3.1. Chemistry
3.1.1. Synthesis of curcumin phosphorylated derivatives.
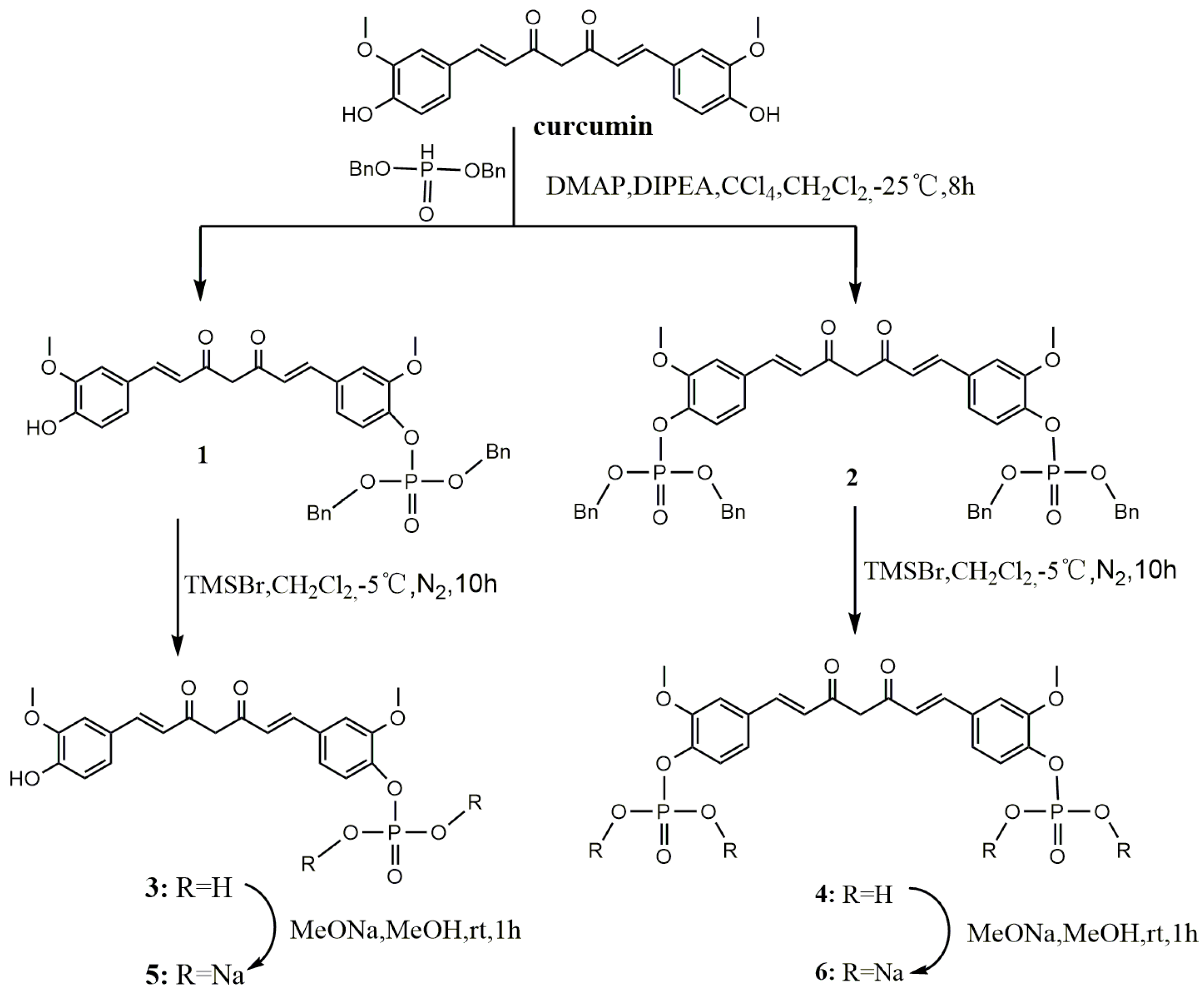
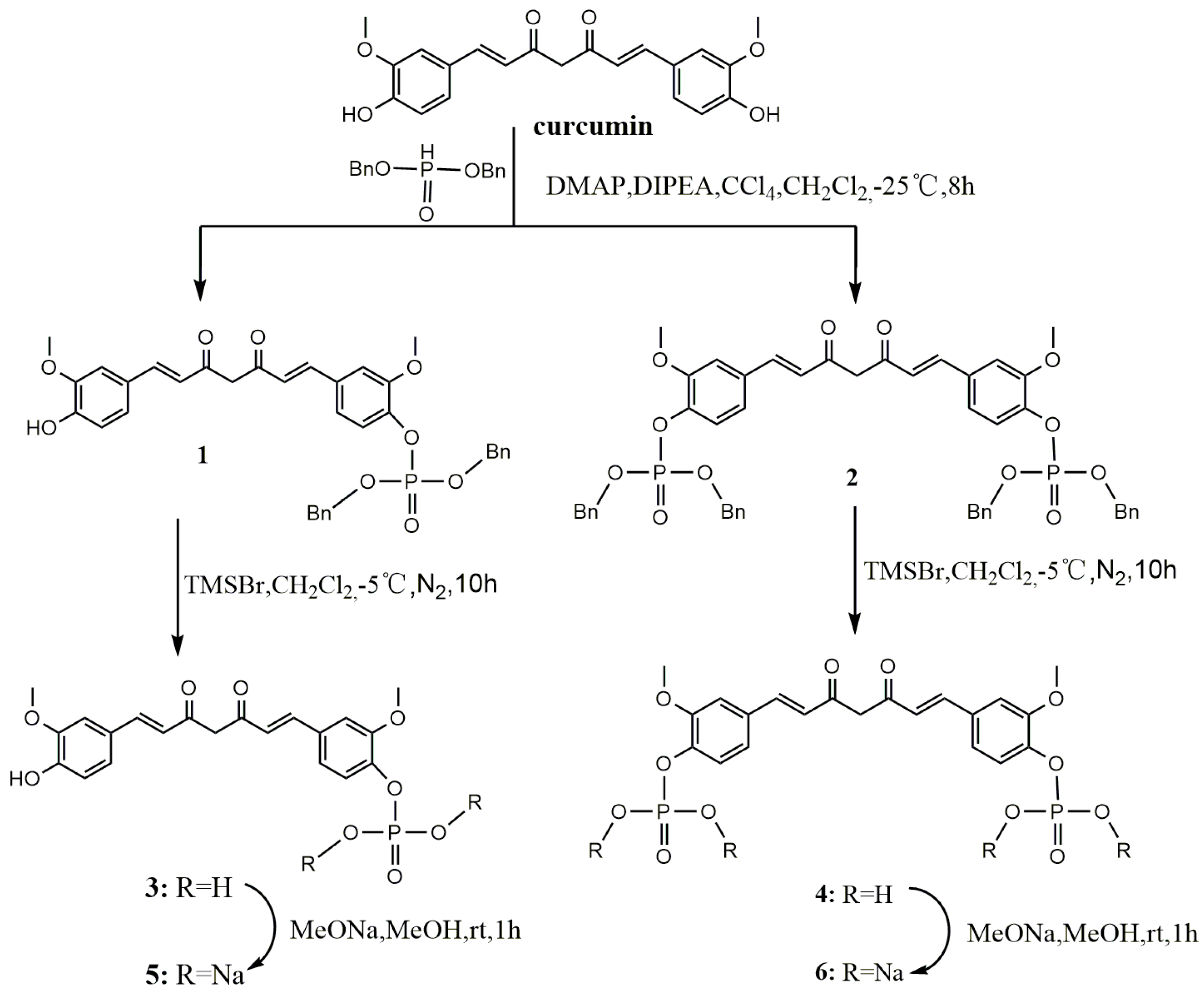
Dibenzyl 4-((1E,6E)-7-(4-hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl phosphate (1). To a solution of curcumin (0.3 g, 0.81 mmol), CCl4 (0.78 mL, 8.10 mmol), DIPEA (0.44 g, 3.42 mmol) and DMAP (19.90 mg, 0.16 mmol) in anhydrous ethyl acetate (30 mL), dibenzyl phosphate (0.641 g, 2.44 mmol) was added dropwise at −25 °C under a N2 atmosphere. The resulting homogeneous mixture was stirred for 8 h at −25 °C and evaporated to dryness under reduced pressure. The residue was diluted to 100 mL with ethyl acetate, successively washed with water (3 × 100 mL) and brine (3 × 100 mL), and dried over anhydrous Na2SO4. After filtration, the filtrate was evaporated under reduced pressure to give crude product, which was purified by silica gel chromatography to give yellow oil (0.1 g, 20%). 1H-NMR (600 MHz, DMSO-d6) δ: 9.74 (s, 1H), 7.60 (d, J = 6.2 Hz, 1H), 7.58 (d, J = 6.2 Hz, 1H), 7.50 (s, 1H), 7.46–7.35 (m, 10H), 7.34 (d, J = 5.2 Hz, 1H), 7.28 (d, J = 8.3 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 7.17 (d, J = 8.1 Hz, 1H), 6.95 (d, J = 15.9 Hz, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.80 (d, J = 15.8 Hz, 1H), 6.12 (d, J = 8.6 Hz, 1H), 5.19 (d, J = 8.1 Hz, 4H), 3.85 (d, J = 10.4 Hz, 6H);13C-NMR (150 MHz, DMSO-d6) δ: 185.3, 181.8, 151.1, 149.9, 148.4, 142.0, 140.8, 139.3, 136.2, 133.4, 128.9 (5C), 128.7, 128.3 (5C), 127.9, 126.6, 124.9, 123.8, 121.8, 121.6, 116.1, 112.8, 111.8, 101.8, 69.8 (2C), 56.5, 56.1; ESI-MS [M − H]− m/z: 627.5.
Dibenzyl 4,4′-((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) diphosphate (2). The synthetic method of 2 was similar to that of compound 1, affording yellow oil (0.28 g, 39%). 1H-NMR (600 MHz, DMSO-d6) δ: 7.64 (d, J = 15.8 Hz, 2H), 7.52 (s, 2H), 7.38 (s, 20H), 7.30 (d, J = 8.4 Hz, 2H), 7.24 (d, J = 8.2 Hz, 2H), 6.99 (d, J = 15.9 Hz, 2H), 6.20 (s, 1H), 5.19 (d, J = 8.0 Hz, 8H), 3.85 (d, J = 10.3 Hz, 6H); 13C-NMR (150 MHz, DMSO-d6) δ: 183.6 (2C), 151.1 (2C), 141.0 (2C), 140.1 (2C), 136.1 (4C), 133.3 (2C), 128.9 (12C), 128.3 (10C), 124.9 (2C), 121.8 (2C), 112.9 (2C), 102.1, 69.8 (4C), 56.4 (2C); ESI-MS [M − H]− m/z: 887.6.
4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl dihydrogen phosphate (3). TMSBr (0.13 mL, 0.982 mmol) was added dropwise to a stirred solution of 1 (0.308 g, 0.491 mmol) in anhydrous dichloromethane (5 mL) at −5 °C under a N2 atmosphere. The reaction mixture was stirred for 10 h at 0 °C. The mixture was poured into methanol (20 mL) and then evaporated to dryness under reduced pressure. The residue was purified by Sephadex LH-20 column chromatography to afford yellow solid (0.153 g, 70%). m.p. 144–146 °C. 1H-NMR (600 MHz, MeOD-d4) δ: 7.57 (d, J = 9.4 Hz, 1H), 7.54 (d, J = 9.4 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.24 (s, 1H), 7.18 (d, J = 1.0 Hz, 1H), 7.13 (d, J = 7.8 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.68 (d, J = 15.8 Hz, 1H), 6.61 (d, J = 15.8 Hz, 1H), 3.89 (d, J = 7.4 Hz, 6H); 13C-NMR (150 MHz, MeOD-d4) δ: 184.5, 181.9, 151.3, 149.1, 147.9, 143.2, 141.2, 139.5, 131.5, 129.8, 127.0, 122.9, 122.8, 121.0, 120.9, 115.1, 111.4, 110.3, 55.1, 55.0; 31P-NMR (243 MHz, MeOD-d4) δ:−4.31 (1P); ESI-MS [M − H]− m/z: 447.4 ; HR-ESI-MS [M − H]− m/z: 447.0854, Calcd. for C21H21O9P (M − H) 447.0923.
4,4′-((1E,6E)-3,5-Dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) bis(dihydrogen phosphate) (4). TMSBr (0.5 mL, 3.7 mmol) was added dropwise to a stirred solution of 2 (0.736 g, 0.83 mmol) in anhydrous dichloromethane (5 mL) at −5 °C under a N2 atmosphere. The reaction mixture was stirred for 10 h at 0 °C. The mixture was poured into methanol (20 mL) and evaporated to dryness under reduced pressure. The residue was purified by Sephadex LH-20 column chromatography to afford yellow solid (0.32 g, 72%). m.p. 163–166 °C; 1H-NMR (600 MHz, MeOD-d4) δ: 7.54 (d, J = 15.9 Hz, 2H), 7.27 (d, J = 8.3 Hz, 2H), 7.24 (s, 2H), 7.12 (dd, J = 8.3, 1.7 Hz, 2H), 6.69 (d, J = 15.9 Hz, 2H), 3.83 (s, 6H); 13C-NMR (150 MHz, MeOD-d4) δ: 183.2 (2C), 151.4 (2C), 142.6 (2C), 139.9 (2C), 132.1 (2C), 123.3 (2C), 121.1 (2C), 120.9 (2C), 111.5 (2C), 55.1 (2C); 31P-NMR (243 MHz, MeOD-d4) δ: −4.96 (2P); ESI-MS [M − 2H/2]− m/z: 263; HR-ESI-MS [M − H]− m/z: 527.0203, Calcd. for C21H22O12P2 (M − H) 527.0586.
Sodium 4-((1E,6E)-7-(4-hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl phosphate (5). Compound 3 (0.049 g, 0.11 mmol) was dissolved in anhydrous methanol (5 mL), and was added to a solution of MeONa (0.012 g, 0.22 mmol) in methanol (1 mL) with stirring for 1 h at room temperature. The mixture was evaporated to dryness and then dissolved in water (1 mL) and acetonitrile (6 mL) solution. The product was crystallized and collected by filtration to give pure dark yellow solid (0.049 g, 91%). 1H-NMR (600 MHz, MeOD-d4) δ: 7.57 (d, J = 9.4 Hz, 1H), 7.54 (d, J = 9.4 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.24 (s, 1H), 7.18 (d, J = 1.0 Hz, 1H), 7.13 (d, J = 7.8 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.68 (d, J = 15.8 Hz, 1H), 6.61 (d, J= 15.8 Hz, 1H), 3.89 (d, J = 7.4 Hz, 6H); 13C-NMR (150 MHz, MeOD-d4) δ: 184.5, 181.9, 151.3, 149.1, 147.9, 143.2, 141.2, 139.5, 131.5, 129.8, 127.0, 122.9, 122.8, 121.0, 120.9, 115.1, 111.4, 110.3, 55.1, 55.0; 31P-NMR (243 MHz, MeOD-d4) δ: −4.31 (1P).
Sodium 4,4′-((1E,6E)-3,5-dioxohepta-1,6-diene-1,7-diyl)bis(2-methoxy-4,1-phenylene) diphosphate (6). The synthetic method of 6 was similar to that of compound 5, giving dark yellow solid (0.063 g, 91%). 1H-NMR (600 MHz, MeOD-d4) δ: 7.54 (d, J = 15.9 Hz, 2H), 7.27 (d, J = 8.3 Hz,2H), 7.24 (s, 2H), 7.12 (dd, J = 8.3, 1.7 Hz, 2H), 6.69 (d, J = 15.9 Hz, 2H), 3.83 (s, 6H); 13C-NMR (150 MHz, MeOD-d4) δ: 183.2 (2C), 151.4 (2C), 142.6 (2C), 139.9 (2C), 132.1 (2C), 123.3 (2C), 121.1 (2C), 120.9 (2C), 111.5 (2C), 55.1 (2C); 31P-NMR (243 MHz, MeOD-d4) δ: −4.96 (2P).
3.1.2. General Procedure for Synthesis of Curcumin Etherified Derivatives
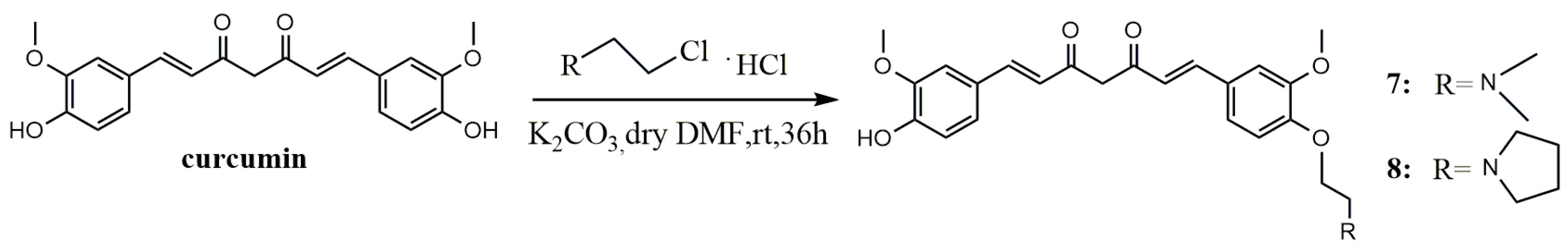
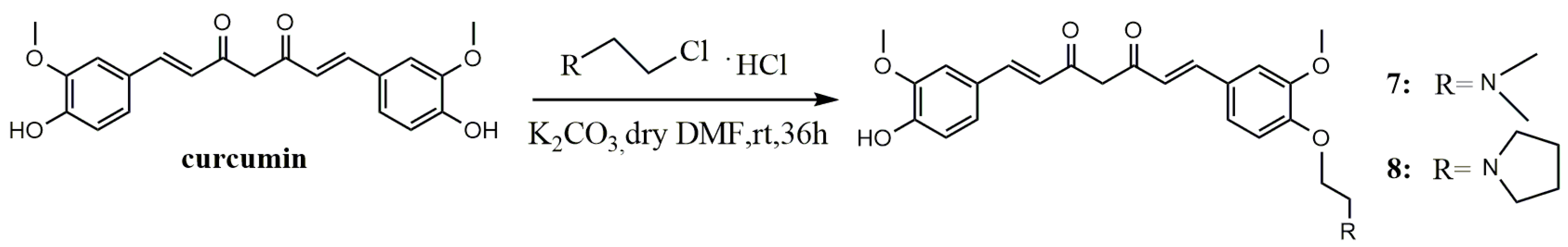
A mixture of curcumin (0.5 g, 1.36 mmol), N,N-Dimethyl-2-chloroethylamine hydrochloride (0.196 g, 1.36 mmol) or 1-(2-ethyl chloride) pyrrolidine hydrochloride (0.23 g, 1.36 mmol) and anhydrous potassium carbonate (0.376 g, 2.72 mmol) was dissolved in anhydrous dimethyl formamide (DMF) (10 mL), stirred for 36 h at room temperature, and evaporated to dryness under reduced pressure. The residue was diluted to 100 mL with dichloromethane, successively washed with water (3 × 100 mL) and brine (3 × 100 mL), and dried over anhydrous MgSO4. By filtration and evaporation of filtrate under reduced pressure, a crude product was obtained, then purified by thin-layer chromatography (TLC) and Sephadex LH-20 column chromatography, respectively, to give the pure product.
(1E,6E)-1-(4-(2-(Dimethylamino)ethoxy)-3-methoxyphenyl)-7-(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione (7). Dark red solid; yield: 18.3%. m.p. 129–132 °C; 1H-NMR (400 MHz, DMSO-d6) δ: 7.58 (d, J = 3.8 Hz, 1H), 7.54 (d, J = 3.8 Hz, 1H), 7.34 (dd, J = 9.0, 1.5 Hz, 2H), 7.25 (d, J = 8.4 Hz, 1H), 7.16 (dd, J = 8.2, 1.6 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 6.84 (d, J = 5.3 Hz, 1H), 6.81 (d, J = 2.1 Hz, 1H), 6.77 (d, J = 15.8 Hz, 1H), 6.08 (s, 1H), 4.09 (t, J = 5.9 Hz, 2H), 3.83 (t, J = 5.7 Hz, 6H), 2.64 (dd, J = 13.5, 7.7 Hz, 2H), 2.22 (s, 6H); 13C-NMR (100 MHz, DMSO-d6) δ: 184.2, 183.2, 150.7, 149.9, 149.7, 148.5, 141.4, 140.6, 128.2, 126.8, 123.7, 123.3, 122.6, 121.6, 116.2, 113.3, 111.9, 111.2, 101.4, 67.1, 58.1, 56.2 (2C), 46.1 (2C); ESI-MS [M + H]+ m/z: 440; HR-ESI-MS [M + H]+ m/z: 440.2068, Calcd. for C25H29NO6 (M + H) 440.2028.
(1E,6E)-1-(4-Hydroxy-3-methoxyphenyl)-7-(3-methoxy-4-(2-(pyrrolidin-1-yl)ethoxy)phenyl)hepta-1,6-diene-3,5-dione (8). Dark red solid; yield:24%. m.p. 128–132 °C; 1H-NMR (600 MHz, MeOD-d4) δ: 7.51 (d, J = 2.8 Hz, 1H), 7.49 (d, J = 2.8 Hz, 1H), 7.16 (s, 1H), 7.13 (s, 1H), 7.10 (d, J = 8.2 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 6.75 (d, J = 8.2 Hz, 1H), 6.60 (d, J = 15.8 Hz, 1H), 6.55 (d, J = 15.8 Hz, 1H), 4.11 (t, J = 5.6 Hz, 2H), 3.83 (d, J = 12.0 Hz, 6H),2.95 (t, J = 5.6 Hz, 2H), 2.71 (d, J = 5.7 Hz, 4H), 1.82–1.77 (m, 4H); 13C-NMR (150 MHz, MeOD-d4) δ: 183.9, 182.6, 150.1, 149.7, 149.4, 148.1, 140.9, 139.9, 128.7, 126.9, 122.9, 122.3, 121.9, 120.7, 115.2, 112.9, 110.3, 110.2, 67.1, 55.0, 54.9, 54.3, 54.2 (2C), 22.8 (2C); ESI-MS [M + H]+ m/z: 466; HR-ESI-MS [M + H]+ m/z: 466.2224, Calcd. for C27H31NO6 (M + H) 466.2185.
3.1.3. General Procedure for Synthesis of Dipeptide Methyl Ester
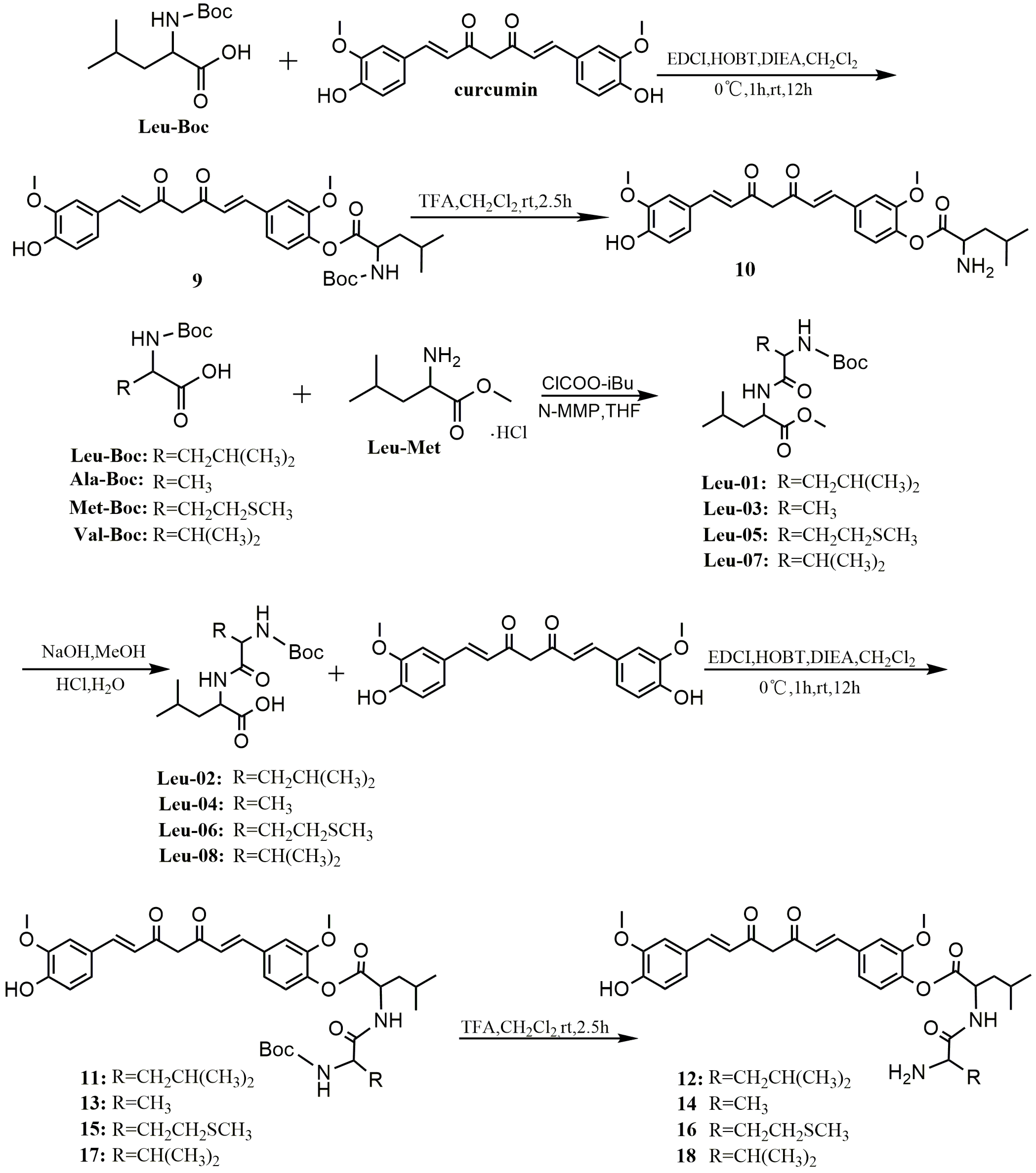
To a stirred solution of N-tert-butoxycarbonyl-l-leucine(or alanine, methionine, valine) (9.3 mmol) and N-methylmorpholine (2.1 mL, 19 mmol) in THF (20 mL), isobutyl chloroformate (1.4 mL, 11 mmol) was added at −5 °C and the mixture was stirred over 30 min. l-leucine methyl ester hydrochloride (1.69 g, 9.3 mmol) was added to the mixture. The stirring was continued for 1 h at −5 °C, and then the mixture was stirred at room temperature for 5 h. The solution was concentrated with a rotary evaporator in vacuo. The residue was dissolved in EtOAc and successively washed with 5% NaHCO3, 10% acetic acid, and brine. The EtOAc solution was dried over anhydrous Na2SO4 and concentrated with a rotary evaporator to afford a product.
Methyl2-(2-(tert-butoxycarbonylamino)-4-methylpentanamido)-4-methylpentanoate (Leu-01). White powder; yield: 92%; 1H-NMR (400 MHz, DMSO-d6) δ: 8.08 (d, J = 7.6 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 4.34–4.21 (m, 1H), 4.07–3.89 (m, 1H), 3.60 (s, 3H), 1.59 (ddd, J = 15.1, 12.6, 5.0 Hz, 3H), 1.47 (ddd, J = 13.8, 9.3, 4.9 Hz, 1H), 1.40–1.38(m, 1H), 1.37 (s, 9H), 0.96–0.74 (m, 13H); 13C-NMR (100 MHz, DMSO-d6) δ: 173.4, 173.1, 155.7, 78.4, 53.0, 52.2, 50.5, 41.1, 40.2,28.62 (3C), 24.6, 24.5, 23.4, 23.3, 22.2, 21.6; ESI-MS [M + Na]+ m/z: 381.
(2S)-Methyl2-(2-(tert-butoxycarbonylamino)propanamido)-4-methylpentanoate (Leu-03). White powder; yield: 92%; 1H-NMR (400 MHz, DMSO-d6) δ: 8.06 (d, J = 7.6 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 4.37–4.20 (m, 1H), 4.14–3.85 (m, 1H), 3.61 (d, J = 3.5 Hz, 3H), 1.70–1.59 (m, 1H), 1.59–1.43 (m, 2H), 1.37 (s,9H), 1.20–1.12 (m, 3H), 0.89–0.79 (m, 6H); 13C-NMR (100 MHz, DMSO-d6) δ: 173.4 (2C), 155.5, 78.5, 52.3, 50.6, 49.9, 40.3, 28.6 (3C), 24.6, 23.2, 21.8, 18.4; ESI-MS [M + Na]+ m/z: 339.
(2S)-Methyl2-(2-(tert-butoxycarbonylamino)-4-(methylthio)butanamido)-4-methylpentanoate (Leu-05). White powder; yield: 92%; 1H-NMR (400 MHz, DMSO-d6) δ: 8.14 (d, J = 7.6 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 4.59–4.13 (m, 1H), 4.13–3.93 (m, 1H), 3.61 (m, 3H), 2.45 (t, J = 7.6 Hz, 2H), 2.01 (d, J = 20.1 Hz, 3H), 1.90–1.71 (m, 2H), 1.71–1.53 (m, 2H), 1.52–1.45 (m, 1H), 1.37 (s, 9H), 0.92–0.80 (m, 6H); 13C-NMR (100 MHz, DMSO-d6) δ: 173.3, 172.3, 155.7, 78.6, 53.8, 52.2, 50.6, 40.2, 32.2, 30.0, 28.6 (3C), 24.6, 23.3, 21.7, 15.1; ESI-MS [M + Na]+ m/z: 399.
(2S)-Methyl2-(2-(tert-butoxycarbonylamino)-3-methylbutanamido)-4-methylpentanoate (Leu-07). White powder; yield: 92%; 1H-NMR (400 MHz, DMSO-d6) δ: 12.47 (s, 1H), 7.97 (d, J = 7.9 Hz, 1H), 6.62 (d, J = 9.1 Hz, 1H), 4.24 (td, J = 9.8, 5.2 Hz, 1H), 3.76 (m, 1H), 1.92 (m, 1H), 1.66 (d, J = 5.7 Hz, 1H), 1.53 (m, 2H), 1.38 (s, 9H), 0.87 (m, 12H); 13C-NMR (100 MHz, DMSO-d6) δ: 173.3, 172.1, 155.8, 78.5, 60.1, 52.2, 50.5, 40.2, 30.8, 28.6 (3C), 24.5, 23.2, 21.6, 19.5, 18.6; ESI-MS [M + Na]+ m/z: 367.
3.1.4. General Procedure for Synthesis of Dipeptide
Leu-01, -03, -05, or -07 (0.005 mmol) was dissolved in MeOH (100 mL), and 10 mL of 1 mol/L NaOH was added over 5 min with stirring. The reaction mixture was continuously stirred for 12 h at room temperature. After the pH value of the resulting solution was adjusted to 2–3 with 1 mol/L hydrochloric acid, a white precipitated solid was produced, collected by filtration, and dried to give the pure product.
2-(2-(Tert-butoxycarbonylamino)-4-methylpentanamido)-4-methylpentanoic acid (Leu-02). Pale yellow oil; yield: 93%; 1H-NMR (400 MHz, DMSO-d6) δ: 12.38 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H), 4.24 (m, 1H), 3.98 (m, 1H), 1.78–1.57 (m, 2H), 1.57–1.48 (m, 2H), 1.40 (m, 1H), 1.38 (s, 9H), 0.95–0.77 (m, 13H); 13C-NMR (100 MHz, DMSO-d6) δ: 174.4, 171.7, 155.7, 78.4, 53.2, 50.5, 43.2, 41.2, 28.6 (3C), 24.6,24.6, 23.4, 23.3,22.1, 21.8. ESI-MS [M − H]− m/z: 343.
(2S)-2-(2-(Tert-butoxycarbonylamino)propanamido)-4-methylpentanoic acid (Leu-04). Pale yellow oil; yield: 93%; 1H-NMR (400 MHz, DMSO-d6) δ: 12.51 (s, 1H), 7.89 (d, J = 7.9 Hz, 1H), 6.86 (d, J = 7.7 Hz, 1H), 4.26–4.20 (m, 1H), 4.03–3.94 (m, 1H), 1.67–1.62 (m, 1H), 1.58–1.44 (m, 2H), 1.37 (s, 9H), 1.16 (d, J = 7.1 Hz, 3H), 0.82–0.9 (m, 6H); 13C-NMR (100 MHz, DMSO-d6) δ: 174.4, 173.1, 155.4, 78.5, 55.3, 50.5, 49.9, 28.6 (3C), 24.6, 23.3, 21.8, 18.5; ESI-MS [M − H]− m/z: 301.
(2S)-2-(2-(Tert-butoxycarbonylamino)-4-(methylthio)butanamido)-4-methylpentanoic acid (Leu-06). Pale yellow oil; yield: 93%; 1H-NMR (400 MHz, DMSO-d6) δ: 12.55 (s, 1H), 8.00 (d, J = 7.7 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 4.25–4.19 (m, 1H), 4.04–3.99 (m,1H), 2.45 (t, J = 7.7 Hz, 1H), 2.03 (s, 3H), 1.88–1.72 (m, 2H), 1.66 (m, 1H), 1.58–1.47 (m, 2H), 1.38 (s, 9H), 0.90–0.83 (m, 6H); 13C-NMR (100 MHz, DMSO-d6) δ: 174.4, 172.1, 155.7, 78.6, 55.4, 53.9, 50.6, 32.2, 30.0, 28.6 (3C), 24.6, 23.4, 21.7, 15.1; ESI-MS [M − H]− m/z: 361.
(2S)-2-(2-(Tert-butoxycarbonylamino)-3-methylbutanamido)-4-methylpentanoic acid (Leu-08). Pale yellow oil; yield: 93%; 1H-NMR (400 MHz, DMSO-d6) δ: 8.15 (d, J = 7.6 Hz, 1H), 6.60 (t, J = 23.2 Hz, 1H), 4.30 (m, 1H), 3.79 (m, 1H), 3.60 (s, 3H), 1.89 (dt, J = 26.8, 10.0 Hz, 1H), 1.66 (m, 1H), 1.56 (m, 1H), 1.48 (ddd, J = 13.8, 9.2, 5.0 Hz, 1H), 1.38 (s, 9H), 0.85 (m, 12H); 13C-NMR (100 MHz, DMSO-d6) δ: 173.3, 172.1, 155.8, 78.5, 60.1, 52.2, 50.5, 30.8, 28.6 (3C), 24.5, 23.3, 21.6, 19.6, 18.7; ESI-MS [M − H]− m/z: 329.
3.1.5. General Procedure of Curcumin Dipeptide Conjugation
A mixture of Leu-Boc, Leu-02, -04, -06, or -08 (1.36 mmol), EDCI (0.244 g, 1.36 mmol), HOBT (0.184 g, 1.36 mmol), and DIEA (240 µL, 1.36 mmol) in anhydrous dichloromethanewas stirred for 1 h at 0 °C. Curcumin (0.5 g, 1.36 mmol) was dissolved in anhydrous dichloromethaneand added dropwise to the above solution and was stirred overnight at room temperature. The mixture was diluted to 200 mL with dichloromethane, successively washed with 1N HCl (3 × 200 mL), water (3 × 200 mL) and brine (3 × 200 mL), and dried over anhydrous MgSO4. After filtration, the filtrate was evaporated under reduced pressure to give crude product, which was purified by preparative TLC and silica gel column chromatography (petroleum ether/ethyl acetate 8:1) to give the pure product.
4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(tert-butoxycarbonylamino)-4-methylpentanoate (9). Yellow powder; yield: 21%; m.p. 93–96 °C; 1H-NMR (600 MHz, MeOD-d4) δ: 7.56 (d, J = 7.9 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.26 (s, 1H), 7.16 (s, 2H), 7.06 (d, J = 8.2 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 6.72 (d, J = 15.8 Hz, 1H), 6.60 (d, J = 15.8 Hz, 1H), 4.35 (dd, J = 10.0, 4.9 Hz, 1H), 3.84 (d, J = 31.8Hz, 6H), 1.84–1.78 (m, 1H), 1.77–1.71 (m, 1H), 1.71.64 (m, 1H), 1.43 (s, 9H), 0.96 (dd, J = 13.6, 6.5 Hz, 6H); 13C-NMR (150 MHz, MeOD-d4) δ: 185.1, 181.2, 171.6, 156.8, 151.5, 149.2, 147.9, 141.5, 141.1, 138.9, 134.4, 126.9, 124.0, 122.9, 122.7, 120.9, 120.7, 115.1, 111.3, 110.3, 101.1, 79.2, 55.1, 55.0, 52.2, 40.1, 27.3 (3C), 24.6, 21.9, 20.5; ESI-MS [M − H]− m/z: 580.
4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-(tert-butoxycarbonylamino)-4-methylpentanamido)-4-methylpentanoate (11). Yellow powder; yield: 28%; m.p. 100–102 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 8.43 (d, J = 7.8 Hz, 1H), 7.60 (d, J = 15.6 Hz, 2H), 7.32 (s, 1H), 7.22 (s, 2H), 7.11 (d, J = 8.3 Hz, 1H), 7.06 (d, J = 7.7 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.73 (d, J = 16.2 Hz, 1H), 6.65 (d, J = 16.1 Hz, 1H), 6.01 (s, 1H), 4.76–4.69 (m, 1H), 4.19–4.05 (m, 1H), 3.88 (d, J = 20.3 Hz, 6H), 1.90–1.79 (m, 3H), 1.77–1.66 (m, 1H), 1.63–1.48 (m, 2H), 1.43 (s, 9H), 1.05–0.89 (m, 12H); 13C-NMR (100 MHz, MeOD-d4) δ: 185.2, 181.2, 173.8, 170.6, 156.4, 151.5, 149.3, 148.1, 141.5, 141.1, 138.9, 134.5, 127.1, 124.2, 122.9, 122.7, 121.0, 120.7, 115.2, 111.4, 110.5, 101.0, 79.1, 55.1 (2C), 52.9, 50.8, 40.8, 40.1, 27.3 (3C), 24.5 (2C), 21.9 (4C); ESI-MS [M − H]− m/z: 693.
(2S)-4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-(tert-butoxycarbonylamino)propanamido)-4-methylpentanoate (13). Yellow powder; yield: 33%; m.p. 100–102 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.49 (dd, J = 15.8, 3.7 Hz, 2H), 7.19 (d, J = 20.1 Hz, 1H), 7.16 (s, 2H), 7.02–6.96 (m, 2H), 6.72 (d, J = 8.1 Hz, 1H), 6.67 (d, J = 15.7 Hz, 1H), 6.54 (d, J = 15.7 Hz, 1H), 4.61 (m, 1H), 4.01 (m, 1H), 3.78 (d, J = 20.1 Hz, 6H) , 1.85–1.62 (m, 3H), 1.34 (s, 9H), 1.23 (dd, J = 7.2, 3.7 Hz, 3H), 0.98–0.84 (m, 6H); 13C-NMR (100 MHz, MeOD-d4) δ: 185.0, 181.2, 174.8, 170.6, 156.1, 151.4, 149.2, 147.9, 141.5, 140.9, 138.9, 134.5, 127.1, 124.2, 122.9, 122.8, 121.0, 120.7, 115.3, 111.5, 110.6, 79.3, 55.1 (2C), 53.4, 50.6, 40.1, 27.4 (3C), 24.6, 22.0, 20.6, 17.2; ESI-MS [M − H]− m/z: 651.
(2S)-4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-(tert-butoxycarbonylamino)-4-(methylthio)butanamido)-4-methylpentanoate (15). Yellow powder; yield: 35%; m.p. 95–97 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.61 (d, J = 11.0 Hz, 2H), 7.32 (s, 1H), 7.22 (s, 2H), 7.11 (m, 2H), 6.84 (d, J = 8.0 Hz, 1H), 6.77 (d, J = 13.5 Hz, 1H), 6.66 (d, J = 15.3 Hz, 1H), 4.78–4.65 (m, 1H), 4.25 (m, 1H), 3.90 (d, J = 20.2 Hz, 6H), 2.57 (m, 2H), 2.07 (m, 2H), 2.01 (s, 3H), 1.94–1.79 (m, 3H), 1.46 (d, J = 1.8 Hz, 9H), 1.02 (dd, J = 15.1, 4.4 Hz, 6H); 13C-NMR (100 MHz, MeOD-d4) δ: 185.1, 181.3, 173.8, 170.6, 156.3, 151.5, 149.3, 148.1, 141.5, 141.0, 138.9, 134.5, 127.1, 124.2, 122.9, 122.8, 121.0, 120.7, 115.2, 111.4, 110.5, 79.3, 55.1 (2C), 53.6, 50.9, 39.9, 31.7, 29.6, 27.3 (3C), 24.6, 21.9, 20.5, 13.9; ESI-MS [M − H]− m/z: 711.
(2S)-4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-(tert-butoxycarbonylamino)-3-methylbutanamido)-4-methylpentanoate (17). Yellow powder; yield: 35%; m.p. 106–109 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.61 (dd, J = 15.8, 6.4 Hz, 2H), 7.32 (s, 1H), 7.22 (d, J = 6.1 Hz, 2H), 7.11 (m, 2H), 6.84 (d, J = 8.2 Hz, 1H), 6.78 (d, J = 15.9 Hz, 1H), 6.66 (d, J = 15.8 Hz, 1H), 6.02 (s, 1H), 4.70 (m, 1H), 3.95 (m, 1H), 3.89 (d, J = 22.4 Hz, 6H), 2.06 (dt, J = 13.6, 6.7 Hz, 1H), 1.86 (m, 3H), 1.46 (d, J = 2.5 Hz, 9H), 1.01 (m, 12H); 13C-NMR (100 MHz, MeOD-d4) δ: 185.2, 181.3, 173.4, 170.6, 156.5, 151.5, 149.3, 148.0, 141.5, 141.0, 138.9, 134.5, 127.1, 124.2, 122.9, 122.7, 121.0, 120.7, 115.2, 111.4, 110.5, 101.1, 79.1, 60.2, 55.1 (2C), 50.8, 39.9, 30.7, 27.3(3C), 24.5, 21.9, 20.4, 18.4, 17.1; ESI-MS [M − H]− m/z: 679.
3.1.6. General Procedure for Deprotection of Boc Group
TFA (200 µL) was added dropwise with stirring to a solution of compounds 9, 11, 13, 15, and 17 (0.136 mmol) in anhydrous dichloromethane at 0 °C. The reaction mixture was stirred for 2.5 h at room temperature. The mixture was evaporated to dryness to give the pure product.
4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-amino-4-methylpentanoate (10). Dark red solid; yield: 89%; m.p. 114–116 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.53 (d, J = 10.7 Hz, 2H), 7.29 (d, J = 13.8 Hz, 1H), 7.16 (d, J = 16.3 Hz, 2H), 7.11–7.00 (m, 2H), 6.74 (d, J = 6.5 Hz, 2H), 6.59 (s, 1H), 4.25 (t, J = 7.0 Hz, 1H), 3.82 (d, J = 4.0 Hz, 6H), 2.03–1.85 (m, 2H), 1.74 (m,1H), 1.00 (t, J = 6.5 Hz, 6H); 13C-NMR (100 MHz, MeOD-d4) δ: 185.4, 180.9, 167.8, 151.1, 149.3, 148.1, 141.7, 140.1, 138.5, 135.3, 127.0, 124.7, 122.9, 122.5, 120.9, 120.7, 115.2, 111.4, 110.5, 55.2, 55.1, 51.1, 39.6, 24.2, 21.1, 21.0; ESI-MS [M − H]− m/z: 480; HR-ESI-MS [M − H]− m/z: 480.2020, Calcd. for C27H31NO7 (M − H) 480.2101.
4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-amino-4-methylpentanamido)-4-methylpentanoate (12). Dark red solid; yield: 89%; m.p. 127–129 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.52 (d, J = 14.1 Hz, 2H), 7.26 (s, 1H), 7.13 (s,2H), 7.06–6.96 (m, 2H), 6.74 (d, J = 7.7 Hz, 2H), 6.56 (d, J = 13.9 Hz, 1H), 4.66–4.70 (m, 1H), 3.91–3.85 (m, 1H), 3.80 (d, J = 16.2 Hz, 6H), 1.77–1.83 (m, 2H), 1.76 -1.65 (m, 3H), 1.66 (s, 1H), 1.00–0.86 (m, 12H); 13C-NMR (100 MHz, MeOD-d4) δ: 187.3, 183.2, 172.3, 171.7, 153.5, 151.3, 150.1, 143.6, 142.9, 140.8, 136.7, 129.1, 126.3, 124.9, 124.6, 123.0, 122.8, 117.3, 113.4, 112.5, 57.2 (2C), 53.5, 53.1, 42.5, 41.9, 26.6, 25.9, 23.9, 23.7, 22.7, 22.5; ESI-MS [M + H]+ m/z: 595; HR-ESI-MS [M + H]+ m/z: 595.3022, Calcd. for C33H42N2O8 (M + H) 595.2975.
(2S)-4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-aminopropanamido)-4-methylpentanoate (14). Dark red solid; yield: 89%; m.p. 126–128 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.51 (d, J = 9.4 Hz, 2H), 7.22 (d, J = 24.9 Hz, 1H), 7.13 (s, 2H), 7.00 (s, 2H), 6.74 (d, J = 6.8 Hz, 2H), 6.57 (s, 1H), 4.68–4.64 (m, 1H), 3.95–3.88 (m, 1H), 3.80 (d, J = 16.4 Hz, 6H), 1.85–1.69 (m, 3H), 1.46 (dd, J = 7.0, 1.6 Hz, 3H), 0.99–0.88 (m, 6H); 13C-NMR (100 MHz, MeOD-d4) δ: 187.2, 183.2, 172.5, 171.9, 153.5, 151.3, 150.1, 143.6, 142.9, 140.8, 136.7, 129.1, 126.3, 124.9, 124.7, 123.0, 122.8, 117.3, 113.4, 112.6, 57.2 (2C), 53.1, 50.8, 42.0, 26.7, 23.8, 22.4, 18.3; ESI-MS [M + H]+ m/z: 553; HR-ESI-MS [M + H]+ m/z: 553.2558, Calcd. for C30H36N2O8 (M + H) 553.2505.
(2S)-4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-amino-4-(methylthio)butanamido)-4-methylpentanoate (16). Dark red solid; yield: 89%; m.p. 112–115 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.52 (d, J = 13.3 Hz, 2H), 7.26 (s, 1H), 7.13 (s, 2H), 7.02 (s, 2H), 6.74 (d, J = 7.6 Hz, 2H), 6.56 (d, J = 12.5 Hz, 1H), 4.65 (m, 1H), 3.99 (m, 1H), 3.80 (d, J = 14.9 Hz, 6H), 2.53 (m, 2H), 2.13 (m, 2H), 2.00 (m, 3H), 1.76 (m, 3H), 0.94 (m,6 H); 13C-NMR (100 MHz, MeOD-d4) δ: 187.3, 183.2, 172.4, 170.6, 153.5, 151.3, 150.1, 143.6, 142.9, 140.8, 136.7, 129.1, 126.4, 124.9, 124.7, 123.0, 122.8, 117.3, 113.4, 112.6, 57.2 (2C), 54.3, 53.1, 41.9, 32.9, 30.3, 26.7, 23.9, 22.4, 15.7; ESI-MS [M+H]+ m/z: 613; HR-ESI-MS [M + H]+ m/z: 613.2577, Calcd. for C32H40N2O8S (M + H) 613.2539.
(2S)-4-((1E,6E)-7-(4-Hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dienyl)-2-methoxyphenyl 2-(2-amino-3-methylbutanamido)-4-methylpentanoate (18). Dark red solid; yield: 89%; m.p. 138–140 °C; 1H-NMR (400 MHz, MeOD-d4) δ: 7.60 (s, 2H), 7.34 (s, 1H), 7.22 (s, 2H), 7.11 (d, J = 7.6 Hz, 2H), 6.84 (d, J = 6.6 Hz, 2H), 6.67 (s, 1H), 4.77 (m, 1H), 3.90 (d, J = 17.0 Hz, 6H), 3.78 (dd, J = 8.3, 5.4 Hz, 1H), 2.26 (m, 1H), 1.87 (m, 3H), 1.05 (m, 12H); 13C-NMR (100 MHz, MeOD-d4) δ: 185.2, 181.2, 170.3, 168.4, 151.4, 149.3, 148.1, 141.6, 140.8, 138.8, 134.6, 127.1, 124.3, 122.9, 122.6, 120.9, 120.7, 115.2, 111.4, 110.5, 99.9, 58.4, 58.2 , 55.1, 51.0, 39.9, 30.3, 24.6, 21.9, 20.3, 17.5, 16.3; ESI-MS [M+H]+ m/z: 581; HR-ESI-MS [M + H]+ m/z: 581.2881, Calcd. for C32H40N2O8 (M + H) 581.2818.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}