
Synthesis, Crystal Structures and Spectroscopic Properties of Triazine-Based Hydrazone Derivatives; A Comparative Experimental-Theoretical Study


Abstract
:
1. Introduction
2. Results and Discussion
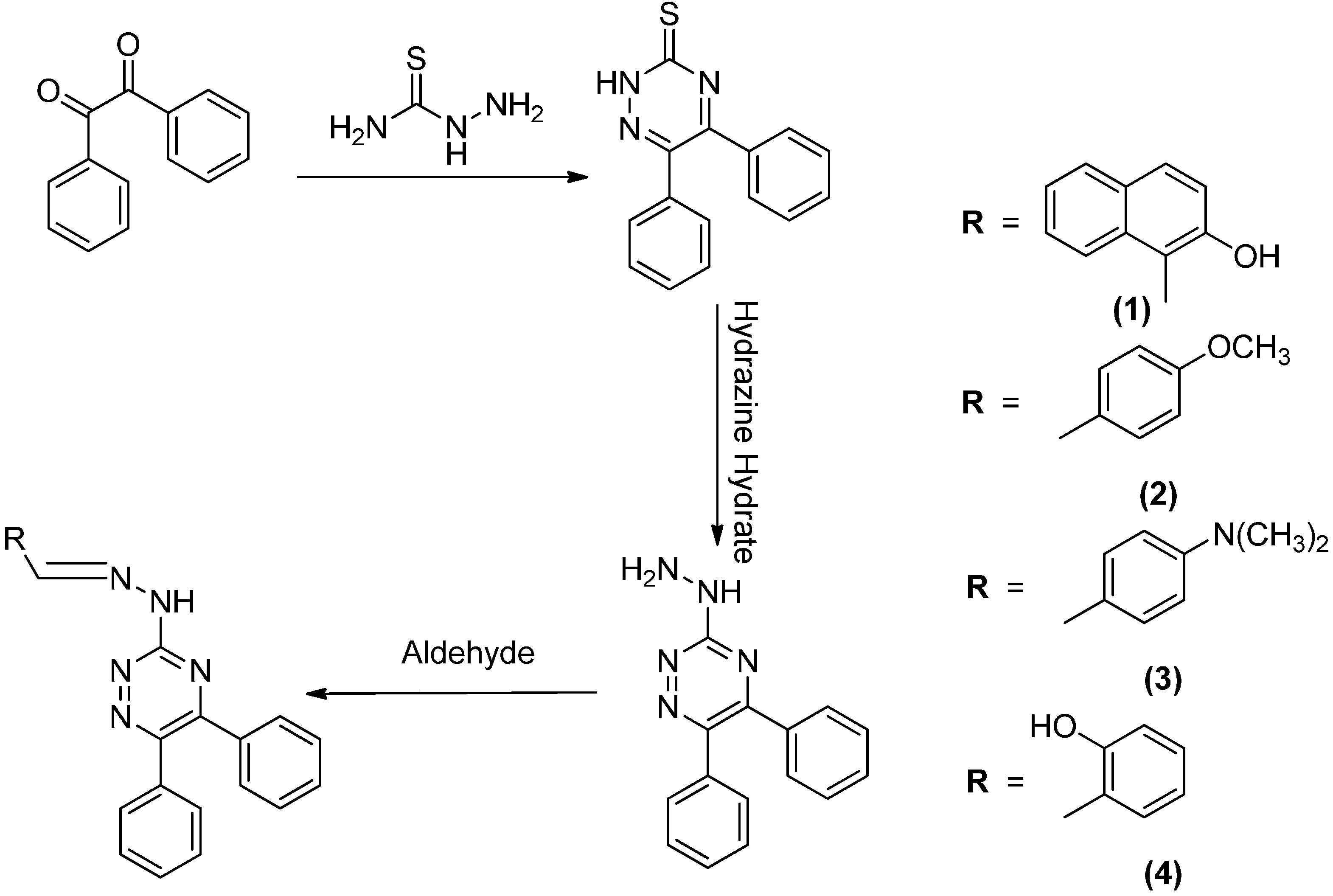
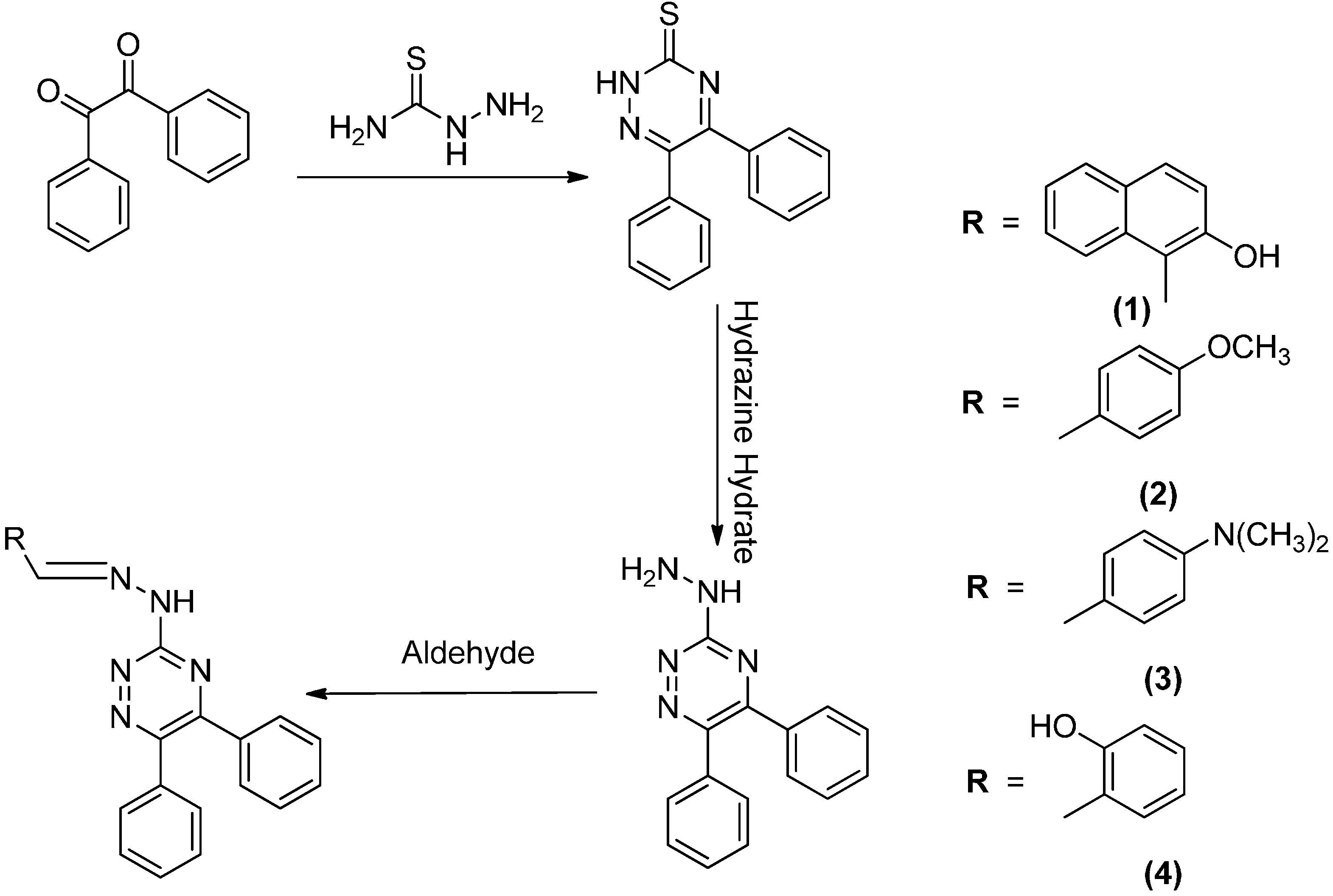
2.1. Synthesis
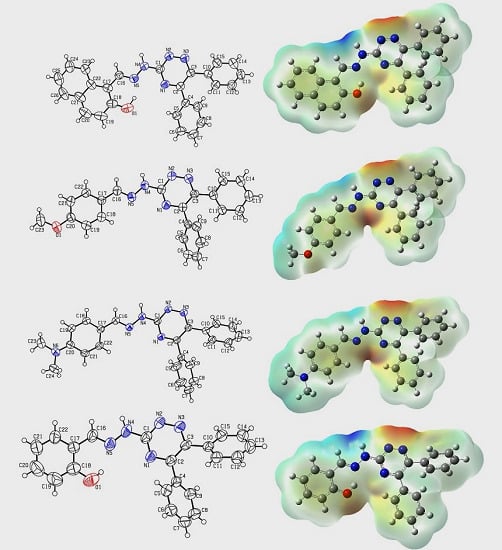
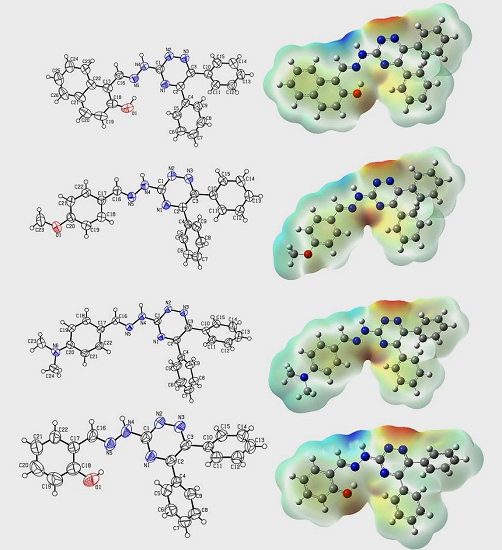
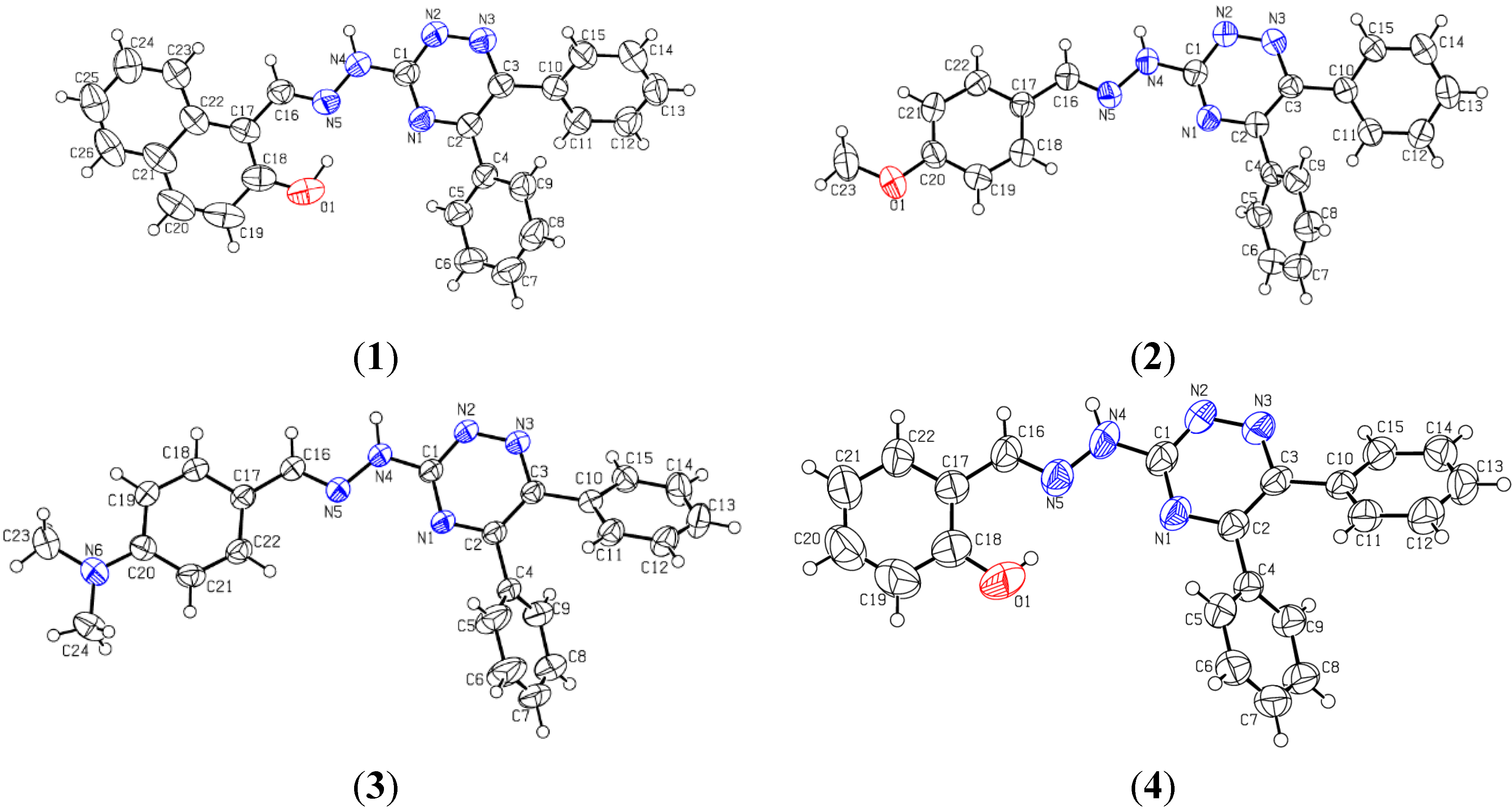
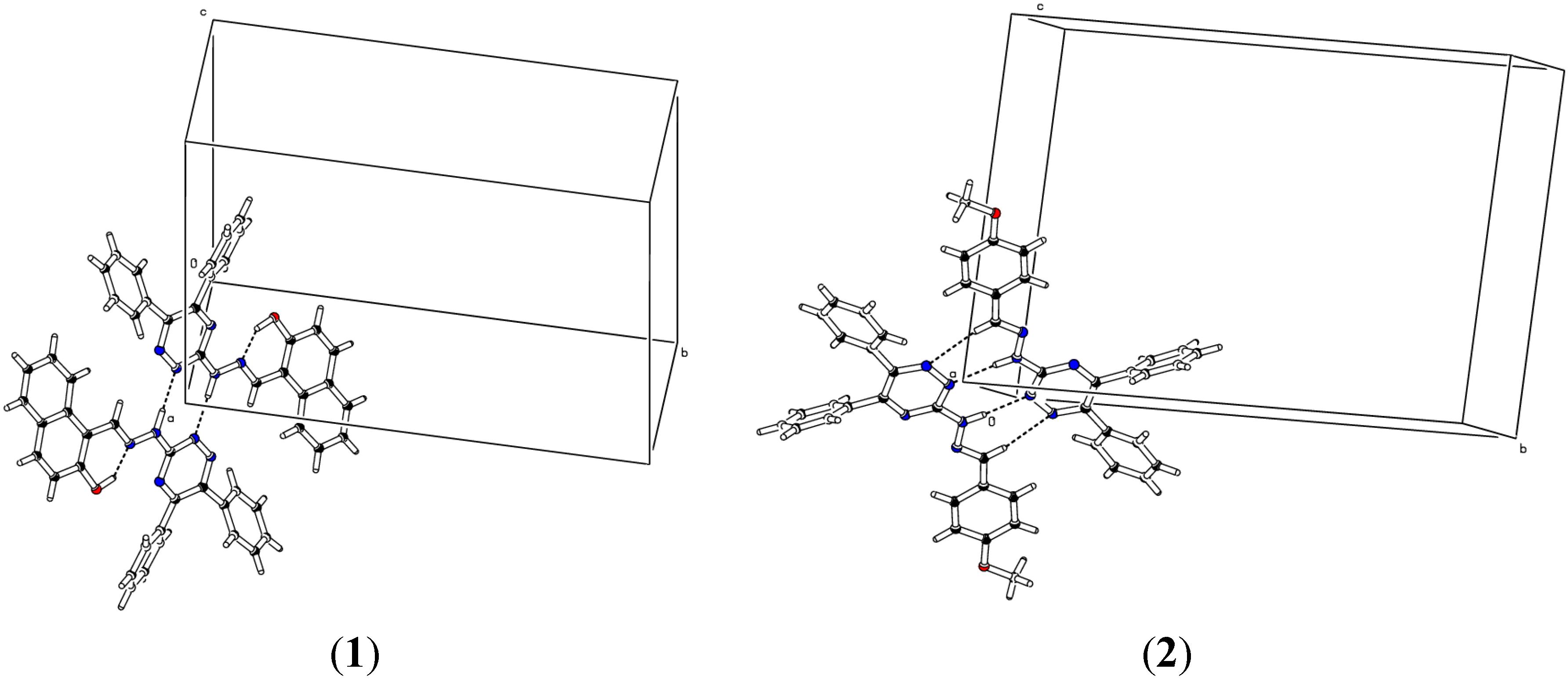
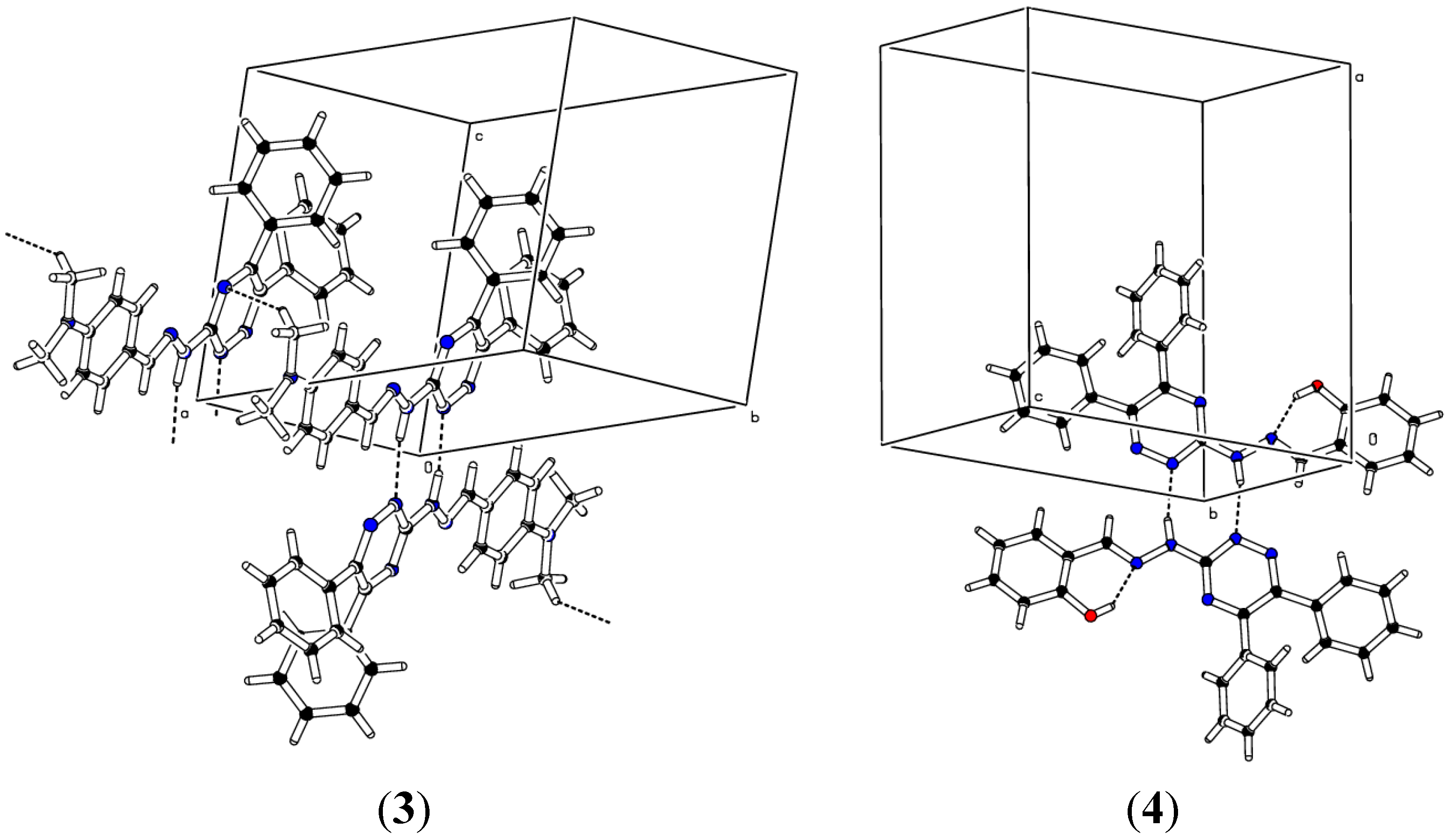
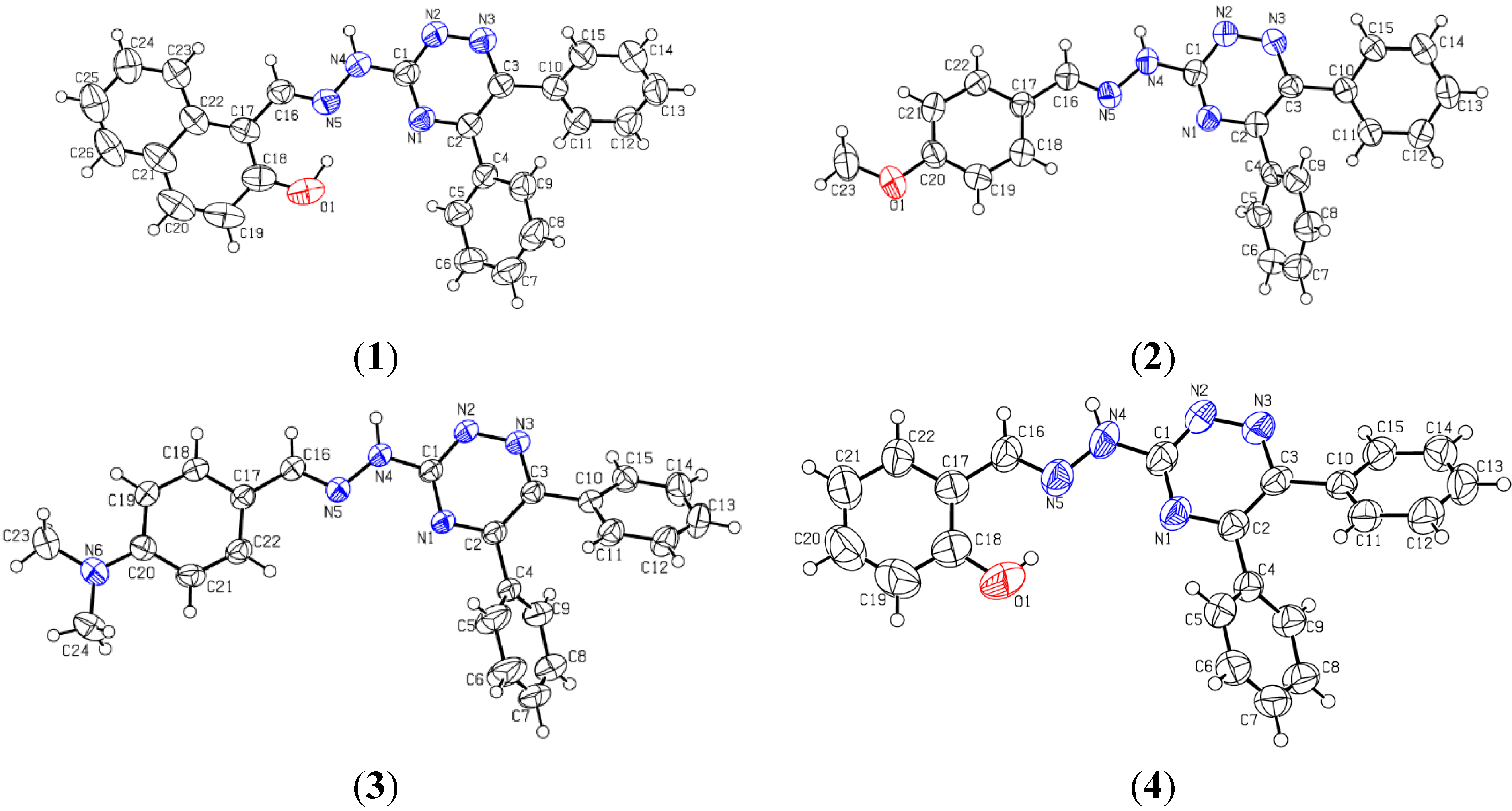
2.2. Crystallographic Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
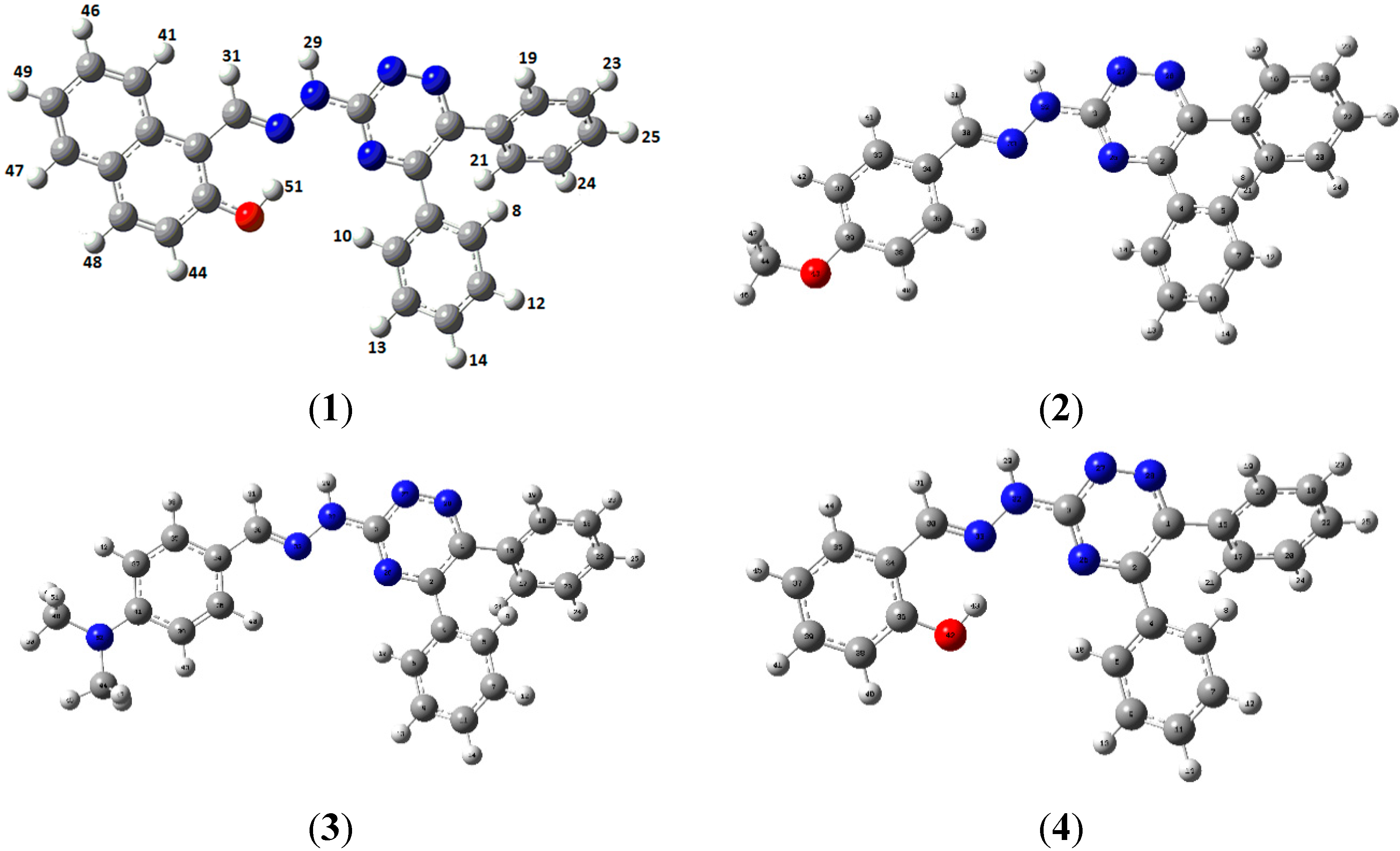{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identification Code | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Empirical formula | C26H19N5O | C23H19N5O | C24H22N6 | C22H17N5O |
| Formula weight | 417.46 | 381.43 | 394.48 | 367.41 |
| Temperature/K | 296.15 | 296.15 | 296.15 | 296.15 |
| Crystal system | monoclinic | monoclinic | triclinic | monoclinic |
| Space group | P21/n | P21/c | P-1 | P21/c |
| a/Å | 6.5745(2) | 5.9756(18) | 8.8933(10) | 14.9753(17) |
| b/Å | 19.9133(6) | 21.573(6) | 11.1477(11) | 6.1220(6) |
| c/Å | 15.9805(5) | 14.980(4) | 11.7484(13) | 21.038(2) |
| α/° | 90.00 | 90.00 | 91.212(9) | 90.00 |
| β/° | 96.369(3) | 93.45(2) | 106.044(10) | 103.992(11) |
| γ/° | 90.00 | 90.00 | 108.519(9) | 90.00 |
| Volume/Å3 | 2079.25(11) | 1927.7(9) | 1053.8(2) | 1871.6(3) |
| Z | 4 | 4 | 2 | 4 |
| ρcalcmg/mm3 | 1.334 | 1.314 | 1.243 | 1.304 |
| m/mm−1 | 0.677 | 0.084 | 0.077 | 0.084 |
| F(000) | 872.0 | 800.0 | 416.0 | 768.0 |
| Crystal size/mm3 | 0.48 × 0.16 × 0.15 | 0.48 × 0.08 × 0.05 | 0.32 × 0.26 × 0.17 | 0.43 × 0.15 × 0.05 |
| 2θ range for data collection | 7.12 to 152° | 5.76 to 58.66° | 5.62 to 58.9° | 5.6 to 59.14° |
| Index ranges | −6 ≤ h ≤ 8, | −7 ≤ h ≤ 8, | −11 ≤ h ≤ 9, | −19 ≤ h ≤ 19, |
| −25 ≤ k ≤ 21, | −27 ≤ k ≤ 24, | −12 ≤ k ≤ 14, | −8 ≤ k ≤ 7, | |
| −20 ≤ l ≤ 19 | −20 ≤ l ≤ 18 | −16 ≤ l ≤ 15 | −29 ≤ l ≤ 26 | |
| Reflections collected | 12339 | 11554 | 9379 | 11939 |
| Independent reflections | 4304[R(int) = 0.0185] | 4608[R(int) = 0.0727] | 5004[R(int) = 0.0252] | 4597[R(int) = 0.0620] |
| Data/restraints/parameters | 4304/0/290 | 4608/1/266 | 5004/0/274 | 4597/1/257 |
| Goodness-of-fit on F2 | 1.036 | 1.012 | 1.038 | 0.961 |
| Final R indexes [I ≥ 2σ (I)] | R1 = 0.0428, | R1 = 0.0626, | R1 = 0.0541, | R1 = 0.0601, |
| wR2 = 0.1156 | wR2 = 0.0925 | wR2 = 0.1221 | wR2 = 0.1236 | |
| Final R indexes [all data] | R1 = 0.0546, | R1 = 0.1988, | R1 = 0.0840, | R1 = 0.1880, |
| wR2 = 0.1273 | wR2 = 0.1358 | wR2 = 0.1423 | wR2 = 0.1670 | |
| Largest diff. peak/hole/e Å−3 | 0.19/−0.20 | 0.16/−0.19 | 0.25/−0.16 | 0.13/−0.16 |
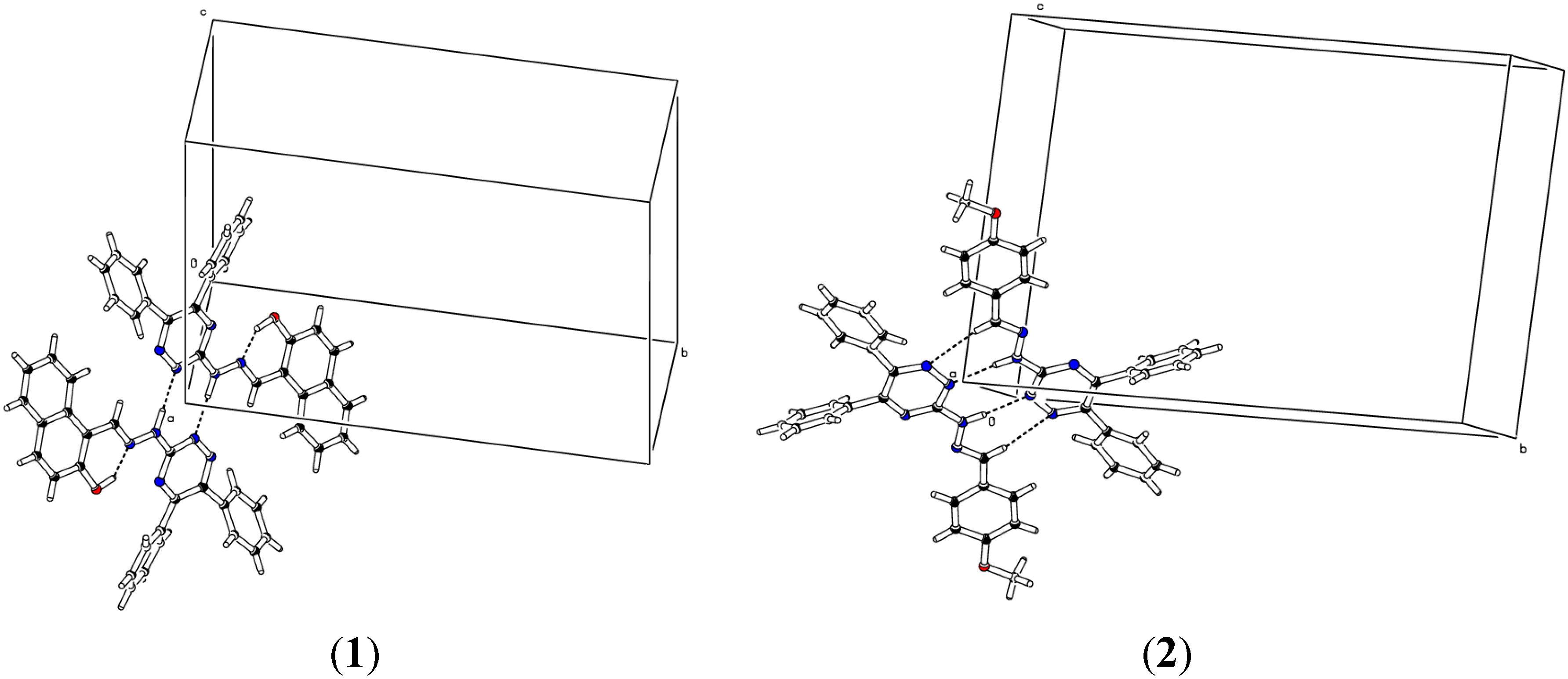
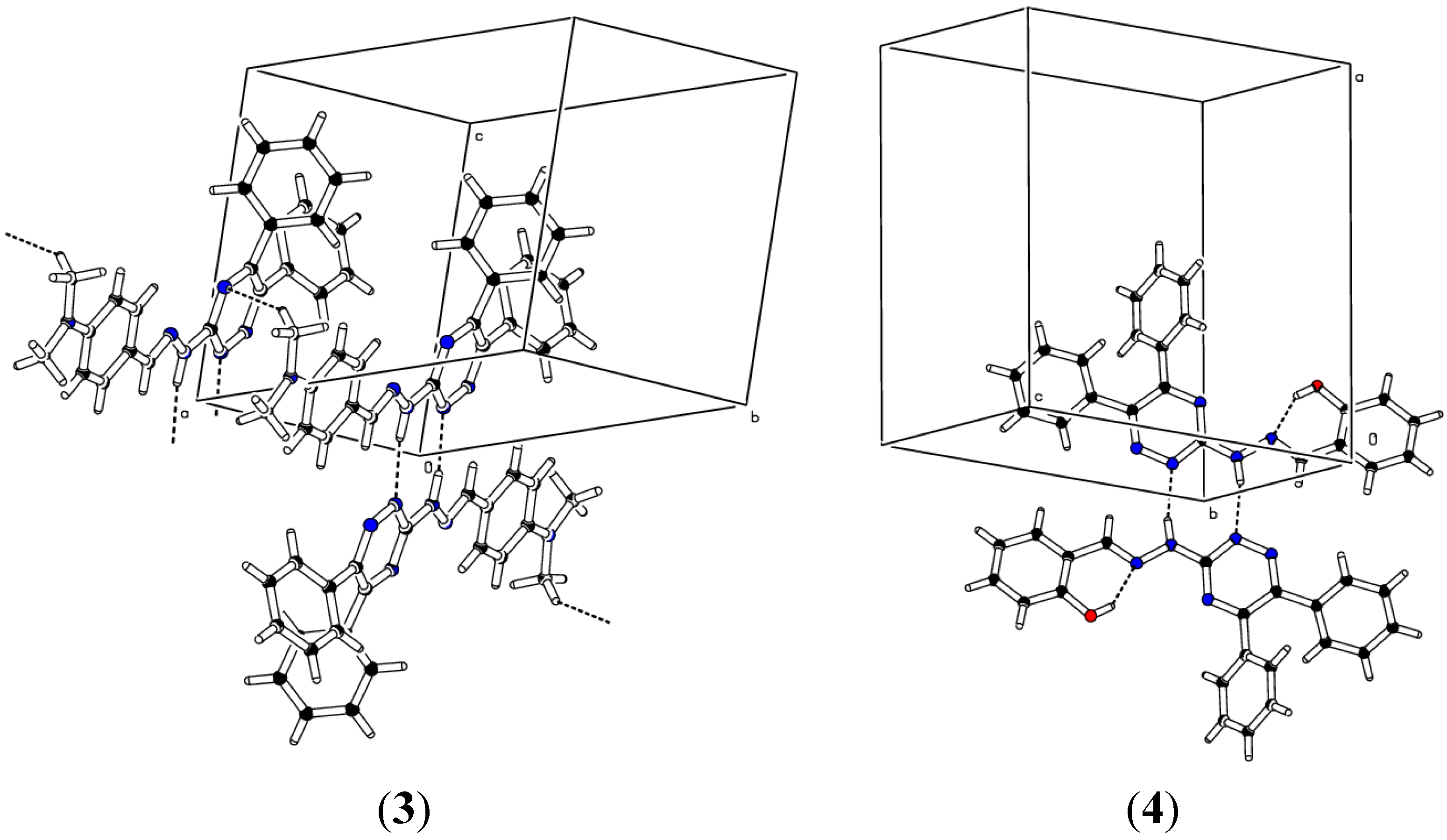
| D | H | A | d(D-H)/Å | d(H-A)/Å | d(D-A)/Å | D-H-A/° |
|---|---|---|---|---|---|---|
| (1) | ||||||
| N4 | H2 | N21 | 0.97 | 1.91 | 2.8750(18) | 171.9 |
| (2) | ||||||
| C16 | H16 | N32 | 0.93 | 2.58 | 3.496(4) | 168.5 |
| N4 | H1 | N22 | 0.893(10) | 2.047(11) | 2.939(3) | 176(3) |
| (3) | ||||||
| N4 | H1 | N23 | 0.97 | 2.03 | 2.995(2) | 168.8 |
| C24 | H24C | N14 | 0.96 | 2.59 | 3.449(3) | 148.7 |
| (4) | ||||||
| O1 | H1O | N5 | 0.82 | 1.93 | 2.646(3) | 146.1 |
| N4 | H2 | N25 | 0.889(10) | 2.033(12) | 2.907(4) | 167(3) |
| 12 − X, −Y, −Z, 21 − X, 1 − Y, 1 − Z, 3−X, −Y, −Z, 41 + X, +Y, +Z, 5−X, 2 − Y, −Z | ||||||
2.3. Geometries Optimization
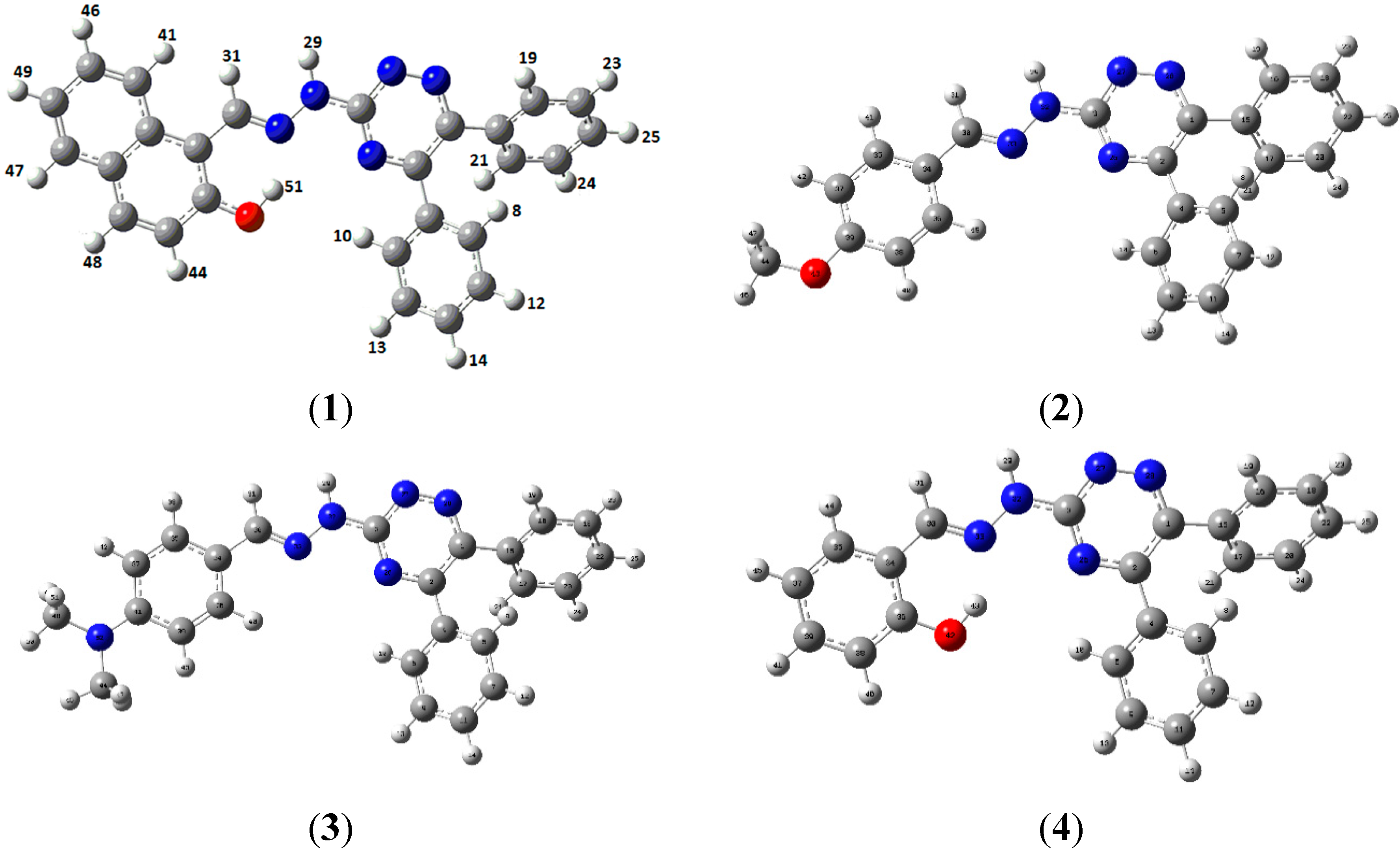
| Bond Lengths (A°) Experimental B3LYP/6-31G (d, p) | ||
| O1-C18 | 1.353(2) | 1.430 |
| N1-C1 | 1.341(18) | 1.335 |
| N1-C2 | 1.327(18) | 1.335 |
| N2-N3 | 1.328(17) | 1.315 |
| N2-C1 | 1.338(19) | 1.354 |
| N3-C3 | 1.336(17) | 1.347 |
| N4-N5 | 1.364(18) | 1.354 |
| N4-C1 | 1.356(19) | 1.368 |
| N5-C16 | 1.286(19) | 1.294 |
| Bond Angles (°) | ||
| C2-N1-C1 | 116.2(12) | 116.3 |
| N3-N2-C1 | 118.1(12) | 116.9 |
| N2-N3-C3 | 120.2(12) | 121.2 |
| C1-N4-N5 | 119.4(12) | 123.1 |
| C16-N5-N4 | 117.9(13) | 116.6 |
| N1-C1-N4 | 119.3(13) | 120.6 |
| N2-C1-N1 | 125.4(13) | 125.7 |
| N2-C1-N4 | 115.2(13) | 113.5 |
| N1-C2-C3 | 120.0(12) | 119.4 |
| N1-C2-C4 | 114.6(12) | 115.1 |
| N3-C3-C2 | 119.4(13) | 118.9 |
| N3-C3-C10 | 115.0(12) | 114.7 |
| O1-C18-C19 | 116.1(16) | 118.9 |
| Bond Lengths (A°) Experimental B3LYP/6-31G (d, p) | ||
| O1-C20 | 1.377(3) | 1.362 |
| O1-C23 | 1.426(3) | 1.419 |
| N1-C1 | 1.332(3) | 1.334 |
| N1-C2 | 1.330(3) | 1.335 |
| N2-N3 | 1.343(3) | 1.316 |
| N2-C1 | 1.353(3) | 1.353 |
| N3-C3 | 1.334(3) | 1.346 |
| N4-N5 | 1.387(3) | 1.351 |
| N4-C1 | 1.372(3) | 1.370 |
| Bond Angles (°) | ||
| C2-N1-C1 | 115.6(2) | 116.3 |
| N3-N2-C1 | 117.3(2) | 116.7 |
| N2-N3-C3 | 120.2(2) | 121.2 |
| C1-N4-N5 | 121.9(2) | 121.9 |
| C16-N5-N4 | 112.5(2) | 119.1 |
| N1-C1-N4 | 122.1(2) | 119.7 |
| N2-C1-N1 | 126.3(2) | 126.1 |
| N2-C1-N4 | 111.6(3) | 114.0 |
| N1-C2-C3 | 120.9(3) | 119.1 |
| N1-C2-C4 | 115.5(2) | 115.3 |
| N3-C3-C2 | 119.4(2) | 119.1 |
| N3-C3-C10 | 113.9(2) | 114.7 |
| C20-O1-C23 | 117.6(3) | 118.5 |
| Bond Lengths (A°) Experimental B3LYP/6-31G (d, p) | ||
| O1-C1 | 1.347(2) | 1.521 |
| N1-C2 | 1.325(2) | 1.402 |
| N2-N3 | 1.333(2) | 1.540 |
| N2-C1 | 1.351(2) | 1.395 |
| N3-C3 | 1.336(2) | 1.402 |
| N4-N5 | 1.381(2) | 1.540 |
| N4-C1 | 1.349(2) | 1.540 |
| N5-C16 | 1.282(2) | 1.540 |
| N6-C20 | 1.372(2) | 1.383 |
| Bond Angles (°) | ||
| C2-N1-C1 | 115.3(14) | 116.3 |
| N3-N2-C1 | 118.3(13) | 119.9 |
| N2-N3-C3 | 119.6(14) | 119.6 |
| C1-N4-N5 | 122.4(14) | 109.4 |
| C16-N5-N4 | 113.8(14) | 109.4 |
| N1-C1-N4 | 120.7(15) | 120.9 |
| N2-C1-N1 | 125.4(15) | 118.1 |
| N2-C1-N4 | 113.7(14) | 120.9 |
| N1-C2-C3 | 121.2(15) | 120.4 |
| N1-C2-C4 | 116.5(15) | 119.1 |
| N3-C3-C2 | 119.7(14) | 120.4 |
| N3-C3-C10 | 116.5(14) | 119.1 |
| Bond Lengths (A°) Experimental B3LYP/6-31G (d) | ||
| O1-C18 | 1.357(4) | 1.341 |
| N1-C1 | 1.336(3) | 1.334 |
| N1-C2 | 1.337(3) | 1.336 |
| N2-N3 | 1.338(3) | 1.317 |
| N2-C1 | 1.338(3) | 1.350 |
| N3-C3 | 1.339(3) | 1.346 |
| N4-N5 | 1.383(3) | 1.351 |
| N4-C1 | 1.360(3) | 1.371 |
| N5-C16 | 1.271(3) | 1.291 |
| Bond Angles (°) | ||
| C2-N1-C1 | 115.9(2) | 116.3 |
| N3-N2-C1 | 117.9(2) | 116.7 |
| N2-N3-C3 | 120.4(2) | 121.2 |
| C1-N4-N5 | 122.5(3) | 121.8 |
| C16-N5-N4 | 115.1(2) | 119.2 |
| N1-C1-N4 | 120.9(3) | 119.6 |
| N2-C1-N1 | 126.0(3) | 126.1 |
| N2-C1-N4 | 113.1(3) | 114.0 |
| N1-C2-C3 | 120.4(2) | 119.0 |
| N1-C2-C4 | 116.0(2) | 115.3 |
| N3-C3-C2 | 119.1(2) | 119.1 |
| N3-C3-C10 | 114.3(2) | 114.6 |
| O1-C18-C19 | 118.3(3) | 115.8 |
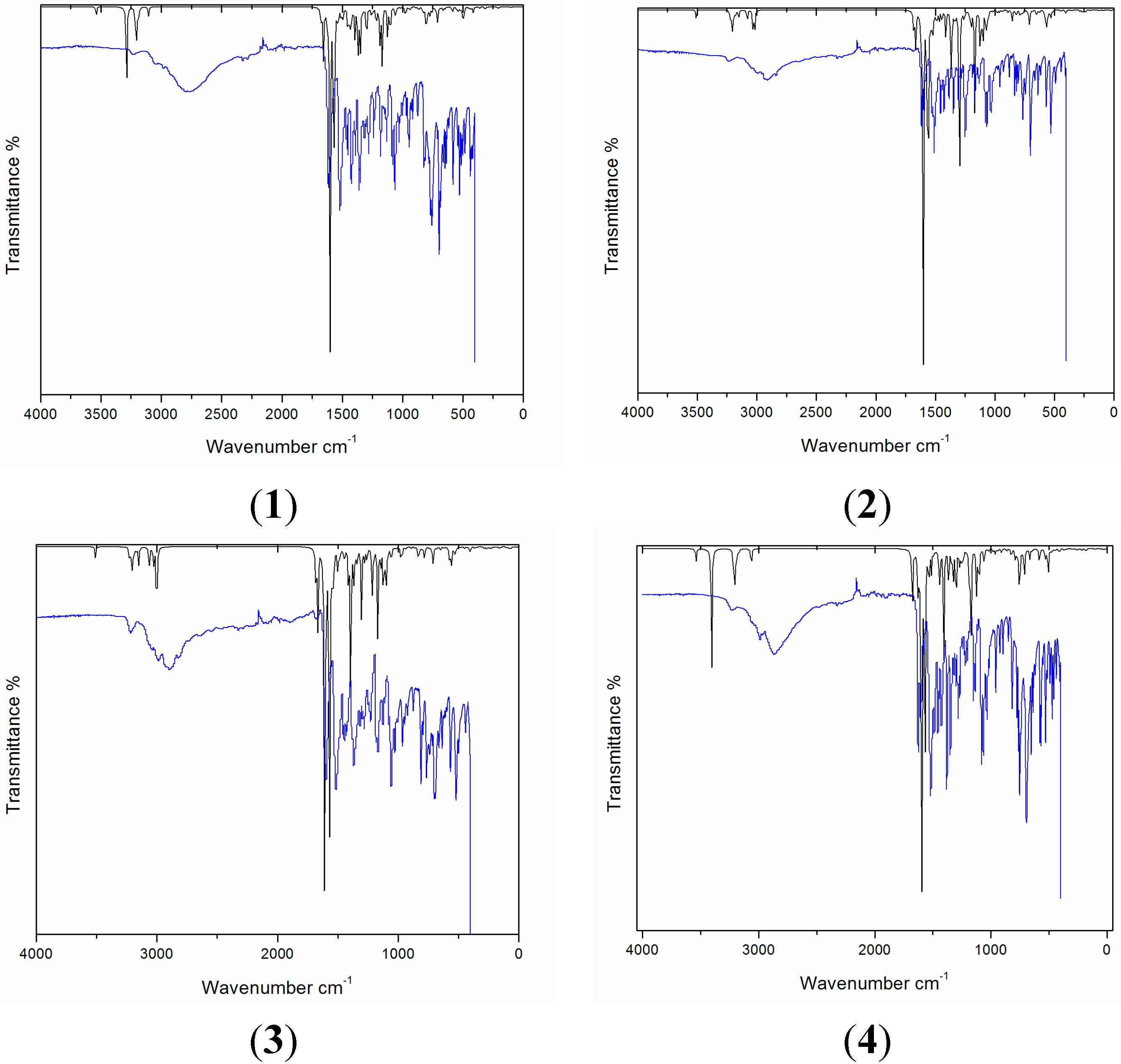
2.4. Vibrational Analysis
| 1 | 2 | 3 | 4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp. B3LYP Assignment | Exp. B3LYP Assignment | Exp. B3LYP Assignment | Exp. B3LYP Assignment | ||||||||
| 1611 1517 | 1595 | ѴC=Carom | 1611 1513 | 1605 | ѴC=C | 1593 1507 | 1605 | ѴC=C | 1621 1515 | 1613 | ѴC=Carom |
| 1542 | ѴC=Narom | 1542 | ѴC=Narom | 1550 | ѴC=N | 1565 | ѴC=Carom | ||||
| 1512 | ѴC=Narom | 1511 | ѴC=Narom | 1538 | ѴC=Narom | 1536 | ѴC=Narom | ||||
| 1367 1280 | 1506 | ѴN=Carom | 1350 1247 | 1499 | ѴC=Carom | 1428 1363 | 1511 | ѴN=Carom | 1375 1279 | 1510 | ѴC=Narom |
| 1319 | ѴC-Carom | 1318 | ѴC-Carom | 1508 | ѴC=Narom | 1353 | βCHarom | ||||
| 1301 | ѴC-Carom | 1250 | ѴO-Ph | 1341 | ѴNPh | 1251 | ѴC-Carom | ||||
| 1239 | 1252 | ѴC-Carom | 1167 | 1127 | βCHarom | 1277 | 1257 | ѴC-Carom | 1133 | 1127 | ѴC=Narom |
| 1179 | 1127 | ѴC=Narom | 1126 | ѴC=Narom | 1171 | βCHarom | 1078 | 1081 | βCHarom | ||
| 1062 | 1061 | βCHarom | 1060 | 1080 | βCHarom | 1160 | 1126 | ѴC=Narom | 1062 | 1061 | βCHarom |
| 756 | 775 | βCHarom | 1060 | βCHarom | 1052 | 1079 | βCHarom | 750 | 724 | γCHarom | |
| 764 | 759 | γCHarom | 807 | 803 | γCHarom | 688 | 686 | γCHarom | |||
| 516 | 540 | γCHarom | |||||||||
2.5. Nuclear Magnetic Resonance Studies (1H-NMR)
| (1) | B3LYP (ppm) | (2) | B3LYP (ppm) | (3) | B3LYP (ppm) | (4) | B3LYP (ppm) |
|---|---|---|---|---|---|---|---|
| 51H | 12.87 | 10H | 8.88 | 10H | 8.87 | 43H | 11.34 |
| 31H | 9.39 | 19H | 8.79 | 19H | 8.82 | 10H | 8.87 |
| 10H | 8.98 | 48H | 8.67 | 40H | 8.64 | 19H | 8.76 |
| 19H | 8.67 | 29H | 8.49 | 29H | 8.42 | 29H | 8.34 |
| 29H | 8.43 | 31H | 8.00 | 31H | 7.93 | 31H | 8.28 |
| 41H | 8.39 | 13H | 7.84 | 13H | 7.82 | 13H | 7.90 |
| 48H | 8.06 | 23H | 7.83 | 23H | 7.80 | 23H | 7.85 |
| 47H | 8.05 | 14H | 7.63 | 14H | 7.62 | 14H | 7.68 |
| 13H | 7.97 | 25H | 7.62 | 8H | 7.60 | 25H | 7.66 |
| 23H | 7.89 | 8H | 7.57 | 25H | 7.59 | 41H | 7.58 |
| 46H | 7.83 | 41H | 7.44 | 38H | 7.37 | 8H | 7.57 |
| 14H | 7.64 | 24H | 7.35 | 24H | 7.33 | 44H | 7.46 |
| 8H | 7.64 | 12H | 7.31 | 12H | 7.31 | 24H | 7.37 |
| 25H | 7.63 | 40H | 7.29 | 21H | 7.20 | 40H | 7.37 |
| 49H | 7.62 | 21H | 7.21 | 43H | 6.87 | 12H | 7.33 |
| 44H | 7.55 | 42H | 6.83 | 42H | 6.74 | 21H | 7.25 |
| 24H | 7.40 | 46H | 4.11 | 49H | 3.41 | 45H | 6.99 |
| 21H | 7.36 | 47H | 3.71 | 46H | 3.40 | ||
| 12H | 7.28 | 45H | 3.71 | 47H | 3.08 | ||
| 51H | 3.06 | ||||||
| 45H | 2.75 | ||||||
| 50H | 2.71 |
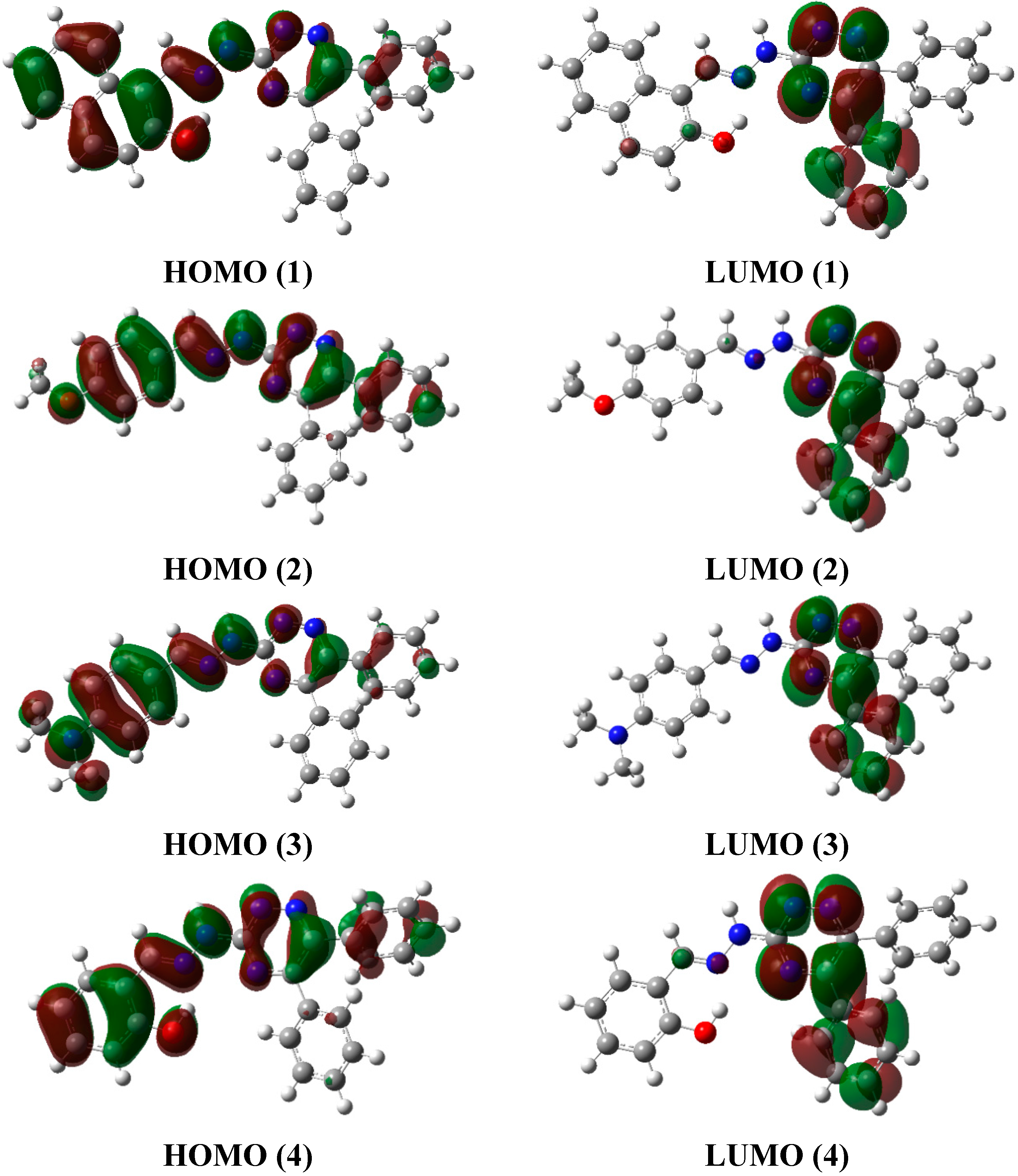
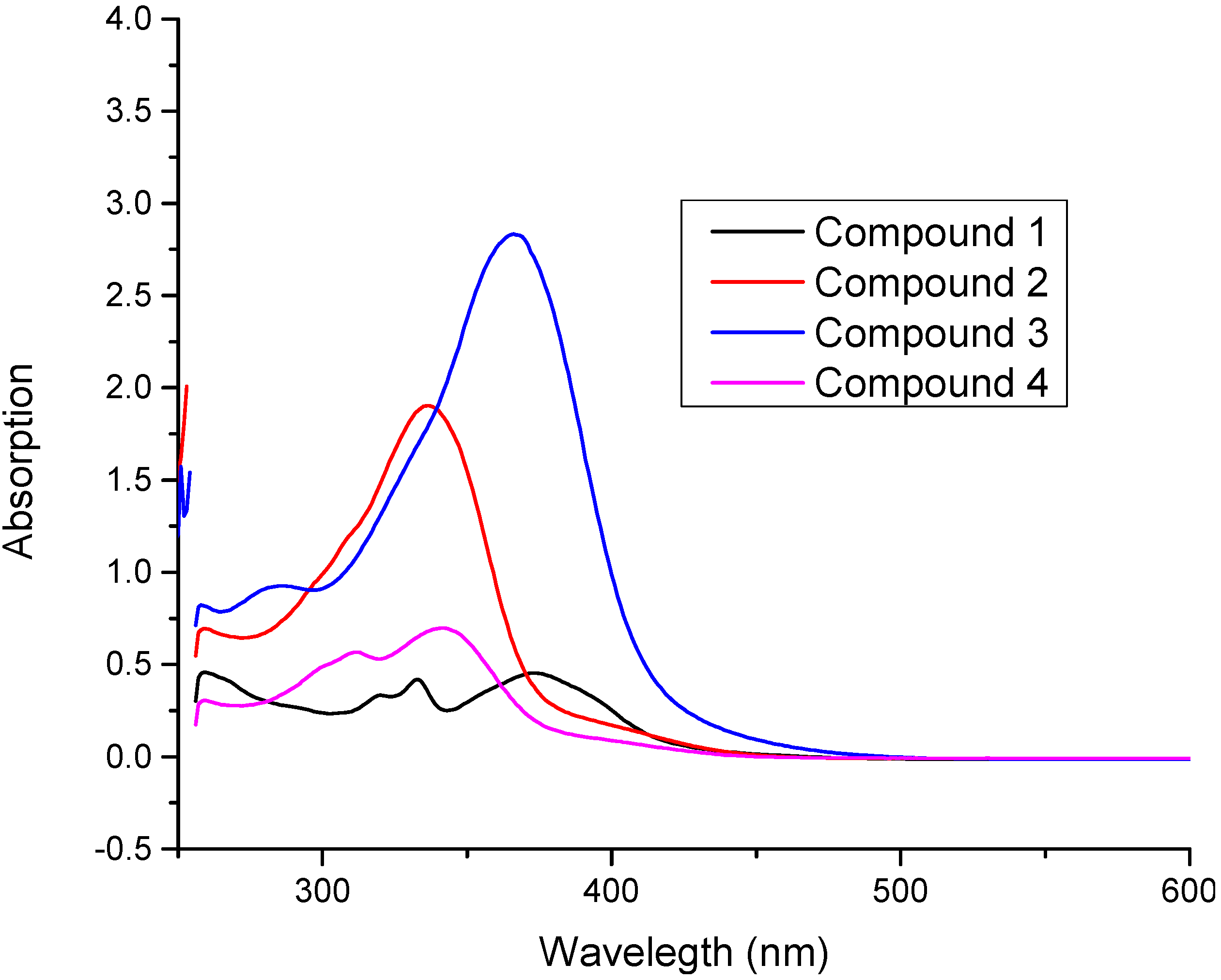
2.6. Frontier Molecular Orbital Analysis (FMO) and UV-Vis. Absorption Studies

| Compound | E (HOMO) | E (LUMO) | ΔE (LUMO‒HOMO) |
|---|---|---|---|
| 1 | −0.196 | −0.0713 | 0.124 |
| 2 | −0.196 | −0.064 | 0.132 |
| 3 | −0.18 | −0.060 | 0.119 |
| 4 | −0.204 | −0.072 | 0.133 |

| Experimental | Theoretical (TD-SCF/B3LYP/6-31G (d, p)) | |||
|---|---|---|---|---|
| Compound | λmax (abs.) (DMSO) | λmax (osc. Strength), Gas Phase | λmax (osc. Strength) DMSO | Assignment |
| (1) | 333 (0.419), 374 (0.454) | 380.8 (0.782) | 390.3 (0.945) | π‒π* |
| (2) | 337 (1.903) | 345.2 (1.189) | 358.2 (1.401) | π‒π* |
| (3) | 366 (2.834) | 370.4 (1.147) | 395.5 (1.196) | π‒π* |
| (4) | 312 (0.567), 342 (0.697) | 350.2 (0.739) | 356.1 (1.007) | π‒π* |
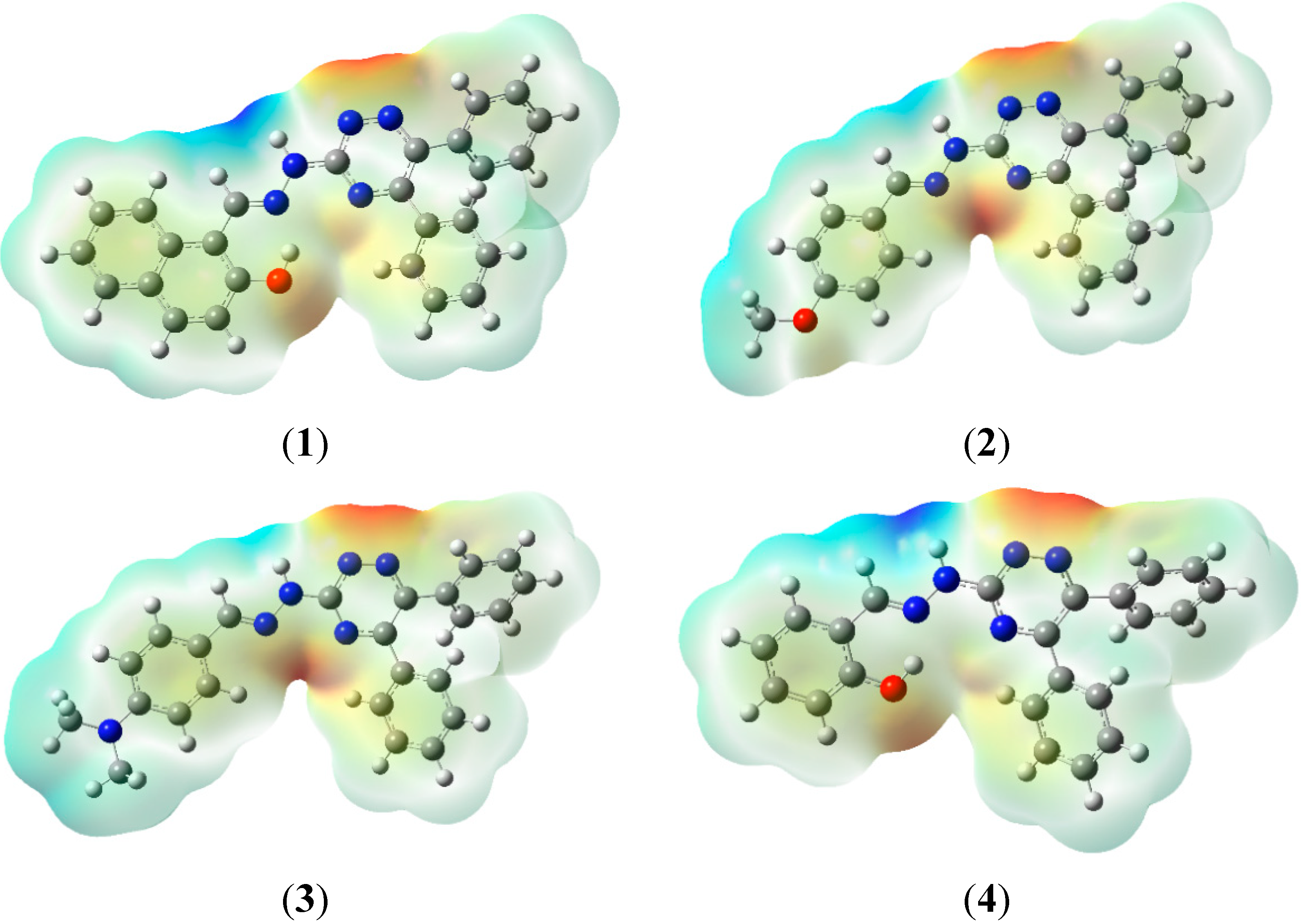
2.7. Molecular Electrostatic Potential (MEP)
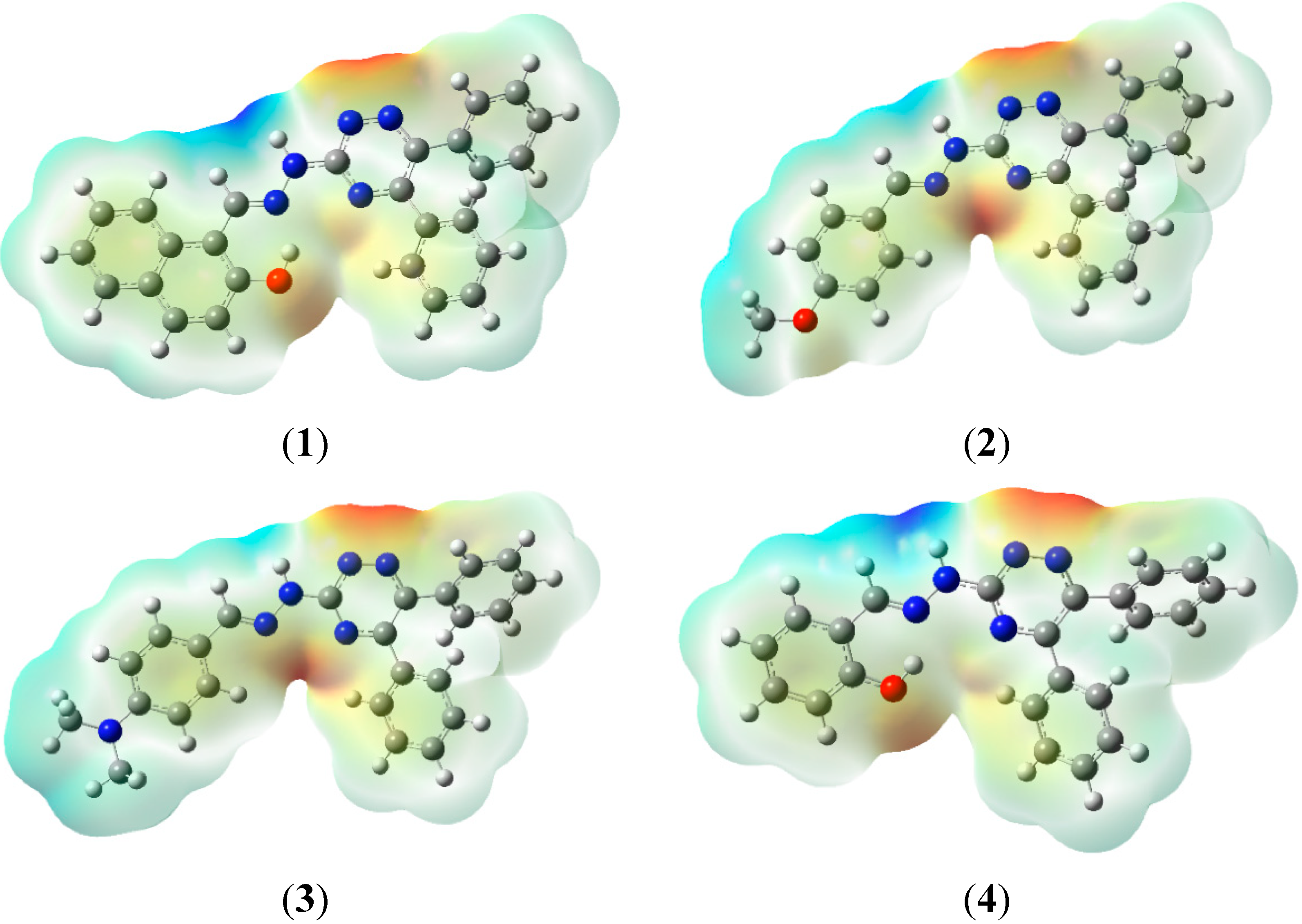
2.8. Hyperpolarizability and Non-Linear Optical Properties
| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| βxxx | 2300.433 | 3639.217 | 8152.091 | 1281.031 |
| βxxy | 6.139 | 41.559 | −559.94 | 223.445 |
| βxyy | −520.79 | 207.2331 | −62.360 | −303.73 |
| βyyy | −244.06 | −402.645 | −357.94 | −459.49 |
| βxxz | 584.743 | 17.3065 | −46.037 | 40.0394 |
| βxyz | 33.763 | 64.606 | 94.261 | 42.973 |
| βyyz | 1.283 | −3.485 | −15.970 | 0.236 |
| βxzz | 39.428 | 1.523 | −8.302 | 34.723 |
| βyzz | 14.681 | 4.147 | 37.649 | 17.0493 |
| βzzz | 9.201 | 9.715 | −8.129 | 7.664 |
| βtot. (esu) | 16.647 | 29.797 | 70.233 | 8.957 |
2.9. Chemical Reactivity
| Properties | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| ETotal (eV) | 0.124 | 0.132 | 0.119 | 0.133 |
| EHOMO | −0.196 | −0.196 | −0.18 | −0.204 |
| ELUMO | −0.0713 | −0.064 | −0.060 | −0.072 |
| η (eV) | 0.062 | 0.066 | 0.059 | 0.066 |
| µ (eV) | −0.133 | −0.13 | −0.120 | −0.138 |
| ω (eV) | 0.142 | 0.128 | 0.119 | 0.143 |
| µ(Debye) | 1.01 | 3.12 | 5.37 | 0.57 |
3. Experimental
3.1. General Information
3.2. Synthesis of 5,6-Diphenyl-4H-[1,2,4]triazine-3-thione
3.3. Synthesis of 3-Hydrazinyl-5,6-diphenyl-1,2,4-triazine
3.4. Synthesis of Compounds 1–4
3.5. Crystallography
3.6. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Smolin, E.M.; Rapoport, L. s-Triazine and Derivatives. In The Chemistry of Heterocyclic Compounds; Weissberger, A., Ed.; Wiley Intercience Publisher Inc.: New York, NY, USA, 1959; Volume 13, pp. 597–626. [Google Scholar]
- Patel, R.V.; Kumari, P.; Rajani, D.P.; Pannecouque, C.; de Clercq, E.; Chikhalia, K.H. Antimicrobial, anti-TB, anticancer and anti-HIV evaluation of new s-triazine-based heterocycles. Future Med. Chem. 2012, 4, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Khoshneviszadeh, M.; Mohammad, H.; Foroumadi, G.A.; Miri, R.; Firuzi, O.; Madadkar-Sobhani, A.; Edraki, N.; Parsa, M.; Shafiee, A. Design, synthesis and biological evaluation of novel anti-cytokine 1,2,4-triazine derivatives. Bioorg. Med. Chem. 2013, 21, 6708–6717. [Google Scholar] [CrossRef] [PubMed]
- El-Brollosy, N.R. Synthesis and antimicrobial evaluation of 6-azauracil non-nucleosides. Monatsh. Chem. 2008, 139, 1483–1490. [Google Scholar] [CrossRef]
- Mary, Y.S.; Abdul-Malek, S.; Al-Tamimi El-Brollosy, N.R.; El-Emam, A.A.; Jojo, P.J.; Panicker, C.Y.; Alsenoy, C.V. Molecular structure and vibrational spectra of 2-Ethoxymethyl-6-ethyl-2,3,4,5-tetrahydro-1,2,4-triazine-3,5-dione, a potential chemotherapeutic agent, by density functional methods. Spectrochim. Acta Part A 2014, 133, 439–448. [Google Scholar] [CrossRef]
- Liu, J.; Wang, K.; Zhang, X.; Li, C.; You, X. Triazine dyes as photosensitizers for dye-sensitized solar cells. Tetrahedron 2013, 69, 190–200. [Google Scholar] [CrossRef]
- Rafat, M.; Fleita, D.H.; Sakka, O.K. Novel Synthesis of Hydrazide-Hydrazone Derivatives and Their Utilization in the Synthesis of Coumarin, Pyridine, Thiazole and Thiophene Derivatives with Antitumor Activity. Molecules 2011, 16, 16–27. [Google Scholar]
- Vicini, P.; Zani, F.; Cozzini, P.; Doytchinova, I. Hydrazones of 1,2-benzisothiazole hydrazides: Synthesis, antimicrobial activity and QSAR investigations. Eur. J. Med. Chem. 2002, 37, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Rahman, V.M.; Mukhtar, S.; Ansari, W.H.; Lemiere, G. Synthesis, stereochemistry and biological activity of some novel long alkyl chain substituted thiazolidin-4-ones and thiazan-4-one from 10-undecenoic acid hydrazide. Eur. J. Med. Chem. 2005, 40, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Jani, G.R.; Vyas, K.B.; Franco, Z. Preparation and Antimicrobial Activity of s-Triazine Hydrazones of 7-Hydroxy Coumarin and their Metal Complexes. Eur. J. Chem. 2009, 6, 1228–1232. [Google Scholar]
- Taib, L.A.; Faidallah, H.M.; Şahin, Z.S.; Asiri, A.M.; Şahin, O.; Arshad, M.N. Synthesis, spectral analysis, X-ray crystal structures and evaluation of chemical reactivity of five new benzoindazole derivatives through experimental and theoretical studies. J. Mol. Struct. 2014, 1076, 272–279. [Google Scholar] [CrossRef]
- Javed, I.; Khurshid, A.; Arshad, M.N.; Wang, Y. Photophysical and electrochemical properties and temperature dependent geometrical isomerism in alkyl quinacridonediimines. New J. Chem. 2014, 38, 752–761. [Google Scholar] [CrossRef]
- Arshad, M.N.; Şahin, O.; Zia-ur-Rehman, M.; Khan, I.U.; Asiri, A.M.; Rafique, H.M. 4-hydroxy-2H-1, 2-benzothiazine-3-carbohydrazide 1, 1-dioxide-oxalohydrazide (1:1): X-ray structure and DFT calculations. J. Struct. Chem. 2013, 54, 437–442. [Google Scholar] [CrossRef]
- Sundaraganesan, N.; Ilakiamani, S.; Saleem, H.; Wojciechowski, P.M.; Michalska, D. FT-Raman and FT-IR spectra, vibrational assignments and density functional studies of 5-bromo-2-nitropyridine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2005, 61, 2995–3001. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.N.; Mahmood, T.; Khan, A.F.; Zia-Ur-Rehman, M.; Asiri, A.M.; Khan, I.U.; Un-Nisa, R.; Ayub, K.; Mukhtar, A.; Saeed, M.T. Synthesis, crystal structure and spectroscopic properties of 1,2-benzothiazine derivatives: An experimental and DFT study. Chin. J. Struct. Chem. 2015, 34, 15–25. [Google Scholar]
- Ebenezar, J.D.; Ramalingam, S.; Raja, C.R.; Helan, V. Precise spectroscopic [IR, Raman and NMR] investigation and gaussian hybrid computational analysis (UV-visible, NIR, MEP Maps and Kubo Gap) on l-valine. J. Theor. Comput. Sci. 2013, 1, 1–13. [Google Scholar] [CrossRef]
- Szabo, L.; Chiş, V.; Pirnau, A.; Leopold, N.; Cozar, O.; Orosz, S. Spectroscopic and theoretical study of amlodipine besylate. J. Mol. Struct. 2009, 924, 385–392. [Google Scholar] [CrossRef]
- Bartošková, M.; Friedl, Z. The Relationship between the heats of formation and the molecular electrostatic potentials of polyazaarenes. Cent. Eur. J. Energy Mater. 2013, 10, 103–112. [Google Scholar]
- Sagadevan, S. Investigation on the optical properties of nonlinear optical (NLO) single crystal: l-Valine zinc hydrochloride. Am. J. Opt. Photonics 2014, 2, 24–27. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, X.; Jiang, Y.; Wang, G.; Qu, C.; Zhao, B. Computational study on the second-order nonlinear optical properties of coumarin derivatives with N-p-vinylphenyl carbazole chromophores. Asian J. Chem. 2014, 26, 7404–7408. [Google Scholar]
- Mebi, C.A. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4-S)2Fe(CO)2]. J. Chem. Sci. 2011, 123, 727–773. [Google Scholar] [CrossRef]
- Sukumar, N. A Matter of Density; Wiley: Hoboken, NJ, USA, 2012; pp. 157–201. [Google Scholar]
- Špirtović-Halilović, S.; Salihovic, M.; Džudžević-Čančar, H.; Trifunovic, S.; Roca, S.; Softic, D.; Završnik, D. DFT study and microbiology of some coumarin-based compounds containing a chalcone moiety. J. Serb. Chem. Soc. 2014, 79, 435–443. [Google Scholar] [CrossRef]
- CrysAlis PRO; Agilent Technologies: Yarnton, UK, 2012.
- Sheldrick, G.M. A short History of SHELX. Acta Crystallogr. 2007, A64, 112–122. [Google Scholar] [CrossRef]
- Barbour, L.J. A software tool for Supramolecular Crystallography. J. Supramol. Chem. 2001, 1, 189–191. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Roy, D.; Todd, K.; John, M. Gauss View; Version 5; Semichem, Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Hung, T.Q.; Hoang, D.H.; Thang, N.N.; Dang, T.T.; Ayub, K.; Villinger, A.; Friedrich, A.; Lochbrunner, S.; Flechsig, G.-U.; Langer, P. Palladium catalyzed synthesis and physical properties of indolo[2,3-b]quinoxalines. Org. Biomol. Chem. 2014, 12, 6151–6166. [Google Scholar] [CrossRef] [PubMed]
- Babar, A.; Khalid, H.; Ayub, K.; Saleem, S.; Waseem, A.; Mahmood, T.; Munawar, M.A.; Abbas, G.; Khan, A.F. Synthesis, characterization and density functional theory study of some new 2-anilinothiazoles. J. Mol. Struct. 2014, 1072, 221–227. [Google Scholar] [CrossRef]
- Ullah, H.; Rauf, A.; Ullah, Z.; Fazl-i-Sattar; Anwar, M.; Shah, A.-H.A.; Uddin, G.; Ayub, K. Density functional theory and phytochemical study of Pistagremic acid. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 118, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Fazl-i-Sattar; Ullah, Z.; Ata-ur-Rahman; Rauf, A.; Tariq, M.; Tahir, A.A.; Ayub, K.; Ullah, H. Phytochemical, spectroscopic and density functional theory study of Diospyrin, and non-bonding interactions of Diospyrin with atmospheric gases. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 141, 71–79. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–4 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arshad, M.N.; Bibi, A.; Mahmood, T.; Asiri, A.M.; Ayub, K. Synthesis, Crystal Structures and Spectroscopic Properties of Triazine-Based Hydrazone Derivatives; A Comparative Experimental-Theoretical Study. Molecules 2015, 20, 5851-5874. https://doi.org/10.3390/molecules20045851
Arshad MN, Bibi A, Mahmood T, Asiri AM, Ayub K. Synthesis, Crystal Structures and Spectroscopic Properties of Triazine-Based Hydrazone Derivatives; A Comparative Experimental-Theoretical Study. Molecules. 2015; 20(4):5851-5874. https://doi.org/10.3390/molecules20045851
Chicago/Turabian StyleArshad, Muhammad Nadeem, Aisha Bibi, Tariq Mahmood, Abdullah M. Asiri, and Khurshid Ayub. 2015. "Synthesis, Crystal Structures and Spectroscopic Properties of Triazine-Based Hydrazone Derivatives; A Comparative Experimental-Theoretical Study" Molecules 20, no. 4: 5851-5874. https://doi.org/10.3390/molecules20045851
APA StyleArshad, M. N., Bibi, A., Mahmood, T., Asiri, A. M., & Ayub, K. (2015). Synthesis, Crystal Structures and Spectroscopic Properties of Triazine-Based Hydrazone Derivatives; A Comparative Experimental-Theoretical Study. Molecules, 20(4), 5851-5874. https://doi.org/10.3390/molecules20045851