
Antibiotic Discovery: Combatting Bacterial Resistance in Cells and in Biofilm Communities


{kind=link}
Abstract
:1. Antibiotic Resistance
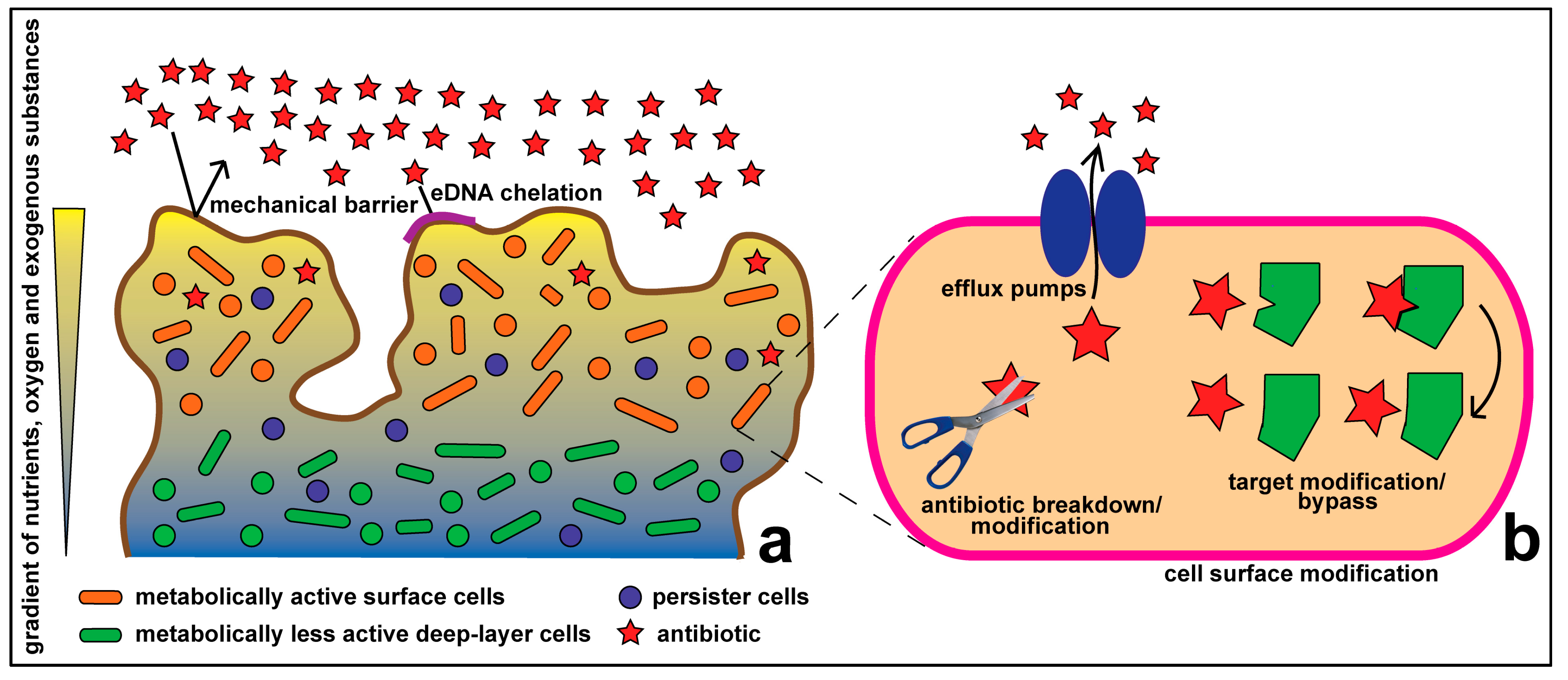
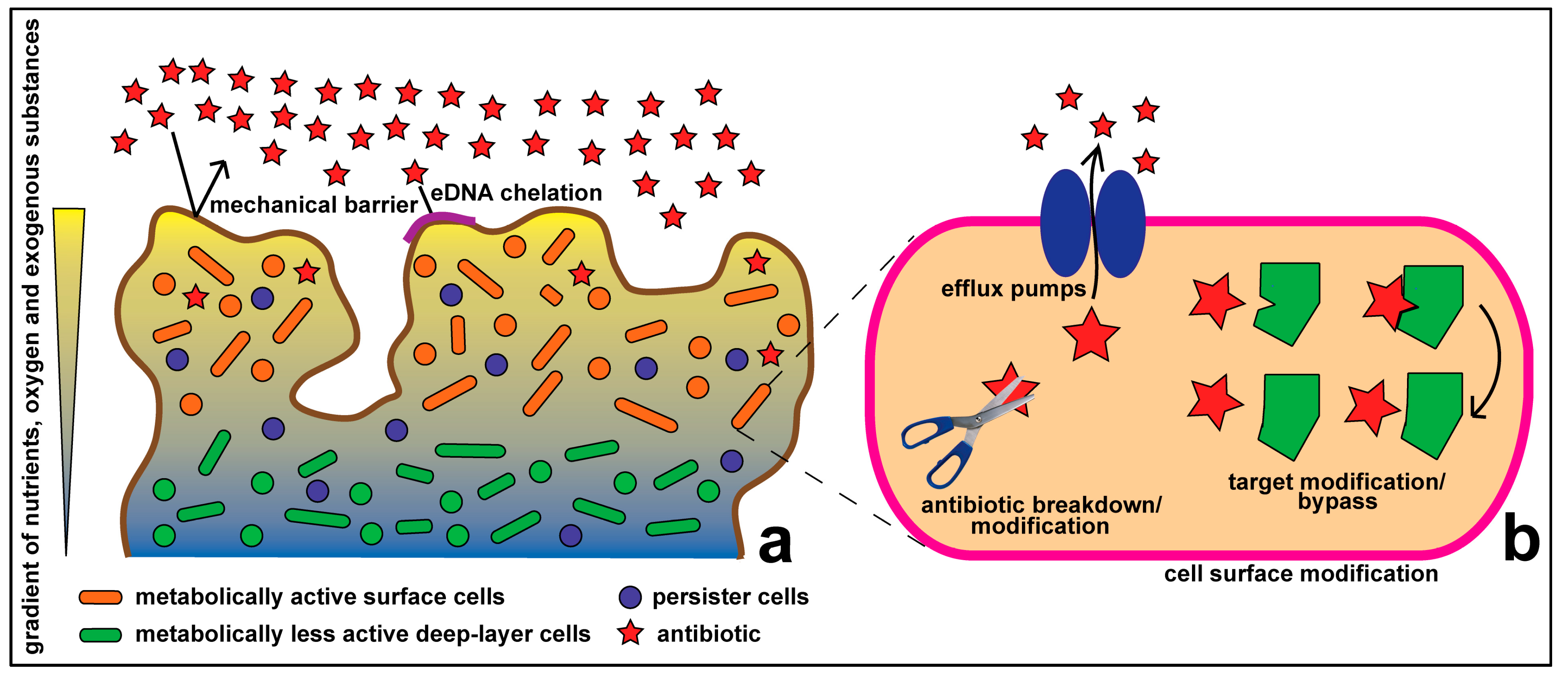
1.1. Resistance at the Cellular Level
1.2. Community Level Resistance
1.3. Synergy between Community and Cellular Level Resistance Mechanisms
2. Antibiotic Discovery
2.1. The Past and the Present
2.2. Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, S.B.; Barrett, J.F. Empirical antibacterial drug discovery—Foundation in natural products. Biochem. Pharmacol. 2006, 71, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Lerner, P.I. Producing penicillin. N. Engl. J. Med. 2004, 351, 524. [Google Scholar] [CrossRef] [PubMed]
- Barriere, S.L. Clinical, economic and societal impact of antibiotic resistance. Expert Opin. Pharmacother. 2015, 16, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Alanis, A.J. Resistance to antibiotics: Are we in the post-antibiotic era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Nadell, C.D.; Xavier, J.B.; Foster, K.R. The sociobiology of biofilms. FEMS Microbiol. Rev. 2009, 33, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Camilli, A.; Bassler, B.L. Bacterial small-molecule signaling pathways. Science 2006, 311, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [PubMed]
- Nikaido, H. Multiple antibiotic resistance and efflux. Curr. Opin. Microbiol. 1998, 1, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T. Multidrug efflux pumps and resistance: Regulation and evolution. Curr. Opin. Microbiol. 2003, 6, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Coque, T.M.; Baquero, F. What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol. 2015, 13, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Wright, G.D. Forces shaping the antibiotic resistome. Bioessays 2014, 36, 1179–1184. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; McGrann, K.M.; Hughes, D.W.; Wright, G.D. Sampling the antibiotic resistome. Science 2006, 311, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.O.A.; Dantas, G.; Church, G.M. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 2009, 325, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. The antibiotic resistome: The nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007, 5, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Antibiotic resistance in the environment: A link to the clinic? Curr. Opin. Microbiol. 2010, 13, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R. Evolutionary consequences of antibiotic use for the resistome, mobilome, and microbial pangenome. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.G.; Balkwill, D.L. Antibiotic resistance in bacteria isolated from the deep terrestrial subsurface. Microb. Ecol. 2009, 57, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Miteva, V.I.; Sheridan, P.P.; Brenchley, J.E. Phylogenetic and physiological diversity of microorganisms isolated from a deep greenland glacier ice core. Appl. Environ. Microbiol. 2004, 70, 202–213. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-dependent multidrug efflux systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [PubMed]
- Debabov, D. Antibiotic resistance: Origins, mechanisms, approaches to counter. Appl. Biochem. Microbiol. 2013, 49, 665–671. [Google Scholar] [CrossRef]
- Ceri, H.; Olson, M.E.; Stremick, C.; Read, R.R.; Morck, D.; Buret, A. The calgary biofilm device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 1999, 37, 1771–1776. [Google Scholar] [PubMed]
- Romero, D.; Aguilar, C.; Losick, R.; Kolter, R. Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc. Natl. Acad. Sci. USA 2010, 107, 2230–2234. [Google Scholar] [CrossRef] [PubMed]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA required for bacterial biofilm formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Reffuveille, F.; Fernández, L.; Hancock, R.E.W. Bacterial biofilm development as a multicellular adaptation: Antibiotic resistance and new therapeutic strategies. Curr. Opin. Microbiol. 2013, 16, 580–589. [Google Scholar]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Annu. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.; Bernier, S.P.; Kuchma, S.L.; Hammond, J.H.; Hasan, F.; O’Toole, G.A. Aminoglycoside resistance of Pseudomonas aeruginosa biofilms modulated by extracellular polysaccharide. Int. Microbiol. 2010, 13, 207–212. [Google Scholar] [PubMed]
- Yang, L.; Hu, Y.; Liu, Y.; Zhang, J.; Ulstrup, J.; Molin, S. Distinct roles of extracellular polymeric substances in Pseudomonas aeruginosa biofilm development. Environ. Microbiol. 2011, 13, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed]
- Chiang, W.C.; Nilsson, M.; Jensen, P.O.; Høiby, N.; Nielsen, T.E.; Givskov, M.; Tolker-Nielsen, T. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2013, 57, 2352–2361. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Qu, F.; Ling, Y.; Mao, P.; Xia, P.; Chen, H.; Zhou, D. Biofilm-associated infections: Antibiotic resistance and novel therapeutic strategies. Future Microbiol. 2013, 8, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Bernier, S.P.; Lebeaux, D.; DeFrancesco, A.S.; Valomon, A.; Soubigou, G.; Coppée, J.Y.; Ghigo, J.M.; Beloin, C. Starvation, together with the sos response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 2013, 9, e1003144. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 2008, 322, 107–131. [Google Scholar] [PubMed]
- Mulcahy, L.R.; Burns, J.L.; Lory, S.; Lewis, K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010, 192, 6191–6199. [Google Scholar] [CrossRef] [PubMed]
- Fridman, O.; Goldberg, A.; Ronin, I.; Shoresh, N.; Balaban, N.Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 2014, 513, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Conibear, T.C.R.; Collins, S.L.; Webb, J.S. Role of mutation in Pseudomonas aeruginosa biofilm development. PLoS One 2009, 4, e6289. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, J. Hypermutation as a factor contributing to the acquisition of antimicrobial resistance. Clin. Infect. Dis. 2003, 37, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Macía, M.D.; Blanquer, D.; Togores, B.; Sauleda, J.; Pérez, J.L.; Oliver, A. Hypermutation is a key factor in development of multiple-antimicrobial resistance in Pseudomonas aeruginosa strains causing chronic lung infections. Antimicrob. Agents Chemother. 2005, 49, 3382–3386. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.P.; Nielsen, K.M.; Fishert, D.S.; Hartl, D.L. Horizontal acquisition of divergent chromosomal DNA in bacteria: Effects of mutator phenotypes. Genetics 2003, 164, 13–21. [Google Scholar] [PubMed]
- Bernard, C.S.; Giraud, C.; Spagnolo, J.; de Bentzmann, S. Biofilms: The secret story of microbial communities. In Bacterial Pathogenesis: Molecular and Cellular Mechanisms; Locht, C., Simonet, M., Eds.; Caister Academic Press: Norfolk, UK, 2012; pp. 129–168. [Google Scholar]
- Gillings, M.R.; Holley, M.P.; Stokes, H.W. Evidence for dynamic exchange of qac gene cassettes between class 1 integrons and other integrons in freshwater biofilms. FEMS Microbiol. Lett. 2009, 296, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Koenig, J.E.; Bourne, D.G.; Curtis, B.; Dlutek, M.; Stokes, H.W.; Doolittle, W.F.; Boucher, Y. Coral-mucus-associated vibrio integrons in the great barrier reef: Genomic hotspots for environmental adaptation. ISME J. 2011, 5, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.G.H.; Verner-Jeffreys, D.W.; Baker-Austin, C. Aquatic systems: Maintaining, mixing and mobilising antimicrobial resistance? Trends Ecol. Evol. 2011, 26, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, S.J.; Bailey, M.; Hansen, L.H.; Kroer, N.; Wuertz, S. Studying plasmid horizontal transfer in situ: A critical review. Nat. Rev. Microbiol. 2005, 3, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Kloesges, T.; Popa, O.; Martin, W.; Dagan, T. Networks of gene sharing among 329 proteobacterial genomes reveal differences in lateral gene transfer frequency at different phylogenetic depths. Mol. Biol. Evol. 2011, 28, 1057–1074. [Google Scholar] [CrossRef] [PubMed]
- Domingues, S.; Harms, K.; Fricke, W.F.; Johnsen, P.J.; da Silva, G.J.; Nielsen, K.M. Natural transformation facilitates transfer of transposons, integrons and gene cassettes between bacterial species. PLoS Pathog. 2012, 8, e1002837. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R.; Stokes, H.W. Are humans increasing bacterial evolvability? Trends Ecol. Evol. 2012, 27, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Gillings, M.R.; Paulsen, I.T. Microbiology of the anthropocene. Anthropocene 2014. [Google Scholar] [CrossRef]
- Zhang, L.; Mah, T.F. Involvement of a novel efflux system in biofilm-specific resistance to antibiotics. J. Bacteriol. 2008, 190, 4447–4452. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Dixon, L.; Benoit, M.R.; Brodie, E.L.; Keyhan, M.; Hu, P.; Ackerley, D.F.; Andersen, G.L.; Matin, A. Role of the rapA gene in controlling antibiotic resistance of Escherichia coli biofilms. Antimicrob. Agents Chemother. 2007, 51, 3650–3658. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, R.J.; Colmer, S.; Tynan, H.; Demain, A.L.; Gullo, V.P. Antimicrobials, drug discovery, and genome mining. Appl. Microbiol. Biotechnol. 2013, 97, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, R.P. The antibiotic pipeline—Challenges, costs, and values. N. Engl. J. Med. 2004, 351, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.L.; Mueller, L.V.; Polyakov, M.; Weinstock, S.F. Where have all the antibiotic patents gone? Nat. Biotechnol. 2006, 24, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W. The end of an era? Nat. Rev. Drug Discov. 2007, 6, 28. [Google Scholar] [CrossRef]
- Projan, S.J. Whither antibacterial drug discovery? Drug Discov. Today 2008, 13, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.; Penesyan, A. Marine bacteria as a source of new antibiotics. In Bioactive Compounds: Types, Biological Activities and Health Effects; Nova Science Publishers: Hauppauge, NY, USA, 2012; pp. 381–412. [Google Scholar]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.O.; Smedsgaard, J.; Nielsen, K.F.; Hansen, M.E.; Frisvad, J.C. Phenotypic taxonomy and metabolite profiling in microbial drug discovery. Nat. Prod. Rep. 2005, 22, 672–695. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. Chemgps-np: Tuned for navigation in biologically relevant chemical space. J. Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Jose, P.A.; Jebakumar, S.R.D. Unexplored hypersaline habitats are sources of novel actinomycetes. Front. Microbiol. 2014, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Penesyan, A.; Kjelleberg, S.; Egan, S. Development of novel drugs from marine surface associated microorganisms. Mar. Drugs 2010, 8, 438–459. [Google Scholar] [CrossRef] [PubMed]
- Penesyan, A.; Marshall-Jones, Z.; Holmstrom, C.; Kjelleberg, S.; Egan, S. Antimicrobial activity observed among cultured marine epiphytic bacteria reflects their potential as a source of new drugs. FEMS Microbiol. Ecol. 2009, 69, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef] [PubMed]
- Bérdy, J. Bioactive microbial metabolites: A personal view. J. Antibiot. (Tokyo) 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Penesyan, A.; Ballestriero, F.; Daim, M.; Kjelleberg, S.; Thomas, T.; Egan, S. Assessing the effectiveness of functional genetic screens for the identification of bioactive metabolites. Mar. Drugs 2013, 11, 40–49. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schaberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.T.; Khosla, C. Combinatorial biosynthesis of polyketides-a perspective. Curr. Opin. Chem. Biol. 2012, 16, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Floss, H.G. Combinatorial biosynthesis-potential and problems. J. Biotechnol. 2006, 124, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, C.J.; Khosla, C. Combinatorial biosynthesis of “unnatural” natural products: The polyketide example. Chem. Biol. 1995, 2, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Cane, D.E. Programming of erythromycin biosynthesis by a modular polyketide synthase. J. Biol. Chem. 2010, 285, 27517–27523. [Google Scholar] [CrossRef] [PubMed]
- Fisch, K.M.; Bakeer, W.; Yakasai, A.A.; Song, Z.; Pedrick, J.; Wasil, Z.; Bailey, A.M.; Lazarus, C.M.; Simpson, T.J.; Cox, R.J. Rational domain swaps decipher programming in fungal highly reducing polyketide synthases and resurrect an extinct metabolite. J. Am. Chem. Soc. 2011, 133, 16635–16641. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. The need for new antibiotics. Clin. Microbiol. Infec. Suppl. 2004, 10, 1–9. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J.; America, T.I.D.S.O. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the infectious diseases society of america. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- McDougald, D.; Rice, S.A.; Barraud, N.; Steinberg, P.D.; Kjelleberg, S. Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 2012, 10, 39–50. [Google Scholar]
- Kolodkin-Gal, I.; Romero, D.; Cao, S.; Clardy, J.; Kolter, R.; Losick, R. d-amino acids trigger biofilm disassembly. Science 2010, 328, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Hochbaum, A.I.; Kolodkin-Gal, I.; Foulston, L.; Kolter, R.; Aizenberg, J.; Losick, R. Inhibitory effects of d-amino acids on Staphylococcus aureus biofilm development. J. Bacteriol. 2011, 193, 5616–5622. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Hassett, D.J.; Hwang, S.H.; Rice, S.A.; Kjelleberg, S.; Webb, J.S. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 7344–7353. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Schleheck, D.; Klebensberger, J.; Webb, J.S.; Hassett, D.J.; Rice, S.A.; Kjelleberg, S. Nitric oxide signaling in Pseudomonas aeruginosa biofilms mediates phosphodiesterase activity, decreased cyclic di-GMP levels, and enhanced dispersal. J. Bacteriol. 2009, 191, 7333–7342. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human host defense peptide ll-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [PubMed]
- De La Fuente-Núñez, C.; Korolik, V.; Bains, M.; Nguyen, U.; Breidenstein, E.B.M.; Horsman, S.; Lewenza, S.; Burrows, L.; Hancock, R.E.W. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar]
- Manefield, M.; de Nys, R.; Kumar, N.; Read, R.; Givskov, M.; Steinberg, P.; Kjelleberg, S. Evidence that halogenated furanones from Delisea pulchra inhibit acylated homoserine lactone (AHL)-mediated gene expression by displacing the AHL signal from its receptor protein. Microbiology 1999, 145, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Manefield, M.; Kjelleberg, S.; Givskov, M. Controlling bacterial infection by inhibiting intercellular signalling. Curr. Med. Chem.: Anti-Infect. Agents 2003, 2, 213–218. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Givskov, M. Quorum sensing inhibitory drugs as next generation antimicrobials: Worth the effort? Curr. Infect. Dis. Rep. 2008, 10, 22–28. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penesyan, A.; Gillings, M.; Paulsen, I.T. Antibiotic Discovery: Combatting Bacterial Resistance in Cells and in Biofilm Communities. Molecules 2015, 20, 5286-5298. https://doi.org/10.3390/molecules20045286
Penesyan A, Gillings M, Paulsen IT. Antibiotic Discovery: Combatting Bacterial Resistance in Cells and in Biofilm Communities. Molecules. 2015; 20(4):5286-5298. https://doi.org/10.3390/molecules20045286
Chicago/Turabian StylePenesyan, Anahit, Michael Gillings, and Ian T. Paulsen. 2015. "Antibiotic Discovery: Combatting Bacterial Resistance in Cells and in Biofilm Communities" Molecules 20, no. 4: 5286-5298. https://doi.org/10.3390/molecules20045286
APA StylePenesyan, A., Gillings, M., & Paulsen, I. T. (2015). Antibiotic Discovery: Combatting Bacterial Resistance in Cells and in Biofilm Communities. Molecules, 20(4), 5286-5298. https://doi.org/10.3390/molecules20045286