
Economical Synthesis of 13C-Labeled Opiates, Cocaine Derivatives and Selected Urinary Metabolites by Derivatization of the Natural Products


Abstract
:1. Introduction
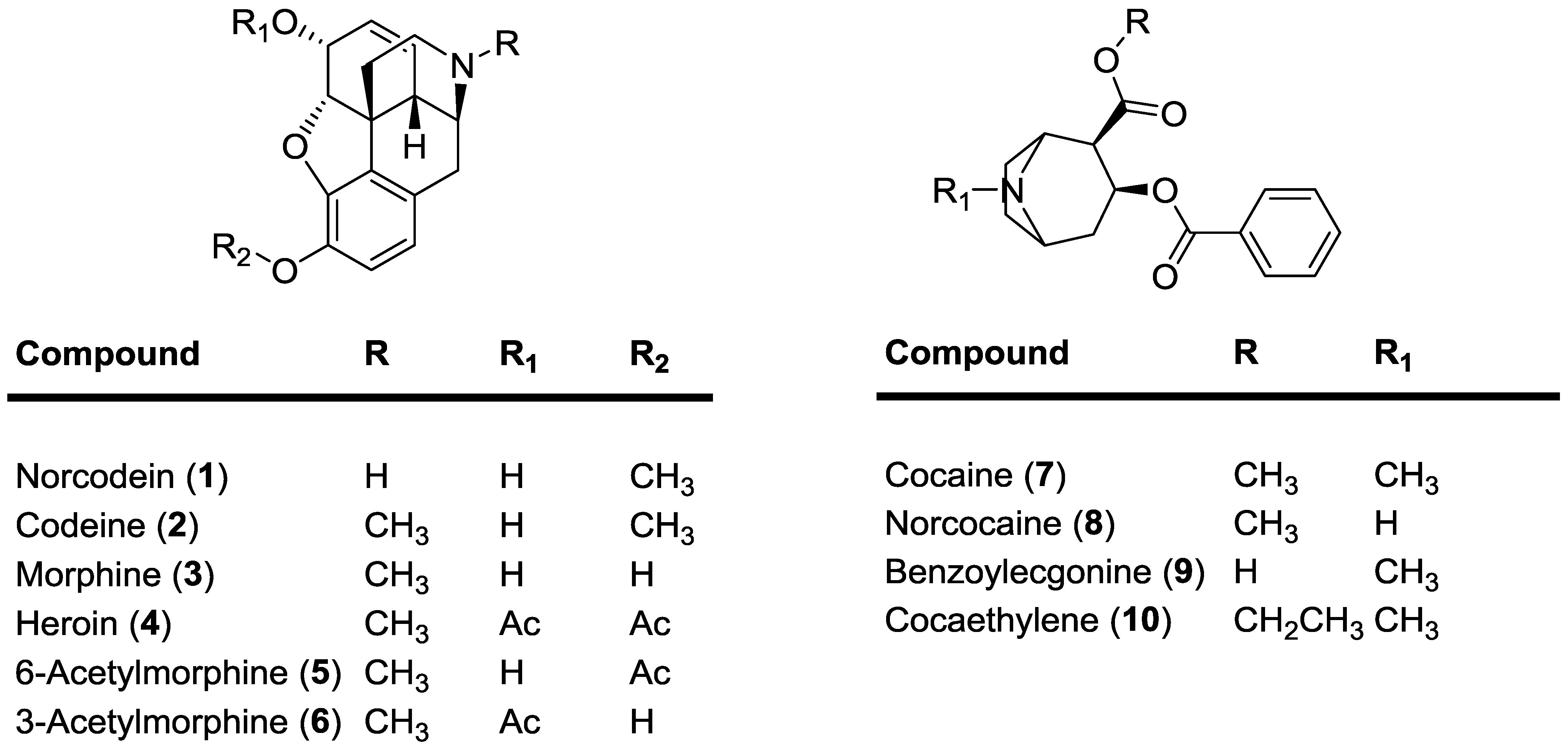
2. Results and Discussion
2.1. Synthesis of 13C-Labeled Opiates

2.2. [Phenyl-13C6]-Labeled Cocaine and Derivatives
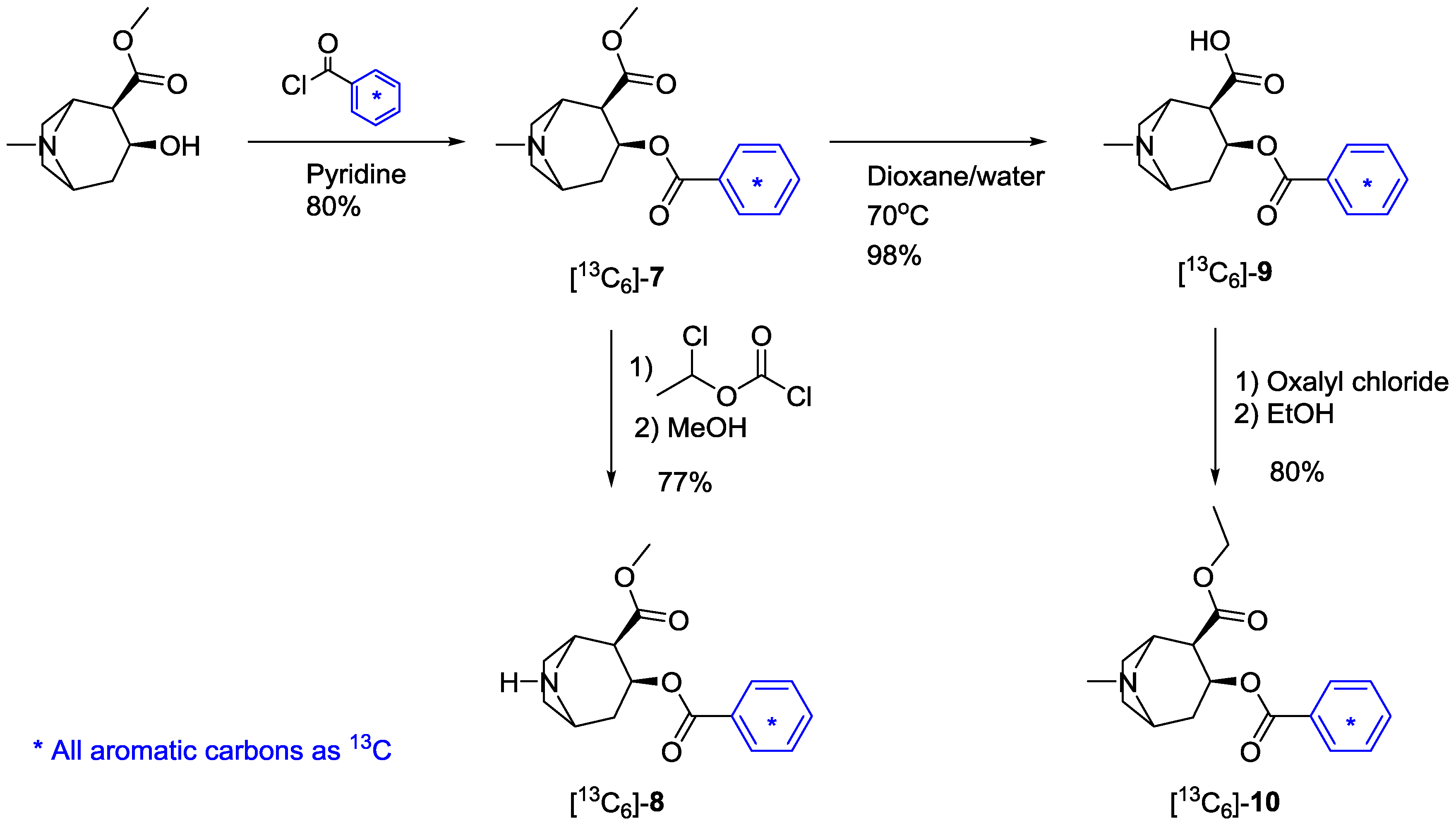
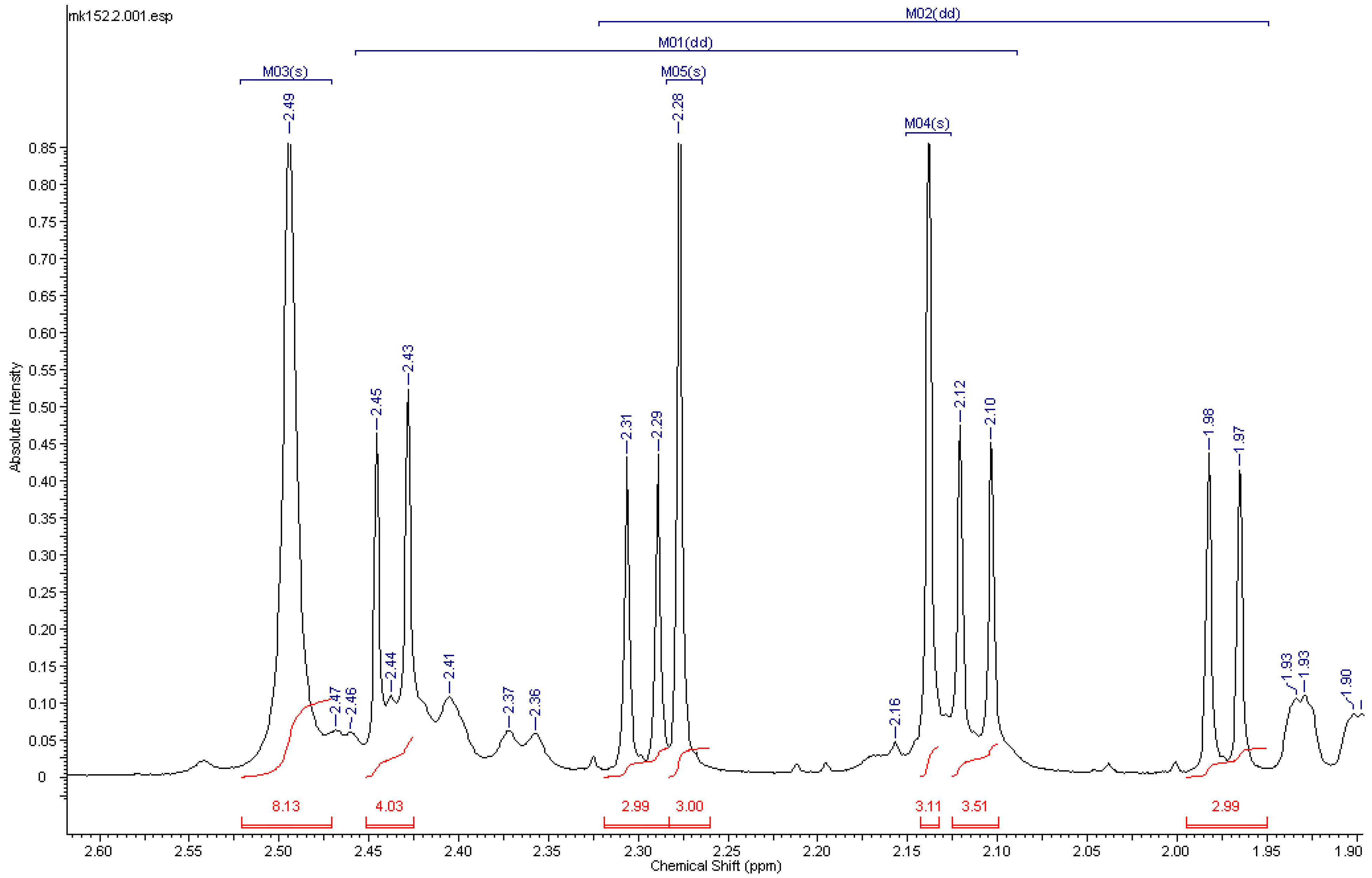
2.3. Analysis of the Products

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Identity | 1H-Shift (ppm)/Multiplicity | 1JC,H (Hz) | 2JC,H (Hz) | 1JC,C (Hz) |
|---|---|---|---|---|---|
| [N-Methyl-13C]codeine ([13C]-2) | N-CH3 | 2.46 (d) | 133.1 | - | - |
| [N-Methyl-13C-O-Methyl-13C]codeine ([13C2]-2) | N-CH3 | 2.46 (d) | 133.1 | - | - |
| 3-O-CH3 | 3.95 (d) | 144.3 | - | - | |
| [Methyl-13C]morphine ([13C]-3) | N-CH3 | 2.46 (d) | 133.1 | - | - |
| [Acetyl-13C4]heroin ([13C4]-4) | 6-OAc | 2.14 (dd) | 129.6 | 7.1 | 60.0 |
| 3-OAc | 2.28 (dd) | 129.9 | 6.8 | 60.7 | |
| [Acetyl-13C4-methyl-13C]heroin ([13C5]-4) | 6-OAc | 2.14 (dd) | 129.7 | 7.0 | 60.1 |
| 3-OAc | 2.28 (dd) | 130.2 | 7.0 | 60.1 | |
| N-CH3 | 2.50 (d) | 133.6 | - | - | |
| [Acetyl-13C2-methyl-13C]6-MAM ([13C3]-5) | 6-OAc | 2.17 (dd) | 130.1 | 6.6 | 59.3 |
| N-CH3 | 2.53 (d) | 134.9 | - | ||
| [Acetyl-13C2-methyl-13C]3-MAM ([13C3]-6) | 3-OAc | 2.30 (dd) | 129.9 | 6.9 | 60.1 |
| N-CH3 | 2.52 (d) | 134.2 | - |
3. Experimental Section
3.1. Chemicals and Analysis
3.2. Synthesis of 13C-Labeled Opiates
3.2.1. Synthesis of Norcodeine (1)
3.2.2. Synthesis of [N-Methyl-13C]codeine ([13C]-2)
3.2.3. Synthesis of [N-Methyl-13C-O-methyl-13C]codeine ([13C2]-2)
3.2.4. Synthesis of [Methyl-13C]morphine ([13C]-3)
3.2.5. Synthesis of [Acetyl-13C4]heroin ([13C4]-4)
3.2.6. Synthesis of [Acetyl-13C4-metyl-13C]heroin ([13C5]-4)
3.2.7. Synthesis of [Acetyl-13C2-metyl-13C]6-acetylmorphine ([13C3]-5)
3.2.8. Synthesis of [Acetyl-13C2-metyl-13C]3-acetylmorphine ([13C3]-6)
3.3. Synthesis of [Phenyl-13C6]-Labeled Cocaine Derivatives
3.3.1. Synthesis of [Phenyl-13C6]cocaine ([13C6]-7) Hydrochloride
3.3.2. Synthesis of [Phenyl-13C6]norcocaine ([13C6]-8)
3.3.3. Synthesis of [Phenyl-13C6]benzoylecgonine ([13C6]-9)
3.3.4. Synthesis of [Phenyl-13C6]cocaethylene (10)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wickizer, T.M. State-level estimates of the economic costs of alcohol and drug abuse. J. Health Care Financ. 2013, 39, 71–84. [Google Scholar]
- Levy, M.H. Pharmacologic treatment of cancer pain. N. Engl. J. Med. 1996, 335, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- De Pinto, M.; Cahana, A. Medical management of acute pain in patients with chronic pain. Expert Rev. Neurother. 2012, 12, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.; Rowett, D.; Allcroft, P.; Abernethy, A.; Currow, D.C. Chronic refractory dyspnoea-evidence based management. Aust. Fam. Phys. 2013, 42, 137–140. [Google Scholar]
- Abernethy, A.P.; Currow, D.C.; Frith, P.; Fazekas, B.S.; McHugh, A.; Bui, C. Randomised, double blind, placebo controlled crossover trial of sustained release morphine for the management of refractory dyspnea. Br. Med. J. 2003, 327, 523–526. [Google Scholar] [CrossRef]
- Gibson, P.G.; Ryan, N.M. Cough pharmacotherapy: Current and future status. Expert Opin. Pharmacother. 2011, 12, 1745–1755. [Google Scholar] [PubMed]
- Baldi, F.; Bianco, M.A.; Nardone, G.; Pilotto, A.; Zamparo, E. Focus on acute diarrhoeal disease. World J. Gastroenterol. 2009, 15, 3341–3348. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.R.A. XLIX. On the action of organic acids and their anhydrides on the natural alkaloids. Part I. J. Chem. Soc. Trans. 1874, 27, 1031–1043. [Google Scholar]
- Strang, J.; Metrebian, N.; Lintzeris, N.; Potts, L.; Carnwath, T.; Mayet, S.; Williams, H.; Zador, D.; Evers, R.; Groshkova, T.; et al. Supervised injectable heroin or injectable methadone versus optimised oral methadone as treatment for chronic heroin addicts in England after persistent failure in orthodox treatment (RIOTT): A randomised trial. Lancet 2010, 375, 1885–1895. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, A.J. Pharmacokinetic Modeling and Pharmacokinetic-Pharmacodynamic Correlations; CRC Press LLC: Boca Raton, FL, USA, 2008; pp. 15–19. [Google Scholar]
- Kraemer, T.; Paul, L.D. Bioanalytical procedures for determination of drugs of abuse in blood. Anal. Bioanal. Chem. 2007, 388, 1415–1435. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- De Castro, A.; Concheiro, M.; Shakleya, D.M.; Huestis, M.A. Simultaneous quantification of methadone, cocaine, opiates, and metabolites in human placenta by liquid chromatography-mass spectrometry. J. Anal. Toxicol. 2009, 33, 243–252. [Google Scholar]
- Dams, R.; Murphy, C.M.; Lambert, W.E.; Huestis, M.A. Urine drug testing for opioids, cocaine, and metabolites by direct injection liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Karlsen, M.; Øiestad, Å.M.L.; Liu, H.; Johansen, J.E. Evaluation of 13C- and 2H-labeled internal standards for the determination of amphetamines in biological samples, by reversed-phase ultra-high performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2014, 1344, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cyronak, M.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response?: A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Bogusz, M.J.; Maier, R.D.; Kruger, K.D.; Kohls, U. Determination of common drugs of abuse in body fluids using one isolation procedure and liquid chromatography-atmospheric-pressure chemical-ionization mass spectromery. J. Anal. Toxicol. 1998, 22, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Edinboro, L.E.; Backer, R.C.; Poklis, A. Direct analysis of opiates in urine by liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2005, 29, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, E.; Andersson, M.; Stephanson, N.; Beck, O. Validation of direct injection electrospray LC-MS/MS for confirmation of opiates in urine drug testing. J. Mass Spectrom. 2007, 42, 881–889. [Google Scholar] [CrossRef] [PubMed]
- Rice, K.C. Synthetic opium alkaloids and derivatives. A short total synthesis of (±)-dihydrothebainone, (±)-dihydrocodeinone, and (±)-nordihydrocodeinone as an approach to a practical synthesis of morphine, codeine, and congeners. J. Org. Chem. 1980, 45, 3135–3137. [Google Scholar] [CrossRef]
- Rinner, U.; Hudlicky, T. Synthesis of morphine alkaloids and derivatives. Top. Curr. Chem. 2012, 309, 33–66. [Google Scholar] [PubMed]
- Willstatter, R.; Wolfes, O.; Mader, H. Synthesis of natural cocaine. Justus Liebigs Ann. Chem. 1923, 434, 111–139. [Google Scholar] [CrossRef]
- Robinson, R. LXIII.-A synthesis of tropinone. J. Chem. Soc. Trans. 1917, 111, 762–768. [Google Scholar] [CrossRef]
- Casale, J.F. A practical total synthesis of cocaine’s enantiomers. Forensic Sci. Int. 1987, 33, 275–298. [Google Scholar] [CrossRef]
- Tufariello, J.J. Alkaloids from nitrones. Acc. Chem. Res. 1979, 12, 396–403. [Google Scholar] [CrossRef]
- Shing, T.K.M.; So, K.H. Facile and enantiospecific syntheses of (6S,7R)-6-chloro-7-benzyloxy-, (7S)-halo-, and (7S)-hydroxy-cocaine and natural (−)-cocaine from d-(−)-ribose. Org. Lett. 2011, 13, 2916–2919. [Google Scholar] [CrossRef] [PubMed]
- Mans, D.M.; Pearson, W.H. Total synthesis of (+)-cocaine via desymmetrization of a meso-dialdehyde. Org. Lett. 2004, 6, 3305–3308. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Lee, K.; Cha, J.K. Enantioselective synthesis of unnatural (S)-(+)-cocaine. J. Org. Chem. 2000, 65, 4773–4775. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.G.; Graves, D.M.; Green, N.J.; Jacobson, R.R.; Nowak, D.M. Photochemistry of structurally-modified morphine alkaloids. J. Am. Chem. Soc. 1994, 116, 10450–10462. [Google Scholar] [CrossRef]
- Coop, A.; Janetka, J.W.; Lewis, J.W.; Rice, K.C. L-Selectride as a general reagent for the O-demethylation and N-decarbomethoxylation of opium alkaloids and derivatives. J. Org. Chem. 1998, 63, 4392–4396. [Google Scholar] [CrossRef]
- Hudlicky, T.; Carroll, R.J.; Lalush, H.; Machara, A.; Werner, L.; Endoma-Arias, M.A. Methods for One-Pot N-Demethylation/N-Functionalization of Morphine and Tropane Alkaloids. US20110313163A1, 22 December 2011. [Google Scholar]
- Rice, K.C.; May, E.L. Procedural refinements in the N-demethylation of morphine and codeine using phenyl chloroformate and hydrazine. J. Heterocycl. Chem. 1977, 14, 665–666. [Google Scholar] [CrossRef]
- Chang, F.N.H.; Oneto, J.F.; Sah, P.P.T.; Tolbert, B.M.; Rapoport, H. The synthesis of codeine labeled in the 3-methoxy group with C14. J. Org. Chem. 1950, 15, 634–636. [Google Scholar] [CrossRef]
- Small, L.F.; Lutz, R.E. Chemistry of the Opium Alkaloids. In Public Health Report; Suppl. No. 103, U.S. Government Printing Office: Washington, DC, USA, 1932; pp. 1–375. [Google Scholar]
- Ai, L.; Chen, X.; Zhang, L. Synthesis of Codeine from Morphine. CN1600784A, 2005. [Google Scholar]
- Ayyangar, N.R.; Choudhary, A.R.; Kalkote, U.R.; Sharma, V.K. Improved Process for the Preparation of Codeine from Morphine. EP268710A1, 1 June 1988. [Google Scholar]
- Klemenc, S. 4-Dimethylaminopyridine as a catalyst in heroin synthesis. Forensic Sci. Int. 2002, 129, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Lawson, J.A. Methods of Synthesis of Diastereomerically Enriched Morphinans. WO2009154673A1, 23 December 2009. [Google Scholar]
- Boerner, U.; Abbott, S.; Roe, R.L. Metabolism of morphine and heroin in man. Drug Metab. Rev. 1975, 4, 39–73. [Google Scholar] [CrossRef] [PubMed]
- Garrido, J.M.P.J.; Delerue-Matos, C.; Borges, F.; Macedo, T.R.A.; Oliveira-Brett, A.M. Voltammetric oxidation of drugs of abuse III. Heroin and metabolites. Electroanalysis 2004, 16, 1497–1502. [Google Scholar] [CrossRef]
- Welsh, L.H. O3-Monoacetylmorphine. J. Org. Chem. 1954, 19, 1409–1415. [Google Scholar] [CrossRef]
- Olofson, R.A.; Martz, J.T.; Senet, J.P.; Piteau, M.; Malfroot, T. A new reagent for the selective, high-yield N-dealkylation of tertiary amines: Improved syntheses of naltrexone and nalbuphine. J. Org. Chem. 1984, 49, 2081–2082. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, A.; Klass, T.; Johnson, K.M.; Wang, C.Z.; Ye, Y.P.; Kozikowski, A.P. Biaryl analogues of conformationally constrained tricyclic tropanes as potent and selective norepinephrine reuptake inhibitors: Synthesis and evaluation of their uptake inhibition at monoamine transporter sites. J. Med. Chem. 2003, 46, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.J.; Lewin, A.H. Novel Tropane Esters and Methods for Producing and Using Them. WO2004052888A2, 24 June 2004. [Google Scholar]
- Jatlow, P.; Elsworth, J.D.; Bradberry, C.W.; Winger, G.; Taylor, J.R.; Russell, R.; Roth, R.H. Cocaethylene: A neuropharmacologically active metabolite associated with concurrent cocaine-ethanol ingestion. Life Sci. 1991, 48, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Everhart, E.T.; Jacob, P., III; Mendelson, J.; Jones, R.T. The synthesis of deuterium-labeled cocaine, cocaethylene and metabolites. J. Label. Compd. Radiopharm. 1999, 42, 1265–1275. [Google Scholar] [CrossRef]
- Linders, J.T.M.; Booth, R.J.; Lie, T.S.; Kieboom, A.P.G.; Maat, L. Chemistry of opium alkaloids. Part XXVII. Diene systems in N-formylmorphinans; formation of a dibenz[d,f]azonine. A new example of molecular acrobatics in morphinans. Recueil des Travaux Chimiques des Pays-Bas 1989, 108, 189–194. [Google Scholar] [CrossRef]
- Zhang, A.; Li, F.; Ding, C.; Yao, Q.; Knapp, B.I.; Bidlack, J.M.; Neumeyer, J.L. Synthesis and Pharmacological Evaluation of 6,7-indolo/thiazolo-morphinans-Further SAR of levorphanol. J. Med. Chem. 2007, 50, 2747–2751. [Google Scholar] [CrossRef] [PubMed]
- Habibi-Yangjeh, A.; Pourbasheer, E.; Danandeh-Jenagharad, M. Prediction of melting point for drug-like compounds using principal component-genetic algorithm-artificial neural network. Bull. Korean Chem. Soc. 2008, 29, 833–841. [Google Scholar] [CrossRef]
- Hughes, L.D.; Palmer, D.S.; Nigsch, F.; Mitchell, J.B.O. Why are some properties more difficult to predict than others? A otudy of QSPR models of solubility, melting point, and log P. J. Chem. Inf. Model. 2008, 48, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Smith, W. Melting point of cocaine hydrochloride. Q. J. Pharm. Allied Sci. 1929, 1, 387–388. [Google Scholar]
- Schmidt, H.L.; Werner, G. Radioactive labeling of tropane alkaloids. II. Synthetic incorporation of C14 into (−)-cocaine, (−)-ecgonine, and derivatives. Justus Liebigs Ann. Chem. 1962, 653, 184–194. [Google Scholar] [CrossRef]
- Lin, R.; Castells, J.; Rapoport, H. Enantiospecific synthesis of natural (−)-cocaine and unnatural (+)-cocaine from d- and l-glutamic acid. J. Org. Chem. 1998, 63, 4069–4078. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the final products, except compound 5, are available from Chiron AS.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karlsen, M.; Liu, H.; Johansen, J.E.; Hoff, B.H. Economical Synthesis of 13C-Labeled Opiates, Cocaine Derivatives and Selected Urinary Metabolites by Derivatization of the Natural Products. Molecules 2015, 20, 5329-5345. https://doi.org/10.3390/molecules20045329
Karlsen M, Liu H, Johansen JE, Hoff BH. Economical Synthesis of 13C-Labeled Opiates, Cocaine Derivatives and Selected Urinary Metabolites by Derivatization of the Natural Products. Molecules. 2015; 20(4):5329-5345. https://doi.org/10.3390/molecules20045329
Chicago/Turabian StyleKarlsen, Morten, Huiling Liu, Jon Eigill Johansen, and Bård Helge Hoff. 2015. "Economical Synthesis of 13C-Labeled Opiates, Cocaine Derivatives and Selected Urinary Metabolites by Derivatization of the Natural Products" Molecules 20, no. 4: 5329-5345. https://doi.org/10.3390/molecules20045329
APA StyleKarlsen, M., Liu, H., Johansen, J. E., & Hoff, B. H. (2015). Economical Synthesis of 13C-Labeled Opiates, Cocaine Derivatives and Selected Urinary Metabolites by Derivatization of the Natural Products. Molecules, 20(4), 5329-5345. https://doi.org/10.3390/molecules20045329