
Synthesis and Antiplatelet Activity of Antithrombotic Thiourea Compounds: Biological and Structure-Activity Relationship Studies


Abstract
:
1. Introduction
2. Results and Discussion
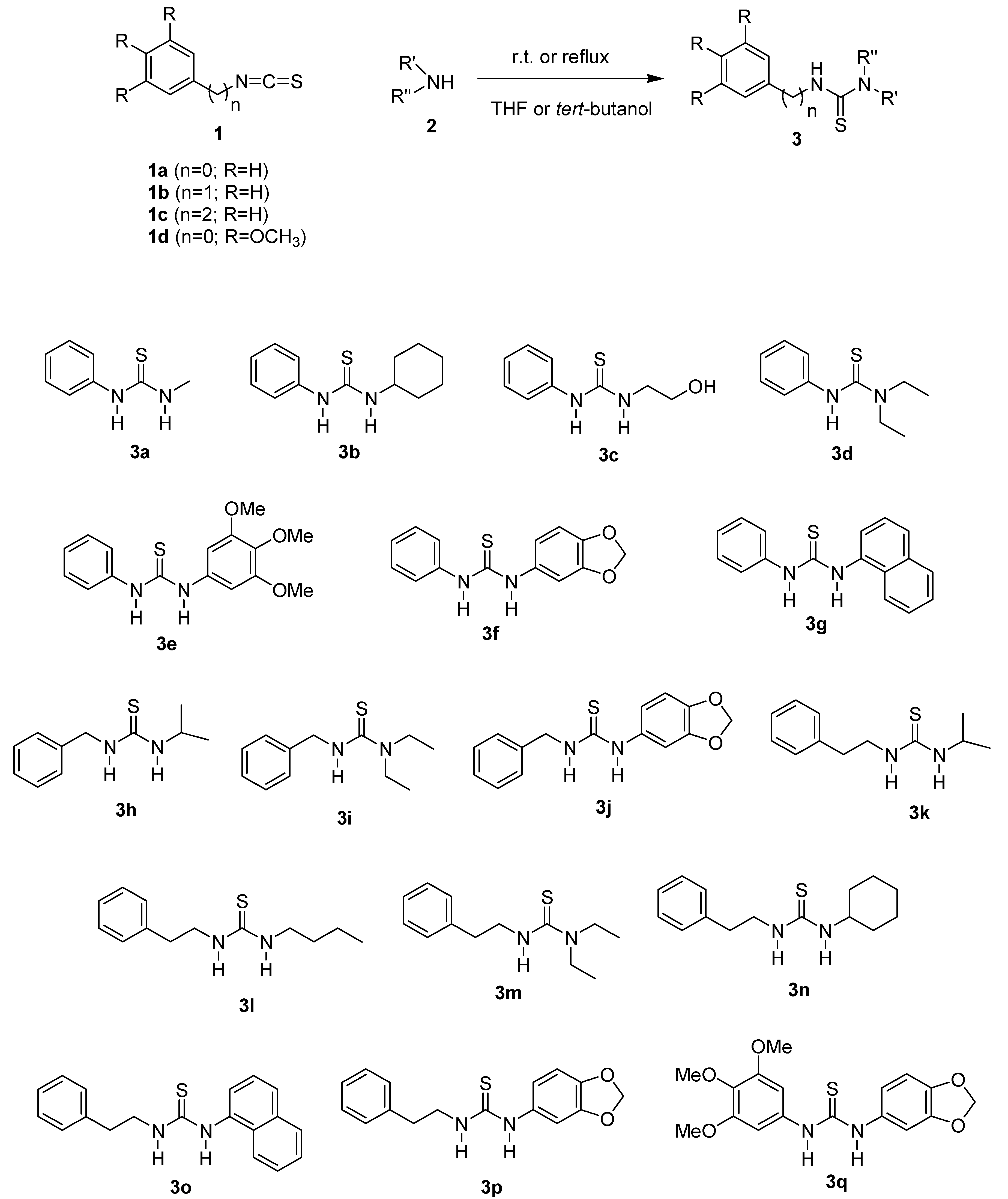
2.1. Chemistry
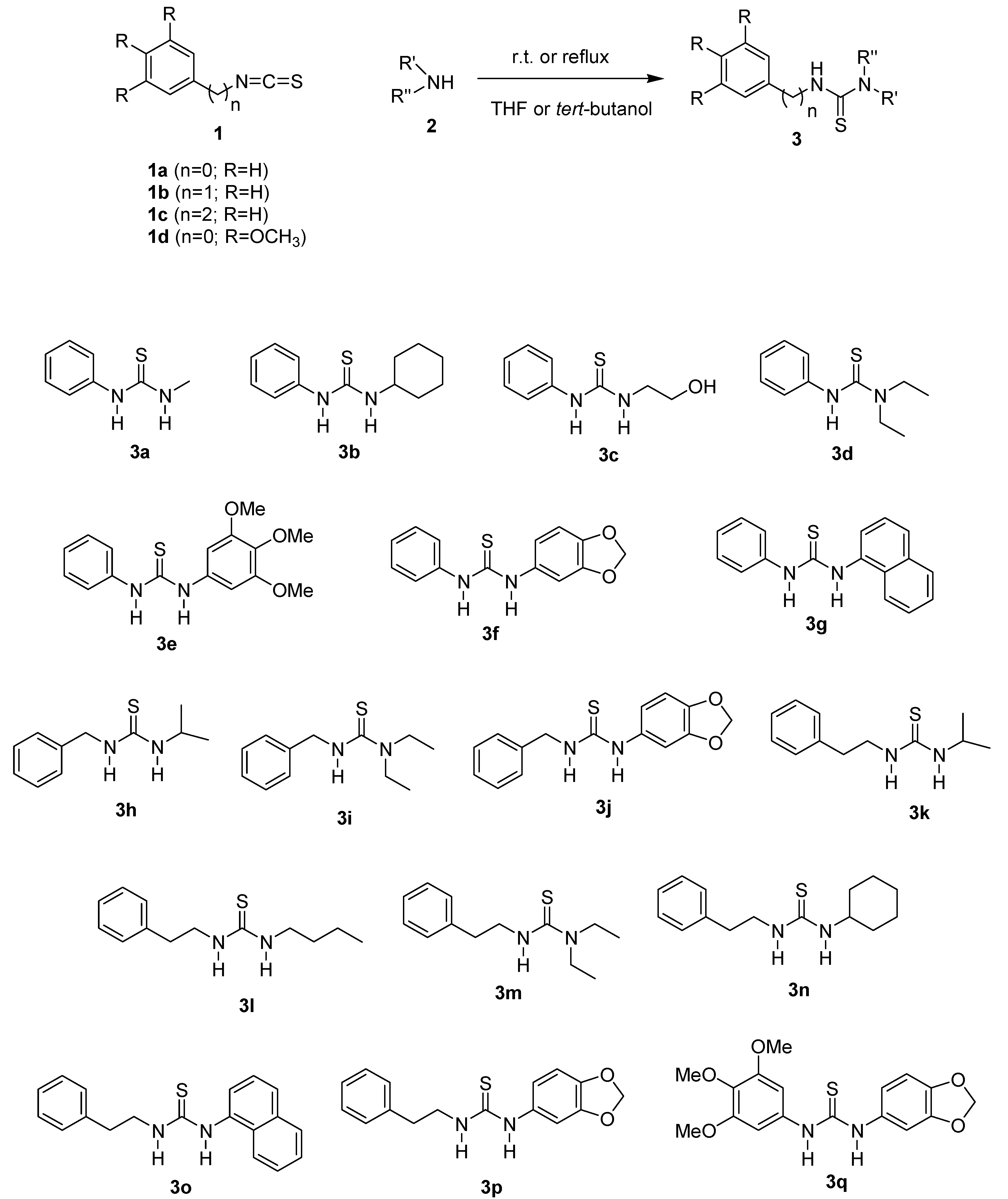

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Thiourea | Solvent | Temperature | Time (h) | Yield % a |
|---|---|---|---|---|
| 3a | THF | r.t. | 4.0 | 97 |
| 3b | THF | r.t. | 8.0 | 90 |
| 3c | THF | r.t. | 6.0 | 89 |
| 3d | THF | r.t. | 6.0 | 96 |
| 3e | t-BuOH | reflux | 8.0 | 82 |
| 3f | t-BuOH | reflux | 10.0 | 92 |
| 3g | t-BuOH | reflux | 7.0 | 86 |
| 3h | THF | r.t. | 5.0 | 85 |
| 3i | THF | r.t. | 6.0 | 95 |
| 3j | t-BuOH | reflux | 10.0 | 85 |
| 3k | THF | r.t. | 5.0 | 89 |
| 3l | THF | r.t. | 4.5 | 89 |
| 3m | THF | r.t. | 4.0 | 88 |
| 3n | THF | r.t. | 6.0 | 88 |
| 3o | t-BuOH | reflux | 8.0 | 87 |
| 3p | t-BuOH | reflux | 12.0 | 94 |
| 3q | t-BuOH | reflux | 8.0 | 84 |
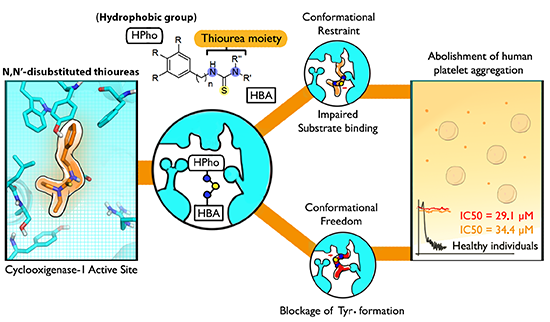
2.2. Biological Activity Evaluation
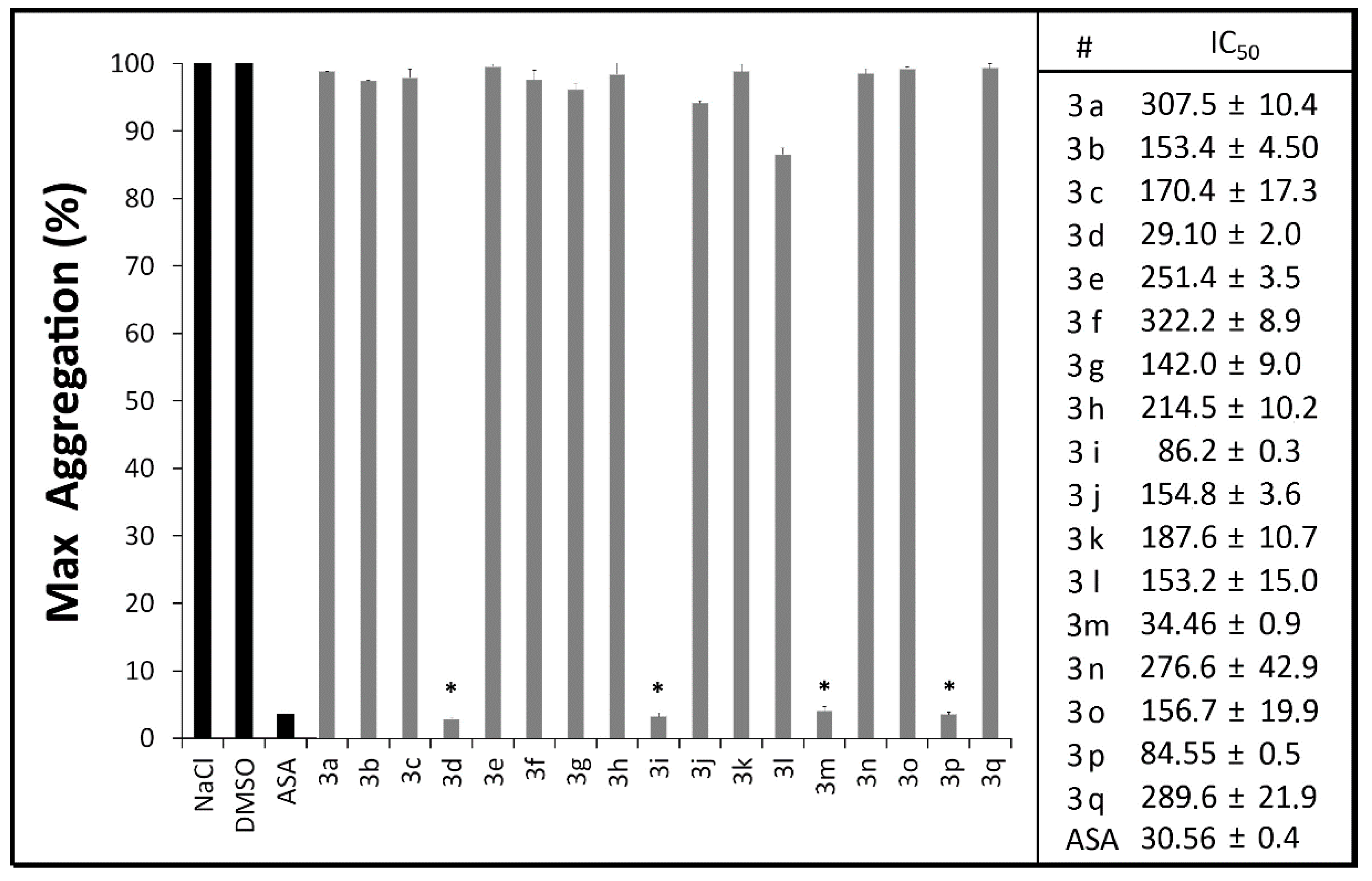
2.2.1. Platelet Aggregation Assays
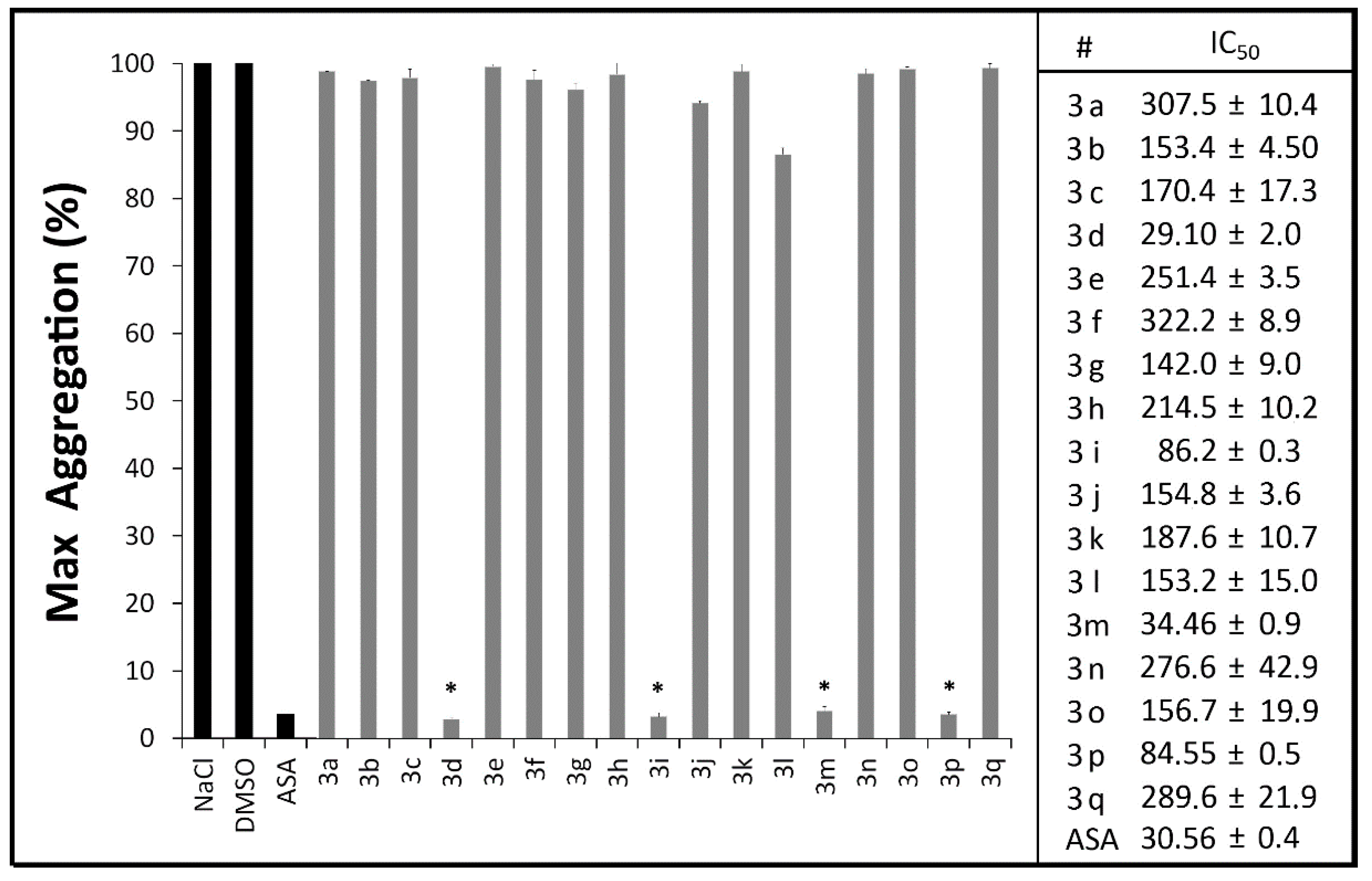
2.2.2. Measurement of Plasma PGE2 and TXB2 Levels
| # | Platelet Inhibition of PGE2 (%) | Platelet Inhibition of TXB2 (%) |
|---|---|---|
| 3d | 25.6 ± 5.9 | 74.2 ± 10.2 |
| 3i | 21.1 ± 5.0 | 74.7 ± 5.5 |
| 3m | 28.2 ± 5.9 | 91.2 ± 0.0 |
| 3p | 7.9 ± 7.9 | 88.1 ± 5.5 |
| Aspirin | 47.5 ± 1.4 | 78.5 ± 5.0 |
| Indomethacin | 28.0 ± 1.6 | 99.8 ± 0.0 |
| Ozagrel | - | 98.0 ± 2.8 |
| DMSO | 0.0 ± 1.6 | 0.0 ± 0.0 |
| Thiourea | Ames Test S. typhimurium | SOS Chromotest E. coli | ||||
|---|---|---|---|---|---|---|
| TA97 | TA98 | TA100 | TA102 | PQ35 | PQ37 | |
| 3a | - | - | - | - | - | - |
| 3b | - | - | - | - | - | - |
| 3c | - | - | - | - | - | - |
| 3d | - | - | - | - | - | - |
| 3e | - | - | - | - | - | - |
| 3f | - | - | - | - | - | - |
| 3g | - | - | - | - | - | - |
| 3h | - | - | - | - | - | - |
| 3i | - | - | - | - | - | - |
| 3j | - | - | - | - | - | - |
| 3k | - | - | - | - | - | - |
| 3l | - | - | - | - | - | - |
| 3m | - | - | - | - | - | - |
| 3n | - | - | - | - | - | - |
| 3o | - | - | - | - | - | - |
| 3p | - | - | - | - | - | - |
| 3q | - | - | - | - | - | - |
| 4-NQO | + | + | + | + | + | + |
| DMSO | - | - | - | - | - | - |
| ASA | - | - | - | - | - | - |
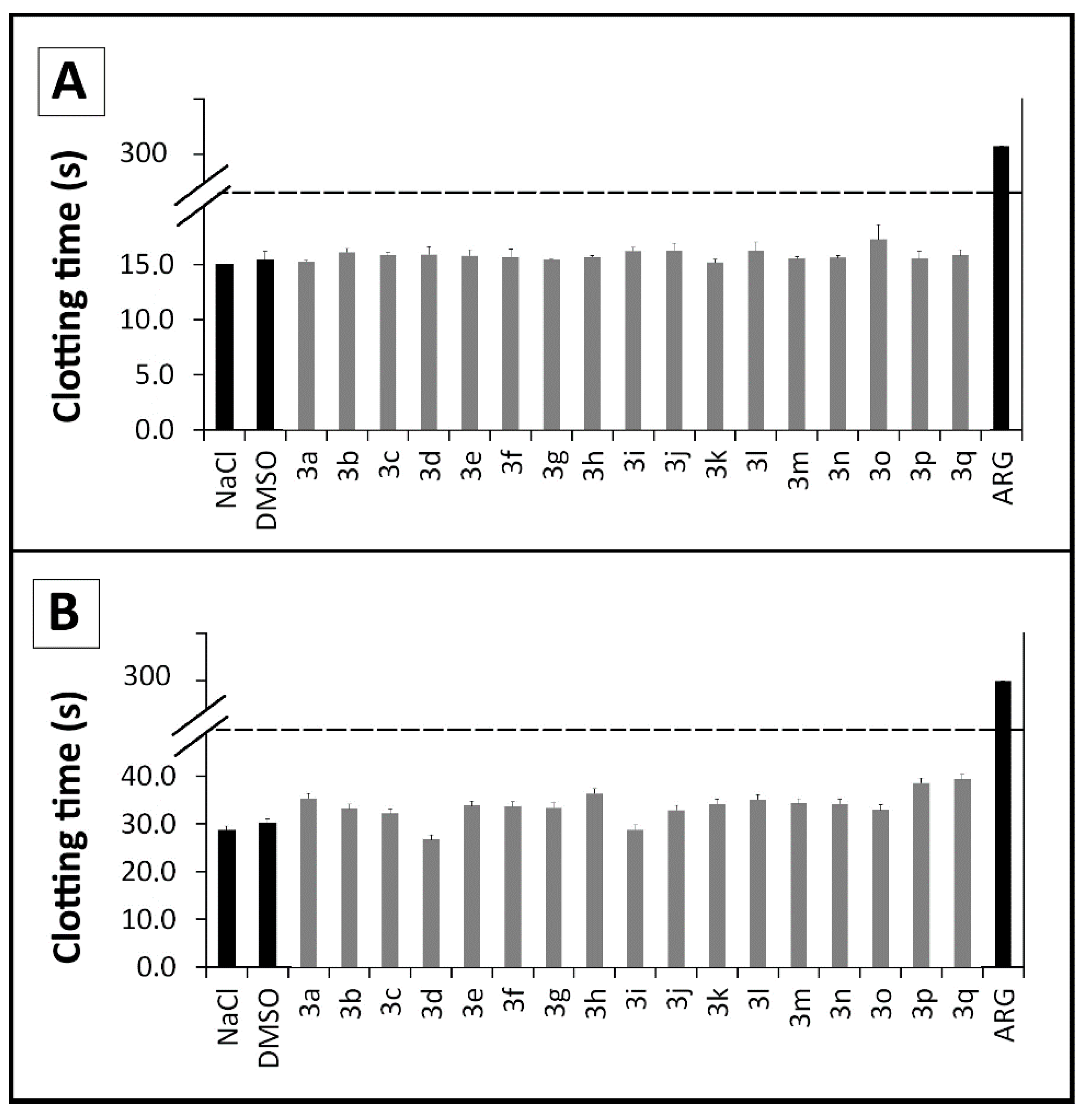
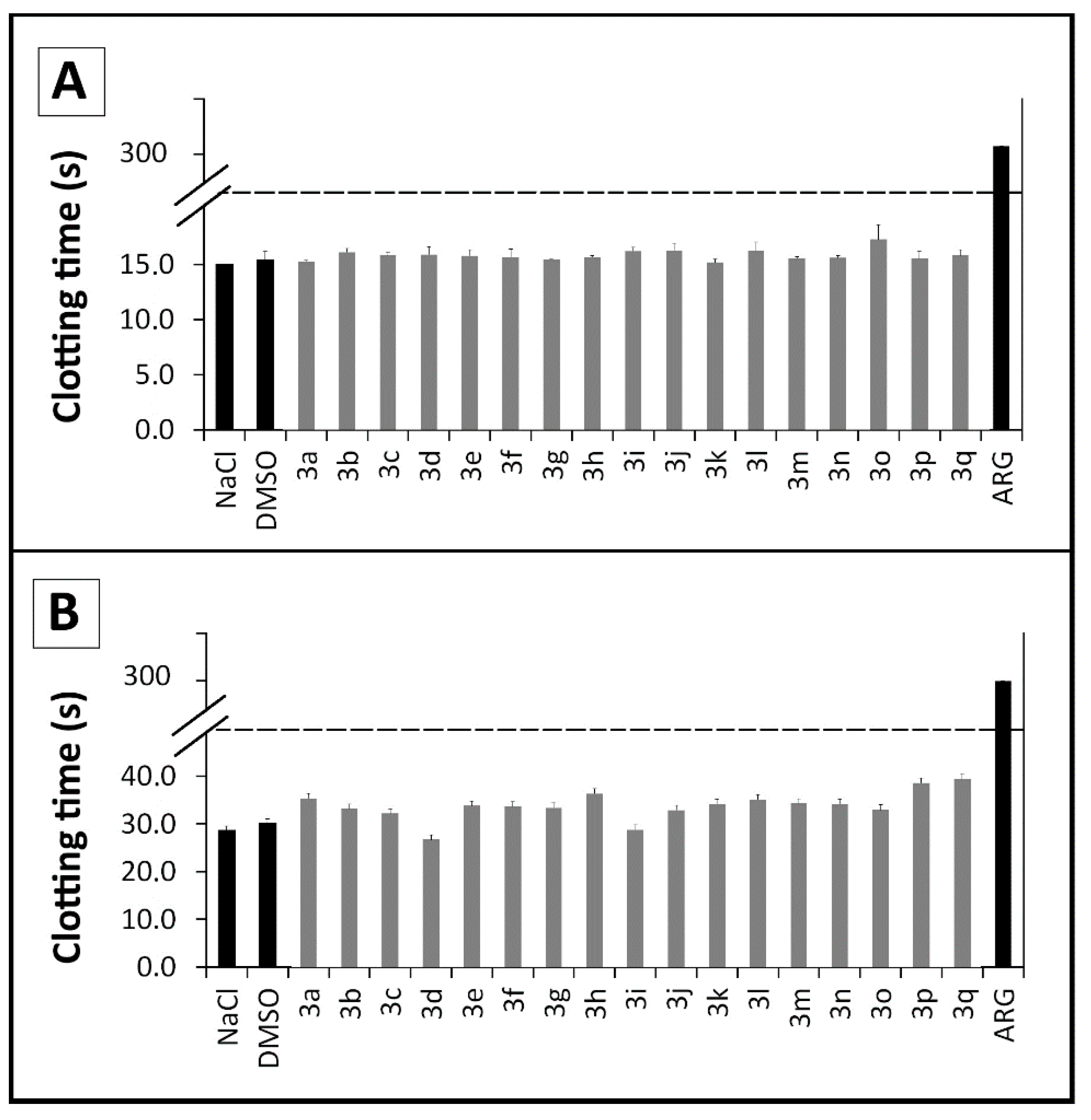
2.2.3. Coagulation Assays
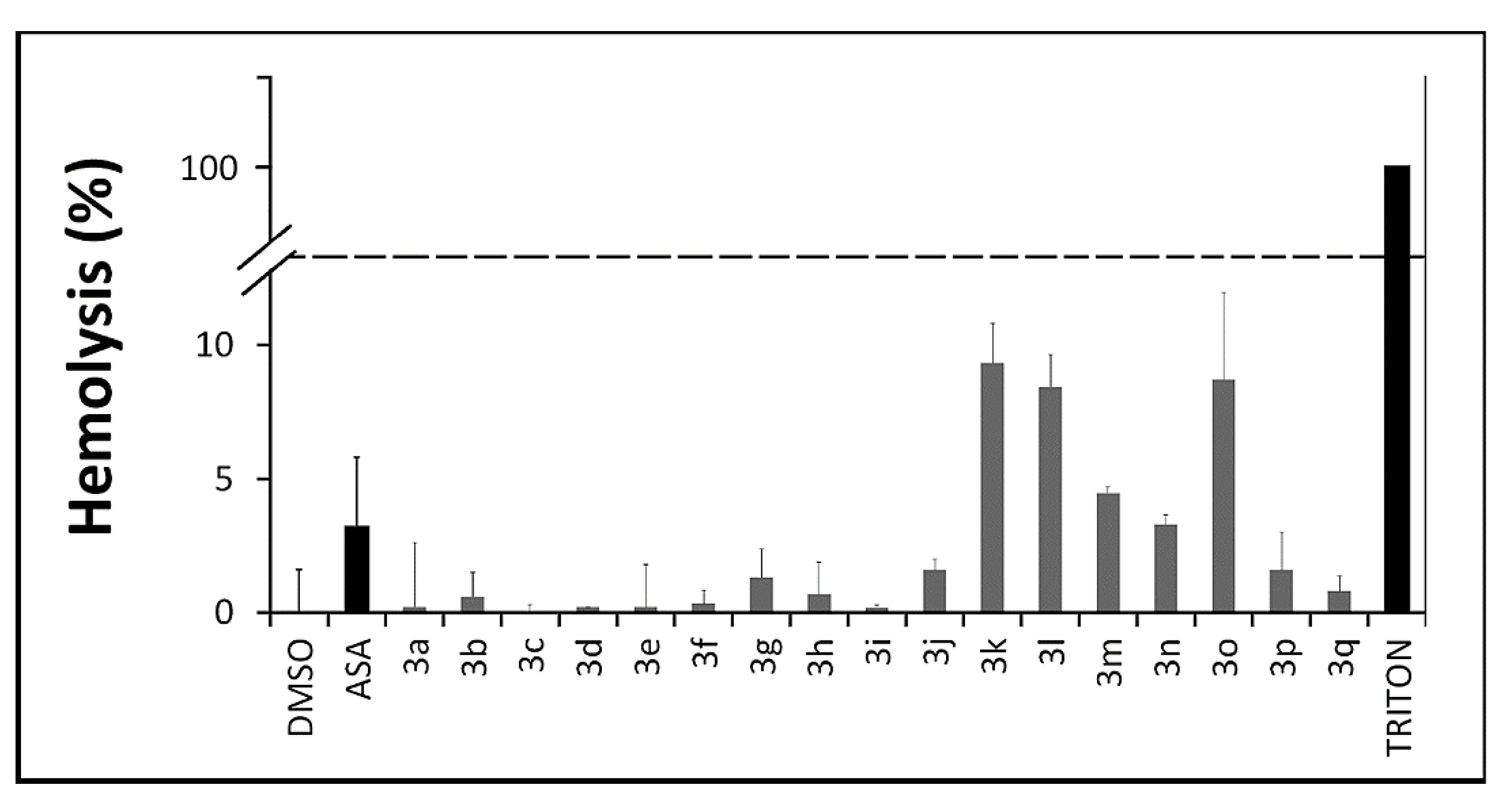
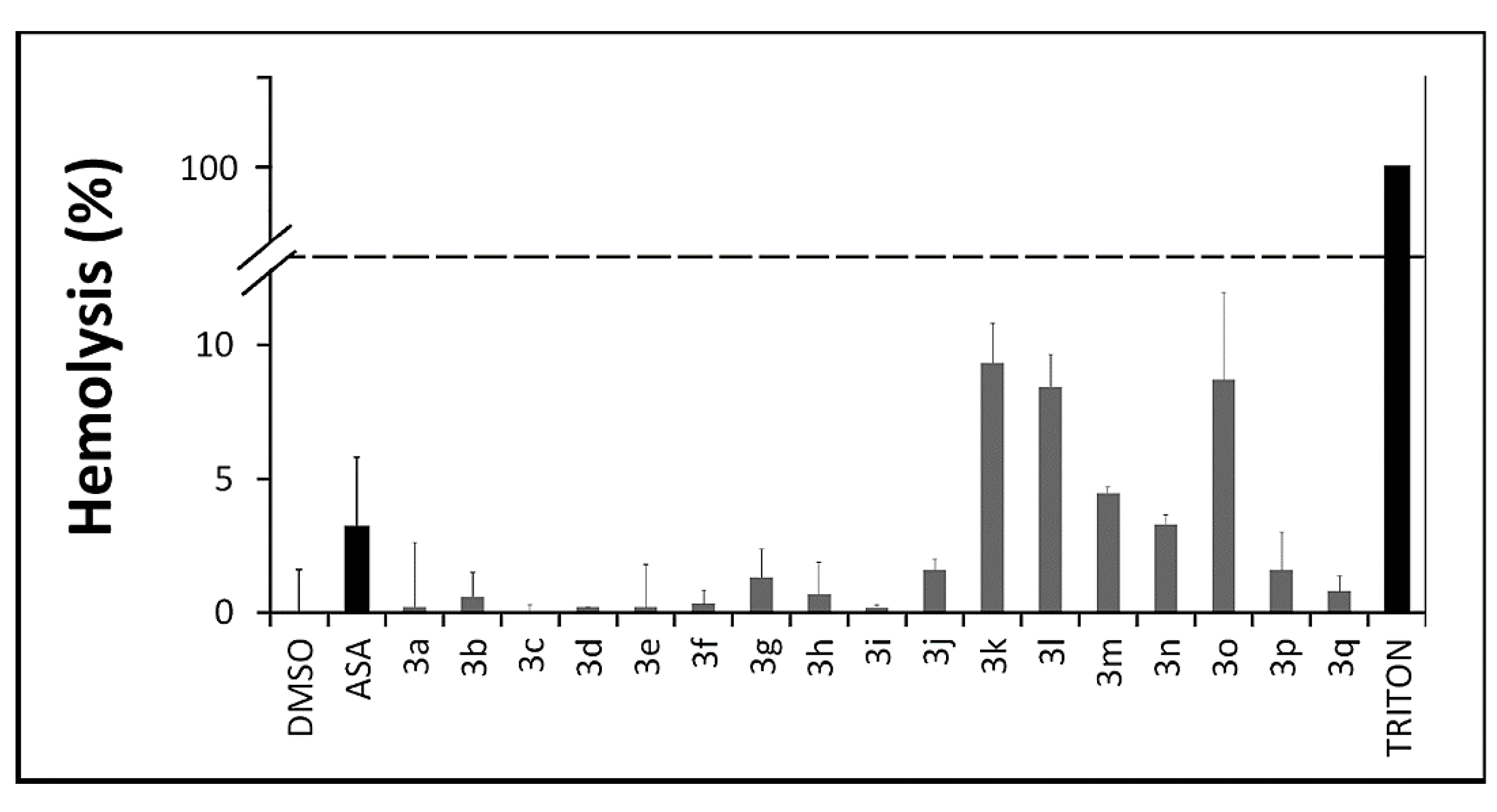
2.2.4. In Vitro Toxicity Assays
2.3. Computational Analysis
2.3.1. Structure-Activity Relationship Studies (SAR)
| # | EHOMO (eV) | ELUMO (eV) | DM | AREA (Å2) | VOLUME (Å3) | PSA | Lipinski Rule of Five | |||
|---|---|---|---|---|---|---|---|---|---|---|
| MW | clog P | HBD | HBA | |||||||
| 3a | −8.34 | 3.12 | 5.82 | 196.34 | 172.56 | 20.237 | 166.25 | 1.99 | 2 | 3 |
| 3b | −8.27 | 3.15 | 5.78 | 272.63 | 251.41 | 18.336 | 234.37 | 3.52 | 2 | 3 |
| 3c | −8.24 | 3.22 | 5.66 | 225.92 | 198.74 | 39.126 | 196.27 | 1.48 | 3 | 4 |
| 3d | −8.19 | 3.01 | 5.21 | 248.09 | 227.35 | 11.734 | 208.33 | 3.05 | 1 | 3 |
| 3e | −7.95 | 2.7 | 6.94 | 331.01 | 315.91 | 40.151 | 318.4 | 3.28 | 2 | 6 |
| 3f | −7.73 | 2.85 | 6.16 | 272.97 | 259.97 | 37.832 | 272.33 | 3.44 | 2 | 5 |
| 3g | −7.64 | 2.13 | 6.43 | 293.95 | 285.88 | 20.889 | 278.38 | 4.66 | 2 | 3 |
| 3h | −8.26 | 3.52 | 6.17 | 257.56 | 228.28 | 19.745 | 208.33 | 2.72 | 2 | 3 |
| 3i | −8.19 | 3.58 | 5.60 | 276.39 | 247.98 | 9.5490 | 222.36 | 3.12 | 1 | 3 |
| 3j | −8.10 | 3.16 | 5.94 | 303.20 | 281.49 | 35.421 | 286.36 | 3.51 | 2 | 5 |
| 3k | −8.26 | 3.65 | 5.78 | 276.49 | 246.67 | 19.462 | 222.36 | 3.00 | 2 | 3 |
| 3l | −8.27 | 3.64 | 5.62 | 295.98 | 265.13 | 20.208 | 236.38 | 3.58 | 2 | 3 |
| 3m | −8.21 | 3.72 | 5.15 | 294.52 | 266.42 | 9.4050 | 236.38 | 3.40 | 1 | 3 |
| 3n | −8.21 | 3.68 | 5.79 | 312.13 | 288.54 | 18.806 | 262.42 | 3.89 | 2 | 3 |
| 3o | −7.92 | 2.18 | 5.66 | 343.86 | 325.92 | 18.886 | 306.43 | 5.00 | 2 | 3 |
| 3p | −8.05 | 3.15 | 5.82 | 320.69 | 299.61 | 34.234 | 300.38 | 3.79 | 2 | 5 |
| 3q | −7.85 | 2.69 | 6.86 | 356.02 | 341.05 | 56.282 | 362.41 | 3.06 | 2 | 8 |
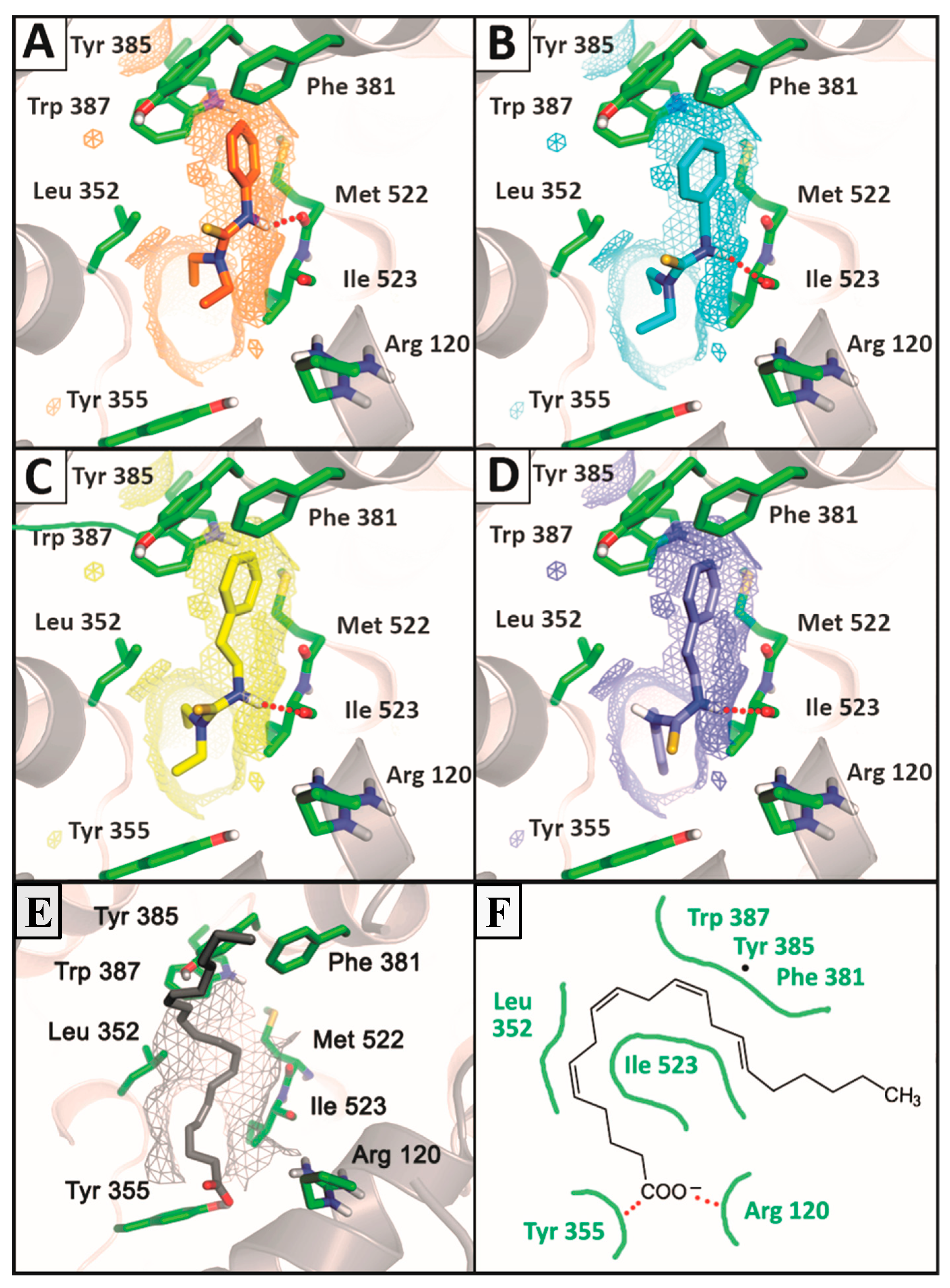
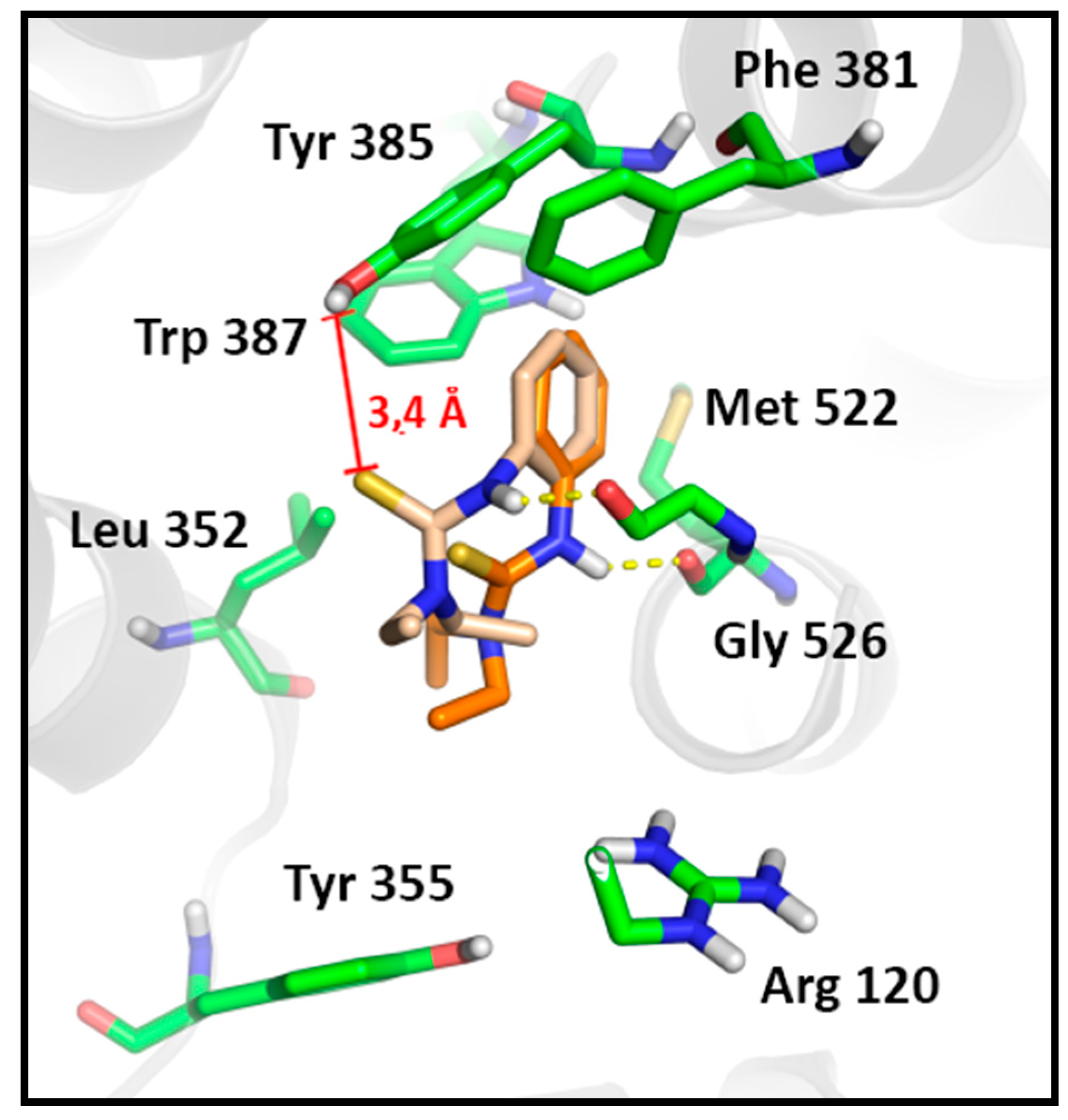
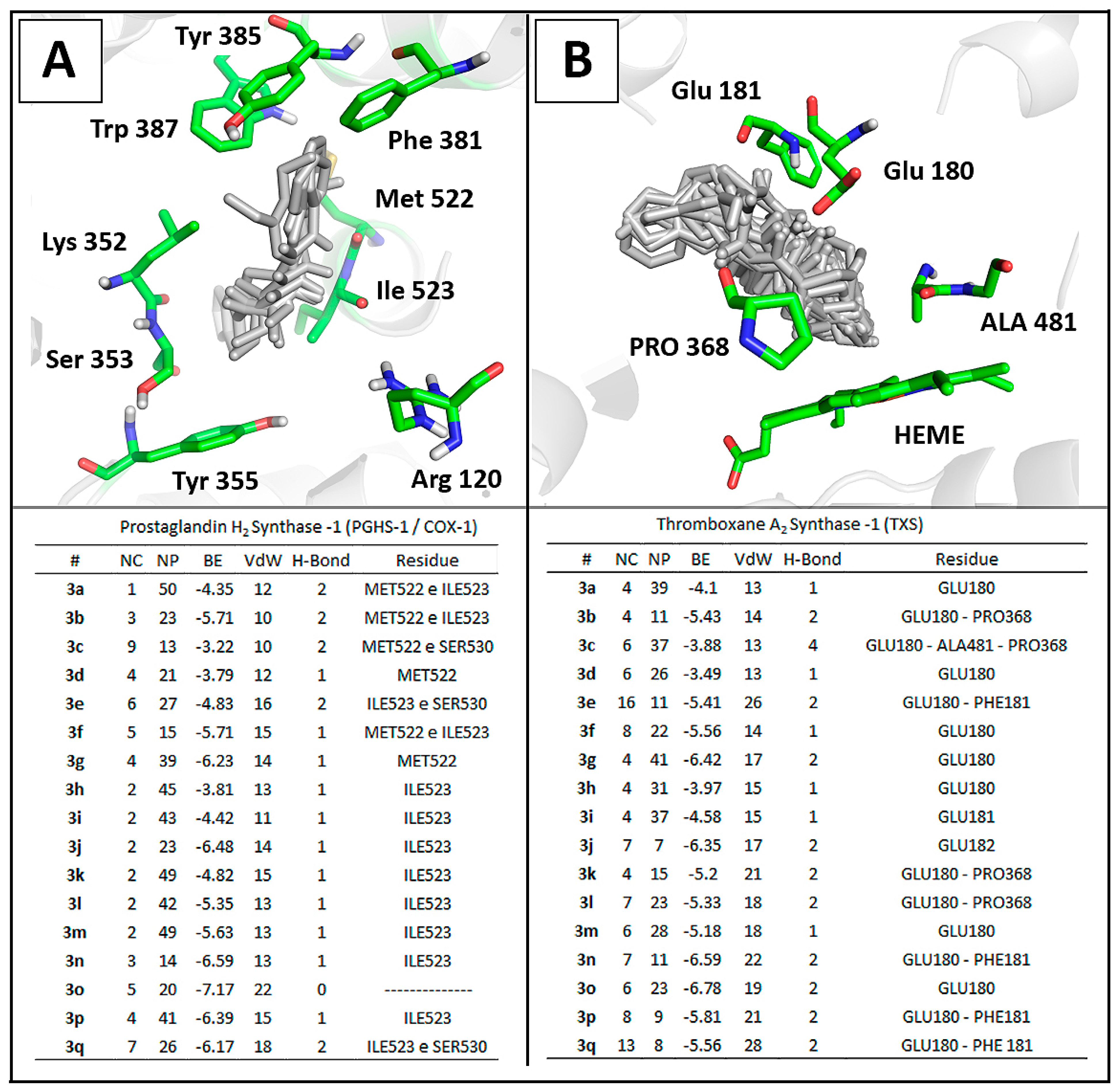
2.3.2. Docking Analysis
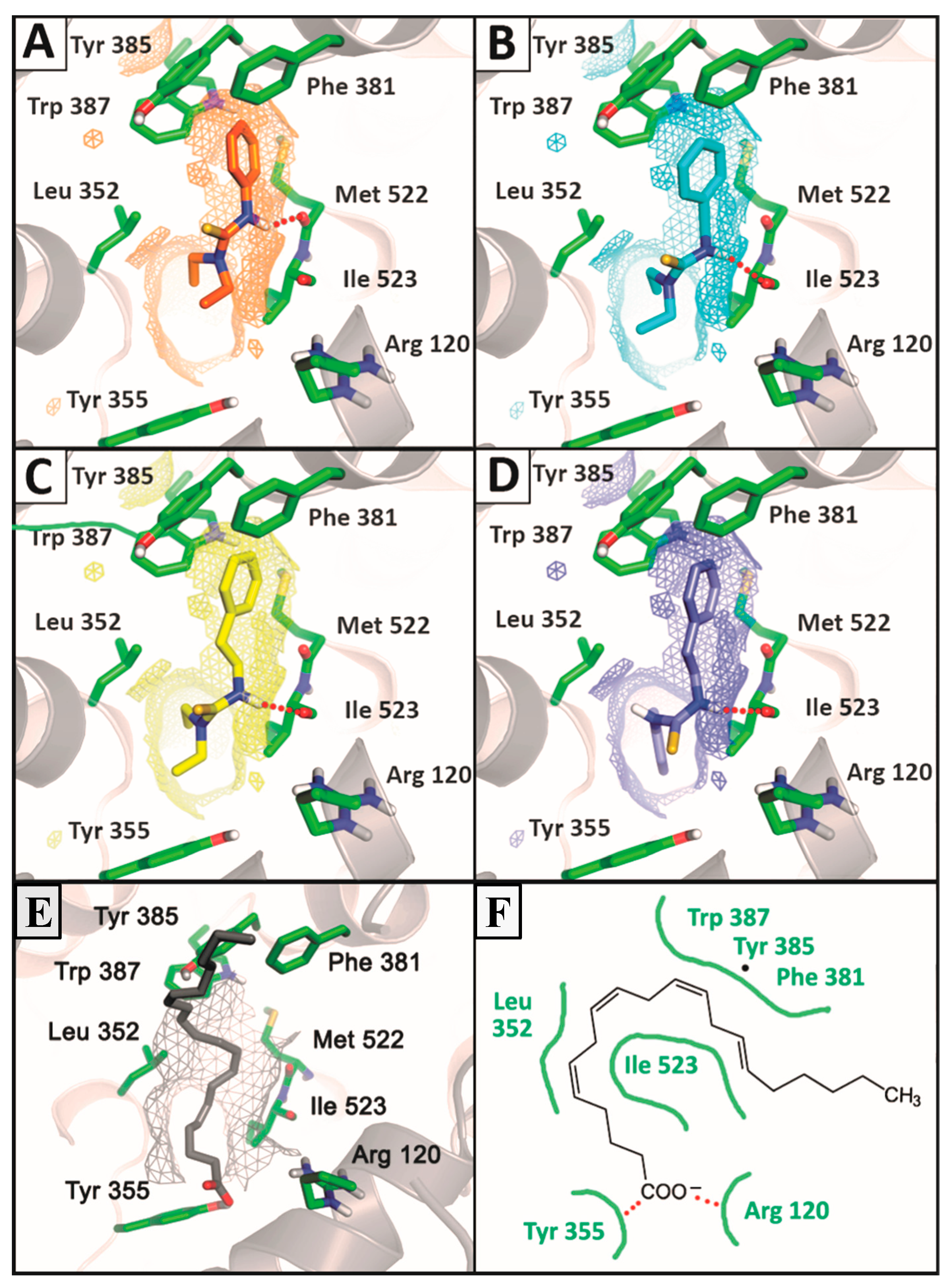
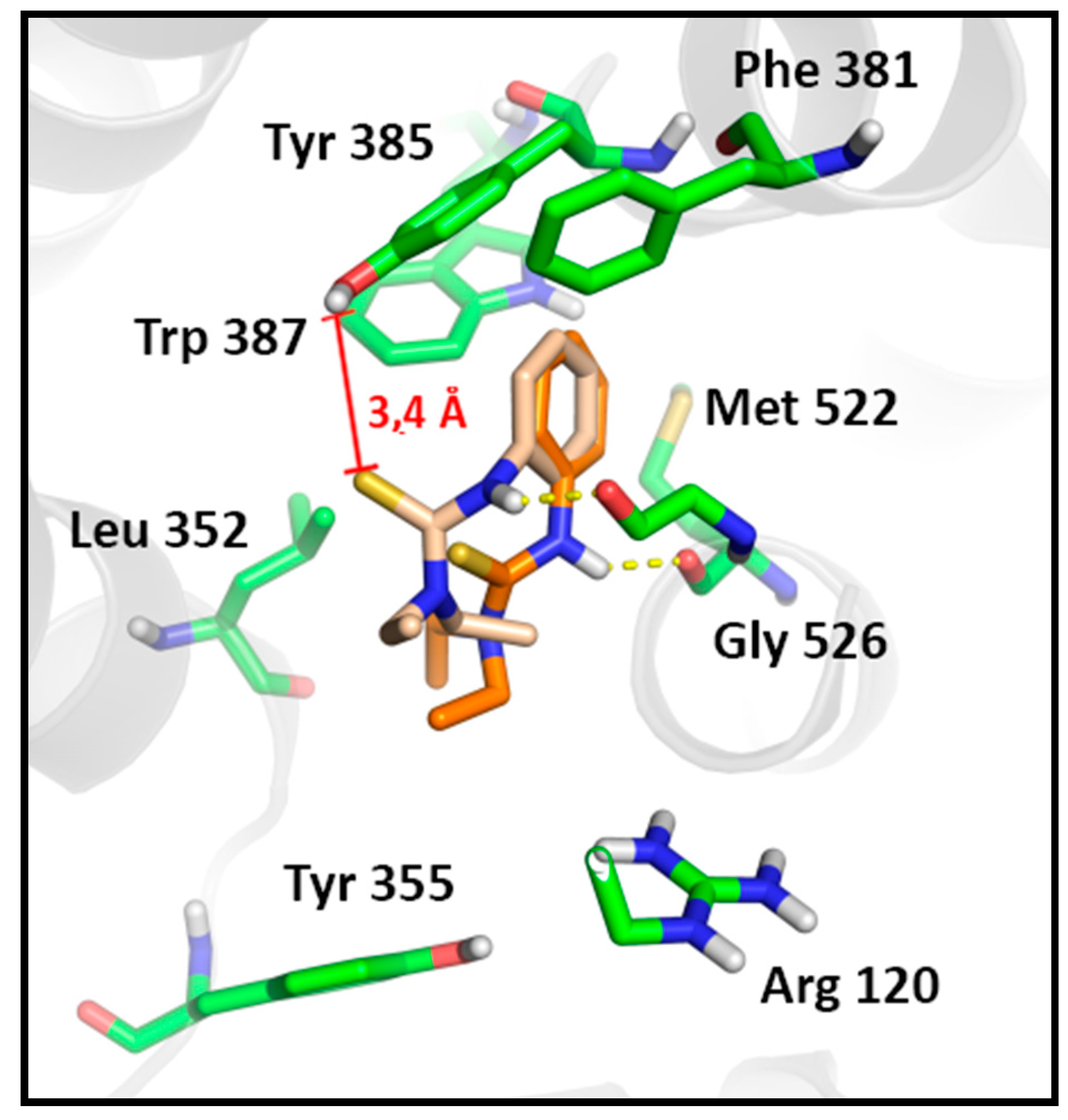
3. Experimental Section
3.1. General Information
General Procedure for the Preparation of Thioureas 3a–q
3.2. Pharmacology
3.2.1. Platelet Aggregation Assays
3.2.2. Clotting Assay
3.2.3. Measurement of Plasma TXB2 Levels
3.2.4. Measurement of Plasma PGE2 Levels
3.2.5. Hemolysis Assay
3.2.6. Reverse Mutagenesis to Histidine Prototrophy (Ames Test)—“Spot Test”
| Designations | Relevant Genotype |
|---|---|
| TA97 | hisD6610/ hisO1242—ΔuvrB rfa pKM101 (ampR) |
| TA98 | hisD3052—ΔuvrB rfa pKM101(ampR) |
| TA100 | hisG46—ΔuvrB rfa pKM101 (ampR) |
| TA102 | hisG428-wild type rfapKM101(ampR) pAQ1 (tetR) |
3.2.7. SOS Chromotest—“Spot Test”
| Designations | Critical Markers | Other Markers |
|---|---|---|
| PQ35 | sfiA::Mud(Aplac) cts lacΔU169 mal+, uvr+, galEgalY, PhoC, rfa | Same markers as GC4436 rpoB |
| PQ37 | sfiA::Mud(Aplac) cts lacΔU169 mal+, uvrA, galEgalY, PhoC, rfa | Same markers as GC4436 rpoB |
3.3. Molecular Modeling
SAR and Docking Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Almeida Chaves, D.S.; Costa, S.S.; de Almeida, A.P.; Frattani, F.; Assafim, M.; Zingali, R.B. Metabólitos secundários de origem vegetal: Uma fonte potencial de fármacos antitrombóticos. Quim Nova 2010, 33, 172–180. [Google Scholar]
- Baker, D.C.; Brassard, J. Review of Continuing Education Course on Hemostasis. Toxicol. Pathol. 2011, 39, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Al Ghumlas, A.K.; Gader, A.G.M.A. The Blood Platelet: An Intriguing Cell. J. Appl. Hematol. 2013, 4, 1–12. [Google Scholar]
- Naik, M.U.; Stalker, T.J.; Brass, L.F.; Naik, U.P. JAM-A protects from thrombosis by suppressing integrin αIIbβ3-dependent outside-in signaling in platelets. Blood 2012, 119, 3352–3360. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Aleman, M.M.; Leiderman, K.; Machlus, K.R. Procoagulant activity in hemostasis and thrombosis: Virchow’s triad revisited. Anesth. Analg. 2012, 114, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kei, A.A.; Florentin, M.; Mikhailidis, D.P.; Elisaf, M.S.; Liberopoulos, E.N. Review: Antiplatelet drugs: What comes next? Clin. Appl. Thromb. Hemost. 2011, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Datto, C.; Raines, S.; Yeomans, N.D. Impact of concomitant low-dose aspirin on the safety and tolerability of naproxen and esomeprazole magnesium delayed-release tablets in patients requiring chronic nonsteroidal anti-inflammatory drug therapy: An analysis from 5 Phase III studies. J. Thromb. Thrombolysis 2013, 38, 11–23. [Google Scholar] [CrossRef]
- Choi, J.-T.; Shin, K.-A.; Kim, Y.-K. Prevalence of Aspirin Resistance and Clinical Characteristics in Patients with Cerebral Infarction. Korean Soc. Biomed. Lab. Sci. 2013, 19, 233–238. [Google Scholar]
- Kunadian, V.; Sinclair, H.; Sutton, A.; Dangas, G.D. Aspirin, Platelet P2Y12 Receptor Inhibitors, and Other Oral Antiplatelets: Comparative Pharmacology and Role in Elective PCI. Interv. Cardiol. Clin. 2013, 2, 527–535. [Google Scholar]
- Shah, R.; Keough, L.A.; Belalcazar-Portacio, A.; Ramanathan, K.B. Ticagrelor as an alternative in clopidogrel-associated neutropenia. Platelets 2014, 26, 80–82. [Google Scholar] [CrossRef] [PubMed]
- Mattiello, T.; Guerriero, R.; Lotti, L.V.; Trifirò, E.; Felli, M.P.; Barbarulo, A.; Pucci, B.; Gazzaniga, P.; Gaudio, C.; Frati, L. Others Aspirin Extrusion From Human Platelets Through Multidrug Resistance Protein-4–Mediated TransportEvidence of a Reduced Drug Action in Patients After Coronary Artery Bypass Grafting. J. Am. Coll. Cardiol. 2011, 58, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Floyd, C.N.; Ferro, A. Mechanisms of aspirin resistance. Pharmacol. Ther. 2014, 141, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Paloscia, L.; Liani, R.; di Nicola, M.; di Marco, M.; Lattanzio, S.; la Barba, S.; Pascale, S.; Mascellanti, M.; Davì, G. Circulating Myeloid-Related Protein–8/14 is Related to Thromboxane-Dependent Platelet Activation in Patients With Acute Coronary Syndrome, With and Without Ongoing Low-Dose Aspirin Treatment. J. Am. Heart Assoc. 2014, 3, e000903. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Tacconelli, S.; Perrone, M.G.; Malerba, P.; Simone, L.; Scilimati, A.; Lavecchia, A.; Dovizio, M.; Marcantoni, E.; Bruno, A. Others Synthesis, pharmacological characterization, and docking analysis of a novel family of diarylisoxazoles as highly selective cyclooxygenase-1 (COX-1) inhibitors. J. Med. Chem. 2013, 56, 4277–4299. [Google Scholar] [CrossRef] [PubMed]
- Roche, V.F. A receptor-grounded approach to teaching nonsteroidal antiinflammatory drug chemistry and structure-activity relationships. Am. J. Pharm. Educ. 2009, 73, 143. [Google Scholar] [CrossRef] [PubMed]
- Khamdang, S.; Takeda, M.; Noshiro, R.; Narikawa, S.; Enomoto, A.; Anzai, N.; Piyachaturawat, P.; Endou, H. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2002, 303, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Hagos, Y.; Wolff, N.A. Assessment of the role of renal organic anion transporters in drug-induced nephrotoxicity. Toxins 2010, 2, 2055–2082. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, A.J.; Crews, B.C.; Daniel, C.M.; Blobaum, A.L.; Kingsley, P.J.; Ghebreselasie, K.; Marnett, L.J. Cyclooxygenase-1-selective inhibitors based on the(E)-2'-des-methyl-sulindac sulfide scaffold. J. Med. Chem. 2012, 55, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, Z.; Tang, S.; Hewlett, I.; Pang, R.; He, M.; He, S.; Tian, B.; Chen, K.; Yang, M. Discovery of dual inhibitors targeting both HIV-1 capsid and human cyclophilin A to inhibit the assembly and uncoating of the viral capsid. Bioorg. Med. Chem. 2009, 17, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Q.; Lv, P.-C.; Yan, T.; Zhu, H.-L. Urea derivatives as anticancer agents. Anticancer Agents Med. Chem. 2009, 9, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, Y.; Wu, G.; Zhou, J.; Huang, W.; Hu, X. Synthesis and biological evaluation of sulfonylurea and thiourea derivatives substituted with benzenesulfonamide groups as potential hypoglycemic agents. Bioorg. Med. Chem. Lett. 2009, 19, 1740–1744. [Google Scholar] [CrossRef] [PubMed]
- Perlovich, G.L.; Proshin, A.N.; Volkova, T.V.; Kurkov, S.V.; Grigoriev, V.V.; Petrova, L.N.; Bachurin, S.O. Novel isothiourea derivatives as potent neuroprotectors and cognition enhancers: Synthesis, biological and physicochemical properties. J. Med. Chem. 2009, 52, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.-J.; Wang, L.-W.; Lee, C.-C.; Lee, Y.-C.; Chao, Y.-S.; Hsu, T.-A.; Chern, J.-H. Design, synthesis, and anti-HCV activity of thiourea compounds. Bioorg. Med. Chem. Lett. 2009, 19, 1950–1955. [Google Scholar] [CrossRef] [PubMed]
- Kazimierczuk, Z.; Chalimoniuk, M.; Laudy, A.E.; Moo-Puc, R.; Cedillo-Rivera, R.; Starosciak, B.J.; Chrapusta, S.J. Synthesis and antimicrobial and nitric oxide synthase inhibitory activities of novel isothiourea derivatives. Arch. Pharm. Res. 2010, 33, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Michaux, C.; de Leval, X.; Julémont, F.; Dogné, J.-M.; Pirotte, B.; Durant, F. Structure-based pharmacophore of COX-2 selective inhibitors and identification of original lead compounds from 3D database searching method. Eur. J. Med. Chem. 2006, 41, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Ventosa-Andrés, P.; Valdivielso, Á.M.; Pappos, I.; García-López, M.T.; Tsopanoglou, N.E.; Herranz, R. Design, synthesis and biological evaluation of new peptide-based ureas and thioureas as potential antagonists of the thrombin receptor PAR1. Eur. J. Med. Chem. 2012, 58, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Okada, Y.; Sabirov, R.Z. Regulation of an ATP-conductive large-conductance anion channel and swelling-induced ATP release by arachidonic acid. J. Physiol. 2002, 542, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Dearman, J.; Abram, S.R.; Carter, C.; Hester, R.L. Insulin resistance and impaired functional vasodilation in obese Zucker rats. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H1658–H1666. [Google Scholar] [CrossRef] [PubMed]
- Sathler, P.C.; Santana, M.; Lourenço, A.L.; Rodrigues, C.R.; Abreu, P.; Cabral, L.M.; Castro, H.C. Human thromboxane synthase: Comparative modeling and docking evaluation with the competitive inhibitors Dazoxiben and Ozagrel. J. Enzym. Inhib. Med. Chem. 2013, 29, 527–531. [Google Scholar] [CrossRef]
- Cathcart, M.C.; Gately, K.; Cummins, R.; Drakeford, C.; Kay, E.W.; O’Byrne, K.J.; Pidgeon, G.P. Thromboxane synthase expression and correlation with VEGF and angiogenesis in non-small cell lung cancer. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2014, 1842, 747–755. [Google Scholar] [CrossRef]
- Sun, J.; Zhong, W.; Gu, Y.; Groome, L.J.; Wang, Y. 1, 25 (OH)<sub> 2 D<sub> 3 suppresses COX-2 up-regulation and thromboxane production in placental trophoblast cells in response to hypoxic stimulation. Placenta 2014, 35, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Cuendet, M.; Mesecar, A.D.; DeWitt, D.L.; Pezzuto, J.M. An ELISA method to measure inhibition of the COX enzymes. Nat. Protoc. 2006, 1, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Iuchi, Y.; Fujii, J.; Sugata, H.; Iijima, K.; Kato, K.; Shimosegawa, T.; Yoshimura, T. In vivo study on cross talk between inducible nitric-oxide synthase and cyclooxygenase in rat gastric mucosa: Effect of cyclooxygenase activity on nitric oxide production. J. Pharmacol. Exp. Ther. 2004, 309, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.P.; Dong, L.; Yuan, C.; Noon, K.R.; Smith, W.L. Asymmetric acetylation of the cyclooxygenase-2 homodimer by aspirin and its effects on the oxygenation of arachidonic, eicosapentaenoic, and docosahexaenoic acids. Mol. Pharmacol. 2010, 77, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jennings, N.L.; Dart, A.M.; Du, X.-J. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J. Exp. Med . 2012, 2, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Sadilkova, L.; Paluch, Z.; Mottlova, J.; Bednar, F.; Alusik, S. The purification step is not crucial in EIA measurements of thromboxane B2 and 11-dehydrothromboxane B2 in human plasma. Clin. Lab. 2012, 58, 177–183. [Google Scholar] [PubMed]
- Jeng, J.-H.; Chen, S.-Y.; Liao, C.-H.; Tung, Y.-Y.; Lin, B.-R.; Hahn, L.-J.; Chang, M.-C. Modulation of platelet aggregation by areca nut and betel leaf ingredients: Roles of reactive oxygen species and cyclooxygenase. Free Radic. Biol. Med. 2002, 32, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Aziz, S.M.; Pauly, T.H.; Gillespie, M.N. Intrinsic microbicidal activity and pulmonary hypertension in isolated newborn piglet lungs. Pediatr. Res. 1993, 34, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.P., Jr.; Klausner, J.M.; Anner, H.; Feingold, H.; Kobzik, L.; Valeri, C.R.; Shepro, D.; Hechtman, H.B. Inhibition of thromboxane (Tx) synthesis by free radical scavengers. J. Trauma-Inj. Infect. Crit. Care 1988, 28, 458–464. [Google Scholar] [CrossRef]
- Araujo, M.C.P.; Antunes, L.M.; Takahashi, C.S. Protective effect of thiourea, a hydroxyl-radical scavenger, on curcumin-induced chromosomal aberrations in an in vitro mammalian cell system. Teratog. Carcinog. Mutagen. 2001, 21, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Ulbrich, H.K.; Dannhardt, G. Investigations concerning the COX/5-LOX inhibiting and hydroxyl radical scavenging potencies of novel 4, 5-diaryl isoselenazoles. Eur. J. Med. Chem. 2008, 43, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.-C.; Lu, J.-F.; Wang, J.-S.; Yang, H.-C.; Pan, T.-A.; Chou, S.C.-W.; Wang, L.-H.; Chou, P.-T. Probing Ligand Binding to Thromboxane Synthase. Biochemistry (Mosc.) 2013, 52, 1113–1121. [Google Scholar] [CrossRef]
- Kontogiorgis, C.; Hadjipavlou-Litina, D. Thromboxane synthase inhibitors and thromboxane A2 receptor antagonists: A quantitative structure activity relationships (QSARs) analysis. Curr. Med. Chem. 2010, 17, 3162–3214. [Google Scholar] [CrossRef] [PubMed]
- Sathler, P.C.; Lourenço, A.L.; Rodrigues, C.R.; da Silva, L.C.R.P.; Cabral, L.M.; Jordão, A.K.; Cunha, A.C.; Vieira, M.C.B.; Ferreira, V.F.; Carvalho-Pinto, C.E.; et al. In vitro and in vivo analysis of the antithrombotic and toxicological profile of new antiplatelets N-acylhydrazone derivatives and development of nanosystems: Determination of novel NAH derivatives antiplatelet and nanotechnological approach. Thromb. Res. 2014, 134, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Curvers, J.; van de Kerkhof, D.; Stroobants, A.K.; van Den Dool, E.-J.; Scharnhorst, V. Measuring Direct Thrombin Inhibitors With Routine and Dedicated Coagulation Assays Which Assay Is Helpful? Am. J. Clin. Pathol. 2012, 138, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Raju, N.C.; Eikelboom, J.W.; Hirsh, J. Platelet ADP-receptor antagonists for cardiovascular disease: Past, present and future. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Palnati, G.R.; Avula, S.R.; Singh, S.; Jain, M.; Dikshit, M. Synthesis and evaluation of anti-thrombotic activity of benzocoumarin amide derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 3115–3121. [Google Scholar] [CrossRef] [PubMed]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, A.L.; Tempesti, T.C.; Spesia, M.B.; Milanesio, M.E.; Durantini, E.N. Synthesis and photodynamic properties of adamantylethoxy Zn(II) phthalocyanine derivatives in different media and in human red blood cells. Eur. J. Med. Chem. 2012, 50, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Ilker, M.F.; Nüsslein, K.; Tew, G.N.; Coughlin, E.B. Tuning the Hemolytic and Antibacterial Activities of Amphiphilic Polynorbornene Derivatives. J. Am. Chem. Soc. 2004, 126, 15870–15875. [Google Scholar] [CrossRef] [PubMed]
- Pennell, A.M.K.; Aggen, J.B.; Sen, S.; Chen, W.; Xu, Y.; Sullivan, E.; Li, L.; Greenman, K.; Charvat, T.; Hansen, D.; et al. 1-(4-Phenylpiperazin-1-yl)-2-(1H-pyrazol-1-yl)ethanones as novel CCR1 antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Hajisharifi, Z.; Piryaiee, M.; Mohammad Beigi, M.; Behbahani, M.; Mohabatkar, H. Predicting anticancer peptides with Chou’s pseudo amino acid composition and investigating their mutagenicity via Ames test. J. Theor. Biol. 2014, 341, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Quillardet, P.; Hofnung, M. The SOS chromotest, a colorimetric bacterial assay for genotoxins: Procedures. Mutat. Res. Mutagen. Relat. Subj. 1985, 147, 65–78. [Google Scholar] [CrossRef]
- Heler, R.; Bell, J.K.; Boland, L.M. Homology model and targeted mutagenesis identify critical residues for arachidonic acid inhibition of Kv4 channels. Channels 2013, 7, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.J.; de La Cruz, J.P.; Muñoz-Marin, J.; Guerrero, A.; Lopez-Villodres, J.A.; Madrona, A.; Espartero, J.L.; Gonzalez-Correa, J.A. Antiplatelet effect of new lipophilic hydroxytyrosol alkyl ether derivatives in human blood. Eur. J. Nutr. 2013, 52, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Aldrovandi, M.; Hammond, V.J.; Podmore, H.; Hornshaw, M.; Clark, S.R.; Marnett, L.J.; Slatter, D.A.; Murphy, R.C.; Collins, P.W.; O’Donnell, V.B. Human platelets generate phospholipid-esterified prostaglandins via cyclooxygenase-1 that are inhibited by low dose aspirin supplementation. J. Lipid Res. 2013, 54, 3085–3097. [Google Scholar] [CrossRef] [PubMed]
- Jedlitschky, G.; Greinacher, A.; Kroemer, H.K. Transporters in human platelets: Physiologic function and impact for pharmacotherapy. Blood 2012, 119, 3394–3402. [Google Scholar] [CrossRef] [PubMed]
- Malkowski, M.G.; Ginell, S.L.; Smith, W.L.; Garavito, R.M. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 2000, 289, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Bonomi, M.; Marinelli, L.; Gervasio, F.L.; Cavalli, A.; Novellino, E.; Parrinello, M. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 5411–5416. [Google Scholar] [CrossRef] [PubMed]
- Furse, K.E.; Pratt, D.A.; Porter, N.A.; Lybrand, T.P. Molecular dynamics simulations of arachidonic acid complexes with COX-1 and COX-2: Insights into equilibrium behavior. Biochemistry (Mosc.) 2006, 45, 3189–3205. [Google Scholar] [CrossRef]
- Kalgutkar, A.S.; Marnett, A.B.; Crews, B.C.; Remmel, R.P.; Marnett, L.J. Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitors. J. Med. Chem. 2000, 43, 2860–2870. [Google Scholar] [CrossRef] [PubMed]
- Selinsky, B.S.; Gupta, K.; Sharkey, C.T.; Loll, P.J. Structural analysis of NSAID binding by prostaglandin H2 synthase: Time-dependent and time-independent inhibitors elicit identical enzyme conformations. Biochemistry (Mosc.) 2001, 40, 5172–5180. [Google Scholar] [CrossRef]
- Duggan, K.C.; Walters, M.J.; Musee, J.; Harp, J.M.; Kiefer, J.R.; Oates, J.A.; Marnett, L.J. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J. Biol. Chem. 2010, 285, 34950–34959. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Balaramnavar, V.M.; Hohlfeld, T.; Saxena, A.K. Drug/drug interaction of common NSAIDs with antiplatelet effect of aspirin in human platelets. Eur. J. Pharmacol. 2013, 721, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I.; Hamberg, M.; Lindh, R.; Oliw, E.H. Novel insights into cyclooxygenases, linoleate diol synthases, and lipoxygenases from deuterium kinetic isotope effects and oxidation of substrate analogs. Biochim. Biophys. Acta 2012, 1821, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Tsai, A.-L.; Kulmacz, R.J. Cyclooxygenase Competitive Inhibitors Alter Tyrosyl Radical Dynamics in Prostaglandin H Synthase-2. Biochemistry (Mosc.) 2009, 48, 11902–11911. [Google Scholar] [CrossRef]
- Alawode, O.E.; Robinson, C.; Rayat, S. Clean Photodecomposition of 1-Methyl-4-phenyl-1H-tetrazole-5(4H)-thiones to Carbodiimides Proceeds via a Biradical. J. Org. Chem. 2011, 76, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, A.; Guo, Y.; Arthanari, H.; Wagner, G.; Halperin, J.A.; Chorev, M. Synthetic Studies toward Aryl-(4-aryl-4H-[1,2,4]triazole-3-yl)-amine from 1,3-Diarylthiourea as Urea Mimetics. J. Org. Chem. 2005, 70, 6362–6368. [Google Scholar] [CrossRef] [PubMed]
- Bernacki, A.L.; Zhu, L.; Hennings, D.D. A Selective and Convenient Method for the Synthesis of 2-Phenylaminothiazolines. Org. Lett. 2010, 12, 5526–5529. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.A.; Leon, E. The Thermal Breakdown of Diaryltetrazoles. J. Am. Chem. Soc. 1958, 80, 4647–4654. [Google Scholar] [CrossRef]
- De Sequeira Aguiar, L.C.; Viana, G.M.; dos Santos Romualdo, M.V.; Costa, M.V.; Bonato, B.S. A Simple and Green Procedure for the Synthesis of N-Benzylthioureas. Lett. Org. Chem. 2011, 8, 540–544. [Google Scholar] [CrossRef]
- Maddani, M.; Prabhu, K.R. A convenient method for the synthesis of substituted thioureas. Tetrahedron Lett. 2007, 48, 7151–7154. [Google Scholar] [CrossRef]
- Maddani, M.R.; Prabhu, K.R. A Concise Synthesis of Substituted Thiourea Derivatives in Aqueous Medium. J. Org. Chem. 2010, 75, 2327–2332. [Google Scholar] [CrossRef] [PubMed]
- Born, G.V.R.; Cross, M.J. The aggregation of blood platelets. J. Physiol. 1963, 168, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Cunha, A.C.; Figueiredo, J.M.; Tributino, J.L.; Miranda, A.L.; Castro, H.C.; Zingali, R.B.; Fraga, C.A.; de Souza, M.C.B.; Ferreira, V.F.; Barreiro, E.J. Antiplatelet properties of novel N-substituted-phenyl-1,2,3-triazole-4-acylhydrazone derivatives. Bioorg. Med. Chem. 2003, 11, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Jordão, A.K.; Ferreira, V.F.; Lima, E.S.; de Souza, M.C.B.V.; Carlos, E.C.L.; Castro, H.C.; Geraldo, R.B.; Rodrigues, C.R.; Almeida, M.C.B.; Cunha, A.C. Synthesis, antiplatelet and in silico evaluations of novel N-substituted-phenylamino-5-methyl-1H-1,2,3-triazole-4-carbohydrazides. Bioorg. Med. Chem. 2009, 17, 3713–3719. [Google Scholar] [CrossRef] [PubMed]
- Faye, E.; Drouet, L.; de Raucourt, E.; Green, A.; Bal-dit -Sollier, C.; Boudaoud, L.; Corcos, O.; Bergmann, J.-F.; Joly, F.; Lloret-Linares, C. Absorption and Efficacy of Acetylsalicylic Acid in Patients With Short Bowel Syndrome. Ann. Pharmacother. 2014, 48, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Parnham, M.J.; Wetzig, H. Toxicity screening of liposomes. Chem. Phys. Lipids 1993, 64, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Lautenschlaeger, C.; Kempe, K.; Tauhardt, L.; Schubert, U.S.; Fischer, D. Poly (2-ethyl-2-oxazoline) as Alternative for the Stealth Polymer Poly (ethylene glycol): Comparison of in vitro Cytotoxicity and Hemocompatibility. Macromol. Biosci. 2012, 12, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Maron, D.M.; Ames, B.N. Revised methods for the Salmonella mutagenicity test. Mutat. Res. Mutagen. Relat. Subj. 1983, 113, 173–215. [Google Scholar] [CrossRef]
- Valencia, A.; Prous, J.; Mora, O.; Sadrieh, N.; Valerio, J. A novel QSAR model of Salmonella mutagenicity and its application in the safety assessment of drug impurities. Toxicol. Appl. Pharmacol. 2013, 273, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Menegatti, R.; Cunha, A.C.; Ferreira, V.F.; Perreira, E.F.; El-Nabawi, A.; Eldefrawi, A.T.; Albuquerque, E.X.; Neves, G.; Rates, S.M.; Fraga, C.A.; et al. Design, synthesis and pharmacological profile of novel dopamine D2 receptor ligands. Bioorg. Med. Chem. 2003, 11, 4807–4813. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual screening with AutoDock: Theory and practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.B.R.; Lobato, C.C.; Braga, F.S.; Morais, S.S.S.; Santos, C.F.; Fernandes, C.P.; Brasil, D.S.B.; Hage-Melim, L.I.S.; Macêdo, W.J.C.; Carvalho, J.C.T. Application of Hartree-Fock Method for Modeling of Bioactive Molecules Using SAR and QSPR. Comput. Mol. Biosci. 2014, 4, 1–24. [Google Scholar] [CrossRef]
- Gervasio, F.L.; Laio, A.; Parrinello, M. Flexible docking in solution using metadynamics. J. Am. Chem. Soc. 2005, 127, 2600–2607. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, S.C.; Cavalcanti, D.F.; de Souza, A.M.; Castro, H.C.; Rodrigues, C.R.; Santos, D.O.; Albuquerque, M.G.; da Silva, G.G.; da Silva, F.C.; Ferreira, V.F. Trypanosoma cruzi: Insights into naphthoquinone effects on growth and proteinase activity. Exp. Parasitol. 2011, 127, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 3a–q are available from the authors via INCT-IF (http://www.inct-if.com.br/portal/).
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, A.L.; Saito, M.S.; Dorneles, L.E.G.; Viana, G.M.; Sathler, P.C.; Aguiar, L.C.d.S.; De Pádula, M.; Domingos, T.F.S.; Fraga, A.G.M.; Rodrigues, C.R.; et al. Synthesis and Antiplatelet Activity of Antithrombotic Thiourea Compounds: Biological and Structure-Activity Relationship Studies. Molecules 2015, 20, 7174-7200. https://doi.org/10.3390/molecules20047174
Lourenço AL, Saito MS, Dorneles LEG, Viana GM, Sathler PC, Aguiar LCdS, De Pádula M, Domingos TFS, Fraga AGM, Rodrigues CR, et al. Synthesis and Antiplatelet Activity of Antithrombotic Thiourea Compounds: Biological and Structure-Activity Relationship Studies. Molecules. 2015; 20(4):7174-7200. https://doi.org/10.3390/molecules20047174
Chicago/Turabian StyleLourenço, André Luiz, Max Seidy Saito, Luís Eduardo Gomes Dorneles, Gil Mendes Viana, Plínio Cunha Sathler, Lúcia Cruz de Sequeira Aguiar, Marcelo De Pádula, Thaisa Francielle Souza Domingos, Aline Guerra Manssour Fraga, Carlos Rangel Rodrigues, and et al. 2015. "Synthesis and Antiplatelet Activity of Antithrombotic Thiourea Compounds: Biological and Structure-Activity Relationship Studies" Molecules 20, no. 4: 7174-7200. https://doi.org/10.3390/molecules20047174
APA StyleLourenço, A. L., Saito, M. S., Dorneles, L. E. G., Viana, G. M., Sathler, P. C., Aguiar, L. C. d. S., De Pádula, M., Domingos, T. F. S., Fraga, A. G. M., Rodrigues, C. R., De Sousa, V. P., Castro, H. C., & Cabral, L. M. (2015). Synthesis and Antiplatelet Activity of Antithrombotic Thiourea Compounds: Biological and Structure-Activity Relationship Studies. Molecules, 20(4), 7174-7200. https://doi.org/10.3390/molecules20047174