
Novel Coumarin-Containing Aminophosphonatesas Antitumor Agent: Synthesis, Cytotoxicity, DNA-Binding and Apoptosis Evaluation

Abstract
:
1. Introduction
2. Results and Discussion
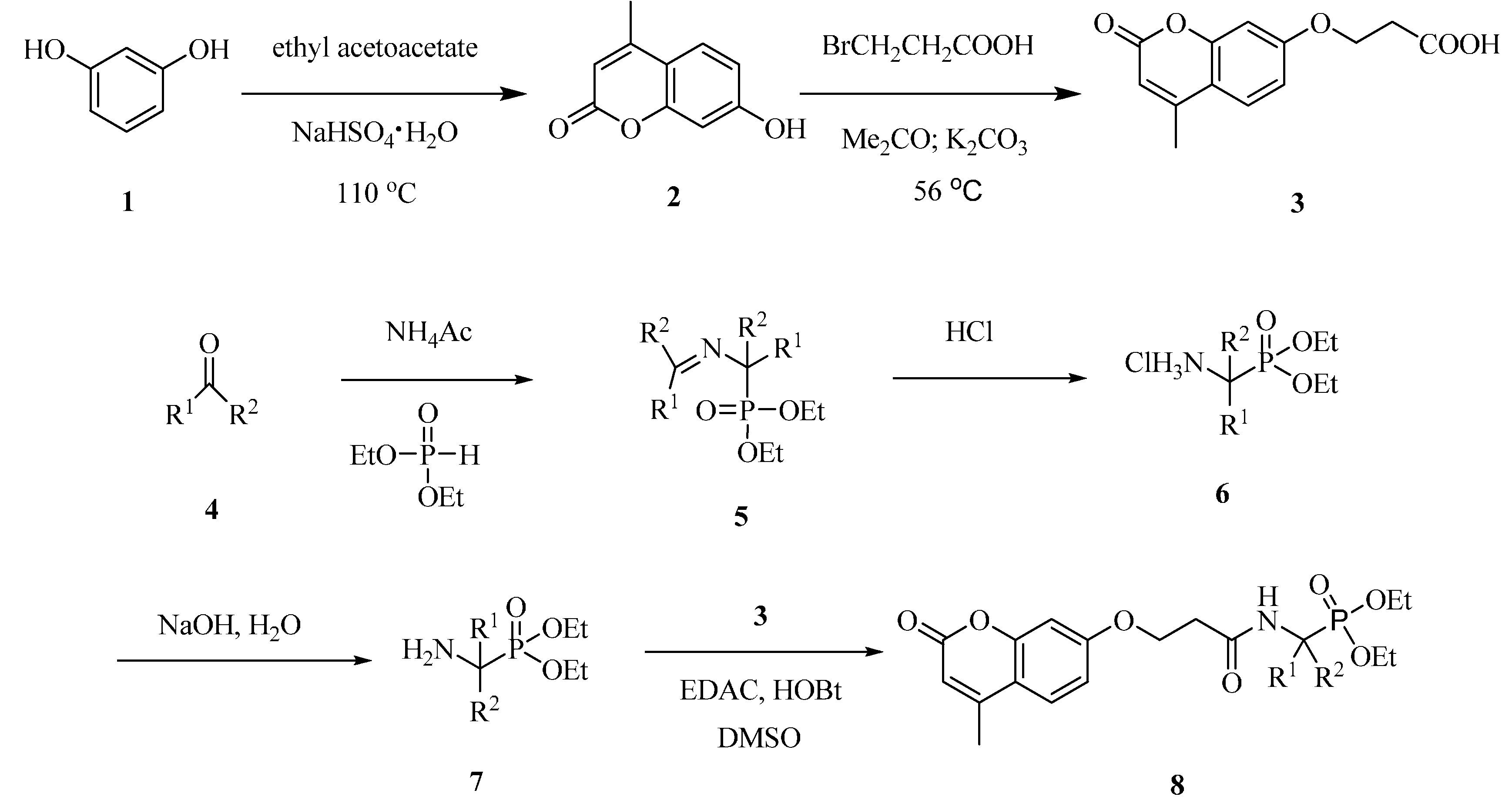
2.1. Synthesis of Coumarin Derivatives
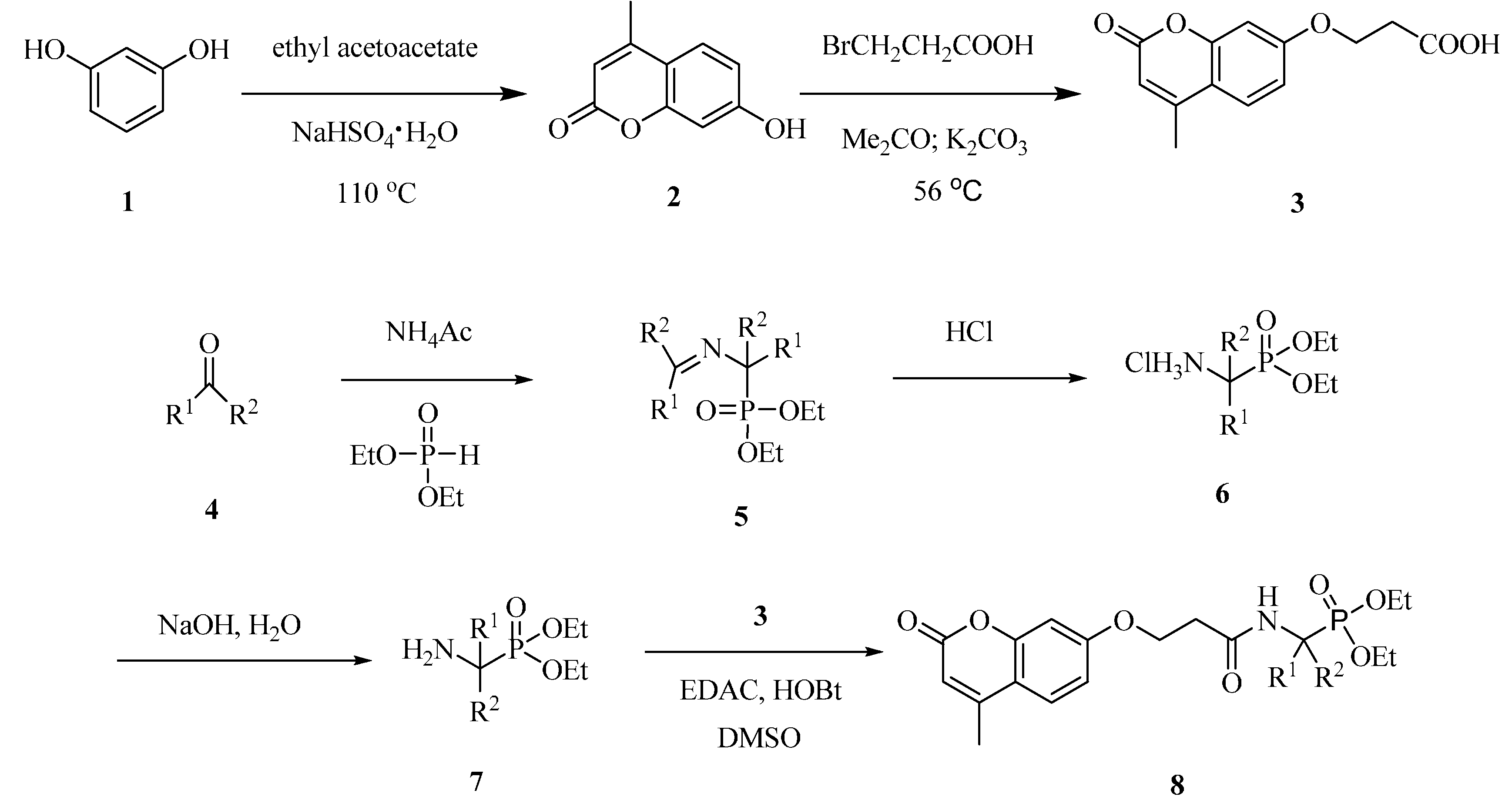
2.2. Cytotoxicity against Tumor Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R1 | R2 | IC50a, μM | |||
|---|---|---|---|---|---|---|
| MGC-803 | HUVEC | HCT-116 | KB | |||
| 2 | - | - | >100 | >100 | >100 | >50 |
| 8a | H | Ph | >100 | >100 | >100 | >50 |
| 8b | H | o-F-Ph | >100 | >100 | >100 | >50 |
| 8c | H | o-Cl-Ph | 79.38 ± 3.82 | >100 | 88.68 ± 1.69 | 33.4 ± 1.20 |
| 8d | H | p-Cl-Ph | 76.85 ± 3.89 | >100 | 86.18 ± 1.86 | 7.8 ± 5.36 |
| 8e | H | o-Br-Ph | 67.49 ± 3.26 | >100 | 90.55 ± 1.09 | 9.55 ± 0.09 |
| 8f | H | m-Br-Ph | 74.16 ± 3.36 | >100 | 25.75 ± 1.21 | 0.50 ± 2.68 |
| 8g | H | p-Br-Ph | 85.63 ± 2.83 | >100 | 88.52 ± 4.62 | 7.27 ± 6.18 |
| 8h | H | o-OCH3-Ph | 36.85 ± 1.96 | >100 | 23.68 ± 5.89 | 0.45 ± 1.03 |
| 8i | H | 2-Na | 41.47 ± 2.86 | >100 | 38.90 ± 0.04 | 0.79 ± 1.70 |
| 8j | CH3 | Ph | 14.55 ± 1.11 | >100 | 8.68 ± 3.20 | 0.08 ± 1.12 |
| 5-Fu | 15.32 ± 1.96 | 56 ± 3.21 | 10.05 ± 2.20 | 1.23 ± 1.09 | ||
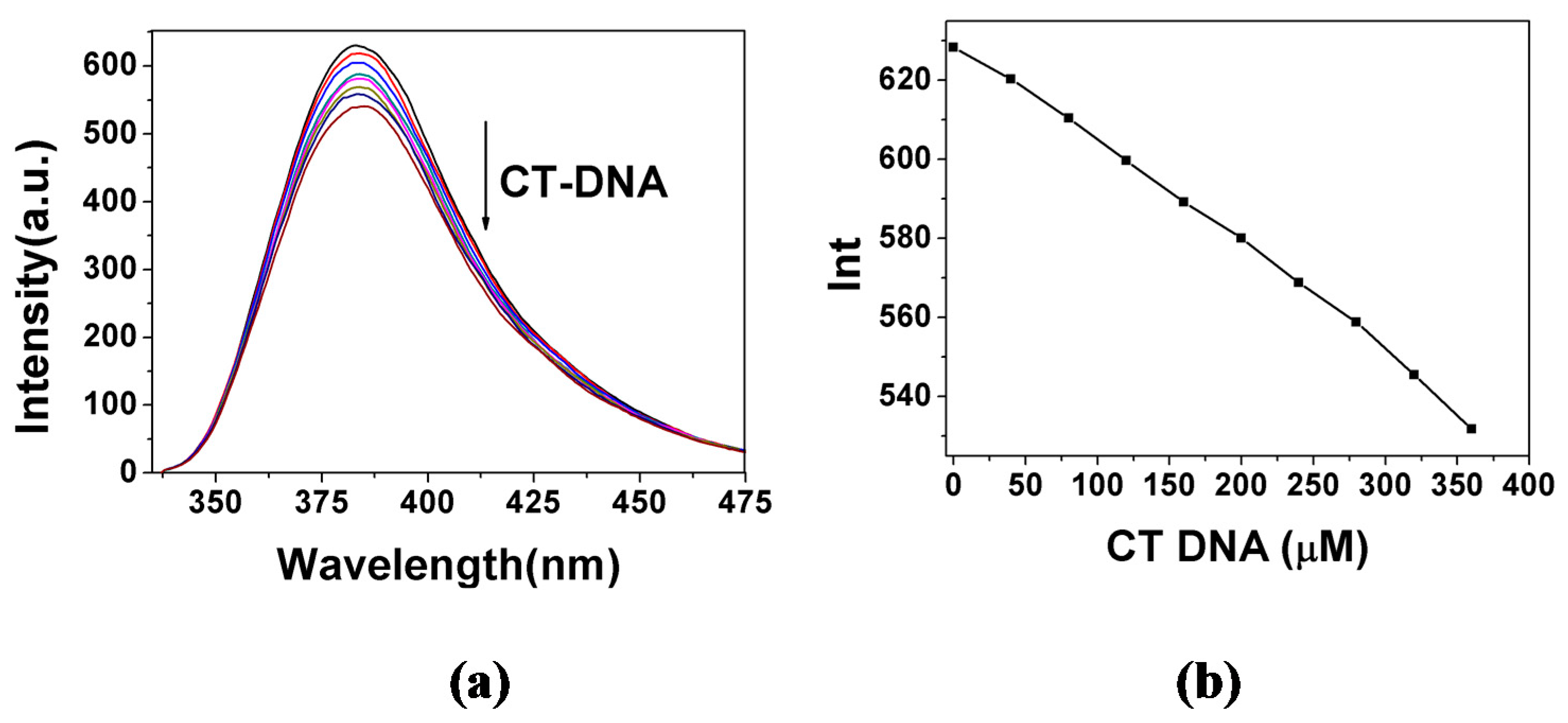
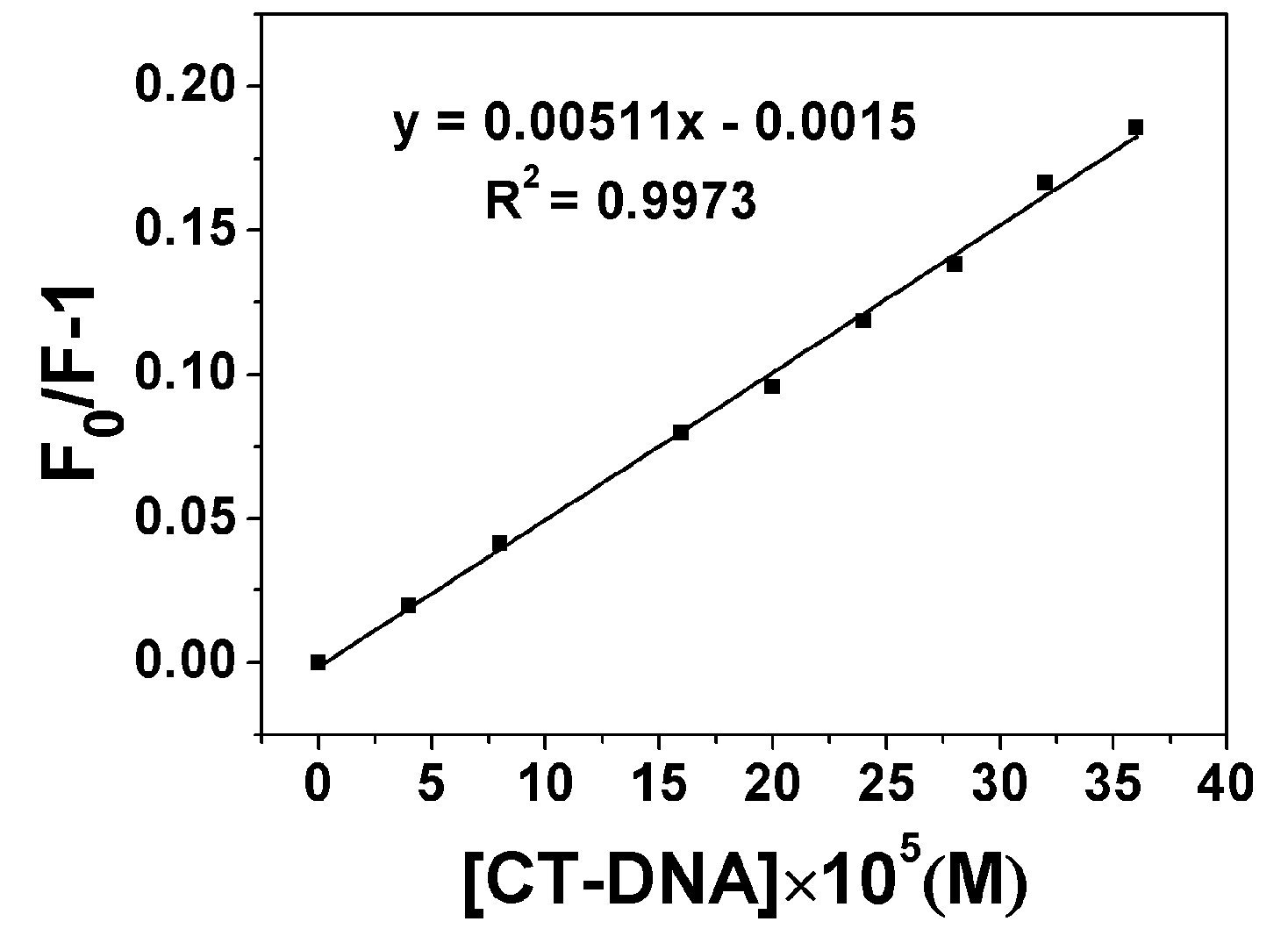
2.3. DNA Binding Properties
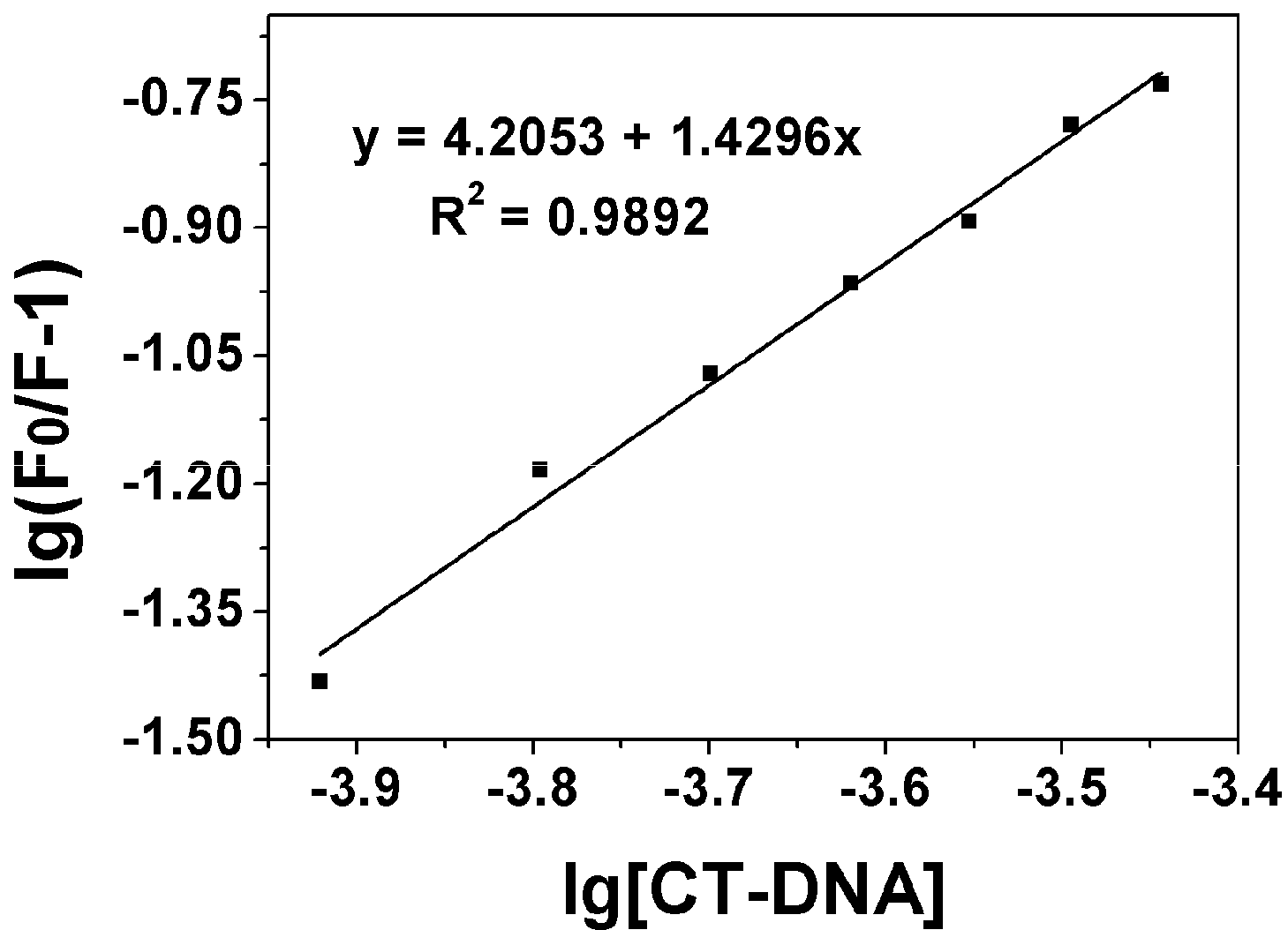
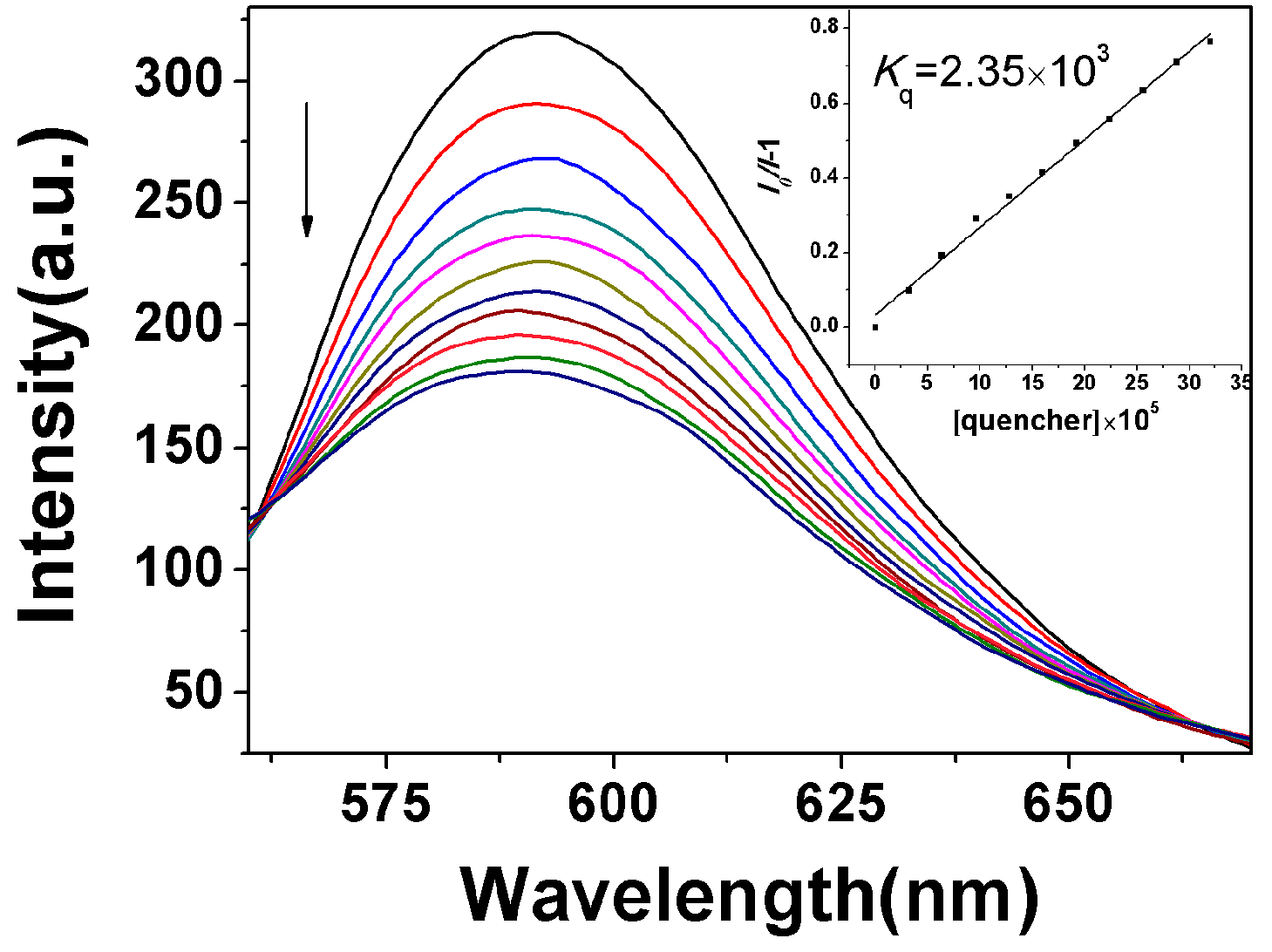
2.3.1. Fluorescence Emission Titration
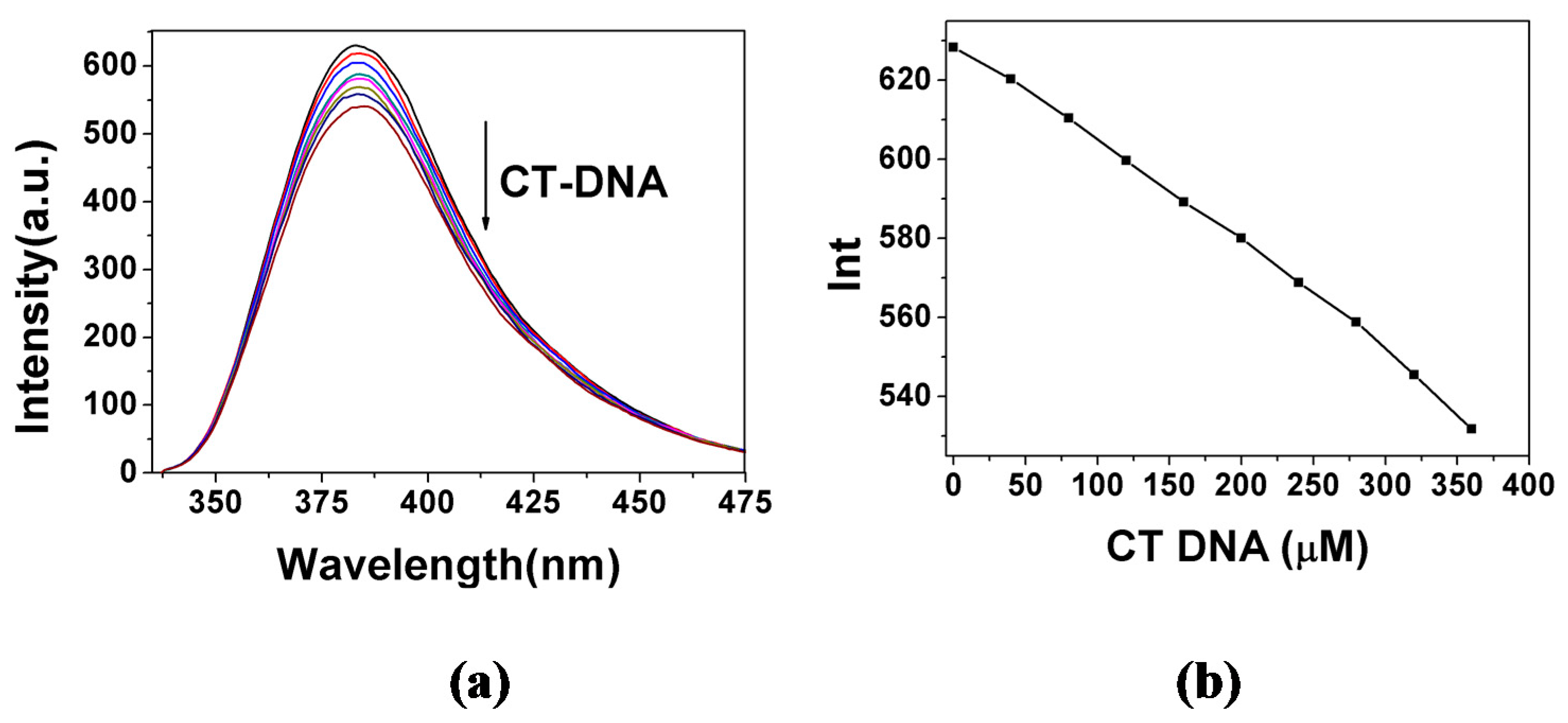
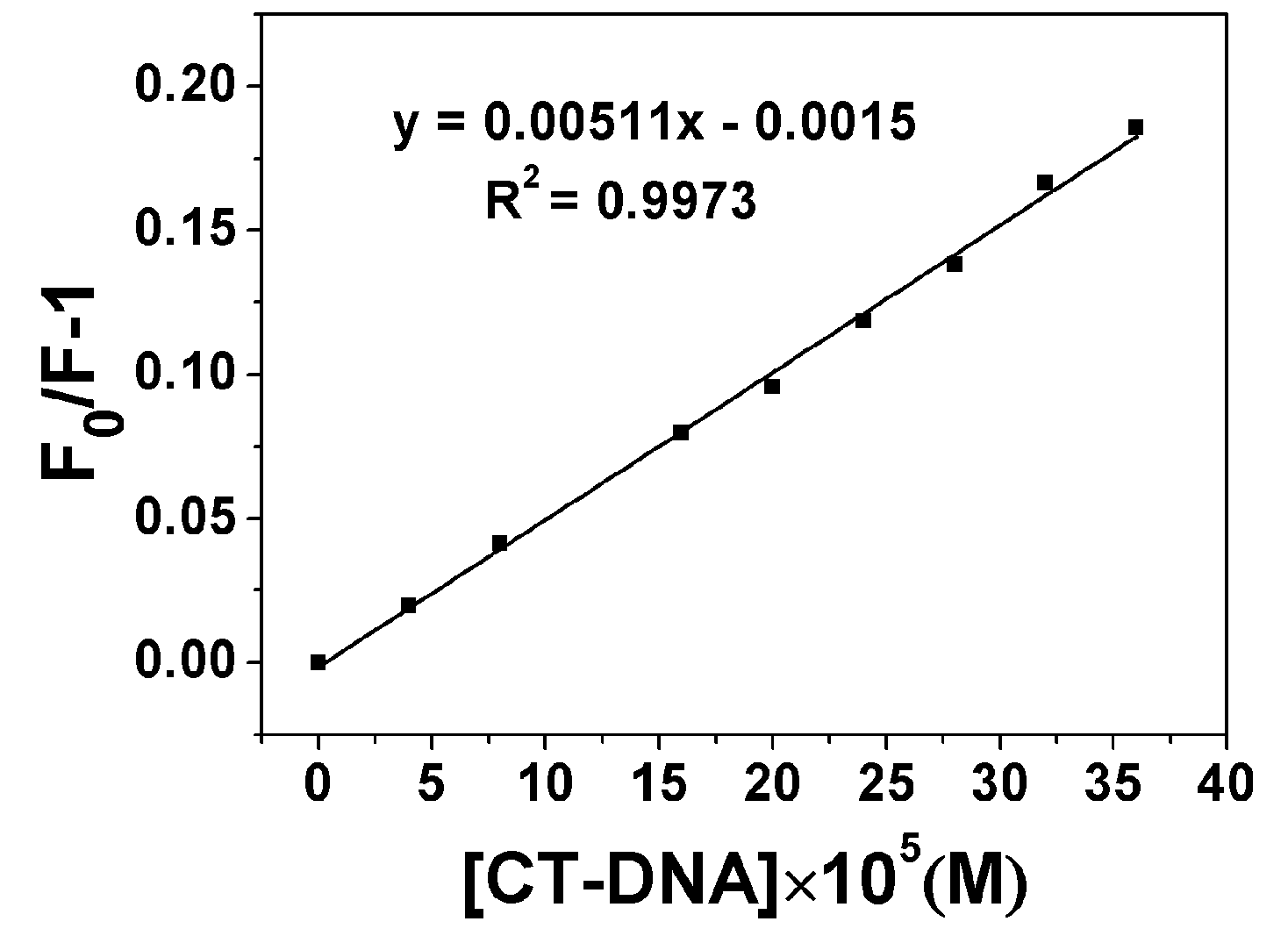
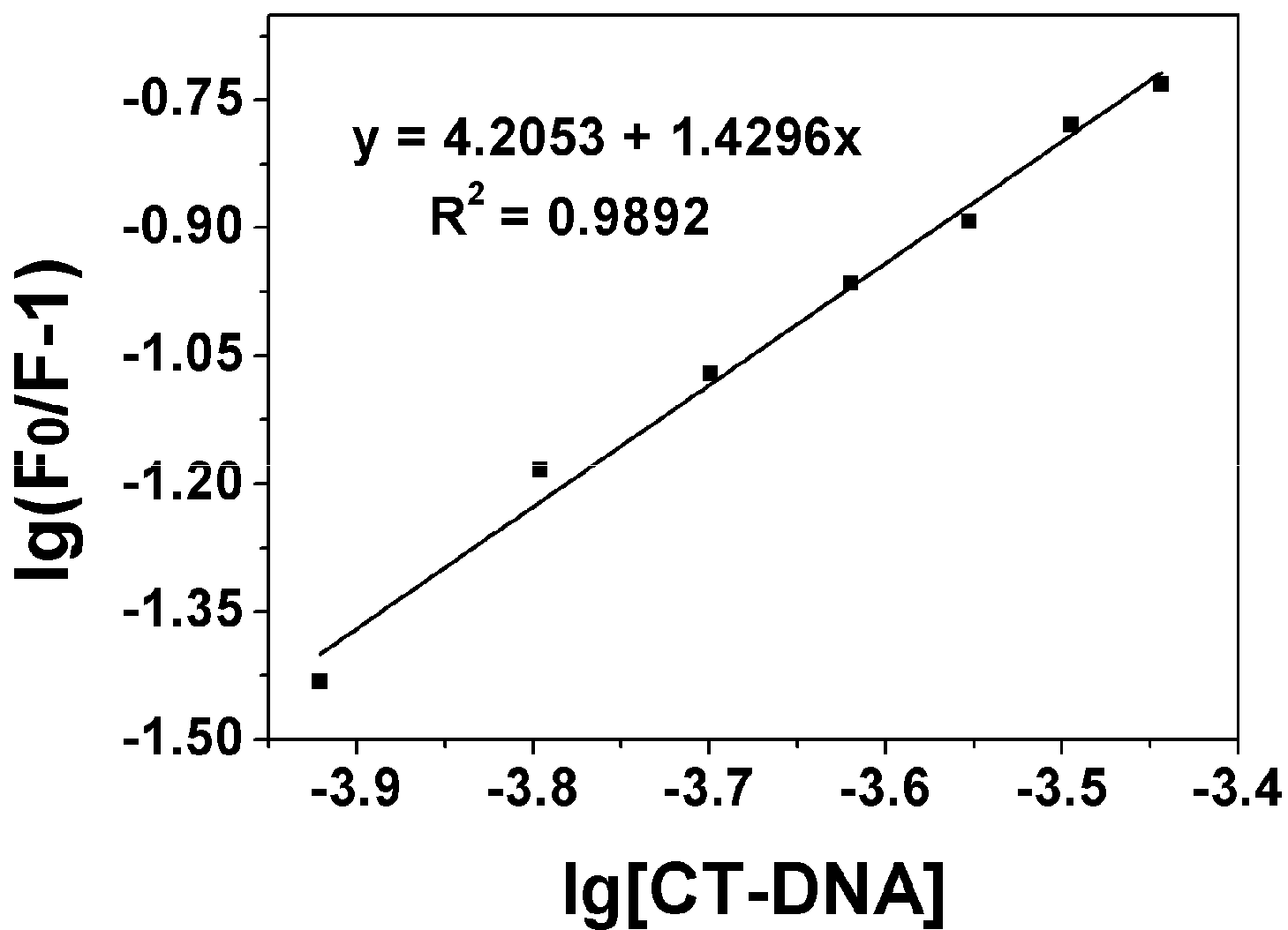
| Compound | Kb (M−1) | n | KSV(M−1) |
|---|---|---|---|
| 2 | 4.56 × 102 | 1.00 | 4.42 × 102 |
| 8a | 2.44 × 102 | 0.96 | 3.4 × 102 |
| 8b | 77.5 | 0.83 | 2.99 × 102 |
| 8c | 1.30 × 102 | 0.83 | 4.81 × 102 |
| 8d | 5.91 × 102 | 1.05 | 4.29 × 102 |
| 8e | 1.37 × 102 | 0.84 | 4.75 × 102 |
| 8f | 1.16 × 104 | 1.42 | 4.12 × 102 |
| 8g | 5.76 × 102 | 1.04 | 4.1 × 102 |
| 8h | 7.38 × 105 | 1.95 | 3.57 × 102 |
| 8i | 1.24 × 104 | 0.96 | 1.81 × 104 |
| 8j | 1.6 × 104 | 1.43 | 5.11 × 102 |
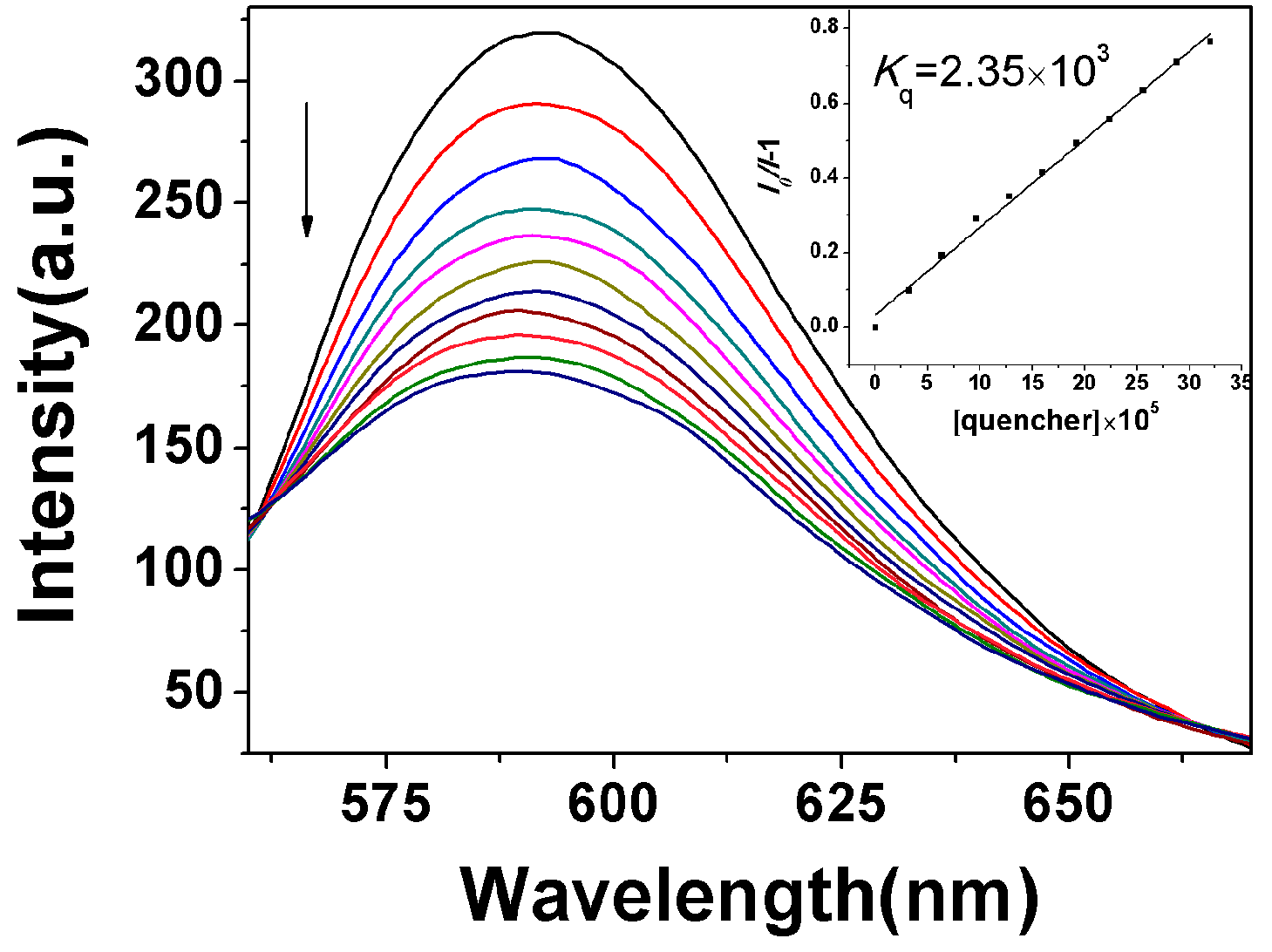
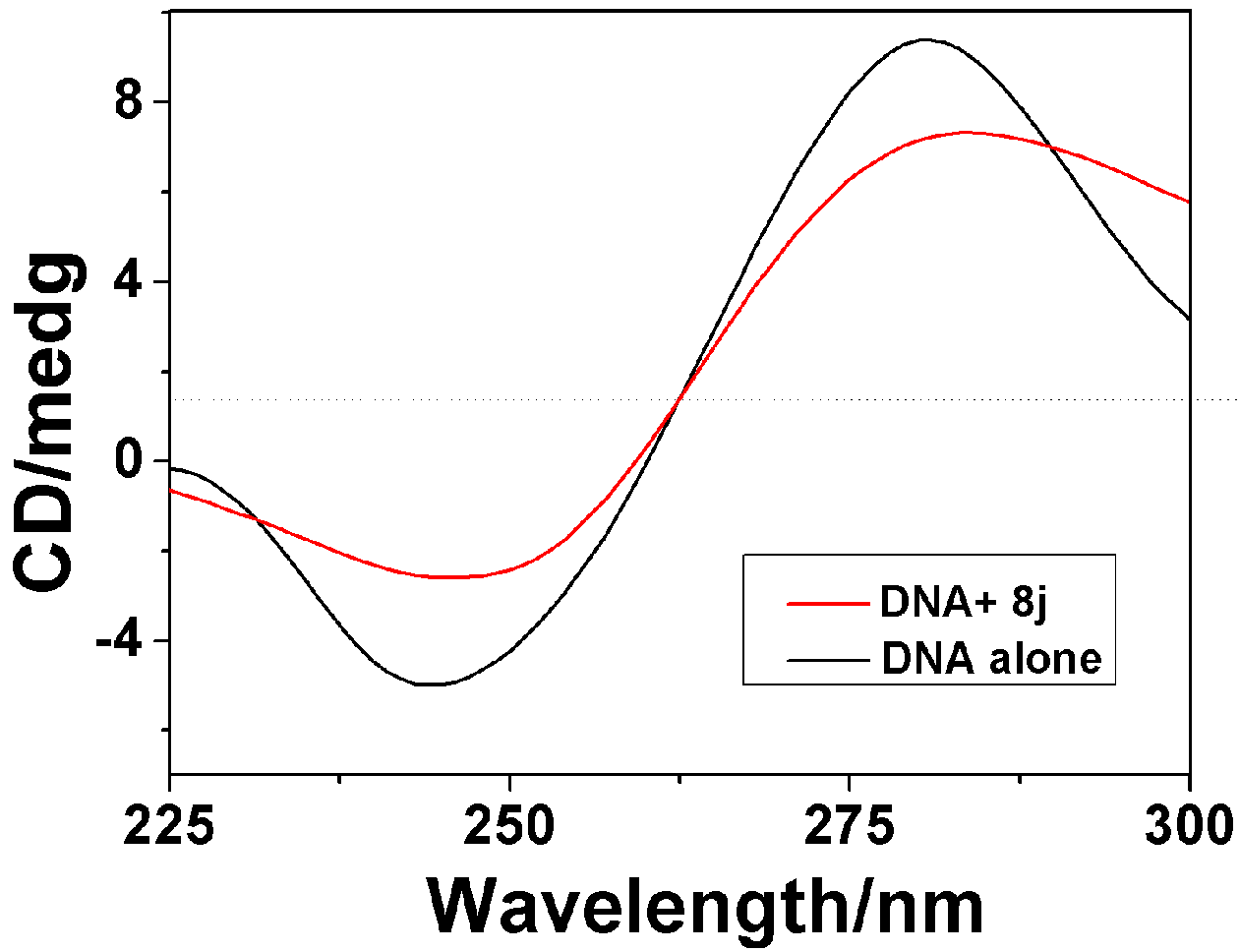
2.3.2. Circular Dichroism Spectra
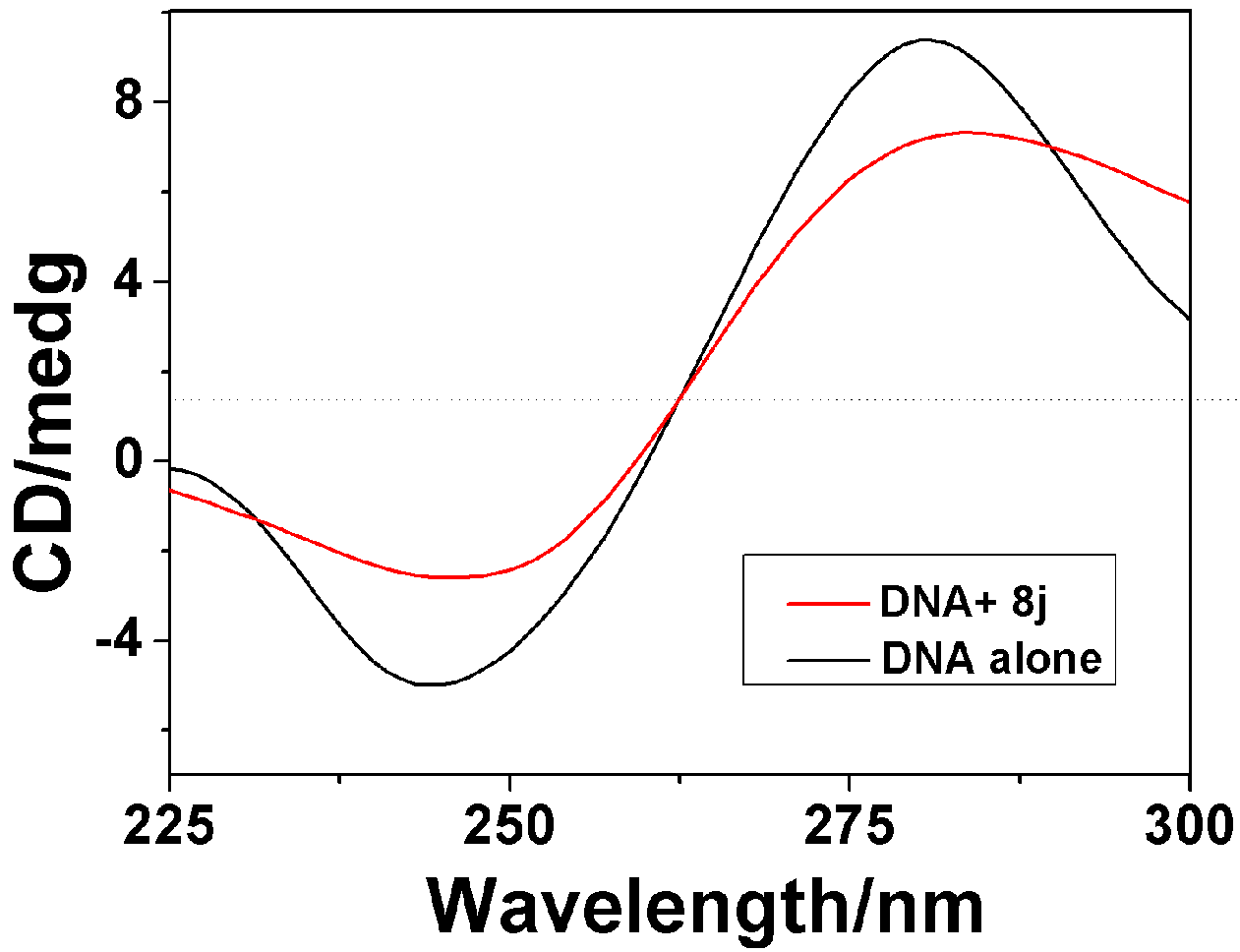
| Compounds | 8f | 8h | 8j |
|---|---|---|---|
| Positive AI decrease(%) a | 23.1 | 34.5 | 26.82 |
| Negative AI decrease(%) a | 26.9 | 28.5 | 30.4 |
2.4. Apoptosis and Cell Cycle Analysis
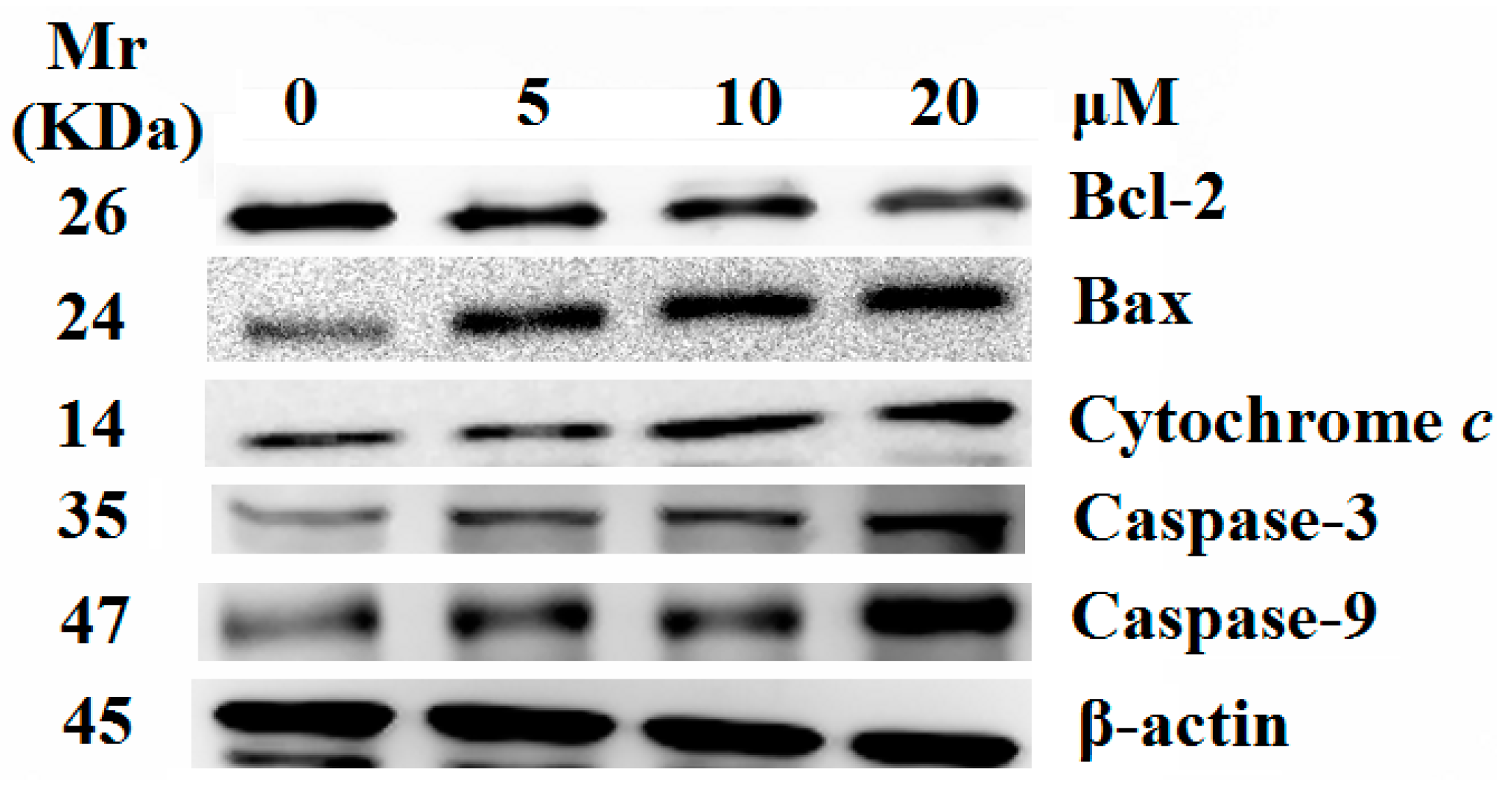
2.5. Release of Cytochrome c and Activation of Caspases were Involved in the Apoptosis Induced by 8j
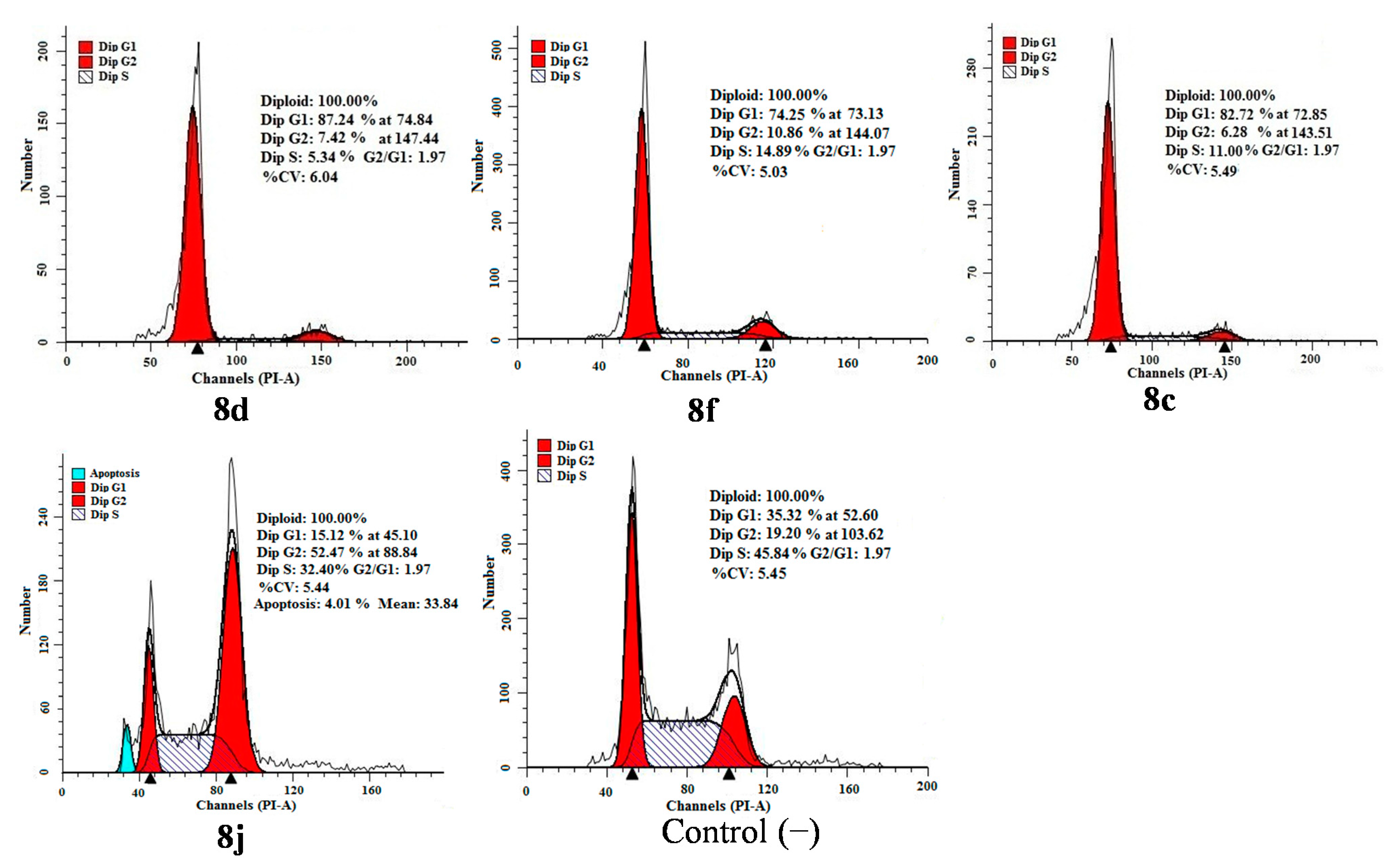
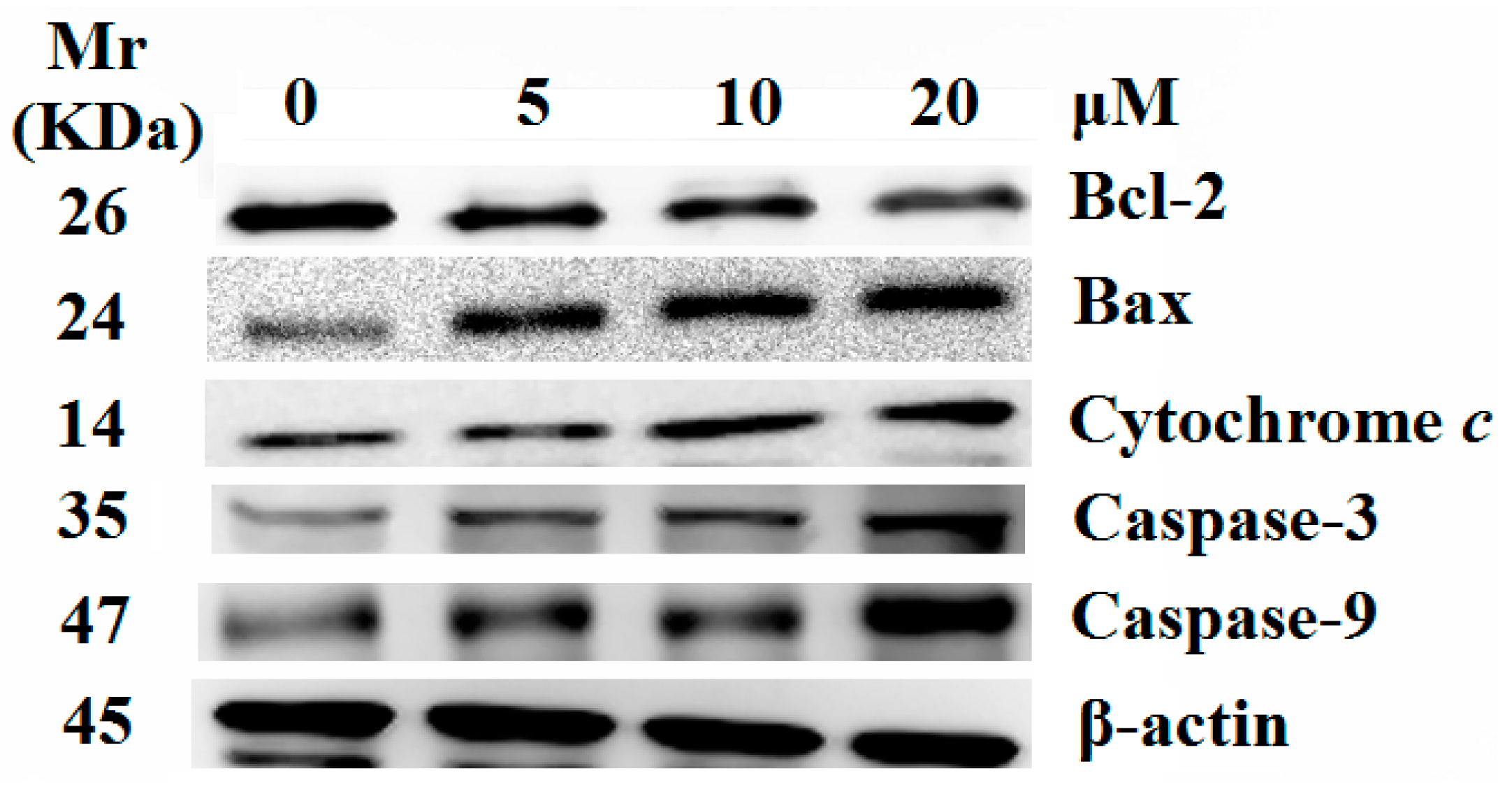
3. Experimental Section
3.1. Chemistry
3.1.1. 7-Hydroxy-4-methyl-2H-chromen-2-one (2)
3.1.2. 3-(4-Methyl-2-oxo-2H-chromen-7-yloxy)propanoic Acid (3)
3.1.3. General Procedure for the Preparation of the O-Substituted Coumarin Derivatives 8a–8j
3.2. Biological Assays
3.2.1. Cytotoxicityassay in Vitro
3.2.2. Apoptosis and Cell Cycle Analysis
3.3. Spectrofluorimetric Titration and DNA Binding Assay
3.4. Statistics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bhattacharyya, S.S.; Mandal, S.K.; Biswas, R.; Paul, S.; Pathak, S.; Boujedaini, N.; Belon, P.; Khuda-Bukhsh, A.R. A syntheticcoumarin (4-methyl-7-hydroxy coumarin) has anti-cancerpotentials against DMBA-induced skin cancer in mice. Eur. J. Pharmacol. 2009, 614, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Arshad, A.; Osman, H.; Bagley, M.C.; Lam, C.K.; Mohamad, S.; Zahariluddin, A.S.M. Synthesis and antimicrobial properties of somenew thiazolyl coumarin derivatives. Eur. J. Med. Chem. 2011, 46, 3788–3794. [Google Scholar] [CrossRef] [PubMed]
- Abdelhafez, O.M.; Amin, K.M.; Batran, R.Z.; Maher, T.J.; Nada, S.A.; Sethumadhavan, S. Synthesis, anticoagulant and PIVKA-IIinduced by new 4-hydroxycoumarin derivatives. Bioorg. Med. Chem. 2010, 18, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Yang, Z.Y.; Xia, P.; Hackl, T.; Hamel, E.; Mauger, A.; Wu, J.H.; Lee, K.H. Antitumor agents. 211. Fluorinated 2-phenyl-4-quinolone derivatives as antimitotic antitumor agents 1. J. Med. Chem. 2001, 44, 3932–3936. [Google Scholar]
- Suzuki, A.Z.; Watanabe, T.; Kawamoto, M.; Nishiyama, K.; Yamashita, H.; Ishii, M.; Iwamura, M.; Furuta, T. Coumarin-4-ylmethoxycarbonylsas phototriggers for alcohols and phenols. Org. Lett. 2003, 5, 4867–4870. [Google Scholar] [CrossRef] [PubMed]
- Sunthitikawinsakul, A.; Kongkathip, N.; Kongkathip, B.; Phonnakhu, S.; Daly, J.W.; Spande, T.F.; Nimit, Y.; Rochanaruangrai, S. Coumarins andcarbazoles fromClausena excavateexhibited antimycobacterial andantifungal activities. Planta Med. 2003, 69, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Park, S.K.; Lim, J.H.; Choi, Y.H.; Kim, W.J.; Moon, S.K. Esculetin inhibits cell proliferation through the Ras/ERK1/2 pathway in human colon cancer cells. Oncol. Rep. 2011, 25, 223–229. [Google Scholar] [PubMed]
- Stanchev, S.; Momekov, G.; Jensen, F.; Manolov, I. Synthesis, computational study and cytotoxic activity of new 4-hydroxycoumarin derivatives. Eur. J. Med. Chem. 2008, 43, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Donnelly, A.C.; Kusuma, B.R.; Brandt, G.E.L.; Brown, D.; Rajewski, R.A.; Vielhauer, G.; Holzbeierlein, J.; Cohen, M.S.; Blagg, B.S.J. Engineering an antibiotic to fight cancer: Optimization ofthe novobiocin scaffold to produce anti-proliferative agents. J. Med. Chem. 2011, 54, 3839–3853. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.S.; Paul, S.; De, A.; Das, D.; Samadder, A.; Boujedaini, N.; Khuda-Bukhsh, A.R. Poly (lactide-co-glycolide) acid nanoencapsulation of a synthetic coumarin: Cytotoxicity and bio-distribution in mice, in cancer cell line and interaction with calf thymus DNA as target. Toxicol. Appl. Pharm. 2011, 253, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.; Wheate, N.J.; Wang, S.Y.; Grant-Collins, J.; Ralph, S.F.; Day, A.I.; Higgins, V.J.; Aldrich-Wright, J.R. Encapsulation of platinum(II)-based DNA intercalators within cucurbit[6,7,8]urils. J. Biol. Inorg. Chem. 2007, 12, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.Y.; Yao, G.Y.; Wei, J.C.; Pan, Y.M.; Liao, Z.X.; Wang, H.S. Synthesis, Cytotoxicity, DNA Binding and Apoptosis of Rhein-Phosphonate Derivatives as Antitumor Agents. Int. J. Mol. Sci. 2013, 14, 9424–9439. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, Y.; Qian, X. Novel antitumor agent family of 1H-benzo [c,d] indol-2-one with flexible basic side chains: Synthesis and biological evaluation. Bioorg. Med. Chem. 2007, 15, 1356–1362. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable potential of thea-aminophosphonatephosphinate structural motif in medicinalchemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.Y.; Ye, M.Y.; Huang, R.Z.; Li, Y.J.; Pan, Y.M.; Xu, Q.; Liao, Z.X.; Wang, H.S. Synthesis and antitumor activities of novel rheina-aminophosphonates conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.Y.; Yao, G.Y.; Pan, Y.M.; Liao, Z.X.; Zhang, Y.; Wang, H.S. Synthesis and antitumor activities of novel a -aminophosp honatederivatives containing an alizar in moiety. Eur. J. Med. Chem. 2014, 83, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Rogers, C.J.; Fisher, A.J.; Toney, M.D. Aminophosphonateinhibitors of dialkylglycine decarboxylase: Structural basis forslow binding inhibition. Biochemistry 2002, 41, 12320–12328. [Google Scholar] [CrossRef] [PubMed]
- Kotsikorou, E.; Oldfield, E. A quantitative structure-activity relationship and pharmacophore modeling investigation of aryl-X and heterocyclic bisphosphonates as bone resorption agents. J. Med. Chem. 2003, 46, 2932–2944. [Google Scholar] [CrossRef] [PubMed]
- Piccinonna, S.; Margiotta, N.; Pacifico, C.; Lopalco, A.; Denora, N.; Fedi, S.; Corsinic, M.; Natile, G. DinuclearPt (II)-bisphosphonate complexes: A scaffold for multinuclear or different oxidation state platinum drugs. Dalton Trans. 2012, 41, 9689–9699. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.J.; Qian, X.H.; Cui, J.G.; Xiao, Y.; Wang, K.W.; Wu, P.C.; Cong, L.Y. Novel angular furoquinolinones bearing flexible chain asantitumor agent: Design, synthesis, cytotoxic evaluation, and DNA-binding studies. Bioorg. Med. Chem. Lett. 2008, 16, 8713–8718. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Ye, M.Y.; Huang, R.Z.; Yao, G.Y.; Pan, Y.M.; Liao, Z.X.; Wang, H.S. Coumarin-containing aminophosphonates bridged with chiralside chain: Synthesis and influence of chirality on cytotoxicity and DNA binding. Med. Chem. Res. 2014, 23, 3144–3156. [Google Scholar] [CrossRef]
- Sobhani, S.; Tashrifi, Z. Al (OTf) 3 as an efficient catalyst forone-pot synthesis of primary diethyl 1-aminophosphonates undersolvent-free conditions. Synth. Commun. 2008, 39, 120–131. [Google Scholar] [CrossRef]
- Sissi, C.; Leo, E.; Moro, S.; Capranico, G.; Mancia, A.; Menta, E.; Krapcho, A.P.; Palumbo, M. Antitumor AZA–anthrapyrazoles: Bio-physical and biochemical studies on 8-and 9-aza regioisomers. Biochem. Pharmacol. 2004, 67, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.V.; Asuncion, E.H. DNA-binding studies and siteselective fluorescence sensitization of an anthryl probe. J. Am. Chem. Soc. 1993, 115, 8547–8553. [Google Scholar] [CrossRef]
- Qiu, B.; Guo, L.; Wang, W.; Chen, G. Synthesis of N-4-butylamine acridone and its use as fluorescent probe for ct-DNA. Biosens. Bioelectron. 2009, 24, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Weber, G. Quenching of fluorescence by oxygen. Probe for structural fluctuations in macromolecules. Biochemistry 1973, 12, 4161–4170. [Google Scholar] [CrossRef] [PubMed]
- Khorasani-Motlagh, M.; Noroozifar, M.; Mirkazehi-Rigi, S. Fluorescence and DNA-binding properties of neodymium(III) and praseodymium(III) complexes containing 1,10-phenanthroline. Spectrochim. Acta A 2011, 79, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Kandagal, P.B.; Ashoka, S.; Seetharamappa, J.; Shaikh, S.M.T.; Jadegoud, Y.; Ijare, O.B. Study of the interaction of an anticancer drug with human and bovine serum albumin: Spectroscopic approach. J. Pharm. Biomed. Anal. 2006, 41, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Guo, J.; Zhao, N.; Wang, J. Study of interaction between kaempferol-Eu3+complex and DNA with the use of the neutra l red dye as a fluorescence probe. Sens. Actuators B Chem. 2010, 144, 239–246. [Google Scholar] [CrossRef]
- Rajendiran, V.; Murali, M.; Suresh, E.; Palaniandavar, M.; Periasamy, V.S.; Akbarsha, M.A. Non-covalent DNA binding and cytotoxicity of certain mixed-ligand ruthenium(II) complexes of 2,2-dipyridylamine and diimines. Dalton Trans. 2008, 16, 2157–2170. [Google Scholar] [CrossRef] [PubMed]
- Schäfe, S.; Gust, I.; Ott, R.; Sheldrick, W.S. Influence of the polypyridyl (pp) ligand size on the DNA binding properties, cytotoxicity and cellular uptake of organoruthenium(II) complexes of the type (η6-C6Me6) Ru(L)(pp)n+ L = Cl, n = 1; L = (NH2)2CS, n = 2. Eur. J. Inorg. Chem. 2007, 19, 3034–3046. [Google Scholar] [CrossRef]
- Xu, H.; Zheng, K.C.; Chen, Y.; Li, Y.Z.; Lin, L.J.; Li, H.; Zhang, P.X.; Ji, L.N. Effects of ligand planarity on the interaction of polypyridyl Ru(II) complexes with DNA. Dalton Trans. 2003, 11, 2260–2268. [Google Scholar] [CrossRef]
- Zhong, W.; Yu, J.S.; Liang, Y.; Fan, K.; Lai, L. Chlorobenzylidine-calf thymus DNA interaction II: Circulardichroism and nuclearmagneticresonancestudies. Spectrochim. Acta A 2004, 60, 2985–2992. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Barve, A.C.; Kumbhar, A.A.; Kumbhar, A.S.; Puranik, V.G.; Datar, P.A.; Sonawane, U.B.; Joshi, R.R. Synthesis, characterization, X-ray structure and DNA photocleavage by cis-dichloro bis(diimine) Co(III) complexes. J. Inorg. Biochem. 2006, 100, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: Samples of compounds 8 are available from the authors upon request.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-J.; Wang, C.-Y.; Ye, M.-Y.; Yao, G.-Y.; Wang, H.-S. Novel Coumarin-Containing Aminophosphonatesas Antitumor Agent: Synthesis, Cytotoxicity, DNA-Binding and Apoptosis Evaluation. Molecules 2015, 20, 14791-14809. https://doi.org/10.3390/molecules200814791
Li Y-J, Wang C-Y, Ye M-Y, Yao G-Y, Wang H-S. Novel Coumarin-Containing Aminophosphonatesas Antitumor Agent: Synthesis, Cytotoxicity, DNA-Binding and Apoptosis Evaluation. Molecules. 2015; 20(8):14791-14809. https://doi.org/10.3390/molecules200814791
Chicago/Turabian StyleLi, Ya-Jun, Cai-Yi Wang, Man-Yi Ye, Gui-Yang Yao, and Heng-Shan Wang. 2015. "Novel Coumarin-Containing Aminophosphonatesas Antitumor Agent: Synthesis, Cytotoxicity, DNA-Binding and Apoptosis Evaluation" Molecules 20, no. 8: 14791-14809. https://doi.org/10.3390/molecules200814791
APA StyleLi, Y.-J., Wang, C.-Y., Ye, M.-Y., Yao, G.-Y., & Wang, H.-S. (2015). Novel Coumarin-Containing Aminophosphonatesas Antitumor Agent: Synthesis, Cytotoxicity, DNA-Binding and Apoptosis Evaluation. Molecules, 20(8), 14791-14809. https://doi.org/10.3390/molecules200814791