
CAPE Analogs Induce Growth Arrest and Apoptosis in Breast Cancer Cells

 and
and 
Abstract
:
1. Introduction
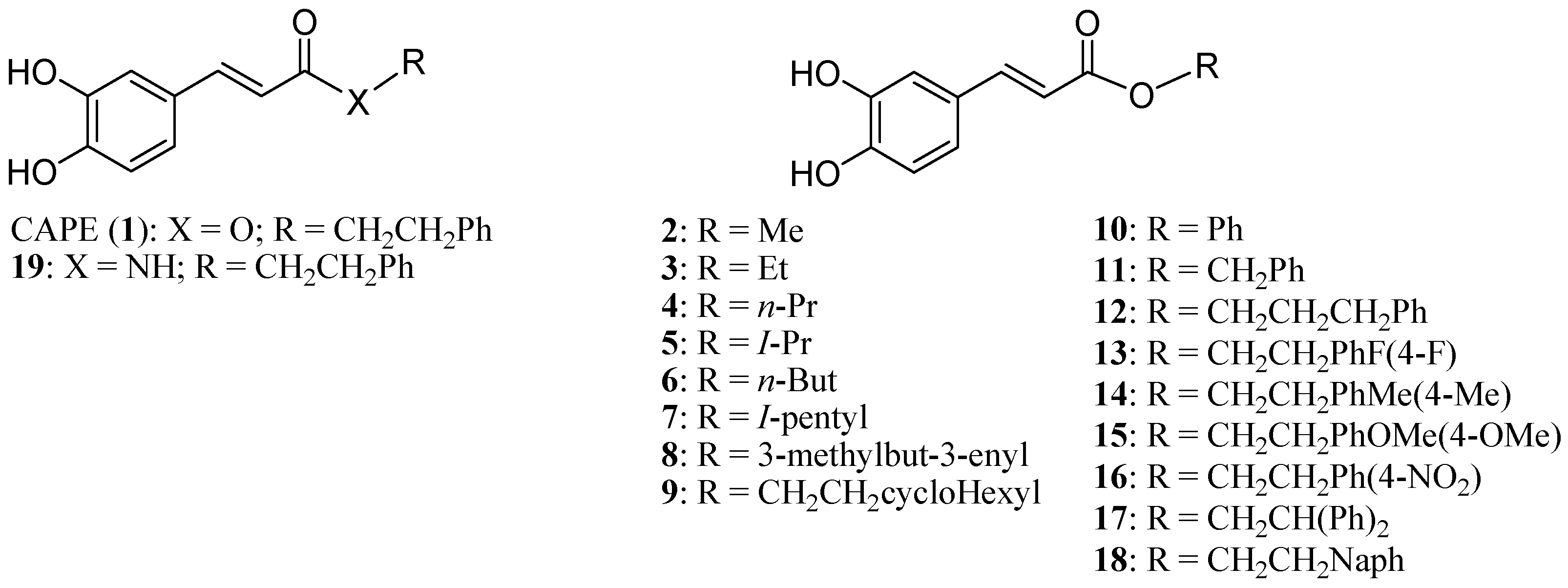
2. Results and Discussion
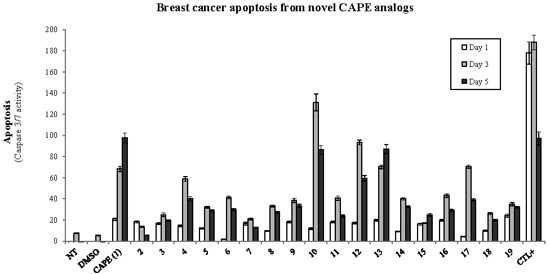
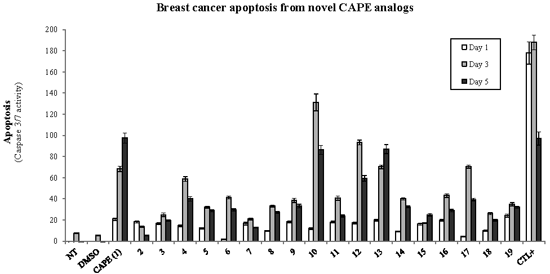
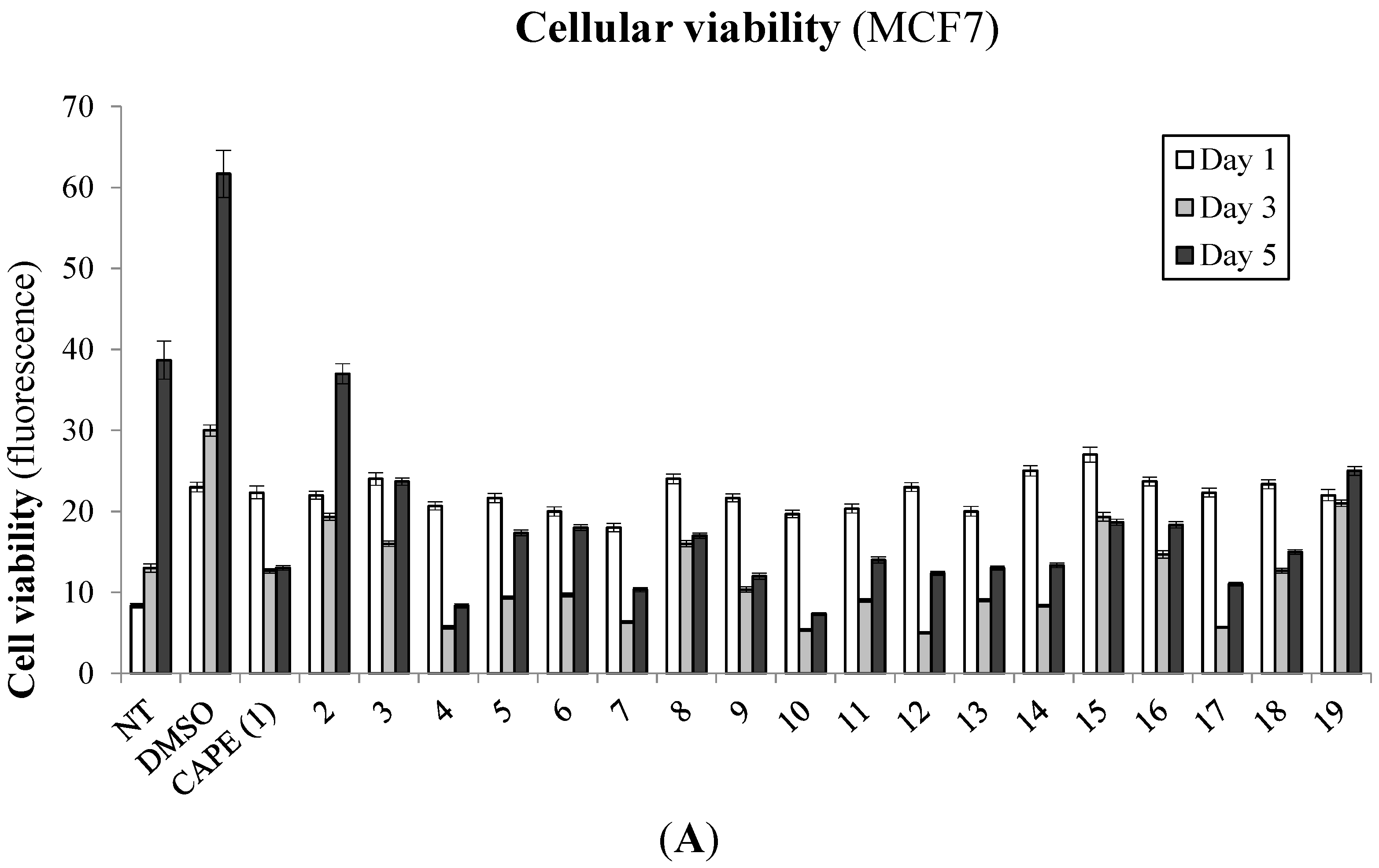
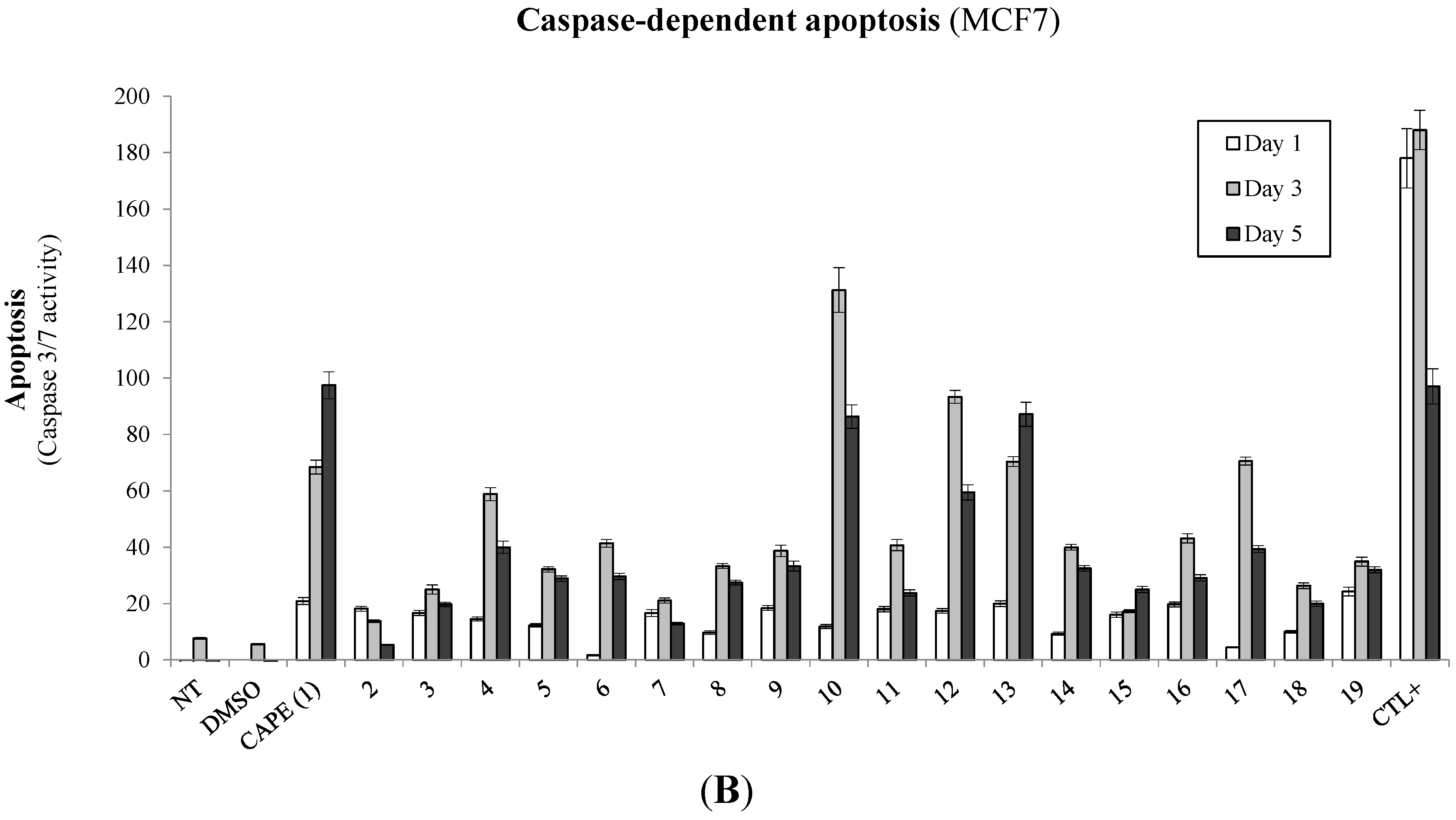
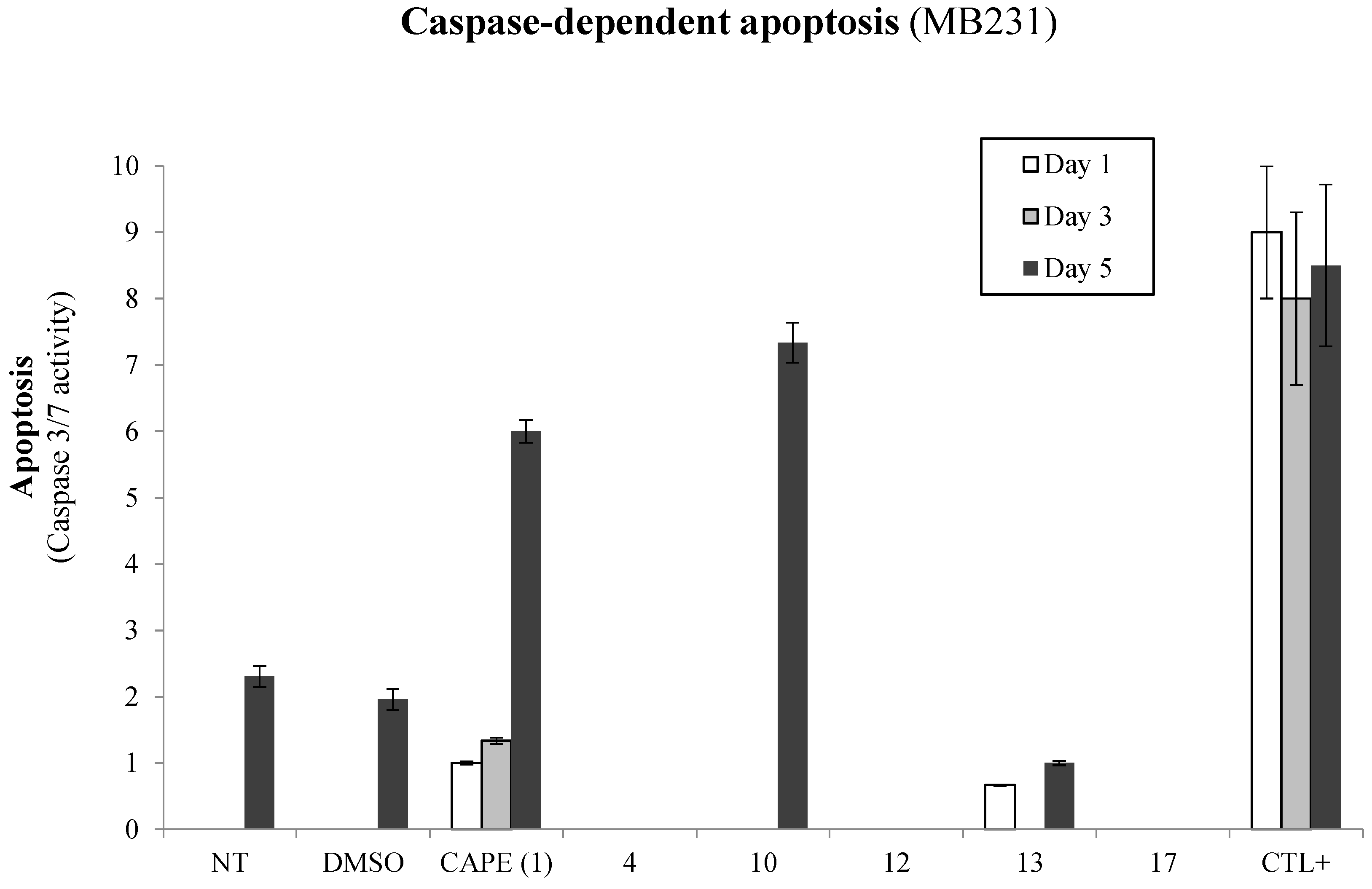
2.1. Biological Evaluation of CAPE Analogs on Breast Cancer Cells
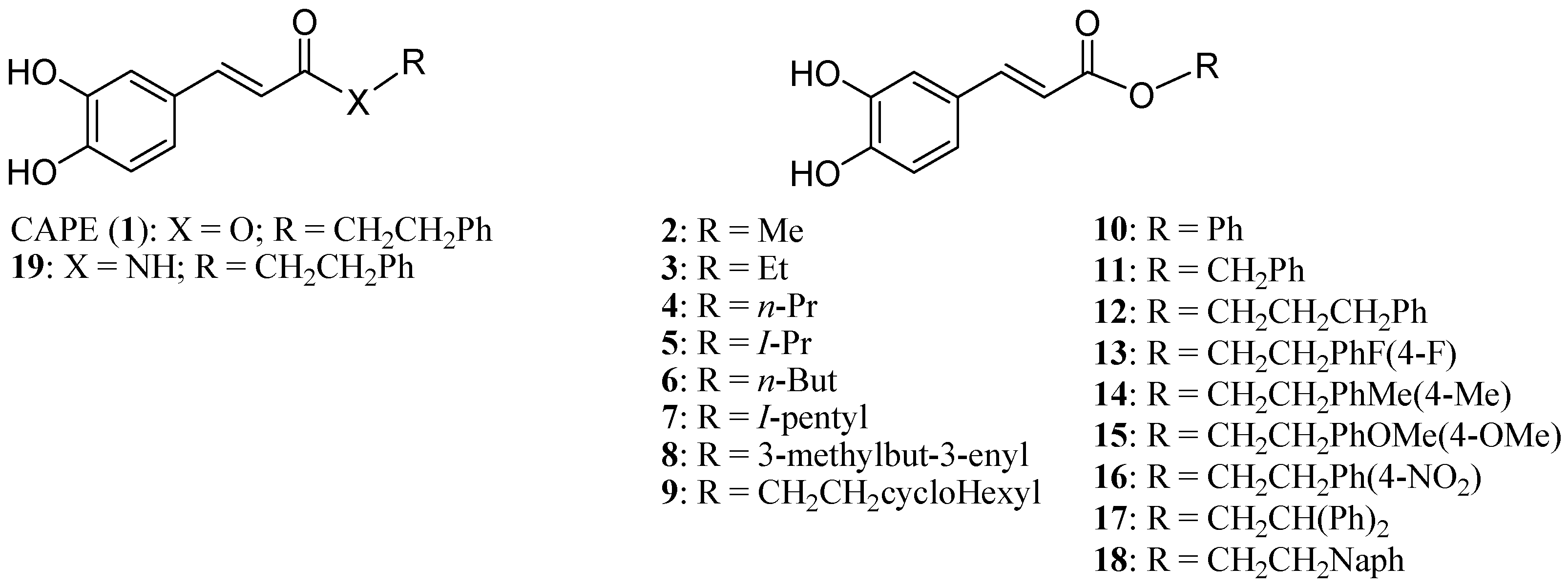
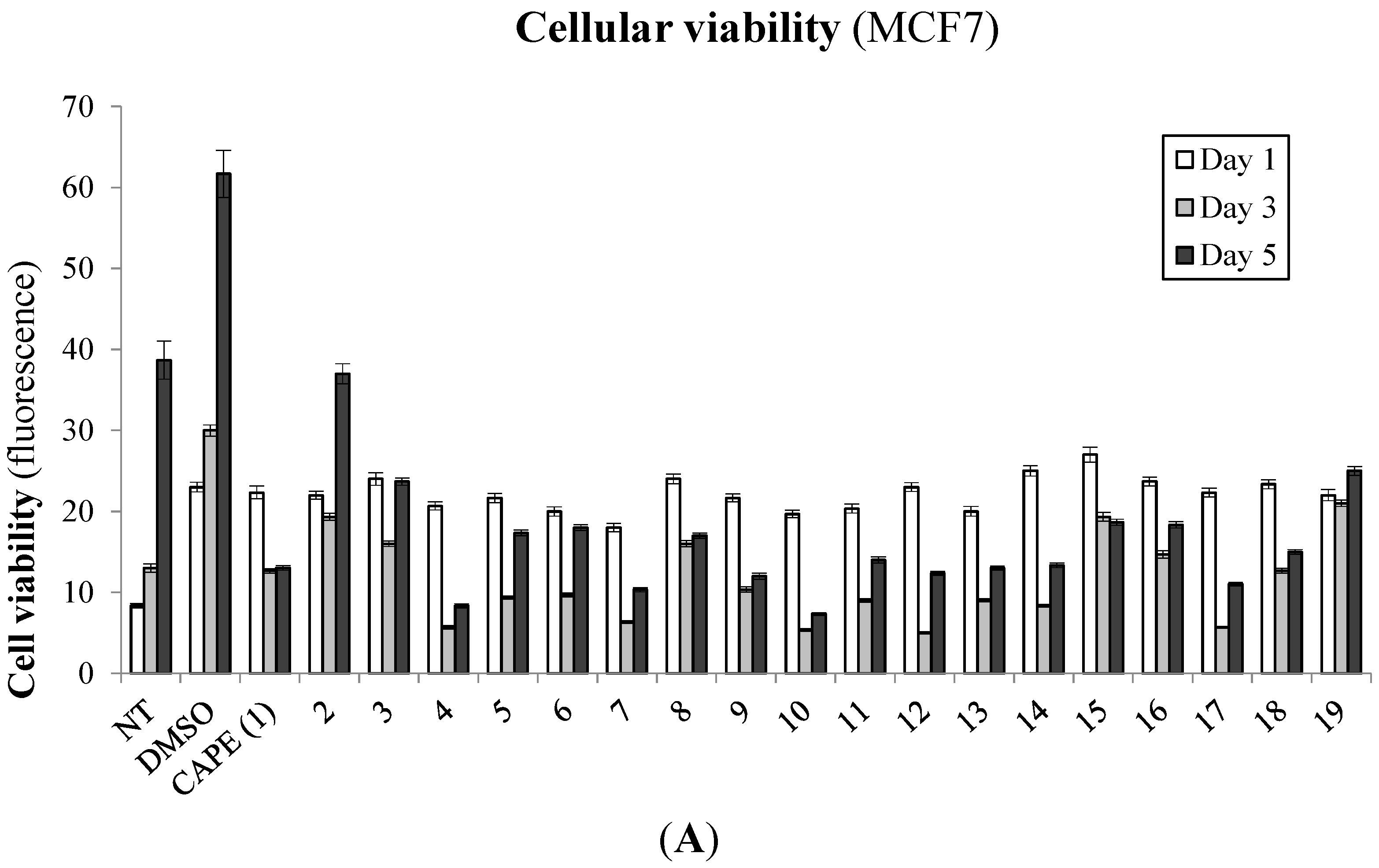
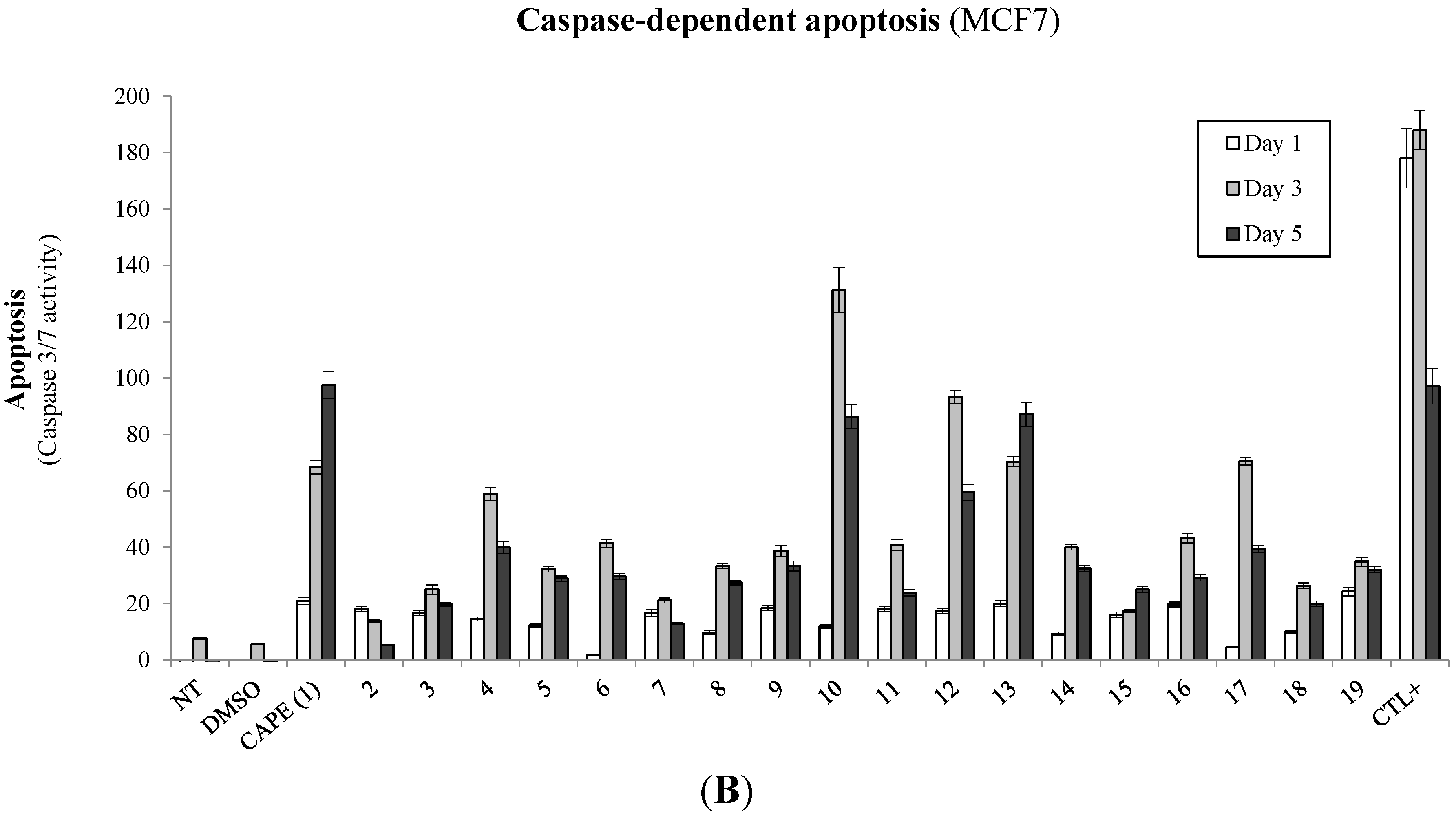
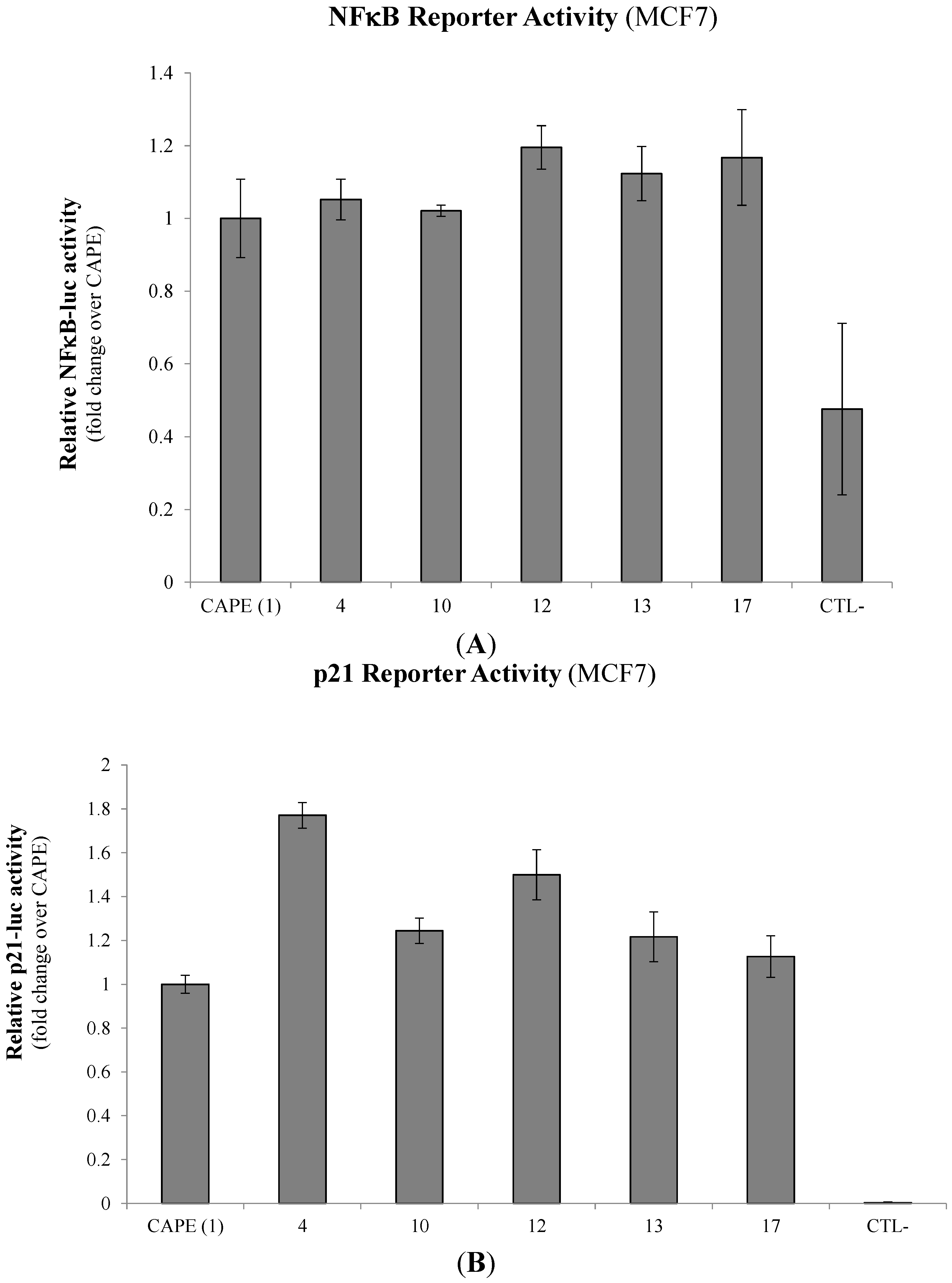
2.2. Modulation of Cell Fate Signaling by Selected CAPE Analogs in Breast Cancer Cells

2.3. Evaluation of p53-Mediated Apoptosis by CAPE Analogs in Breast Cancer Cells
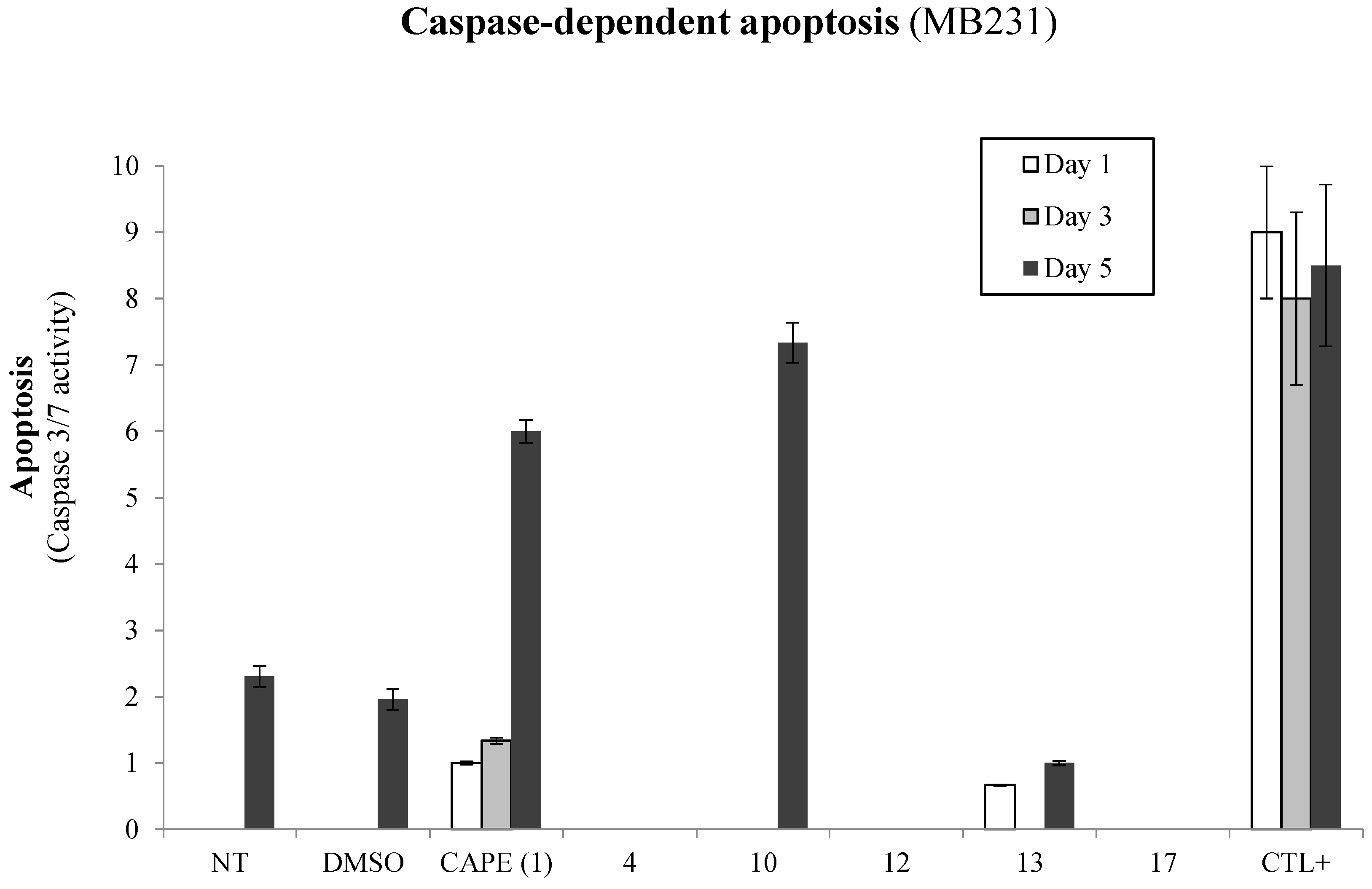
2.4. Antioxidative Potential of CAPE Analogs in Breast Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 ± SEM (µM) |
|---|---|
| CAPE 1 | 16.5 ± 4.0 |
| 4 | 14.2 ± 1.7 |
| 10 | 12.5 ± 2.6 |
| 12 | 11.9 ± 0.6 |
| 13 | 12.8 ± 1.1 |
| 17 | 18.5 ± 2.1 |
2.5. Discussion
3. Experimental Section
3.1. CAPE Analog Synthesis
3.2. Cell Culture and Treatments
3.3. Cell Viability and Apoptosis Assays
3.4. Cell Transfections and Luciferase-Based Reporter Assay
3.5. Radical Scavenging Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CAPE: | Caffeic Acid Phenylethyl Ester |
| NFκB: | Nuclear Factor of kappa B Cells |
| p53: | tumor suppressor gene p53 |
| IκB: | NFκB inhibitor |
| IKK: | IκB kinases |
| DMSO: | Dimethylsulfoxide |
| µM: | micromolar |
| IC50: | Half maximal inhibitory concentration |
| TNF: | tumor necrosis factor |
| TNFR: | tumor necrosis factor receptor |
| Apo/DR: | Trail receptor or Death receptor |
| ROS: | reactive oxygen species |
References
- Jia, L.T.; Zhang, R.; Shen, L.; Yang, A.G. Regulators of carcinogenesis: Emerging roles beyond their primary functions. Cancer Lett. 2015, 357, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappaB in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef] [PubMed]
- Siebenlist, U.; Franzoso, G.; Brown, K. Structure, regulation and function of NF-kappaB. Annu. Rev. Cell. Biol. 1994, 10, 405–455. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109 (Suppl. 109), S81–S96. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Thornborrow, E.C.; Manfredi, J.J. The tumor suppressor protein p53 requires a cofactor to activate transcriptionally the human bax promoter. J. Biol. Chem. 2001, 276, 15598–15608. [Google Scholar] [CrossRef] [PubMed]
- Toshiyuki, M.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [PubMed]
- Yap, J.L.; Worlikar, S.; MacKerell, A.D., Jr.; Shapiro, P.; Fletcher, S. Small-molecule inhibitors of the erk signaling pathway: Towards novel anticancer therapeutics. ChemMedChem 2011, 6, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann. Med. 2006, 38, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hu, Y. Small molecule agents targeting the p53-mdm2 pathway for cancer therapy. Med. Res. Rev. 2012, 32, 1159–1196. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Khan, N.; Mukhtar, H. Apoptosis and autophagy induction as mechanism of cancer prevention by naturally occurring dietary agents. Curr. Drug Targets 2012, 13, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Beutler, J.A. Natural Products and Cancer Drug Discovery; Springer: New York, NY, USA, 2013; p. 244. [Google Scholar]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Natural antioxidants: Therapeutic prospects for cancer and neurological diseases. Mini Rev. Med. Chem. 2009, 9, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Anticancer antioxidant regulatory functions of phytochemicals. Curr. Med. Chem. 2011, 18, 2315–2338. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (cape), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Watabe, M.; Hishikawa, K.; Takayanagi, A.; Shimizu, N.; Nakaki, T. Caffeic acid phenethyl ester induces apoptosis by inhibition of nfkappab and activation of fas in human breast cancer MCF-7 cells. J. Biol. Chem. 2004, 279, 6017–6026. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Shiao, M.S.; Wang, S.Y. The antioxidant caffeic acid phenethyl ester induces apoptosis associated with selective scavenging of hydrogen peroxide in human leukemic HL-60 cells. Anticancer Drugs 2001, 12, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Ulasli, S.S.; Celik, S.; Gunay, E.; Ozdemir, M.; Hazman, O.; Ozyurek, A.; Koyuncu, T.; Unlu, M. Anticancer effects of thymoquinone, caffeic acid phenethyl ester and resveratrol on a549 non-small cell lung cancer cells exposed to benzo(a)pyrene. Asian Pac. J. Cancer Prev. 2013, 14, 6159–6164. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Kaji, A.; Ma, W.; Miyamoto, K.; Dong, Z. Suppression of cell transformation and induction of apoptosis by caffeic acid phenethyl ester. Mol. Carcinog. 2001, 31, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Onori, P.; DeMorrow, S.; Gaudio, E.; Franchitto, A.; Mancinelli, R.; Venter, J.; Kopriva, S.; Ueno, Y.; Alvaro, D.; Savage, J.; et al. Caffeic acid phenethyl ester decreases cholangiocarcinoma growth by inhibition of NF-kappaB and induction of apoptosis. Int. J. Cancer 2009, 125, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, L.R.; Wang, J.; Le, T. Caffeic acid phenethyl ester, an inhibitor of nuclear factor-kappaB, attenuates bacterial peptidoglycan polysaccharide-induced colitis in rats. J. Pharmacol Exp. Ther. 2001, 299, 915–920. [Google Scholar] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ros) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.L.; Liang, W.H.; Lee, Y.J.; Chuang, S.K.; Tseng, T.H. Antitumor progression potential of caffeic acid phenethyl ester involving p75(NTR) in c6 glioma cells. Chem. Biol. Interact. 2010, 188, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Chen, J.H.; Chang, C.; Shieh, C.J. Optimization of ultrasound-accelerated synthesis of enzymatic caffeic acid phenethyl ester by response surface methodology. Ultrason. Sonochem. 2011, 18, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Chu, K.H.; Liang, Y.C.; Lin, Y.L.; Chiang, B.L. Caffeic acid phenethyl ester inhibits nuclear factor-kappab and protein kinase b signalling pathways and induces caspase-3 expression in primary human CD4+ T cells. Clin. Exp. Immunol. 2010, 160, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Orban, Z.; Mitsiades, N.; Burke, T.R., Jr.; Tsokos, M.; Chrousos, G.P. Caffeic acid phenethyl ester induces leukocyte apoptosis, modulates nuclear factor-kappa B and suppresses acute inflammation. Neuroimmunomodulation 2000, 7, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Wang, D.; He, Y.; Xie, J.; Zhong, Z.; Li, Z. Caffeic acid phenethyl ester induces growth arrest and apoptosis of colon cancer cells via the beta-catenin/T-cell factor signaling. Anticancer Drugs 2006, 17, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, D.H.; Kim, J.H.; Hong, S.; Choi, D.; Kim, Y.J.; Kwak, M.K.; Jung, Y. Caffeic acid phenethyl ester-mediated NRF2 activation and ikappab kinase inhibition are involved in NFkappaB inhibitory effect: Structural analysis for nfkappab inhibition. Eur J. Pharmacol. 2010, 643, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kuo, H.C.; Chu, C.Y.; Wang, C.J.; Lin, W.C.; Tseng, T.H. Involvement of tumor suppressor protein p53 and p38 mapk in caffeic acid phenethyl ester-induced apoptosis of c6 glioma cells. Biochem. Pharmacol. 2003, 66, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Junk, D.J.; Vrba, L.; Watts, G.S.; Oshiro, M.M.; Martinez, J.D.; Futscher, B.W. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia 2008, 10, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Chiang, M.F.; Chen, Y.J. Role of WW domain proteins WWOX in development, prognosis, and treatment response of glioma. Exp. Biol Med. 2015, 240, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Steller, H. Mechanisms and genes of cellular suicide. Science 1995, 267, 1445–1449. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S.; Golstein, P. The fas death factor. Science 1995, 267, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Apoptosis control by death and decoy receptors. Curr. Opin. Cell. Biol. 1999, 11, 255–260. [Google Scholar] [CrossRef]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: Trail-R1 and trail-R2 as candidate dosage-dependent tumor suppressor genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Holland, P.; Eckhardt, S.G. Ligand-based targeting of apoptosis in cancer: The potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhapo2l/trail). J. Clin. Oncol. 2008, 26, 3621–3630. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The REL/Nf-kappa B family: Friend and foe. Trends Biochem. Sci. 2000, 25, 434–440. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Grémy, O.; Benderitter, M.; Linard, C. Caffeic acid phenethyl ester modifies the TH1/TH2 balance in ileal mucosa after gamma-irradiation in the rat by modulating the cytokine pattern. World J. Gastroenterol. 2006, 12, 4996–5004. [Google Scholar] [PubMed]
- Kim, E.Y.; Ryu, J.H.; Kim, A.K. Cape promotes trail-induced apoptosis through the upregulation of trail receptors via activation of p38 and suppression of JNK in SK-HEP1 hepatocellular carcinoma cells. Int. J. Oncol. 2013, 43, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Chiao, C.; Carothers, A.M.; Grunberger, D.; Solomon, G.; Preston, G.A.; Barrett, J.C. Apoptosis and altered redox state induced by caffeic acid phenethyl ester (cape) in transformed rat fibroblast cells. Cancer Res. 1995, 55, 3576–3583. [Google Scholar] [PubMed]
- Chen, Y.J.; Shiao, M.S.; Hsu, M.L.; Tsai, T.H.; Wang, S.Y. Effect of caffeic acid phenethyl ester, an antioxidant from propolis, on inducing apoptosis in human leukemic HL-60 cells. J. Agric. Food Chem. 2001, 49, 5615–5619. [Google Scholar] [CrossRef] [PubMed]
- Gocmez, C.; Celik, F.; Kamasak, K.; Kaplan, M.; Uzar, E.; Arikanoglu, A.; Evliyaoglu, O. Effects of intrathecal caffeic acid phenethyl ester and methylprednisolone on oxidant/antioxidant status in traumatic spinal cord injuries. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2014, 76, 20–24. [Google Scholar] [PubMed]
- Choi, K.; Han, Y.H.; Choi, C. N-acetyl cysteine and caffeic acid phenethyl ester sensitize astrocytoma cells to FAS-mediated cell death in a redox-dependent manner. Cancer Lett 2007, 257, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, J.T.; Clabault, H.; Patton, C.; Lassalle-Claux, G.; Jean-François, J.; Paré, A.F.; Hébert, M.J.G.; Surette, M.E.; Touaibia, M. Antiproliferative, antiandrogenic and cytotoxic effects of novel caffeic acid derivatives in lncap human androgen-dependent prostate cancer cells. Bioorg. Med. Chem. 2013, 21, 7182–7193. [Google Scholar] [CrossRef] [PubMed]
- Cormier, K.; Harquail, J.; Ouellette, R.J.; Tessier, P.A.; Guerrette, R.; Robichaud, G.A. Intracellular expression of inflammatory proteins s100a8 and s100a9 leads to epithelial-mesenchymal transition and attenuated aggressivity of breast cancer cells. Anticancer Agents Med. Chem. 2014, 14, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Benzina, S.; Harquail, J.; Jean, S.; Beauregard, A.P.; Colquhoun, C.D.; Carroll, M.; Bos, A.; Gray, C.A.; Robichaud, G.A. Deoxypodophyllotoxin isolated from juniperus communis induces apoptosis in breast cancer cells. Anticancer Agents Med. Chem. 2014, 15, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Kalita, A.; Labrecque, S.; Pim, D.; Banks, L.; Matlashewski, G. Two polymorphic variants of wild-type p53 differ biochemically and biologically. Mol. Cell. Biol. 1999, 19, 1092–1100. [Google Scholar] [PubMed]
- Doiron, J.A.; Metayer, B.; Richard, R.R.; Desjardins, D.; Boudreau, L.H.; Levesque, N.A.; Jean-Francois, J.; Poirier, S.J.; Surette, M.E.; Touaibia, M. Clicked cinnamic/caffeic esters and amides as radical scavengers and 5-lipoxygenase inhibitors. Int. J. Med. Chem. 2014, 2014, 931756–931768. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.L.; Wu, X.; Schaich, K. Standardized methods for the determination of antioxidant capacity and phenolics in foods and dietary supplements. J. Agric. Food Chem. 2005, 53, 4290–4302. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 2–18 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beauregard, A.-P.; Harquail, J.; Lassalle-Claux, G.; Belbraouet, M.; Jean-Francois, J.; Touaibia, M.; Robichaud, G.A. CAPE Analogs Induce Growth Arrest and Apoptosis in Breast Cancer Cells. Molecules 2015, 20, 12576-12589. https://doi.org/10.3390/molecules200712576
Beauregard A-P, Harquail J, Lassalle-Claux G, Belbraouet M, Jean-Francois J, Touaibia M, Robichaud GA. CAPE Analogs Induce Growth Arrest and Apoptosis in Breast Cancer Cells. Molecules. 2015; 20(7):12576-12589. https://doi.org/10.3390/molecules200712576
Chicago/Turabian StyleBeauregard, Annie-Pier, Jason Harquail, Grégoire Lassalle-Claux, Mehdi Belbraouet, Jacques Jean-Francois, Mohamed Touaibia, and Gilles A. Robichaud. 2015. "CAPE Analogs Induce Growth Arrest and Apoptosis in Breast Cancer Cells" Molecules 20, no. 7: 12576-12589. https://doi.org/10.3390/molecules200712576
APA StyleBeauregard, A.-P., Harquail, J., Lassalle-Claux, G., Belbraouet, M., Jean-Francois, J., Touaibia, M., & Robichaud, G. A. (2015). CAPE Analogs Induce Growth Arrest and Apoptosis in Breast Cancer Cells. Molecules, 20(7), 12576-12589. https://doi.org/10.3390/molecules200712576