
Three-Dimensional Compound Comparison Methods and Their Application in Drug Discovery



Abstract
:1. Introduction
2. 3D Shape-Based Compound Descriptors
2.1. Atomic Distance-Based Methods
2.2. Gaussian Function-Based Molecular Shape Description Methods
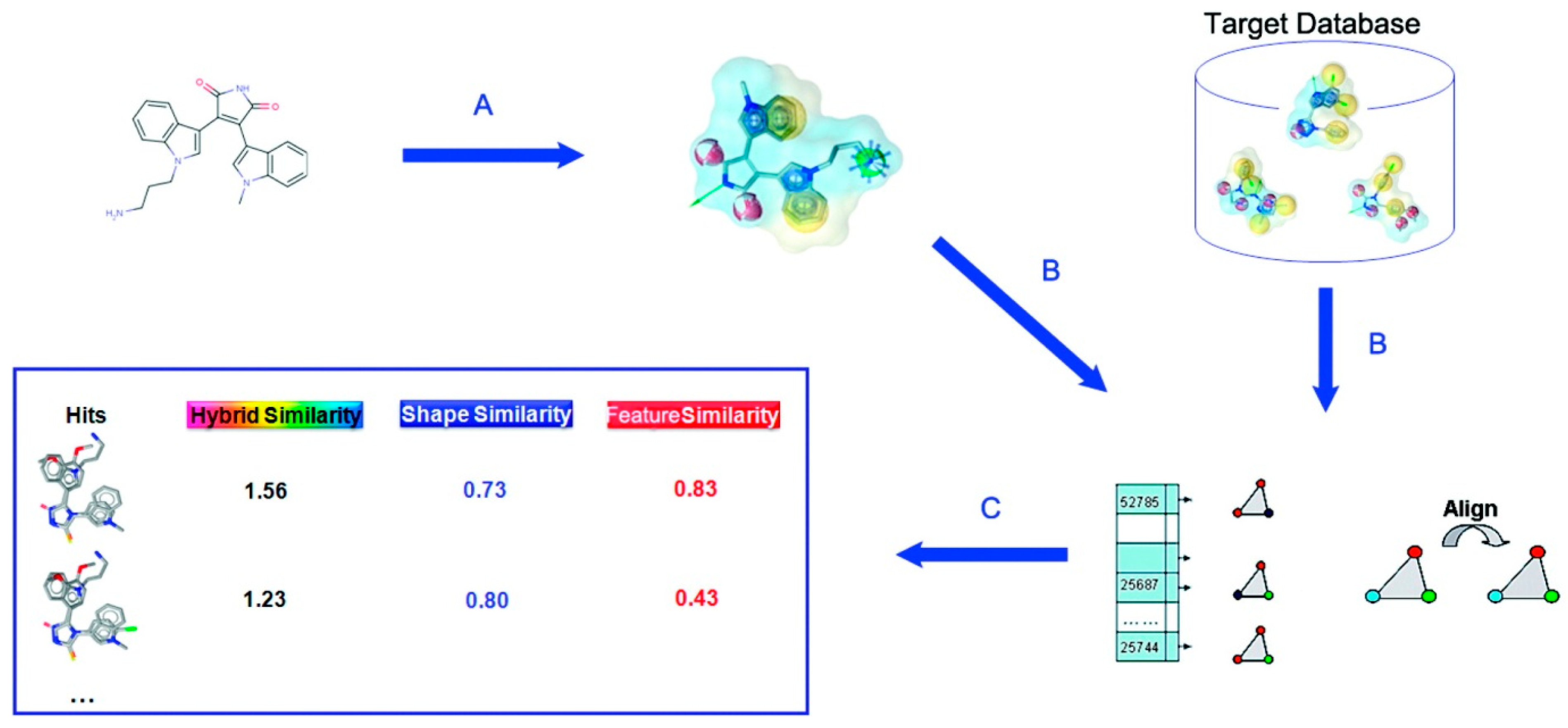
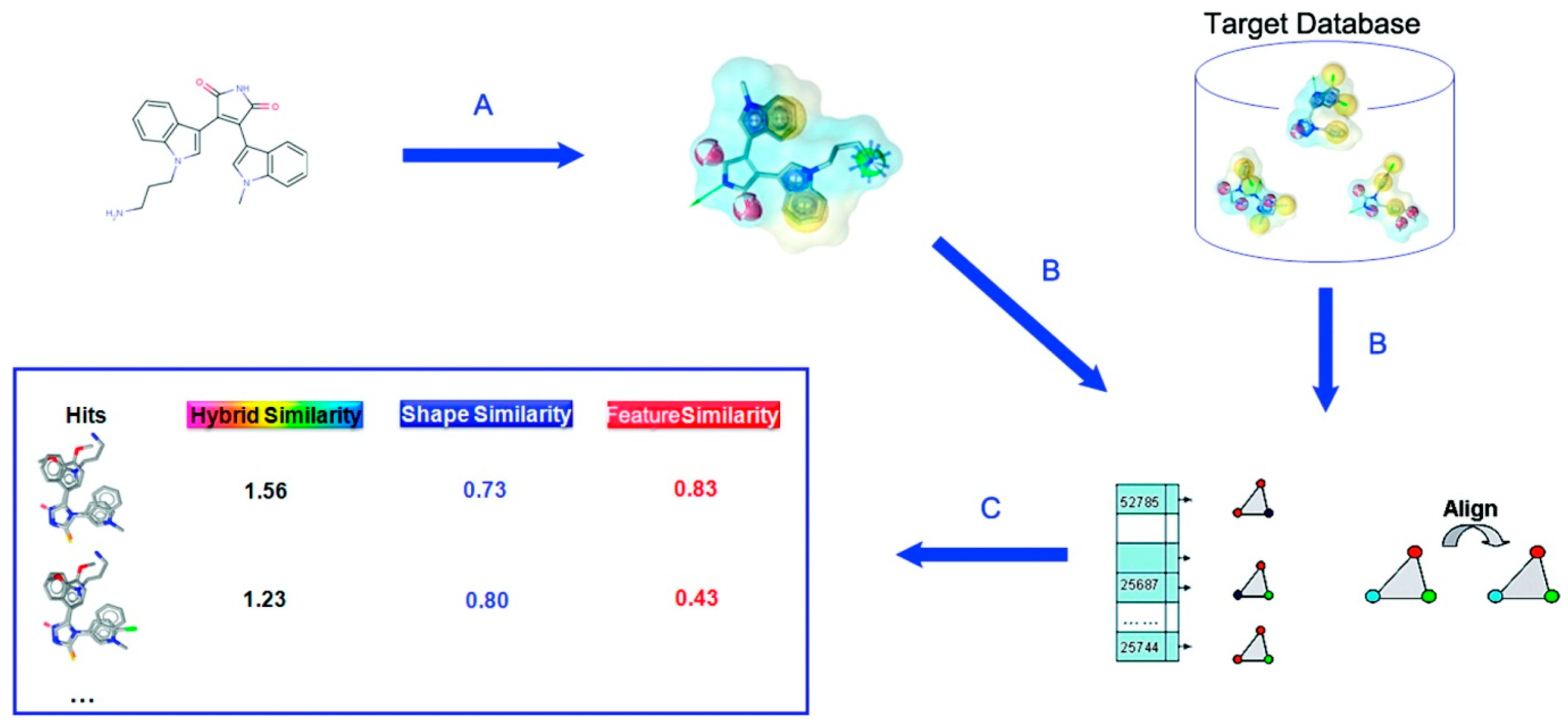
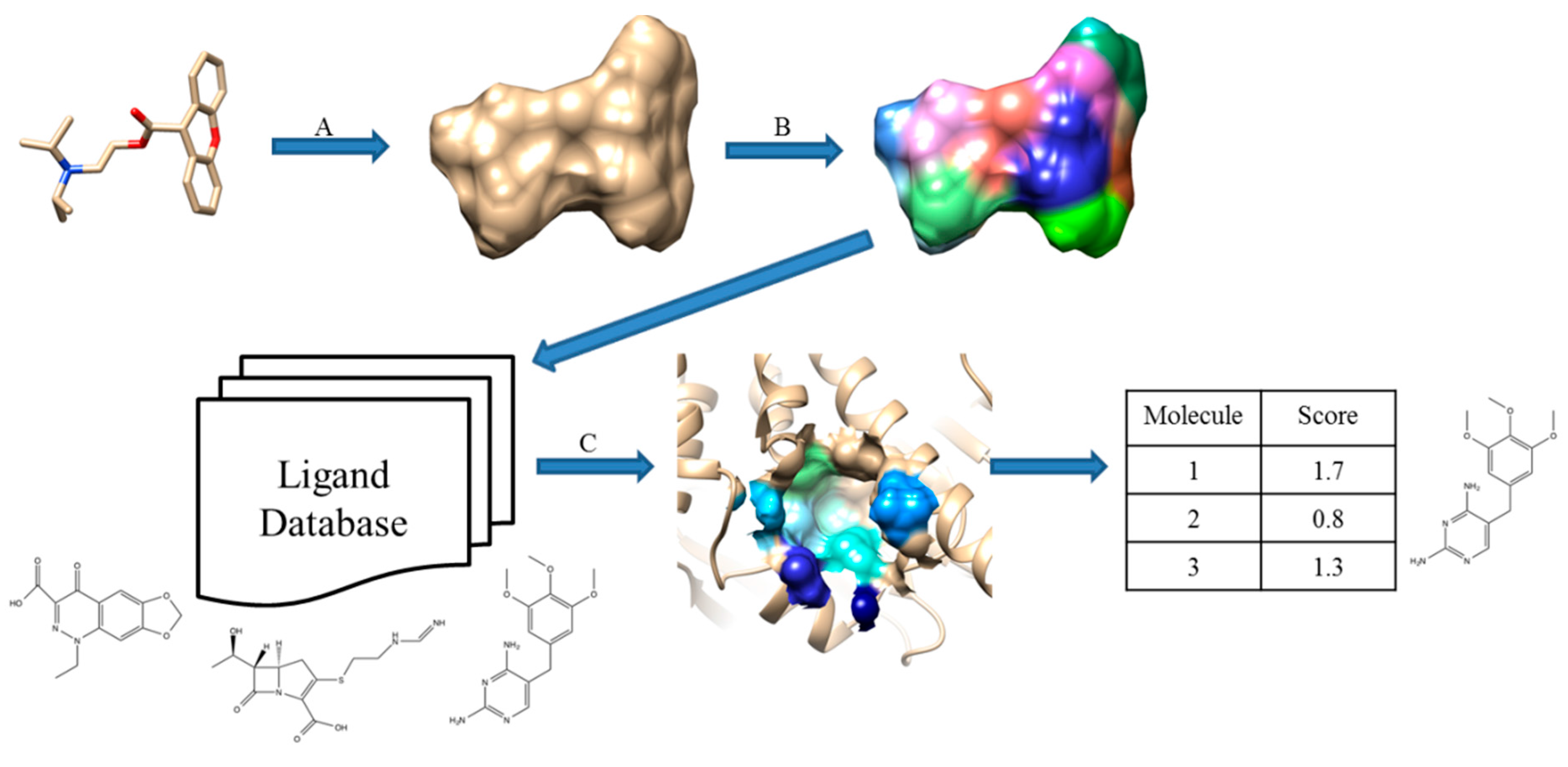
2.3. Surface-Based Molecular Shape Description
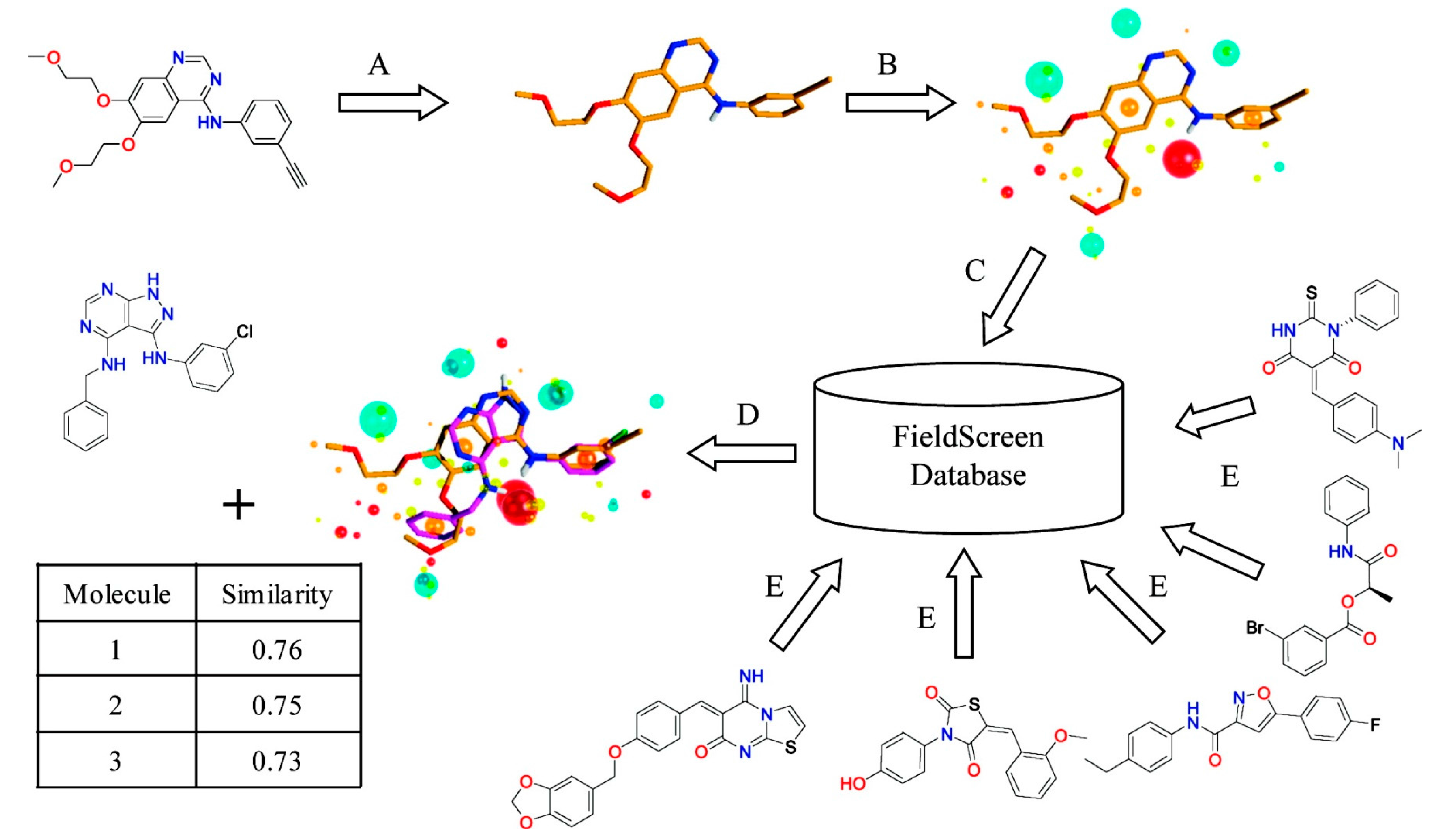
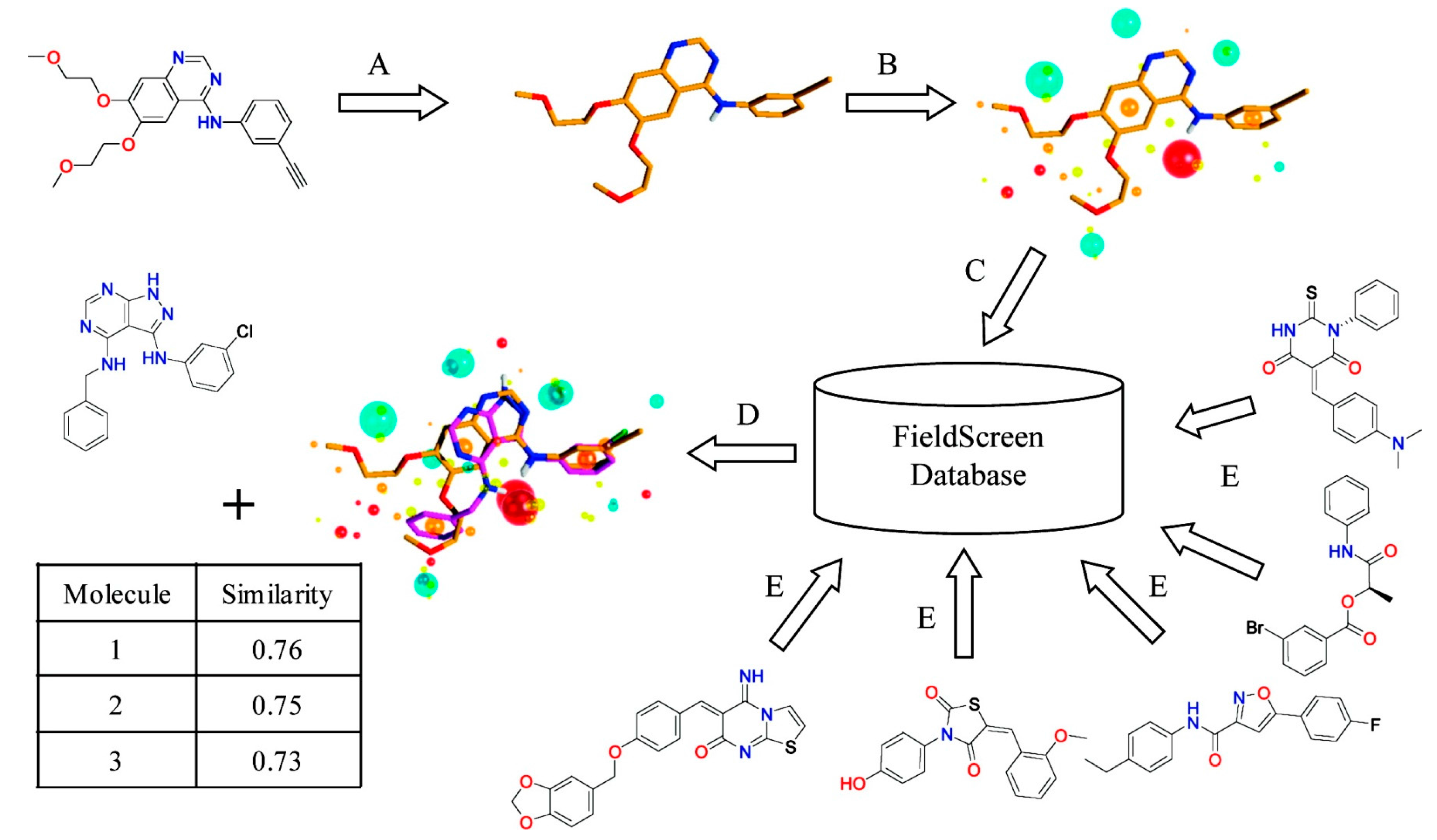
2.4. Field-Based Methods
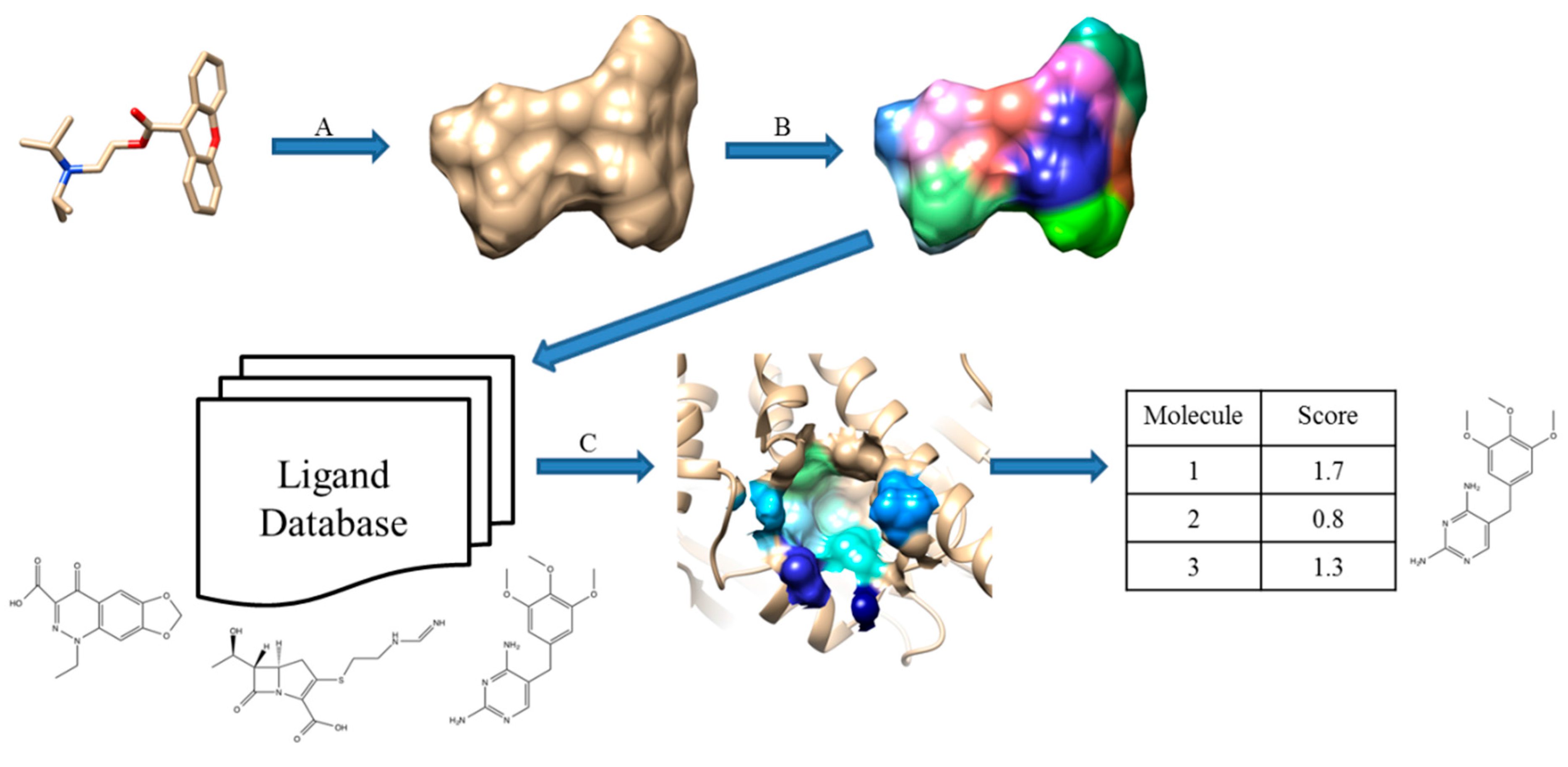
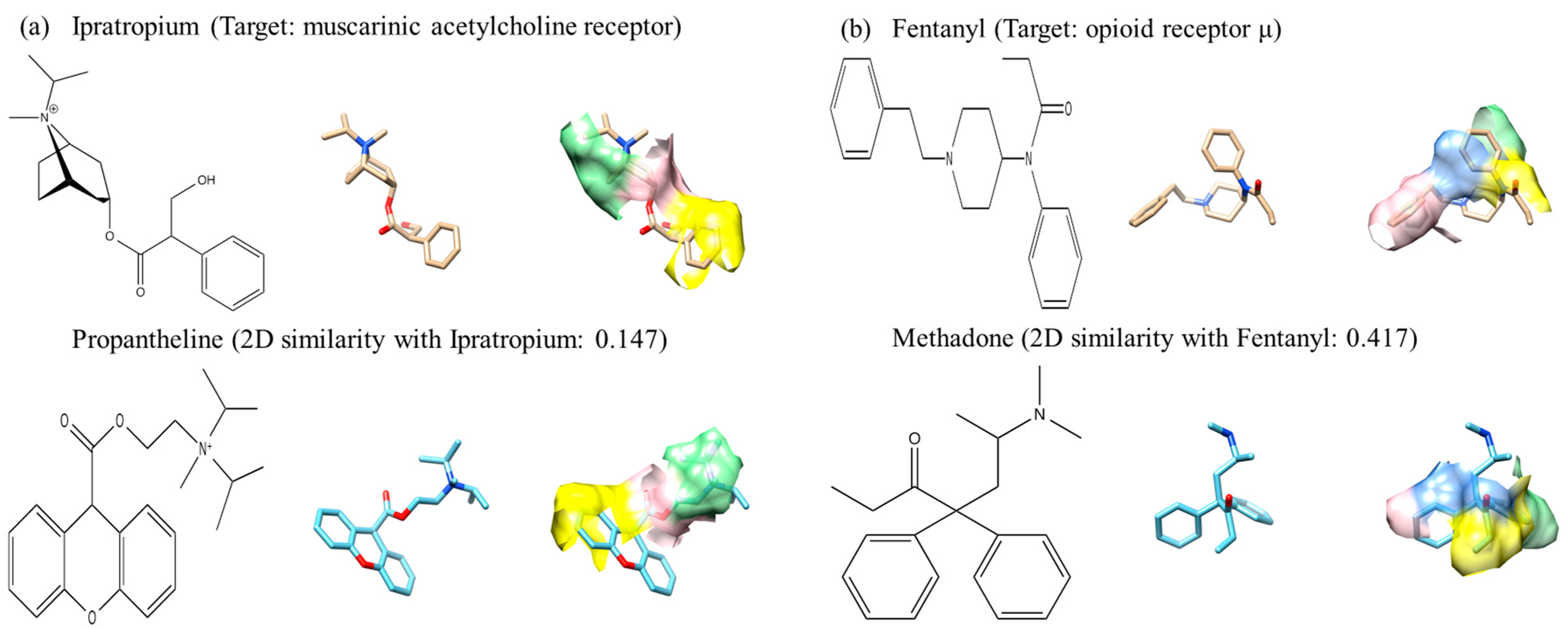
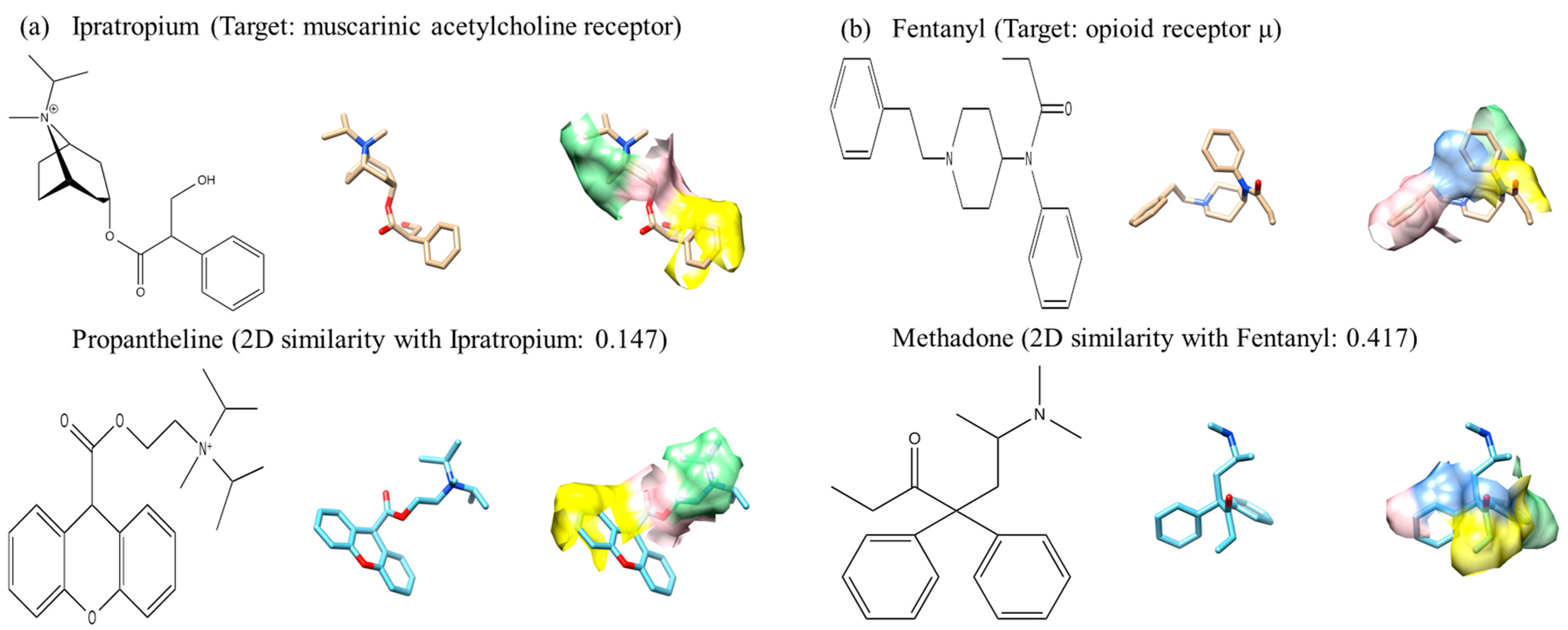
2.5. Pharmacophore-Based Methods
3. Benchmark Study
3.1. Benchmark Set
3.2. OMEGA
3.3. Programs Benchmarked
4. Results and Discussion
4.1. Overall Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EF2% | EF5% | EF10% | AUC | |
|---|---|---|---|---|
| 50 Conformations | ||||
| USR | 10.0 | 6.2 | 4.1 | 0.76 |
| GZD | 13.4 | 8.0 | 5.3 | 0.81 |
| PS | 10.7 | 6.6 | 4.9 | 0.78 |
| ROCS | 20.1 | 10.7 | 6.2 | 0.83 |
| 10 Conformations | ||||
| USR | 9.6 | 6.3 | 4.1 | 0.75 |
| GZD | 13.5 | 7.9 | 5.0 | 0.78 |
| PS | 10.6 | 6.5 | 4.9 | 0.78 |
| ROCS | 18.8 | 9.7 | 6.0 | 0.81 |
| 5 Conformations | ||||
| USR | 9.6 | 6.1 | 4.1 | 0.75 |
| GZD | 12.9 | 7.3 | 4.9 | 0.75 |
| PS | 10.3 | 6.5 | 4.8 | 0.77 |
| ROCS | 18.2 | 9.4 | 5.9 | 0.80 |
| 1 Conformation | ||||
| USR | 8.8 | 5.8 | 4.0 | 0.70 |
| GZD | 12.1 | 7.4 | 4.9 | 0.75 |
| PS | 10.3 | 6.4 | 4.7 | 0.77 |
| ROCS | 15.9 | 8.5 | 5.6 | 0.79 |
| ROCS | GZD | PS | |
|---|---|---|---|
| GZD | 2.319 | - | - |
| PS | 3.544 | 1.118 | - |
| USR | 3.750 | 1.403 | 0.360 |
4.2. Change of Performance after Removing Similar Compounds
| EF2% | EF5% | EF10% | AUC | |
|---|---|---|---|---|
| All Active Compounds | ||||
| USR | 10.0 | 6.2 | 4.1 | 0.76 |
| GZD | 13.4 | 8.0 | 5.3 | 0.81 |
| PS | 10.7 | 6.6 | 4.9 | 0.78 |
| ROCS | 20.1 | 10.7 | 6.2 | 0.83 |
| Similarity < 0.75 | ||||
| USR | 5.3 | 4.7 | 3.4 | 0.721 |
| GZD | 8.5 | 6.4 | 4.7 | 0.775 |
| PS | 8.2 | 5.2 | 4.2 | 0.758 |
| ROCS | 15.6 | 9.4 | 5.6 | 0.801 |
| Similarity < 0.66 | ||||
| USR | 5.9 | 3.9 | 3.0 | 0.652 |
| GZD | 7.5 | 5.6 | 4.4 | 0.740 |
| PS | 7.9 | 4.8 | 3.9 | 0.736 |
| ROCS | 13.6 | 8.6 | 5.3 | 0.764 |
| Similarity < 0.50 | ||||
| USR | 3.5 | 3.0 | 2.0 | 0.621 |
| GZD | 6.0 | 4.3 | 3.5 | 0.719 |
| PS | 6.2 | 4.2 | 3.5 | 0.710 |
| ROCS | 10.1 | 7.9 | 4.3 | 0.727 |
4.3. Consensus Methods
| Combined Programs | 2% | 5% | 10% |
|---|---|---|---|
| USR + GZD | 13.7 | 7.7 | 4.7 |
| USR + PS | 13.1 | 7.9 | 5.0 |
| USR + ROCS | 17.1 | 9.1 | 5.4 |
| GZD + PS | 16.0 | 9.1 | 5.9 |
| GZD + ROCS | 20.3 | 10.8 | 5.3 |
| PS + ROCS | 20.5 | 10.7 | 6.4 |
| Combined Programs | Single Program | t-Value | Single Program | t-Value |
|---|---|---|---|---|
| USR + GZD | USR | 1.414 | GZD | 0.150 |
| USR + PS | USR | 1.373 | PS | 1.369 |
| USR + ROCS | USR | 2.489 | ROCS | 1.409 |
| GZD + PS | GZD | 1.409 | PS | 2.014 |
| GZD + ROCS | GZD | 2.402 | ROCS | 0.137 |
| PS + ROCS | PS | 3.547 | ROCS | 0.452 |
4.4. Computational Speed Comparison
| Programs | Time (s) |
|---|---|
| USR | 2.1 |
| GZD | 2.3 |
| PS | 4.4 |
| ROCS | 5.1 |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cavasotto, C.N.; Orry, A.J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Lemmen, C.; Lengauer, T. Computational methods for the structural alignment of molecules. J. Comput. -Aided Mol. Des. 2000, 14, 215–232. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking: Current status and future challenges. Proteins 2006, 65, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.; Awale, M.; Reymond, J.L. SMIfp (SMILES fingerprint) Chemical Space for Virtual Screening and Visualization of Large Databases of Organic Molecules. J. Chem. Inf. Model. 2013, 53, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Barnard, J.M.; Downs, G.M. Chemical Fragment Generation and Clustering Software. J. Chem. Inf. Comput. Sci. 1997, 37, 141–142. [Google Scholar] [CrossRef]
- Raymond, J.W.; Gardiner, E.J.; Willett, P. RASCAL: Calculation of graph similarity using maximum common edge subgraphs. Comput. J. 2002, 45, 631–644. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Glen, R.C. Similarity Searching of Chemical Databases Using Atom Environment Descriptors (MOLPRINT2D): Evaluation of Performance. J. Chem. Inf. Comput. Sci. 2004, 44, 1708–1718. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Okuno, Y.; Goto, S.; Kanehisa, M. Development of a Chemical Structure Comparison Method for Integrated Analysis of Chemical and Genomic Information in the Metabolic Pathways. J. Am. Chem. Soc. 2003, 125, 11853–11865. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Skillman, A.G.; Nicholls, A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Richards, W.G. Ultrafast Shape Recognition to Search Compound Databases for Similar Molecular Shapes. J. Comput. Chem. 2007, 28, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhu, X.; Monroe, L.; Bures, M.G.; Kihara, D. PL-PatchSurfer: A Novel Molecular Local Surface-Based Method for Exploring Protein-Ligand Interactions. Int. J. Mol. Sci. 2014, 15, 15122–15145. [Google Scholar] [CrossRef] [PubMed]
- Cheeseright, T.J.; Mackey, M.D.; Melville, J.L.; Vinter, J.G. FieldScreen: Virtual Screening Using Molecular Fields. Application to DUD Data Set. J. Chem. Inf. Model. 2008, 48, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Mavridis, L.; Hudson, B.D.; Ritchie, D.W. Toward High Throughput 3D Virtual Screening Using Spherical Harmonic Surface Representations. J. Chem. Inf. Model. 2007, 47, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R.; Gillet, V.J. An Introduction to Chemoinformatics, Revised ed.; Springer: Dordrecht, The Netherlands, 2007; p. 27. [Google Scholar]
- Cleves, A.E.; Jain, A.N. Robust Ligand-Based Modeling of the Biological Targets of Known Drugs. J. Med. Chem. 2006, 49, 2921–2938. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Richards, W.G. Ultrafast shape recognition for similarity search in molecular database. Proc. R. Soc. A. 2007, 463, 1307–1321. [Google Scholar] [CrossRef]
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Cheminform. 2012, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.A.; Gallardo, M.A.; Pickup, B.T. A Fast Method of Molecular Shape Comparison: A Simple Application of a Gaussian Description of Molecular Shape. J. Comput. Chem. 1996, 17, 1653–1666. [Google Scholar] [CrossRef]
- Mills, J.E.J.; Dean, P.M. Three-dimensional hydrogen-bond geometry and probability information from a crystal survey. J. Comput. -Aided Mol. Des. 1996, 10, 607–622. [Google Scholar] [CrossRef] [PubMed]
- Vaz de Lima, L.A.C.; Nascimento, A.S. MolShaCS: A free and open source tool for ligand similarity identification based on Gaussian descriptors. Eur. J. Med. Chem. 2013, 50, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Vainio, M.J.; Puranen, J.S.; Johnson, M.S. ShaEP: Molecular Overlay Based on Shape and Electrostatic Potential. J. Chem. Inf. Model. 2009, 49, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, H.; Li, H. SHAFTS: A Hybrid Approach for 3D Molecular Similarity Calculation. 1. Method and Assessment of Virtual Screening. J. Chem. Inf. Model. 2011, 51, 2372–2385. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular Complexity and Its Impact on the Probability of Finding Leads for Drug Discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surface. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Bender, A.; Mussa, H.Y.; Gill, G.S.; Glen, R.C. Molecular Surface Point Environment for Virtual Screening and the Elucidation of Binding Patterns (MOLPRINT3D). J. Med. Chem. 2004, 47, 6569–6583. [Google Scholar] [CrossRef] [PubMed]
- Goodford, P.J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Reid, D.; Sadjad, B.S.; Zsoldos, Z.; Simon, A. LASSO-ligand activity by similarity order: A new tool for ligand based virtual screening. J. Comput. -Aided Mol. Des. 2008, 22, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking Sets for Molecular Docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]
- Aurenhammer, F. Voronoi Diagrams—A Survey of a Fundamental Geometric Data Structure. ACM Comput. Surv. 1991, 23, 345–405. [Google Scholar] [CrossRef]
- Wilson, J.A.; Bender, A.; Kaya, T.; Clemons, P.A. Alpha Shapes Applied to Molecular Shape Characterization Exhibit Novel Properties Compared to Established Shape Descriptors. J. Chem. Inf. Model. 2009, 49, 2231–2241. [Google Scholar] [CrossRef] [PubMed]
- Rubner, Y.; Guibas, L.J.; Tomasi, C. The earth mover’s distance as a metric for image retrieval. Int. J. Comput. Vis. 2000, 40, 99–121. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, C.M.; Won, C.I.; Kim, J-K.; Ryu, J.; Cho, Y.; Lee, C.; Bhak, J. BetaDock: Shape-Priority Docking Method Based on Beta-Complex. J. Biomol. Struct. Dyn. 2011, 29, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Seo, J.; Kim, D.; Ryu, J.; Cho, C.H. Three-dimensional beta shapes. Comput. Aided Des. 2006, 38, 1179–1191. [Google Scholar] [CrossRef]
- Kihara, D.; Lee, S.; Chikhi, R.; Esquivel-Rodriguez, J. Molecular surface representation using 3D Zernike descriptors for protein shape comparison and docking. Curr. Protein Pept. Sci. 2011, 12, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, V.; Lee, S.; Kihara, D. Potential for protein surface shape analysis using spherical harmonics and 3D Zernike descriptors. Cell Biochem. Biophys. 2009, 54, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.L. Analytical molecular surface calculation. J. Appl. Cryst. 1983, 16, 548–558. [Google Scholar] [CrossRef]
- Venkatraman, V.; Chakravarthy, P.; Kihara, D. Application of 3D Zernike descriptors to shape-based ligand similarity searching. J. Cheminform. 2009, 1, 19. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kihara, D. Detecting local ligand-binding site similarity in nonhomologous proteins by surface patch comparison. Proteins 2012, 80, 1177–1195. [Google Scholar]
- Zhu, X.; Xiong, Y.; Kihara, D. Large-scale binding ligand prediction by improved patch-based method Patch-Surfer2.0. Bioinformatics 2015, 31, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Demange, G.; Gale, D.; Sotomayor, M. Multi-item auctions. J. Polit. Econ. 1986, 94, 863–872. [Google Scholar] [CrossRef]
- Cheersight, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular Field Extrema as Descriptors of Biological Activity: Definition and Validation. J. Chem. Inf. Model. 2006, 46, 605–676. [Google Scholar]
- Tervo, A.J.; Ronkko, T.; Nyronen, T.H.; Poso, A. BRUTUS: Optimization of a Grid-Based Similarity Function for Rigid-Body Molecular Superposition. 1. Alignment and Virtual Screening Applications. J. Med. Chem. 2005, 48, 4076–4086. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Parretti, M.F.; Kromer, R.T.; Rothman, J.H.; Richards, W.G. Alignment of Molecules by the Monte Carlo Optimization of Molecular Similarity Indices. J. Comput. Chem. 1997, 18, 1344–1353. [Google Scholar] [CrossRef]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.; Baroni, M.; Carosati, E.; Benedetti, P.; Clementi, S. FLAP: GRID Molecular Interaction Fields in Virtual Screening. Validation using the DUD Data Set. J. Chem. Inf. Model. 2010, 50, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Abrahamian, E.; Fox, P.C.; Naerum, L.; Christensen, I.T.; Thøgersen, H.; Clark, R.D. Efficient Generation, Storage, and Manipulation of Fully Flexible Pharmacophore Multiplets and Their Use in 3-D Similarity Searching. J. Chem. Inf. Sci. 2003, 43, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Fox, P.C.; Wolohan, P.R.N.; Abrahamian, E.; Clark, R.D. Parameterization and Conformational Sampling Effects in Pharmacophore Multiplet Searching. J. Chem. Inf. Model. 2008, 48, 2326–2334. [Google Scholar] [CrossRef] [PubMed]
- Qing, X.; Lee, X.Y.; de Raeymaeker, J.; Tame, J.R.H.; Zhang, K.Y.J.; de Maeyer, M.; Voet, R.A.D. Pharmacophore modeling: Advances, limitations, and current utility in drug discovery. J. Recept. Ligand Channel Res. 2014, 7, 81–92. [Google Scholar]
- De Luca, L.; Barreca, M.L.; Ferro, S.; Christ, F.; Iraci, N.; Gitto, R.; Monforte, A.M.; Debyser, Z.; Chimirri, A. Pharmacophore-based discovery of small-molecule inhibitors of protein-protein interactions between HIV-1 integrase and cellular cofactor LEDGF/p75. ChemMedChem 2009, 4, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; van der Veken, N.J.; van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem. 2000, 43, 4759–4767. [Google Scholar] [CrossRef] [PubMed]
- Howkins, P.C.; Nicholis, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Kim, J.K.; Kim, D.S.; Seok, C. GalaxyDock2: Protein-Ligand Docking Using Beta-Complex and Global Optimization. J. Comput. Chem. 2013, 34, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.; Hessler, G.; Matter, H.; Klabunde, T. Virtual Screening of Biogenic Amine-Binding G-Protein Coupled Receptors: Comparative Evaluation of Protein- and Ligand-Based Virtual Screening Protocols. J. Med. Chem. 2005, 48, 5448–5465. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Springael, J.-Y.; Parmentier, M.; Hachet-Haas, M.; Galzi, J.-L.; Rognan, D. Identification of Nonpeptide CCR5 Receptor Agonists by Structure-based Virtual Screening. J. Med. Chem. 2007, 50, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Compton, J.R.; AbdulHameed, M.D.M.; Marchand, C.L.; Robertson, K.L.; Leary, D.H.; Jadhav, A.; Hershfield, J.R.; Wallqvist, A.; Friedlander, A.M.; et al. 3-Substituted Indole Inhibitors against Francisella tularensis FabI Identified by Structure-based Virtual Screening. J. Med. Chem. 2013, 56, 5275–5287. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Yu, H.; Li, Y.; Li, P.; Pan, P.; Zhou, S.; Zhang, L.; Li, S.; Lee, S.M.-Y.; Hou, T. Discovery of Rho-Kinase inhibitors by docking-based virtual screening. Mol. BioSyst. 2013, 9, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Truchon, J.F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A. Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 2002, 6, 447–452. [Google Scholar] [CrossRef]
- Venkatraman, V.; Perez-Nueno, V.I.; Mavridis, L.; Ritchie, D.W. Comprehensive Comparison of Ligand-Based Virtual Screening Tools Against the DUD Data set Reveals Limitations of 3D Methods. J. Chem. Inf. Model. 2010, 50, 2079–2093. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: The original compound library is available from jainlab.org.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, W.-H.; Zhu, X.; Bures, M.G.; Kihara, D. Three-Dimensional Compound Comparison Methods and Their Application in Drug Discovery. Molecules 2015, 20, 12841-12862. https://doi.org/10.3390/molecules200712841
Shin W-H, Zhu X, Bures MG, Kihara D. Three-Dimensional Compound Comparison Methods and Their Application in Drug Discovery. Molecules. 2015; 20(7):12841-12862. https://doi.org/10.3390/molecules200712841
Chicago/Turabian StyleShin, Woong-Hee, Xiaolei Zhu, Mark Gregory Bures, and Daisuke Kihara. 2015. "Three-Dimensional Compound Comparison Methods and Their Application in Drug Discovery" Molecules 20, no. 7: 12841-12862. https://doi.org/10.3390/molecules200712841
APA StyleShin, W.-H., Zhu, X., Bures, M. G., & Kihara, D. (2015). Three-Dimensional Compound Comparison Methods and Their Application in Drug Discovery. Molecules, 20(7), 12841-12862. https://doi.org/10.3390/molecules200712841