
Direct Aminolysis of Ethoxycarbonylmethyl 1,4-Dihydropyridine-3-carboxylates

 ,
,
Abstract
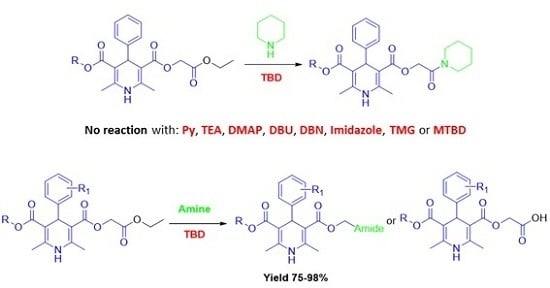
:
1. Introduction
2. Results and Discussion
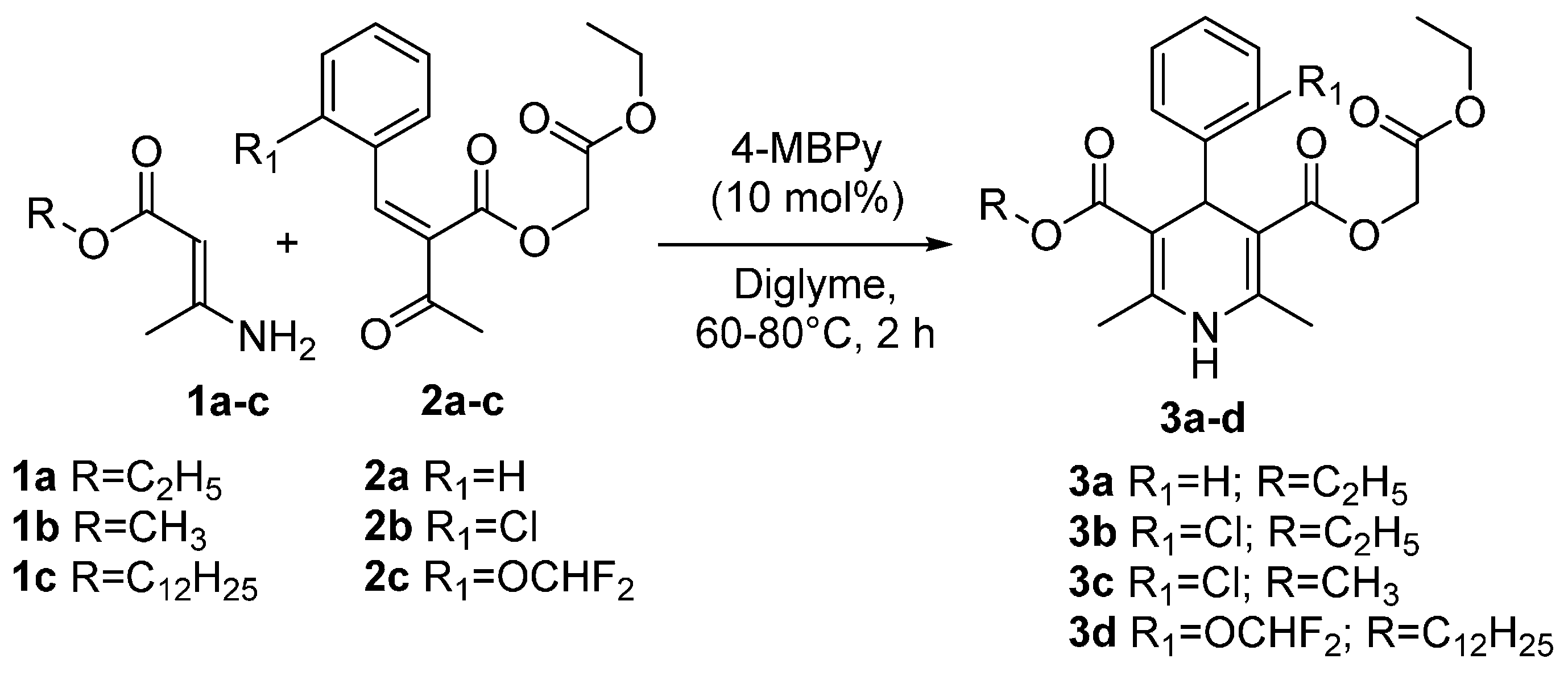
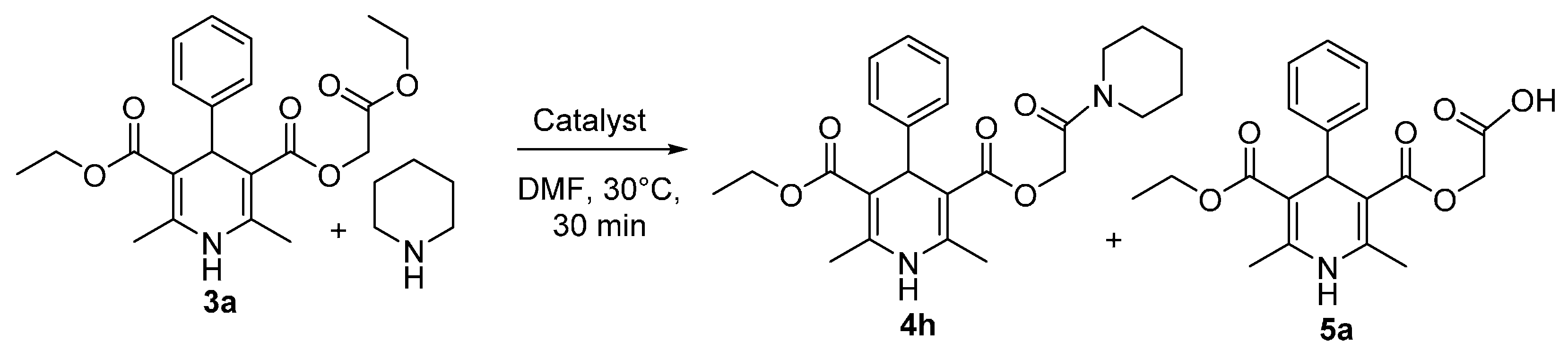

{kind=link}
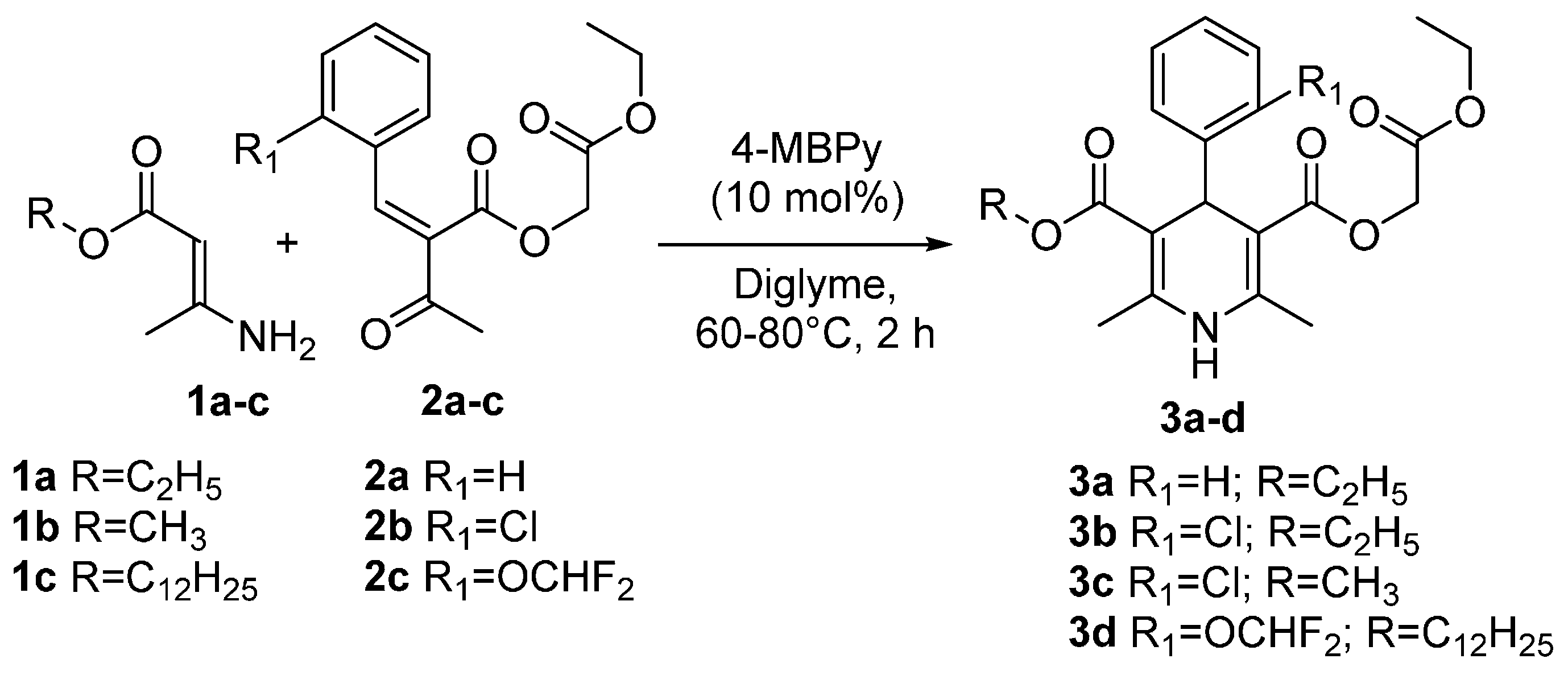{kind=link}
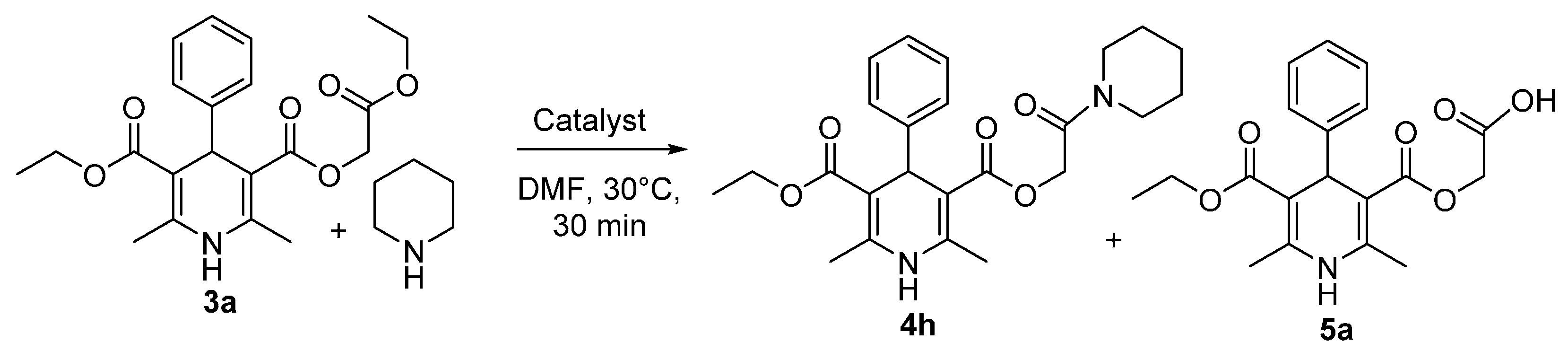{kind=link}
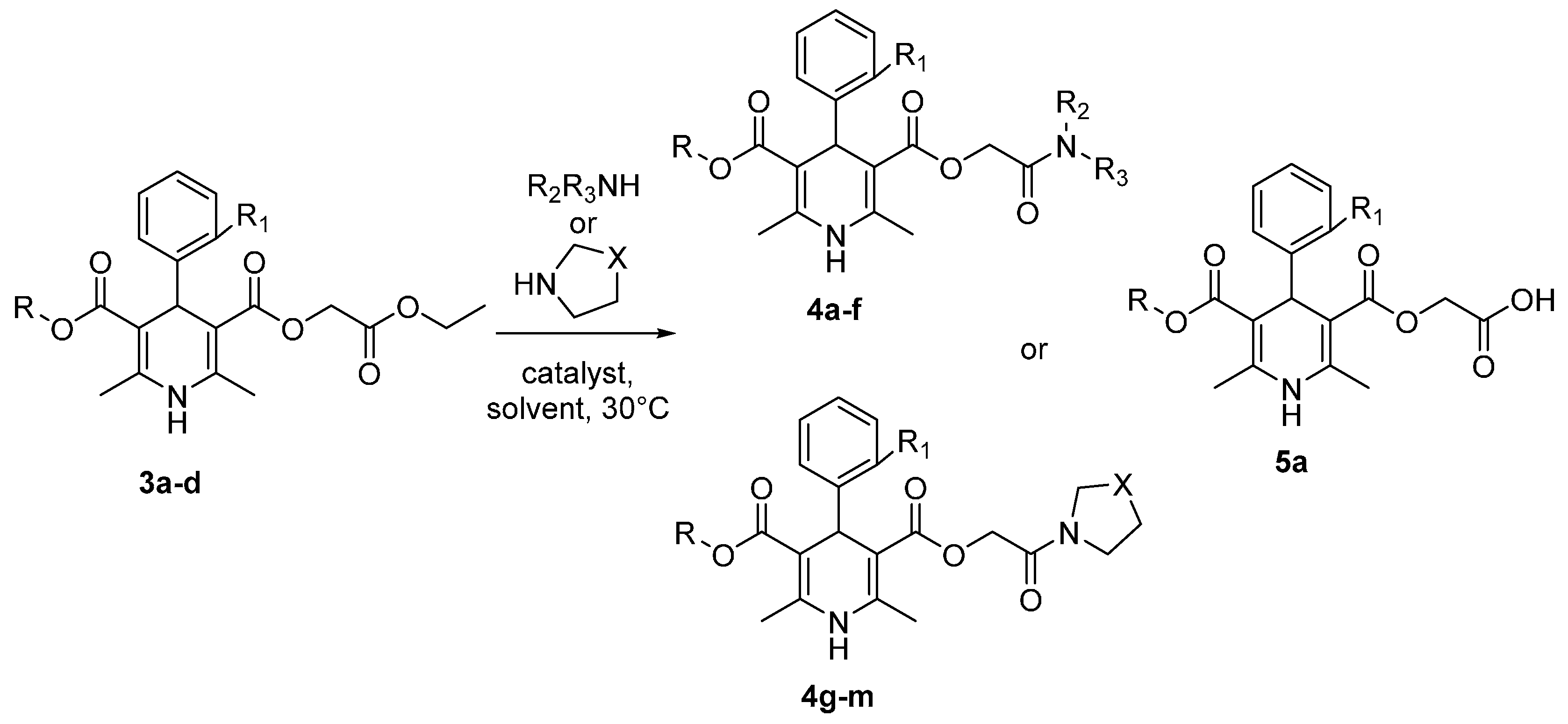{kind=link}
| Entry | Catalyst | Time, h | Conversion Rate of 3a, % * | Selectivity to 4 h, % * | Selectivity to 5a, % * |
|---|---|---|---|---|---|
| 1 | - | 0.5 | <1 | - | >99 |
| 2 |  | 0.5 | <1 | - | >99 |
| (pyridine) | |||||
| 3 |  | 0.5 | <1 | - | >99 |
| (TEA) | |||||
| 4 | 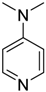 | 0.5 | <1 | - | >99 |
| (DMAP) | |||||
| 5 | 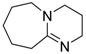 | 0.5 | <1 | - | >99 |
| (DBU) | |||||
| 6 | 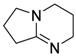 | 0.5 | <1 | - | >99 |
| (DBN) | |||||
| 7 | 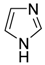 | 0.5 | <1 | - | >99 |
| (imidazole) | |||||
| 8 | 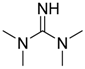 | 0.5 | <1 | - | >99 |
| (TMG) | |||||
| 9 | 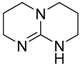 | 0.5 | 85 | 97 | 3 |
| (TBD) | |||||
| 10 | 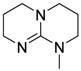 | 0.5 | <1 | - | >99 |
| (MTBD) |
| Entry | TBD, mol % | Time, h | Temp, °C | Solvent | Conversion Rate of 3a, % * | Selectivity to 4 h, % * | Selectivity to 5a, % * |
|---|---|---|---|---|---|---|---|
| 1 | 20 | 4 | 30 | DMF | 98 | 99 | 1 |
| 2 | 10 | 4 | 30 | DMF | 54 | 88 | 12 |
| 3 | 5 | 4 | 30 | DMF | 41 | 27 | 73 |
| 4 | 20 | 4 | 4 | DMF | 77 | 97 | 3 |
| 5 | 20 | 0.5 | 70 | DMF | 98 | 89 | 11 |
| 6 | 20 | 4 | 30 | THF | 94 | 96 | 4 |
| 7 | 20 | 4 | 30 | MeCN | 97 | 79 | 21 |
| 8 | 20 | 4 | 30 | CH2Cl2 | 79 | 61 | 39 |
| 9 | 20 | 4 | 30 | Dioxane | 65 | 56 | 44 |
| 10 | 20 | 4 | 30 | MeOH | 99 | 74 | 26 |
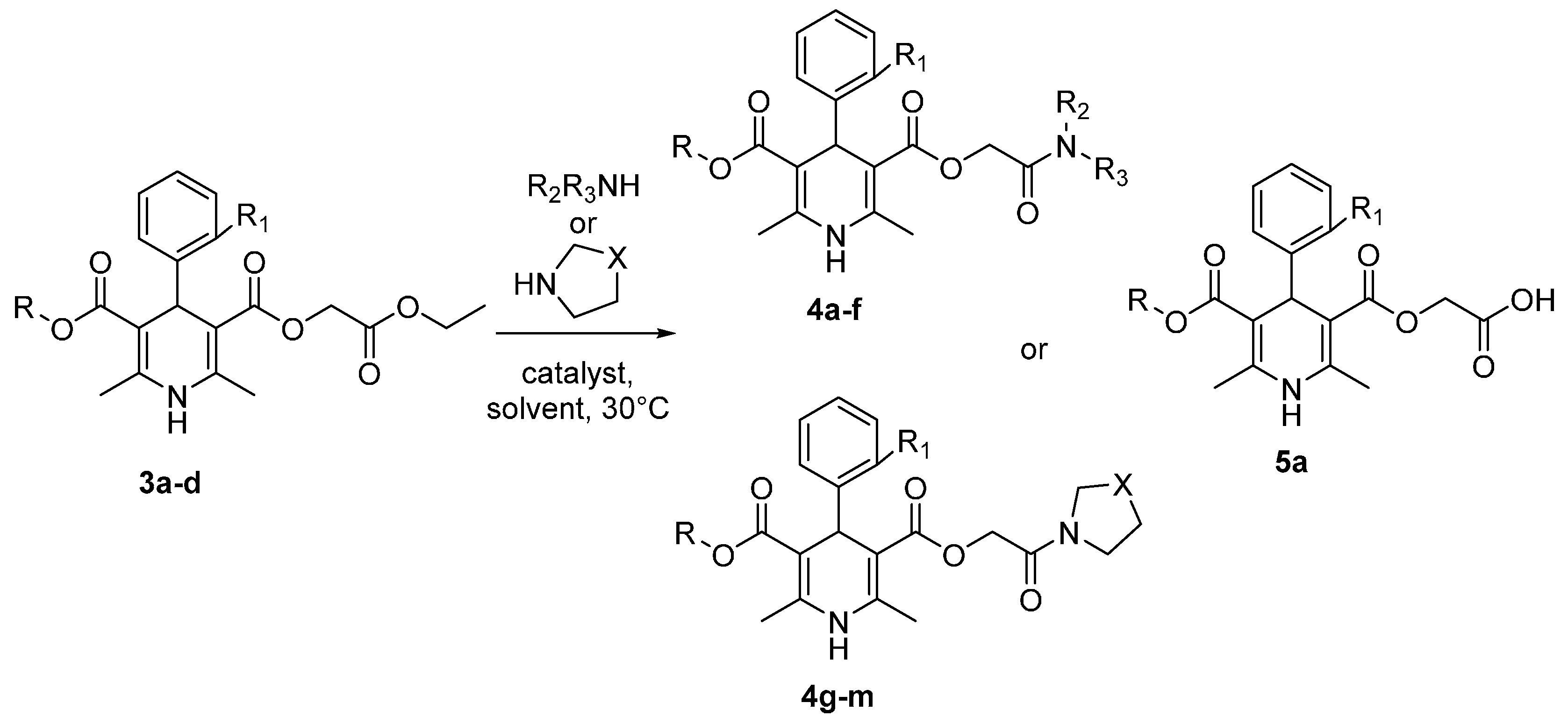
| Entry | Amine | R | R1 | Substrate | Solvent | Catalyst | R2 | R3 | X | Time, h | Product | Yield, % |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1-Propylamine | Et | H | 3a | DMF | TBD | n-Pr | H | - | 2 | 4a | 78 |
| 2 | 1-Propylamine | Et | H | 3a | DMF | - | n-Pr | H | - | 2 | 4a | 2 * |
| 3 | 2-Propylamine | Et | H | 3a | DMF | TBD | i-Pr | H | - | 4 | 4b | 79 |
| 4 | 2-Propylamine | Et | H | 3a | DMF | - | i-Pr | H | - | 4 | 4b | 4 * |
| 5 | 2-Propylamine | Et | H | 3a | 2-Propylamine | TBD | i-Pr | H | - | 4 | 4b | 76 |
| 6 | 1-Butylamine | Et | H | 3a | DMF | TBD | n-Bu | H | - | 4 | 4c | 98 |
| 7 | 1-Butylamine | Et | H | 3a | DMF | - | n-Bu | H | - | 4 | 4c | 3 * |
| 8 | Diethylamine | Et | H | 3a | DMF | TBD | Et | Et | - | 4 | 4d | 75 |
| 9 | Diisopropylamine | Et | H | 3a | DMF | TBD | - | - | - | 4 | 5a | 70 |
| 10 | Diphenylamine | Et | H | 3a | DMF | TBD | - | - | - | 4 | - | - |
| 11 | N-Methylbutylamine | Et | H | 3a | DMF | TBD | Me | n-Bu | - | 4 | 4e | 89 |
| 12 | N-Methyloctylamine | Et | H | 3a | DMF | TBD | Me | n-C8H17 | - | 4 | 4f | 87 |
| 13 | Pyrrolidine | Et | H | 3a | DMF | TBD | - | - | CH2 | 3 | 4g | 94 |
| 14 | Piperidine | Et | H | 3a | DMF | TBD | - | - | CH2CH2 | 3 | 4h | 89 |
| 15 | Piperidine | Et | H | 3a | Piperidine | TBD | - | - | CH2CH2 | 3 | 4h | 85 |
| 16 | Morpholine | Et | H | 3a | DMF | TBD | - | - | CH2O | 4 | 4i | 97 |
| 17 | Morpholine | Et | H | 3a | Morpholine | TBD | - | - | CH2O | 4 | 4i | 94 |
| 18 | Thiomorpholine | Et | H | 3a | DMF | TBD | - | - | CH2S | 3 | 4j | 80 |
| 19 | 1-Naphthylamine | Et | H | 3a | DMF | TBD | - | - | - | 4 | - | - |
| 20 | Thiomorpholine | Et | Cl | 3b | DMF | TBD | - | - | CH2S | 3 | 4k | 85 |
| 21 | Pyrrolidine | Me | Cl | 3c | DMF | TBD | - | - | CH2 | 3 | 4l | 96 |
| 22 | Piperidine | C12H25 | OCHF2 | 3d | DMF | TBD | - | - | CH2CH2 | 3 | 4m | 87 |
| 23 | Piperidine | C12H25 | OCHF2 | 3d | Piperidine | TBD | - | - | CH2CH2 | 3 | 4m | 87 |
3. Experimental Section
3.1. General Information
3.2. General Procedure for the Synthesis of Ethoxycarbonylmethyl Esters of 1,4-Dihydropyridines 3a–d
3.3. General Procedure for the Aminolysis of Ethoxycarbonylmethyl Esters of 1,4-Dihydropyridines 3a–d in DMF
3.4. General Procedure for the Aminolysis of Ethoxycarbonylmethyl 1,4-Dihydropyridine-3-Carboxylates 3a,d when Amine is Used as Solvent/Nucleophile
3.5. 3-Carboxymethyl 5-Ethyl 2,6-dimethyl-4-phenyl-1,4-dihydropyridine 3,5-dicarboxylate (5a)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Triggle, D.J. The 1,4-dihydropyridine nucleus: A pharmacophoric template part 1. Actions at ion channels. Mini-Rev. Med. Chem. 2003, 3, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Rucins, M.; Kaldre, D.; Pajuste, K.; Fernandes, M.A.S.; Vicente, J.A.F.; Klimaviciusa, L.; Jaschenko, E.; Kanepe-Lapsa, I.; Shestakova, I.; Plotniece, M.; et al. Synthesis and studies of calcium channel blocking and antioxidant activities of novel 4-pyridinium and/or N-propargyl substituted 1,4-dihydropyridine derivatives. C. R. Chim. 2014, 17, 69–80. [Google Scholar] [CrossRef]
- Carosati, E.; Ioan, P.; Micucci, M.; Broccatelli, F.; Cruciani, G.; Zhorov, B.S.; Chiarini, A.; Budriesi, R. 1,4-Dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 2): Action in other targets and antitargets. Curr. Med. Chem. 2012, 19, 4306–4323. [Google Scholar] [CrossRef] [PubMed]
- Sobolev, A.; Franssen, M.C.R.; Duburs, G.; de Groot, Ae. Chemoenzymatic synthesis of enantiopure 1,4-dihydropyridine derivatives. Biocatal. Biotransform. 2004, 22, 231–252. [Google Scholar] [CrossRef]
- Chekavichus, B.S.; Sausin, A.É.; Dubur, G.Y. Effect of substituents in the dihydropyridine ring on the reactivity of the ester group of 3,5-dialkoxycarbonyl-1,4-dihydropyridines. Chem. Heterocycl. Compd. 1982, 18, 818–823. [Google Scholar] [CrossRef]
- Sobolev, A.; Franssen, M.C.R.; Poikans, J.; Duburs, G.; de Groot, Ae. Enantioselective lipase-catalysed kinetic resolution of acyloxymethyl and ethoxycarbonylmethyl esters of 1,4-dihydroisonicotinic acid derivatives. Tetrahedron Asymmetry 2002, 13, 2389–2397. [Google Scholar] [CrossRef]
- Sobolev, A.; Franssen, M.C.R.; Makarova, N.; Duburs, G.; de Groot, Ae. Candida antarctica lipase-catalyzed hydrolysis of 4-substituted bis(ethoxycarbonylmethyl) 1,4-dihydropyridine-3,5-dicarboxylates as the key step in the synthesis of optically active dihydropyridines. Tetrahedron Asymmetry 2000, 11, 4559–4569. [Google Scholar] [CrossRef]
- Neidere, Z.; Poikans, J.; Zuka, L.; Uldrikis, J.; Bruvere, I.; Vigante, B.; Kalvins, I.; Bisenieks, E.; Jansone, I.; Jonane-Osa, I.; et al. Antiviral Efficacy of Disodium 2,6-Dimethyl-1,4-dihydropyridine-3,5-bis(carbonyloxyacetate) and itS Derivatives. WO 2013/050625 A1, 11 April 2013. [Google Scholar]
- Stonans, I.; Jansone, I.; Jonane-Osa, I.; Bisenieks, E.; Duburs, G.; Kalvins, I.; Vigante, B.; Uldrikis, J.; Bruvere, I.; Zuka, L.; et al. Derivatives of 1,4-Dihydropyridine Possessing Antiviral Efficacy. WO 2012/010276 A1, 26 January 2013. [Google Scholar]
- Yamamoto, T.; Niwa, S.; Ohno, S.; Onishi, T.; Matsueda, H.; Koganei, H.; Uneyama, H.; Fujita, S.; Takeda, T.; Kito, M.; et al. Structure-activity relationship study of 1,4-dihydropyridine derivatives blocking N-type calcium channels. Bioorg. Med. Chem. Lett. 2006, 16, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Hisayuki, U.; Seiji, N.; Tomoyuki, O. Dihydropyridine Derivative. U.S. Patent 6,350,766 B1, 26 February 2002. [Google Scholar]
- Meguro, K.; Aizawa, M.; Sohda, T.; Kawamatsu, Y.; Nagaoka, A. New 1, 4-Dihydropyridine derivatives with potent and long-lasting hypotensive effect. Chem. Pharm. Bull. 1985, 33, 3787–3797. [Google Scholar] [CrossRef] [PubMed]
- Vilskersts, R.; Vigante, B.; Neidere, Z.; Krauze, A.; Domracheva, I.; Bekere, L.; Shestakova, I.; Duburs, G.; Dambrova, M. Calcium level controlling activities of novel derivatives of amlodipine, riodipine and cerebrocrast. Lett. Drug Des. Discov. 2012, 9, 322–328. [Google Scholar] [CrossRef]
- Li, Y.; Yongye, A.; Giulianotti, M.; Martinez-Mayorga, K.; Yu, Y.; Houghten, R.A. Synthesis of cyclic peptides through direct aminolysis of peptide thioesters catalyzed by imidazole in aqueous organic solutions. J. Comb. Chem. 2009, 11, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Sung, K.; Zhuang, B.-R.; Huang, P.-M.; Jhong, S.-W. Kinetic and mechanistic studies of net3-catalyzed intramolecular aminolysis of carbamate. J. Org. Chem. 2008, 73, 4027–4033. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Q.; Jin, L.; Kim, C.K.; Xue, Y. Role of bifunctional catalyst 2-pyridone in the aminolysis of p-nitrophenyl acetate with n-butylamine: A computational study. J. Mol. Catal. A Chem. 2012, 355, 102–112. [Google Scholar] [CrossRef]
- Sabot, C.; Kumar, K.A.; Meunier, S.; Mioskowski, C. A convenient aminolysis of esters catalyzed by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) under solvent-free conditions. Tetrahedron Lett. 2007, 48, 3863–3866. [Google Scholar] [CrossRef]
- de Lima, E.C.; de Souza, C.C.; de O. Soares, R.; Vaz, B.G.; Eberlin, M.N.; Dias, A.G.; Costa, P.R.R. DBU as a catalyst for the synthesis of amides via aminolysis of methyl esters. J. Braz. Chem. Soc. 2011, 22, 2186–2190. [Google Scholar] [CrossRef]
- Li, Y.; Yang, F.; Yuan, M.; Jiang, L.; Yuan, L.; Zhang, X.; Li, Y.; Dong, L.; Bao, X.; Yin, S. Synthesis and evaluation of asiatic acid derivatives as anti-fibrotic agents: Structure/activity studies. Steroids 2015, 96, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Abouabdellah, A.; Almario Garcia, A.; Hoornaert, C.; Lardenois, P.; Marguet, F. Aryl- and Heteroarylpiperidinecarboxylate-Derivatives Methods for Their Preparation and Use Thereof as Fatty Acid Amido Hydrolase Enzyme Inhibitors. U.S. Patent 2007/0021405 A1, 25 January 2007. [Google Scholar]
- Abouabdellah, A.; Burnier, P.; Hoornaert, C.; Jeunesse, J.; Puech, F. Derivatives of Piperidinyl-and Piperazinyl-alkyl Carbamates, Preparation Methods Thereof and Application of Same in Therapeutics. U.S. Patent 2006/0089344 A1, 27 April 2006. [Google Scholar]
- Sobolev, A.; Franssen, M.C.R.; Vigante, B.; Cekavicus, B.; Zhalubovskis, R.; Kooijman, H.; Spek, A.L.; Duburs, G.; de Groot, Ae. Effect of acyl chain length and branching on the enantioselectivity of Candida rugosa lipase in the kinetic resolution of 4-(2-difluoromethoxyphenyl)-substituted 1,4-dihydropyridine 3,5-diesters. J. Org. Chem. 2002, 67, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Pajuste, K.; Plotniece, A.; Kore, K.; Intenberga, L.; Cekavicus, B.; Kaldre, D.; Duburs, G.; Sobolev, A. Use of pyridinium ionic liquids as catalysts for the synthesis of 3,5-bis(dodecyloxycarbonyl)-1,4-dihydropyridine derivative. Cent. Eur. J. Chem. 2011, 9, 143–148. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: Unification of different basicity scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Štrukil, V.; Antol, I.; Glasovac, Z. The utility of 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a hydrogen bond acceptor in the design of novel superbasic guanidines—A computational study. Croat. Chem. Acta 2014, 87, 423–430. [Google Scholar] [CrossRef]
- Fu, X.; Tan, C.-H. Mechanistic considerations of guanidine-catalyzed reactions. Chem. Commun. 2011, 47, 8210–8222. [Google Scholar] [CrossRef] [PubMed]
- Jennings, W.B.; Watson, S.P.; Boyd, D.R. N-acyloxaziridines: Characterization of both nitrogen inversion and N-C(O) bond rotation in an amido system. J. Chem. Soc. Chem. Commun. 1992, 1078–1079. [Google Scholar] [CrossRef]
- Jin, L.; Wu, Y.; Kim, C.; Xue, Y. Theoretical study on the aminolysis of ester catalyzed by TBD: Hydrogen bonding or covalent bonding of the catalyst? J. Mol. Struct. THEOCHEM 2010, 942, 137–144. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3a–d, 4a, 4d, 4e, 4g, 4h, 4m, 4i are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vigante, B.; Rucins, M.; Plotniece, A.; Pajuste, K.; Luntena, I.; Cekavicus, B.; Bisenieks, E.; Smits, R.; Duburs, G.; Sobolev, A. Direct Aminolysis of Ethoxycarbonylmethyl 1,4-Dihydropyridine-3-carboxylates. Molecules 2015, 20, 20341-20354. https://doi.org/10.3390/molecules201119697
Vigante B, Rucins M, Plotniece A, Pajuste K, Luntena I, Cekavicus B, Bisenieks E, Smits R, Duburs G, Sobolev A. Direct Aminolysis of Ethoxycarbonylmethyl 1,4-Dihydropyridine-3-carboxylates. Molecules. 2015; 20(11):20341-20354. https://doi.org/10.3390/molecules201119697
Chicago/Turabian StyleVigante, Brigita, Martins Rucins, Aiva Plotniece, Karlis Pajuste, Iveta Luntena, Brigita Cekavicus, Egils Bisenieks, Rufus Smits, Gunars Duburs, and Arkadij Sobolev. 2015. "Direct Aminolysis of Ethoxycarbonylmethyl 1,4-Dihydropyridine-3-carboxylates" Molecules 20, no. 11: 20341-20354. https://doi.org/10.3390/molecules201119697
APA StyleVigante, B., Rucins, M., Plotniece, A., Pajuste, K., Luntena, I., Cekavicus, B., Bisenieks, E., Smits, R., Duburs, G., & Sobolev, A. (2015). Direct Aminolysis of Ethoxycarbonylmethyl 1,4-Dihydropyridine-3-carboxylates. Molecules, 20(11), 20341-20354. https://doi.org/10.3390/molecules201119697