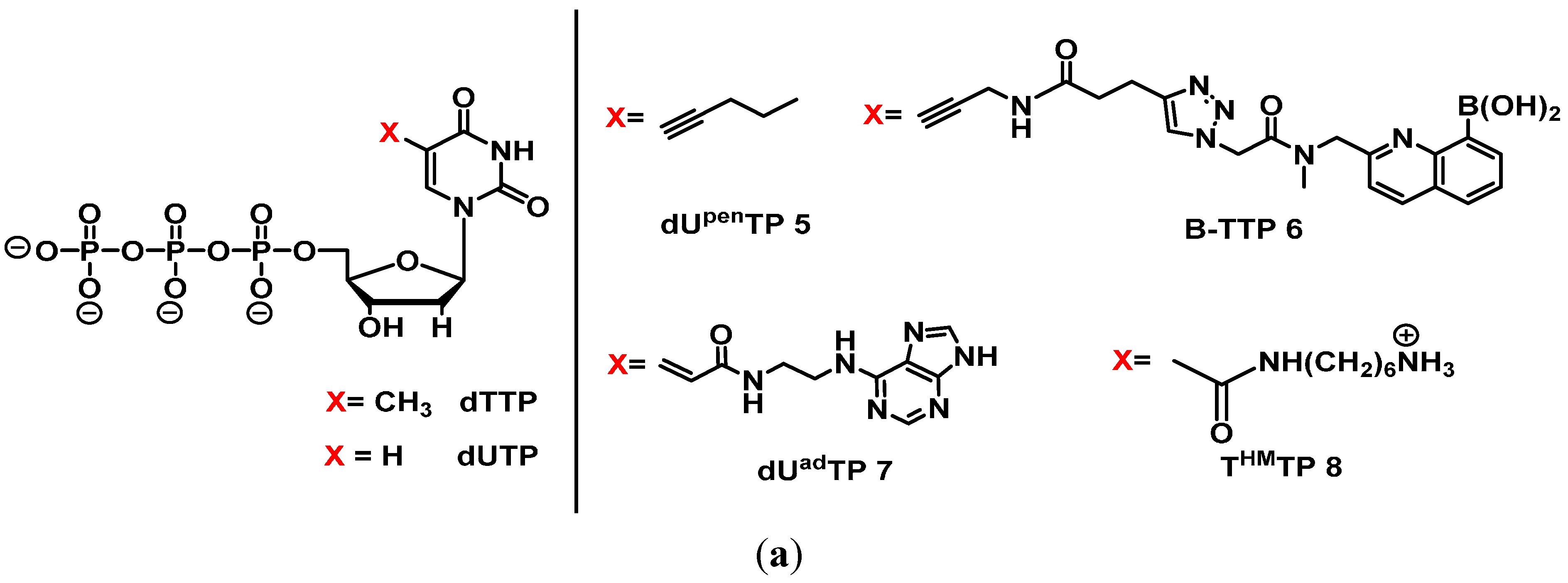
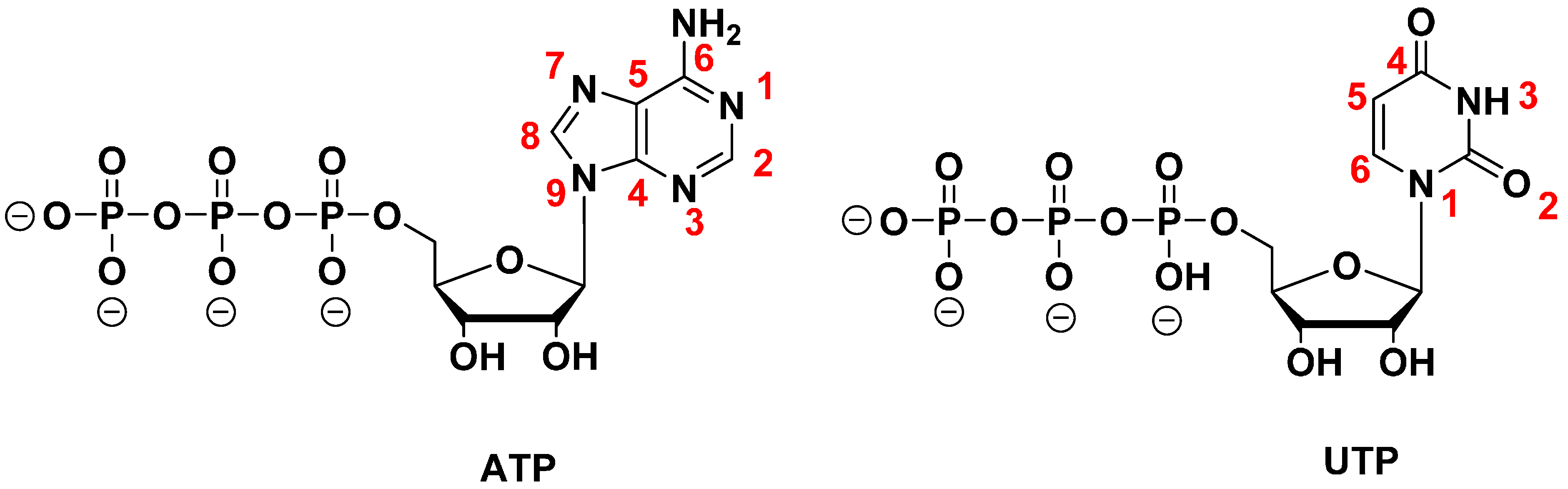
2.3. Base Modifications
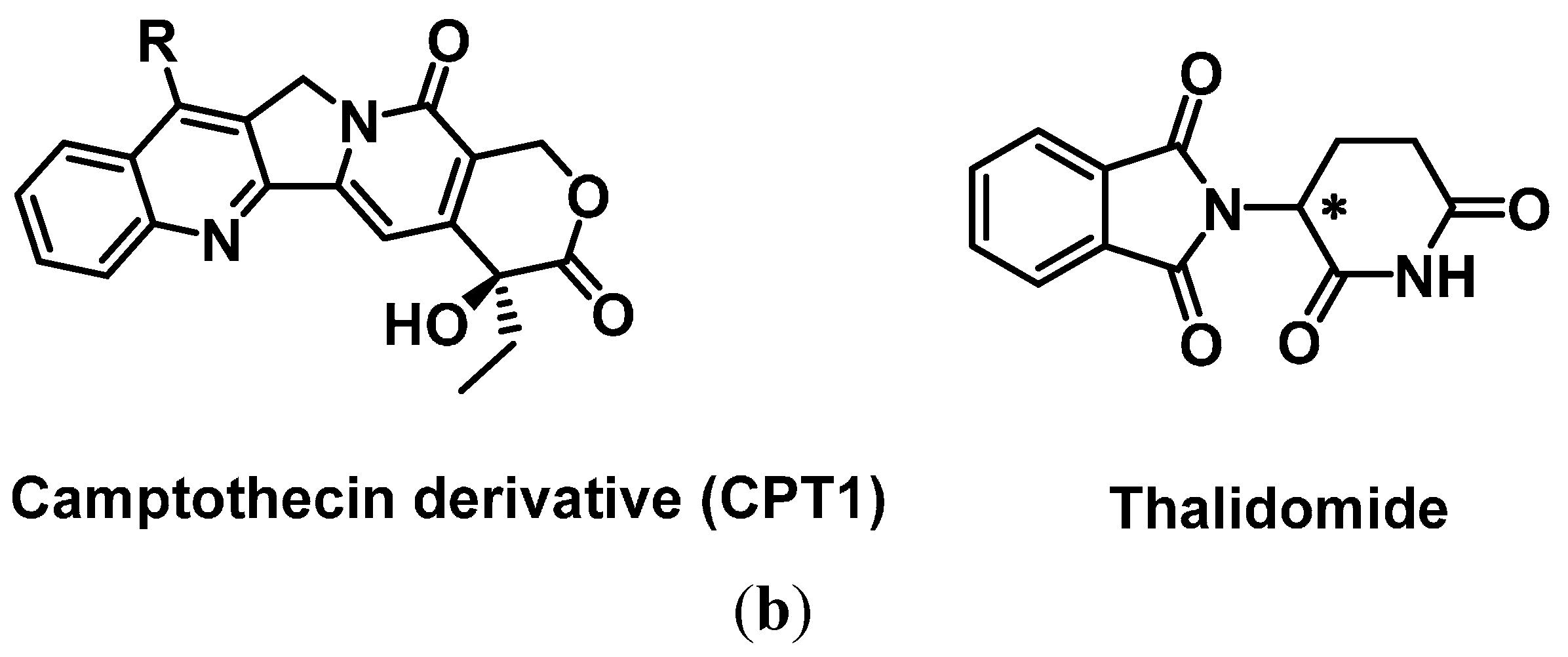
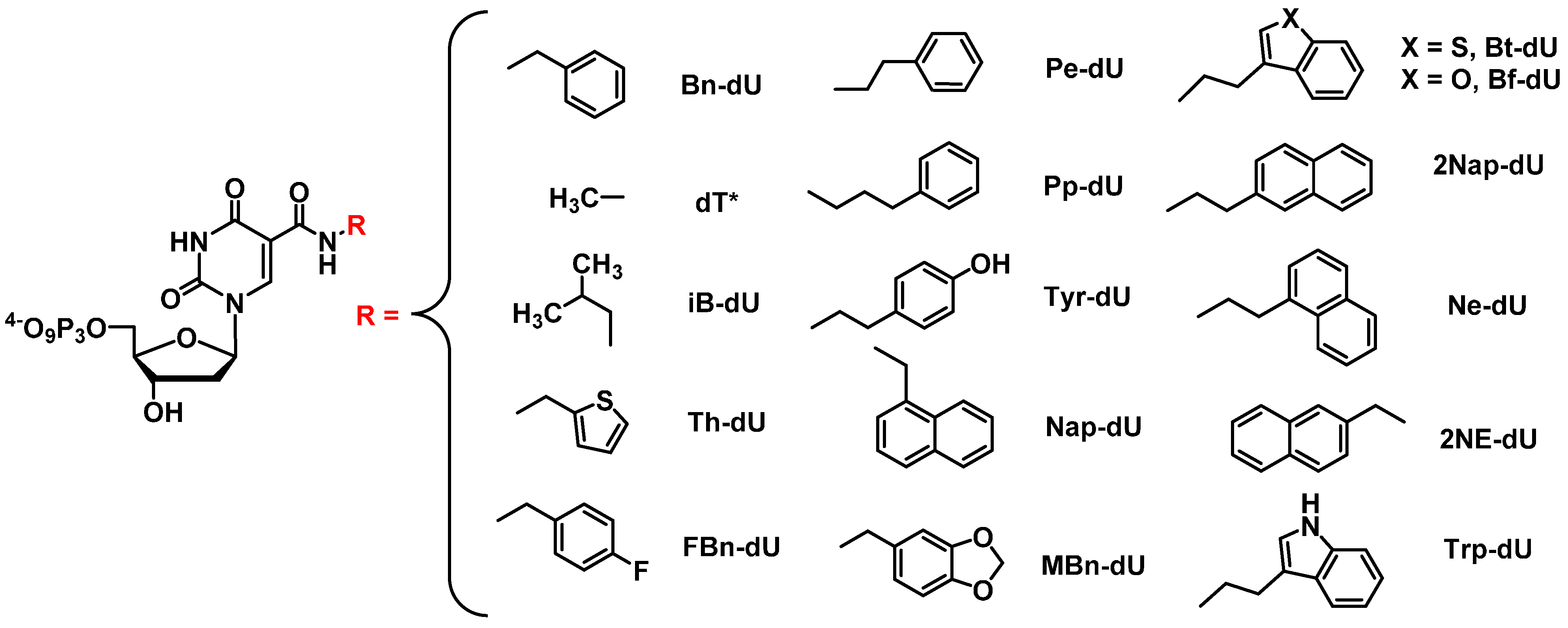
2.3.1. General Base-Modifications

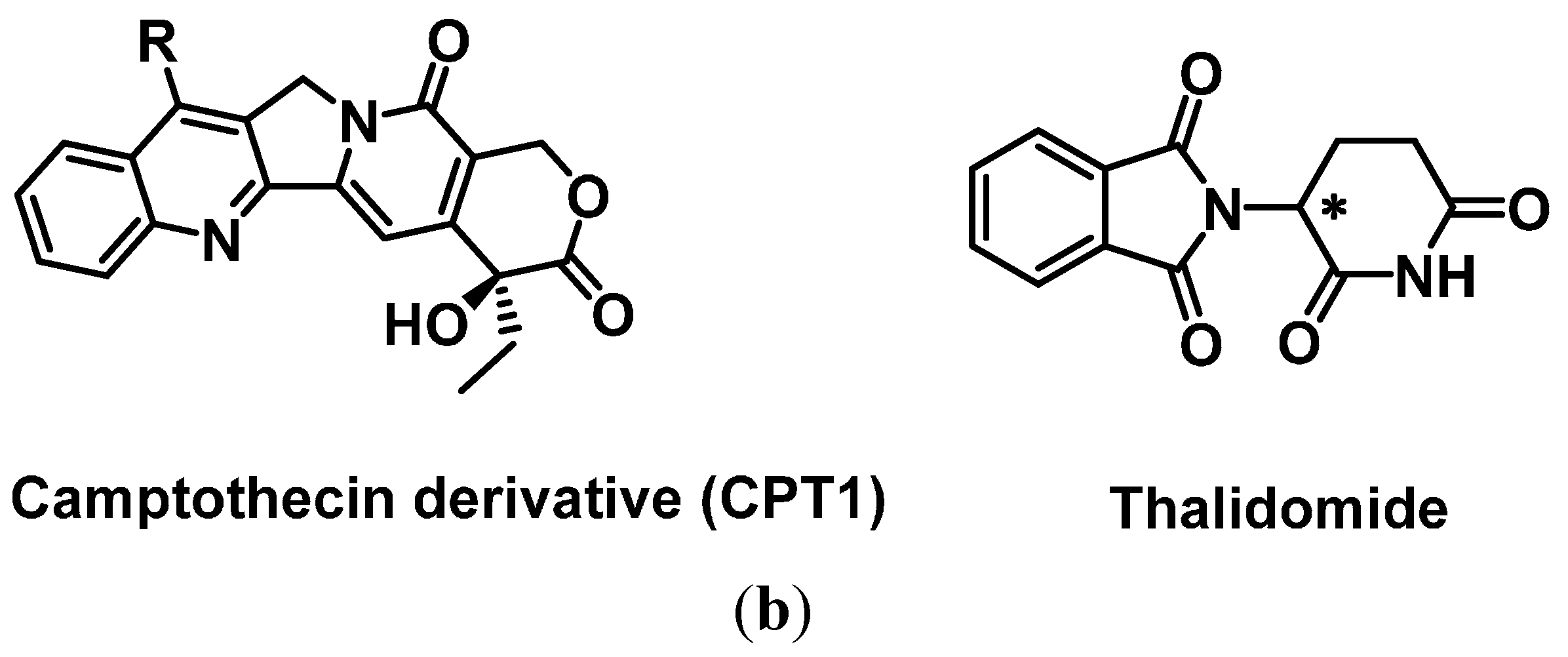
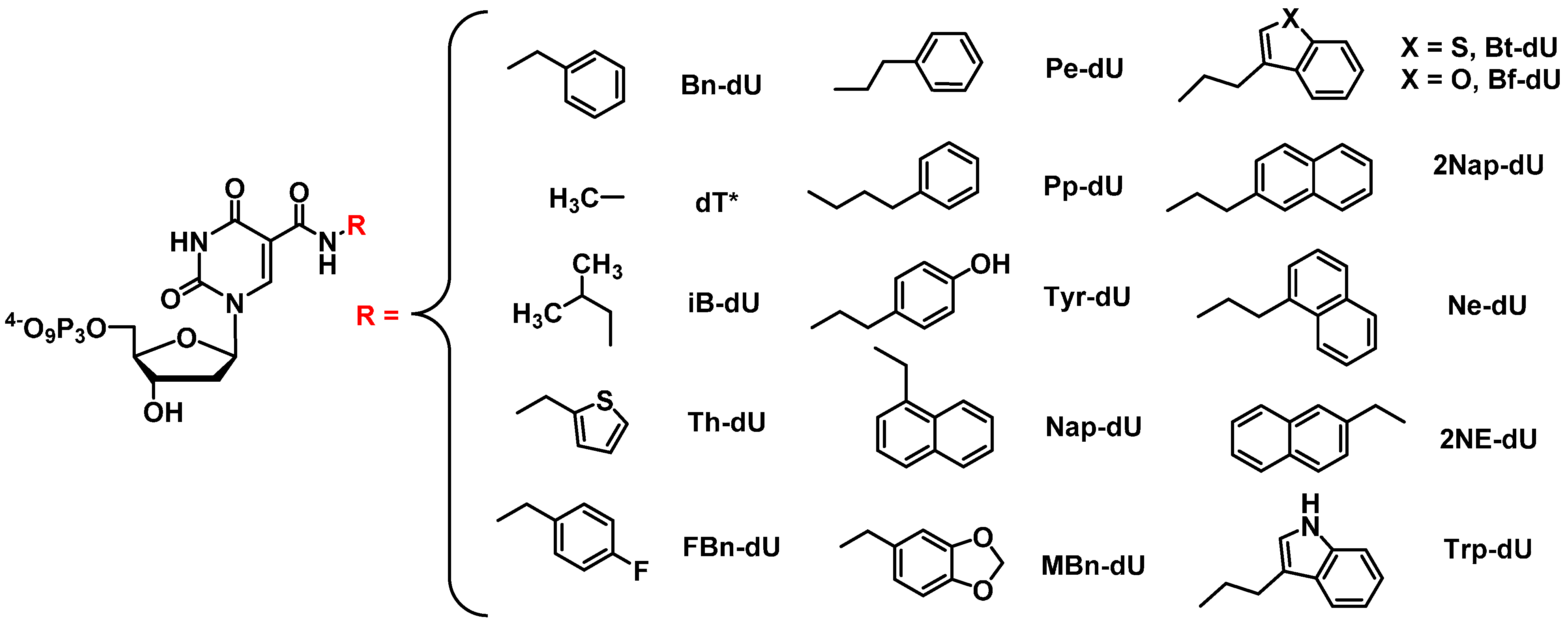
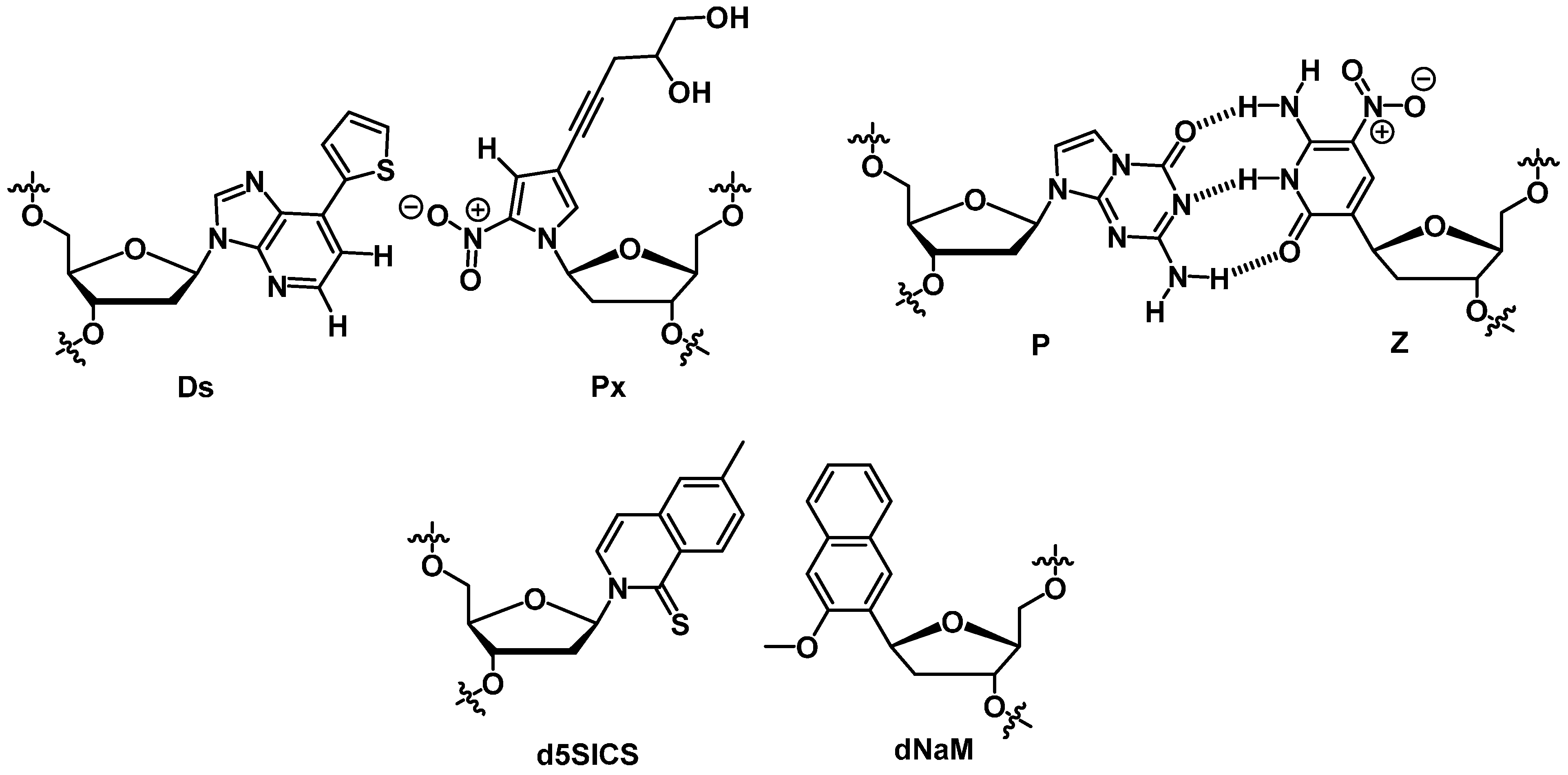
2.3.2. SOMAmers
2.4. Spiegelmers
3. XNAs and Expanded Genetic Systems
4. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mayer, G. The Chemical Biology of Aptamers. Angew. Chem. Int. Ed. 2009, 48, 2672–2689. [Google Scholar] [CrossRef] [PubMed]
- Tolle, F.; Mayer, G. Dressed for success—Applying chemistry to modulate aptamer functionality. Chem. Sci. 2013, 4, 60–67. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Joyce, G.F. Forty Years of in Vitro Evolution. Angew. Chem. Int. Ed. 2007, 46, 6420–6436. [Google Scholar] [CrossRef] [PubMed]
- Silverman, S.K. Catalytic DNA (deoxyribozymes) for synthetic applications—Current abilities and future prospects. Chem. Commun. 2008, 30, 3467–3485. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zu, Y. A Highlight of Recent Advances in Aptamer Technology and Its Application. Molecules 2015, 20, 11959–11980. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Pagano, J.M.; Lis, J.T. New Technologies Provide Quantum Changes in the Scale, Speed, and Success of SELEX Methods and Aptamer Characterization. Mol. Ther. Nucleic Acids 2014, 3, e183. [Google Scholar] [CrossRef] [PubMed]
- Darmostuk, M.; Rimpelová, S.; Gbelcová, H.; Ruml, T. Current approaches in SELEX: An update to aptamer selection technology. Biotechnol. Adv. 2015, 33. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Cload, S.T. SELEX with modified nucleotides. Curr. Opin. Chem. Biol. 2008, 12, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Breaker, R.R. Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2′-Hydroxyl Group. J. Am. Chem. Soc. 1999, 121, 5364–5372. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M. Nucleoside Triphosphates—Building Blocks for the Modification of Nucleic Acids. Molecules 2012, 17, 13569–13591. [Google Scholar] [CrossRef] [PubMed]
- Jellinek, D.; Green, L.S.; Bell, C.; Lynott, C.K.; Gill, N.; Vargeese, C.; Kirschenheuter, G.; McGee, D.P.C.; Abesinghe, P.; Picken, W.A.; et al. Potent 2′-Amino-2′-deoxypyrimidine RNA Inhibitors of Basic Fibroblast Growth Factor. Biochemistry 1995, 34, 11363–11372. [Google Scholar] [CrossRef] [PubMed]
- Pagratis, N.C.; Bell, C.; Chang, Y.F.; Jennings, S.; Fitzwater, T.; Jellinek, D.; Dang, C. Potent 2′-amino-, and 2′-fluoro-2′-deoxyribonucleotide RNA inhibitors of keratinocyte growth factor. Nat. Biotechnol. 1997, 15, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, M.; Sugimoto, N. Molecular Evolution of Functional Nucleic Acids with Chemical Modifications. Molecules 2010, 15, 5423–5444. [Google Scholar] [CrossRef] [PubMed]
- Herdewijn, P.; Marlière, P. Toward Safe Genetically Modified Organisms through the Chemical Diversification of Nucleic Acids. Chem. Biodivers. 2009, 6, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Holliger, P. The XNA world: Progress towards replication and evolution of synthetic genetic polymers. Curr. Opin. Chem. Biol. 2012, 16, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Holliger, P. Towards XNA nanotechnology: New materials from synthetic genetic polymers. Trends Biotechnol. 2014, 32, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jin, C.; Wang, Y.; Fang, X.; Zhang, X.; Chen, Z.; Tan, W. Aptamer-conjugated nanomaterials for specific cancer cell recognition and targeted cancer therapy. NPG Asia Mater. 2014, 6, e95. [Google Scholar] [CrossRef]
- Ray, P.; Viles, K.D.; Soule, E.E.; Woodruff, R.S. Application of Aptamers for Targeted Therapeutics. Arch. Immunol. Ther. Exp. 2013, 61, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.G. Predicting the Uncertain Future of Aptamer-Based Diagnostics and Therapeutics. Molecules 2015, 20, 6866–6887. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.G. A Review of Therapeutic Aptamer Conjugates with Emphasis on New Approaches. Pharmaceuticals 2013, 6, 340–357. [Google Scholar] [CrossRef] [PubMed]
- Nimjee, S.M.; Rusconi, C.P.; Sullenger, B.A. Aptamers: An Emerging Class of Therapeutics. Annu. Rev. Med. 2005, 56, 555–583. [Google Scholar] [CrossRef] [PubMed]
- Lao, Y.-H.; Phua, K.K.L.; Leong, K.W. Aptamer Nanomedicine for Cancer Therapeutics: Barriers and Potential for Translation. ACS Nano 2015, 9, 2235–2254. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, C.P.; Scardino, E.; Layzer, J.; Pitoc, G.A.; Ortel, T.L.; Monroe, D.; Sullenger, B.A. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature 2002, 419, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Rusconi, C.P.; Roberts, J.D.; Pitoc, G.A.; Nimjec, S.M.; White, R.R.; Quick, G., Jr.; Scardino, E.; Fay, W.P.; Sullenger, B.A. Antidote-mediated control of an anticoagulant aptamer in vivo. Nat. Biotechnol. 2004, 22, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tong, R.; Chu, H.; Wang, W.; Langer, R.; Kohane, D.S. Aptamer photoregulation in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 17099–17103. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Parekh, P.; Kim, Y.; Morey, T.E.; Sefah, K.; Gravenstein, N.; Dennis, D.M.; Tan, W. Selection of an Aptamer Antidote to the Anticoagulant Drug Bivalirudin. PLoS ONE 2013, 8, e57341. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- McNamara, J.O.I.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamersiRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hou, J.; Liu, X.; Guo, Y.; Wu, Y.; Zhang, L.; Yang, Z. Nucleolin-targeting liposomes guided by aptamer AS1411 for the delivery of siRNA for the treatment of malignant melanomas. Biomaterials 2014, 35, 3840–3850. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Y.; Wang, W.Y.; Chang, Y.C.; Chang, C.J.; Yang, P.C.; Peck, K. Synergistic inhibition of lung cancer cell invasion, tumor growth and angiogenesis using aptamer-siRNA chimeras. Biomaterials 2014, 35, 2905–2914. [Google Scholar] [CrossRef] [PubMed]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA Aptamers That Inhibit CTLA-4 and Enhance Tumor Immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar] [PubMed]
- Herrmann, A.; Priceman, S.J.; Kujawski, M.; Xin, H.; Cherryholmes, G.A.; Zhang, W.; Zhang, C.; Lahtz, C.; Kowolik, C.; Forman, S.J.; et al. CTLA4 aptamer delivers STAT3 siRNA to tumor-associated and malignant T cells. J. Clin. Investig. 2014, 124, 2977–2987. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Liu, J.; Ali, M.M.; Mahmood, M.A.I.; Labanieh, L.; Lu, M.; Iqbal, S.M.; Zhang, Q.; Zhao, W.; Wan, Y. Nucleic acid aptamers in cancer research, diagnosis and therapy. Chem. Soc. Rev. 2015, 44, 1240–1256. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Tang, Z.; Kim, Y.; Nie, H.; Huang, Y.F.; He, X.; Deng, K.; Wang, K.; Tan, W. In vivo Fluorescence Imaging of Tumors using Molecular Aptamers Generated by Cell-SELEX. Chem. Asian J. 2010, 5, 2209–2213. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Guo, S.; Xie, M.; Luo, W.; Yuan, C.; Huang, W.; Zhou, Y.; Zhang, X.L.; Zhou, X. Diagnostic applications of gastric carcinoma cell aptamers in vitro and in vivo. Talanta 2015, 134, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Hipolito, C.J.; Hollenstein, M.; Lam, C.H.; Perrin, D.M. Protein-inspired modified DNAzymes: Dramatic effects of shortening side-chain length of 8-imidazolyl modified deoxyadenosines in selecting RNaseA mimicking DNAzymes. Org. Biomol. Chem. 2011, 9, 2266–2273. [Google Scholar] [CrossRef] [PubMed]
- Ghadessy, F.J.; Ong, J.L.; Holliger, P. Directed evolution of polymerase function by compartmentalized self-replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4552–4557. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.S.; Liu, D.R. Methods for the directed evolution of proteins Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Kranaster, R.; Marx, A. Engineered DNA Polymerases in Biotechnology. ChemBioChem 2010, 11, 2077–2084. [Google Scholar] [CrossRef] [PubMed]
- Laos, R.; Thomson, J.M.; Benner, S.A. DNA polymerases engineered by directed evolution to incorporate non-standard nucleotides. Front. Microbiol. 2014, 5, 565. [Google Scholar] [CrossRef]
- Lauridsen, L.H.; Rothnagel, J.A.; Veedu, R.N. Enzymatic Recognition of 2′-Modified Ribonucleoside 5′-Triphosphates: Towards the Evolution of Versatile Aptamers. ChemBioChem 2012, 13, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Qiu, Q.; Gill, S.C.; Jayasena, S.D. Modified RNA sequence pools for in vitro selection. Nucleic Acids Res. 1994, 22, 5229–5234. [Google Scholar] [CrossRef] [PubMed]
- Green, L.S.; Jellinek, D.; Bell, C.; Beebe, L.A.; Feistner, B.D.; Gill, S.C.; Jucker, F.M.; Janjić, N. Nuclease-resistant nucleic acid ligands to vascular permeability factor/vascular endothelial growth factor. Chem. Biol. 1995, 2, 683–695. [Google Scholar] [CrossRef]
- Lin, Y.; Nieuwlandt, D.; Magallanez, A.; Feistner, B.; Jayasena, S.D. High-affinity and specific recognition of human thyroid stimulating hormone (hTSH) by in vitro selected 2′-amino-modified RNA. Nucleic Acids Res. 1996, 24, 3407–3414. [Google Scholar] [CrossRef] [PubMed]
- Kubik, M.F.; Bell, C.; Fitzwater, T.; Watson, S.R.; Tasset, D.M. Isolation and characterization of 2′-fluoro-, 2′-amino-, and 2′-fluoro-/amino-modified RNA ligands to human IFN-gamma that inhibit receptor binding. J. Immunol. 1997, 159, 259–267. [Google Scholar] [PubMed]
- Proske, D.; Gilch, S.; Wopfner, F.; Schätzl, H.M.; Winnacker, E.L.; Famulok, M. Prion-Protein-Specific Aptamer Reduces PrPSc Formation. ChemBioChem 2002, 3, 717–725. [Google Scholar] [CrossRef]
- Proske, D.; Höfliger, M.; Söll, R.M.; Beck-Sickinger, A.G.; Famulok, M. A Y2 Receptor Mimetic Aptamer Directed against Neuropeptide Y. J. Biol. Chem. 2002, 277, 11416–11422. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Craven, R.C.; Qiu, Q.; Wilson, C.B.; Wills, J.W.; Golovine, S.; Wang, J.F. Isolation of virus-neutralizing RNAs from a large pool of random sequences. Proc. Natl. Acad. Sci. USA 1995, 92, 11509–11513. [Google Scholar] [CrossRef] [PubMed]
- Ruckman, J.; Green, L.S.; Beeson, J.; Waugh, S.; Gillette, W.L.; Henninger, D.D.; Claesson-Welsh, L.; Janjic, N. 2′-Fluoropyrimidine RNA-based Aptamers to the 165-Amino Acid Form of Vascular Endothelial Growth Factor (VEGF165). J. Biol. Chem. 1998, 273, 20556–20567. [Google Scholar] [CrossRef] [PubMed]
- Khati, M.; Schüman, M.; Ibrahim, J.; Sattentau, Q.; Gordon, S.; James, W. Neutralization of Infectivity of Diverse R5 Clinical Isolates of Human Immunodeficiency Virus Type 1 by gp120-Binding 2′F-RNA Aptamers. J. Virol. 2003, 77, 12692–12698. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, P.E.; Lewis, S.D.; Silva, R.F.; Preiss, J.R.; Horwitz, L.R.; Pendergrast, P.S.; McCauley, T.G.; Kurz, J.C.; Epstein, D.M.; Wilson, C.; et al. Direct in Vitro Selection of a 2′-O-Methyl Aptamer to VEGF. Chem. Biol. 2005, 12, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, P.E.; Wang, C.; Killough, J.R.; Lewis, S.D.; Horwitz, L.R.; Ferguson, A.; Thompson, K.M.; Pendergrast, P.S.; McCauley, T.G.; Kurz, M.; et al. 2′-Deoxy Purine, 2′-O-Methyl Pyrimidine (dRmY) Aptamers as Candidate Therapeutics. Oligonucleotides 2006, 16, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Rohloff, J.C.; Gelinas, A.D.; Jarvis, T.C.; Ochsner, U.A.; Schneider, D.J.; Gold, L.; Janjic, N. Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol. Ther. Nucleic Acids 2014, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, A.; Toulmé, J.J.; Rayner, B. SELEX and dynamic combinatorial chemistry interplay for the selection of conjugated RNA aptamers. Org. Biomol. Chem. 2006, 4, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Nguyen, H.H.; Byrom, M.; Ellington, A.D. Inhibition of Cell Proliferation by an Anti-EGFR Aptamer. PLoS ONE 2011, 6, e20299. [Google Scholar] [CrossRef] [PubMed]
- Esposito, C.L.; Passaro, D.; Longobardo, I.; Condorelli, G.; Marotta, P.; Affuso, A.; de Franciscis, V.; Cerchia, L. A Neutralizing RNA Aptamer against EGFR Causes Selective Apoptotic Cell Death. PLoS ONE 2011, 6, e24071. [Google Scholar] [CrossRef] [PubMed]
- Layzer, J.M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Svobodova, M.; Bunka, D.H.J.; Nadal, P.; Stockley, P.G.; O’Sullivan, C.K. Selection of 2′F-modified RNA aptamers against prostate-specific antigen and their evaluation for diagnostic and therapeutic applications. Anal. Bioanal. Chem. 2013, 405, 9149–9157. [Google Scholar] [CrossRef] [PubMed]
- Lupold, S.E.; Hicke, B.J.; Lin, Y.; Coffey, D.S. Identification and Characterization of Nuclease-stabilized RNA Molecules That Bind Human Prostate Cancer Cells via the Prostate-specific Membrane Antigen. Cancer Res. 2002, 62, 4029–4033. [Google Scholar] [PubMed]
- Mi, J.; Liu, Y.; Rabbani, Z.N.; Yang, Z.; Urban, J.H.; Sullenger, B.A.; Clary, B.M. In vivo selection of tumor-targeting RNA motifs. Nat. Chem. Biol. 2010, 6, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Chen, Y.H.; Lennox, K.A.; Behlke, M.A.; Davidson, B.L. In vivo SELEX for Identification of Brain-penetrating Aptamers. Mol. Ther. Nucleic Acids 2013, 2, e67. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, Y.J.; Kim, J.H.; Lim, J.H.; Kim, J.H.; Han, W.; Lee, S.H.; Noh, G.J.; Lee, S.W. Inhibition of Hepatitis C Virus (HCV) Replication by Specific RNA Aptamers against HCV NS5B RNA Replicase. J. Virol. 2013, 87, 7064–7074. [Google Scholar] [CrossRef] [PubMed]
- Camorani, S.; Esposito, C.L.; Rienzo, A.; Catuogno, S.; Iaboni, M.; Condorelli, G.; De Franciscis, V.; Cerchia, L. Inhibition of Receptor Signaling and of Glioblastoma-derived Tumor Growth by a Novel PDGFR beta Aptamer. Mol. Ther. 2014, 22, 828–841. [Google Scholar] [PubMed]
- Cerchia, L.; Esposito, C.L.; Camorani, S.; Rienzo, A.; Stasio, L.; Insabato, L.; Affuso, A.; de Franciscis, V. Targeting Axl With an High-affinity Inhibitory Aptamer. Mol. Ther. Nucleic Acids 2012, 20, 2291–2303. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Satheesan, S.; Li, H.; Weinberg, M.S.; Morris, K.V.; Burnett, J.C.; Rossi, J.J. Cell-Specific RNA Aptamer against Human CCR5 Specifically Targets HIV-1 Susceptible Cells and Inhibits HIV-1 Infectivity. Chem. Biol. 2015, 22, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; MacLean, D.M.; Ulrich, H.; Zhao, X.; Aronowski, J.; Jayaraman, V. RNA Based Antagonist of NMDA Receptors. ACS Chem. Neurosci. 2014, 5, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, N.; Alshaer, W.; Allozi, O.; Mahafzah, A.; El-Khateeb, M.; Hillaireau, H.; Noiray, M.; Fattal, E.; Ismail, S. In Vitro Selection of Modified RNA Aptamers against CD44 Cancer Stem Cell Marker. Nucleic Acid Ther. 2013, 23, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Swiderski, P.; Li, H.; Zhang, J.; Neff, C.P.; Akkina, R.; Rossi, J.J. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009, 37, 3094–3109. [Google Scholar] [CrossRef] [PubMed]
- Cerchia, L.; Esposito, C.L.; Jacobs, A.H.; Tavitian, B.; De Franciscis, V. Differential SELEX in Human Glioma Cell Lines. PLoS ONE 2009, 4, e7971. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Berg, K.; Eydeler-Haeder, K.; Lorenzen, I.; Groetziger, J.; Rose-John, S.; Hahn, U. Stabilized Interleukin-6 receptor binding RNA aptamers. RNA Biol. 2014, 11, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Soldevilla, M.M.; Villanueva, H.; Kolonias, D.; Inoges, S.; de Cerio, A.L.; Kandzia, R.; Klimyuk, V.; Gleba, Y.; Gilboa, E.; et al. CD28 Aptamers as Powerful Immune Response Modulators. Mol. Ther. Nucleic. Acids 2013, 2, e98. [Google Scholar] [CrossRef] [PubMed]
- Pratico, E.D.; Sullenger, B.A.; Nair, S.K. Identification and Characterization of an Agonistic Aptamer against the T Cell Costimulatory Receptor, OX40. Nucleic Acid Ther. 2013, 23, 35–43. [Google Scholar] [PubMed]
- Gong, Q.; Wang, J.F.; Ahmad, K.M.; Csordas, A.T.; Zhou, J.; Nie, J.; Stewart, R.; Thomson, J.A.; Rossi, J.J.; Soh, H.T. Selection Strategy to Generate Aptamer Pairs that Bind to Distinct Sites on Protein Targets. Anal. Chem. 2012, 84, 5365–5371. [Google Scholar] [CrossRef] [PubMed]
- Padilla, R.; Sousa, R. A Y639F/H784A T7 RNA polymerase double mutant displays superior properties for synthesizing RNAs with non-canonical NTPs. Nucleic Acids Res. 2002, 30, e138. [Google Scholar] [CrossRef] [PubMed]
- Fa, M.; Radeghieri, A.; Henry, A.A.; Romesberg, F.E. Expanding the Substrate Repertoire of a DNA Polymerase by Directed Evolution. J. Am. Chem. Soc. 2004, 126, 1748–1754. [Google Scholar] [CrossRef] [PubMed]
- Chelliserrykattil, J.; Ellington, A.D. Evolution of a T7 RNA polymerase variant that transcribes 2′-O-methyl RNA. Nat. Biotechnol. 2004, 22, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, V.; Santner, T.; Micura, R.; Marx, A. Screening mutant libraries of T7 RNA polymerase for candidates with increased acceptance of 2′-modified nucleotides. Chem. Commun. 2012, 48, 9870–9872. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.K.; Genga, R.M.; Schwartz, M.C.; Nelson, J.A.; Schaub, R.G.; Olson, K.A.; Kurz, J.C.; McGinness, K.E. Aptamer ARC19499 mediates a procoagulant hemostatic effect by inhibiting tissue factor pathway inhibitor. Blood 2011, 117, 5514–5522. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Levy-Nissenbaum, E.; Alexis, F.; Lupták, A.; Teply, B.A.; Chan, J.M.; Shi, J.; Digga, E.; Cheng, J.; Langer, R.; Farokhzad, O.C. Engineering of Targeted Nanoparticles for Cancer Therapy Using Internalizing Aptamers Isolated by Cell-Uptake Selection. ACS Nano 2012, 6, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sefah, K.; Liu, H.; Wang, R.; Tan, W. DNA aptamer–micelle as an efficient detection/delivery vehicle toward cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Minakawa, N.; Komatsu, Y.; Kamiya, H.; Ogawa, N.; Harashima, H.; Matsuda, A. New NTP analogs: The synthesis of 4′-thioUTP and 4′-thioCTP and their utility for SELEX. Nucleic Acids Res. 2005, 33, 2942–2951. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, N.; Sanji, M.; Kato, Y.; Matsuda, A. Investigations toward the selection of fully-modified 4′-thioRNA aptamers: Optimization of in vitro transcription steps in the presence of 4′-thioNTPs. Bioorg. Med. Chem. 2008, 16, 9450–9456. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Shionoya, A.; Minakawa, N.; Kawakami, A.; Ogawa, N.; Matsuda, A. Amplification of 4′-ThioDNA in the Presence of 4′-Thio-dTTP and 4′-Thio-dCTP, and 4′-ThioDNA-Directed Transcription in Vitro and in Mammalian Cells. J. Am. Chem. Soc. 2007, 129, 15424–15425. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Furukawa, K.; Maruyama, H.; Inoue, N.; Tarashima, N.; Matsuda, A.; Minakawa, N. PCR Amplification of 4′-ThioDNA Using 2′-Deoxy-4′-thionucleoside 5′-triphosphates. ACS Synth. Biol. 2013, 2, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, V.; Santner, T.; Micura, R.; Marx, A. Enzymatic synthesis of 2′-methylseleno-modified RNA. Chem. Sci. 2011, 2, 2224–2231. [Google Scholar] [CrossRef]
- Santner, T.; Siegmund, V.; Marx, A.; Micura, R. The synthesis of 2′-methylseleno adenosine and guanosine 5′-triphosphates. Bioorg. Med. Chem. 2012, 20, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Tarashima, N.; Sumitomo, T.; Ando, H.; Furukawa, K.; Ishida, T.; Minakawa, N. Synthesis of DNA fragments containing 2′-deoxy-4′-selenonucleoside units using DNA polymerases: Comparison of dNTPs with O, S and Se at the 4′-position in replication. Org. Biomol. Chem. 2015, 13, 6949–6952. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, F. Phosphorothioate Oligodeoxynucleotides: What Is Their Origin and What Is Unique about Them? Antisense Nucleic Acid Drug Dev. 2000, 10, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Mann, A.P.; Somasunderam, A.; Nieves-Alicea, R.; Li, X.; Hu, A.; Sood, A.K.; Ferrari, M.; Gorenstein, D.G.; Tanaka, T. Identification of Thioaptamer Ligand against E-Selectin: Potential Application for Vasculature Targeting. PLoS ONE 2010, 5, e13050. [Google Scholar] [CrossRef] [PubMed]
- King, D.J.; Bassett, S.E.; Li, X.; Fennewald, S.A.; Herzog, N.K.; Luxon, B.A.; Shope, R.; Gorenstein, D.G. Combinatorial Selection and Binding of Phosphorothioate Aptamers Targeting Human NF-κB RelA(p65) and p50. Biochemistry 2002, 41, 9696–9706. [Google Scholar] [CrossRef] [PubMed]
- Somasunderam, A.; Ferguson, M.R.; Rojo, D.R.; Thiviyanathan, V.; Li, X.; O′Brien, W.A.; Gorenstein, D.G. Combinatorial Selection, Inhibition, and Antiviral Activity of DNA Thioaptamers Targeting the RNase H Domain of HIV-1 Reverse Transcriptase. Biochemistry 2005, 44, 10388–10395. [Google Scholar] [CrossRef] [PubMed]
- Somasunderam, A.; Thiviyanathan, V.; Tanaka, T.; Li, X.; Neerathilingam, M.; Lokesh, G.L.R.; Mann, A.; Peng, Y.; Ferrari, M.; Klostergaard, J.; et al. Combinatorial Selection of DNA Thioaptamers Targeted to the HA Binding Domain of Human CD44. Biochemistry 2010, 49, 9106–9112. [Google Scholar] [CrossRef] [PubMed]
- Gandham, S.H.A.; Volk, D.E.; Lokesh, G.L.R.; Neerathilingam, M.; Gorenstein, D.G. Thioaptamers targeting dengue virus type-2 envelope protein domain III. Biochem. Biophys. Res. Commun. 2014, 453, 309–315. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Elizondo-Riojas, M.A.; Li, X.; Lokesh, G.L.R.; Somasunderam, A.; Thiviyanathan, V.; Volk, D.E.; Durland, R.H.; Englehardt, J.; Cavasotto, C.N.; et al. X-Aptamers: A Bead-Based Selection Method for Random Incorporation of Druglike Moieties onto Next-Generation Aptamers for Enhanced Binding. Biochemistry 2012, 51, 8321–8323. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, N.; Huang, Z. Enzymatic Synthesis of Phosphoroselenoate DNA Using Thymidine 5′-(α-P-seleno)triphosphate and DNA Polymerase for X-ray Crystallography via MAD. J. Am. Chem. Soc. 2004, 126, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Victorova, L.S.; Dyatkina, N.B.; Mozzherin, D.J.; Atrazhev, A.M.; Krayevsky, A.A.; Kukhanova, M.K. Formation of phosphonester bonds catalyzed by DNA polymerase. Nucleic Acids Res. 1992, 20, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Dineva, M.A.; Ivanov, I.G.; Petkov, D.D. P-alpha-methyl deoxynucleoside triphosphates as substrates for E. coli DNA polymerase I in a template-directed synthesis of DNA. Nucleosides Nucleotides 1997, 16, 1875–1882. [Google Scholar] [CrossRef]
- Lin, J.; Shaw, B.R. Synthesis of a novel triphosphate analogue: Nucleoside α-P-borano,α-P-thiotriphosphate. Chem. Commun. 2000, 2115–2116. [Google Scholar] [CrossRef]
- Lato, S.M.; Ozerova, N.D.S.; He, K.; Sergueeva, Z.; Shaw, B.R.; Burke, D.H. Boron-containing aptamers to ATP. Nucleic Acids Res. 2002, 30, 1401–1407. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Rasched, G.; Kornreich-Leshem, H.; Engeser, M.; Thum, O.; Famulok, M. A versatile toolbox for variable DNA functionalization at high density. J. Am. Chem. Soc. 2005, 127, 15071–15082. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M. Synthesis of deoxynucleoside triphosphates that include proline, urea, or sulfamide groups and their polymerase incorporation into DNA. Chem. Eur. J. 2012, 18, 13320–13330. [Google Scholar] [CrossRef] [PubMed]
- Hollenstein, M. Deoxynucleoside triphosphates bearing histamine, carboxylic acid, and hydroxyl residues—Synthesis and biochemical characterization. Org. Biomol. Chem. 2013, 11, 5162–5172. [Google Scholar] [CrossRef] [PubMed]
- Hocek, M. Synthesis of Base-Modified 2′-Deoxyribonucleoside Triphosphates and Their Use in Enzymatic Synthesis of Modified DNA for Applications in Bioanalysis and Chemical Biology. J. Org. Chem. 2014, 79, 9914–9921. [Google Scholar] [CrossRef] [PubMed]
- Latham, J.A.; Johnson, R.; Toole, J.J. The application of a modified nucleotide in aptamer selection: Novel thrombin aptamers containing-(1-pentynyl)-2′-deoxyuridine. Nucleic Acids Res. 1994, 22, 2817–2822. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.B.; Atkinson, B.L.; Willis, M.C.; Koch, T.H.; Gold, L. Using in vitro selection to direct the covalent attachment of human immunodeficiency virus type 1 Rev protein to high-affinity RNA ligands. Proc. Natl. Acad. Sci. USA 1995, 92, 12220–12224. [Google Scholar] [CrossRef] [PubMed]
- Battersby, T.R.; Ang, D.N.; Burgstaller, P.; Jurczyk, S.C.; Bowser, M.T.; Buchanan, D.D.; Kennedy, R.T.; Benner, S.A. Quantitative Analysis of Receptors for Adenosine Nucleotides Obtained via In Vitro Selection from a Library Incorporating a Cationic Nucleotide Analog. J. Am. Chem. Soc. 1999, 121, 9781–9789. [Google Scholar] [CrossRef] [PubMed]
- Vaish, N.K.; Larralde, R.; Fraley, A.W.; Szostak, J.W.; McLaughlin, L.W. A Novel, Modification-Dependent ATP-Binding Aptamer Selected from an RNA Library Incorporating a Cationic Functionality. Biochemistry 2003, 42, 8842–8851. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lin, N.; Huang, Z.; Lupei, D.; Altier, C.; Fang, H.; Wang, B. Selecting Aptamers for a Glycoprotein through the Incorporation of the Boronic Acid Moiety. J. Am. Chem. Soc. 2008, 130, 12636–12638. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, Y.; Kasahara, Y.; Fujita, H.; Kitadume, S.; Ozaki, H.; Endoh, T.; Kuwahara, M.; Sugimoto, N. Efficacy of Base-Modification on Target Binding of Small Molecule DNA Aptamers. J. Am. Chem. Soc. 2013, 135, 9412–9419. [Google Scholar] [CrossRef] [PubMed]
- Shoji, A.; Kuwahara, M.; Ozaki, H.; Sawai, H. Modified DNA aptamer that binds the (R)-Isomer of a thalidomide derivative with high enantioselectivity. J. Am. Chem. Soc. 2007, 129, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
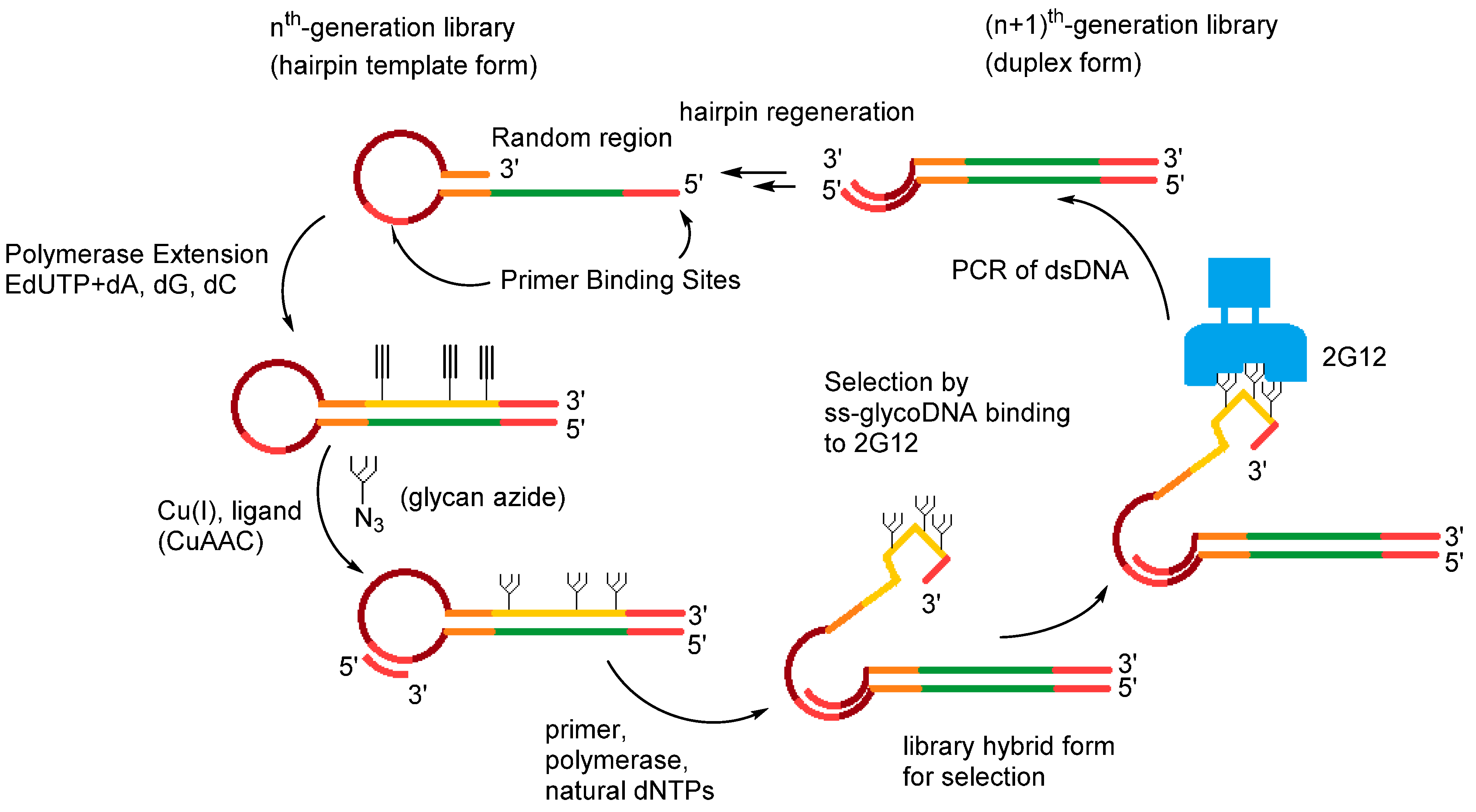
- Horiya, S.; MacPherson, I.S.; Krauss, I.J. Recent strategies targeting HIV glycans in vaccine design. Nat. Chem. Biol. 2014, 10, 990–999. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, I.S.; Temme, J.S.; Habeshian, S.; Felczak, K.; Pankiewicz, K.; Hedstrom, L.; Krauss, I.J. Multivalent Glycocluster Design through Directed Evolution. Angew. Chem. Int. Ed. 2011, 50, 11238–11242. [Google Scholar] [CrossRef]
- Temme, J.S.; Drzyzga, M.G.; MacPherson, I.S.; Krauss, I.J. Directed Evolution of 2G12-Targeted Nonamannose Glycoclusters by SELMA. Chem. Eur. J. 2013, 19, 17291–17295. [Google Scholar] [CrossRef] [PubMed]
- Temme, J.S.; MacPherson, I.S.; DeCourcey, J.F.; Krauss, I.J. High Temperature SELMA: Evolution of DNA-Supported Oligomannose Clusters Which Are Tightly Recognized by HIV bnAb 2G12. J. Am. Chem. Soc. 2014, 136, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Masud, M.M.; Kuwahara, M.; Ozaki, H.; Sawai, H. Sialyllactose-binding modified DNA aptamer bearing additional functionality by SELEX. Bioorg. Med. Chem. 2004, 12, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, K.; Kasamatsu, T.; Nagashima, J.-I.; Hanawa, K.; Kuwahara, M.; Ozaki, H.; Sawai, H. Arginine-modified DNA Aptamers That Show Enantioselective Recognition of the Dicarboxylic Acid Moiety of Glutamic Acid. Anal. Sci. 2008, 24, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Vaught, J.D.; Bock, C.; Carter, J.; Fitzwater, T.; Otis, M.; Schneider, D.; Rolando, J.; Waugh, S.; Wilcox, S.K.; Eaton, B.E. Expanding the Chemistry of DNA for in Vitro Selection. J. Am. Chem. Soc. 2010, 132, 4141–4151. [Google Scholar] [CrossRef] [PubMed]
- Morihiro, K.; Hoshino, H.; Hasegawa, O.; Kasahara, Y.; Nakajima, K.; Kuwahara, M.; Tsunoda, S.I.; Obika, S. Polymerase incorporation of a 2′-deoxynucleoside-5′-triphosphate bearing a 4-hydroxy-2-mercaptobenzimidazole nucleobase analogue. Bioorg. Med. Chem. Lett. 2015, 25, 2888–2891. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Hollenstein, M.; Leumann, C.J. The synthesis and application of a diazirine-modified uridine analogue for investigating RNA-protein interactions. RSC Adv. 2014, 4, 48228–48235. [Google Scholar] [CrossRef]
- Brody, E.; Gold, L.; Mehan, M.; Ostroff, R.; Rohloff, J.; Walker, J.; Zichi, D. Life’s Simple Measures: Unlocking the Proteome. J. Mol. Biol. 2012, 422, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.R.; Gelinas, A.D.; Zhang, C.; Rohloff, J.C.; Carter, J.D.; O’Connell, D.; Gold, L.; Janjic, N.; Jarvis, T.C. Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. USA 2012, 109, 19971–19976. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, J. Quantitative, High-Resolution Proteomics for Data-Driven Systems Biology. Annu. Rev. Biochem. 2011, 80, 273–299. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-Based Multiplexed Proteomic Technology for Biomarker Discovery. PLoS ONE 2010, 5, e15004. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, A.D.; Davies, D.R.; Edwards, T.E.; Rohloff, J.C.; Carter, J.D.; Zhang, C.; Gupta, S.; Ishikawa, Y.; Hirota, M.; Nakaishi, Y.; et al. Crystal Structure of Interleukin-6 in Complex with a Modified Nucleic Acid Ligand. J. Biol. Chem. 2014, 289, 8720–8734. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hirota, M.; Waugh, S.M.; Murakami, I.; Suzuki, T.; Muraguchi, M.; Shibamori, M.; Ishikawa, Y.; Jarvis, T.C.; Carter, J.D.; et al. Chemically-Modified DNA Aptamers Bind Interleukin-6 with High Affinity and Inhibit Signaling by Blocking its Interaction with Interleukin-6 Receptor. J. Biol. Chem. 2014, 289, 8706–8719. [Google Scholar] [CrossRef] [PubMed]
- Hopfield, J.J. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci. USA 1974, 71, 4135–4139. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Brody, E.; Clemens, P.R.; Cripe, L.; DeLisle, R.K.; Furlong, P.; Gordish-Dressman, H.; Hache, L.; Henricson, E.; Hoffman, E.P.; et al. Large-scale serum protein biomarker discovery in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 7153–7158. [Google Scholar] [CrossRef] [PubMed]
- Nolte, A.; Klussmann, S.; Bald, R.; Erdmann, V.A.; Fuerste, J.P. Mirror-design of l-oligonucleotide ligands binding to l-arginine. Nat. Biotechnol. 1996, 14, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, S.; Nolte, A.; Bald, R.; Erdmann, V.A.; Fuerste, J.P. Mirror-image RNA that binds d-adenosine. Nat. Biotechnol. 1996, 14, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- Sooter, L.J.; Ellington, A.D. Reflections on a Novel Therapeutic Candidate. Chem. Biol. 2002, 9, 857–858. [Google Scholar] [CrossRef]
- Semizarov, D.G.; Arzumanov, A.A.; Dyatkina, N.B.; Meyer, A.; Vichier-Guerre, S.; Gosselin, G.; Rayner, B.; Imbach, J.L.; Krayevsky, A.A. Stereoisomers of Deoxynucleoside 5′-Triphosphates as Substrates for Template-dependent and -independent DNA Polymerases. J. Biol. Chem. 1997, 272, 9556–9560. [Google Scholar] [PubMed]
- Forsman, J.J.; Leino, R. L-Pentoses in Biological and Medicinal Applications. Chem. Rev. 2011, 111, 3334–3357. [Google Scholar] [CrossRef] [PubMed]
- Loakes, D.; Holliger, P. Polymerase engineering: Towards the encoded synthesis of unnatural biopolymers. Chem. Commun. 2009, 4619–4631. [Google Scholar] [CrossRef] [PubMed]
- Sczepanski, J.T.; Joyce, G.F. A cross-chiral RNA polymerase ribozyme. Nature 2014, 515, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Vater, A.; Klussmann, S. Turning mirror-image oligonucleotides into drugs: The evolution of Spiegelmer therapeutics. Drug Discov Today 2015, 20, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Vater, A.; Sell, S.; Kaczmarek, P.; Maasch, C.; Buchner, K.; Pruszynska-Oszmalek, E.; Kolodziejski, P.; Purschke, W.G.; Nowak, K.W.; Strowski, M.Z.; et al. A Mixed Mirror-image DNA/RNA Aptamer Inhibits Glucagon and Acutely Improves Glucose Tolerance in Models of Type 1 and Type 2 Diabetes. J. Biol. Chem. 2013, 288, 21136–21147. [Google Scholar] [CrossRef] [PubMed]
- Purschke, W.G.; Hoehlig, K.; Buchner, K.; Zboralski, D.; Schwoebel, F.; Vater, A.; Klussmann, S. Identification and characterization of a mirror-image oligonucleotide that binds and neutralizes sphingosine 1-phosphate, a central mediator of angiogenesis. Biochem. J. 2014, 462, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Sczepanski, J.T.; Joyce, G.F. Binding of a Structured D-RNA Molecule by an L-RNA Aptamer. J. Am. Chem. Soc. 2013, 135, 13290–13293. [Google Scholar] [CrossRef] [PubMed]
- Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure. Science 1999, 284, 2118–2124. [Google Scholar] [CrossRef] [PubMed]
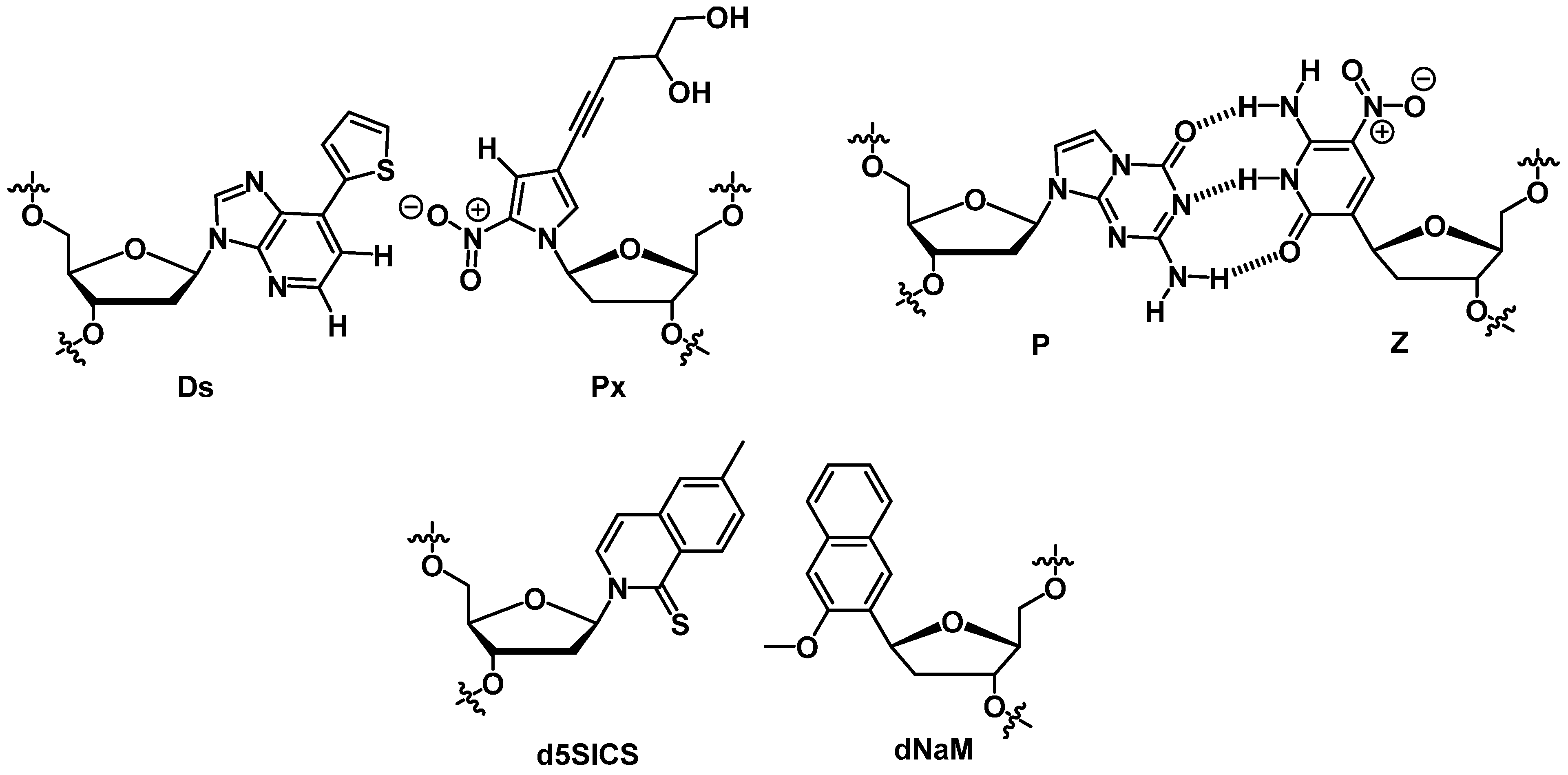
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.; McLaughlin, S.H.; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Kimoto, M.; Yamashige, R.; Matsunaga, K.; Yokoyama, S.; Hirao, I. Generation of high-affinity DNA aptamers using an expanded genetic alphabet. Nat. Biotechnol. 2013, 31, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.I.; Pinheiro, V.B.; Smola, M.J.; Morgunov, A.S.; Peak-Chew, S.; Cozens, C.; Weeks, K.M.; Herdewijn, P.; Holliger, P. Catalysts from synthetic genetic polymers. Nature 2015, 518, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Nasertorabi, F.; Choi, S.-H.; Han, G.W.; Reed, S.A.; Stevens, R.C.; Schultz, P.G. Exploring the potential impact of an expanded genetic code on protein function. Proc. Natl. Acad. Sci. USA 2015, 112, 6961–6966. [Google Scholar] [CrossRef] [PubMed]
- Attwater, J.; Holliger, P. A synthetic approach to abiogenesis. Nat. Methods 2014, 11, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Malyshev, D.A.; Dhami, K.; Lavergne, T.; Chen, T.; Dai, N.; Foster, J.M.; Corrêa, I.R., Jr.; Romesberg, F.E. A semi-synthetic organism with an expanded genetic alphabet. Nature 2014, 509, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, M.M.; Singh, I.; Kellett, W.F.; Hoshika, S.; Benner, S.A.; Richards, N.G.J. Structural Basis for a Six Nucleotide Genetic Alphabet. J. Am. Chem. Soc. 2015, 137, 6947–6955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Q.; Yang, Z.Y.; Sefah, K.; Bradley, K.M.; Hoshika, S.; Kim, M.J.; Kim, H.J.; Zhu, G.Z.; Jimenez, E.; Cansiz, S.; et al. Evolution of Functional Six-Nucleotide DNA. J. Am. Chem. Soc. 2015, 137, 6734–6737. [Google Scholar] [CrossRef] [PubMed]
- Herdewijn, P. Nucleic Acids with a Six-Membered “Carbohydrate” Mimic in the Backbone. Chem. Biodivers. 2010, 7, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Horhota, A.; Zou, K.; Ichida, J.K.; Yu, B.; McLaughlin, L.W.; Szostak, J.W.; Chaput, J.C. Kinetic Analysis of an Efficient DNA-Dependent TNA Polymerase. J. Am. Chem. Soc. 2005, 127, 7427–7434. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.R.; Larsen, A.C.; Zahurancik, W.J.; Fahmi, N.E.; Meyers, M.; Suo, Z.; Chaput, J.C. DNA Polymerase-Mediated Synthesis of Unbiased Threose Nucleic Acid (TNA) Polymers Requires 7-Deazaguanine To Suppress G:G Mispairing during TNA Transcription. J. Am. Chem. Soc. 2015, 137, 4014–4017. [Google Scholar] [CrossRef] [PubMed]
- Ichida, J.K.; Zou, K.; Horhota, A.; Yu, B.; McLaughlin, L.W.; Szostak, J.W. An in Vitro Selection System for TNA. J. Am. Chem. Soc. 2005, 127, 2802–2803. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, S.; Chaput, J.C. Darwinian evolution of an alternative genetic system provides support for TNA as an RNA progenitor. Nat. Chem. Biol. 2012, 4, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Obika, S.; Nanbu, D.; Hari, Y.; Andoh, J.; Morio, K.; Doi, T.; Imanishi, T. Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methyleneribonucleosides. Tetrahedron Lett. 1998, 39, 5401–5404. [Google Scholar] [CrossRef]
- Singh, S.K.; Koshkin, A.A.; Wengel, J.; Nielsen, P. LNA (locked nucleic acids): Synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998, 455–456. [Google Scholar] [CrossRef]
- Jespen, J.S.; Sørensen, M.D.; Wengel, J. Locked Nucleic Acid: A Potent Nucleic Acid Analog in Therapeutics and Biotechnology. Oligonucleotides 2004, 14, 130–146. [Google Scholar]
- Lundin, K.E.; Hojland, T.; Hansen, B.R.; Persson, R.; Bramsen, J.B.; Kjems, J.; Koch, T.; Wengel, J.; Smith, C. Biological activity and biotechnological aspects of locked nucleic acids. Adv. Genet. 2013, 82, 47–107. [Google Scholar] [PubMed]
- Astakhova, I.K.; Wengel, J. Scaffolding along Nucleic Acid Duplexes Using 2′-Amino-Locked Nucleic Acids. Acc. Chem. Res. 2014, 47, 1768–1777. [Google Scholar] [CrossRef] [PubMed]
- Veedu, R.N.; Vester, B.; Wengel, J. Polymerase Chain Reaction and Transcription Using Locked Nucleic Acid Nucleotide Triphosphates. J. Am. Chem. Soc. 2008, 130, 8124–8125. [Google Scholar] [CrossRef] [PubMed]
- Crouzier, L.; Dubois, C.; Edwards, S.L.; Lauridsen, L.H.; Wengel, J.; Veedu, R.N. Efficient Reverse Transcription Using Locked Nucleic Acid Nucleotides towards the Evolution of Nuclease Resistant RNA Aptamers. PLoS ONE 2012, 7, e35990. [Google Scholar] [CrossRef] [PubMed]
- Doessing, H.; Hansen, L.H.; Veedu, R.N.; Wengel, J.; Vester, B. Amplification and Re-Generation of LNA-Modified Libraries. Molecules 2012, 17, 13087–13097. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, Y.; Irisawa, Y.; Ozaki, H.; Obika, S.; Kuwahara, M. 2′,4′-BNA/LNA aptamers: CE-SELEX using a DNA-based library of fulllength 2′-O,4′-C-methylene-bridged/linked bicyclic ribonucleotides. Bioorg. Med. Chem. Lett. 2013, 23, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.L.; Poongavanam, V.; Kanwar, J.R.; Roy, K.; Hillman, K.M.; Prasad, N.; Leth-Larsen, R.; Petersen, M.; Marusic, M.; Plavec, J.; et al. Targeting VEGF with LNA-stabilized G-rich oligonucleotide for efficient breast cancer inhibition. Chem. Commun. 2015, 51, 9499–9502. [Google Scholar] [CrossRef] [PubMed]
- Elle, I.C.; Karlsen, K.K.; Terp, M.G.; Larsen, N.; Nielsen, R.; Derbyshire, N.; Mandrup, S.; Ditzel, H.J.; Wengel, J. Selection of LNA-containing DNA aptamers against recombinant human CD73. Mol. BioSyst. 2015, 11, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Fujita, H.; Kasahara, Y.; Irisawa, Y.; Obika, S.; Kuwahara, M. In vitro selection of DNA-based aptamers that exhibit RNA-like conformations using a chimeric oligonucleotide library that contains two different xeno-nucleic acids. Mol. BioSyst. 2015, 11, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Bande, O.; El Asrar, R.A.; Braddick, D.; Dumbre, S.; Pezo, V.; Schepers, G.; Pinheiro, V.B.; Lescrinier, E.; Holliger, P.; Marlière, P.; et al. Isoguanine and 5-Methyl-Isocytosine Bases, in vitro and in vivo. Chem. Eur. J. 2015, 21, 5009–5022. [Google Scholar] [CrossRef] [PubMed]
- Veedu, R.N.; Burri, H.V.; Kumar, P.; Sharma, P.K.; Hrdlicka, P.J.; Vester, B.; Wengel, J. Polymerase-directed synthesis of C5-ethynyl locked nucleic acids. Bioorg. Med. Chem. Lett. 2010, 20, 6565–6568. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Yang, Z.Y.; Bradley, K.M.; Hoshika, S.; Jimenez, E.; Zhang, L.Q.; Zhu, G.Z.; Shanker, S.; Yu, F.H.; Turek, D.; et al. In vitro selection with artificial expanded genetic information systems. Proc. Natl. Acad. Sci. USA 2014, 111, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Renders, M.; Miller, E.; Hollenstein, M.; Perrin, D.M. A Method for Selecting Modified DNAzymes without the Use of Modified DNA as a Template in PCR. Chem. Commun. 2015, 51, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diafa, S.; Hollenstein, M. Generation of Aptamers with an Expanded Chemical Repertoire. Molecules 2015, 20, 16643-16671. https://doi.org/10.3390/molecules200916643
Diafa S, Hollenstein M. Generation of Aptamers with an Expanded Chemical Repertoire. Molecules. 2015; 20(9):16643-16671. https://doi.org/10.3390/molecules200916643
Chicago/Turabian StyleDiafa, Stella, and Marcel Hollenstein. 2015. "Generation of Aptamers with an Expanded Chemical Repertoire" Molecules 20, no. 9: 16643-16671. https://doi.org/10.3390/molecules200916643
APA StyleDiafa, S., & Hollenstein, M. (2015). Generation of Aptamers with an Expanded Chemical Repertoire. Molecules, 20(9), 16643-16671. https://doi.org/10.3390/molecules200916643