
Dual Radiolabeling as a Technique to Track Nanocarriers: The Case of Gold Nanoparticles

Abstract
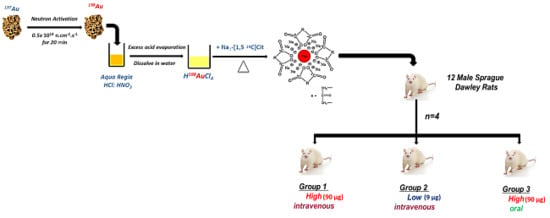
:
1. Introduction
2. Results
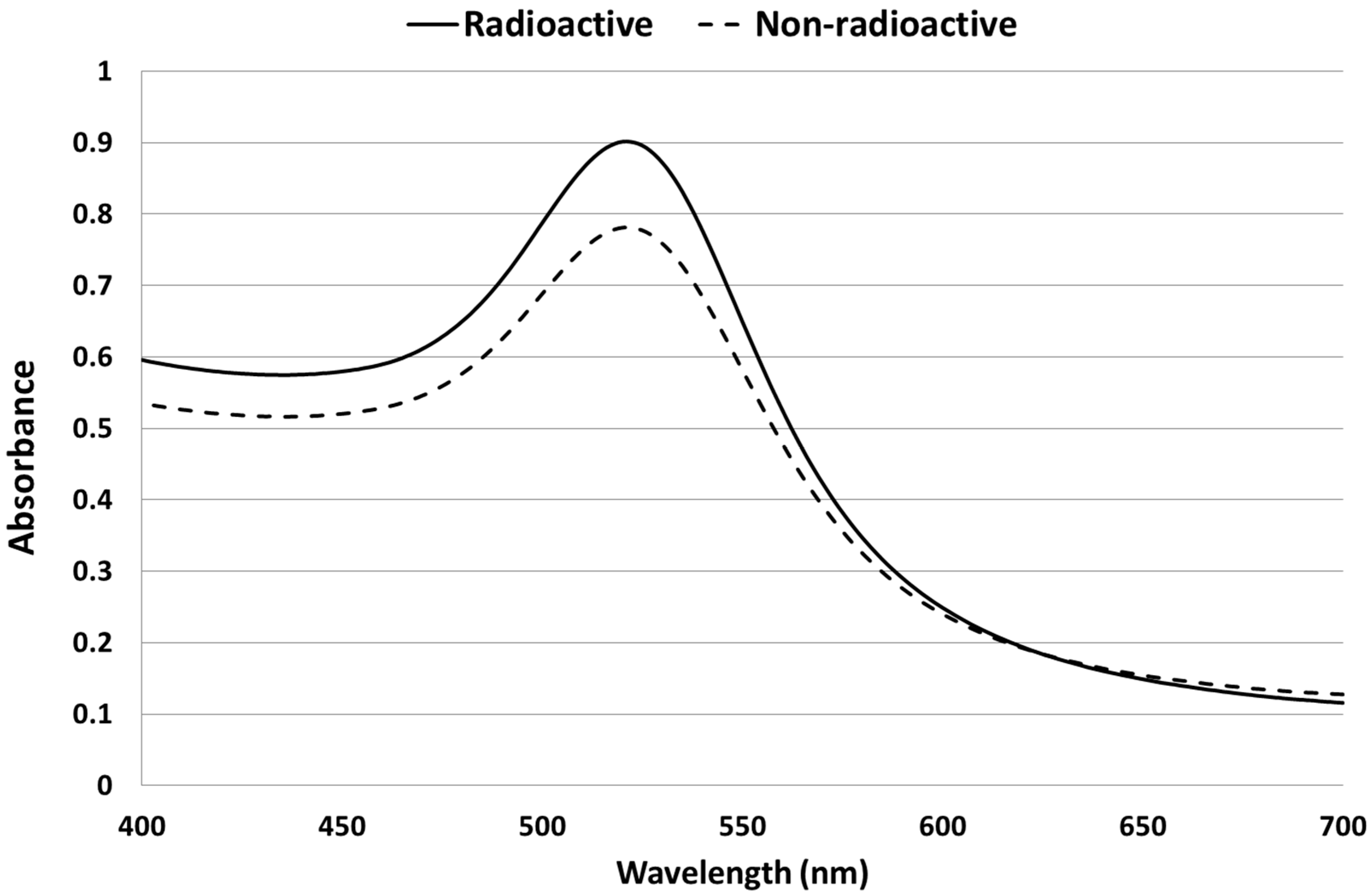
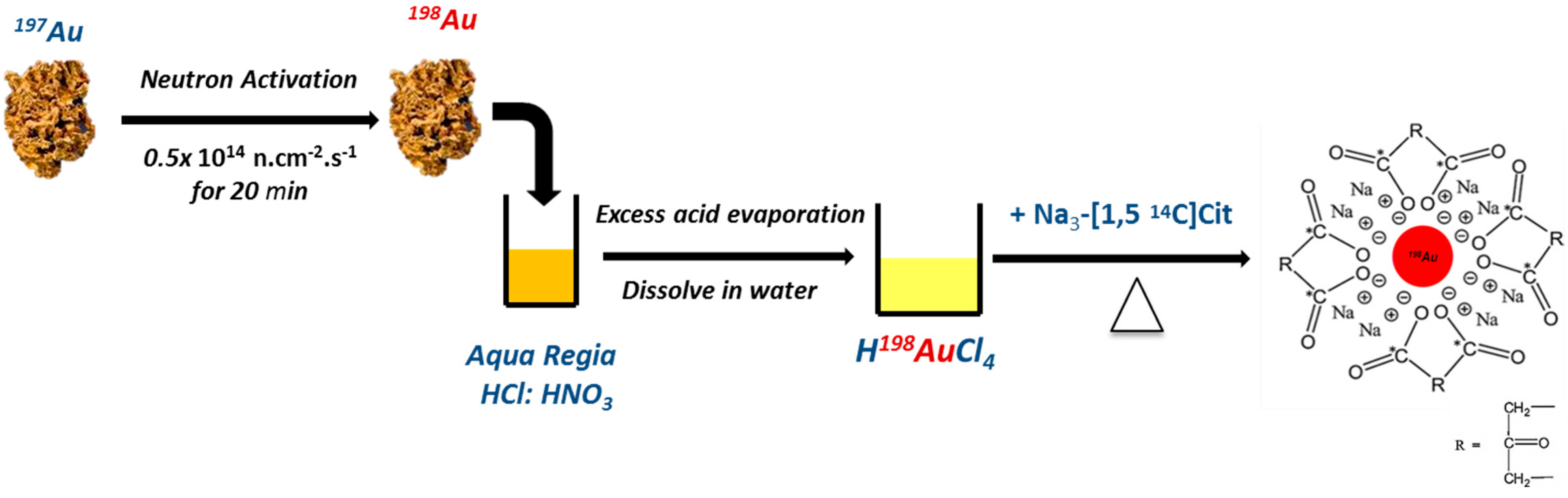
2.1. Synthesis and Characterization of AuNPs
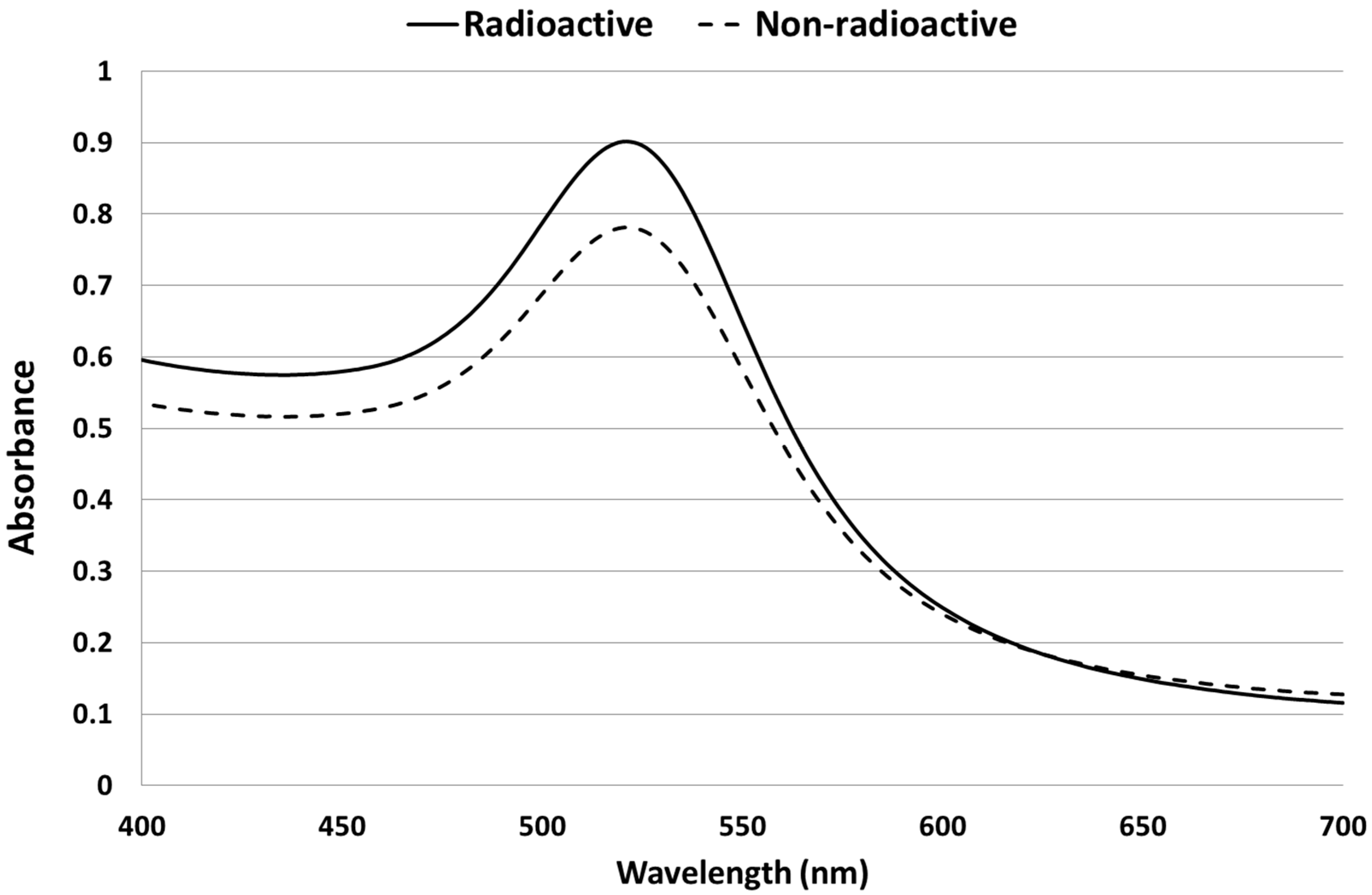
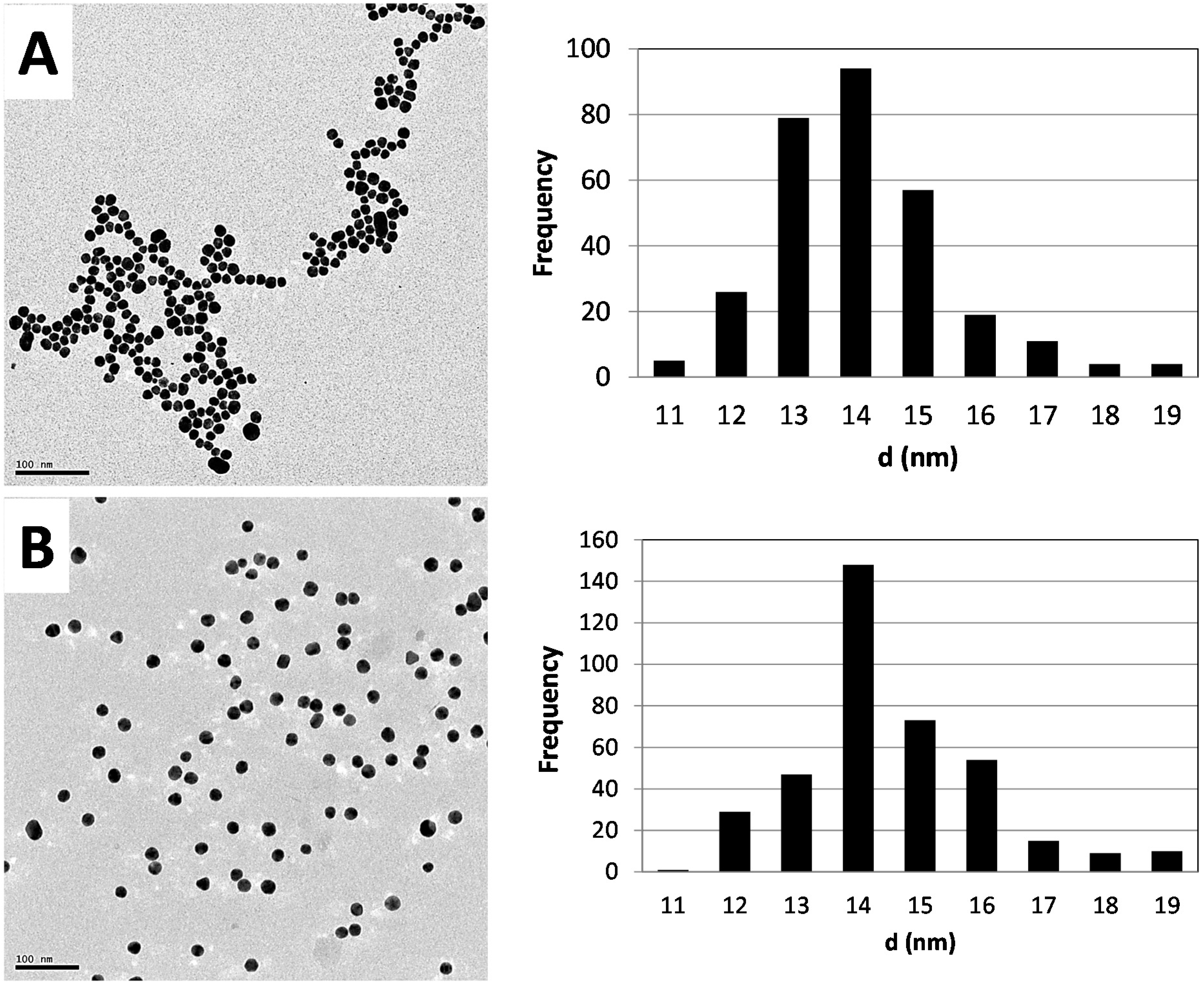
2.2. Biodistribution of Gold vs. Citrate in the Rat
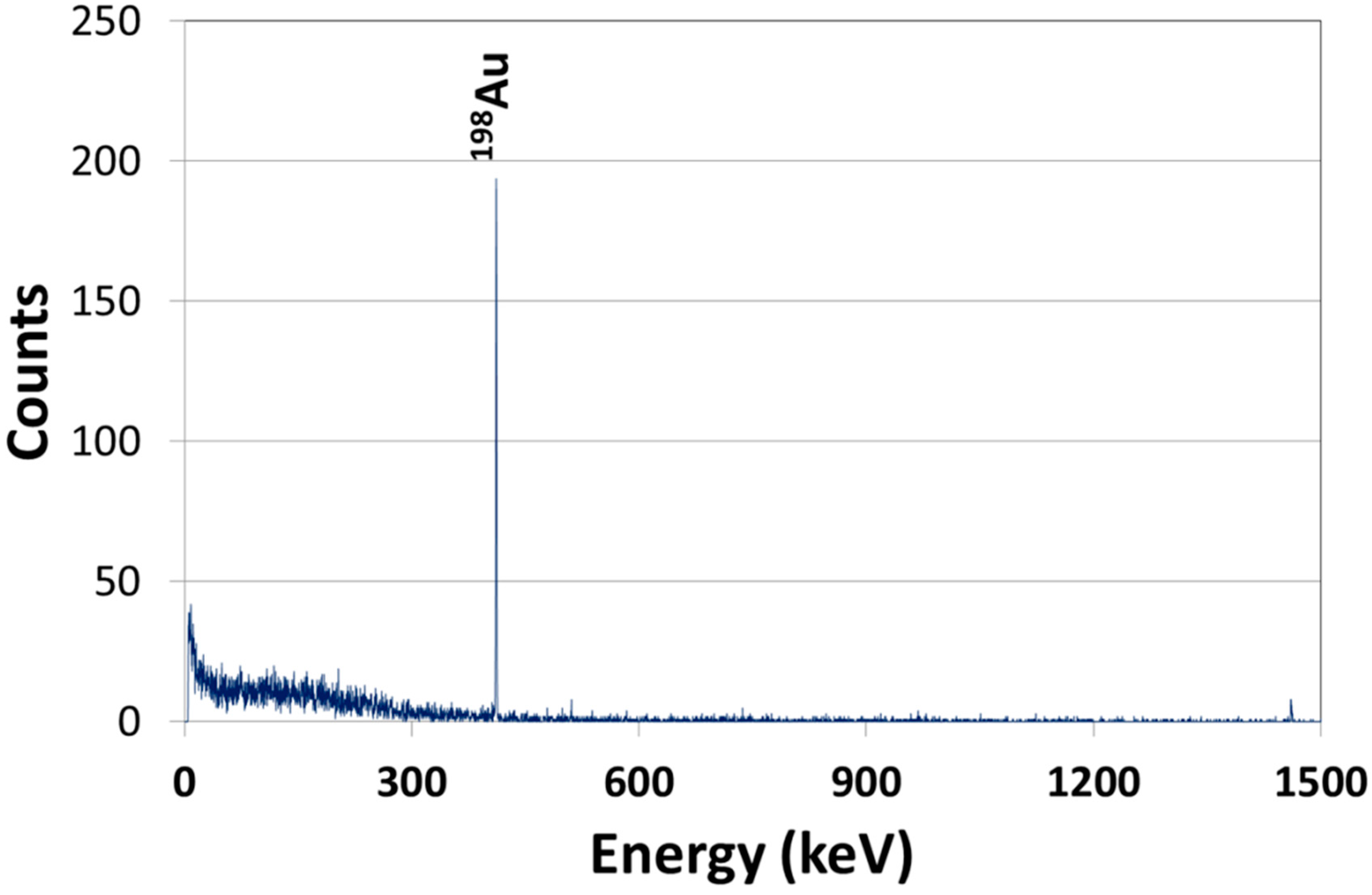
2.2.1. Dosimetry

{kind=link}
{kind=link}
{kind=link}
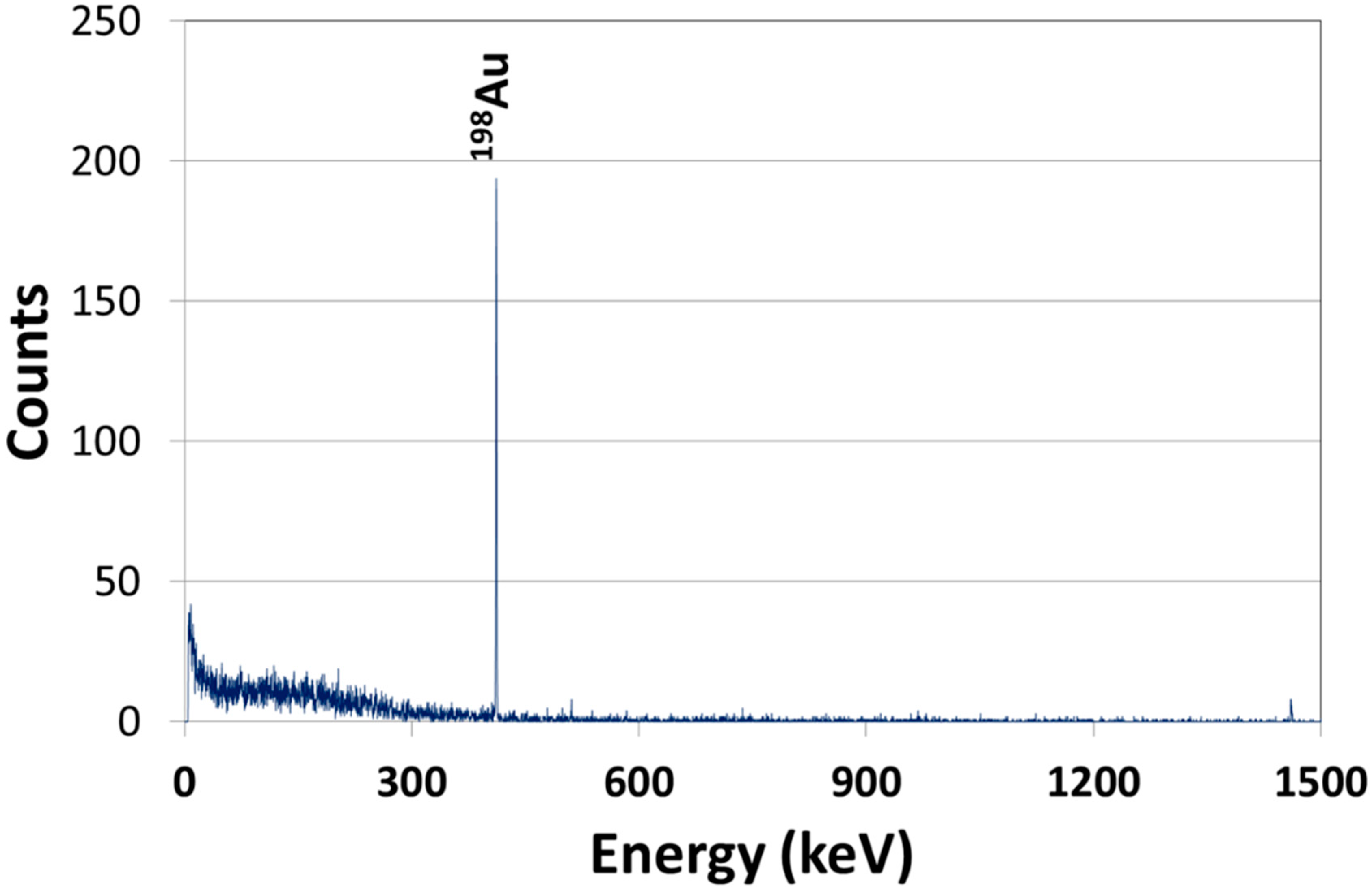{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose | ||||
|---|---|---|---|---|
| High | Low | |||
| Administered radioactivity per rat (MBq) | 198Au | 12.95 | 1.22 | |
| 14C | 0.027 | 0.0027 | ||
| Administered mass per rat (μg) | Au | 90 | 9 | |
| Citrate | 520 | 52 | ||
| Administered number of AuNPs per rat | 3.27 × 1012 | 3.327 × 1011 | ||
| Administered surface area (cm2) of AuNPs per rat | 20.16 | 2.02 | ||
2.2.2. Biodistribution Profiles
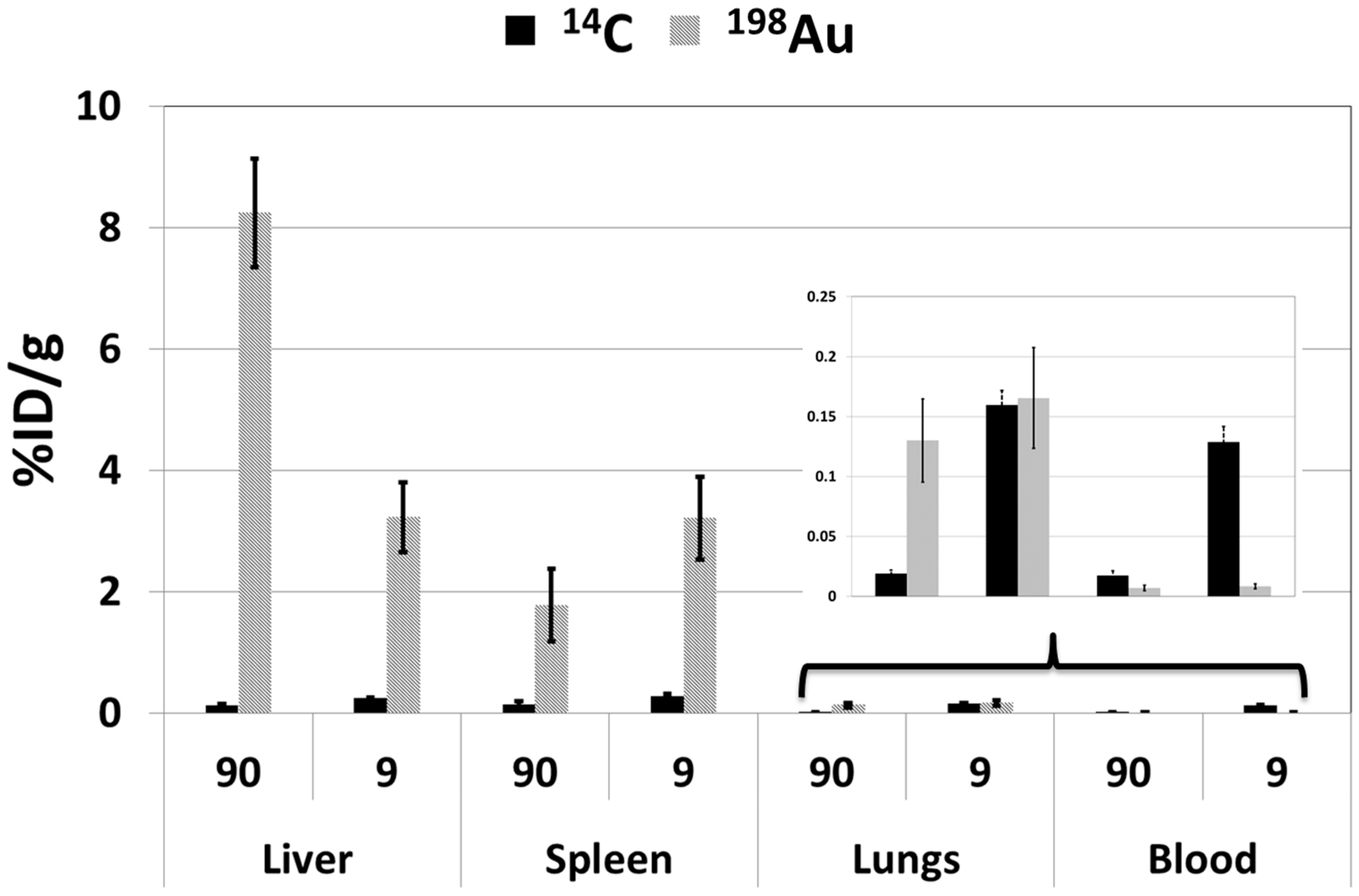
Liver
Spleen
Lungs
Blood
Summary of Biodistribution Profiles
3. Discussion
4. Experimental Section
4.1. Preparation of AuNPs and Dual-Radiolabeled AuNPs
4.2. Characterization of Dual-Radiolabeled AuNPs
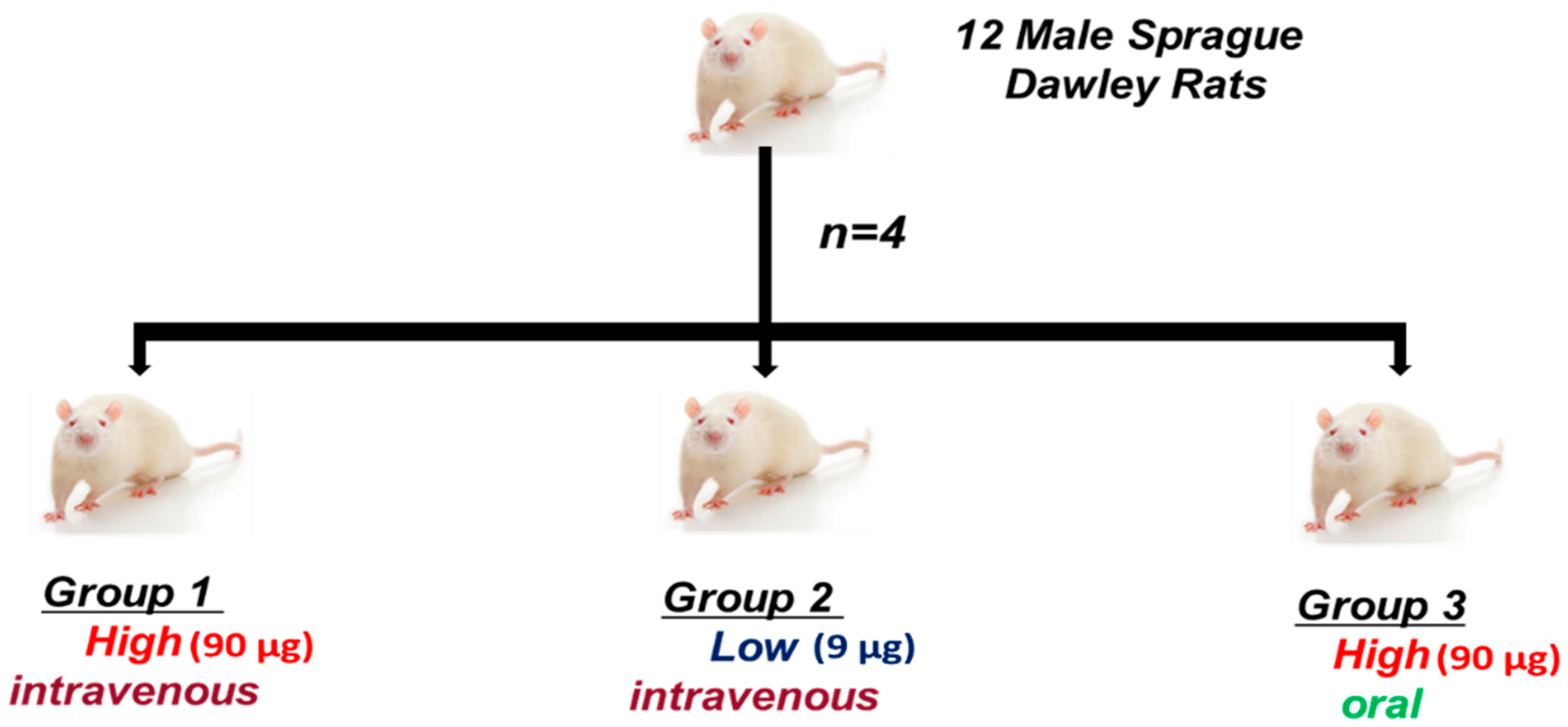
4.3. In Vivo Study
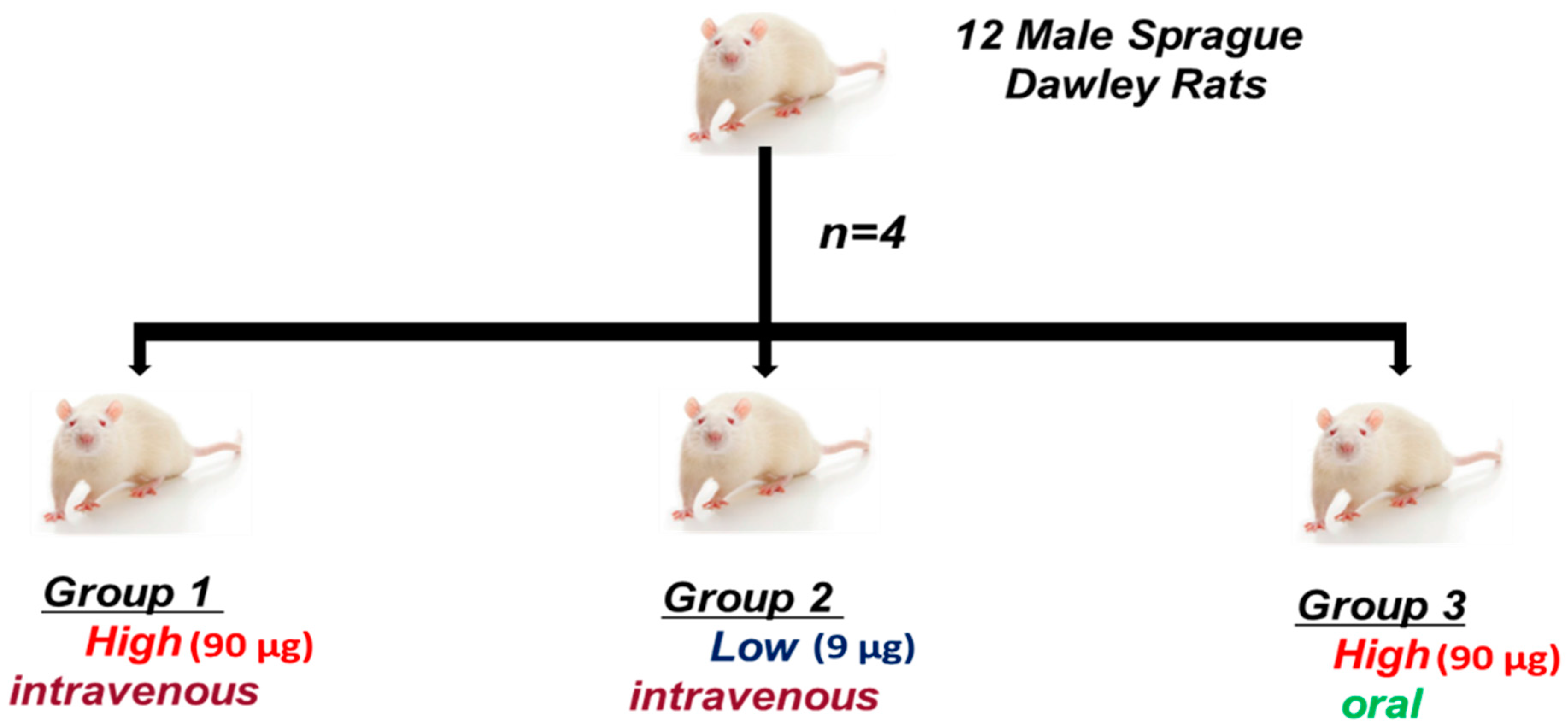
4.3.1. Animals
4.3.2. Experimental Design
4.4. Quantification of Gold and Citrate in Samples
4.4.1. Gold
4.4.2. Citrate
4.5. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Paciotti, G.F.; Myer, L.; Weinreich, D.; Goia, D.; Pavel, N.; McLaughlin, R.E.; Tamarkin, L. Colloidal gold: A novel nanoparticle vector for tumor directed drug delivery. Drug Deliv. 2004, 11, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Eustis, S.; El-Sayed, M.A. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem. Soc. Rev. 2006, 35, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Boisselier, E.; Astruc, D. Gold nanoparticles in nanomedicine: Preparations, imaging, diagnostics, therapies and toxicity. Chem. Soc. Rev. 2009, 38, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E.; Mwamuka, J.; Gole, A.; Murphy, C.J.; Wyatt, M.D. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small 2005, 1, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Papasani, M.R.; Wang, G.; Hill, R.A. Gold nanoparticles: The importance of physiological principles to devise strategies for targeted drug delivery. Nanomedicine 2012, 8, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Bajaj, A.; Mout, R.; Rotello, V.M. Monolayer coated gold nanoparticles for delivery applications. Adv. Drug Deliv. Rev. 2012, 64, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, X.; Liang, X.-J. Gold nanoparticles: Emerging paradigm for targeted drug delivery system. Biotechnol. Adv. 2013, 31, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Vigderman, L.; Zubarev, E.R. Therapeutic platforms based on gold nanoparticles and their covalent conjugates with drug molecules. Adv. Drug Deliv. Rev. 2013, 65, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.H.; Kim, S.J.; Kim, Y.H.; Noh, J.R.; Gang, G.T.; Chung, B.H.; Song, N.W.; Lee, C.H. Susceptibility to gold nanoparticle-induced hepatotoxicity is enhanced in a mouse model of nonalcoholic steatohepatitis. Toxicology 2012, 294, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.A.; Salleng, K.J.; Cliffel, D.E.; Feldheim, D.L. In vivo toxicity, biodistribution, and clearance of glutathione-coated gold nanoparticles. Nanomedicine 2013, 9, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Alkilany, A.; Murphy, C. Toxicity and cellular uptake of gold nanoparticles: What we have learned so far? J. Nanopart. Res. 2010, 12, 2313–2333. [Google Scholar] [CrossRef] [PubMed]
- Khlebtsov, N.; Dykman, L. Biodistribution and toxicity of engineered gold nanoparticles: A review of in vitro and in vivo studies. Chem. Soc. Rev. 2011, 40, 1647–1671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Wu, D.; Shen, X.; Liu, P.X.; Yang, N.; Zhao, B.; Zhang, H.; Sun, Y.M.; Zhang, L.A.; Fan, F.Y. Size-dependent in vivo toxicity of peg-coated gold nanoparticles. Int. J. Nanomed. 2011, 6, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.H.; Hagens, W.I.; Krystek, P.; Burger, M.C.; Sips, A.N.J.A.M.; Geertsma, R.E. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials 2008, 29, 1912–1919. [Google Scholar] [CrossRef] [PubMed]
- Sonavane, G.; Tomoda, K.; Makino, K. Biodistribution of colloidal gold nanoparticles after intravenous administration: Effect of particle size. Colloids Surf. B Biointerfaces 2008, 66, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.S.; Cho, M.; Jeong, J.; Choi, M.; Cho, H.Y.; Han, B.S.; Kim, S.H.; Kim, H.O.; Lim, Y.T.; Chung, B.H.; et al. Acute toxicity and pharmacokinetics of 13 nm-sized peg-coated gold nanoparticles. Toxicol. Appl. Pharmacol. 2009, 236, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Sadauskas, E.; Danscher, G.; Stoltenberg, M.; Vogel, U.; Larsen, A.; Wallin, H. Protracted elimination of gold nanoparticles from mouse liver. Nanomedicine 2009, 5, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.K.; Jittiwat, J.; Manikandan, J.; Ong, C.N.; Yu, L.E.; Ong, W.Y. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials 2010, 31, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.; Gonzalez-Romero, D.; Barria, M.A.; Olmedo, I.; Clos, A.; Urayama, A.; Sadagopa Ramanujam, V.M.; Vergara, L.; Kogan, M.J.; Soto, C. Bioaccumulation and toxicity of gold nanoparticles after repeated administration in mice. Biochem. Biophys. Res. Commun. 2010, 393, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Hirn, S.; Semmler-Behnke, M.; Schleh, C.; Wenk, A.; Lipka, J.; Schäffler, M.; Takenaka, S.; Möller, W.; Schmid, G.; Simon, U.; et al. Particle size-dependent and surface charge-dependent biodistribution of gold nanoparticles after intravenous administration. Eur. J. Pharm. Biopharm. 2011, 77, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Lipka, J.; Semmler-Behnke, M.; Sperling, R.A.; Wenk, A.; Takenaka, S.; Schleh, C.; Kissel, T.; Parak, W.J.; Kreyling, W.G. Biodistribution of peg-modified gold nanoparticles following intratracheal instillation and intravenous injection. Biomaterials 2010, 31, 6574–6581. [Google Scholar] [CrossRef] [PubMed]
- Semmler-Behnke, M.; Kreyling, W.G.; Lipka, J.; Fertsch, S.; Wenk, A.; Takenaka, S.; Schmid, G.; Brandau, W. Biodistribution of 1.4- and 18-nm gold particles in rats. Small 2008, 4, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Schleh, C.; Semmler-Behnke, M.; Lipka, J.; Wenk, A.; Hirn, S.; Schäffler, M.; Schmid, G.; Simon, U.; Kreyling, W.G. Size and surface charge of gold nanoparticles determine absorption across intestinal barriers and accumulation in secondary target organs after oral administration. Nanotoxicology 2012, 6, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Hillyer, J.F.; Albrecht, R.M. Gastrointestinal persorption and tissue distribution of differently sized colloidal gold nanoparticles. J. Pharm. Sci. 2001, 90, 1927–1936. [Google Scholar] [CrossRef] [PubMed]
- Balogh, L.; Nigavekar, S.S.; Nair, B.M.; Lesniak, W.; Zhang, C.; Sung, L.Y.; Kariapper, M.S.T.; El-Jawahri, A.; Llanes, M.; Bolton, B.; et al. Significant effect of size on the in vivo biodistribution of gold composite nanodevices in mouse tumor models. Nanomedicine 2007, 3, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Henriksen-Lacey, M.; Bramwell, V.; Perrie, Y. Radiolabelling of antigen and liposomes for vaccine biodistribution studies. Pharmaceutics 2010, 2, 91–104. [Google Scholar] [CrossRef]
- Black, K.C.L.; Akers, W.J.; Sudlow, G.; Xu, B.; Laforest, R.; Achilefu, S. Dual-radiolabeled nanoparticle spect probes for bioimaging. Nanoscale 2015, 7, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Freund, B.; Tromsdorf, U.I.; Bruns, O.T.; Heine, M.; Giemsa, A.; Bartelt, A.; Salmen, S.C.; Raabe, N.; Heeren, J.; Ittrich, H.; et al. A simple and widely applicable method to 59Fe-radiolabel monodisperse superparamagnetic iron oxide nanoparticles for in vivo quantification studies. ACS Nano 2012, 6, 7318–7325. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kumar, R.; Nagesha, D.; Duclos, R.I., Jr.; Sridhar, S.; Gatley, S.J. Integrity of 111In-radiolabeled superparamagnetic iron oxide nanoparticles in the mouse. Nucl. Med. Biol. 2015, 42, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.Y.; Gupta, V.K. Reversible aggregation of gold nanoparticles induced by pH dependent conformational transitions of a self-assembled polypeptide. J. Colloid Interface Sci. 2007, 316, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Simpson, C.A.; Kim, G.; Carter, C.J.; Feldheim, D.L. Gastrointestinal bioavailability of 2.0 nm diameter gold nanoparticles. ACS Nano 2013, 7, 3991–3996. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.; Usenko, C.; Hutchison, J.E.; Maddux, B.L.S.; Tanguay, R.L. In vivo biodistribution and toxicity depends on nanomaterial composition, size, surface functionalisation and route of exposure. J. Exp. Nanosci. 2008, 3, 195–206. [Google Scholar] [CrossRef]
- Terentyuk, G.S.; Maslyakova, G.N.; Suleymanova, L.V.; Khlebtsov, B.N.; Kogan, B.Y.; Tuchin, V.V.; Akchurin, G.G.; Shantrocha, A.V.; Maksimova, I.L.; Khlebtsov, N.G. Circulation and distribution of gold nanoparticles and induced alterations of tissue morphology at intravenous particle delivery. J. Biophotonics 2009, 2, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yang, Z.; Lu, W.; Zhang, R.; Huang, Q.; Tian, M.; Li, L.; Liang, D.; Li, C. Influence of anchoring ligands and particle size on the colloidal stability and in vivo biodistribution of polyethylene glycol-coated gold nanoparticles in tumor-xenografted mice. Biomaterials 2009, 30, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Casals, E.; Puntes, V.F. Inorganic nanoparticle biomolecular corona: Formation, evolution and biological impact. Nanomedicine 2012, 7, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.Y.; Setyawati, M.I.; Xie, J.; Parak, W.J.; Leong, D.T. Back to basics: Exploiting the innate physico-chemical characteristics of nanomaterials for biomedical applications. Adv. Funct. Mater. 2014, 24, 5936–5955. [Google Scholar] [CrossRef]
- Lynch, I.; Cedervall, T.; Lundqvist, M.; Cabaleiro-Lago, C.; Linse, S.; Dawson, K.A. The nanoparticle-protein complex as a biological entity; a complex fluids and surface science challenge for the 21st century. Adv. Colloid Interface Sci. 2007, 134–135, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Aggarwal, P.; Hall, J.B.; McNeil, S.E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharm. 2008, 5, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Patri, A.K.; Zheng, J.; Clogston, J.D.; Ayub, N.; Aggarwal, P.; Neun, B.W.; Hall, J.B.; McNeil, S.E. Interaction of colloidal gold nanoparticles with human blood: Effects on particle size and analysis of plasma protein binding profiles. Nanomedicine 2009, 5, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, P.; Hall, J.B.; McLeland, C.B.; Dobrovolskaia, M.A.; McNeil, S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv. Drug Deliv. Rev. 2009, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.E.; Madler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding biophysicochemical interactions at the nano-bio interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, M.P.; Walczyk, D.; Campbell, A.; Elia, G.; Lynch, I.; Baldelli Bombelli, F.; Dawson, K.A. Physical-chemical aspects of protein corona: Relevance to in vitro and in vivo biological impacts of nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Hung, Y.C.; Liau, I.; Huang, G. Assessment of the in vivo toxicity of gold nanoparticles. Nanoscale Res. Lett. 2009, 4, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Katti, K.; Kannan, R.; Katti, K.; Kattumori, V.; Pandrapraganda, R.; Rahing, V.; Cutler, C.; Boote, E.; Casteel, S.; Smith, C.; et al. Hybrid gold nanoparticles in molecular imaging and radiotherapy. Czechoslov. J. Phys. 2006, 56, D23–D34. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. The formation of colloidal gold. J. Phys. Chem. 1953, 57, 670–673. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV-Vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rambanapasi, C.; Barnard, N.; Grobler, A.; Buntting, H.; Sonopo, M.; Jansen, D.; Jordaan, A.; Steyn, H.; Zeevaart, J.R. Dual Radiolabeling as a Technique to Track Nanocarriers: The Case of Gold Nanoparticles. Molecules 2015, 20, 12863-12879. https://doi.org/10.3390/molecules200712863
Rambanapasi C, Barnard N, Grobler A, Buntting H, Sonopo M, Jansen D, Jordaan A, Steyn H, Zeevaart JR. Dual Radiolabeling as a Technique to Track Nanocarriers: The Case of Gold Nanoparticles. Molecules. 2015; 20(7):12863-12879. https://doi.org/10.3390/molecules200712863
Chicago/Turabian StyleRambanapasi, Clinton, Nicola Barnard, Anne Grobler, Hylton Buntting, Molahlehi Sonopo, David Jansen, Anine Jordaan, Hendrik Steyn, and Jan Rijn Zeevaart. 2015. "Dual Radiolabeling as a Technique to Track Nanocarriers: The Case of Gold Nanoparticles" Molecules 20, no. 7: 12863-12879. https://doi.org/10.3390/molecules200712863
APA StyleRambanapasi, C., Barnard, N., Grobler, A., Buntting, H., Sonopo, M., Jansen, D., Jordaan, A., Steyn, H., & Zeevaart, J. R. (2015). Dual Radiolabeling as a Technique to Track Nanocarriers: The Case of Gold Nanoparticles. Molecules, 20(7), 12863-12879. https://doi.org/10.3390/molecules200712863