
Synthesis and Biological Evaluation of Novel Water-Soluble Poly-(ethylene glycol)-10-hydroxycamptothecin Conjugates

Abstract
:1. Introduction
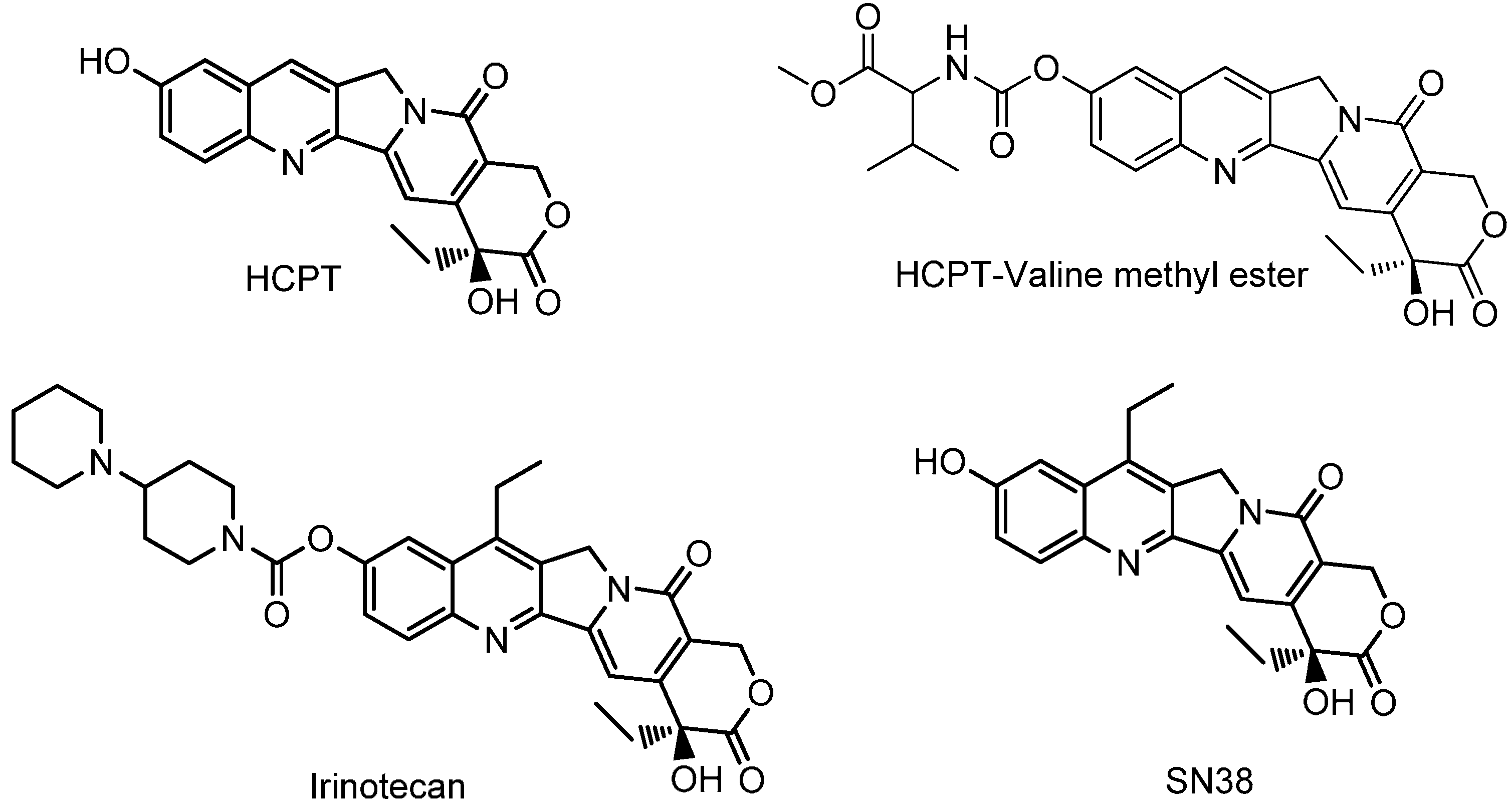
2. Results and Discussion
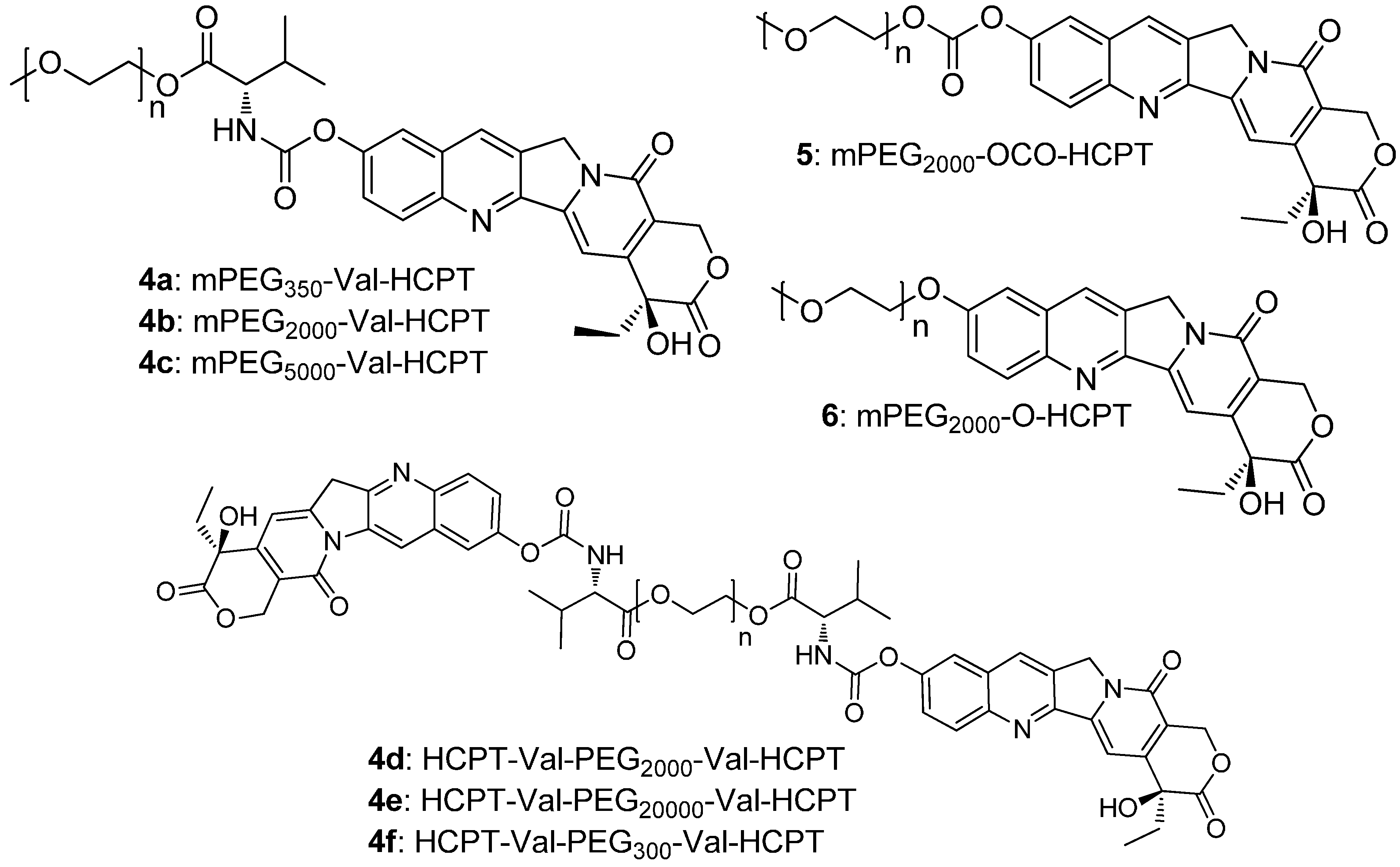
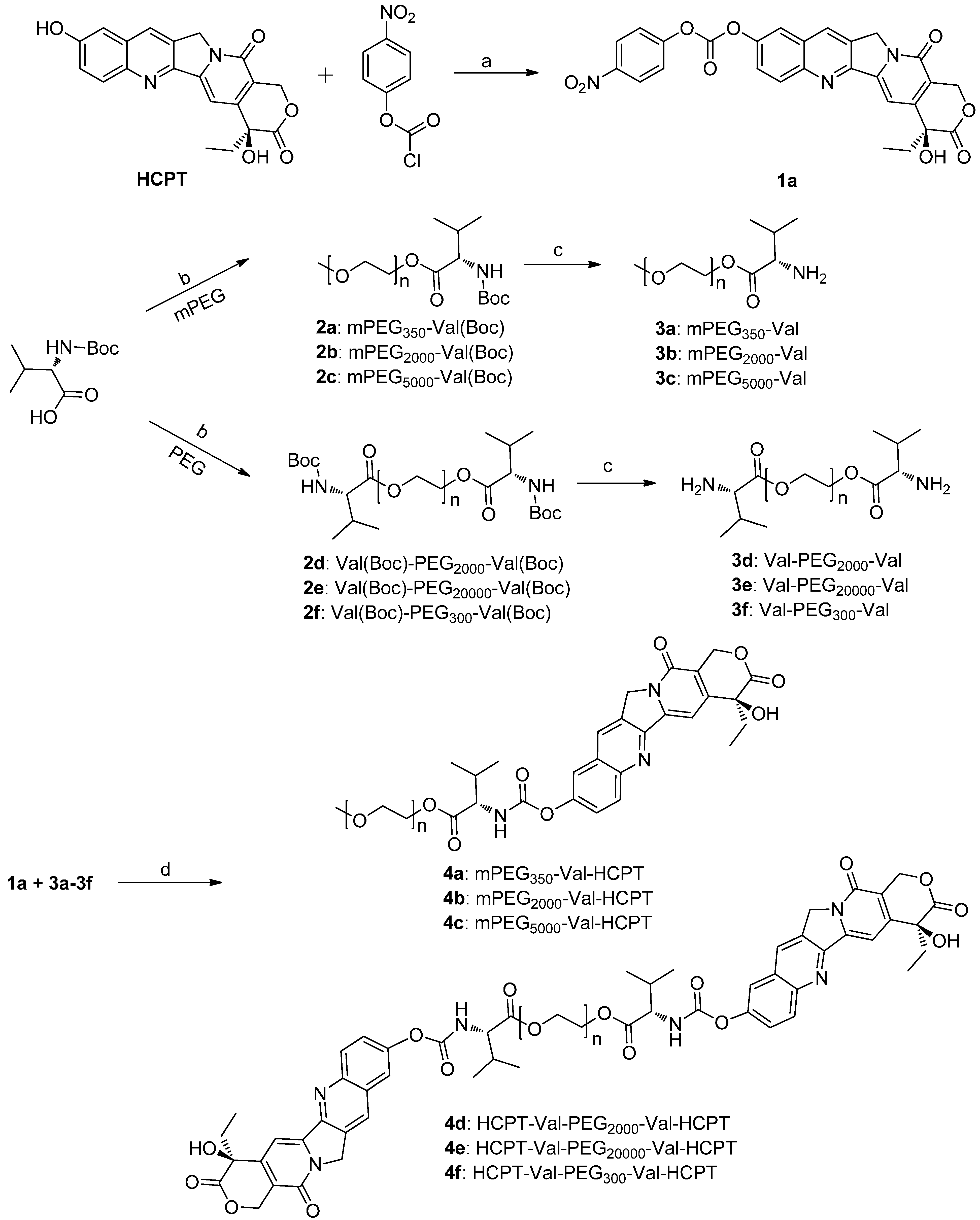
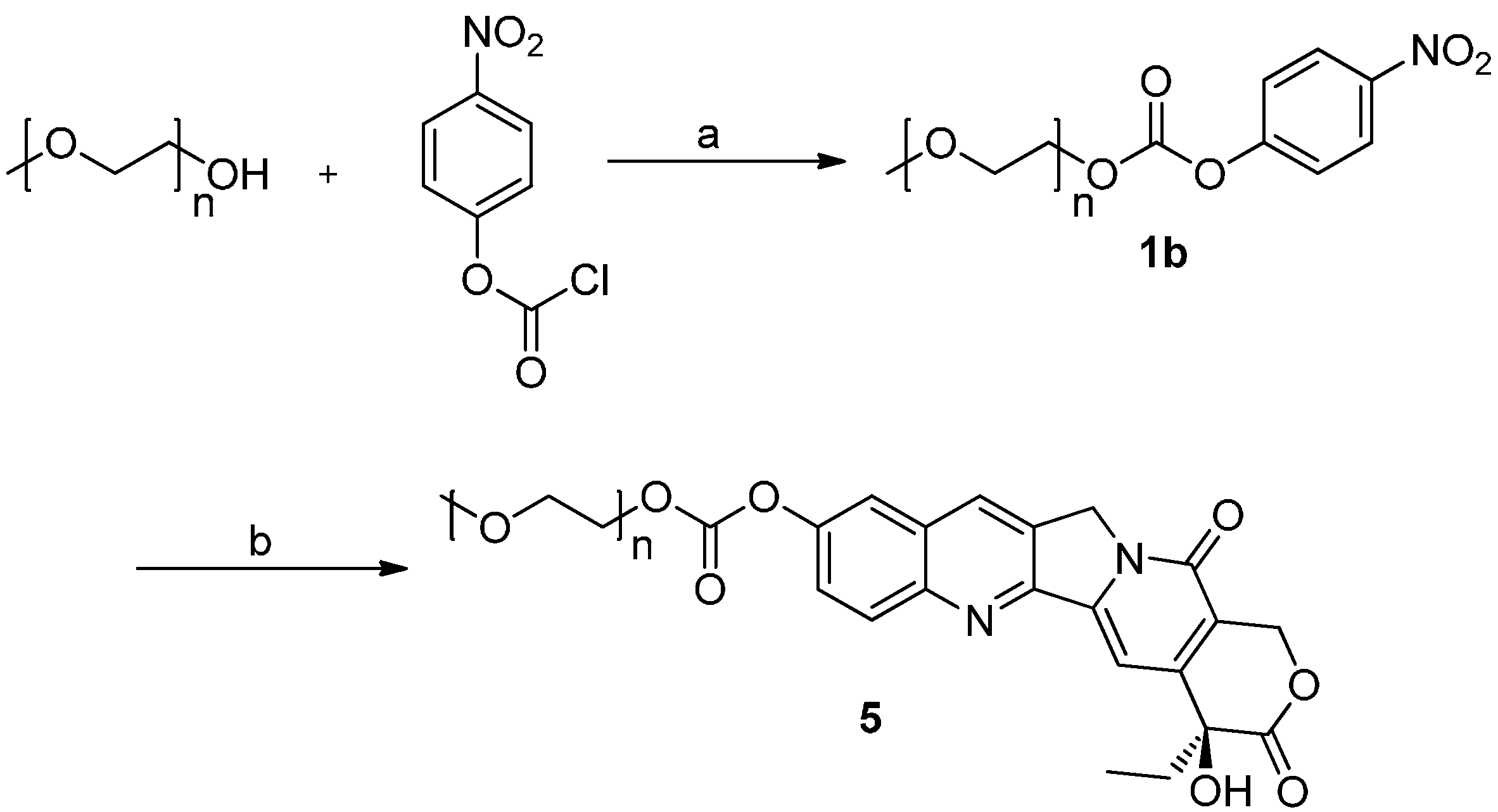
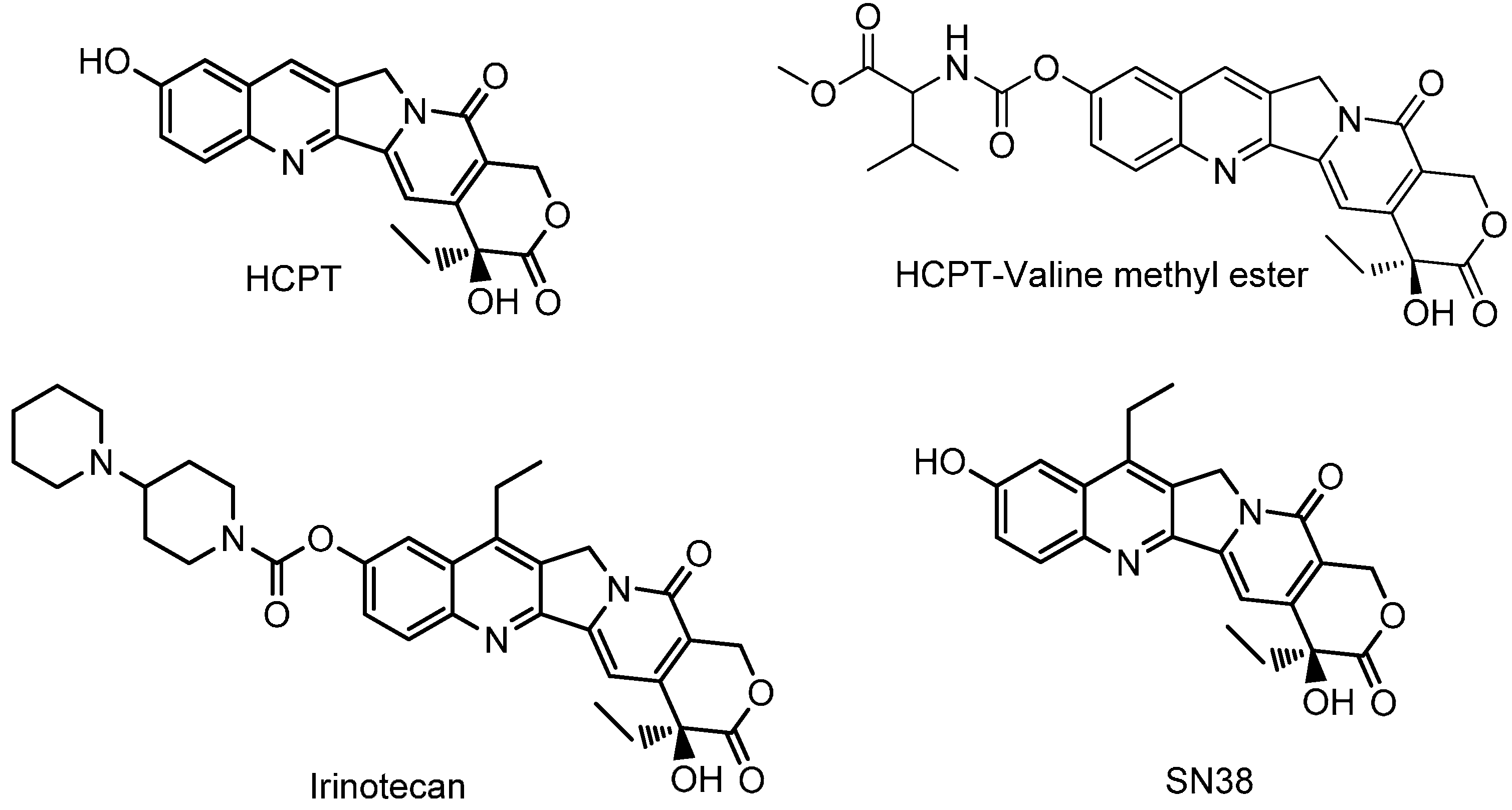
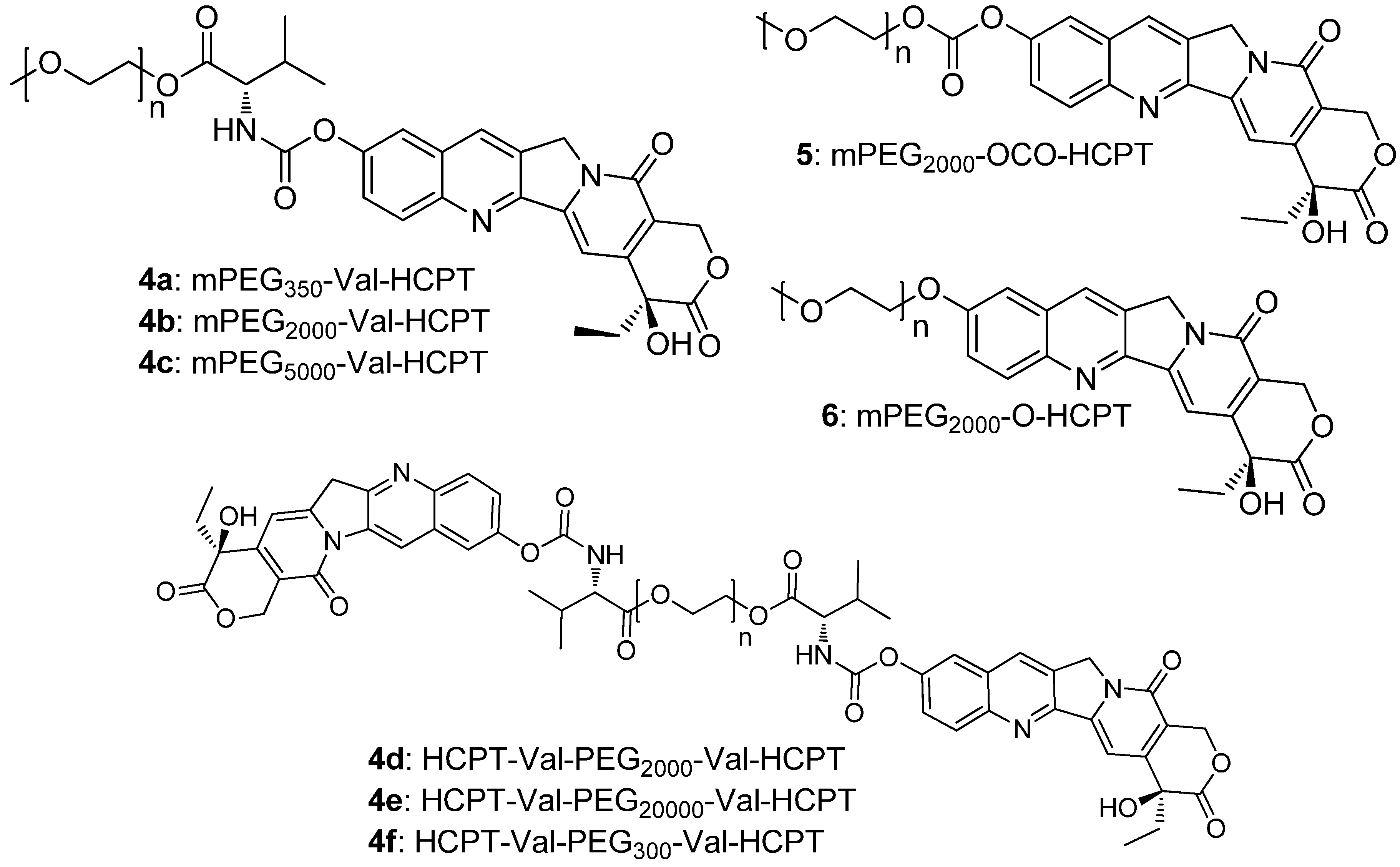
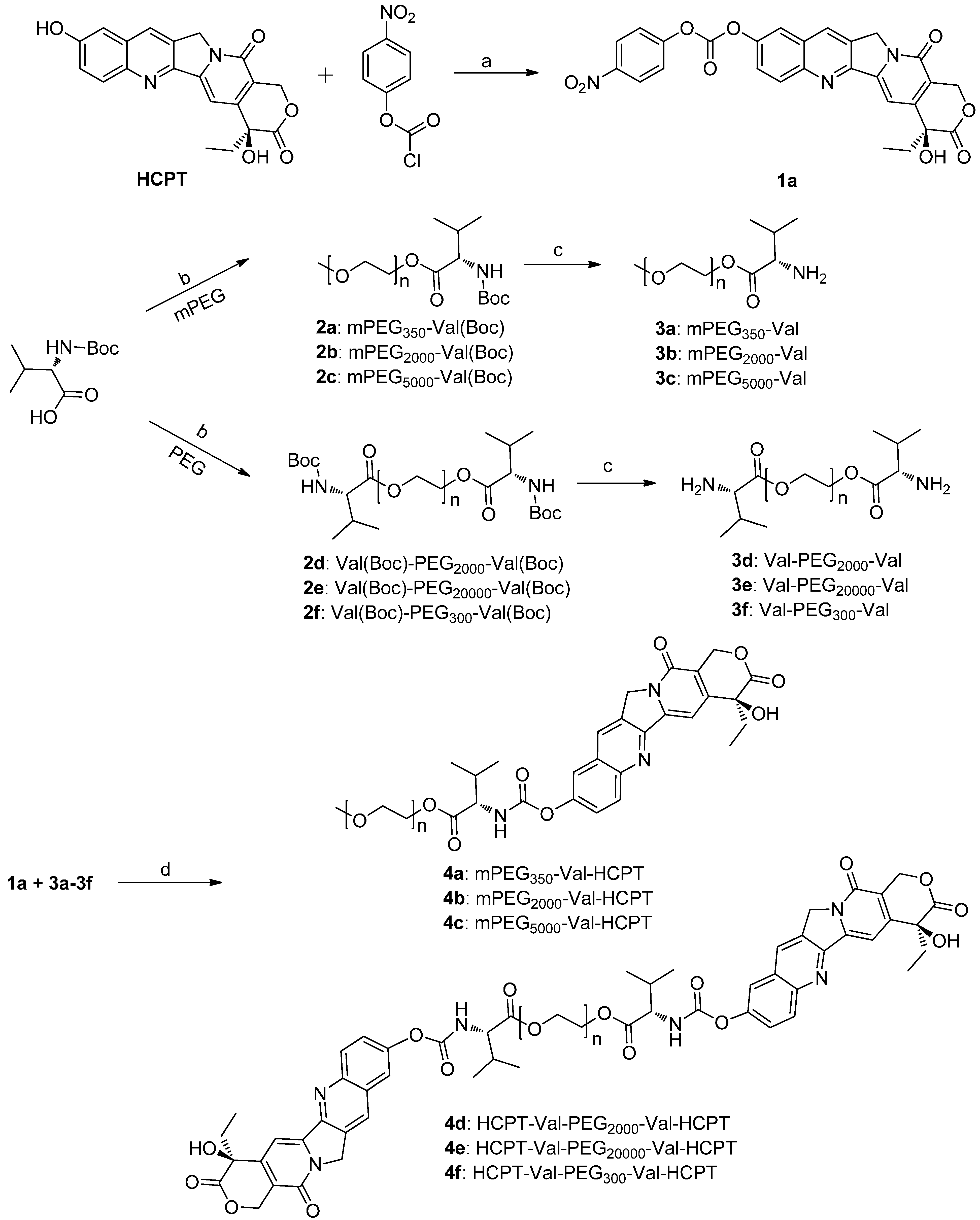
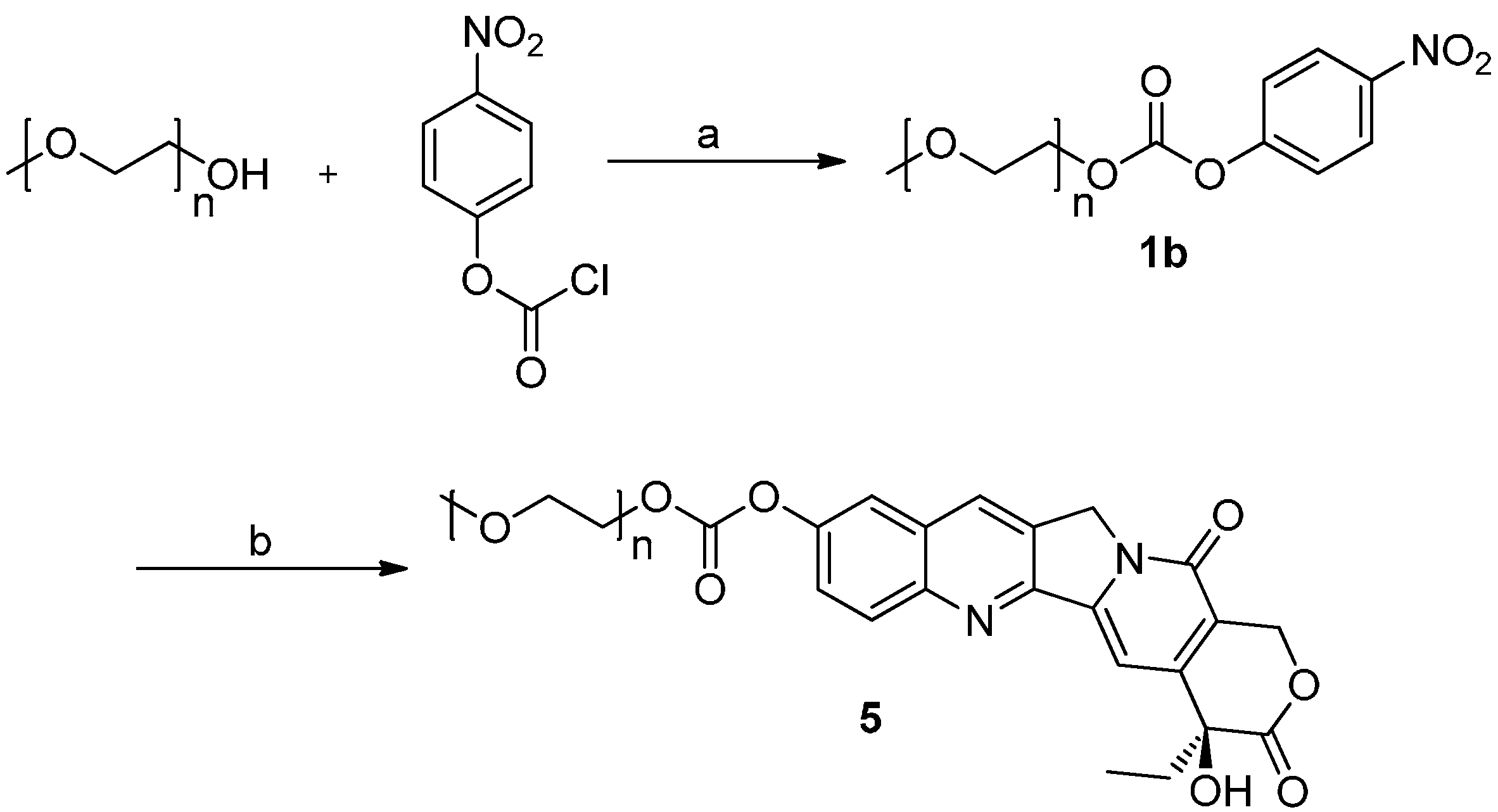
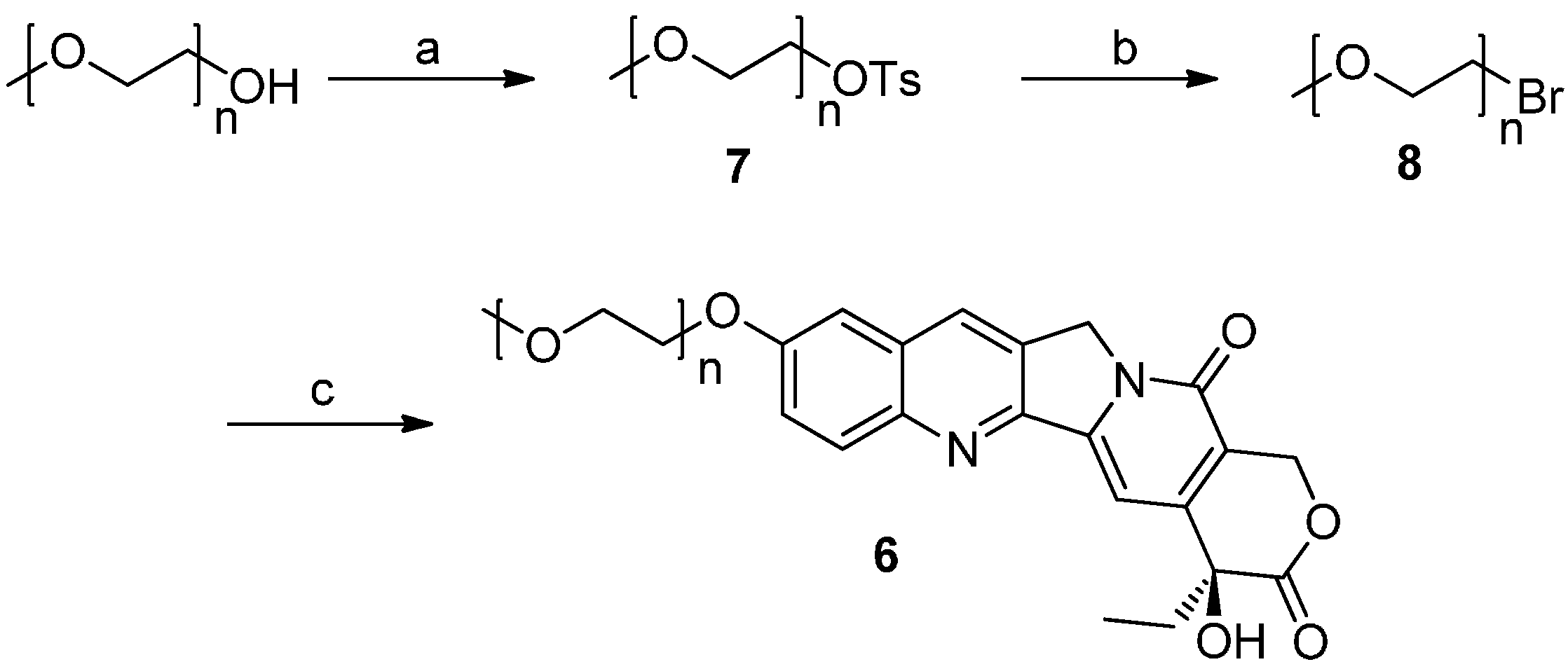
2.1. Chemistry
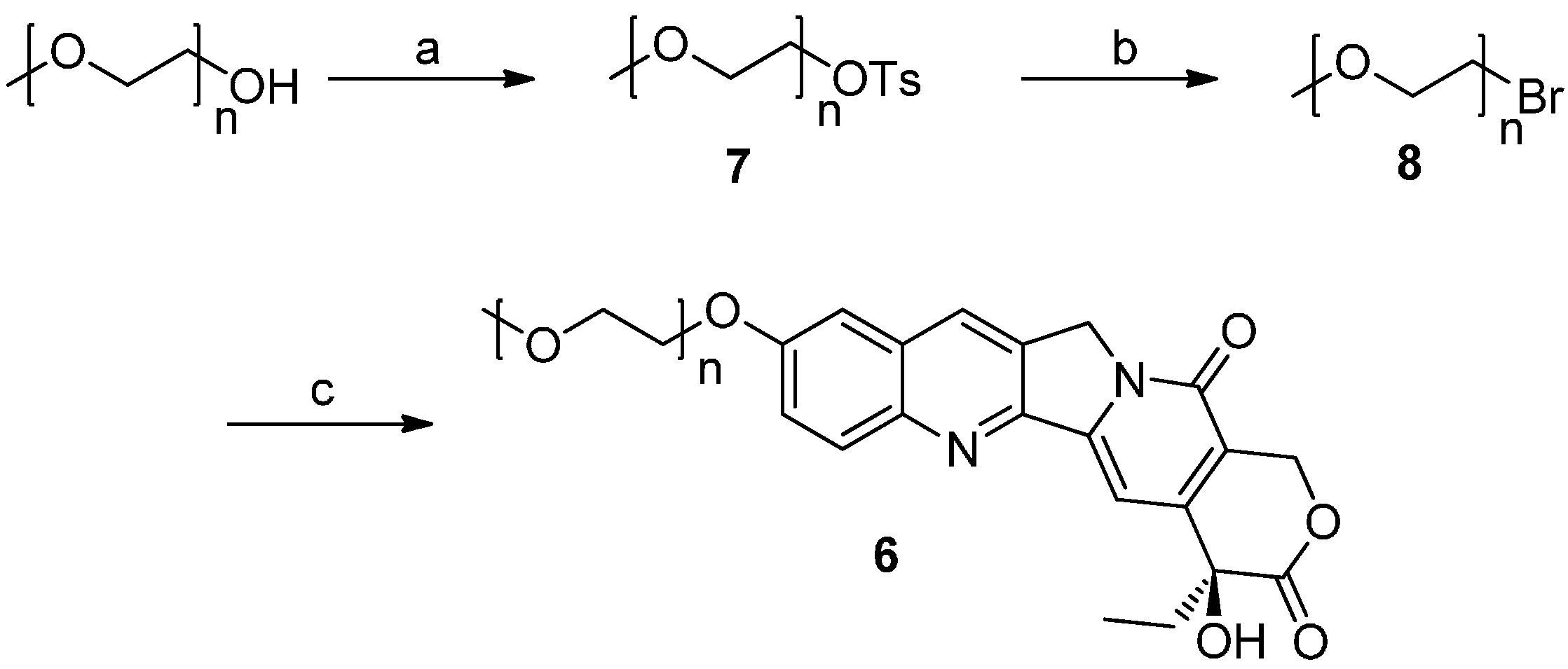
2.2. Cytotoxicity
2.3. Stability and Conversion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | ||
|---|---|---|---|
| K562 | HepG2 | HT-29 | |
| HCPT | 0.11 ± 0.07 | 0.08 ± 0.02 | 0.22 ± 0.09 |
| 4a | 0.02 ± 0.01 | 0.04 ± 0.01 | 0.04 ± 0.01 * |
| 4b | 0.12 ± 0.03 | 0.11 ± 0.06 | 0.19 ± 0.10 |
| 4c | 0.06 ± 0.03 | 0.12 ± 0.08 | 0.21 ± 0.10 |
| 4d | 0.02 ± 0.01 | 0.04 ± 0.02 | 0.04 ± 0.01 * |
| 4e | 0.03 ± 0.01 | 0.05 ± 0.03 * | 0.14 ± 0.09 |
| 4f | 0.02 ± 0.01 | 0.05 ± 0.01 | 0.04 ± 0.01 * |
| 5 | 0.24 ± 0.05 * | 0.39 ± 0.23 | 0.42 ± 0.06 * |
| 6 | 26.99 ± 0.74 ** | 32.30 ± 1.48 ** | 42.97 ± 16.67 ** |
| Compound | Released HCPT (%) | |||
|---|---|---|---|---|
| 1 h | 2 h | 3 h | 4 h | |
| 4a | 75 | 80 | 82 | 82 |
| 4b | 96 | 100 | - | - |
| 4c | 100 | - | - | - |
| 4d | 70 | 90 | 94 | 96 |
| 4e | 100 | - | - | - |
| 4f | 20 | 32 | 35 | 38 |
| 5 | 54 | 65 | 68 | 68 |
| 6 | 33 | 45 | 54 | 56 |
2.4. Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis
3.3. Cytotoxicity Study
3.4. Stability Test
3.5. Statistics
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ohwada, J.; Ozawa, S.; Kohchi, M. Synthesis and biological activities of a pH-dependently activated water-soluble prodrug of a novel hexacyclic camptothecin analog. Bioorg. Med. Chem. Lett. 2009, 19, 2772–2776. [Google Scholar]
- Basili, S.; Moro, S. Novel camptothecin derivatives as topoisomerase I inhibitors. Expert Opin. Ther. Patents. 2009, 19, 555–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Sun, X.R.; Mao, W.W.; Sun, W.L.; Tang, J.B.; Sui, M.H.; Shen, Y.Q.; Gu, Z.W. Tumor redox heterogeneity-responsive prodrug nanocapsules for cancer chemotherapy. Adv. Mater. 2013, 25, 3670–3676. [Google Scholar] [CrossRef] [PubMed]
- Shamay, Y.; Paulin, D.; Ashkenasy, G.; David, A. E-selectin binding peptide-polymer-drug conjugates and their selective cytotoxicity against vascular endothelial cells. Biomaterials 2009, 30, 6460–6468. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.G.; Wang, D.D.; Xu, S.; Liu, X.Y.; Zhang, X.Y.; Zhang, H.X. Preparation of camptothecin prodrug with glutathione-responsive disulfide linker for anticancer drug delivery. Chem. Asian J. 2014, 9, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kim, J.Y.; Han, J.H.; Bhuniya, S.; Sessler, J.L.; Kang, C.; Kim, J.S. Direct fluorescence monitoring of the delivery and cellular uptake of a cancer-targeted RGD peptide-appended naphthalimide theragnostic prodrug. J. Am. Chem. Soc. 2012, 134, 12668–12674. [Google Scholar] [CrossRef] [PubMed]
- Conover, C.D.; Greenwald, R.B.; Pendri, A.; Shum, K.L. Camptothecin delivery systems: The utility of amino acid spacers for the conjugation of camptothecin with polyethylene glycol to create prodrugs. Anti-Cancer Drug Des. 1999, 14, 499–506. [Google Scholar]
- Fleming, A.B.; Haverstick, K.; Saltzman, W.M. In vitro cytotoxicity and in vivo distribution after direct delivery of PEG-camptothecin conjugates to the rat brain. Bioconjugate Chem. 2004, 15, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Beam, K.S.; Mixan, B.; Wahl, A.F. Identification and activities of human carboxylesterases for the activation of CPT-11, a clinically approved anticancer drug. Bioconjugate Chem. 2001, 12, 1074–1080. [Google Scholar] [CrossRef]
- De Jong, F.A.; Mathijssen, R.H.J.; Xie, R.; Verweij, J.; Sparreboom, A. Flat-fixed dosing of irinotecan: Influence on pharmacokinentic and pharmacodynamics variability. Clin. Cancer Res. 2004, 10, 4068–4071. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Ohwada, J.; Ogawa, K.; Yamada-Okabe, H. A water soluble prodrug of a novel camptothecin analog is efficacious against breast cancer resistance protein-expressing tumor xenografts. Cancer Chemother. Pharmacol. 2010, 65, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Guo, N.; Wen, S.P.; Teng, Y.O.; Ma, M.X.; Yu, P. Synthesis and antitumor activity evaluation of a novel series of camptothecin analogs. J. Asian Nat. Prod. Res. 2013, 15, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 4a–4f are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, N.; Jiang, D.; Wang, L.; You, X.; Teng, Y.-O.; Yu, P. Synthesis and Biological Evaluation of Novel Water-Soluble Poly-(ethylene glycol)-10-hydroxycamptothecin Conjugates. Molecules 2015, 20, 9393-9404. https://doi.org/10.3390/molecules20059393
Guo N, Jiang D, Wang L, You X, Teng Y-O, Yu P. Synthesis and Biological Evaluation of Novel Water-Soluble Poly-(ethylene glycol)-10-hydroxycamptothecin Conjugates. Molecules. 2015; 20(5):9393-9404. https://doi.org/10.3390/molecules20059393
Chicago/Turabian StyleGuo, Na, Du Jiang, Luyao Wang, Xing You, Yu-Ou Teng, and Peng Yu. 2015. "Synthesis and Biological Evaluation of Novel Water-Soluble Poly-(ethylene glycol)-10-hydroxycamptothecin Conjugates" Molecules 20, no. 5: 9393-9404. https://doi.org/10.3390/molecules20059393
APA StyleGuo, N., Jiang, D., Wang, L., You, X., Teng, Y.-O., & Yu, P. (2015). Synthesis and Biological Evaluation of Novel Water-Soluble Poly-(ethylene glycol)-10-hydroxycamptothecin Conjugates. Molecules, 20(5), 9393-9404. https://doi.org/10.3390/molecules20059393