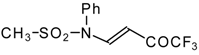
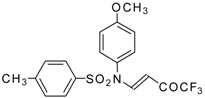
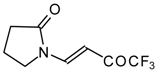
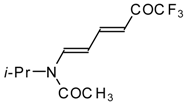
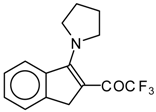
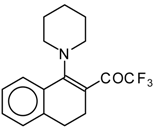
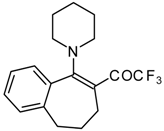
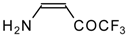
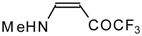
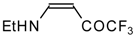
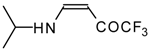
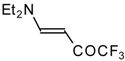
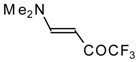
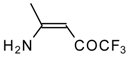
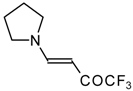
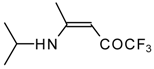
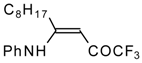
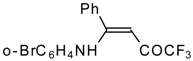
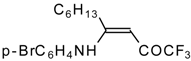
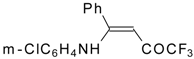
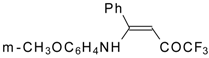
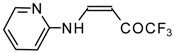
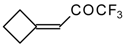
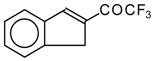
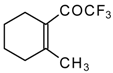
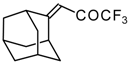
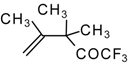
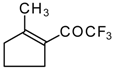
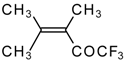
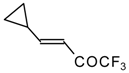
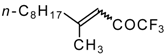
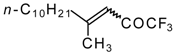
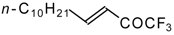
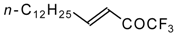
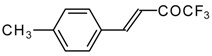
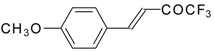
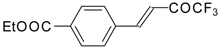
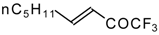
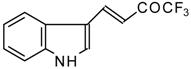
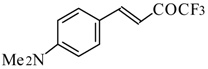
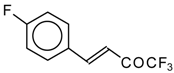
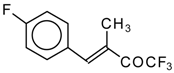
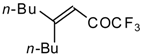
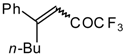
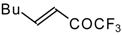
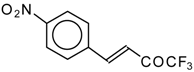
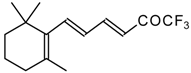
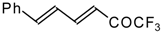
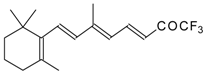
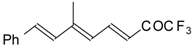
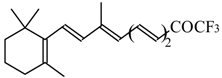
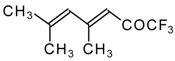
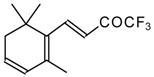
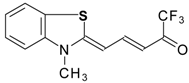
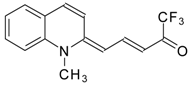
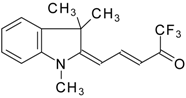
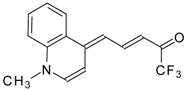
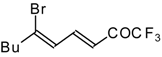
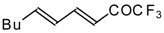
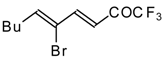
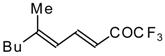
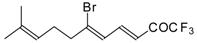
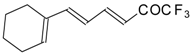
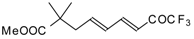
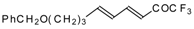
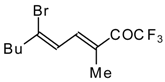
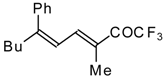
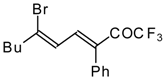
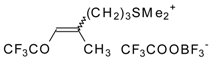
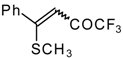
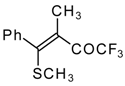
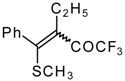
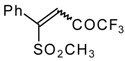
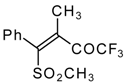
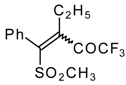
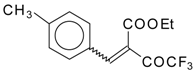
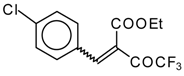
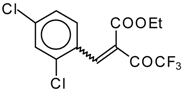
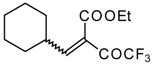
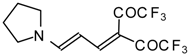
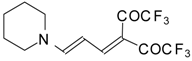
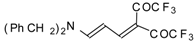
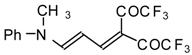
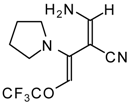
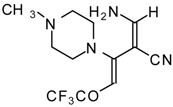
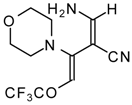
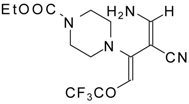
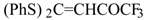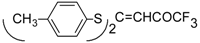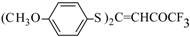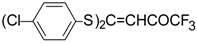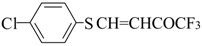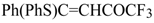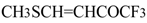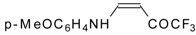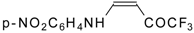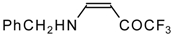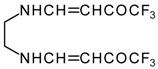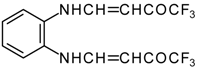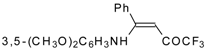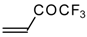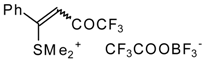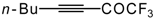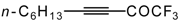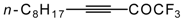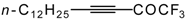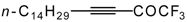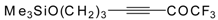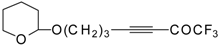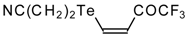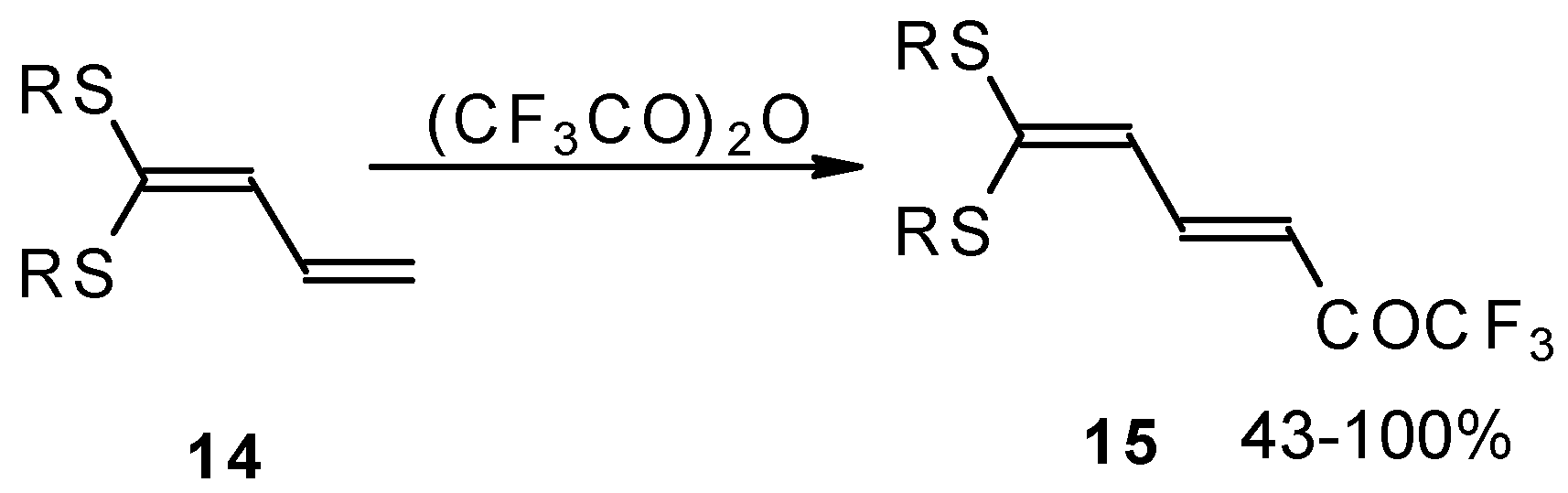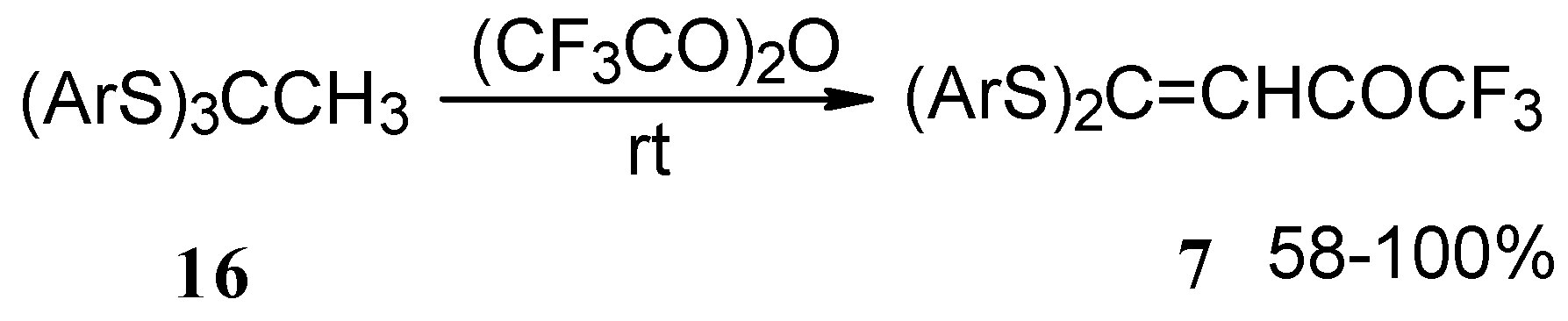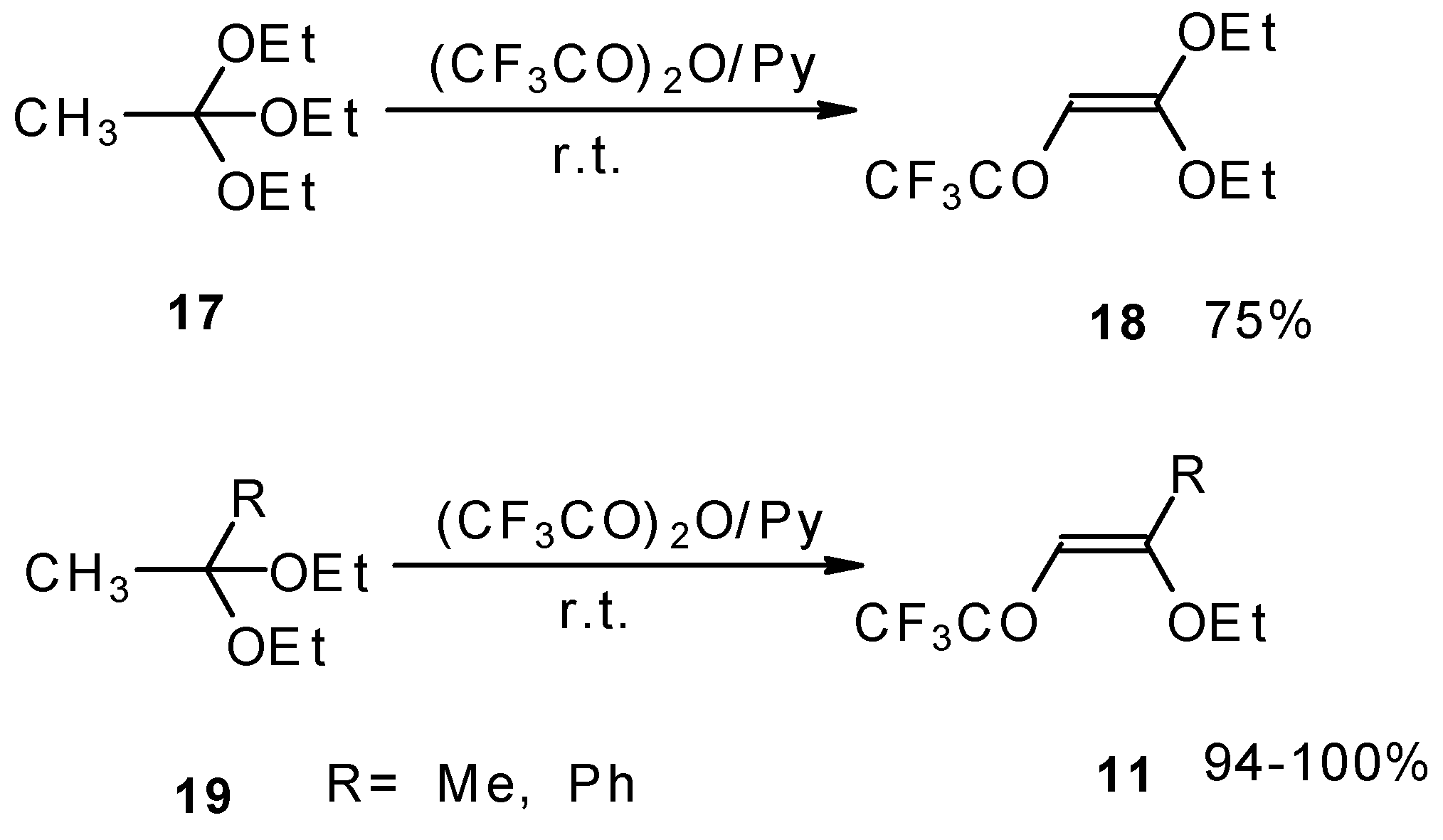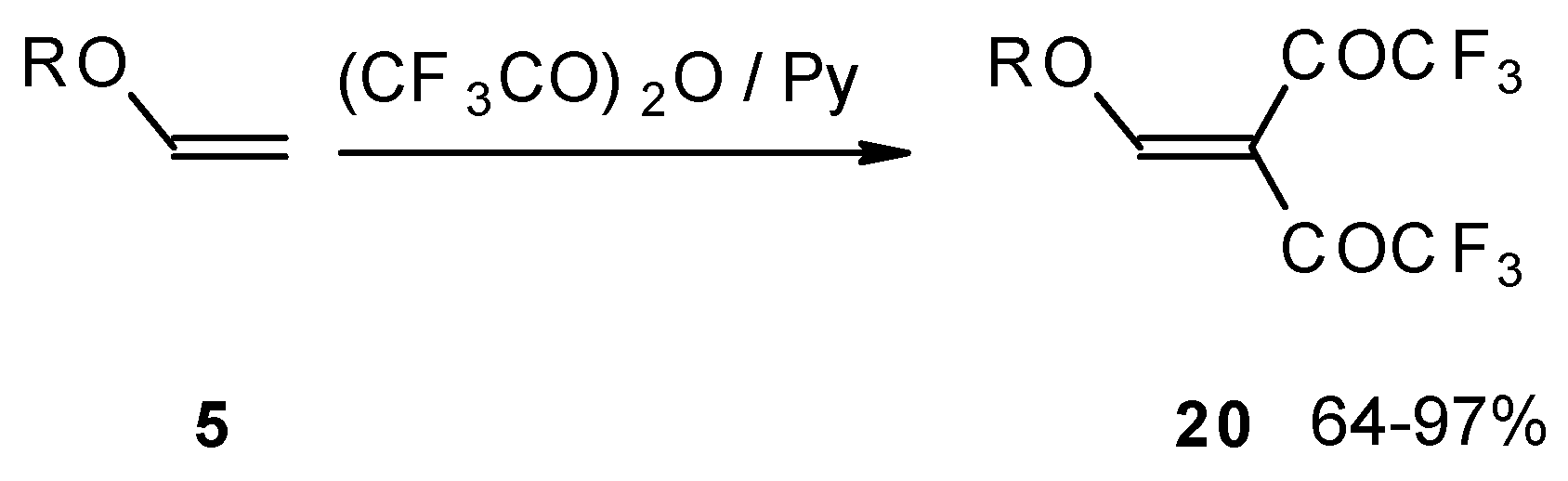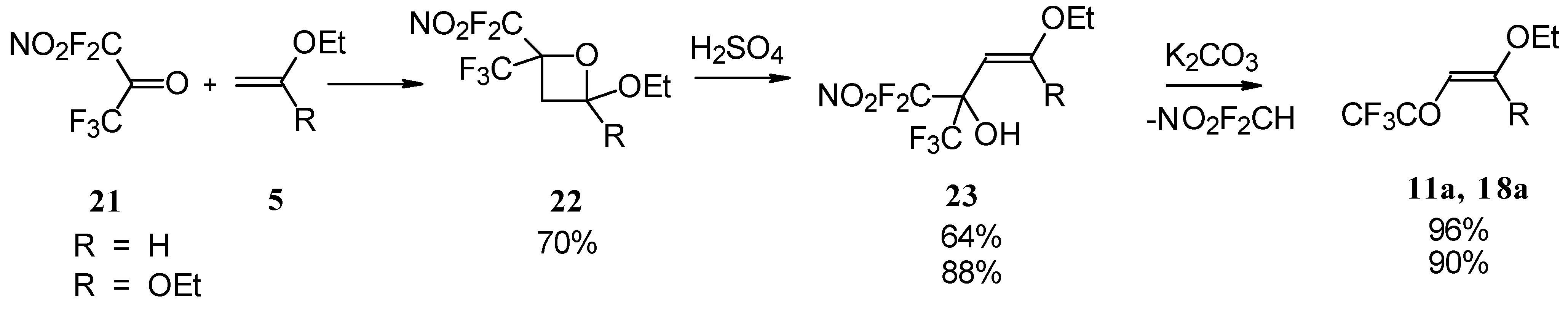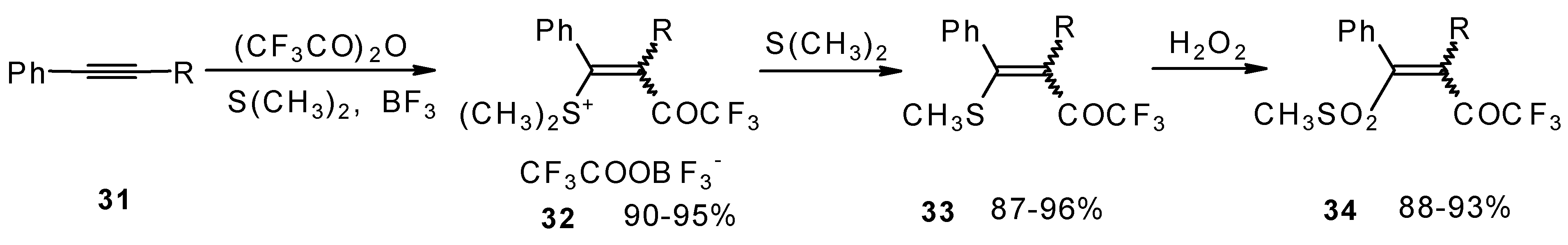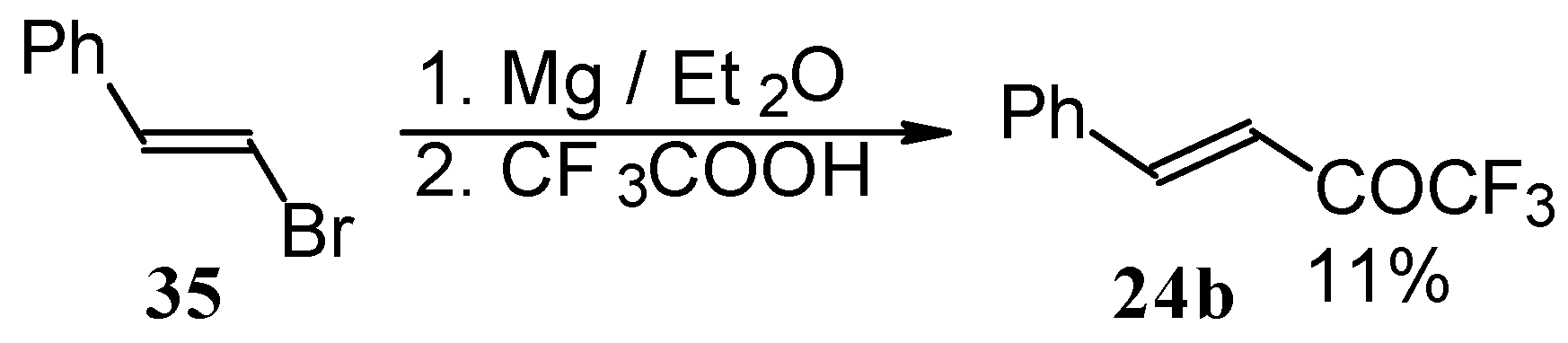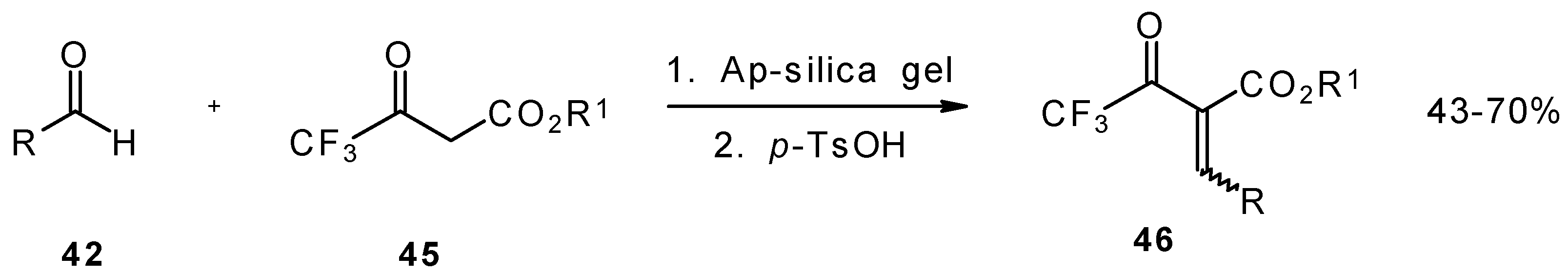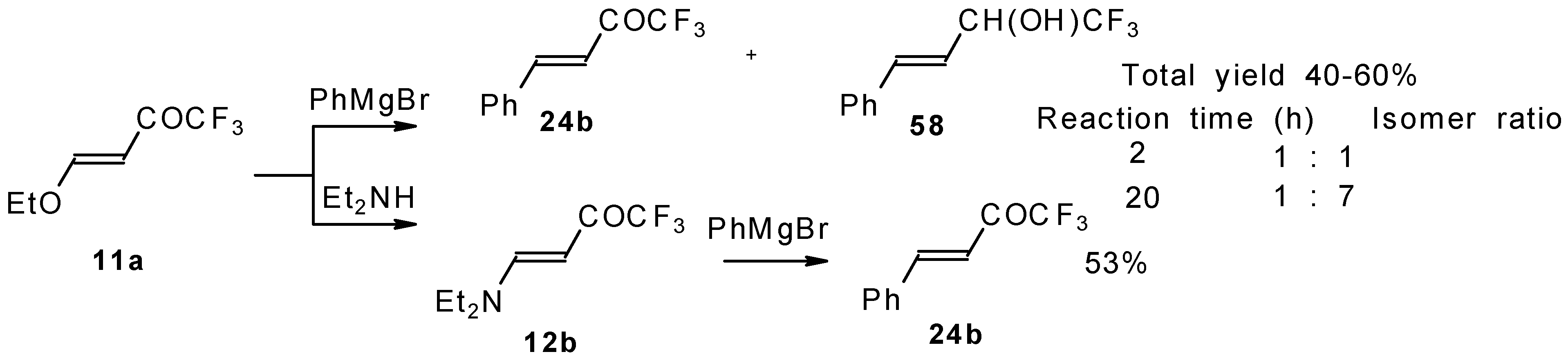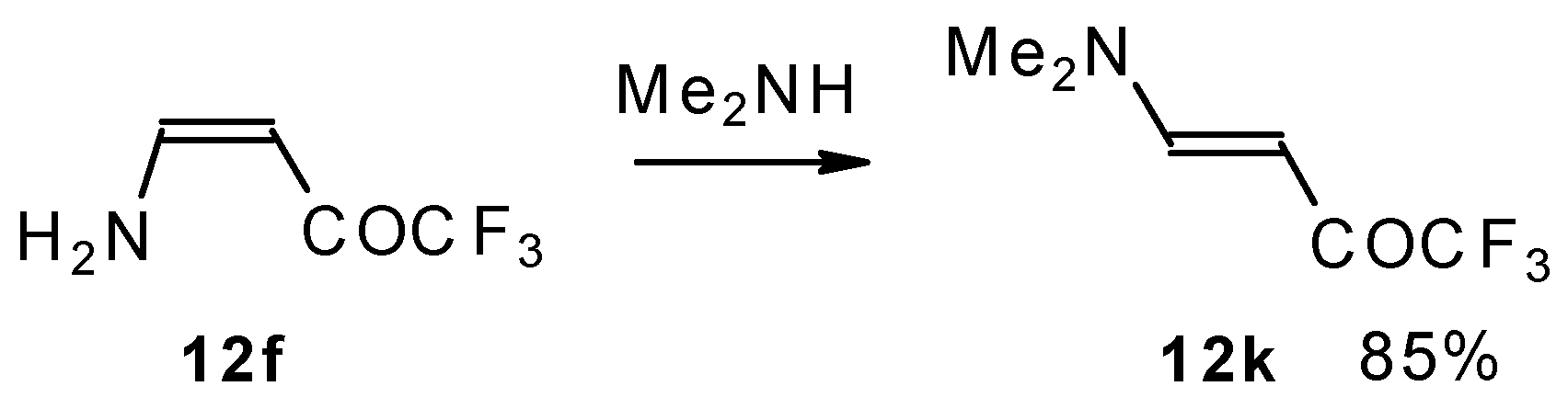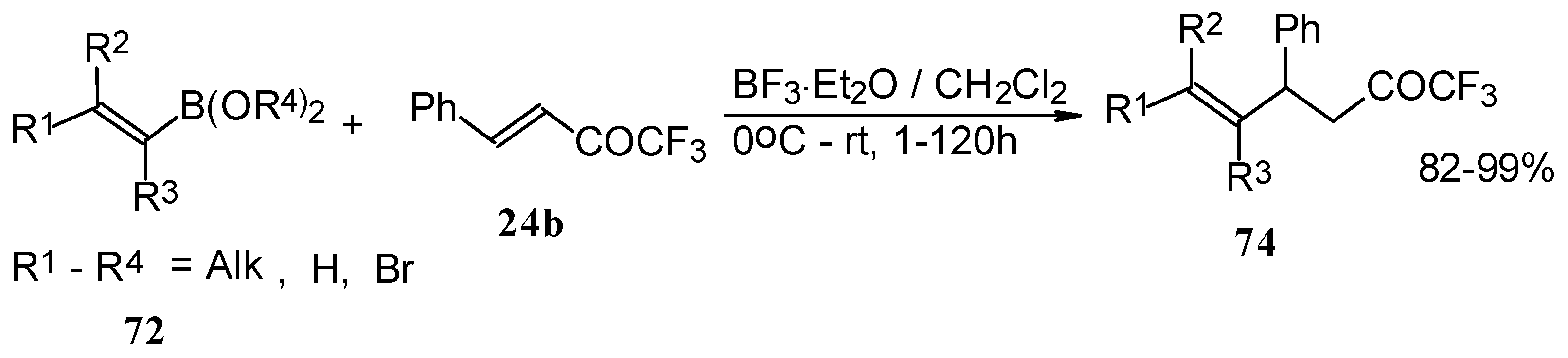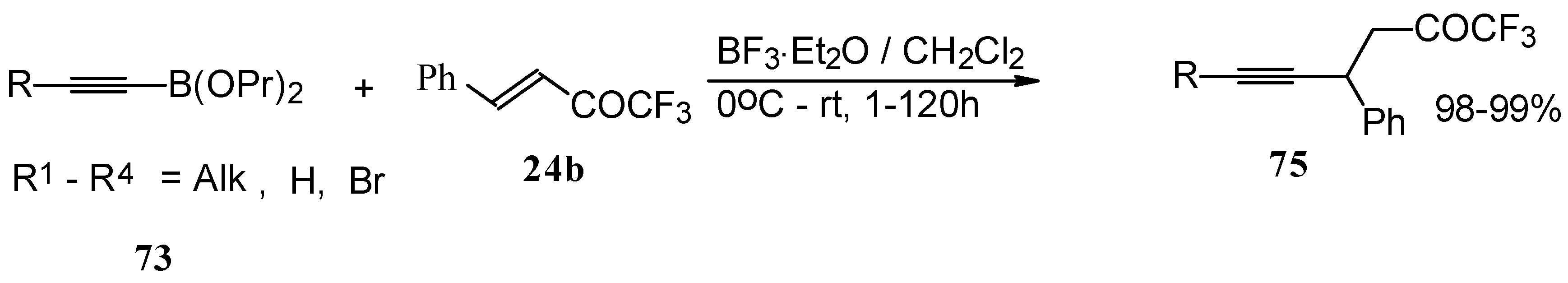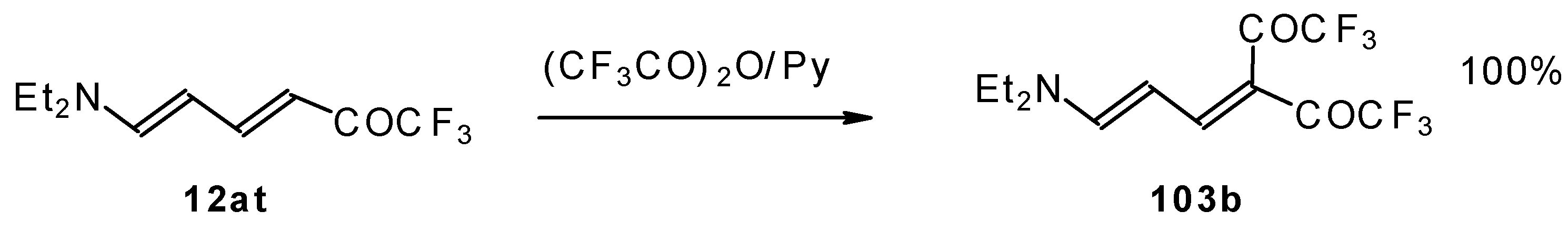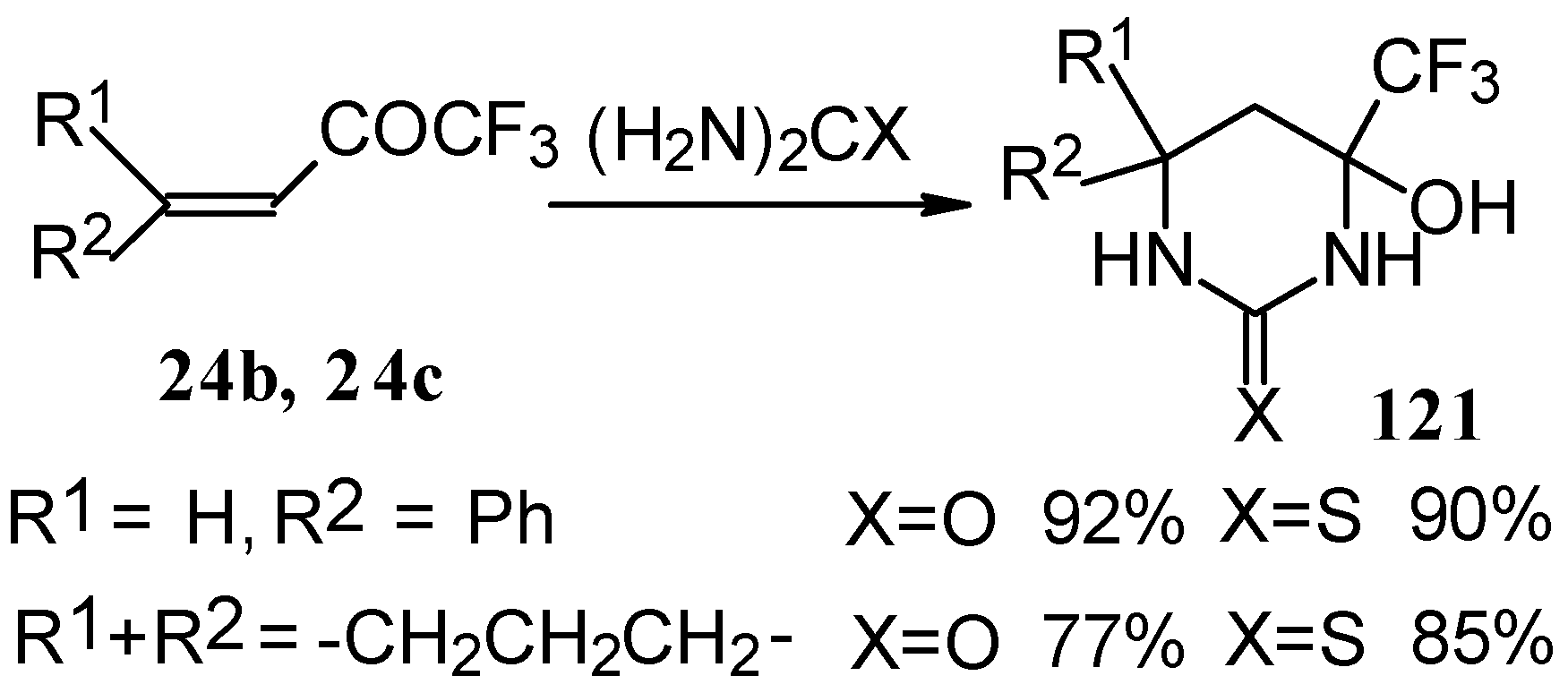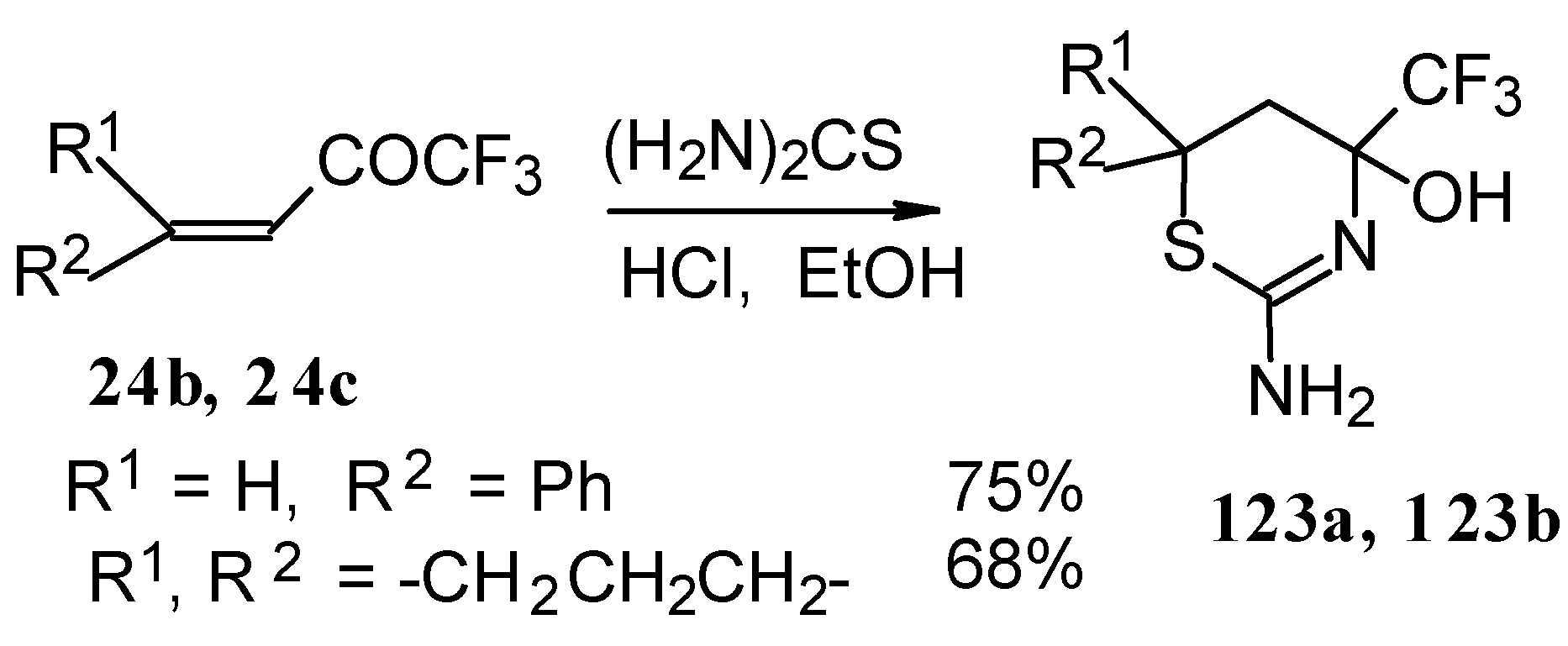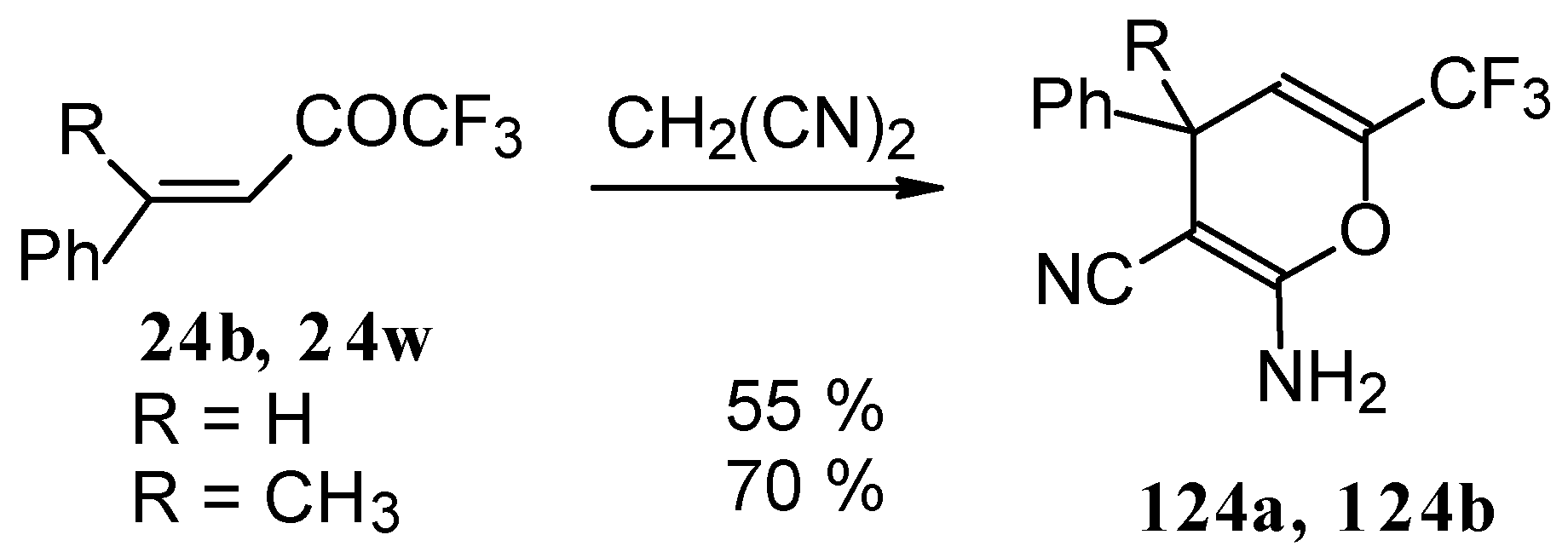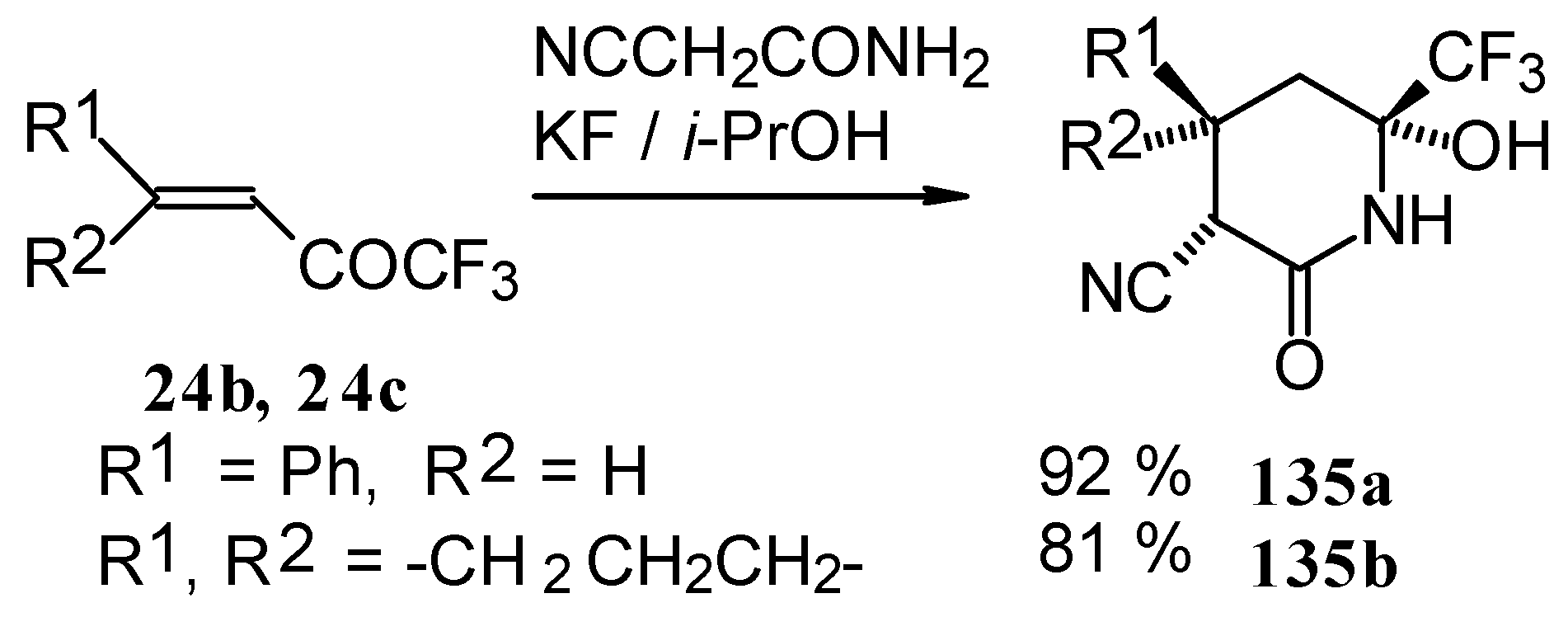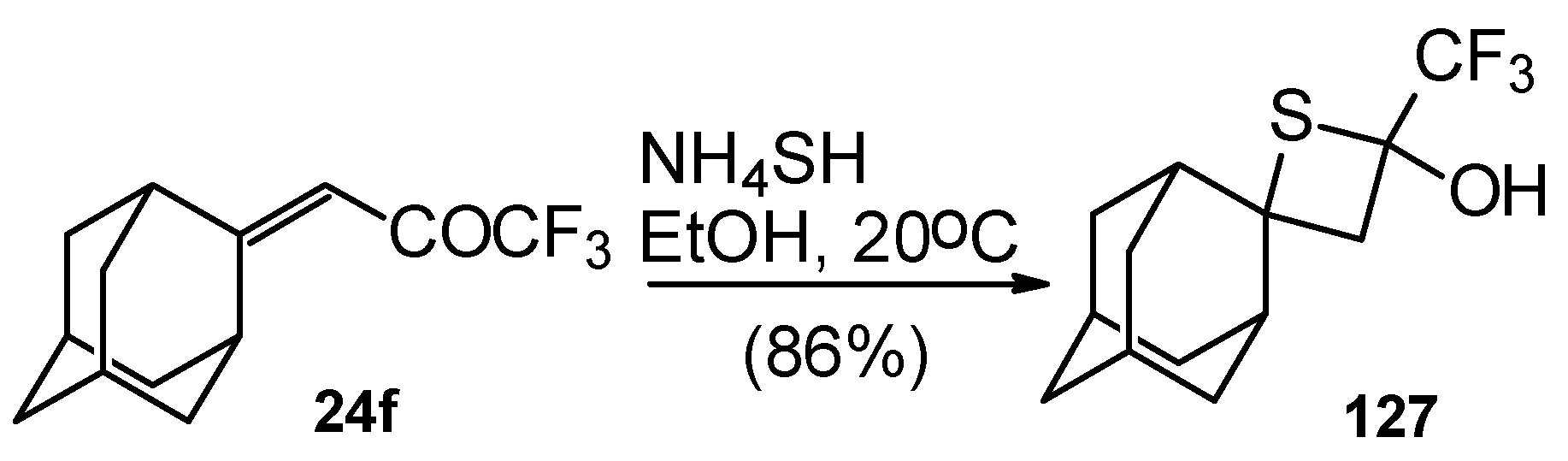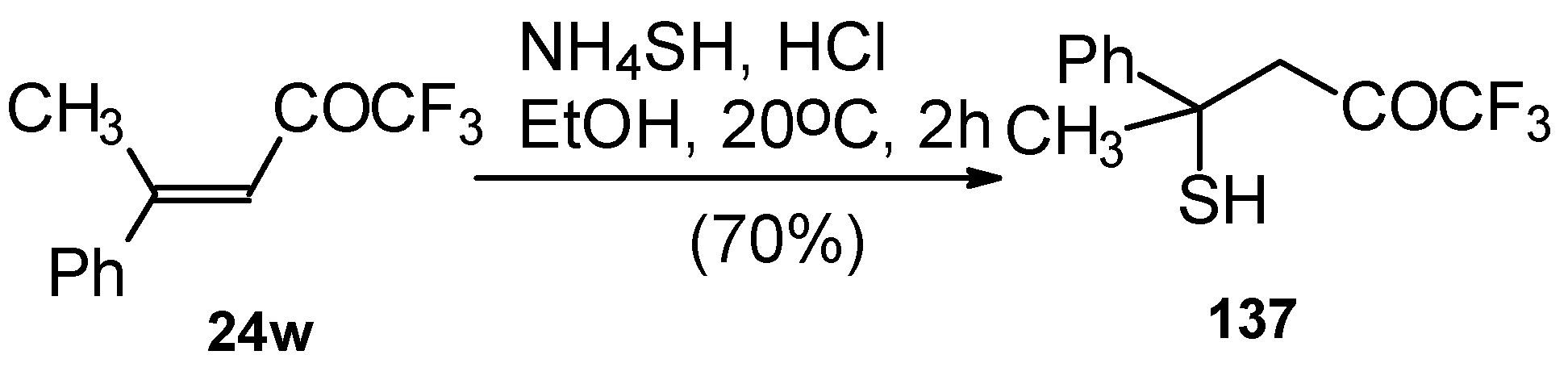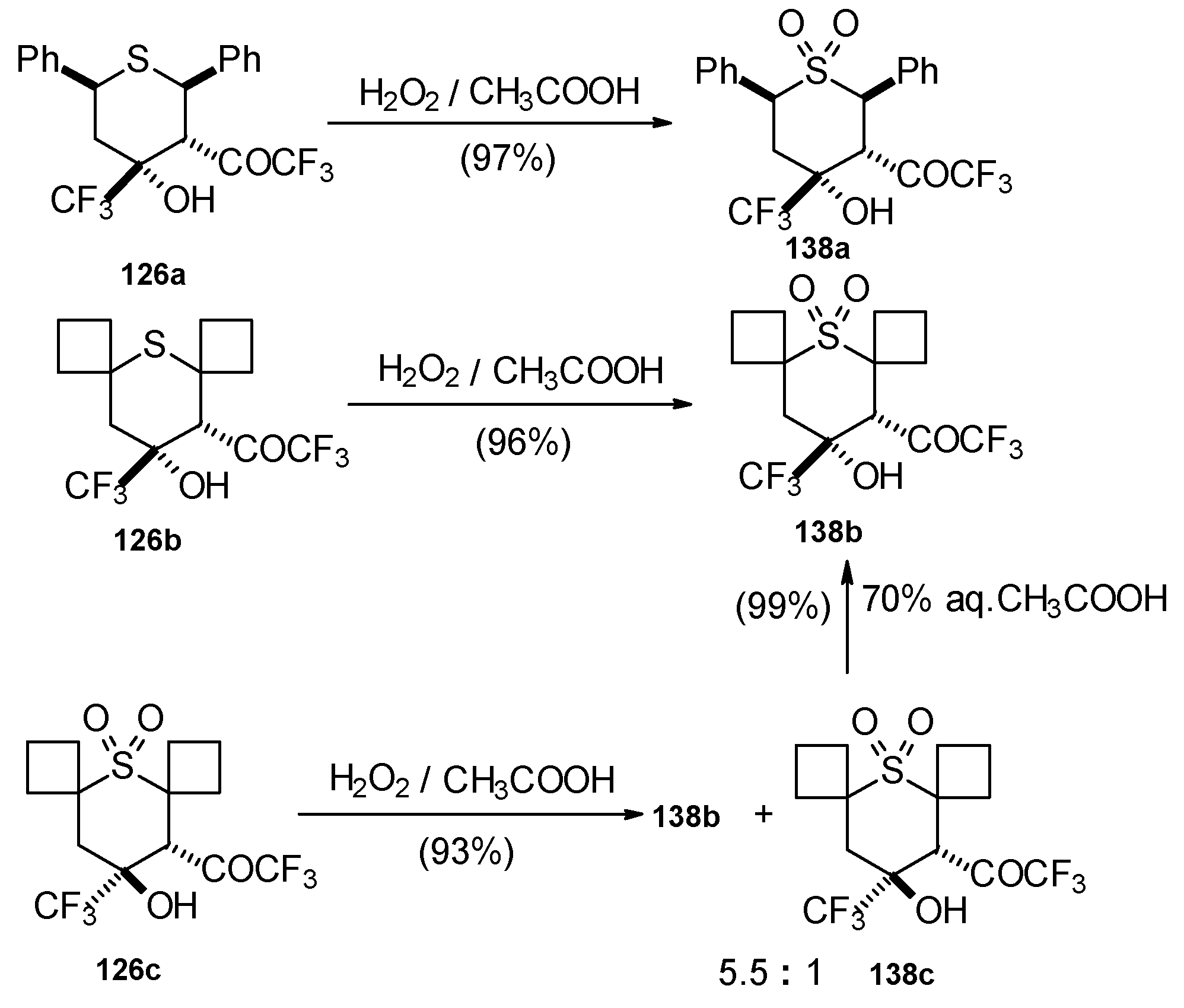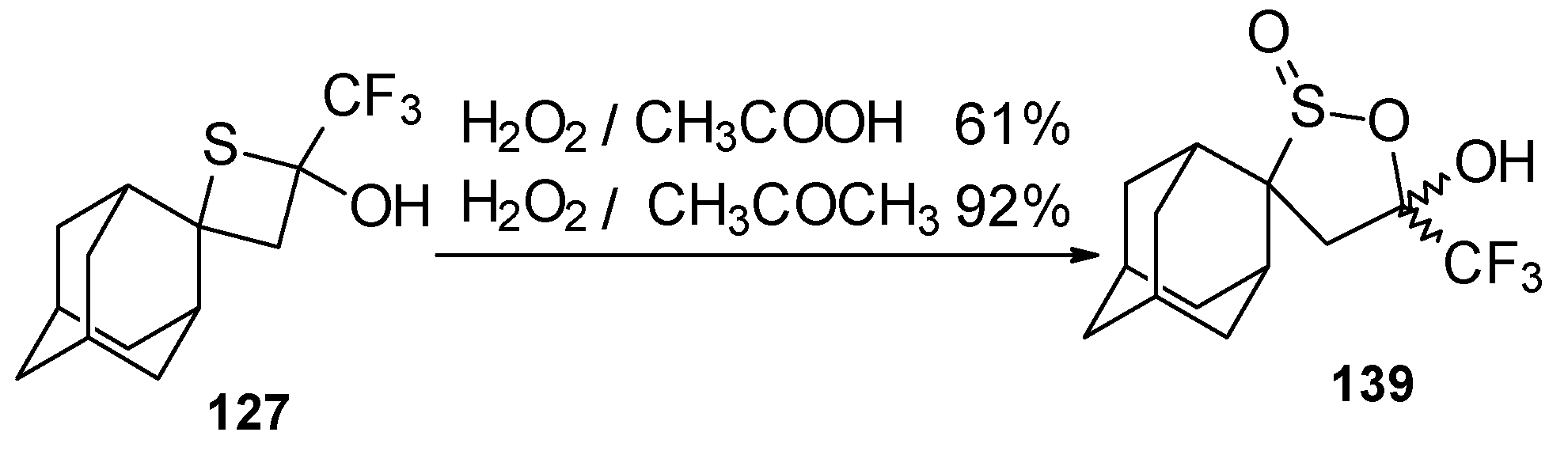Preparation of α,β-Unsaturated Ketones Bearing a Trifluoromethyl Group
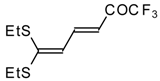
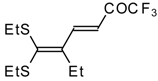
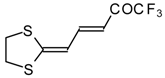
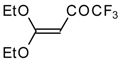
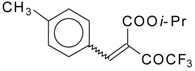
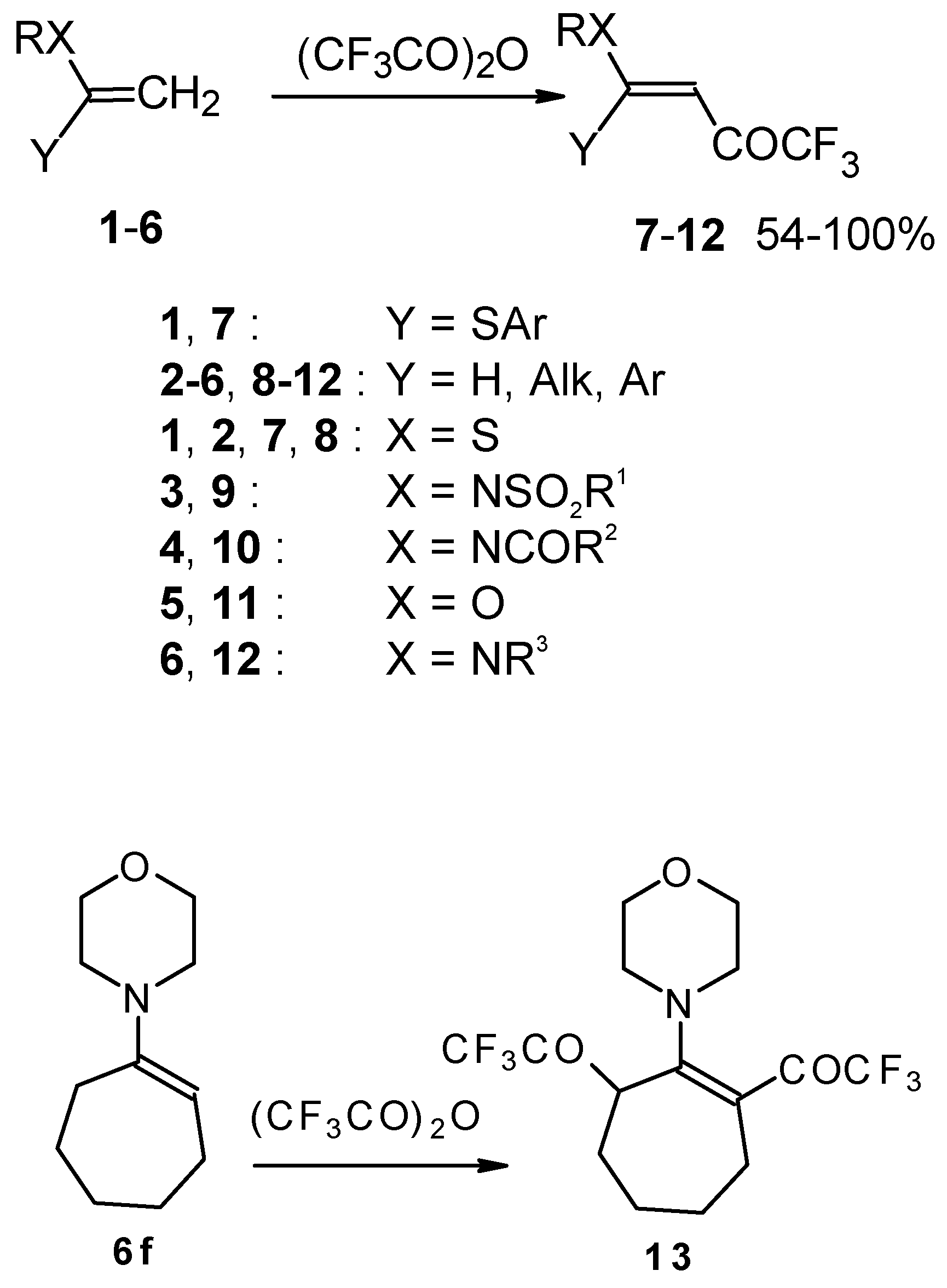
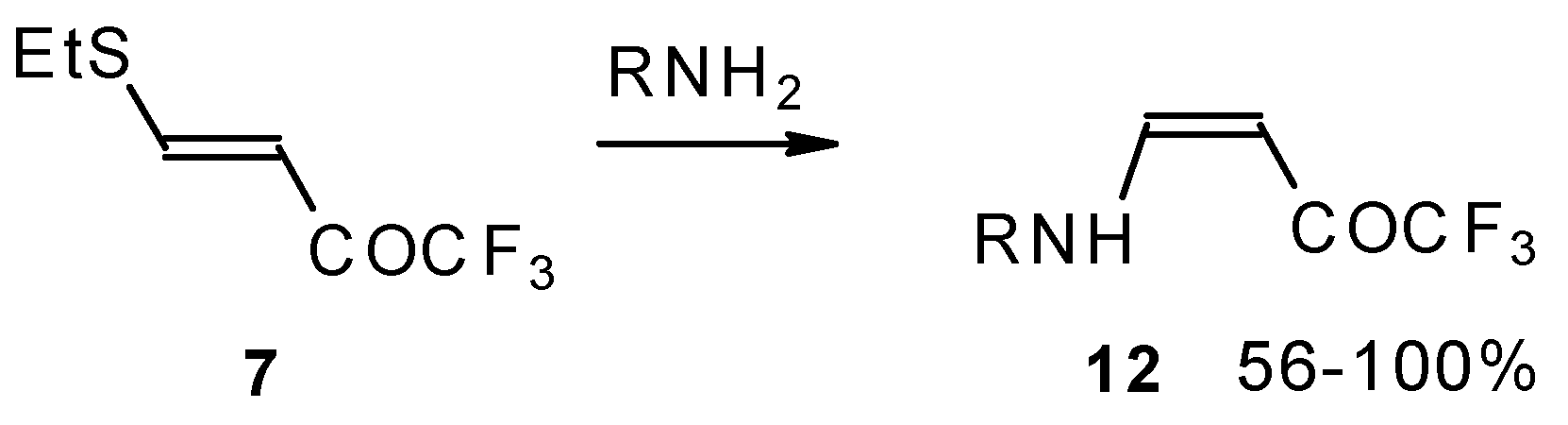
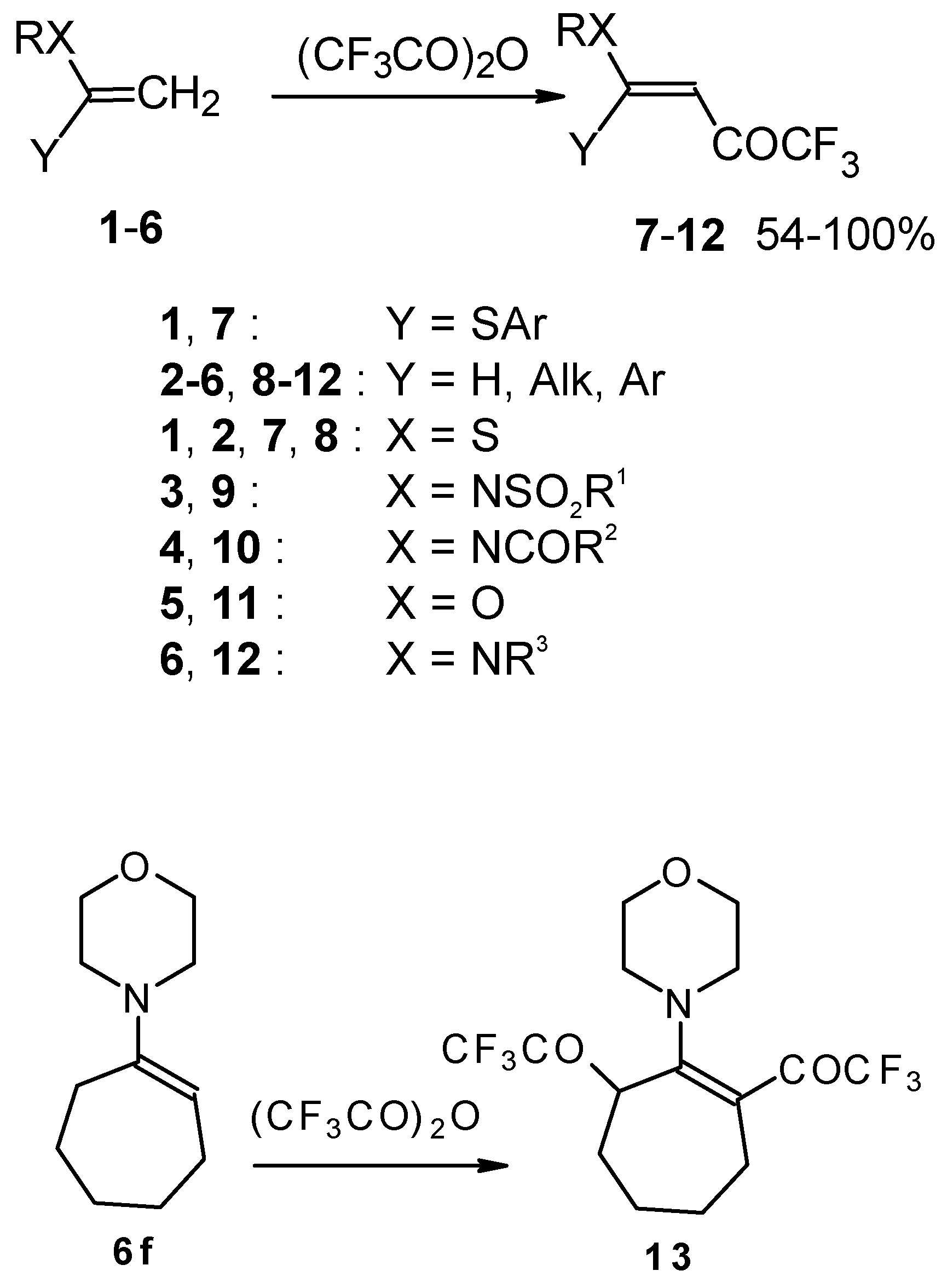
Some trifluoromethyl-containing ketones are readily available by acylation of electron reach alkenes with trifluoroacetic anhydride (or other derivatives of trifluoroacetic acid). Trifluoroacetylation of ketene dithioacetals [
16], vinyl sulfides [
16], vinyl amides [
17], vinyl ethers [
17,
18] (including cyclic vinyl ethers [
18,
19]), enamines [
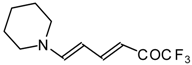
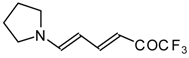
20,
21] proceeds at room temperature to give the corresponding β-trifluoroacetylated compounds in high yields (
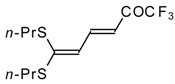
Scheme 2). Activated dienes,
e.g. 1,1-bisalkylthio-1,3-alkadienes, react quite easily also (
Scheme 3) [
22]. The addition-elimination mechanism of trifluoroacylation of electron-reach alkenes was confirmed by
1H NMR and kinetic study [
23,
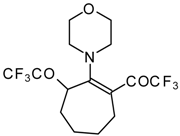
24]. More reactive enamines such as 1-morpholino-1-cyclopentene or 1-morpholino-1-cyclohexene give very complicated reaction mixtures. In the case of 1-morpholino-1-cyclohexene the product of bis trifluoroacylation was isolated (
Scheme 2) [
20].
Trifluoroacetylation of trithioorthoacetates results in the acylated ketene thioacetals in high yield (
Scheme 4) [
25]. The products seem to result from trifluoroacylation of ketene dithioacetals formed by elimination of thiols from trithioorthoacetates.
Trifluoroacylation of triethylorthoacetate and the diethylacetal of some methyl ketones proceeds similarly (
Scheme 5). It is very interesting that this reaction takes place only in the case of trifluoro(trichloro)acetic anhydride. In the case of acetic anhydride this reaction does not proceed [
26].
Recently the trifluoroacylation of vinyl ethers was successfully extended to the diacylation of alkyl vinyl ethers with the use of excess trifluoroacetic anhydride (3 equivalents) in the presence of pyridine (
Scheme 6) [
27].
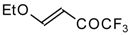
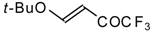
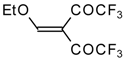
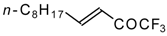
4-Ethoxy-1,1,1-trifluoro-3-buten-2-one
11a was firstly prepared by the three step procedure from nitropentafluoroacetone. Cycloaddition of nitropentafluoroacetone
21 with ethylvinyl ether gives rise to the formation of oxetane
22, subsequent cleavage of the four-membered ring in the presence of sulphuric acid leads to the formation of the corresponding carbinol
23. The thermolysis of this carbinol in the presence of K
2CO
3 gives rise to the target CF
3 ketone. A similar transformation was also investigated for ketene diethylacetal (
Scheme 7) [
28,
29].
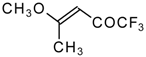
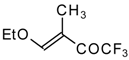
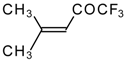
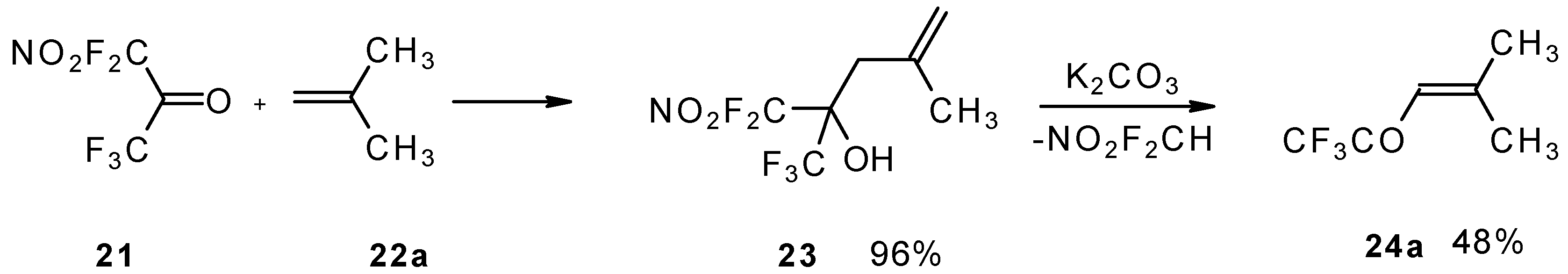
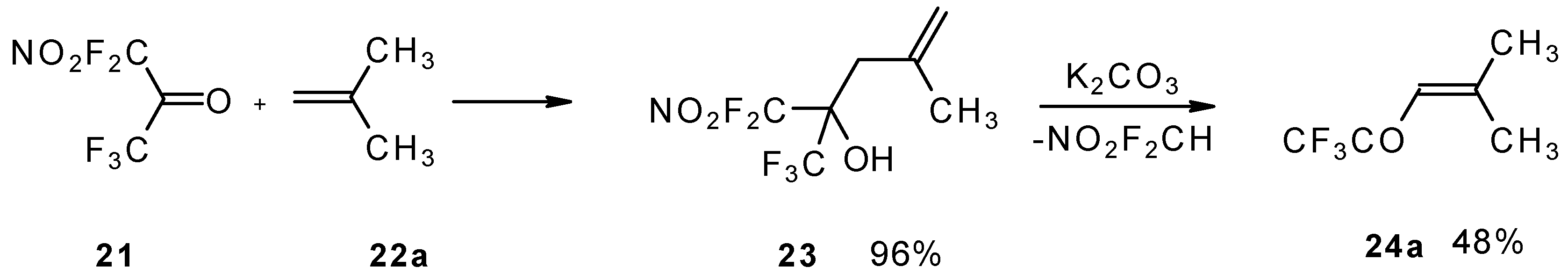
The reaction of nitropentafluoroacetone
21 with isobutylene gives rise to the corresponding carbinol
23 (
Scheme 8). Subsequent elimination of difluoronitromethane in the presence of K
2CO
3 leads to trifluoromethyl-β,β-dimethylvinyl ketone
24a [
30].
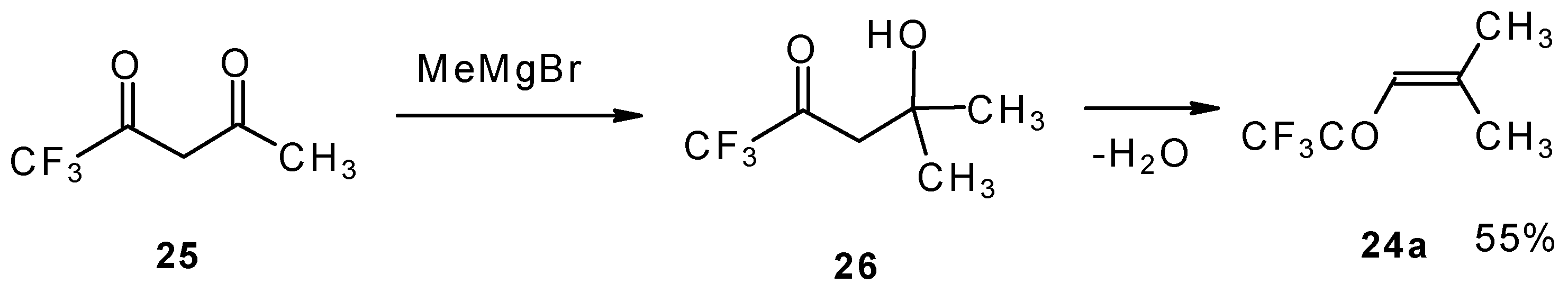
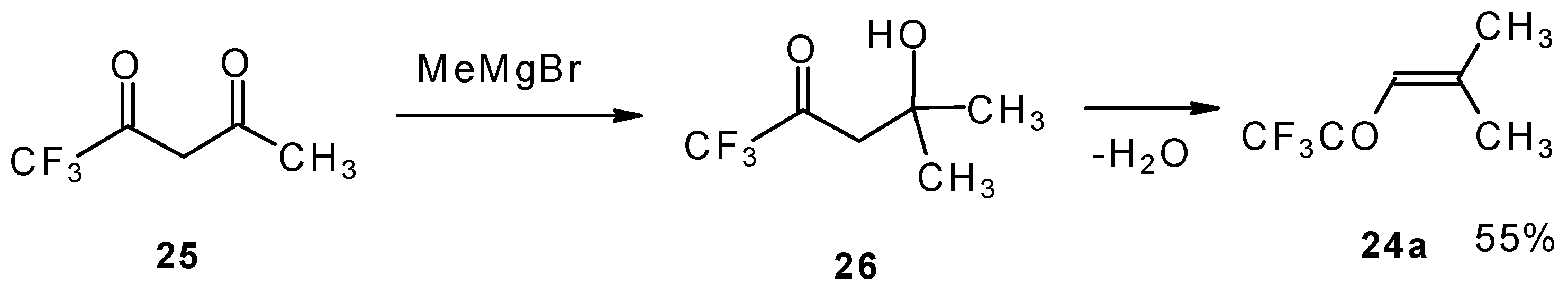
The same ketone was previously prepared by Henne from trifluoroacetylacetone (
Scheme 9). Addition of methylmagnesium bromide with subsequent spontaneous dehydration of water gives this ketone in 55% yield [
31].
Perfluoroacylation of non-activated alkenes was not described before 1992. Trifluoroacylating reagents are restricted to trifluoroacetic anhydride and other derivatives of trifluoroacetic acid. However, the electrophilicity of these reagents is insufficient for acylation of alkenes. Attempts to improve their activity with Lewis acids (as in the case of aromatic hydrocarbons' trifluoroacylation [
32,
33,
34]) lead to cationic polymerization of the unsaturated substrates. Perfluorinated acylium salts which can be used for the perfluoracylation of unsaturated hydrocarbons are unstable and decompose readily with decarbonylation (Eq.1) [
35,
36].
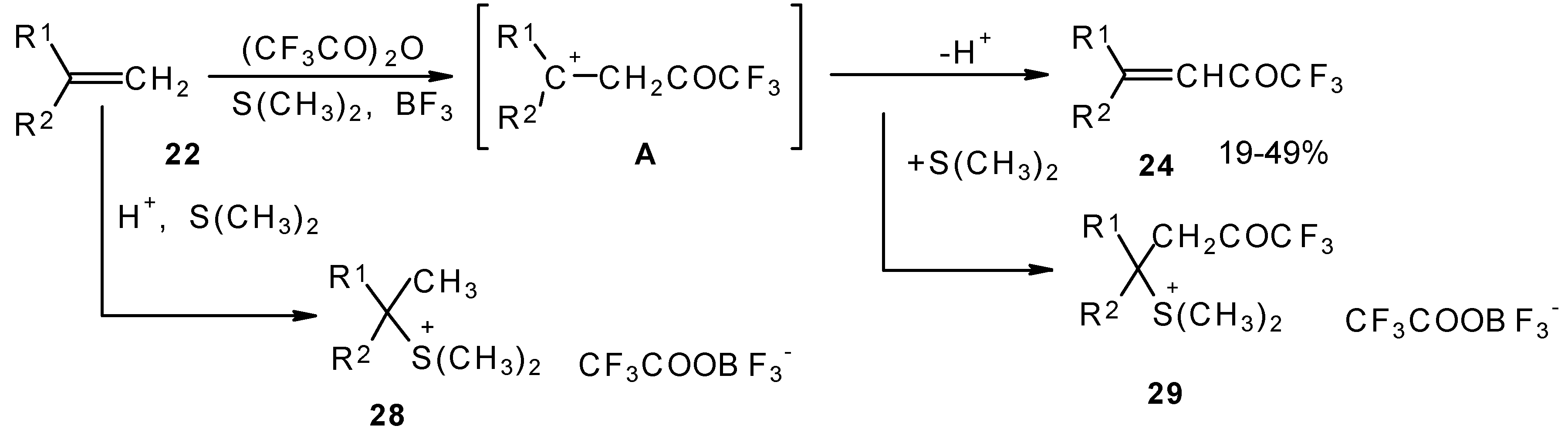
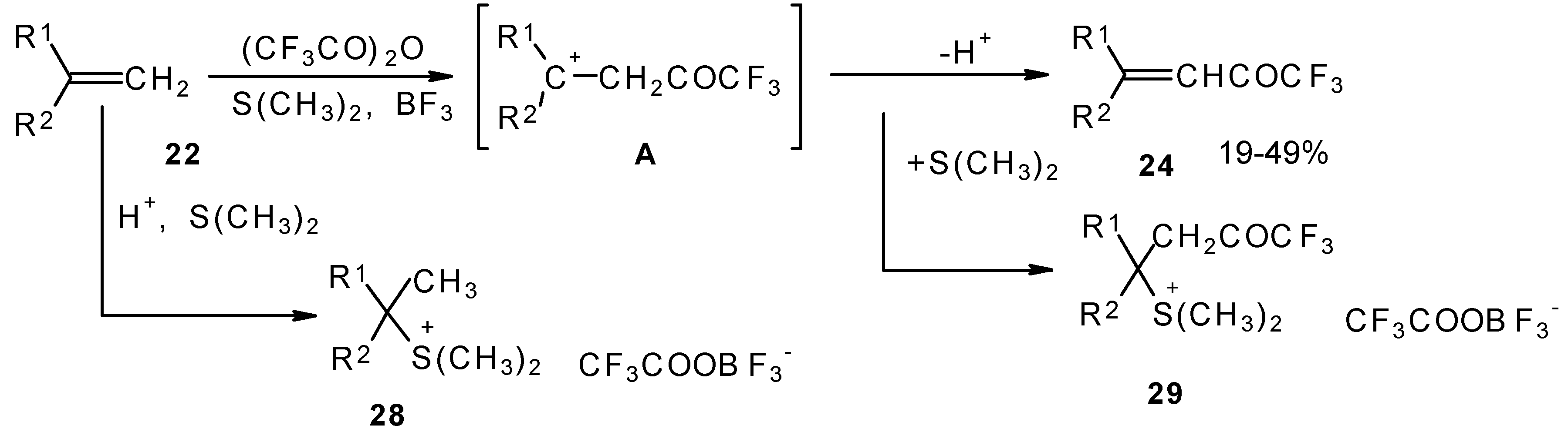
A novel method of direct electrophilic perfluoroacylation of alkenes was proposed, which is based on the use of trifluoroacetic anhydride (or other anhydrides of perfluorinated acids) in the presence of a dimethyl sulphide-borontrifluoride complex (
Scheme 10) [
37].
This method enables unsaturated ketones containing perfluoroalkyl groups without β-heteroalkyl substituents to be obtained in moderate yields (
Scheme 10) [
37,
38,
39,
40,
41,
42]. Recently it was found that this complex is more appropriate for the trifluoroacylation of various aromatic compounds then other trifluoroacylating reagents [
43].
The reaction takes place only for alkenes which give cations with stabilized groups - phenyl, allyl or cyclopropyl or tertiary cations. The electrophilicity of this reagent exceeds the electrophilicity of trifluoroacetic anhydride, which reacts only with electron rich alkenes having heteroatoms at a double bond. In spite of a considerable raising of synthetic possibilities of this method compared with other perfluoroacylating reagents the reactivity of the new reagent is not sufficient to react with all alkenes. The essential disadvantage of this reaction is also the low yields of ketones due to the specific chemistry of the acylation which restricts yield to 50%.
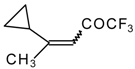
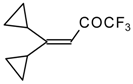
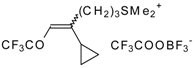
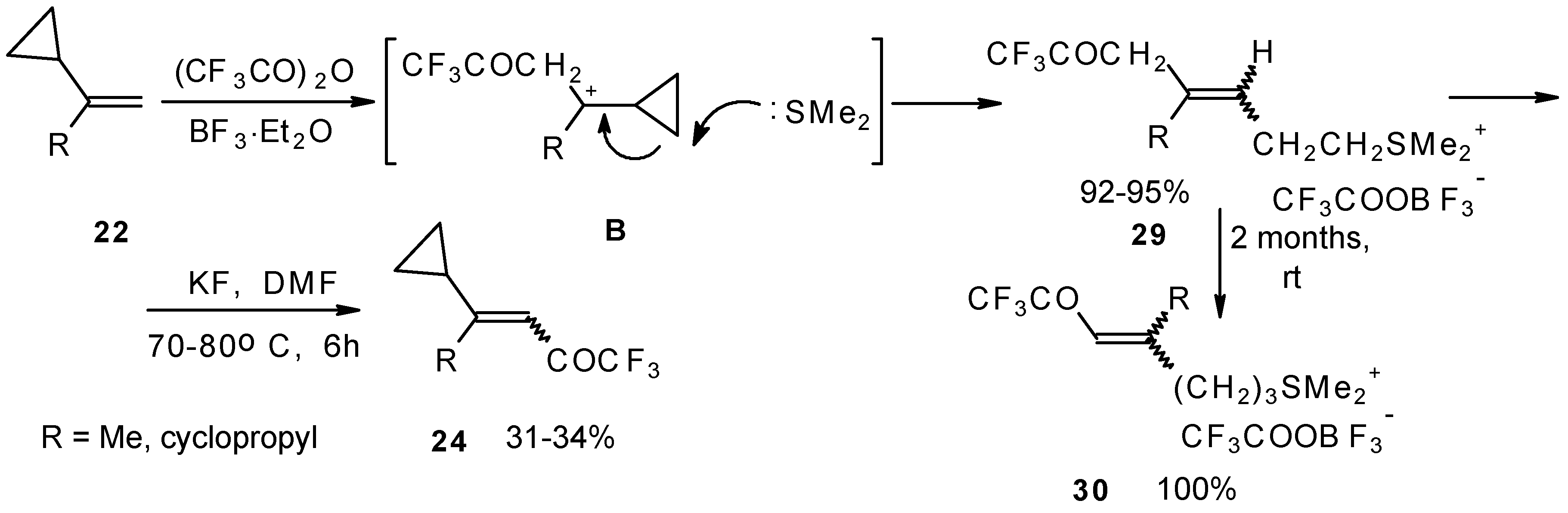
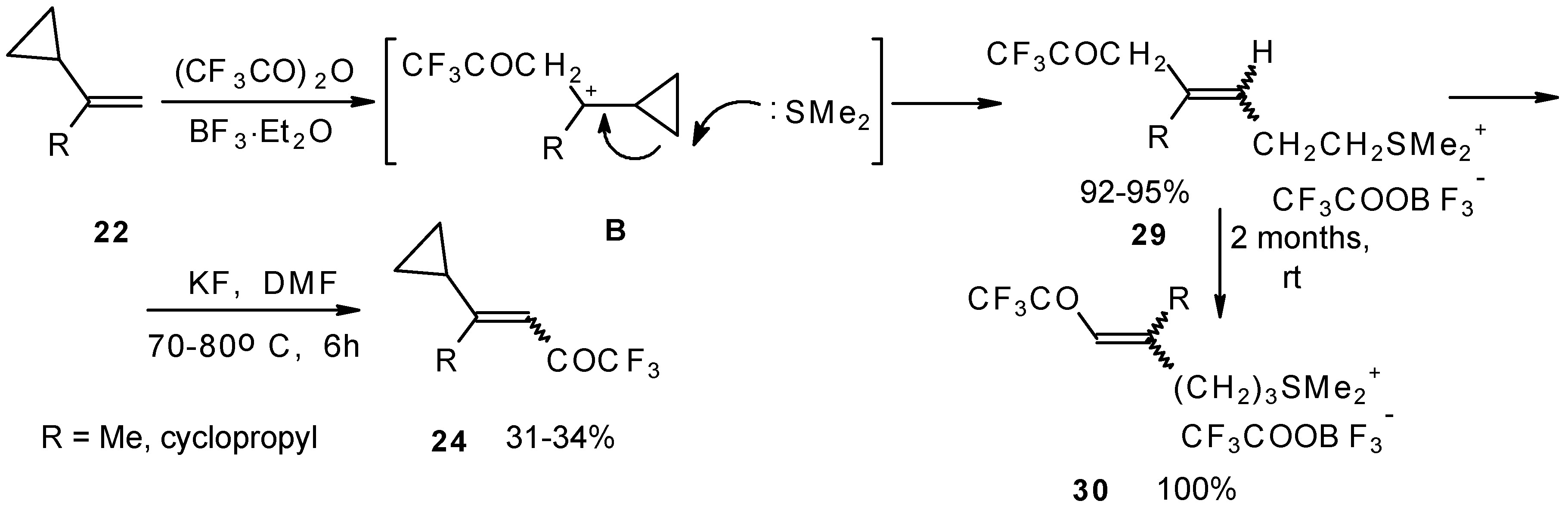
The trifluoroacylation of cyclopropyl containing alkenes such as isopropenylcyclopropane and 1,1-dicyclopropylethylene by trifluoroacetic anhydride in the presence of the dimethyl sulphide-borontrifluoride complex proceeds in an unusual manner (
Scheme 11). This reaction leads to the formation of the corresponding sulphonium salts in high yields [
40], which are the products of the trapping of the homoallyl cation, formed in the process of cyclopropane ring opening. Since these compounds are β,γ-unsaturated ketones, they are prone to spontaneous transformation to the corresponding α,β-unsaturated ketones. The rearrangement of the compounds proceeds over
ca. two months at room temperature with no catalyst in quantitative yield.
The action of potassium fluoride, as a base, on these sulfonium salts furnishes intramolecular nucleophilic substitution of the dimethyl sulphide group, which results in α,β-unsaturated ketones containing a perfluorinated group (
Scheme 11).
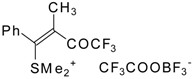
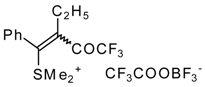
The trifluoroacylation of phenyl-substituted acetylenes having both terminal and internal triple bonds using the complex (CF
3CO)
2O-BF
3-SMe
2 was studied. This reaction leads to the formation of the corresponding sulfonium salts
32, which are the products of conjugate addition of the CF
3CO group and dimethyl sulfide to the acetylene molecule (
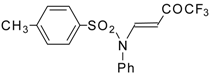
Scheme 12). Reaction of these sulfonium salts with the excess of dimethyl sulphide yields the corresponding trifluoroacetylated vinyl sulphides
33 in high yields. Oxidation of the sulphides by hydrogen peroxide in acetic acid gives rise to the corresponding unsaturated sulfones
34 with the CF
3CO moiety in the β-position [
42].
Some other existing methods for the preparation for unsaturated ketones with trifluoromethyl group are based on the acylation of organoelement compounds. The first synthesis of 1,1,1-trifluoro-4-phenylbut-3-en-2-one by the reaction of phenylvinylmagnesium bromide with trifluoroacetic acid was described in 1959, but the yield was very low (
Scheme 13) [
44].
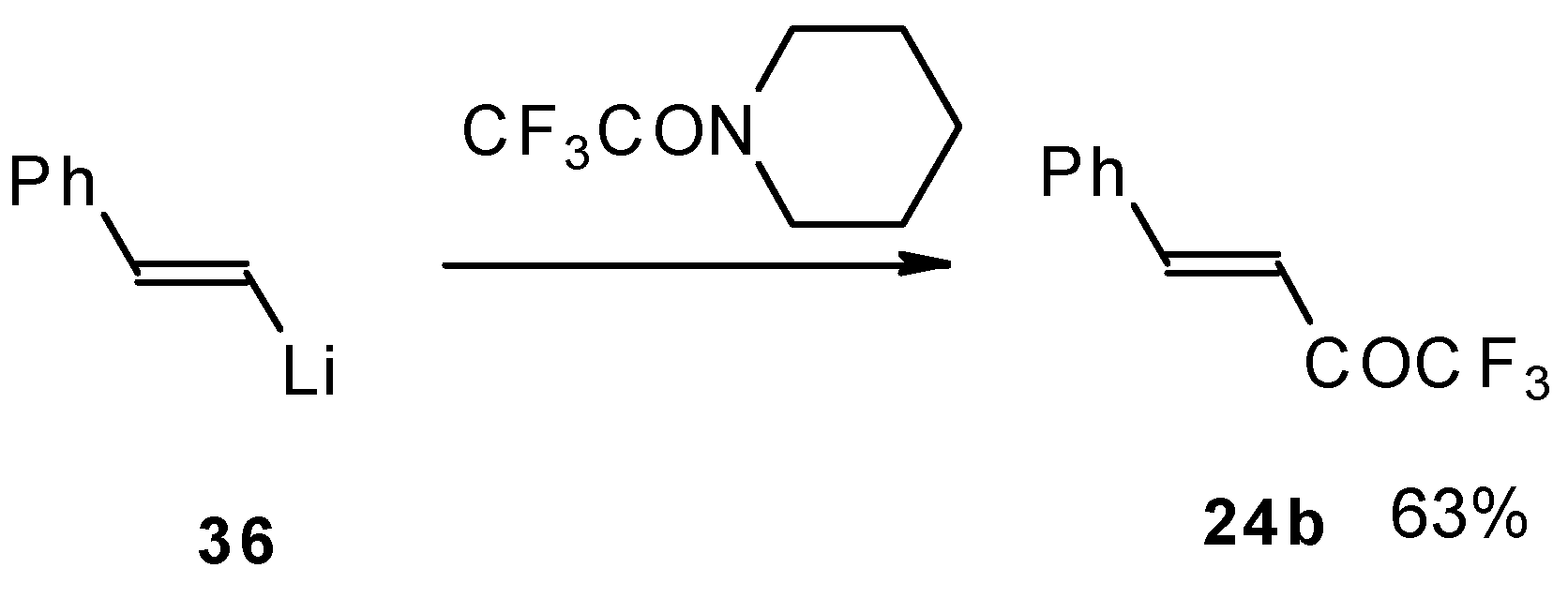
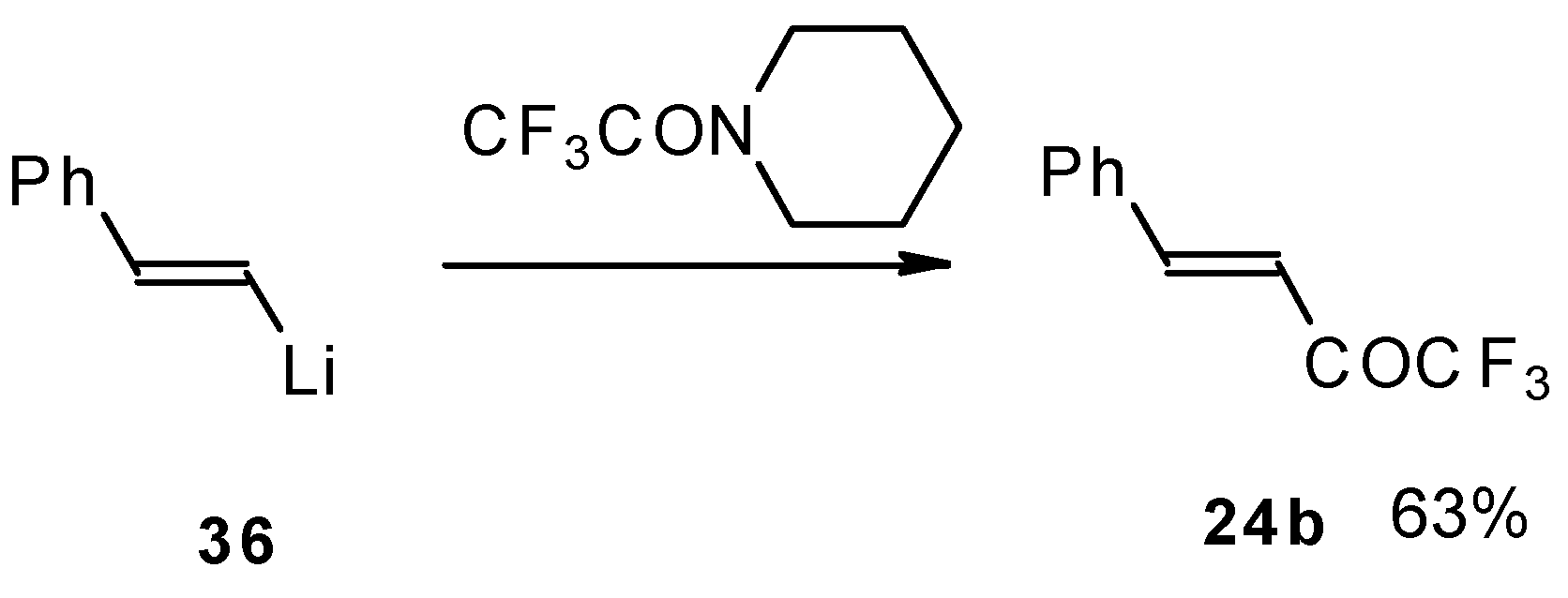
It was found in subsequent investigations that organolithium compounds were more appropriate for the synthesis of trifluoromethyl containing ketones and gave higher yields of the target products (
Scheme 14) [
45].
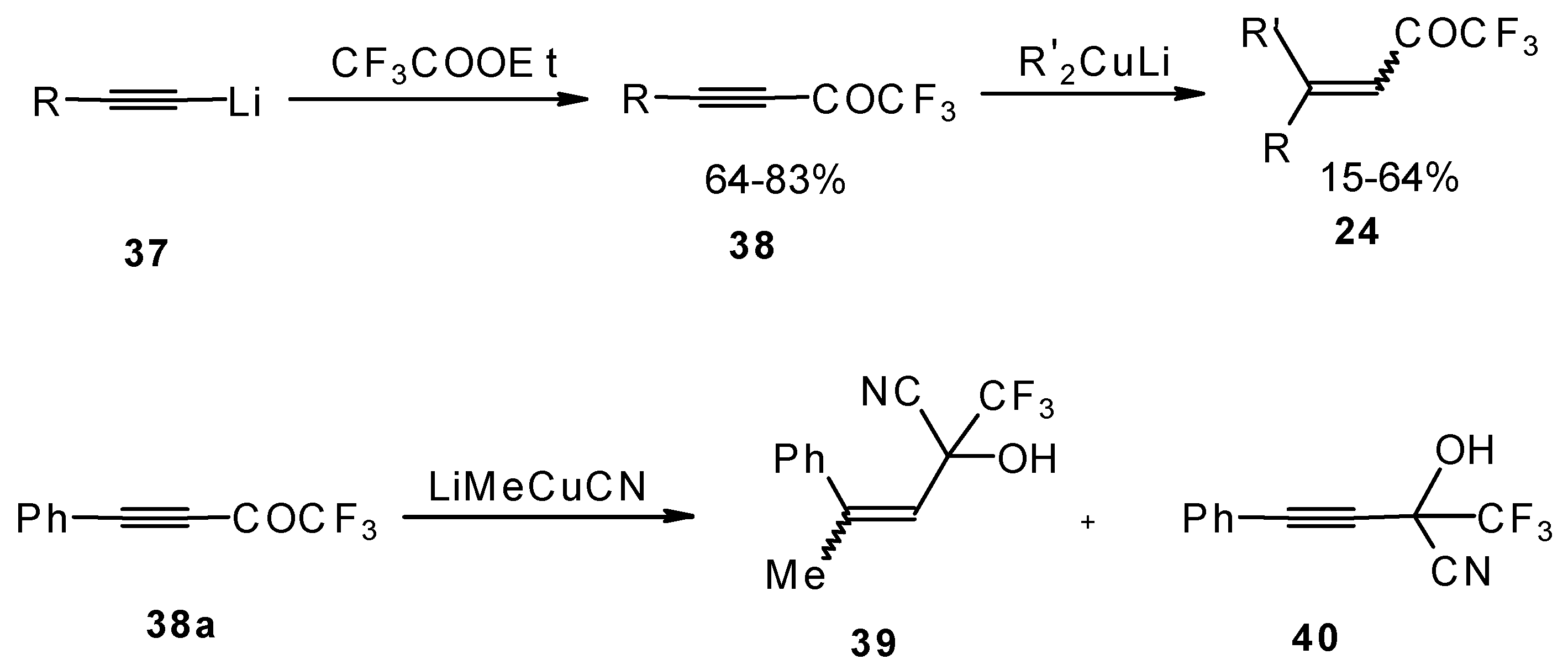
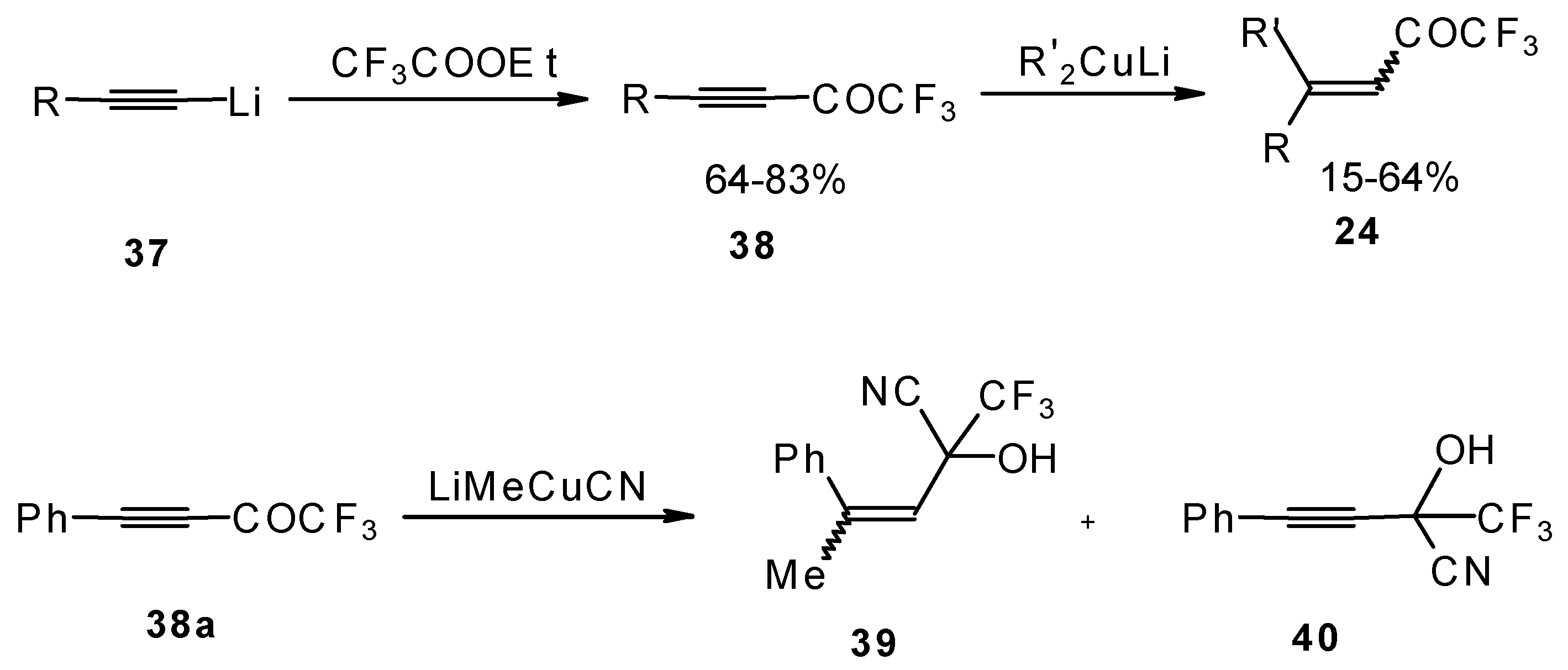
A good route for the synthesis of fluorinated ketones involves 1, 4-addition of an organocuprate to the acetylenic ketones, which are readily obtained by acylation of acetylides (
Scheme 15). This method affords ethylenic ketones with high regioselectivity, however stereoselectivity is variable and yields are moderate [
46,
47,
48,
49,
50].
It should be noted that in the case of some cuprates regioselectivity is low and the products of both 1,2- and 1,4-additions are formed, but use of cyano and higher order cuprates results in only unsaturated CF
3 ketones [
48] . In some cases the formation of cyanohydrins takes place (
Scheme 15).
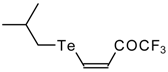
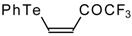
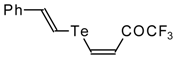
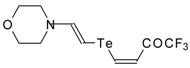
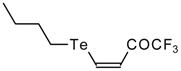
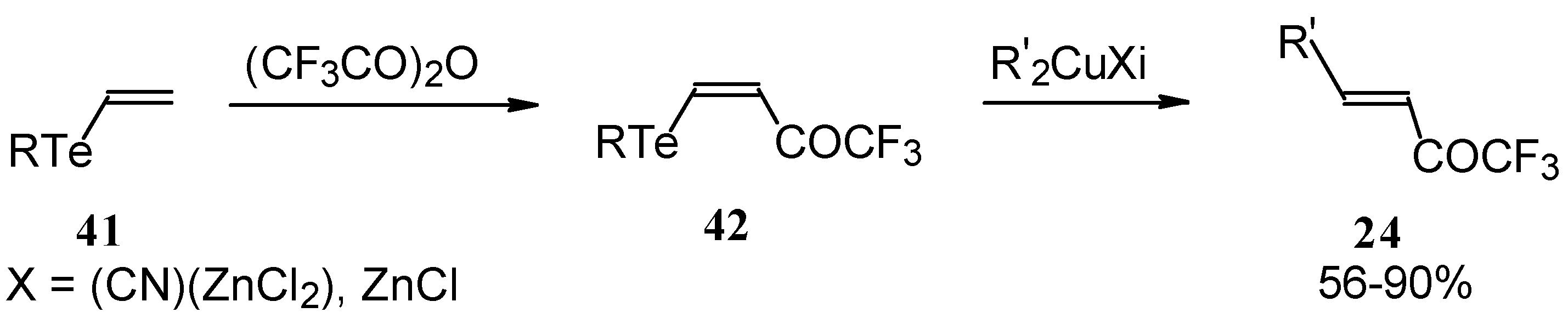
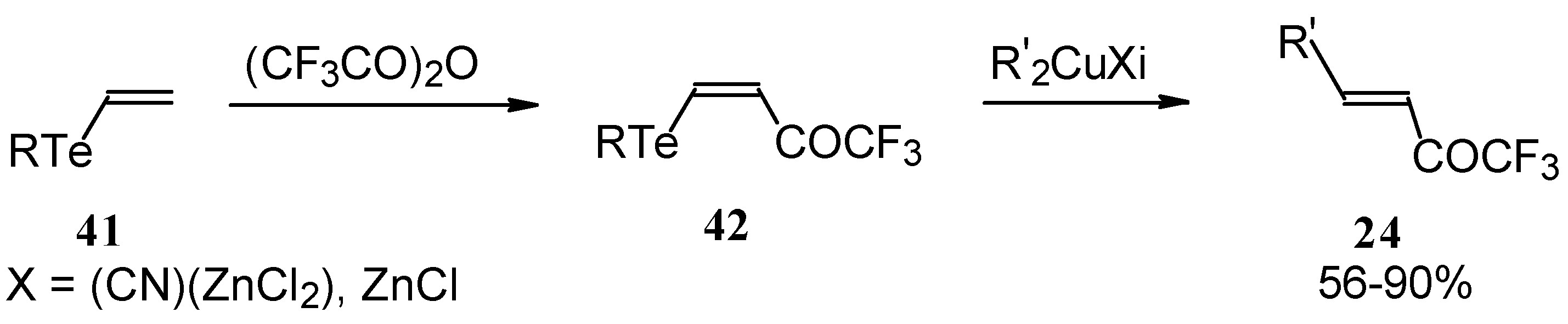
A novel method of the synthesis of trifluromethyl containing unsaturated ketones was described recently [
51]. The first step in this approach involves trifluoroacylation of the corresponding vinyltellurides with trifluoroacetic anhydride (
Scheme 16) [
52]. The reaction proceeds stereospecifically to give only Z isomers of β-trifluoroacetylvinyltellurides in good yields. The subsequent reaction of β-trifluoroacetylvinyltellurides with different zinc cuprates
via a conjugate addition-elimination process affords the corresponding α,β-unsaturated trifluoromethyl ketones. In some cases competitive double addition of zinc cuprates takes place [
51].
Although the required ketones were obtained in good yields, utilization of organotellurium compounds seems to be a big disadvantage of this method.
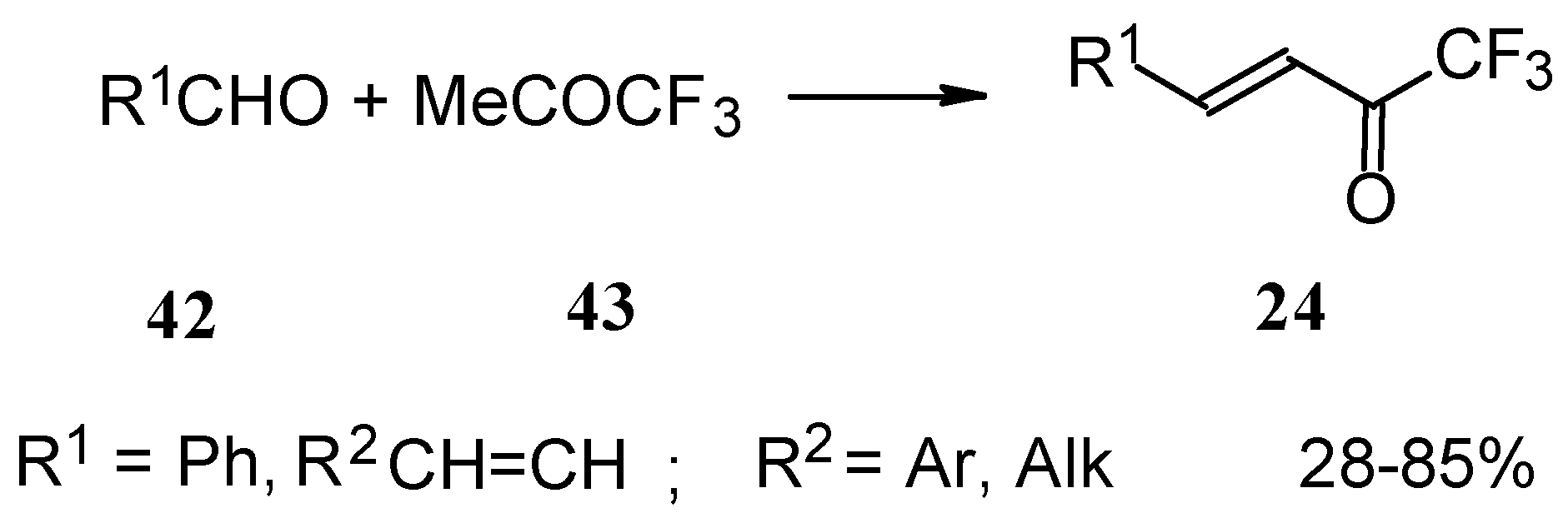
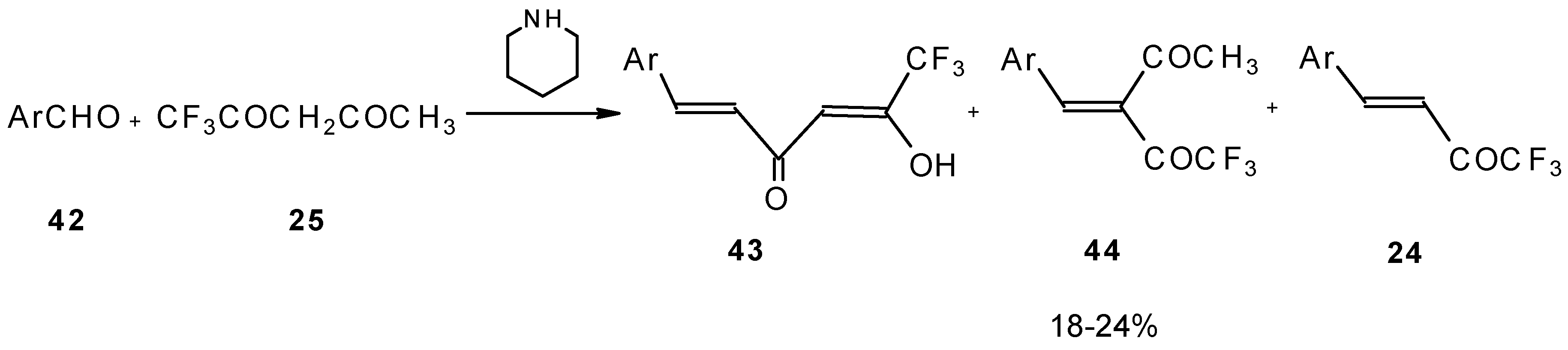
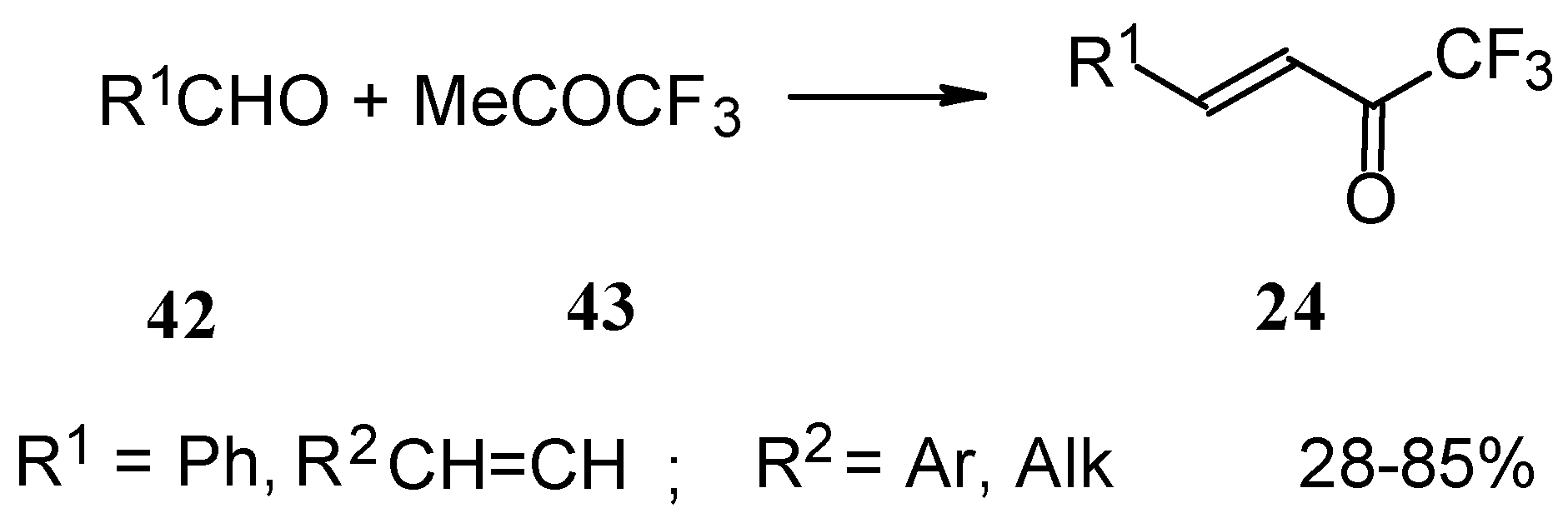
Other described methods for the syntheses of these ketones include crotonic condensation of trifluoroacetone with unsaturated or aryl aldehydes (
Scheme 17) [
53,
54]. However, since competitive self-condensation of trifluoroacetone takes place, it is necessary to use a 10 fold excess of trifluoroacetone.
The reaction of aryl aldehydes with 1,1,1-trifluoroacetylacetone results in the corresponding unsaturated CF
3-containing ketones, but yields of the target ketones are very low [
55].
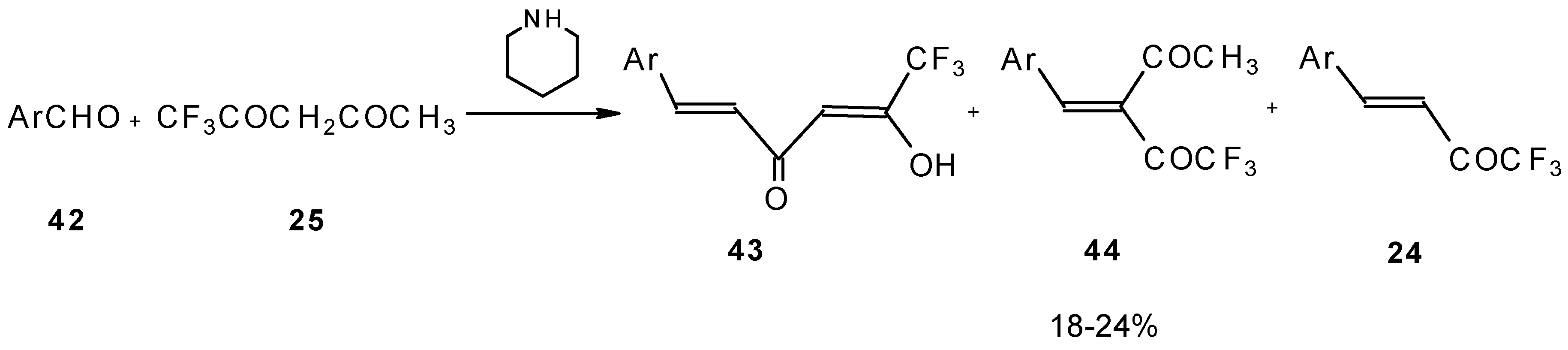
Quite recently a novel method for the synthesis of α-alkoxycarbonyl-α,β-unsaturated trifluoromethyl ketones was found involving the Knoevenagel condensation of aldehydes with trifluoroacetoacetates in the presence of silica gel functionalized with amino groups and
p-toluenesulfonic acid (
Scheme 19). It should be noted that the use of such a catalyst as piperidine/acetic acid gives raise to very low yields of products [
56].
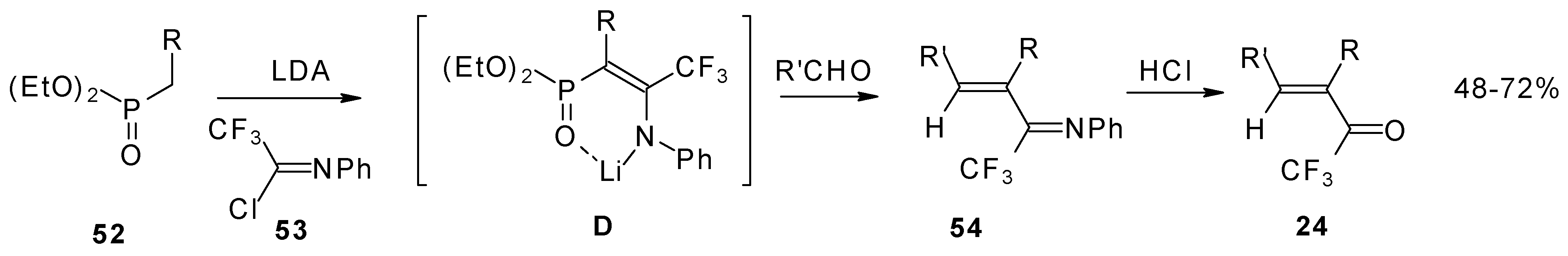
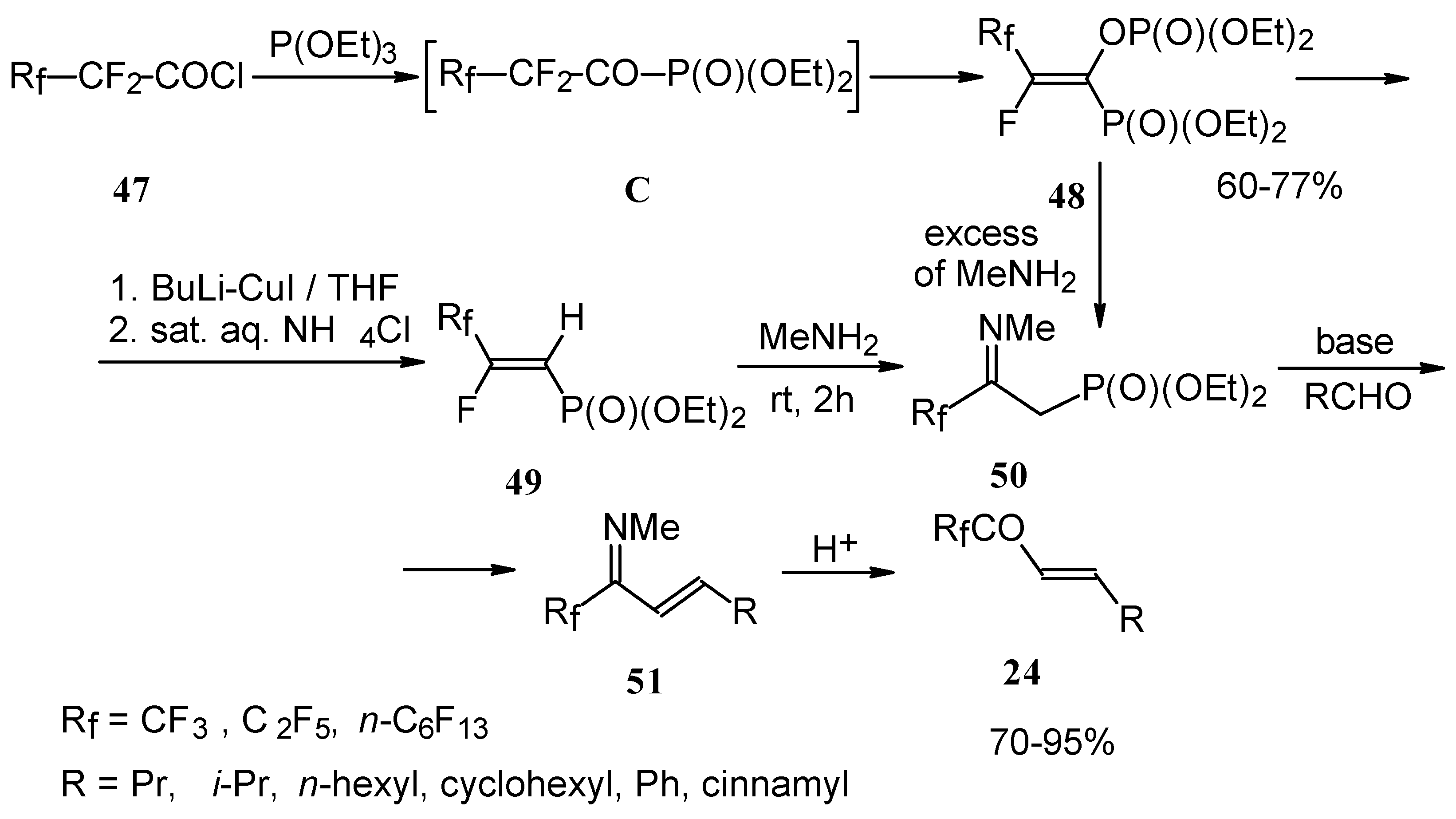
Polyfluoroalkyl α,β-enones may be also prepared from β-iminophosphonates, accessible in four steps from perfluoroalkanoyl chlorides. The β-iminophosphonate anions condensed with an aldehyde lead to the corresponding trifluoromethyl or perfluoroalkyl enones after acidic hydrolysis (
Scheme 20) [
57].
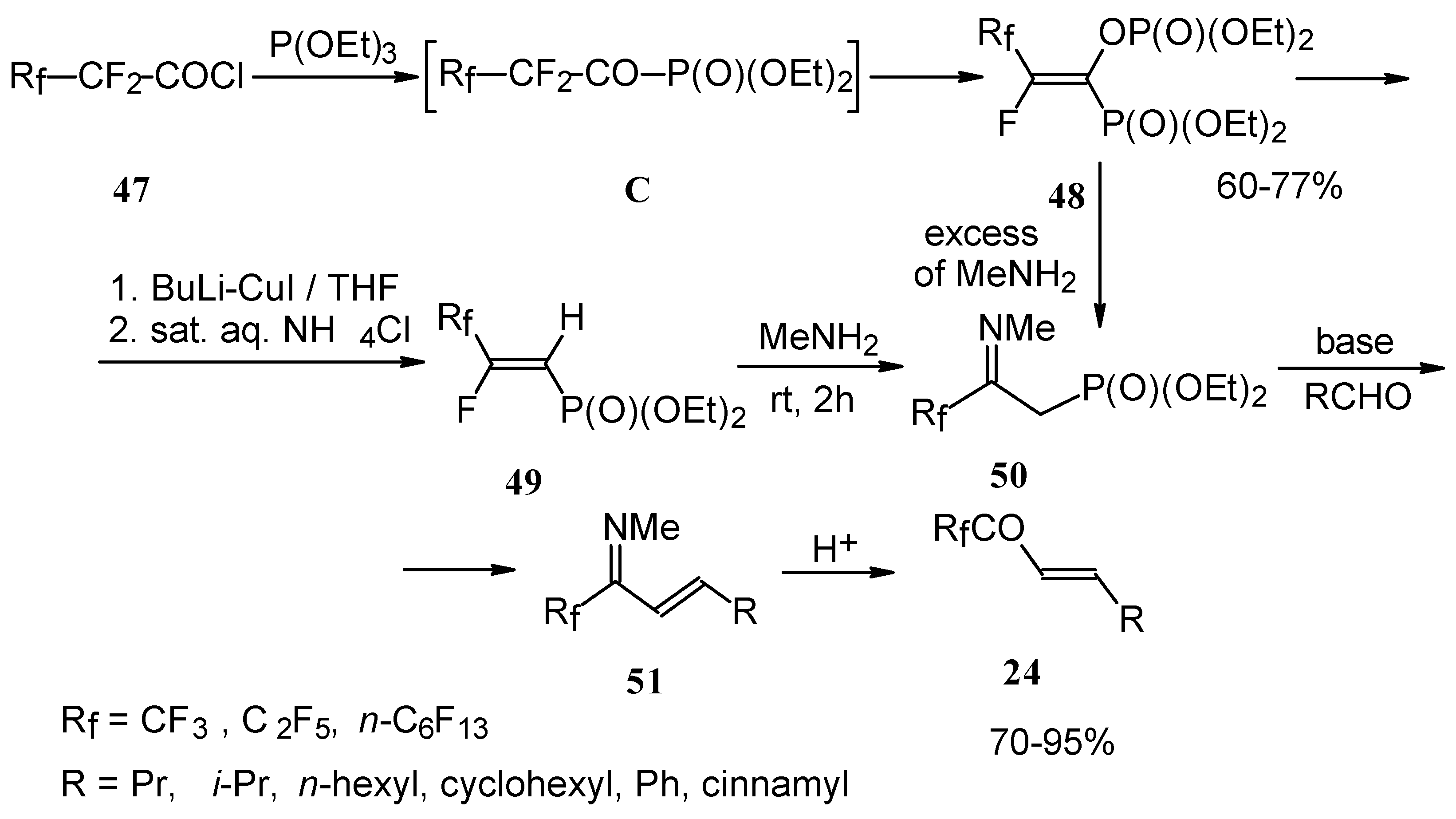
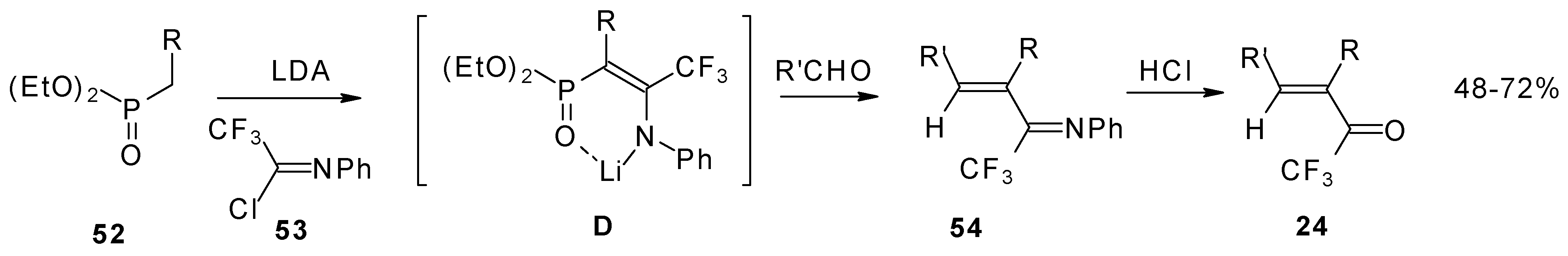
Quite recently a novel one pot procedure based on the Horner-Emmons olefination was elaborated. N-phenylrifluoroacetimidoyl chloride reacts with diethoxyphosphorylmethyl (or ethyl) lithium, generated in situ from diethylmethylphosphonate and lithium diisopropylamide (
Scheme 21). Subsequent addition of the aldehyde gives rise to 1-aza-1,3-diene readily converted by HCl hydrolysis to the corresponding unsaturated ketone bearing a CF
3 group [
58].
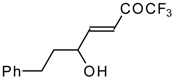
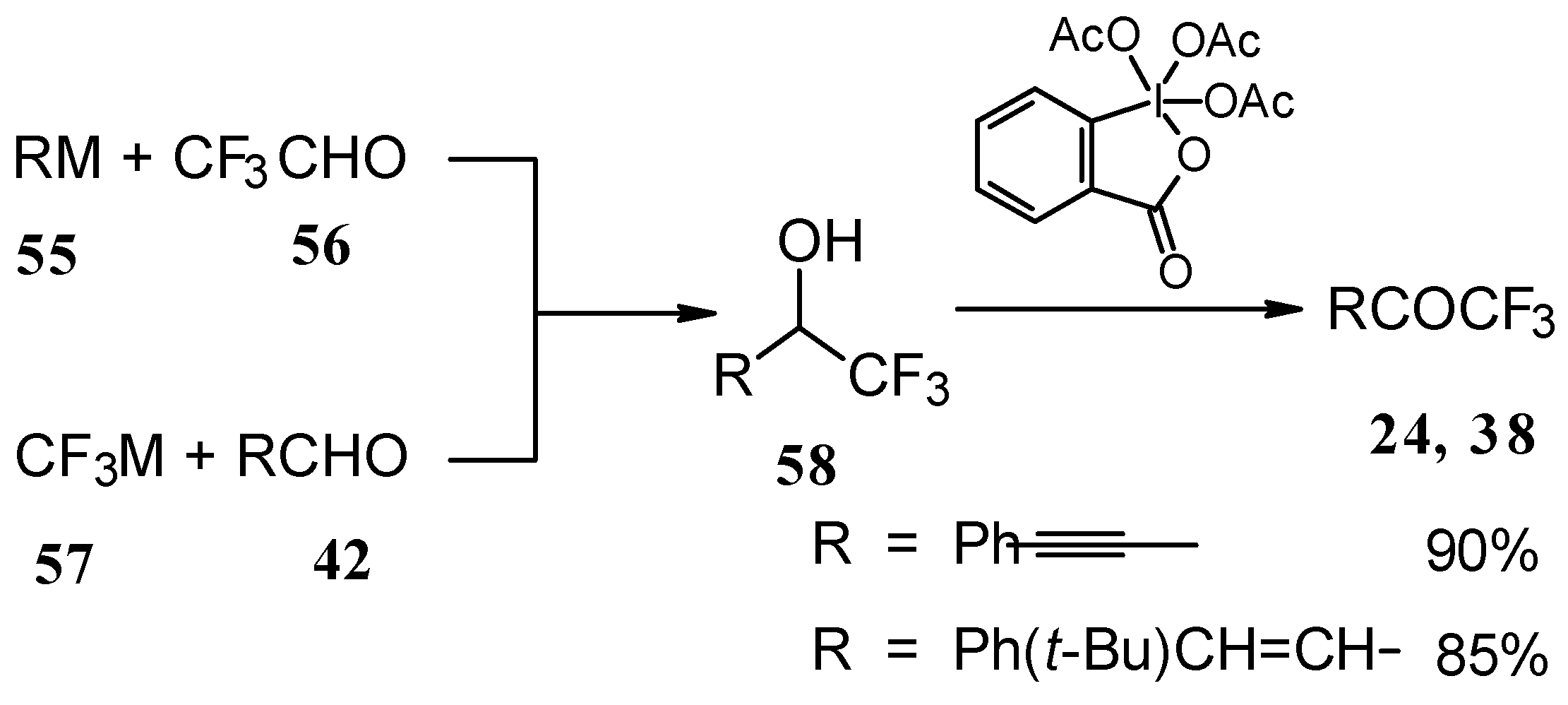
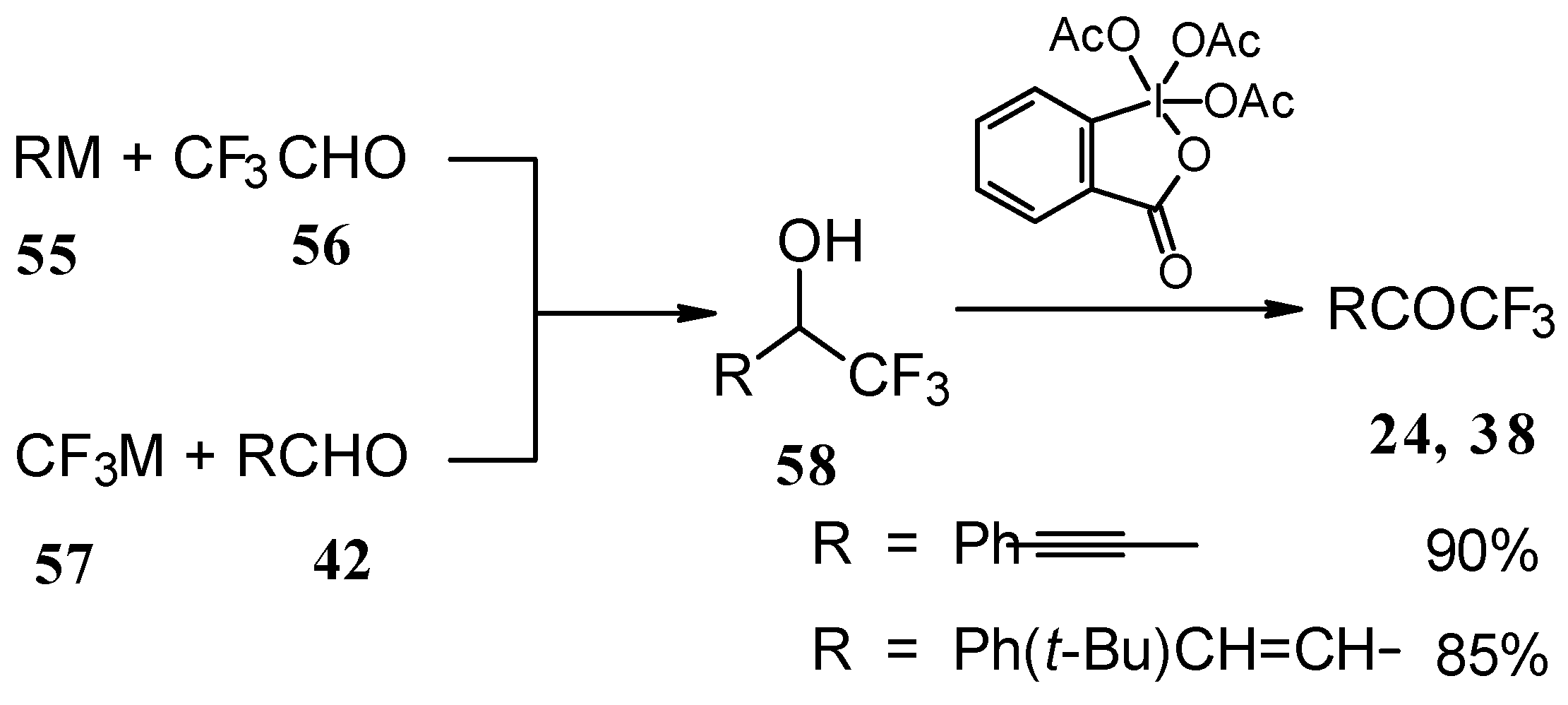
Secondary carbinols, obtained by the addition of vinyl organometallics to trifluoroacetaldehyde [
59] or perfluoroalkyl organometallics to unsaturated aldehydes (
Scheme 22) [
60,
61,
62] can be oxidized by Dess-Martin [
63,
64] or Swern [
65] reagents or using MnO
2 [
66] in dichloromethane to the α,β-ethylenic trifluoromethyl ketones in good yields. [
66,
67] It should be noted that the reduction of acetylenic CF
3 ketones using LiAlH
4 or NaBH
4 does not give CF
3 enones but the corresponding allyl alcohols [
50,
64,
68].
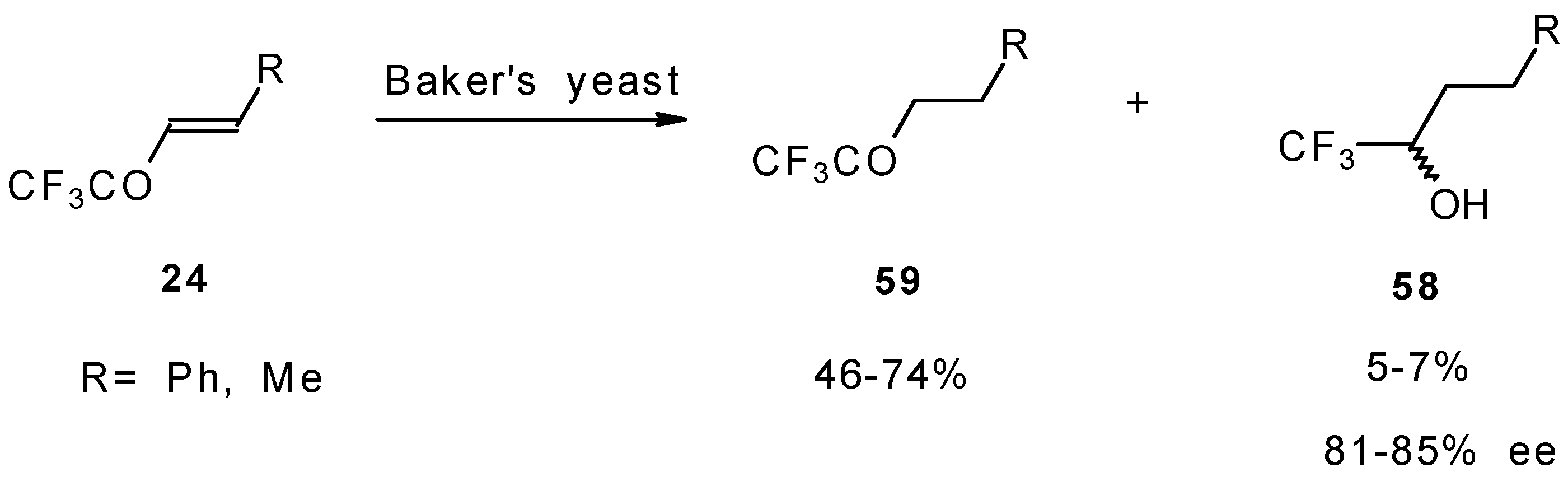
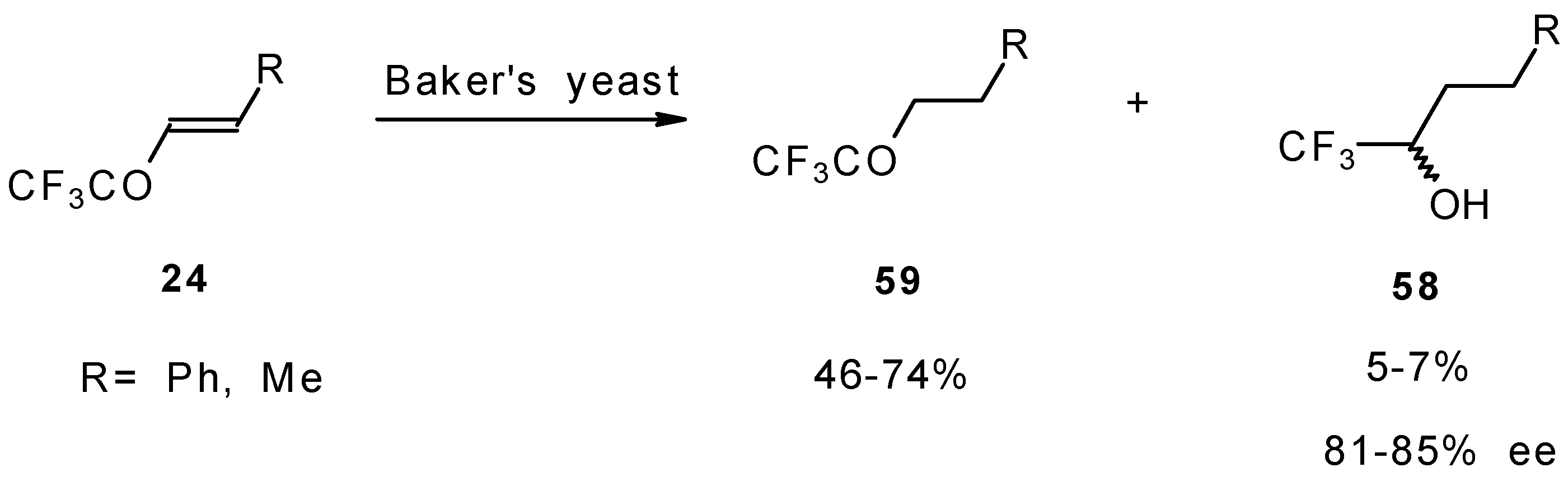
The diastereoselective reduction of CF
3 enones with baker’s yeast was also investigated (
Scheme 23). However, the main reaction product is a CF
3 ketone rather then an alcohol. The optical purity of alcohols prepared by this procedure is 81-85 %
ee [
69].
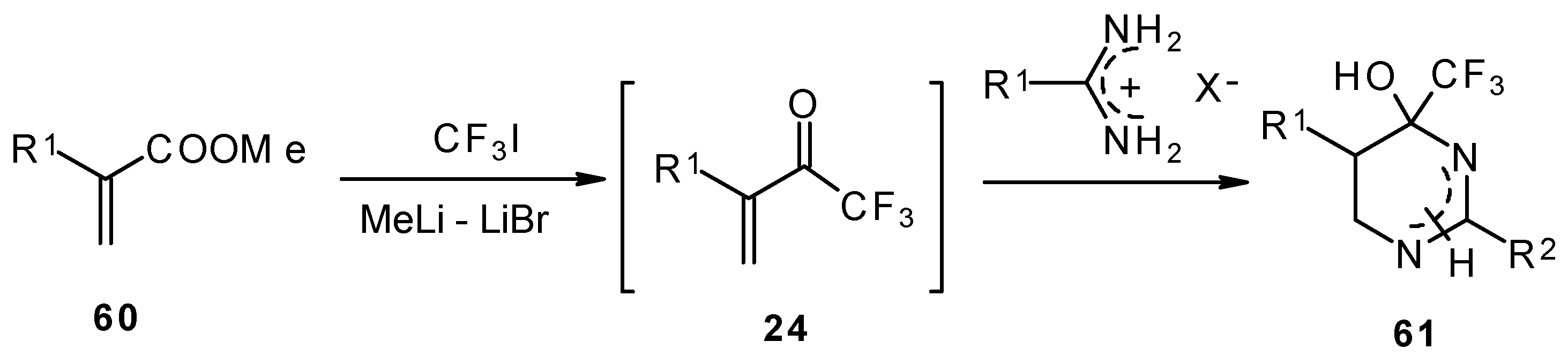
Vinyl perfluoroalkyl ketones are formed
in situ from the reactions of perfluoroalkyllithiums with ethyl acrylate. They can be converted to 4-(perfluoroalkyl)tetrahydropyrimidines in moderate to good yields [
70].
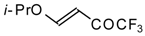
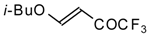
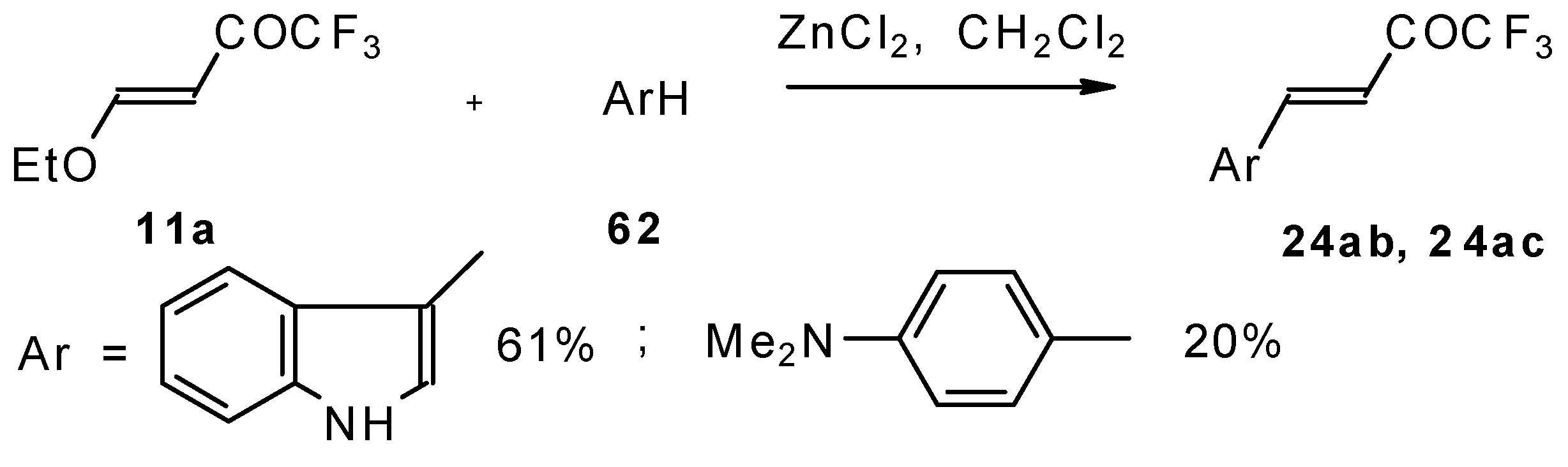
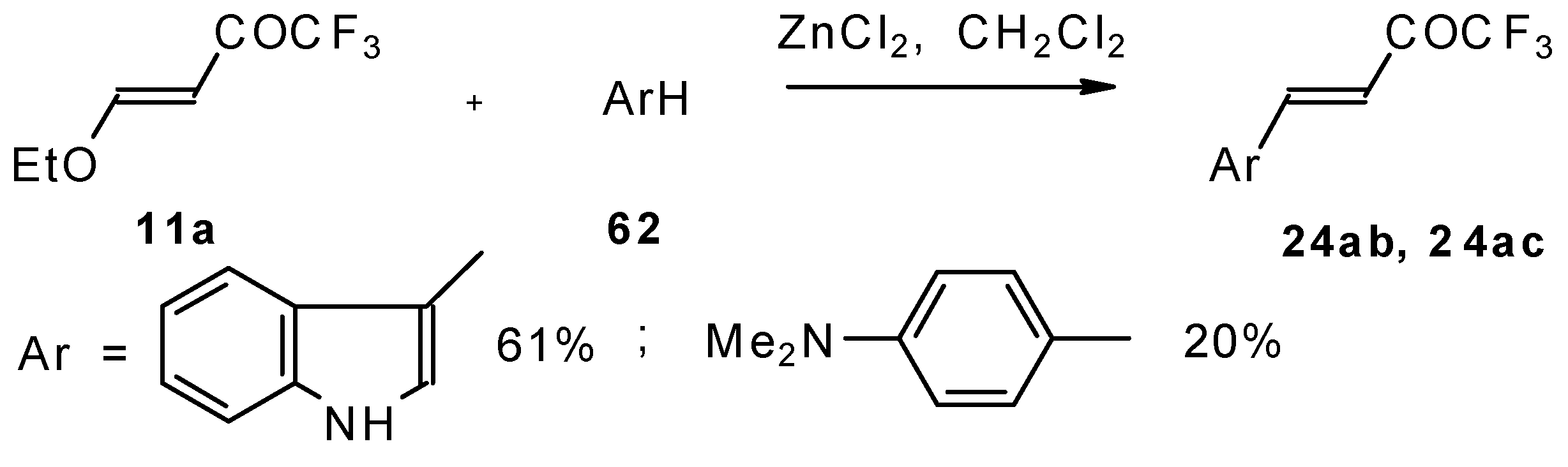
4-Ethoxy-1,1,1-trifluoro-3-buten-2-one
11a is readily obtained by the trifluoroacylation of ethylvinyl ether with trifluoroacetic anhydride [
71]. The reactions of this ketone with electron-rich aromatics (
Scheme 25) or phenyl magnesium bromide (
Scheme 26) results in the formation of trifluoromethyl unsaturated ketones [
72]. However, only very active aromatic compounds such as indole and dimethylaniline take part in the reaction. Reaction with anisole does not take place. The application of stronger Lewis acids such as BF
3 or TiCl
4 at room temperature or on heating was unsuccessful.
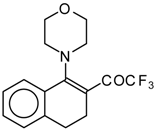
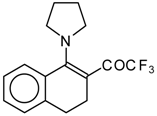
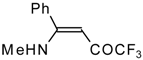
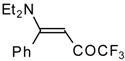
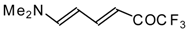
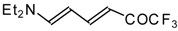
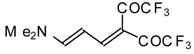
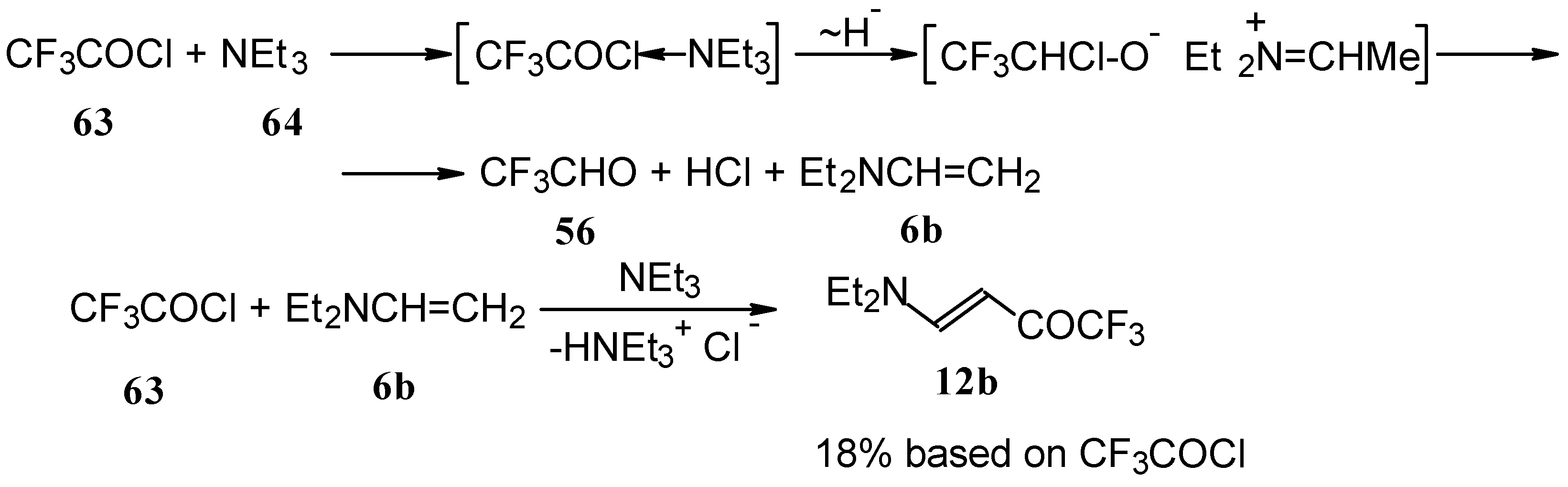
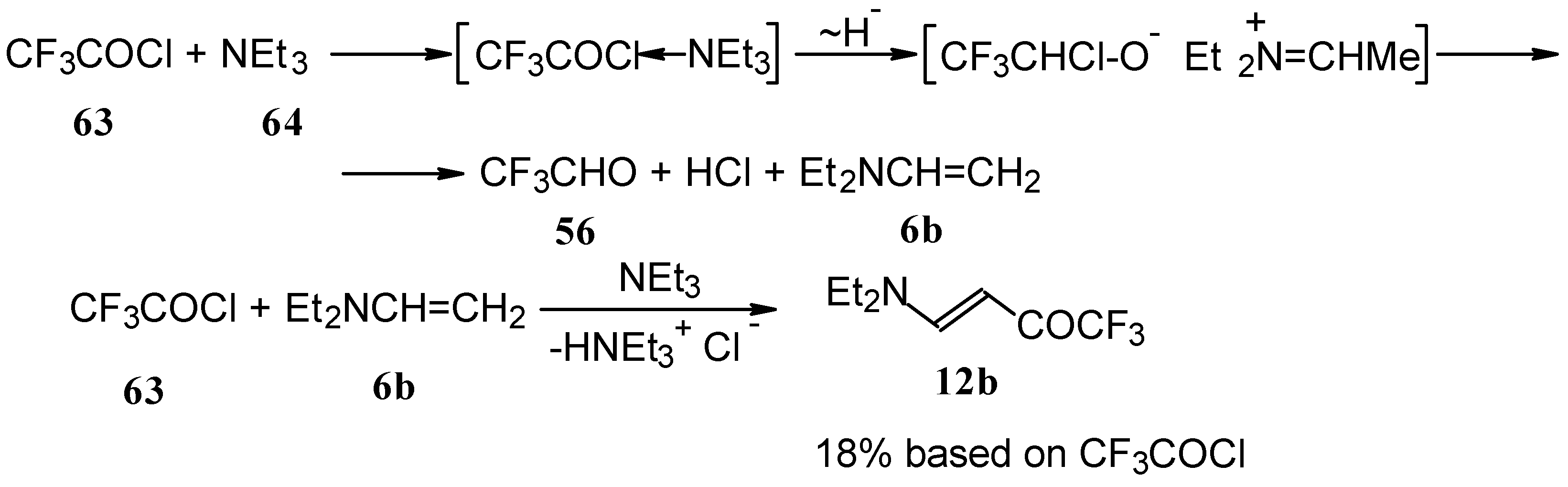
β-Diethylamino-substituted trifluoromethyl-containing enones can be obtained by the reaction of triethylamine with trifluoroacetyl chloride, trifluoroacetyl fluoride or trifluoroacetic anhydride. Reaction of trifluoroacetyl chloride with two equivalents of triethylamine results in the 4-diethylamino-1,1,1-trifluoro-3-buten-2-one
12b. A proposed reaction mechanism involves the oxidation of triethylamine to diethylvinylamine by one equivalent of trifluoroacetyl chloride which is itself reduced to floural (
Scheme 27). Diethylvinylamine immediately reacts with another equivalent of trifluoroacetyl chloride to give rise to the corresponding enone [
73].
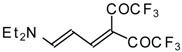
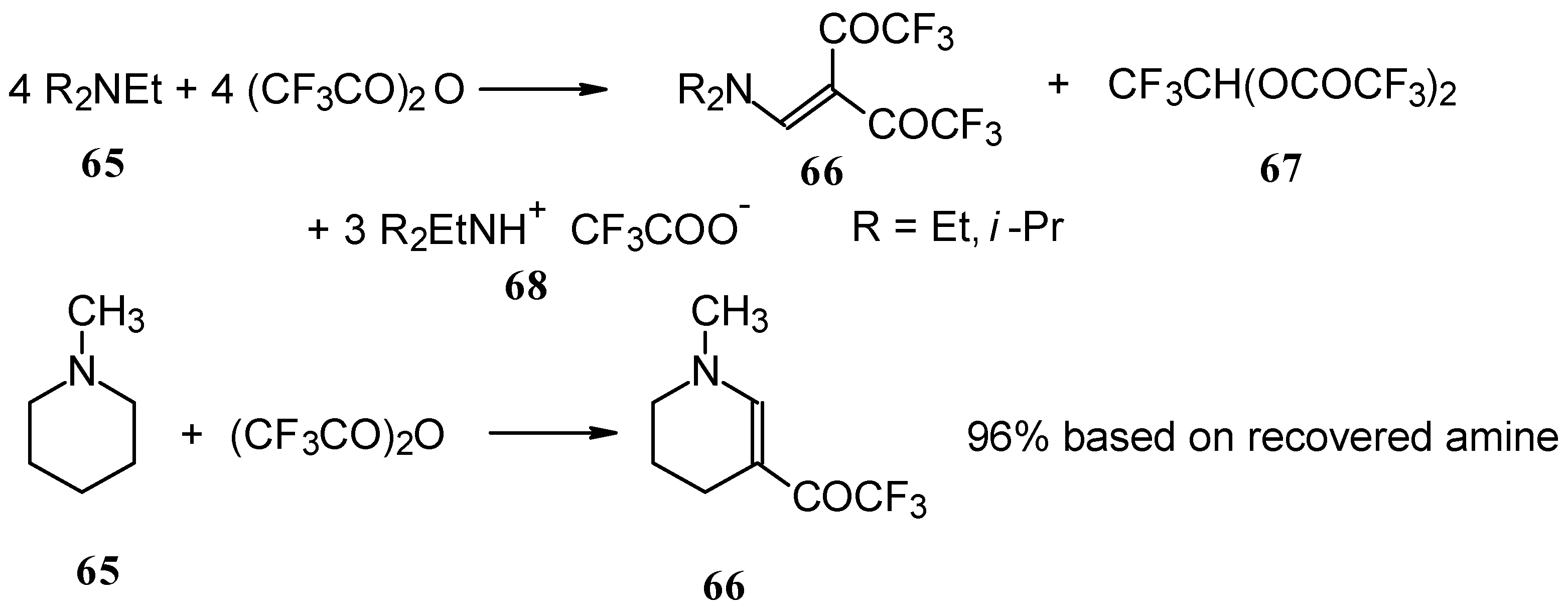
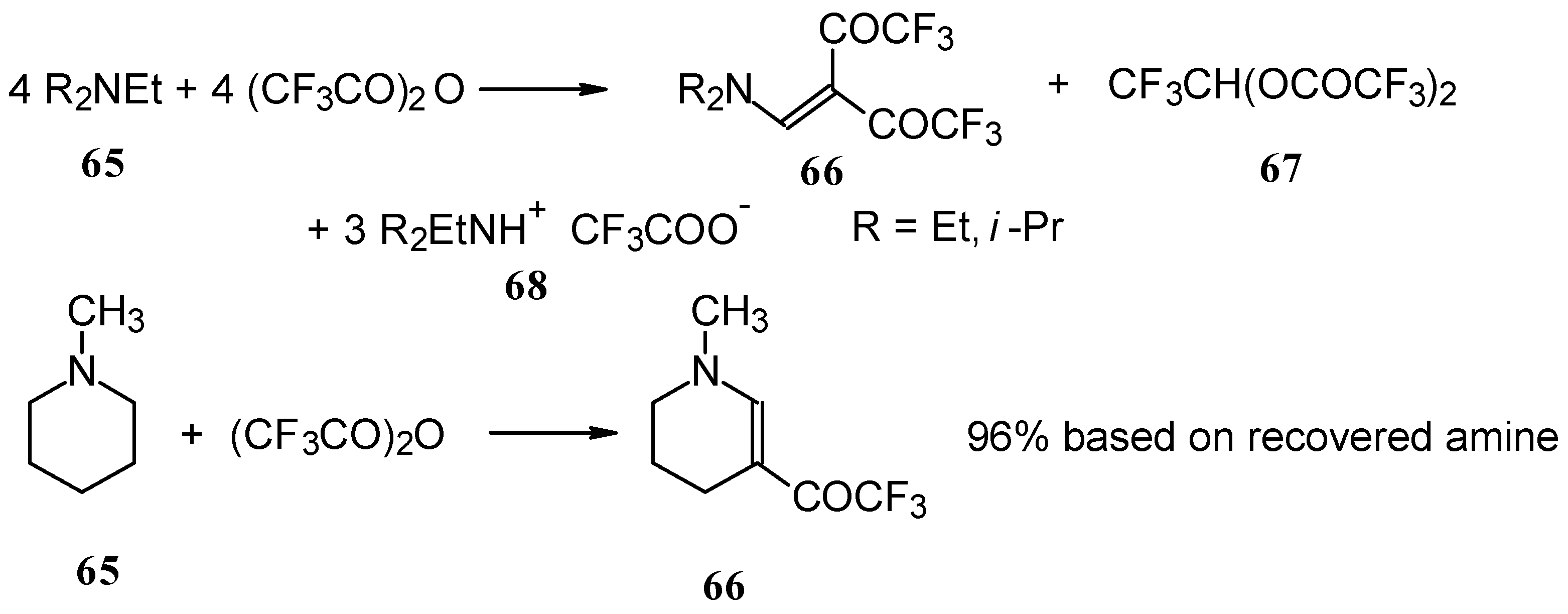
In a subsequent investigation it was found that the reaction of triethylamine with an equimolar amount of trifluoroacetic anhydride proceeds almost quantitatively and leads to the product of double trifluoroacetylation of diethylvinylamine (
Scheme 28) [
74].
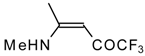
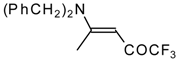
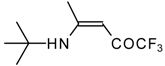
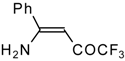
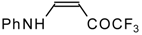
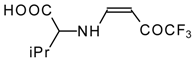
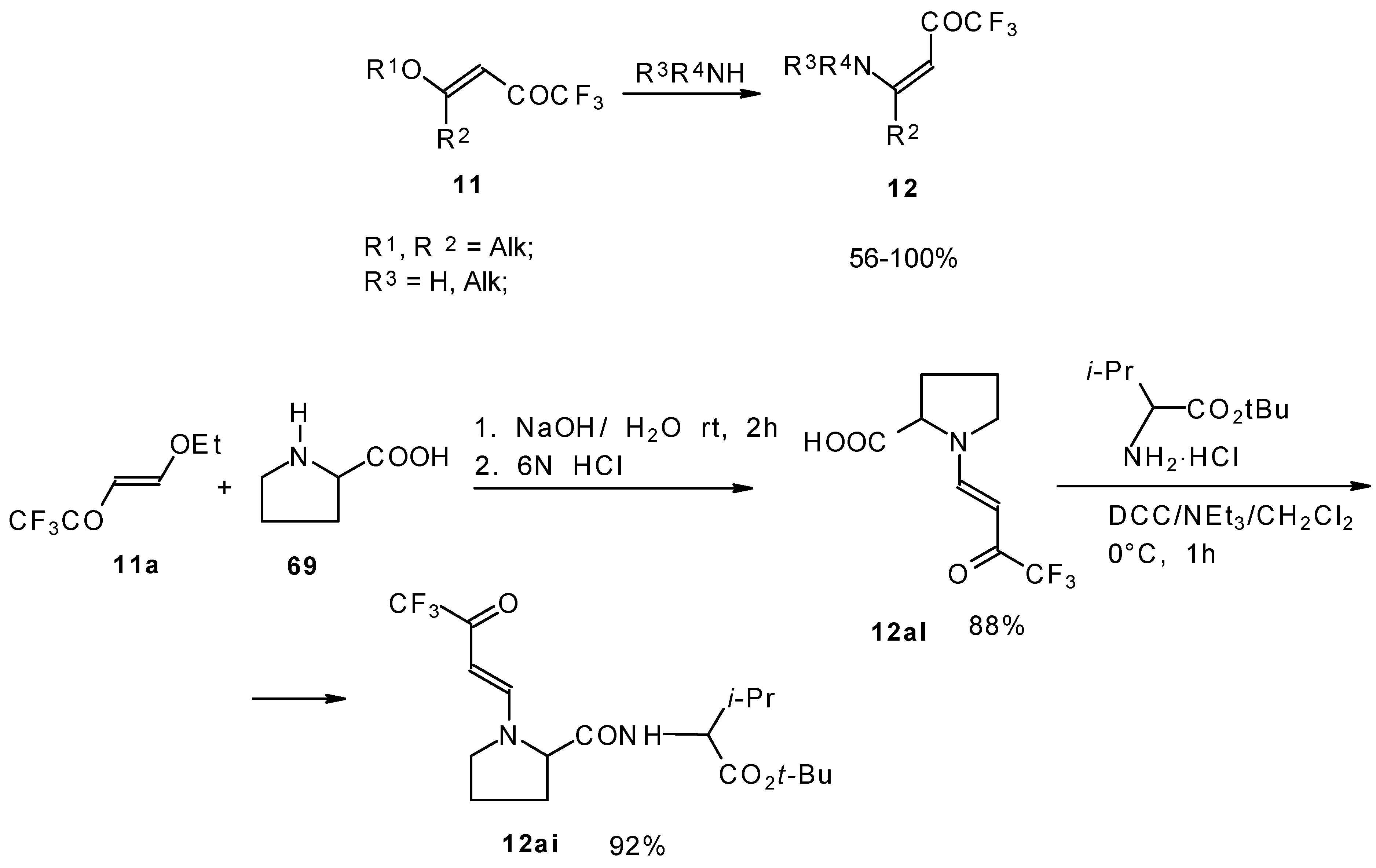
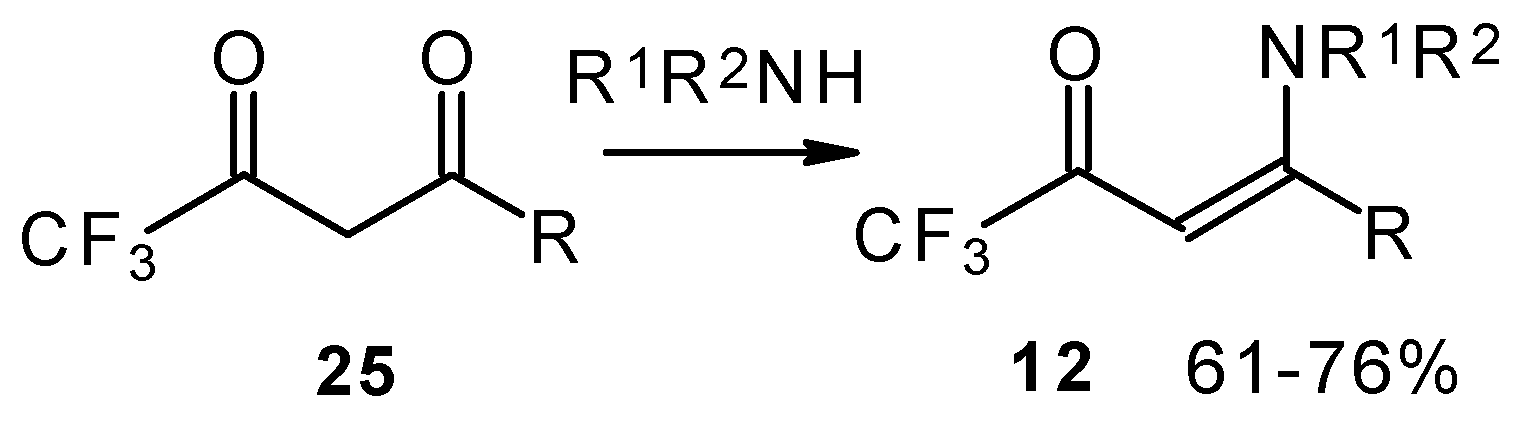
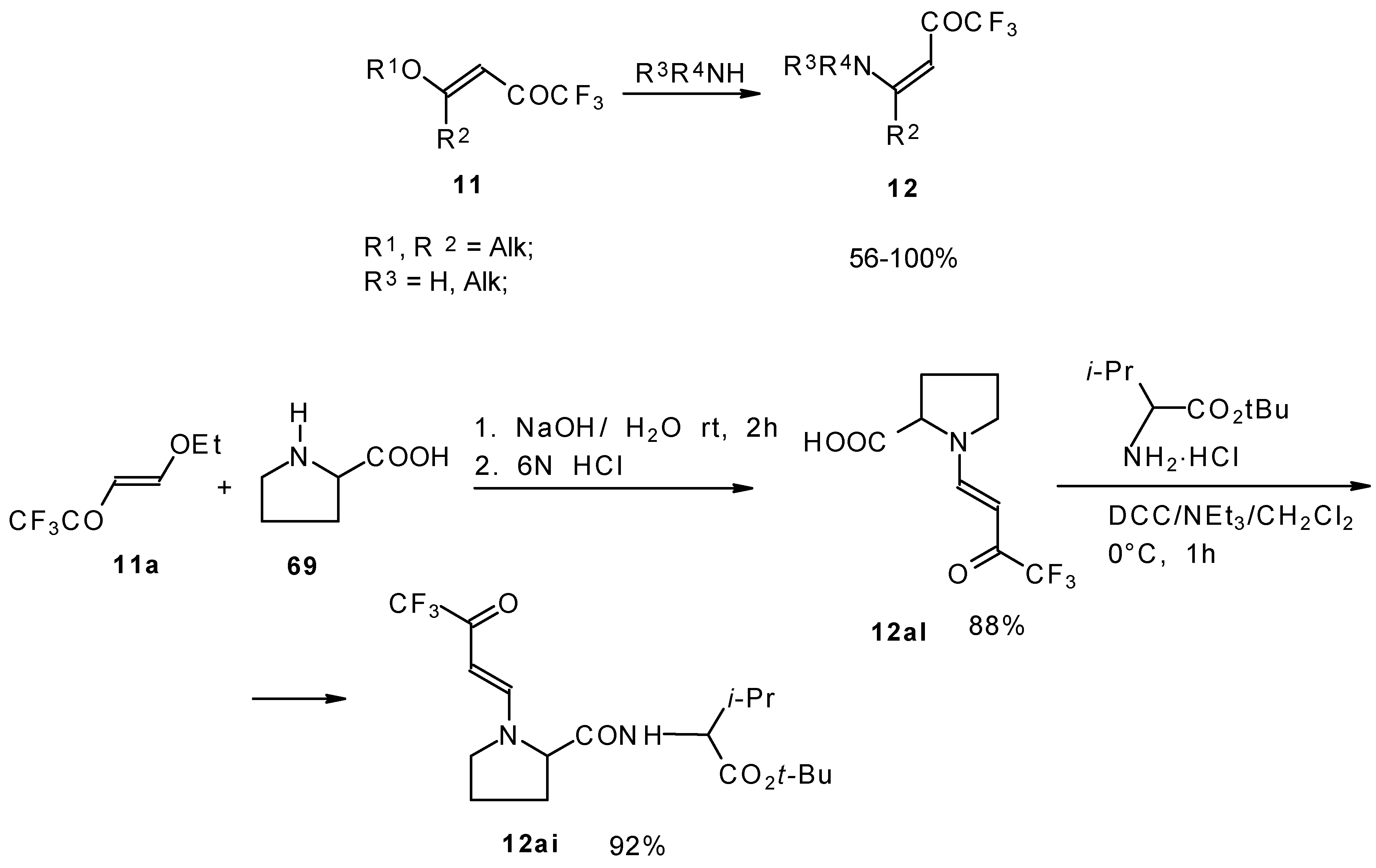
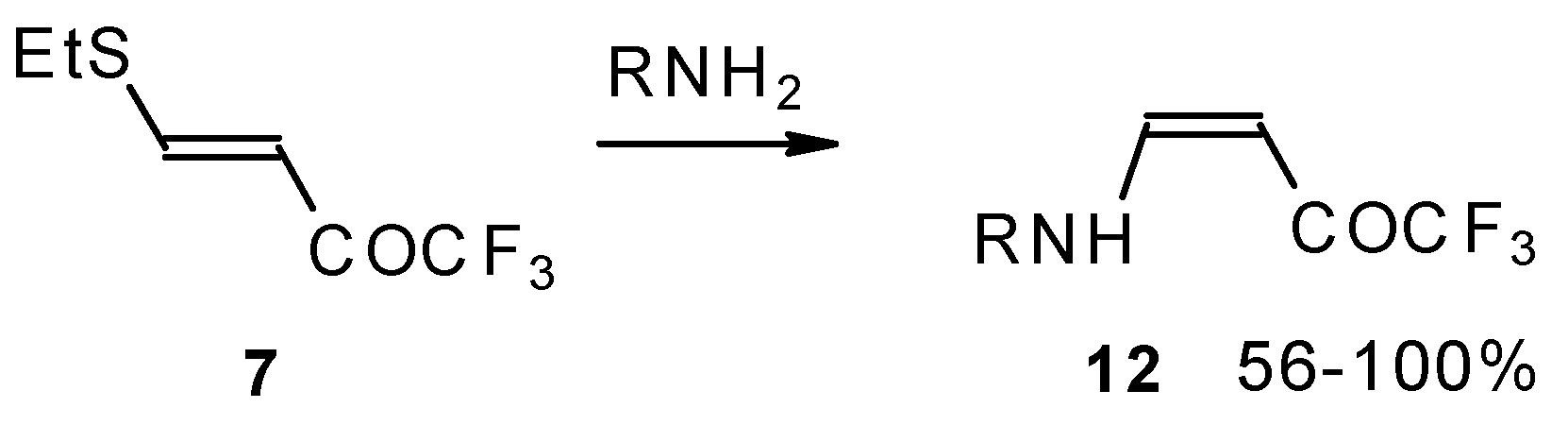
β-Alkyl- or dialkylamino substituted enones bearing a CF
3 group are a very interesting type of unsaturated ketone. These compounds are readily available from the reaction of secondary or primary amines (including aromatic ones) with 4-ethoxy-1,1,1-trifluoro-3-buten-2-one or other trifluoroacylated vinyl ethers. The alkoxy group in these enones is easily substituted by ammonia and mono- or dialkylamines with the formation of β-aminosubstituted CF
3-bearing enones [
75,
76]. 4-Ethoxy-1,1,1-trifluoro-3-buten-2-one can be also used as a protecting reagent in peptide synthesis [
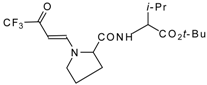
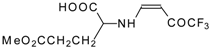
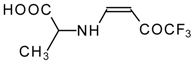
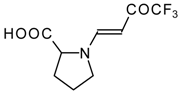
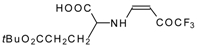
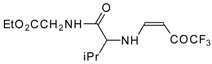
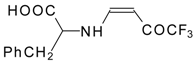
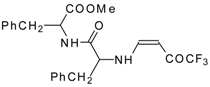
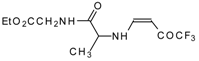
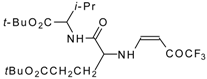
77]. The reaction with amino acids proceeds readily at room temperature to give protected amino acids without racemization. The amino acids protected by this procedure were used for dipeptide preparation (
Scheme 29). The removal of the N-4,4,4-trifluoro-3-oxo-butenyl protecting group occurs under mild conditions in the presence of 3
N HCl.
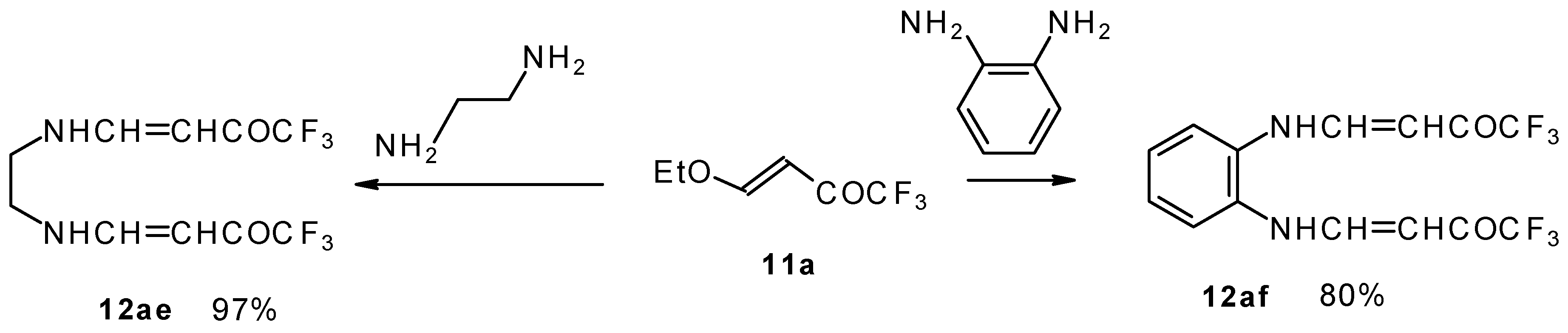
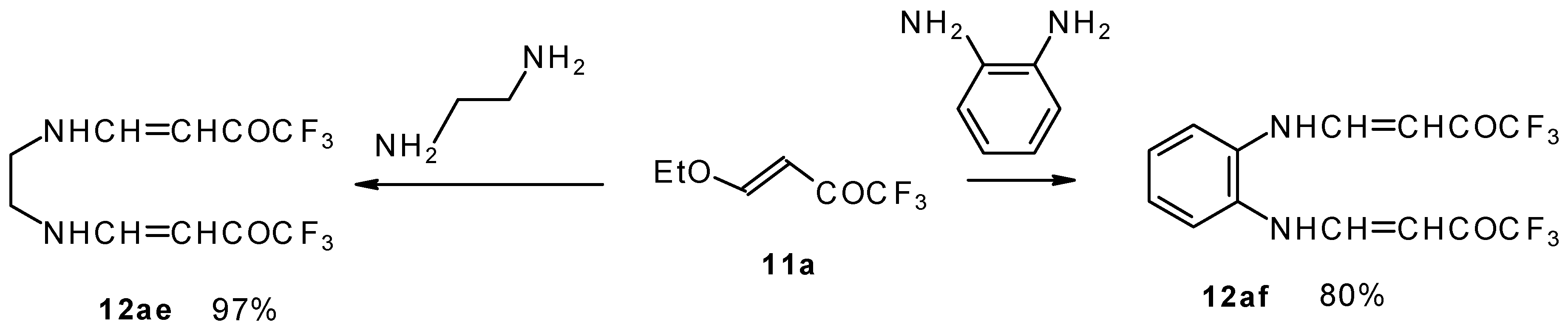
It is also possible to undergo reaction with diamines. The reaction of 4-ethoxy-1,1,1-trifluoro-3-buten-2-one with ethylenediamine and o-phenylenediamine gives rise to the linking of two molecules of a ketone (
Scheme 30) [
75].
The same transformation proceeds readily also with unsaturated trifluoroacetyl ketones having a thioalkyl group in the β-position (
Scheme 31) [
76].
A very interesting possibility is the transaminolisis of β-aminosubstituted ketones. N-N exchange proceeds readily at room temperature without a large excess of the reagent amine. Usually a 1-3 times excess is enough. However the reaction depends on the relative basicities of the leaving and entering amine. As a result the (CH
3)
2N moiety can not be replaced by ammonia, but NH
3 can readily be replaced by dimethylamine (
Scheme 32) [
76].
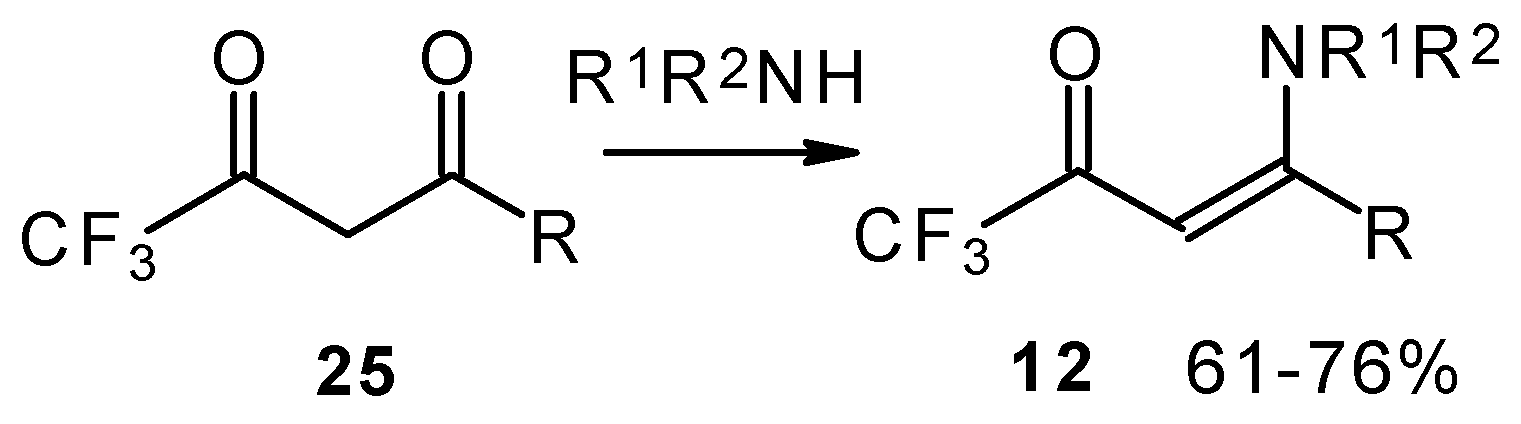
Another approach to the synthesis of β-aminosubstituted CF
3 ketones was elaborated in the reaction of amines with β-diketones having a CF
3 group (
Scheme 33) [
78,
79].
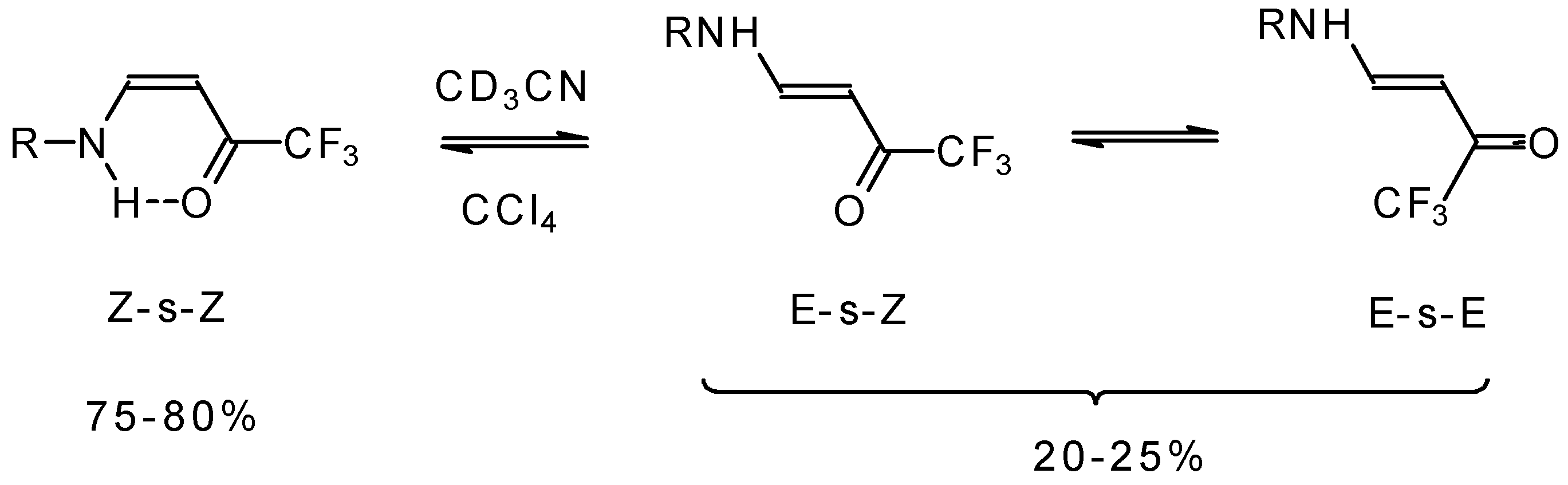
The structure and stereochemistry of these type of enones were investigated by NMR and IR spectroscopy [
75,
76,
80]. It was found that the configuration of the ketone depends on the structure of the amine substituent. In the case of NH
2 and AlkNH or ArNH only the Z configuration of ketones exists in low polar solvents or in the pure form (
Scheme 34). This stereochemistry is due to the possibility of an intramolecular H-bond. More polar solvents such as acetonitrile give rise to transformation to the E-isomer. The
19F NMR spectroscopy shows the presence of an equilibrium among three forms of the ketone. IR spectra give similar data [
75].
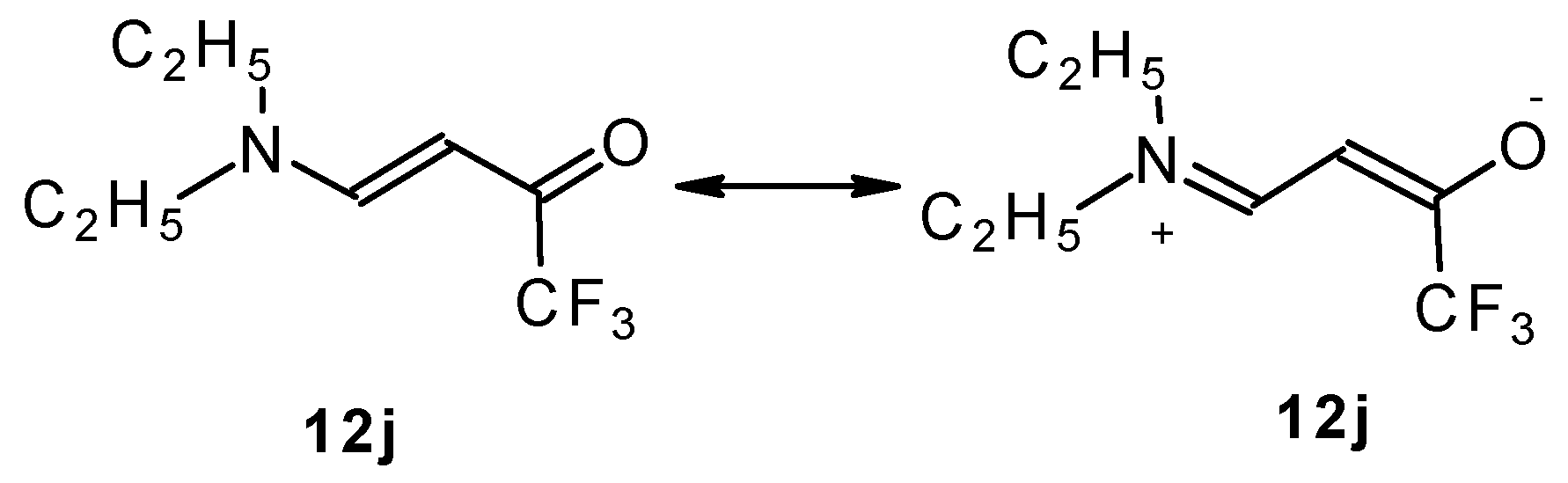
β-Dialkylaminosubstituted ketones as shown by
1H NMR spectra have hindered rotation around the N-C bond due to the interaction of the nitrogen lone pair with the carbonyl group (
Scheme 35).
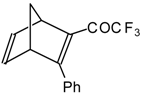
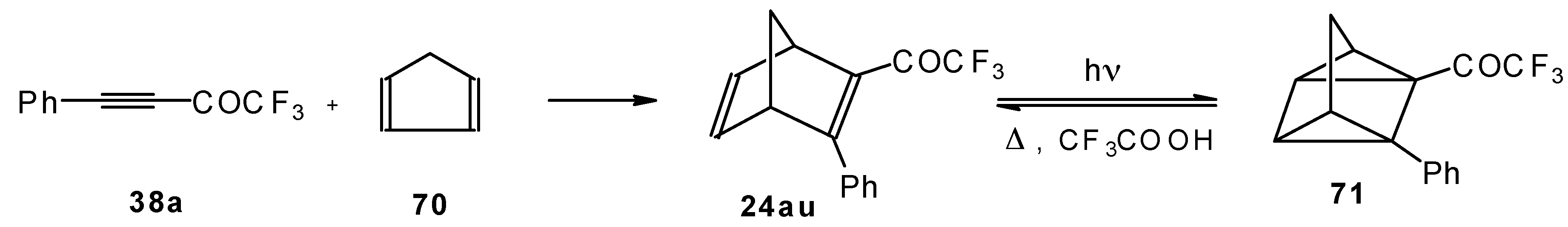
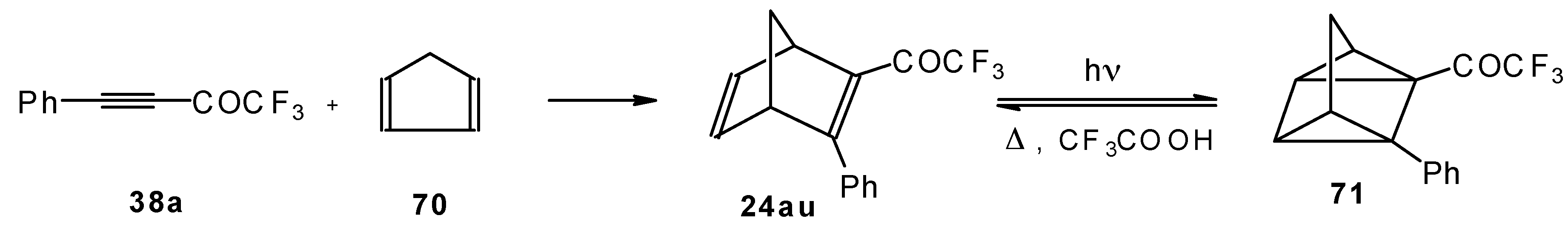
Acetylenic ketones with a CF
3 group are good dienophiles in the Diels-Alder reaction giving with cyclopentadiene the corresponding bicyclic CF
3 enone. Zefirov at al [
81] investigated reversible photoinitiated cycloaddition of this ketone to the quadricyclane skeleton (
Scheme 36).
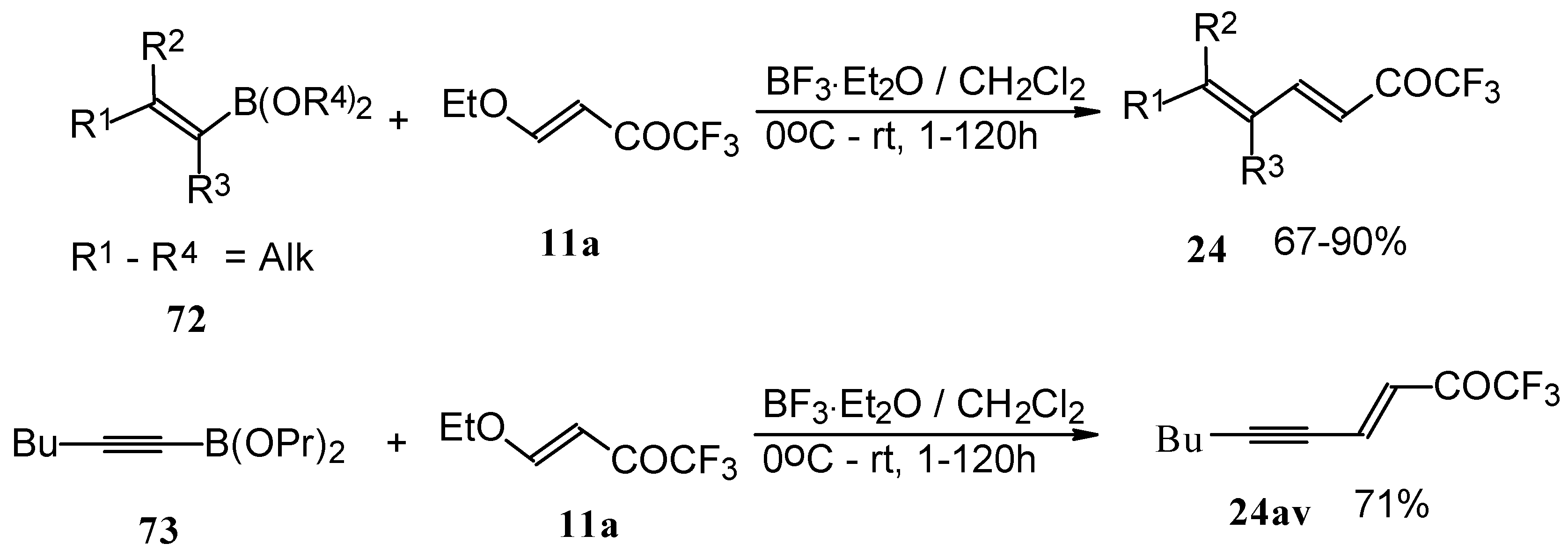
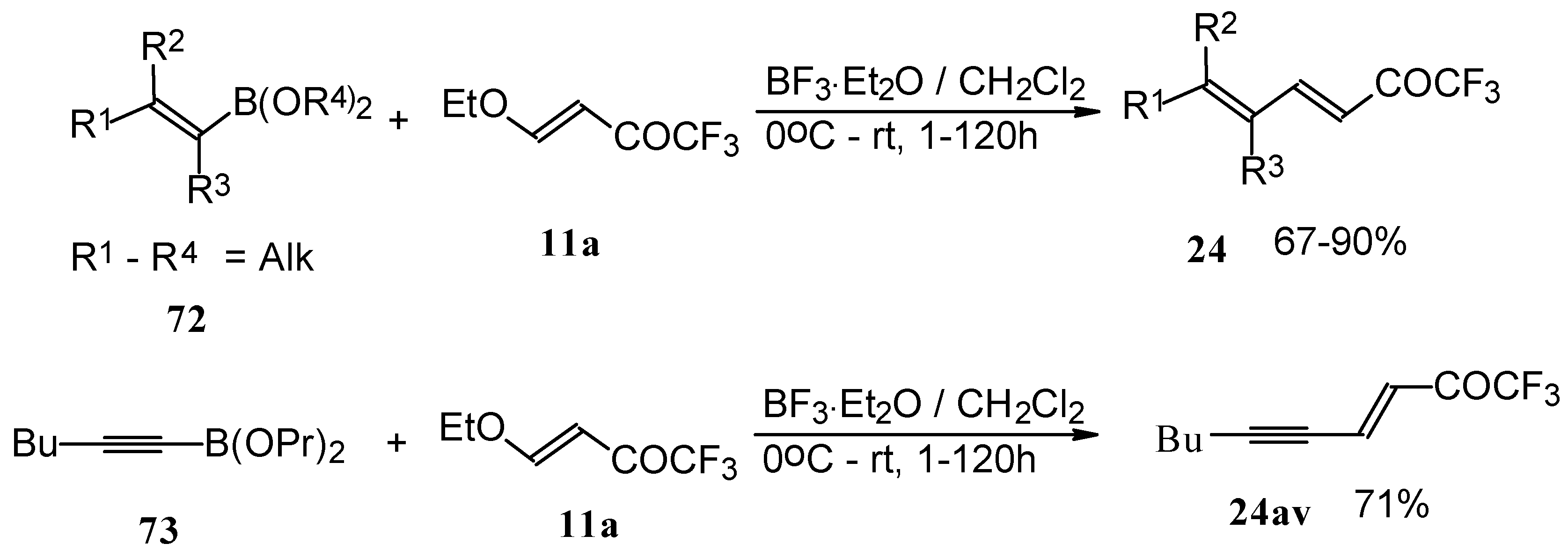
Alkadienyl trifluoromethyl ketones were stereoselectively prepared by the reaction of 4-ethoxy-1,1,1-trifluoro-3-buten-2-one with alkenyldialkoxyboranes in the presence of borontrifluoride etherate (
Scheme 37) [
82].
Similar reactions proceed also with ketones without heteroatom substituents to give 3-alkynyl substituted (
Scheme 38) or γ,δ−unsaturated CF
3 ketones (
Scheme 39) [
83,
84].
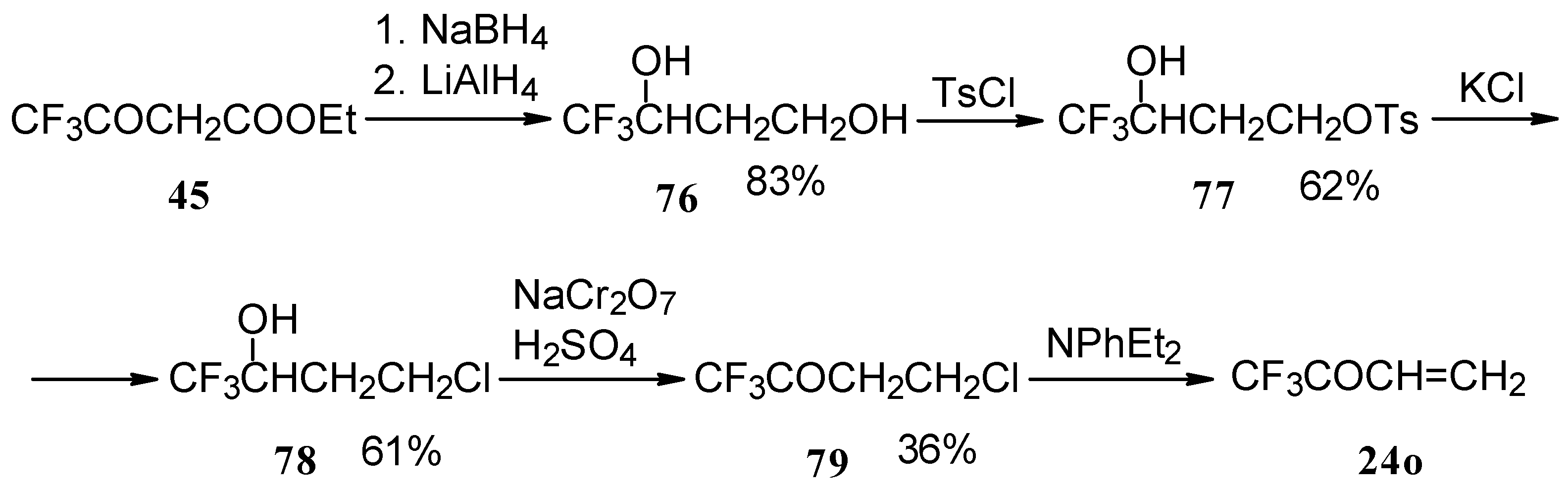
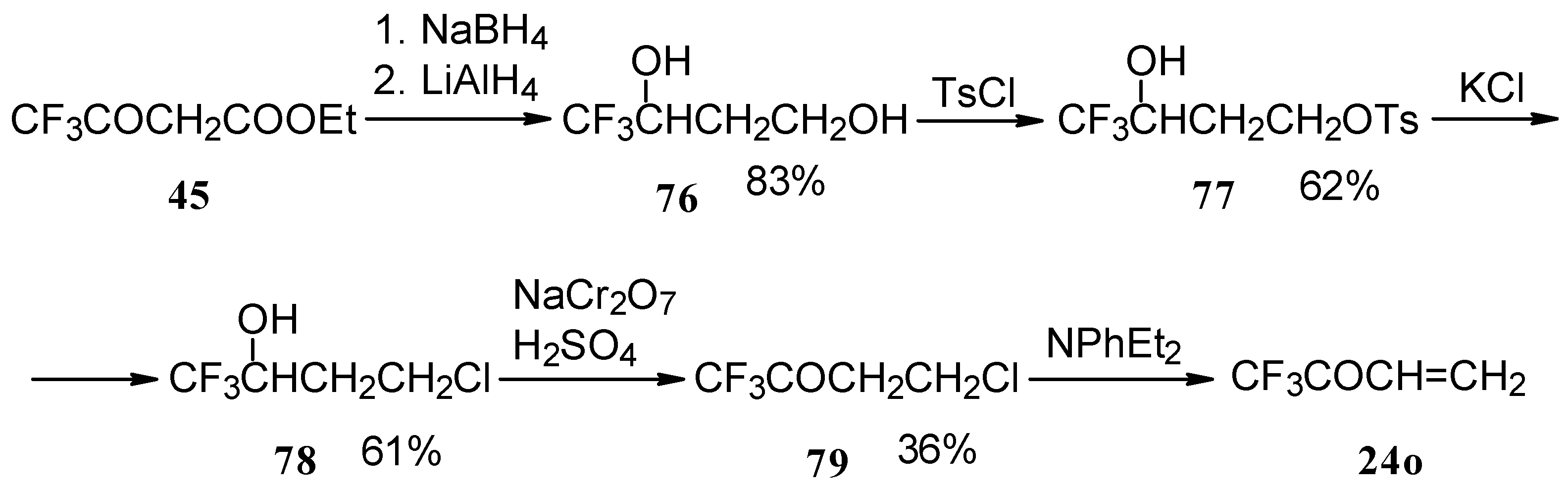
The parent CF
3 bearing enone - trifluoromethyl vinyl ketone
24o is not accessible with the procedures described above. This ketone has been prepared by a five stage procedure from trifluoracetoacetate (
Scheme 40) [
85].
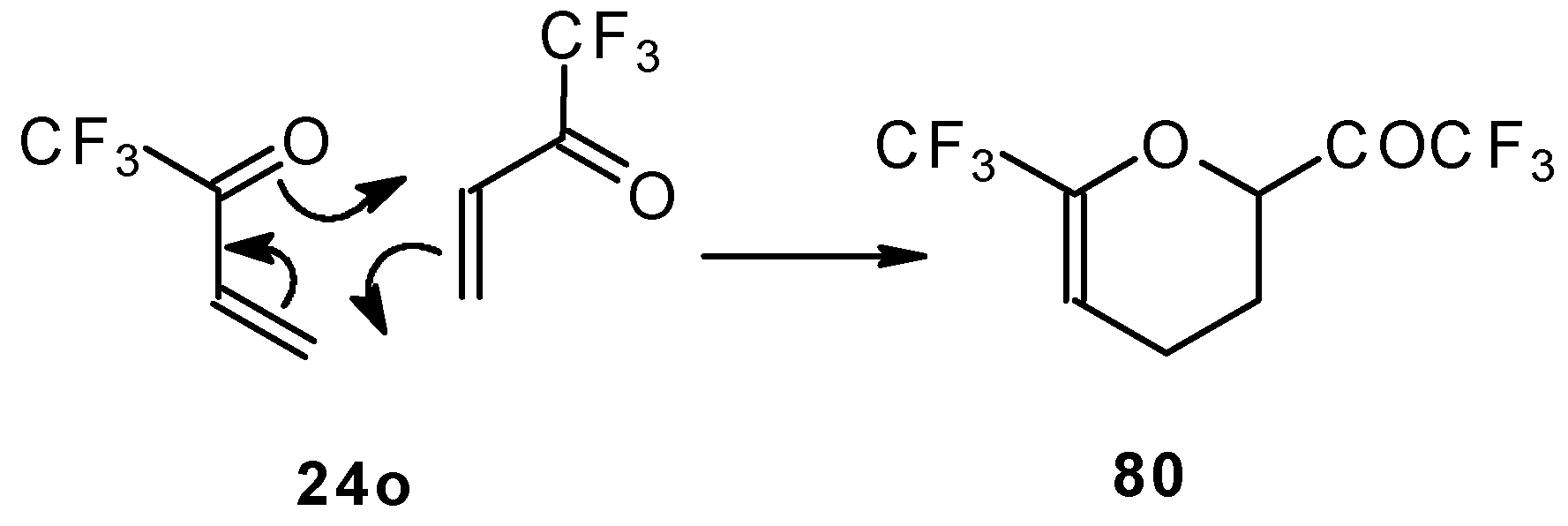
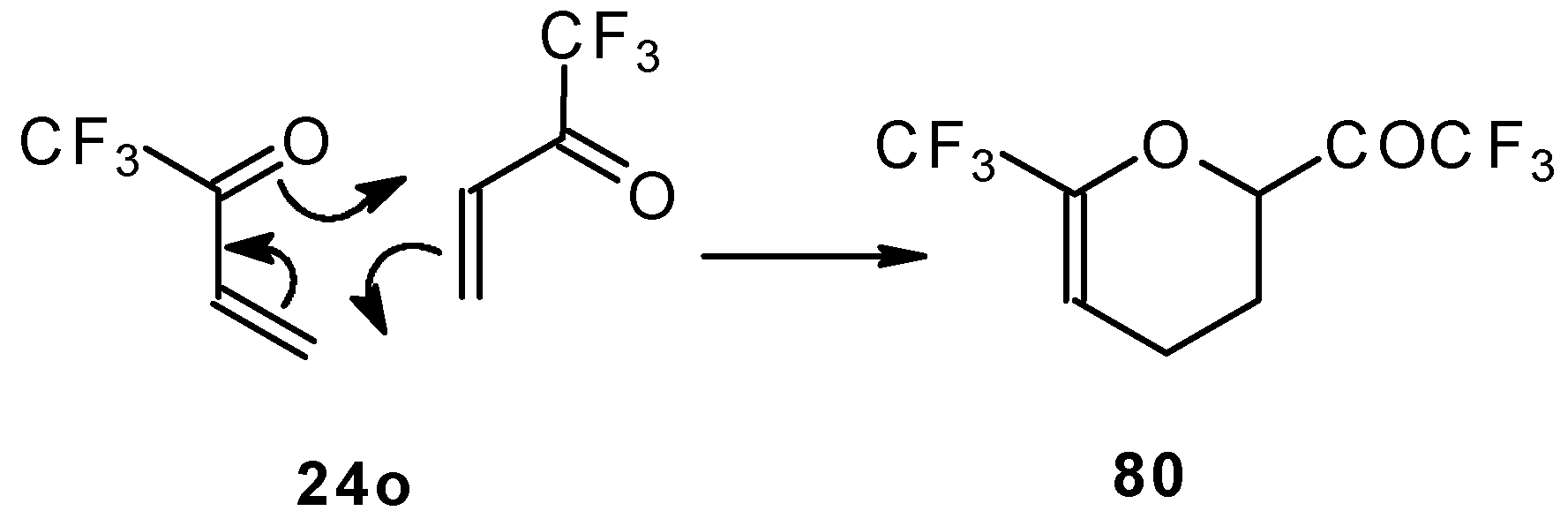
This ketone is spontaneously dimerized to the corresponding pyran (
Scheme 41) [
85].
Table 1.
Synthesis of α,β−unsaturated trifluoromethyl ketones.
Synthetic application of α,β-unsaturated ketones bearing a trifluoromethyl group
It seems to be very fruitful to use unsaturated ketones with a trifluoromethyl group for the synthesis of trifluoromethyl-containing heterocycles. But the use of these enones in synthetic organic chemistry can be considered as unexploited. The first application of unsaturated ketones with CF
3 for the preparation of the corresponding heterocycles was dated 1959 [
44]. However, really extensive utilization of trifluoroacetyl ketones in organic synthesis was started only quite recently.
There are two general approaches: 1) syntheses based on acetylenic CF
3 ketones or enones having an heteroalkyl substituent in the β−position to the CF
3CO group, for example, trifluoroacylated vinyl ethers; 2) syntheses based on ketones without a heteroatom substituent. These two approaches differ in oxidation states of both reagents and the resulting products. It is obvious that the first way gives rise to aromatic heterocycles and the second one leads to the corresponding hydrogenated heterocycles. Investigation of the latter approach was started only quite recently. The first publication is dated 1994 [
50,
114]. Possibly this is due to the fact that the ketones without a heteroatom substituent in the β-position to CF
3CO group are less available and their preparation is more complex.
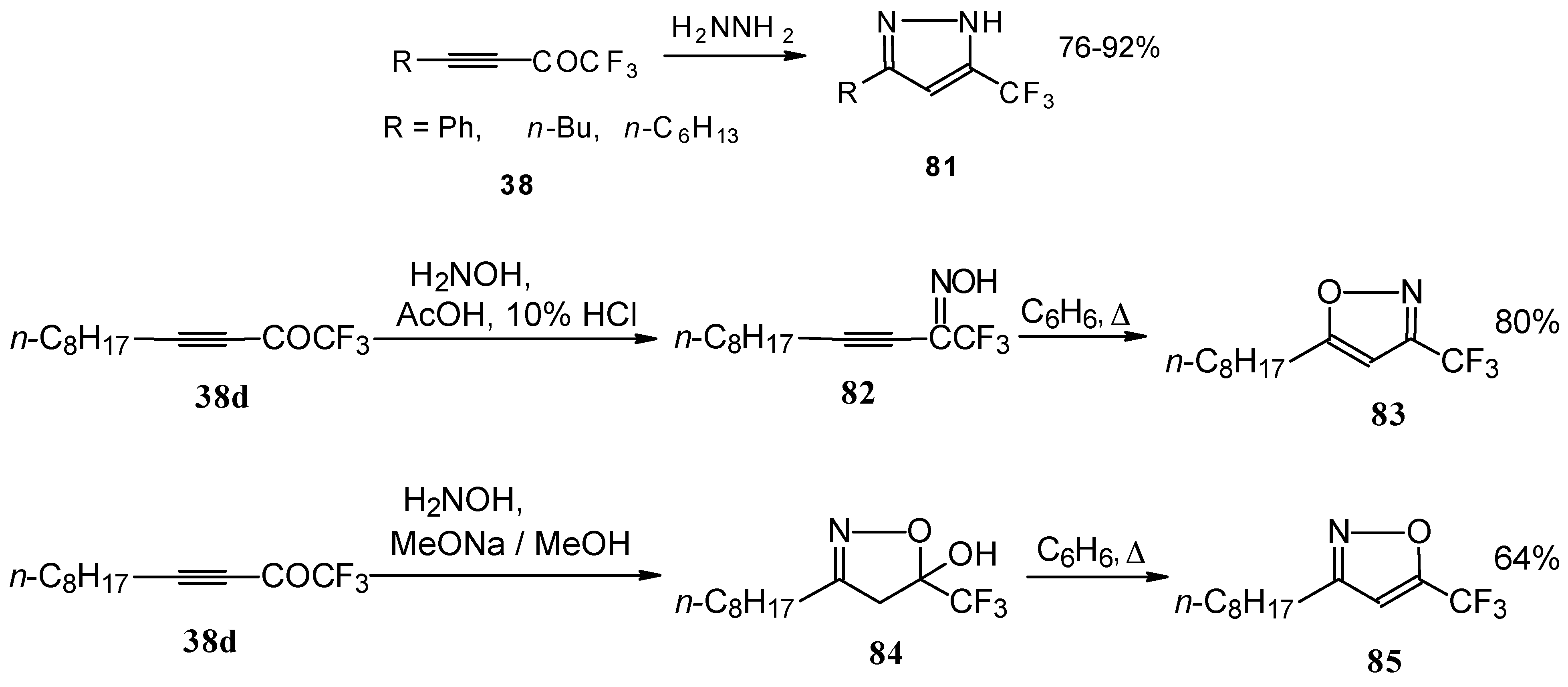
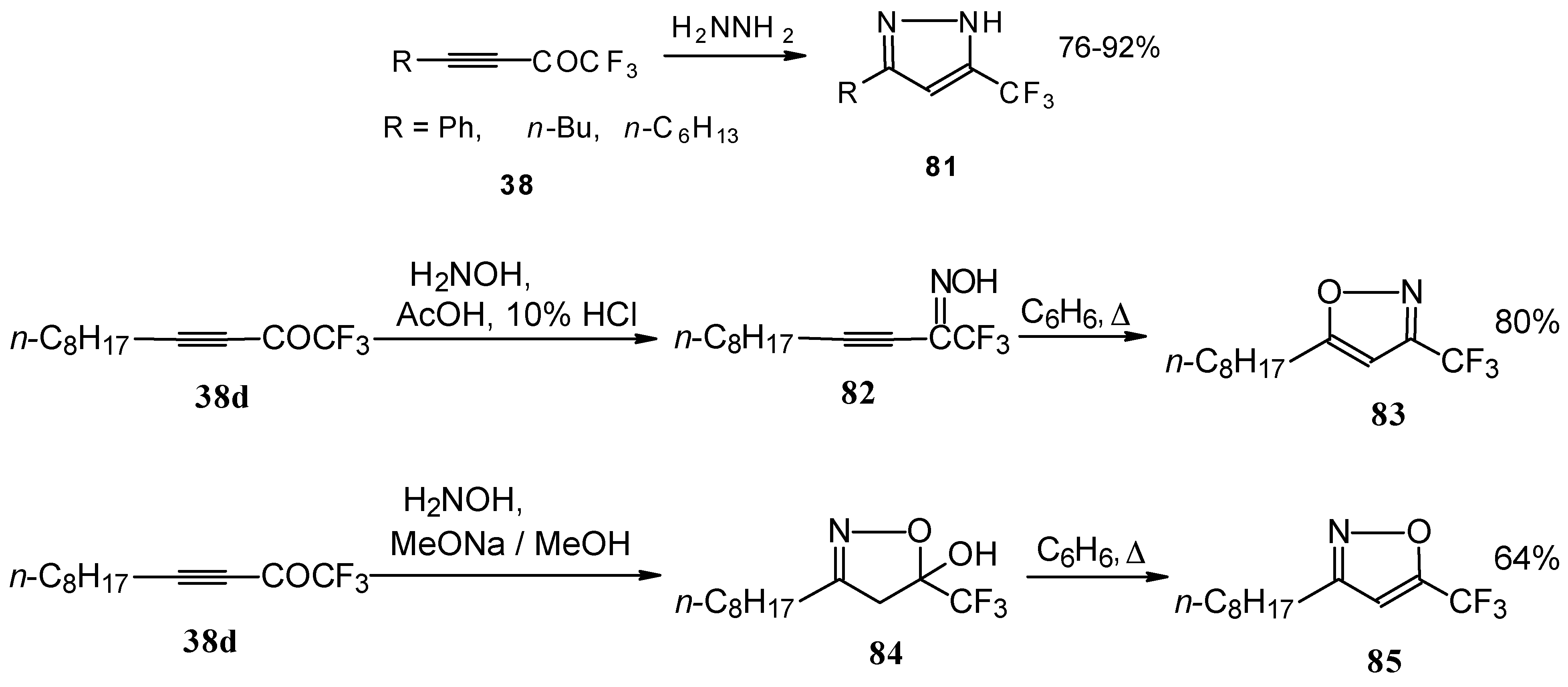
Syntheses of trifluoromethyl-containing heterocycles based on the reactions of acetylenic ketones with some bifunctional nucleophiles were described by Linderman [
86,
87]. The reaction with hydrazine results in the formation of the corresponding pyrazoles (
Scheme 42). In the case of reactions with hydroxylamine under basic or acidic conditions isomeric, the 3-CF
3 or 5-CF
3 isoxazoles
83 and
85 were formed [
86]. It is very interesting that in the case of the reaction of acetylenic ketones with hydroxylamine under basic conditions (MeONa / MeOH) the corresponding
4,5-dihydro-5-isoxazolol was isolated (
Scheme 42). Subsequent elimination of water by reflux in benzene gave rise to the corresponding aromatic CF
3 substituted heterocycle.
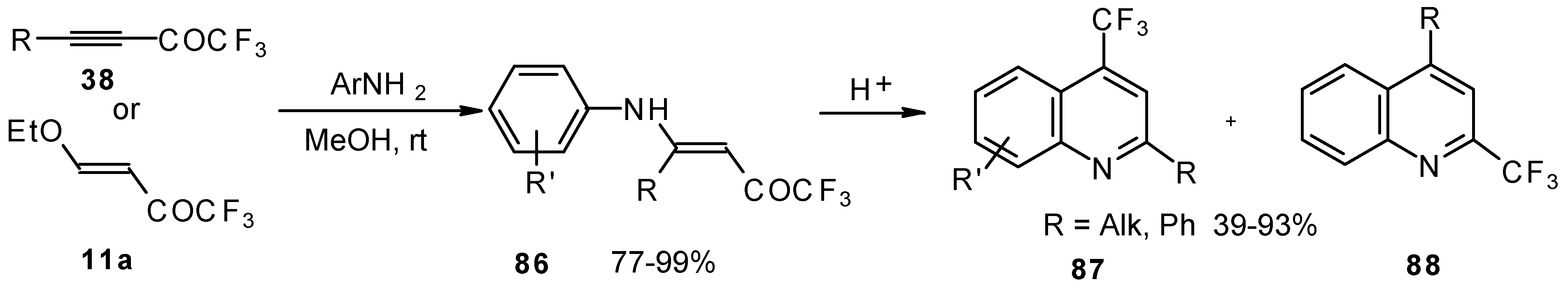
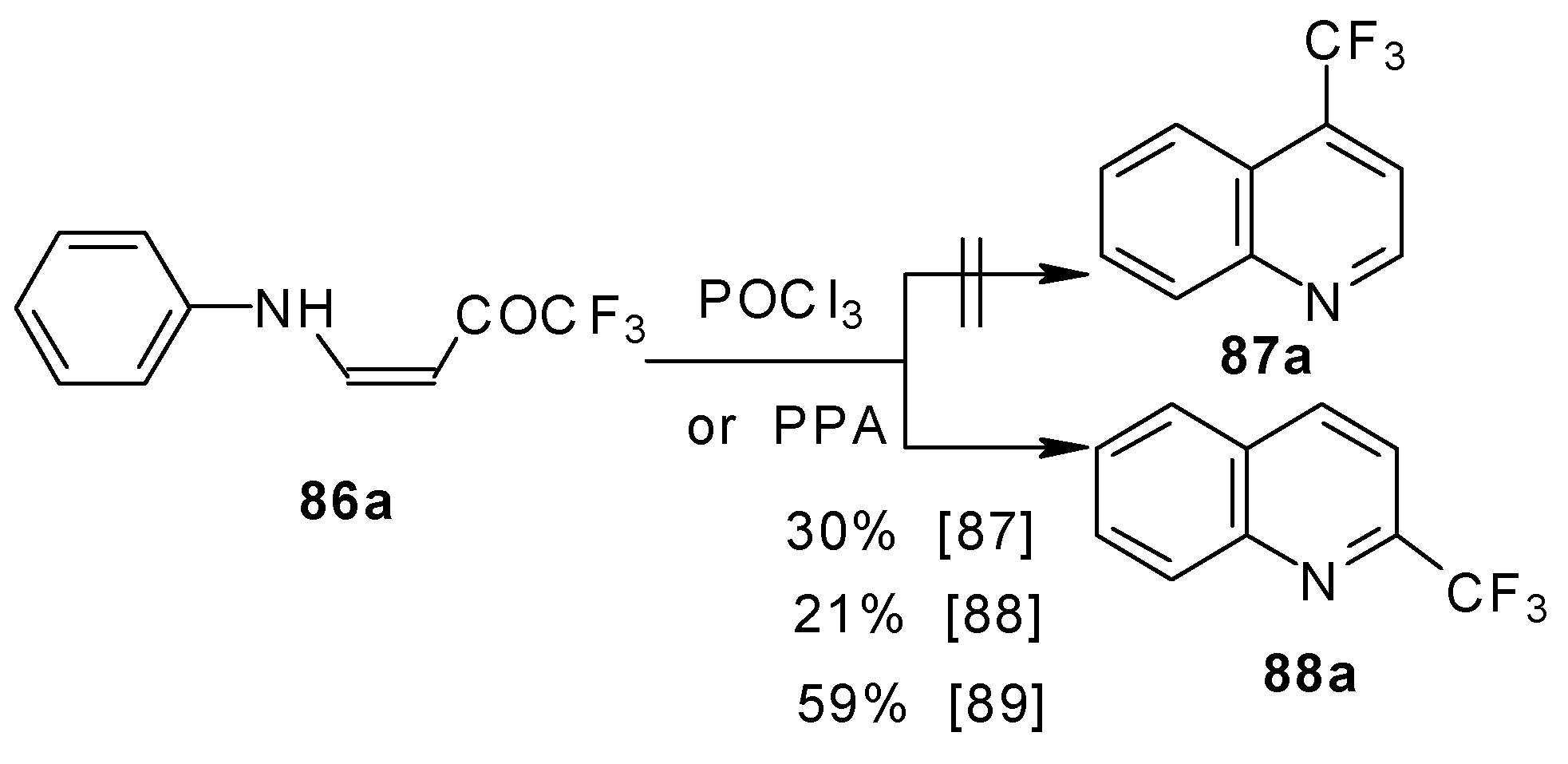
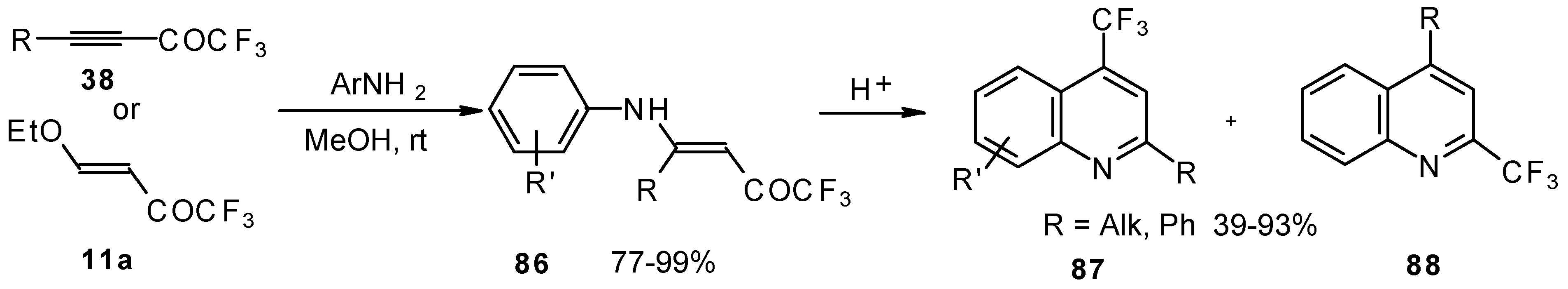
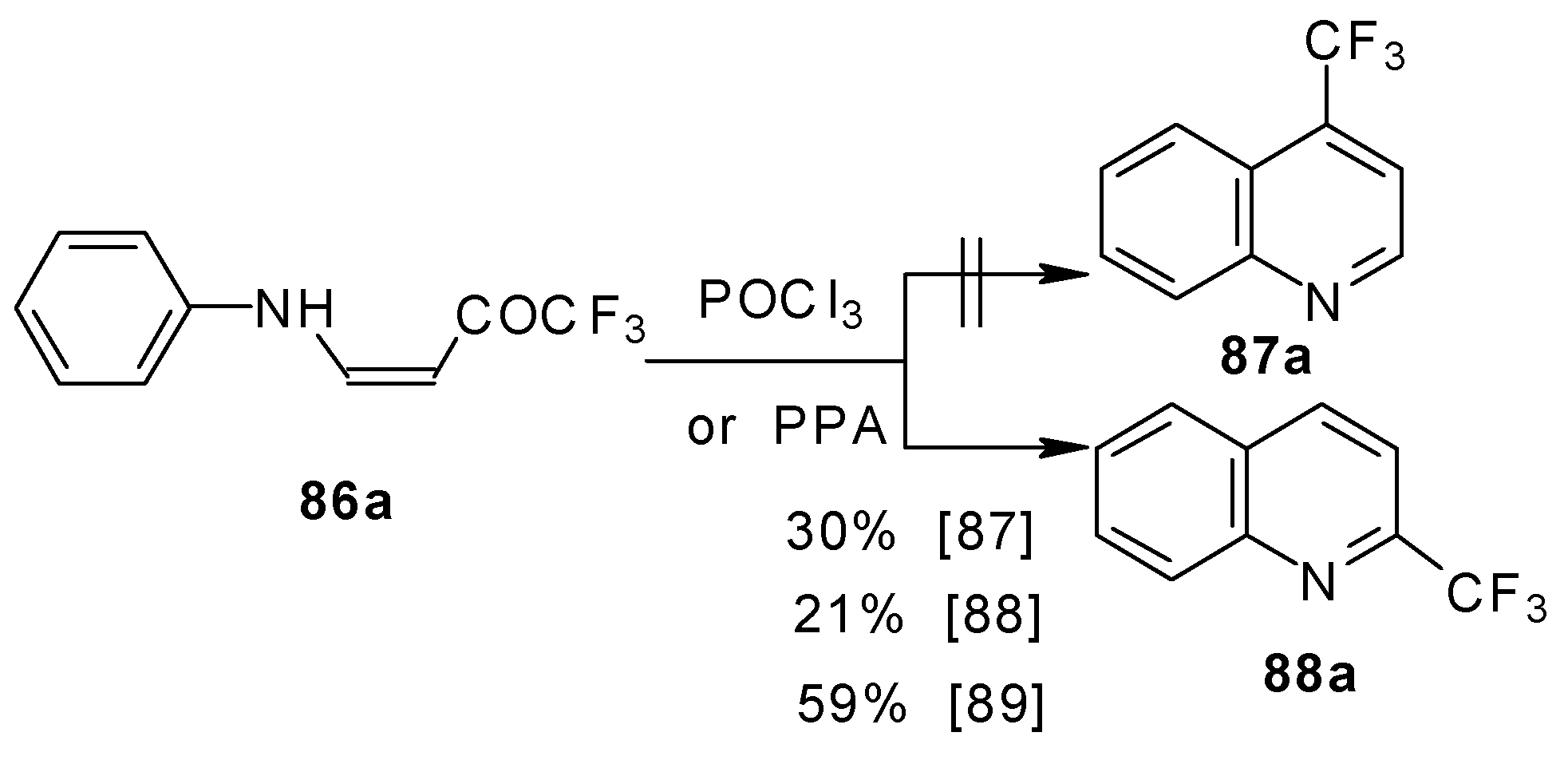
The cyclization of adducts of acetylenic ketones
38 (or 4-ethoxy-1,1,1-trifluoro-3-buten-2-one
11a) with aromatic amines under acidic conditions leads to 2-CF
3 or 4-CF
3 substituted quinolines
87 and
88 in moderate yields (
Scheme 43) [
87,
88,
89].
It should be noted that in the case of the aniline derivative only the 2-CF
3 substituted quinoline was obtained probably
via retro-1,4 addition (
Scheme 44) [
88,
89].
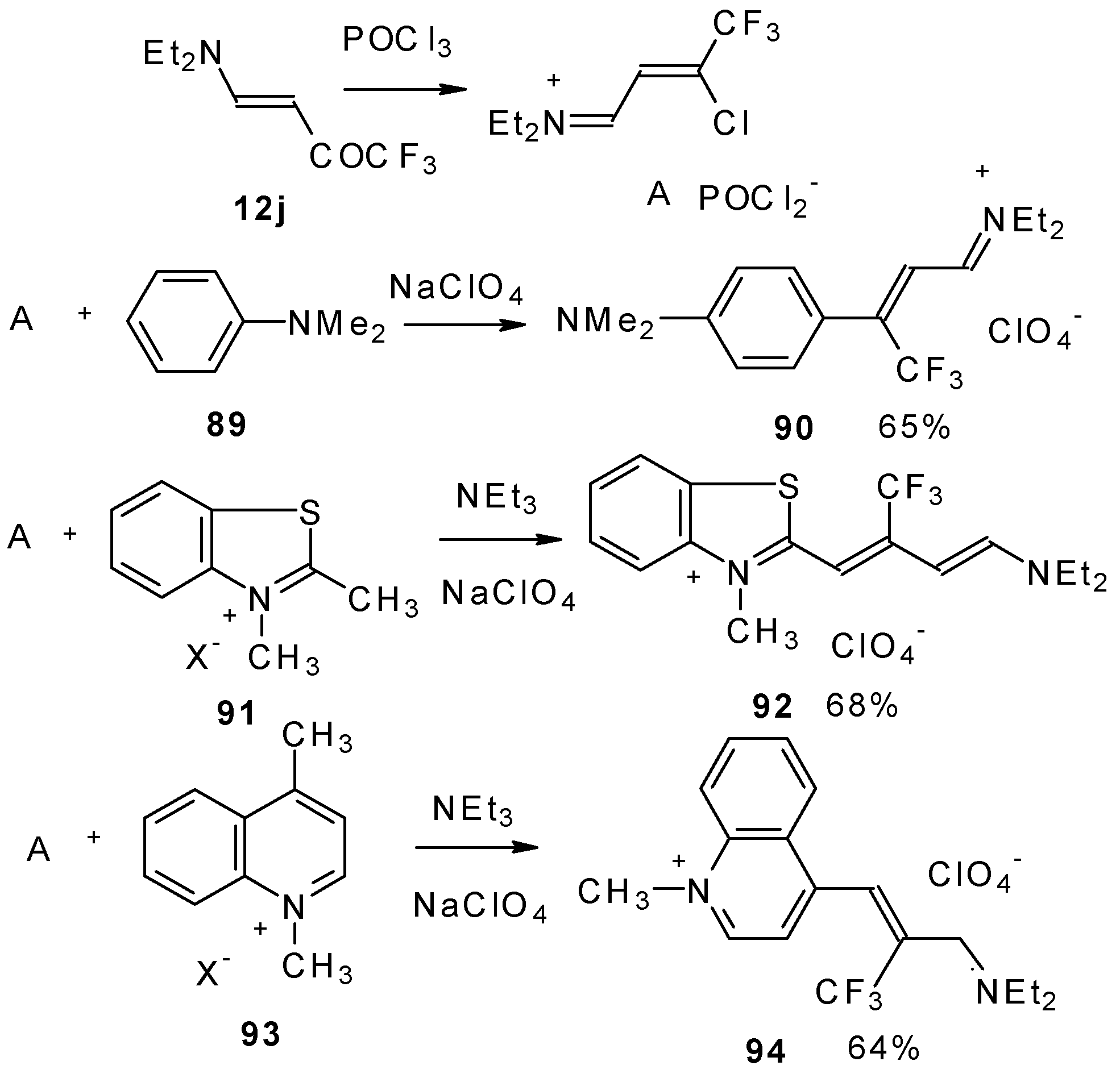
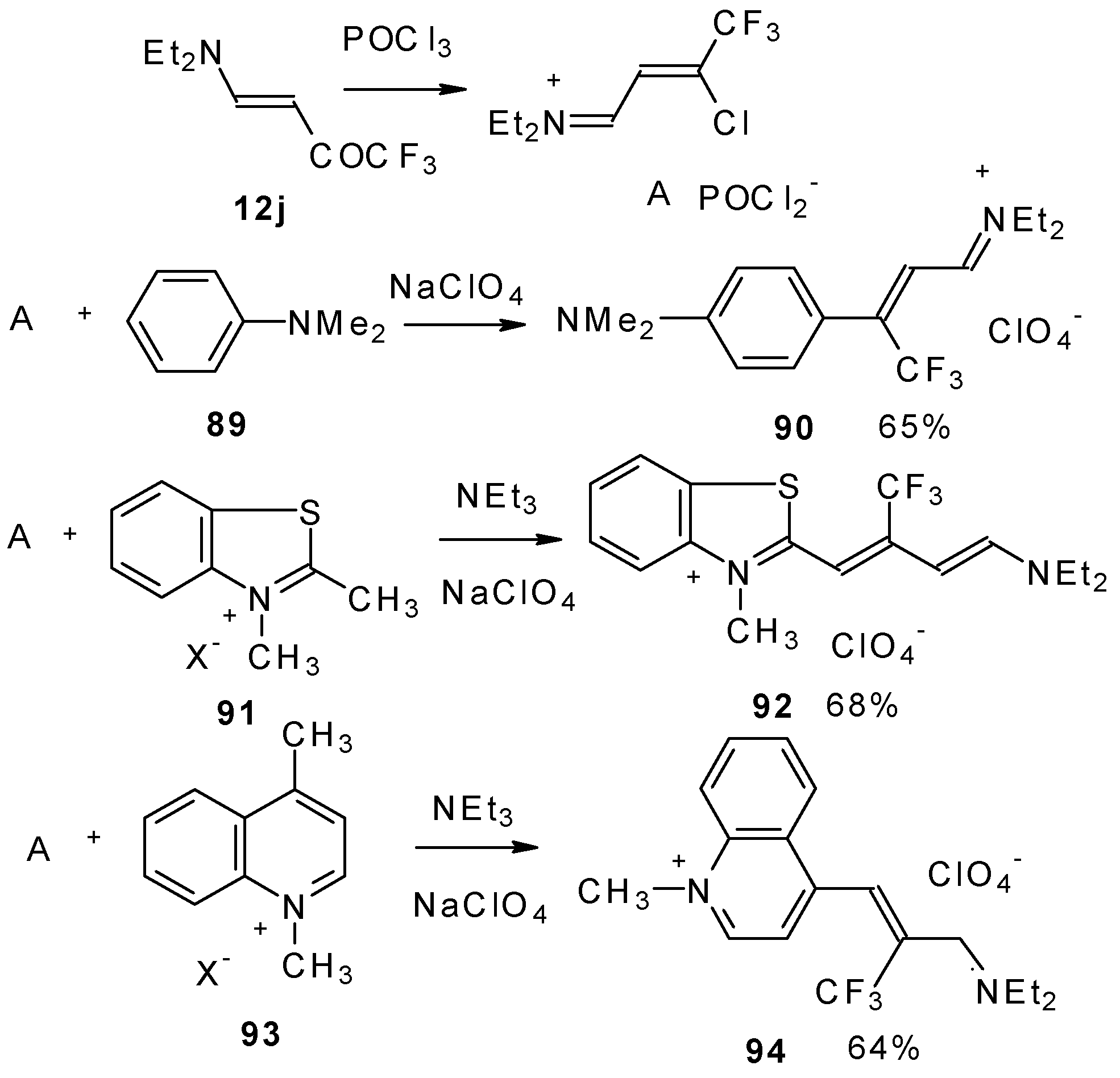
β-Dialkylamino substituted CF
3 ketones react with phosphorous oxychloride to give Vilsmeier-like iminium salts (
Scheme 45) [
90]. These salts are less reactive electrophiles than the DMFA-POCl
3 complex. They react with dimethylaniline and some active methylene bases to give hemicyanines which are valuable for the synthesis of cyanine dyes [
90].
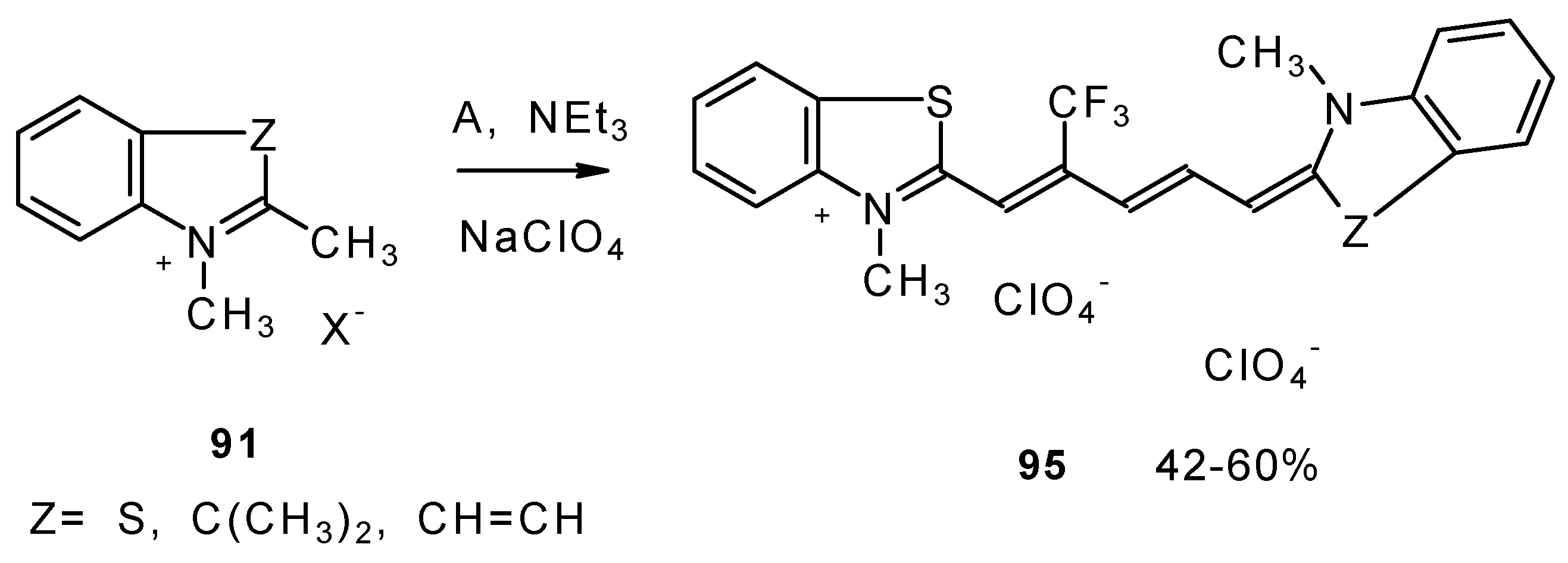
Under more rigorous conditions complex A reacts with two molecules of an ammonium salt to give β-trifluoromethyldicarbocyanines (
Scheme 46).
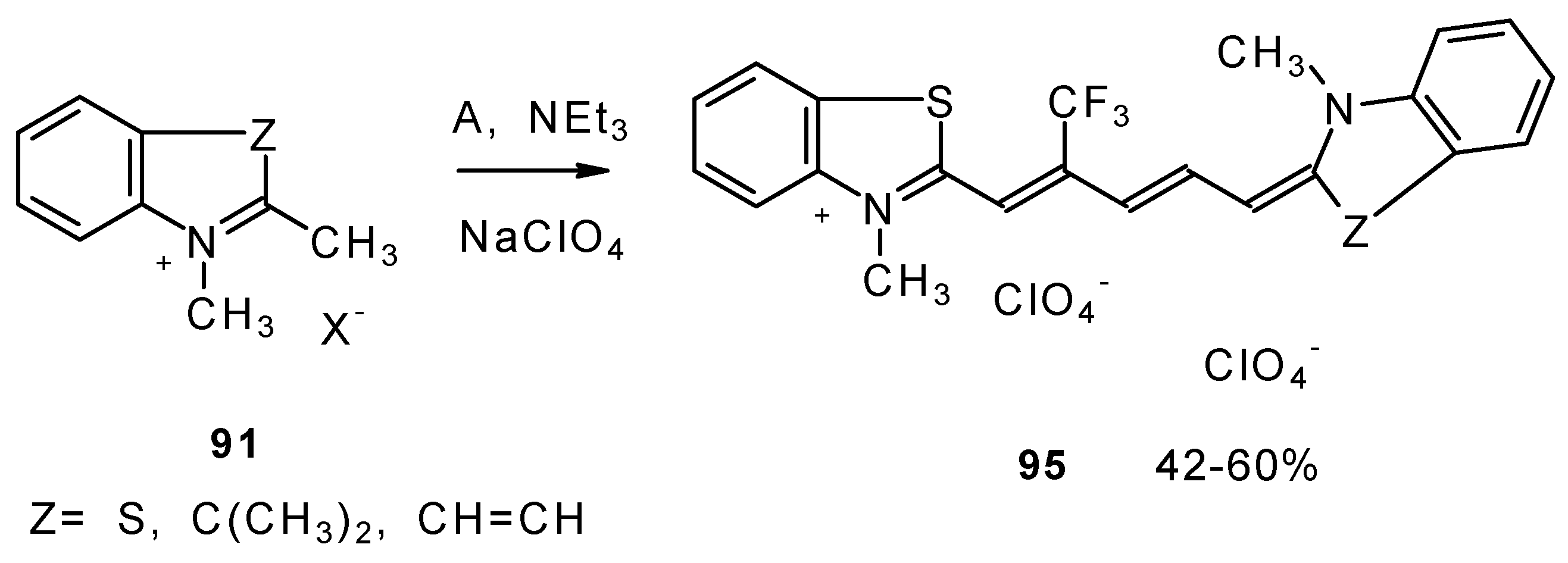
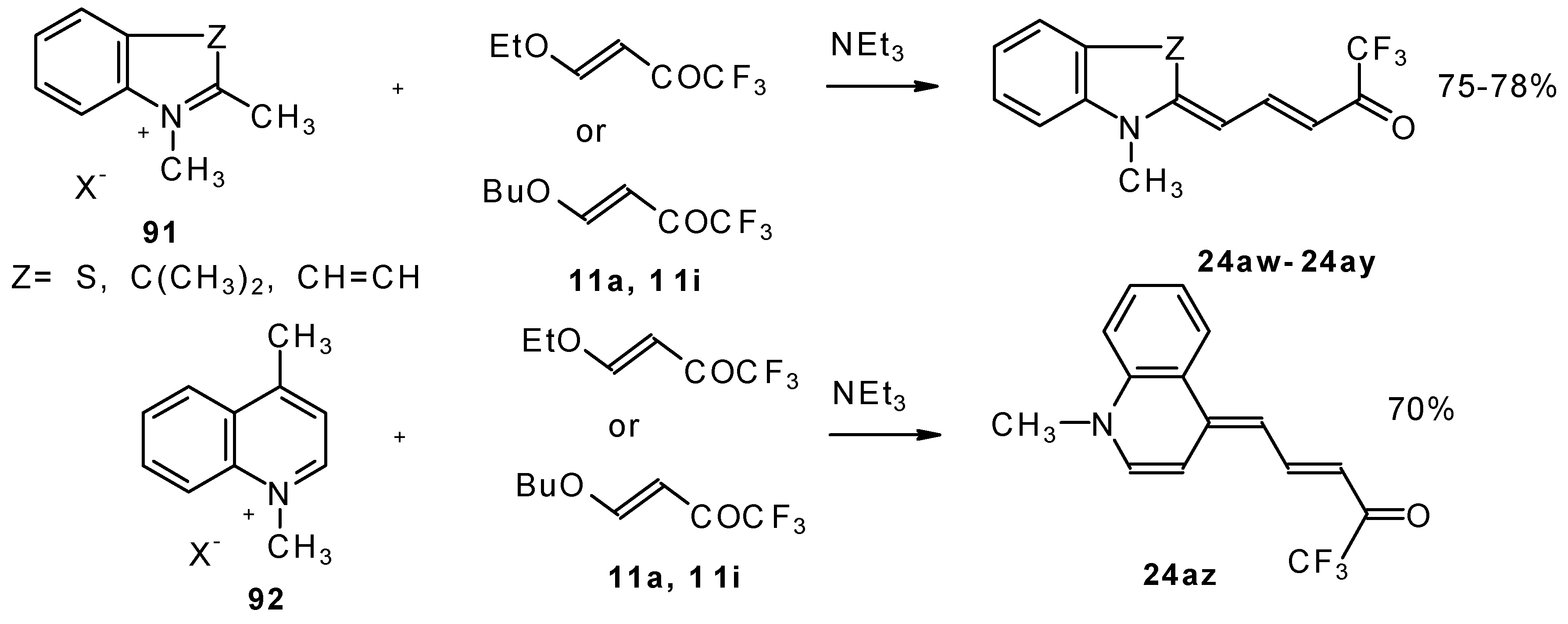
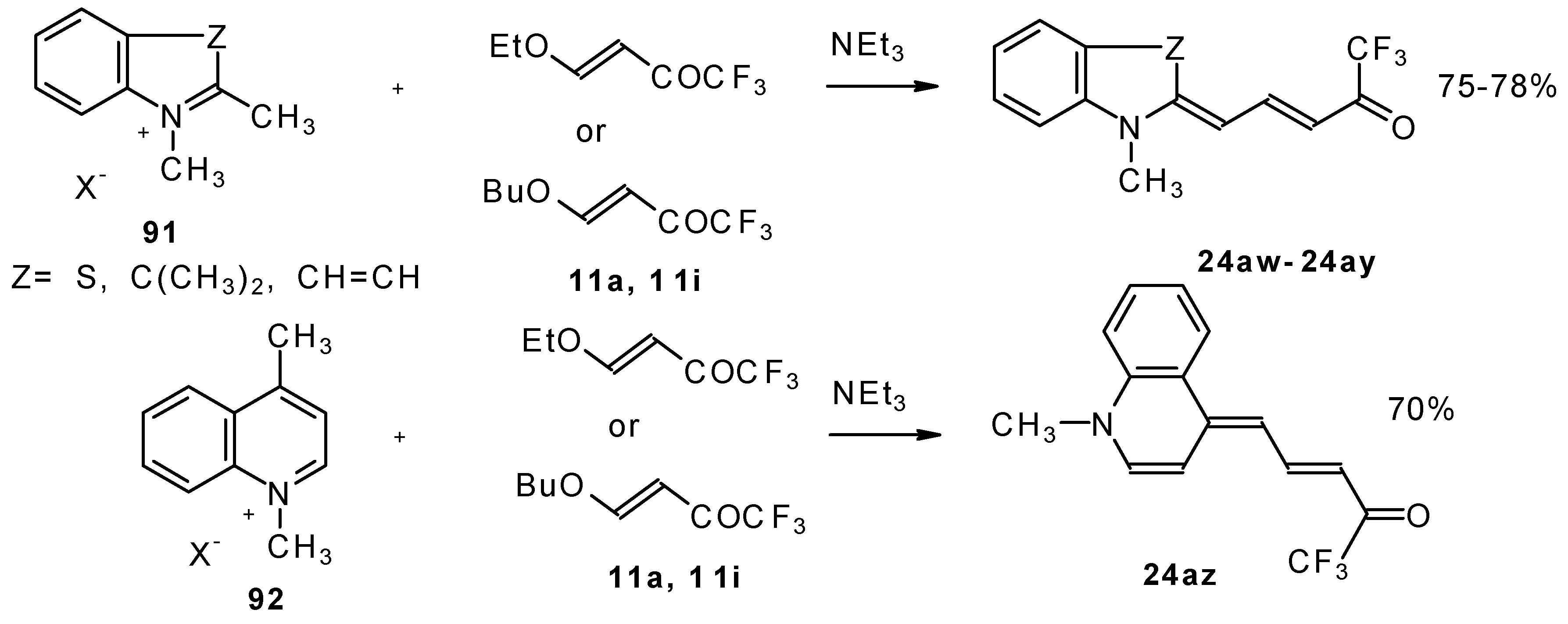
β-Alkoxy substituted CF
3 ketones
11a and
11i also react readily with heterocyclic ammonium salts to give δ-trifluoromethylbutadienylmerocyanines in high yield (
Scheme 47).
Trifluoromethyl containing cyanine dyes had not been described previously. The same authors also investigated spectroscopic data of these dyes [
90].
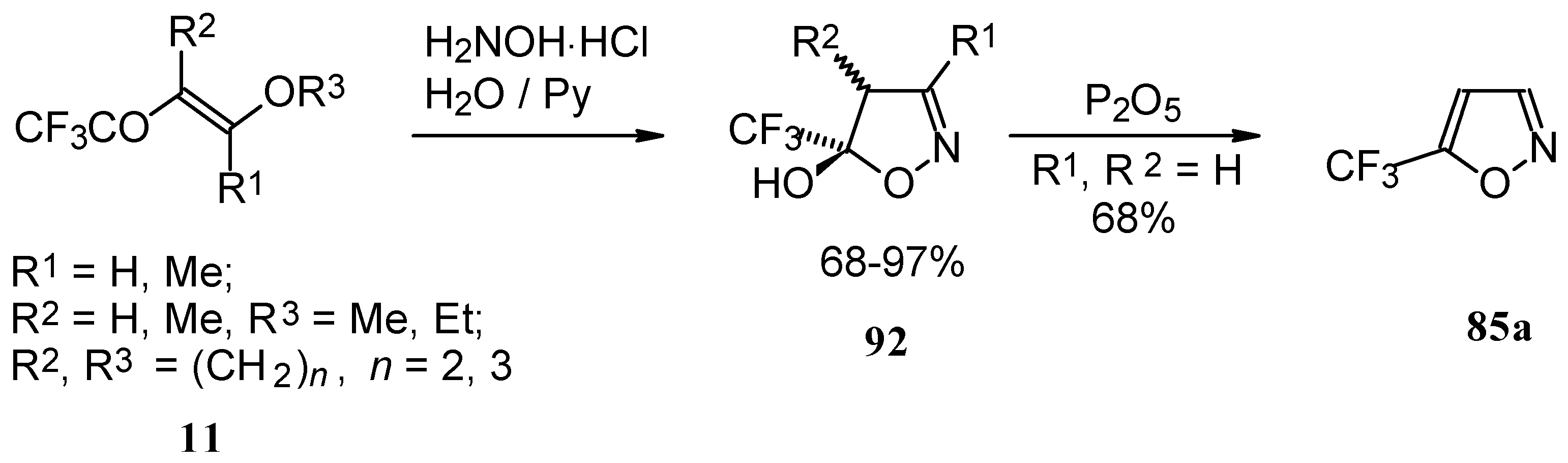
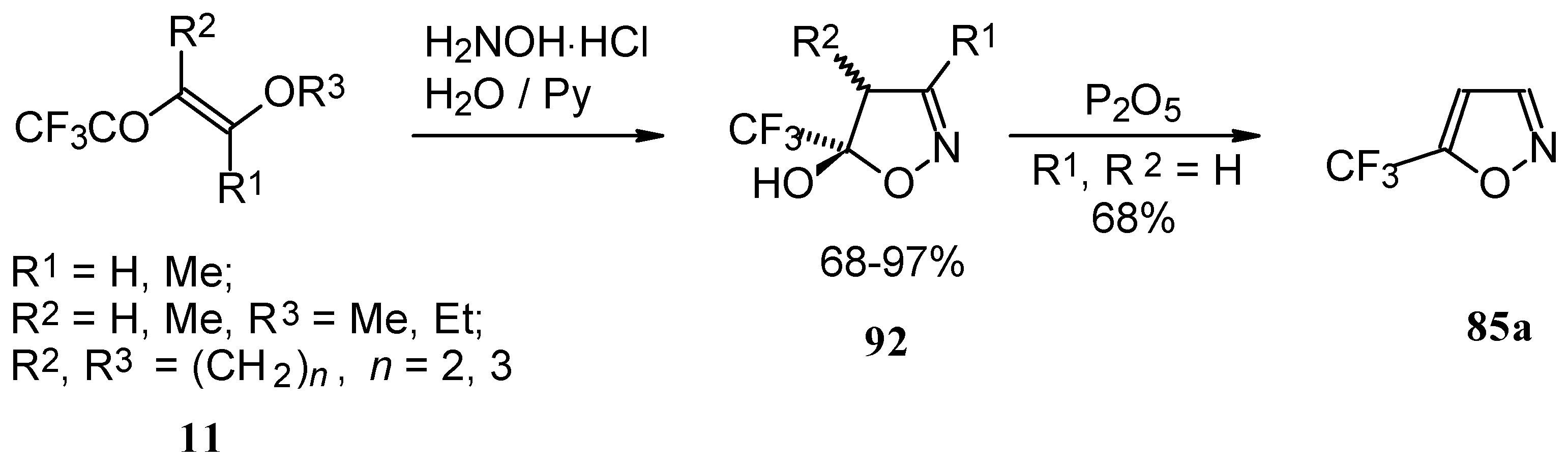
Trifluoroacylated enol ethers are convenient precursors for 5- and 6-membered heterocycles. The reaction of β-alkoxy enones, having a CF
3 group, with hydroxylamine proceeds analogously to the reaction of acetylenic ketones to give a trifluoromethyl-containing 4,5-dihydro-5-isoxazolol (applicable also to condensed rings) in good to excellent yield (
Scheme 48) [
18,
75]. The dehydration of these dihydroisoxazolols with phosphorous pentoxide or sulphuric acid results in the corresponding 5-CF
3 substituted isoxazole [
18,
75,
91].
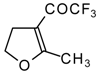
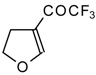
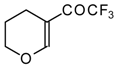
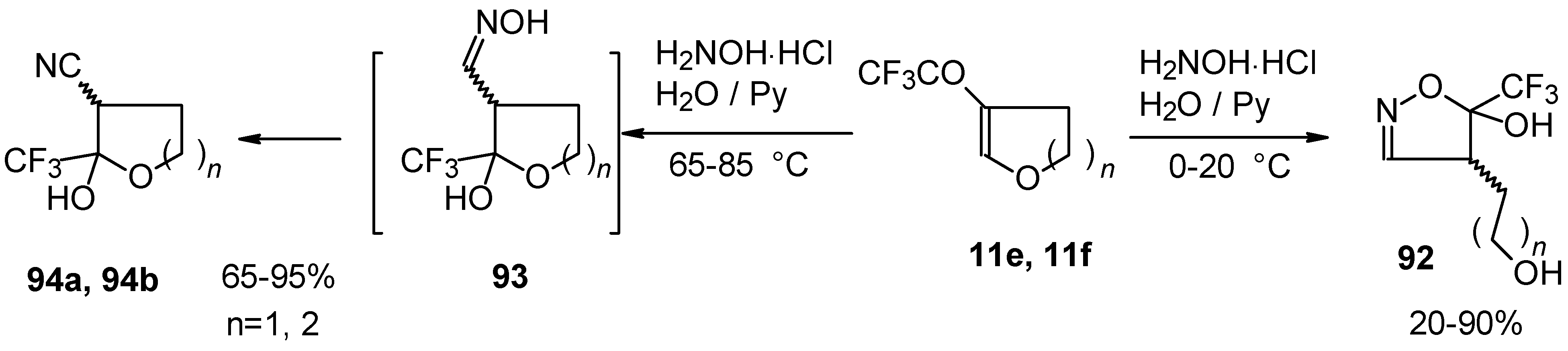
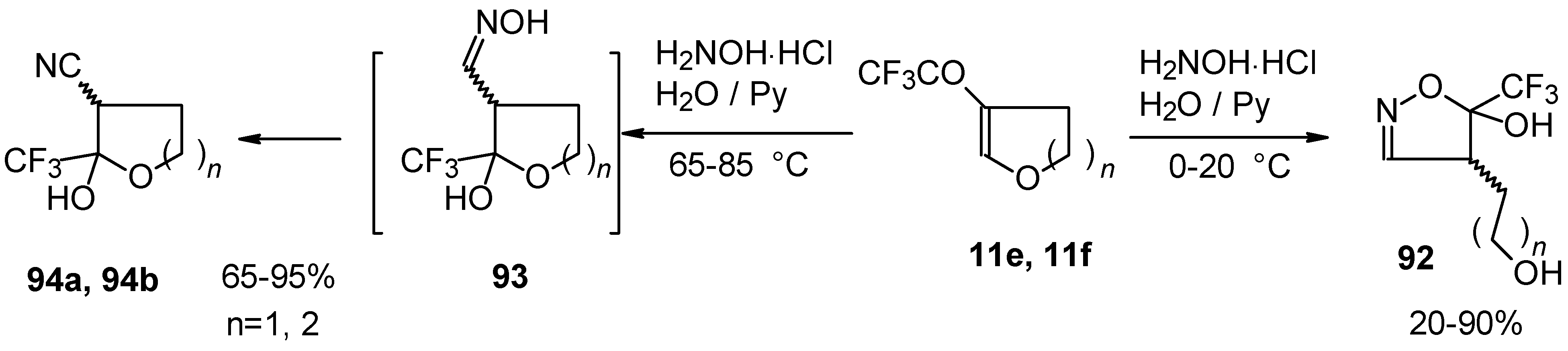
In the case of trifluoroacylated 2,3-dihydrofuran and 3,4-dihydro-2
H-pyran the reaction with hydroxylamine proceeds unexpectedly (
Scheme 49). At a reaction temperature of 0-20 °C the corresponding dihydroisoxazolols which are the products of furan or pyran ring opening were isolated. However, raising of the reaction temperature gave rise to formation of rearranged products [
18].
It seems likely that this rearrangement proceeds via elimination of water from the oximes of the aldehydes forming after recyclization of the ketones to furan or pyrans [
18].
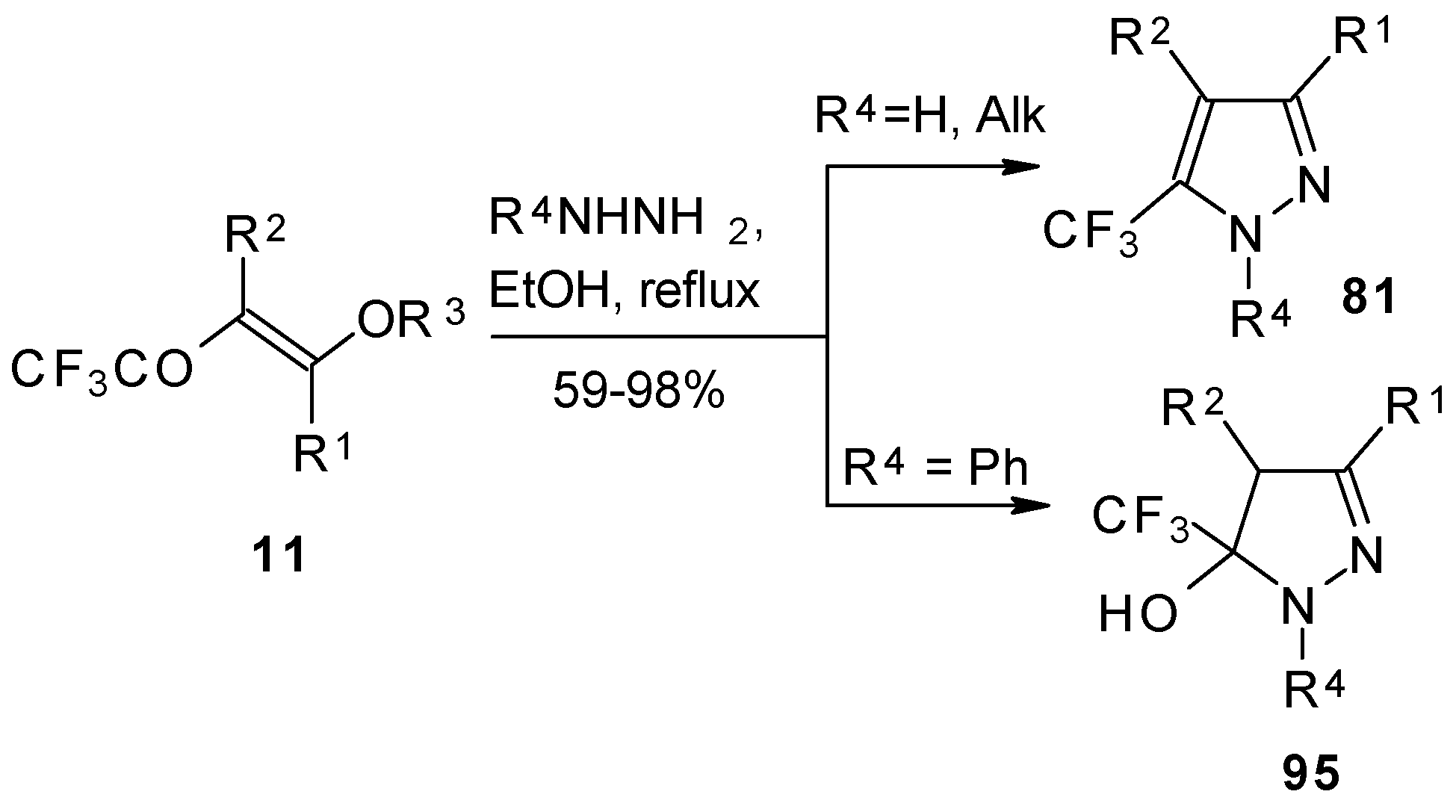
Hydrazine and alkylhydrazines react with trifluoroacetylated vinyl ethers to afford the corresponding pyrazoles in good yield [
75,
92]. Reaction with phenylhydrazine yields the corresponding pyrazoline [
92].
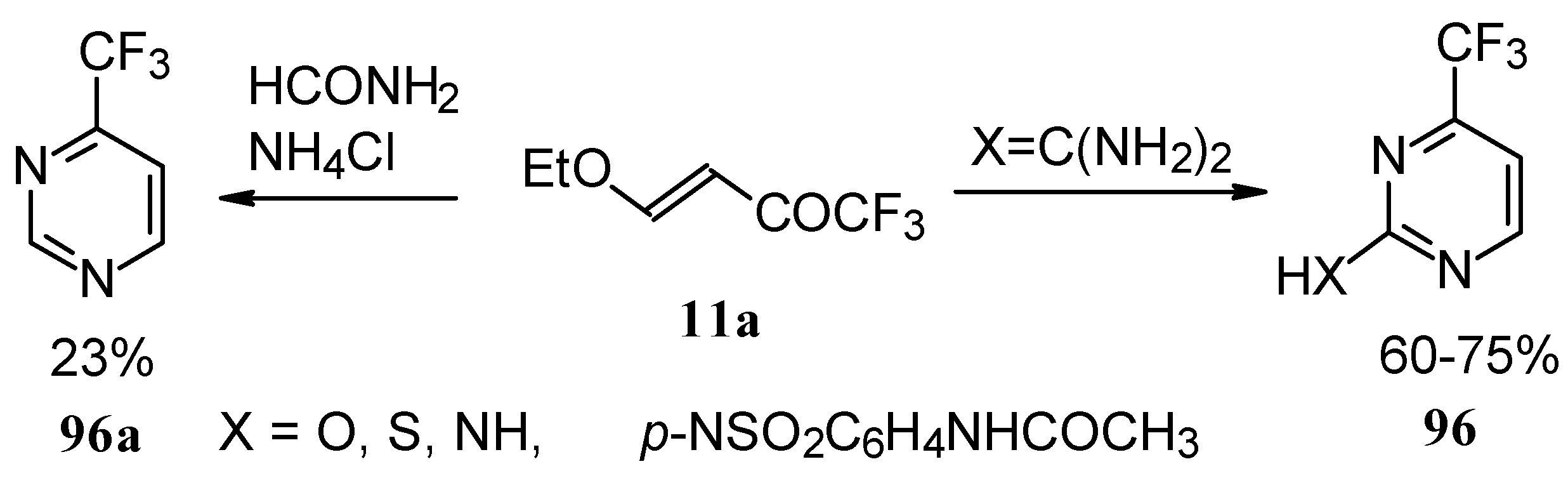
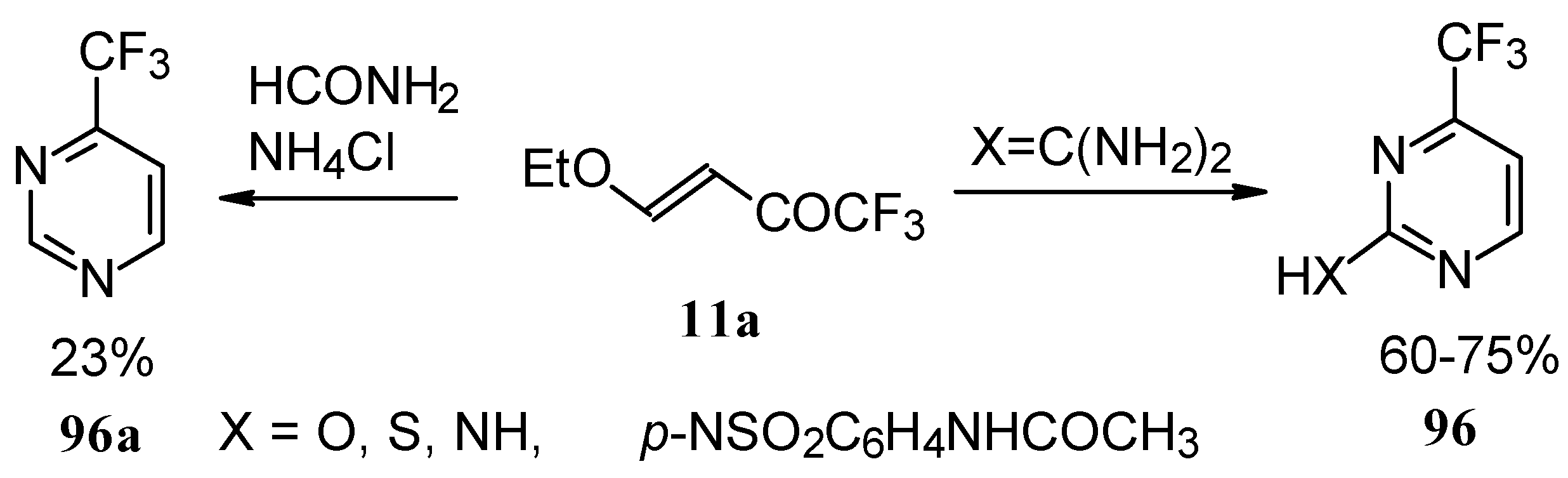
Trifluoromethyl-containing pyrimidines were prepared by reaction of 4-ethoxy-1,1,1-trifluoro-3-buten-2-one
11a with urea derivatives or formamide in the presence of NH
4Cl [
93].
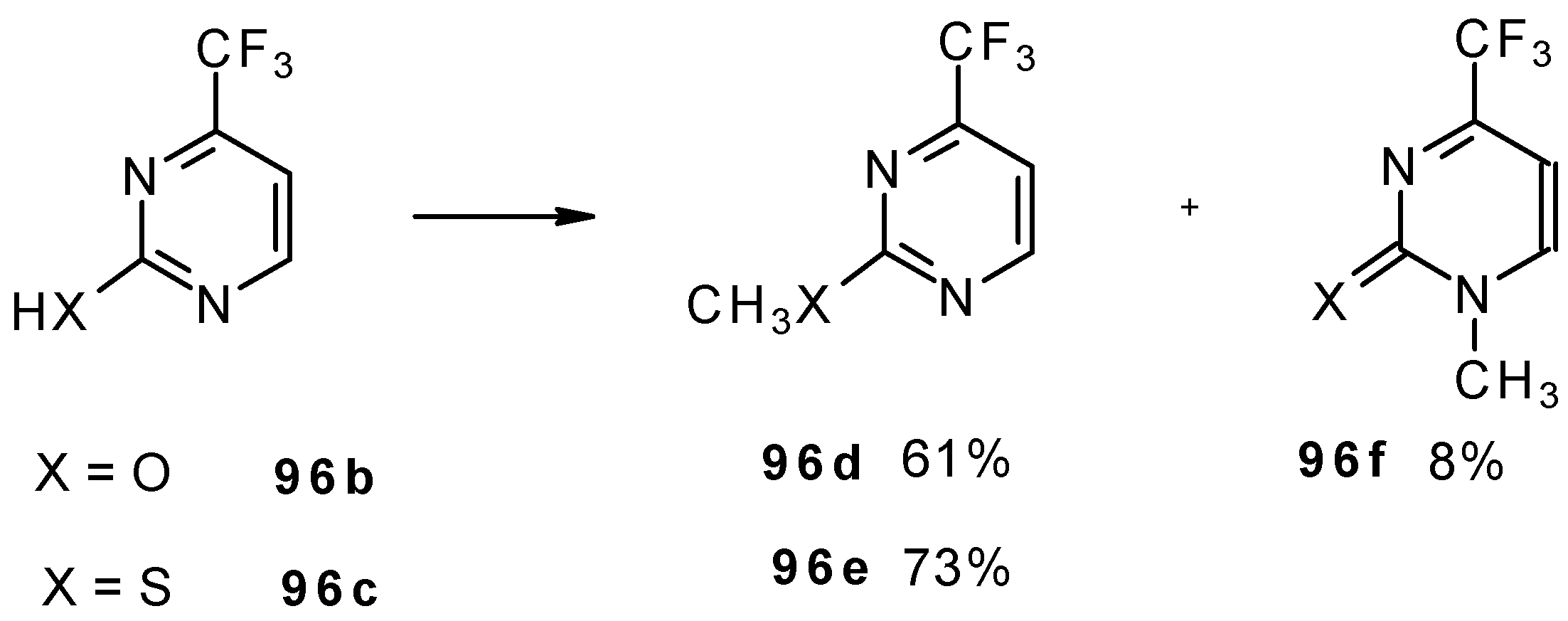
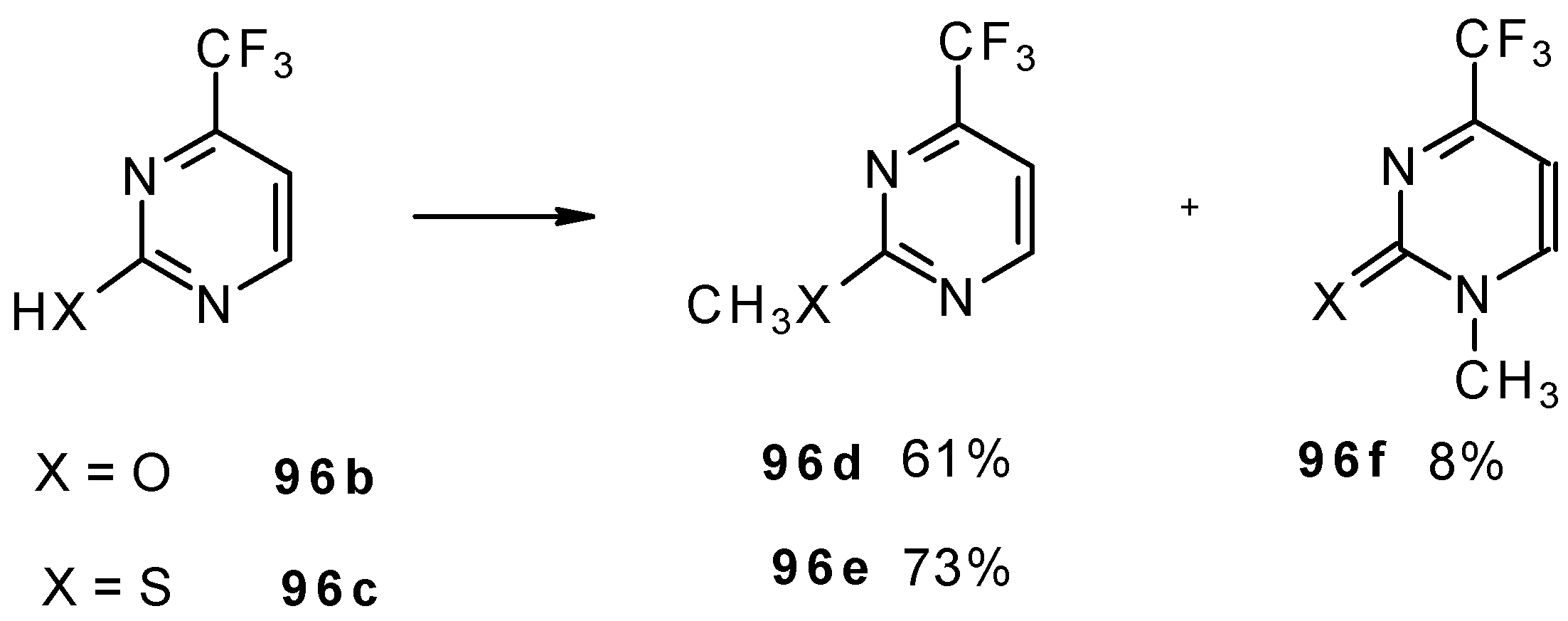
Alkylation of 4-CF
3 pyrimidinone or 4-CF
3 pyrimidinethione with diazomethane gives rise to a mixture of products of methylation at O (or S) and N atoms (
Scheme 52). The same authors also investigated the tautomeric equilibrium of 4-CF
3 pyrimidines in different solvents [
93].
The synthesis of thiomethyl substituted CF
3 pyrimidines was studied by reaction of 2-methyl-2-thiopseudourea sulphate with trifluoroacetylated vinyl ethers. The target products were prepared in 14-94% yield [
94].
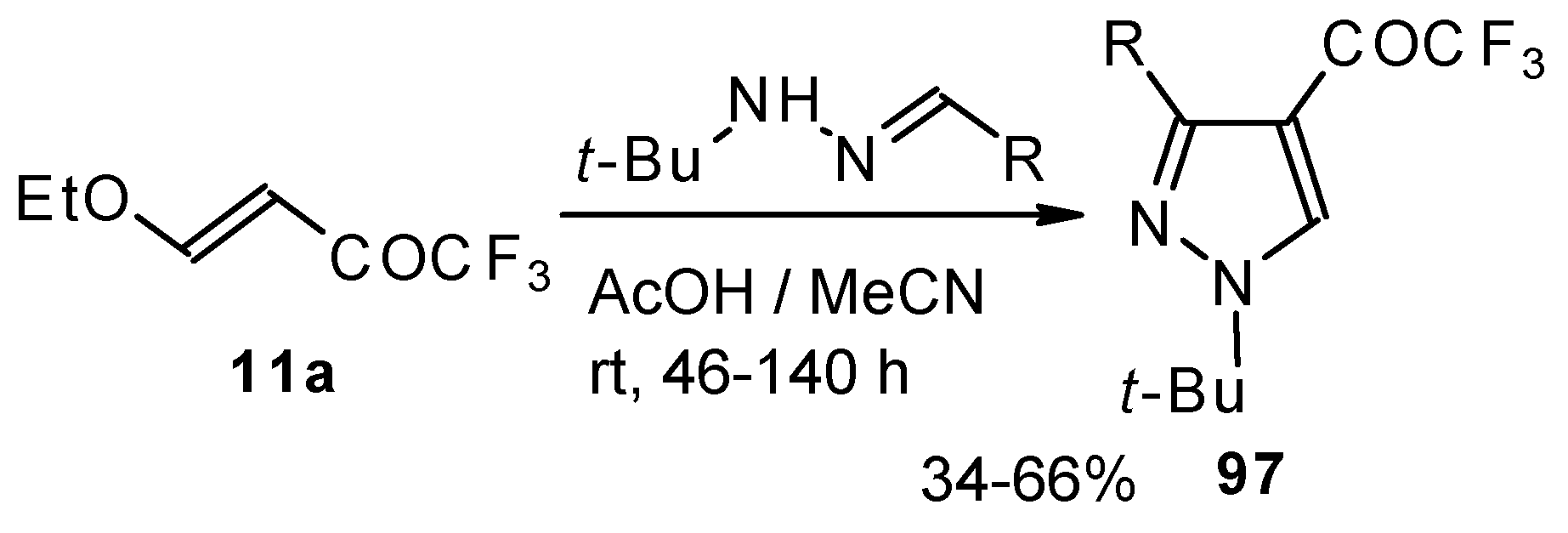
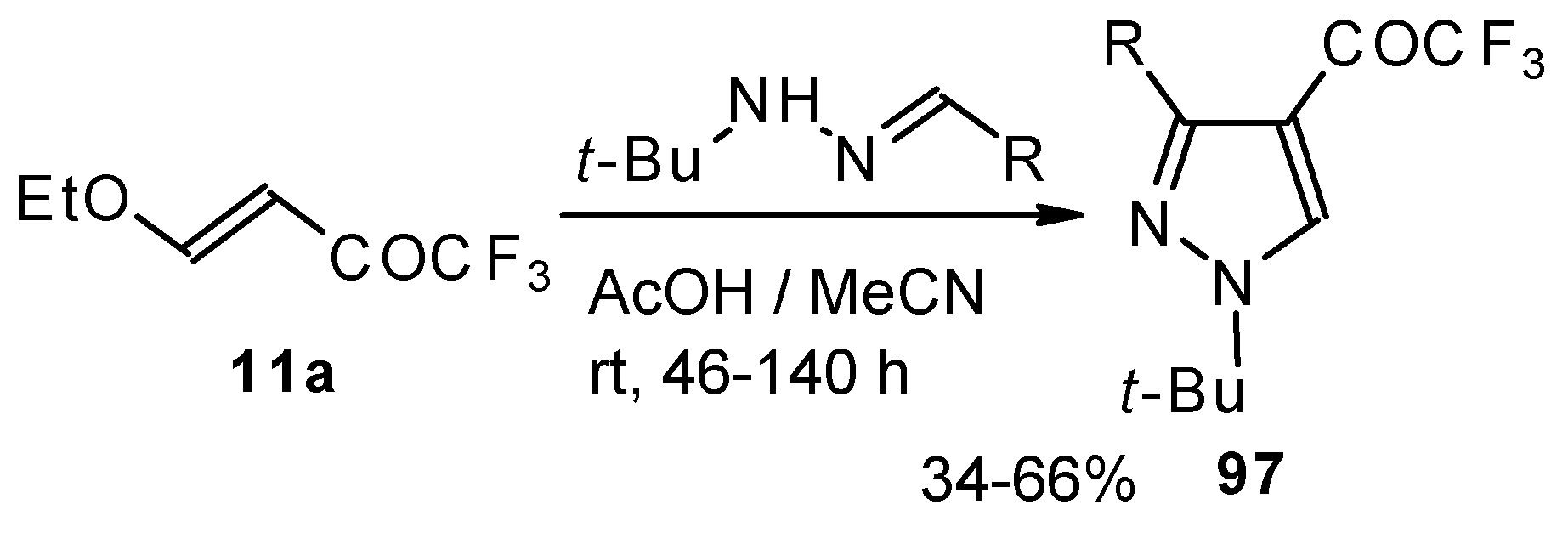
The reaction of
tert-butylhydrazones with trifluoroacetylated vinyl ether results in the corresponding 4-trifluoroacetylpyrazoles (
Scheme 53) [
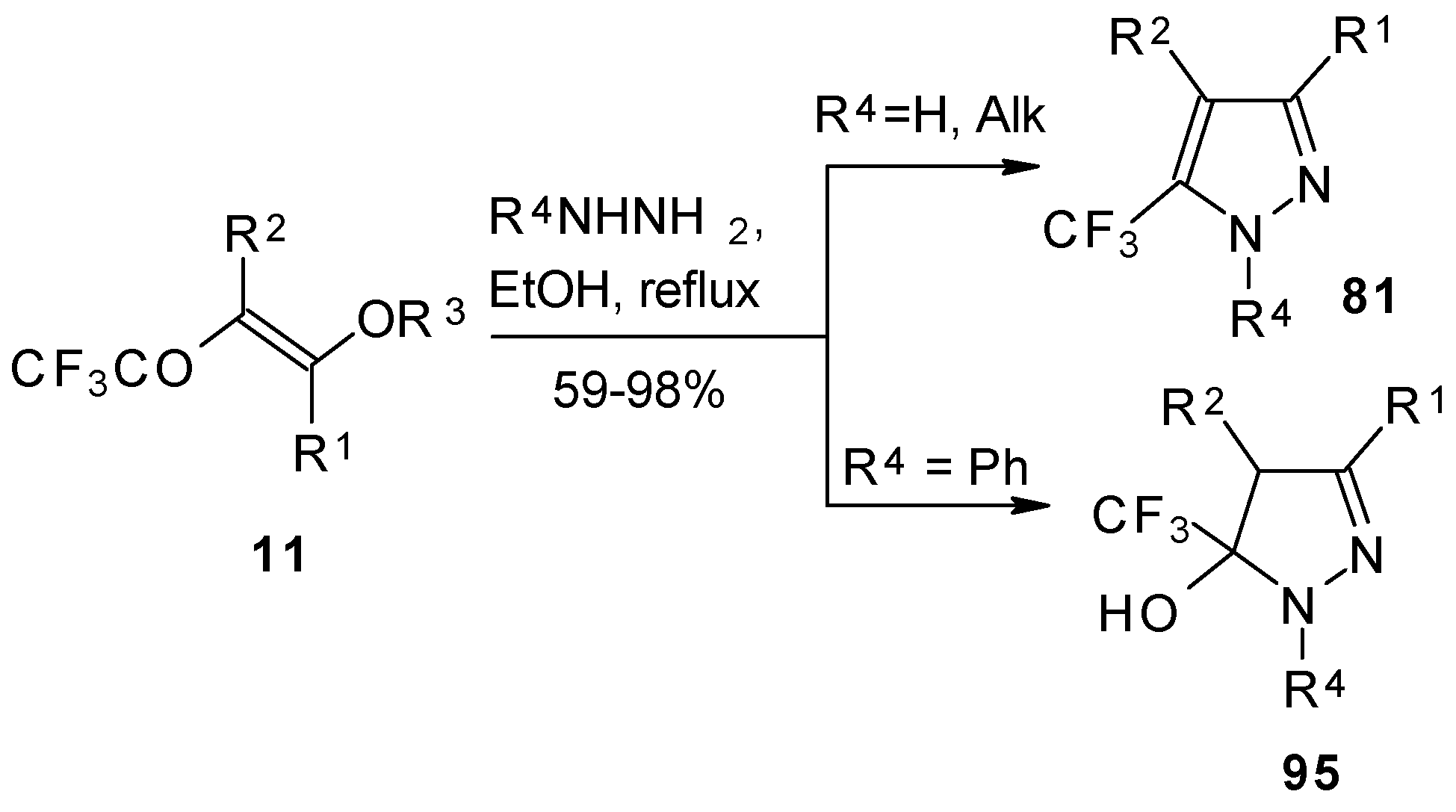
95]. This reaction proceeds by a nucleophilic displacement of the ethoxy group by hydrazone followed by cyclization, initiated by protonation on the azomethine nitrogen. Air oxidation of the intermediate dihydropyrazoles gives the corresponding 4-trifluoroacetylpyrazoles in good yield.
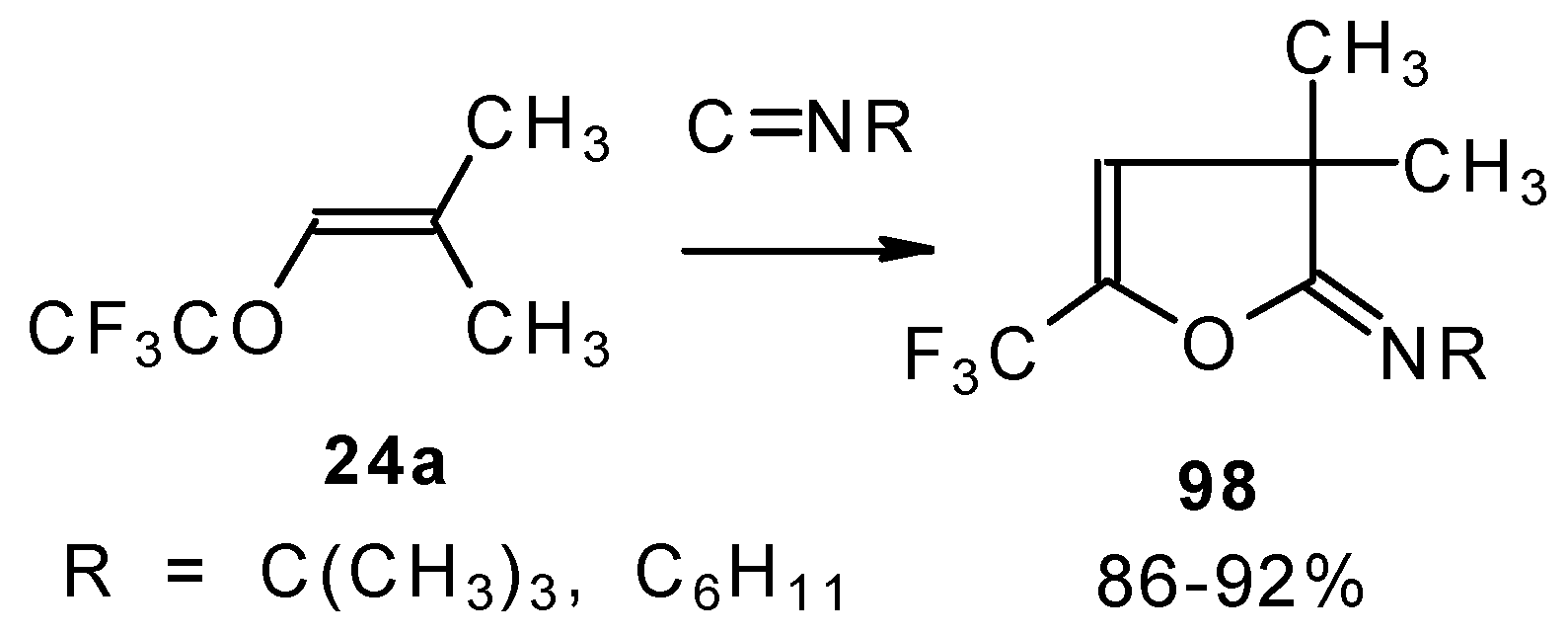
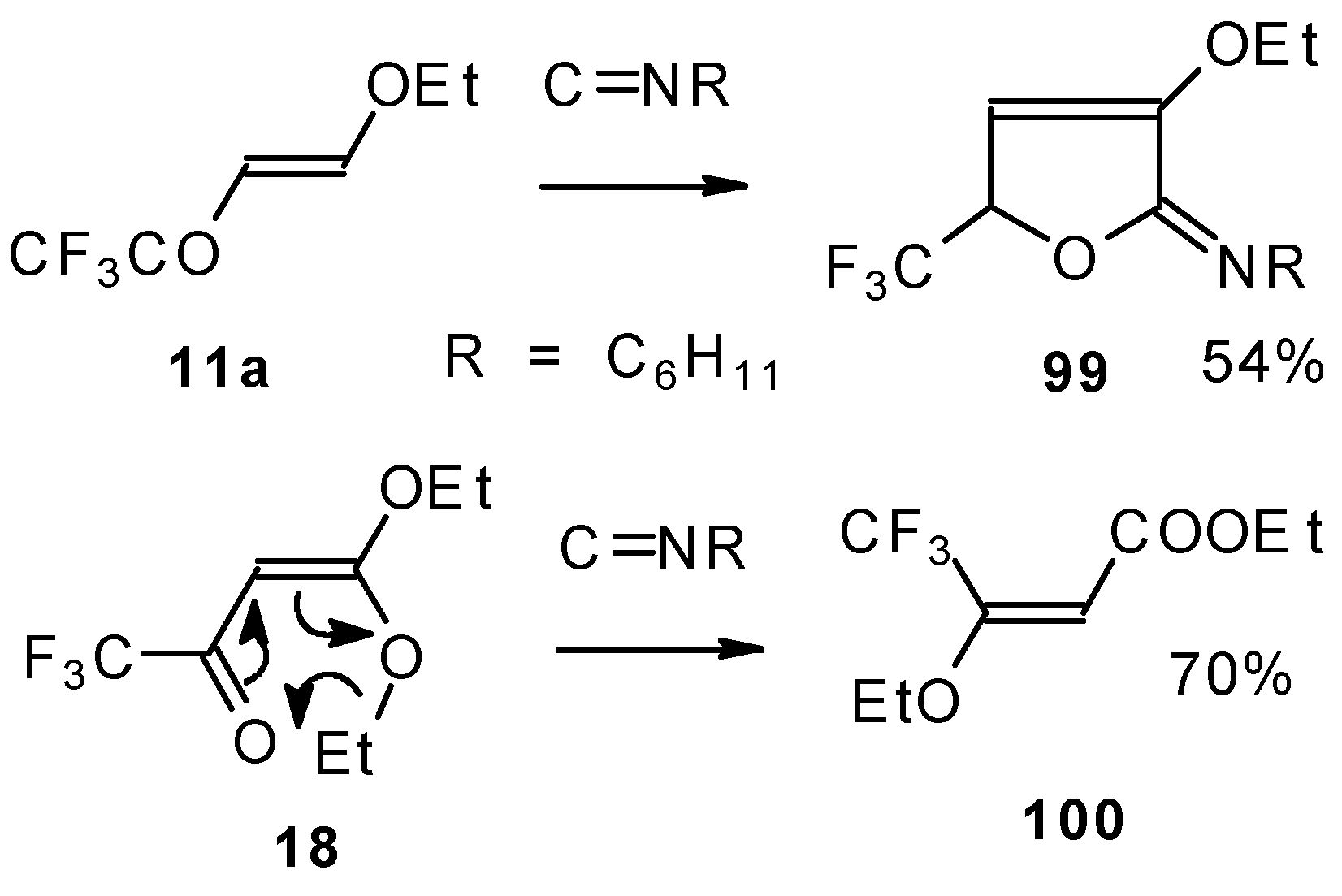
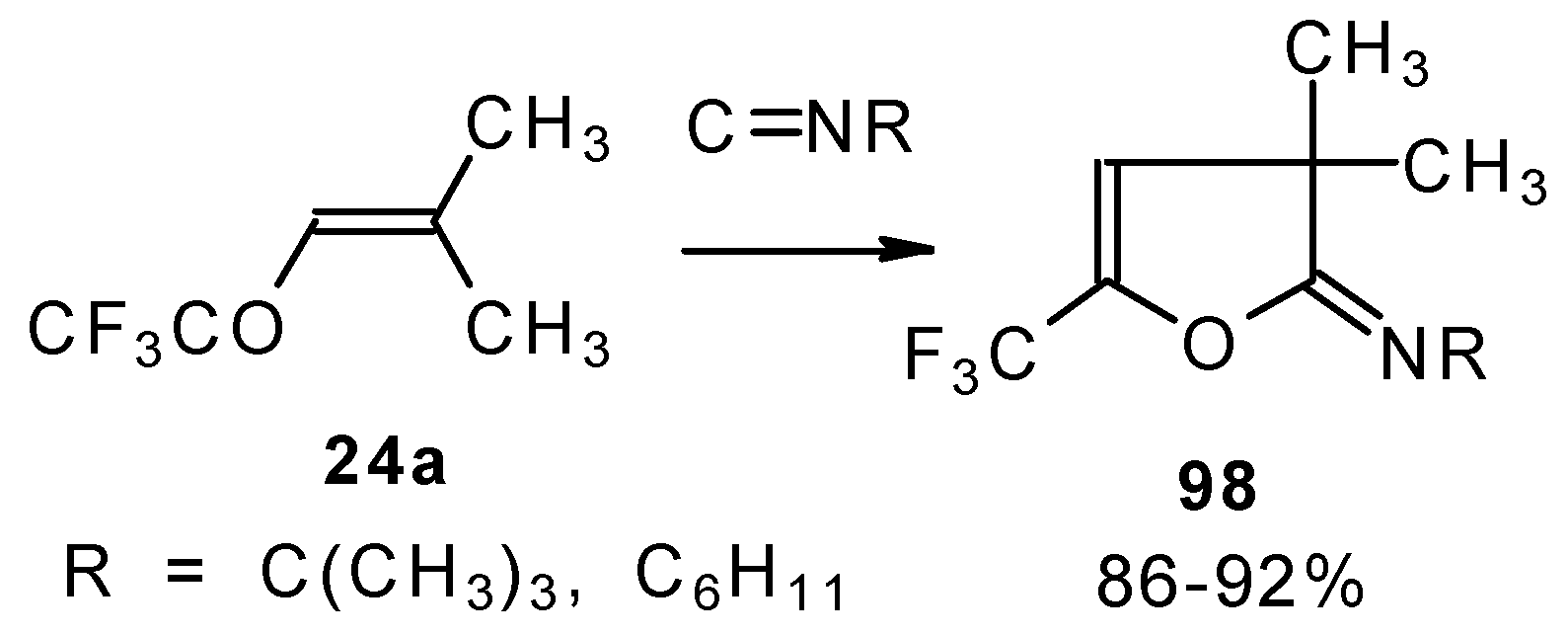
Ketones bearing a CF
3 group enter into the reaction with isonitriles to give the corresponding dihydrofurans, products of 1,4-cycloaddition (
Scheme 54). This reaction proceeds readily both with ketones having a β-alkoxy group and with ketones without a heteroatom substituent. This reaction proceeds below room temperature. Ketones without a CF
3 group do not enter into this reaction even on prolonged heating [
96,
97].
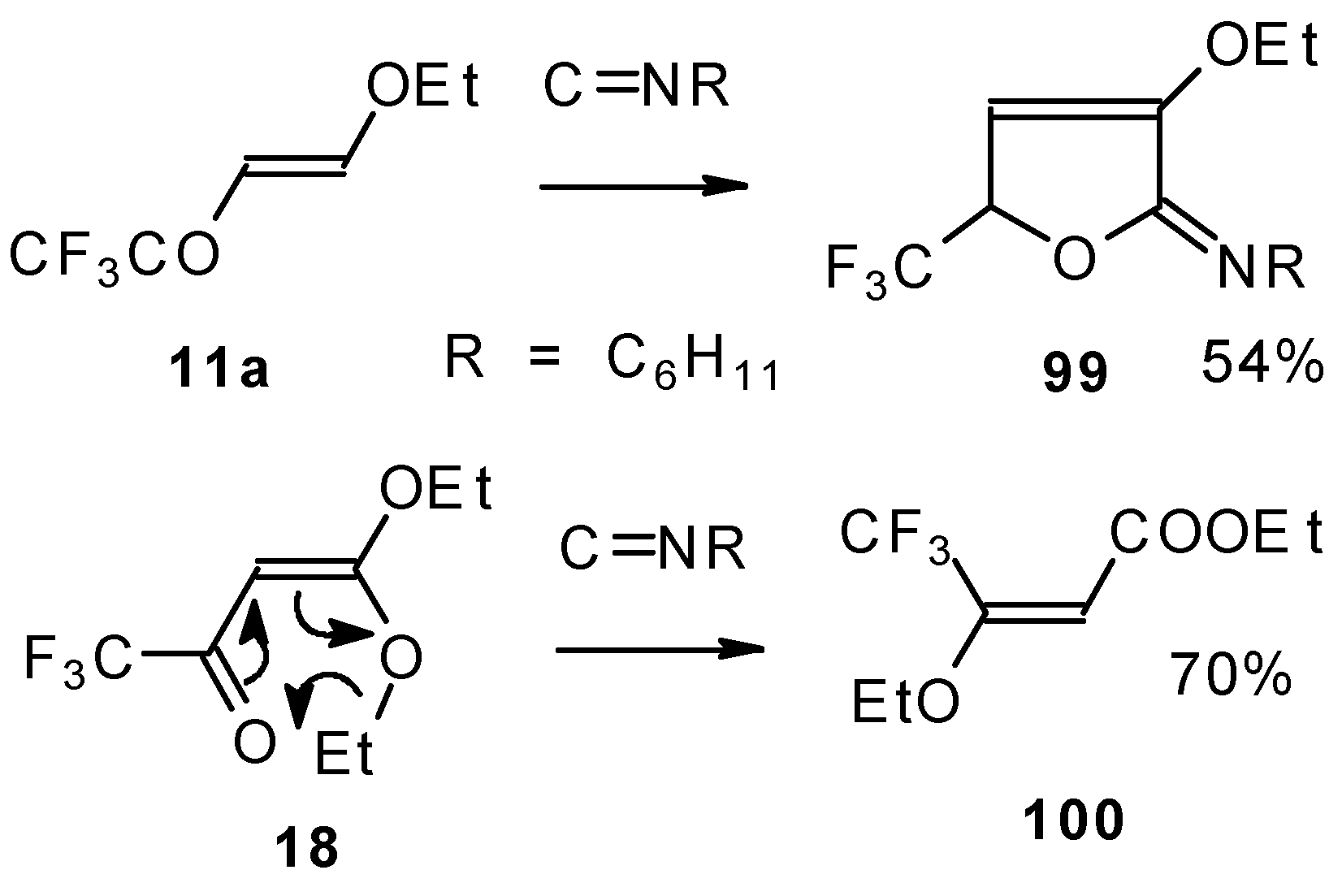
Under the same conditions 4-ethoxy-1,1,1-trifluoro-3-buten-2-one
11a gives the more stable 3-oxolene. When the trifluoroacetyl derivative of ketenediethylacetal
18 was reacted with isonitriles no cycloaddition was observed. Only isomerisation of the ketone to the ester
100 takes place (
Scheme 55) [
98].
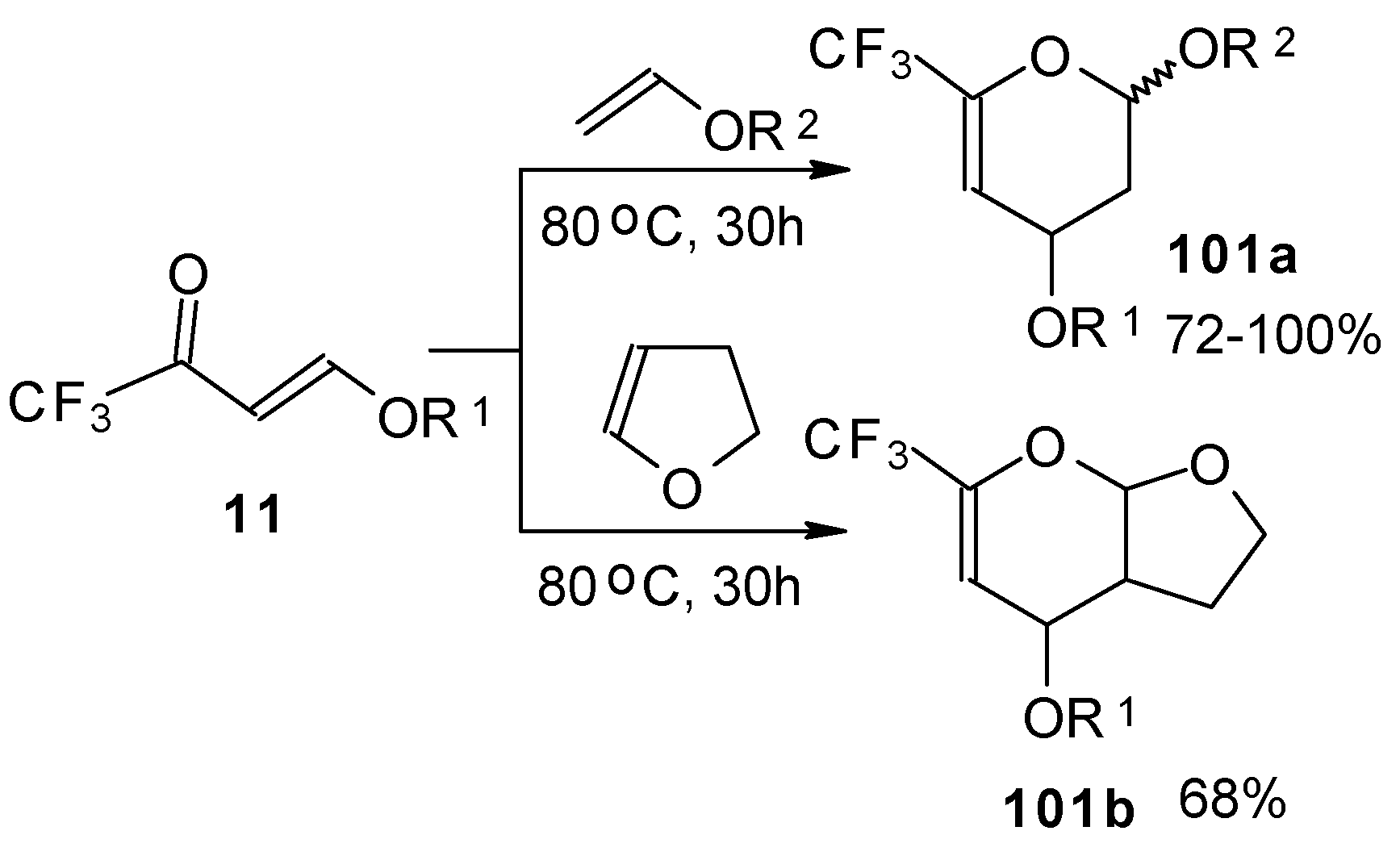
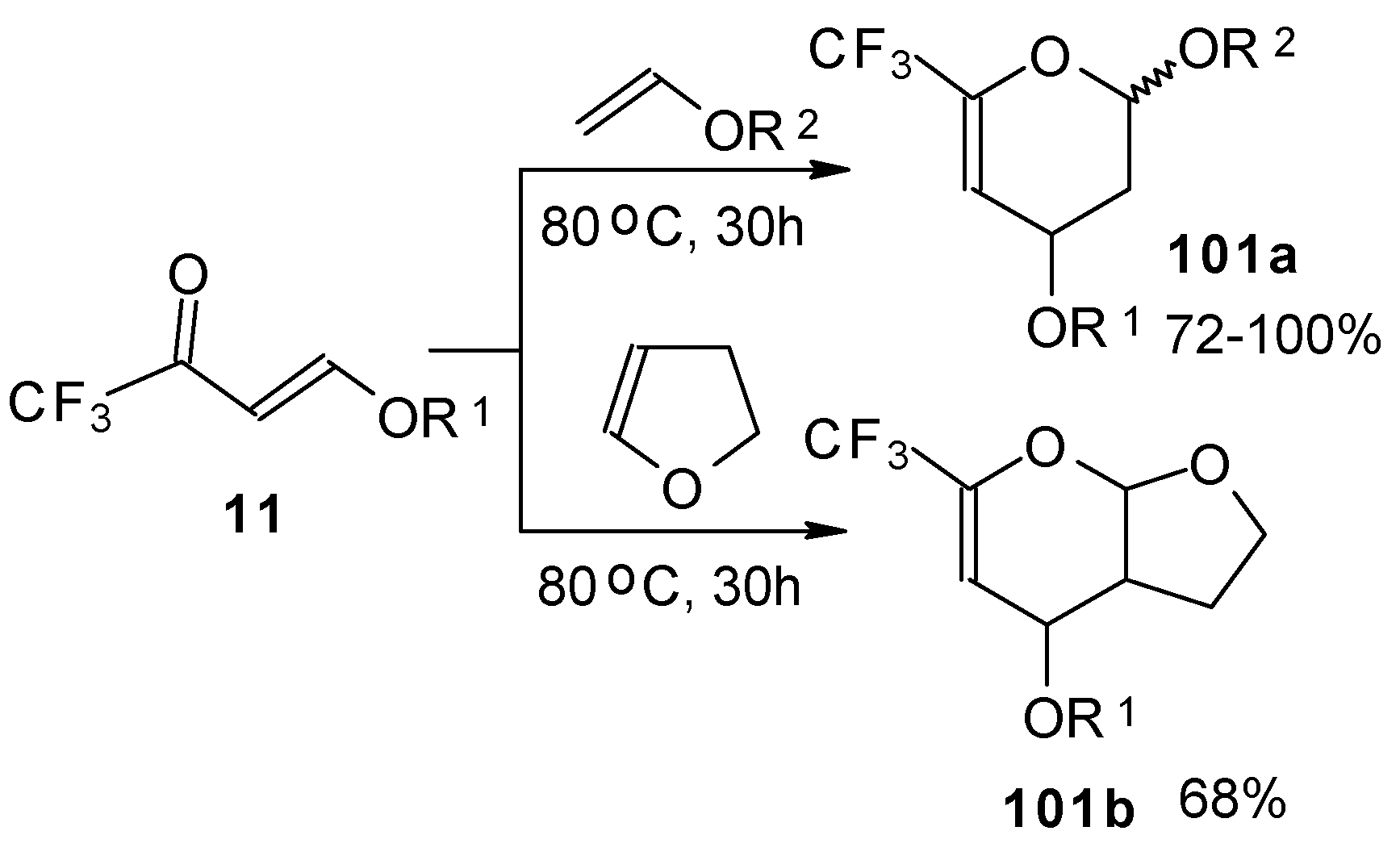
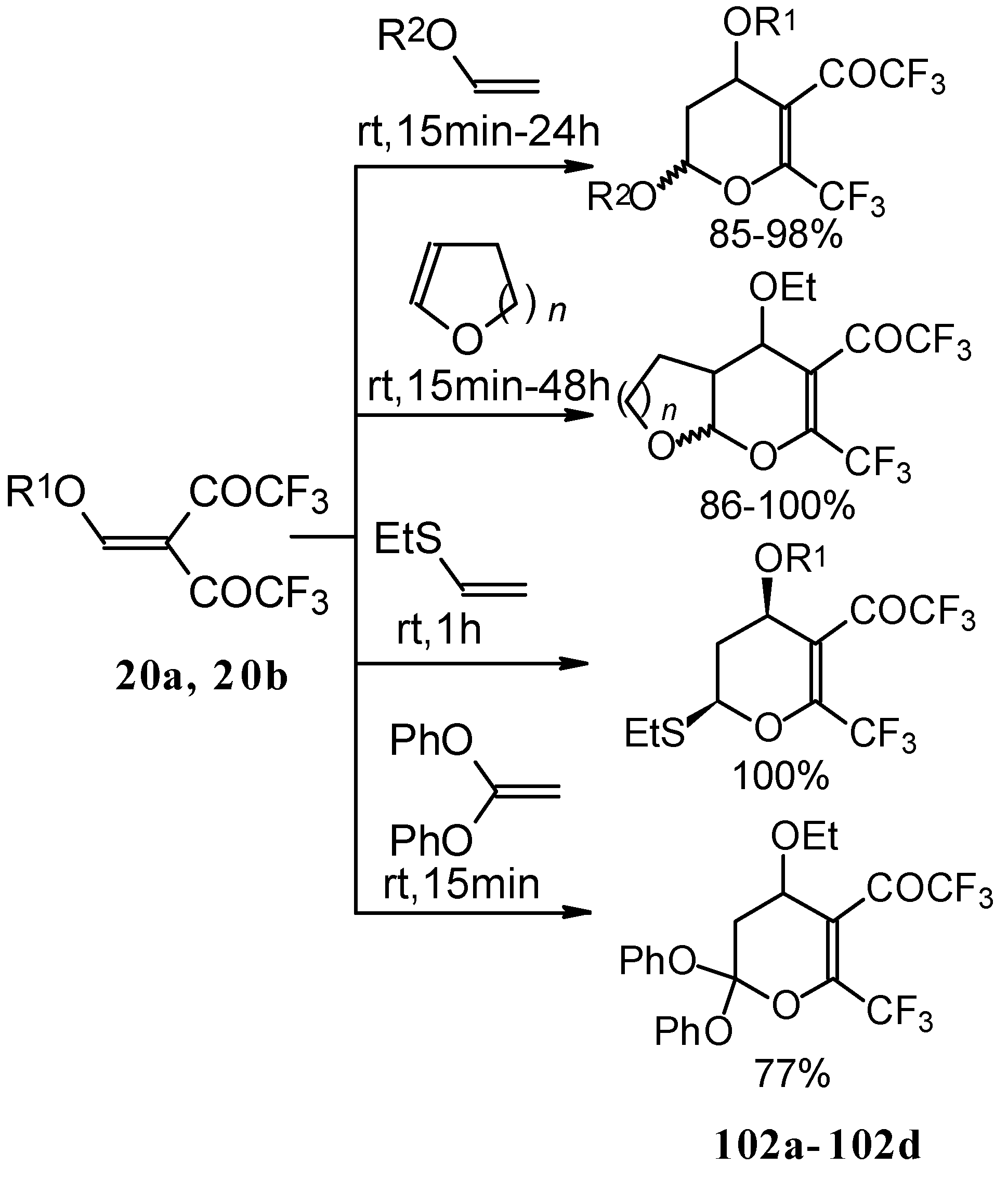
Trifluoroacetylated vinyl ether
11 represents a 1-oxa-1,3-butadiene system bearing an electronwithdrawing CF
3 substituent in the 2-position. It was found that this ketone behaves as an effective heterodiene. Hetero-Diels-Alder reaction with vinyl ethers leads to the corresponding dihydro-2
H-pyrans in good yield (
Scheme 56) [
99].
The same reaction proceeds readily with β,β-bis(trifluoroacetyl)vinyl ethers and various electron rich alkenes under very mild conditions to provide functionalized 5-trifluoroacetyl-6-trifluoromethyl-3,4-dihydro-2
H-pyrans in excellent yields (
Scheme 57) [
25]. ββ-Bis(trifluoroacetyl)vinyl alkyl ethers are much more active dienes than the corresponding β-monotrifluoroacetylvinyl ethers for which vigorous reaction conditions (80 °C, 30h) are required for cycloaddition. The presence of the electron withdrawing COCF
3 group at the C-3 of the 1-oxa-1,3-butadiene system, decreases the energy of the LUMO.
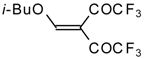
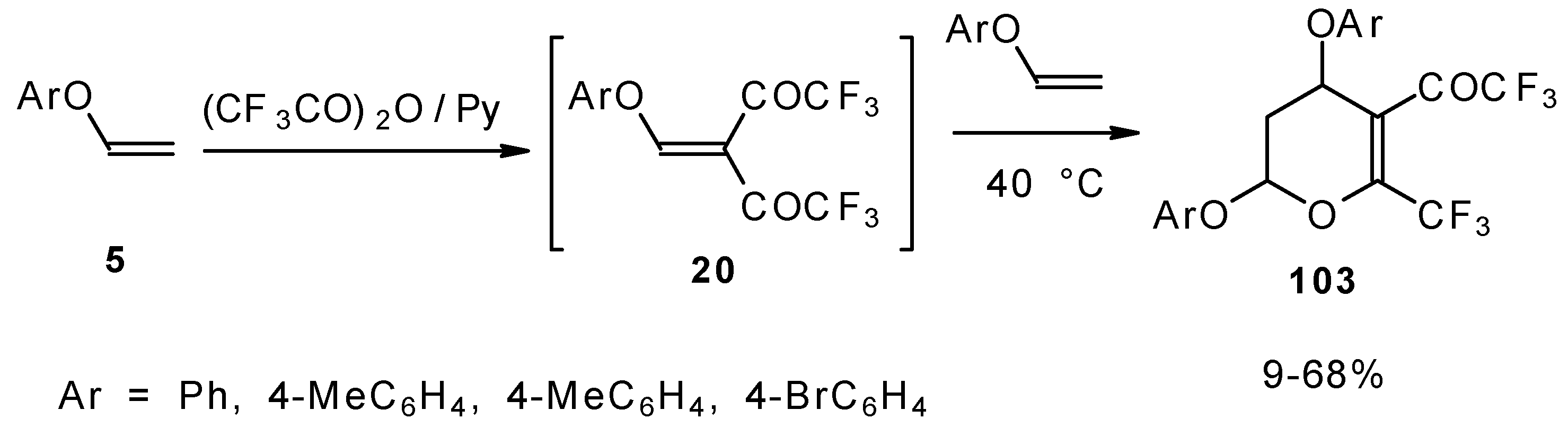
It was found by Hojo and coworkers [
27] that the corresponding β-aryloxy substituted enones with a CF
3 group, which are the products of double trifuoroacylation of aryl vinyl ethers decompose readily and could not be isolated in pure form. However, these authors found it possible to generate the ketones
in situ by diacylation of aryl vinyl ethers with trifluoroacetic anhydride followed by a Hetero-Diels-Alder reaction with an excess of substrate at 40 °C (
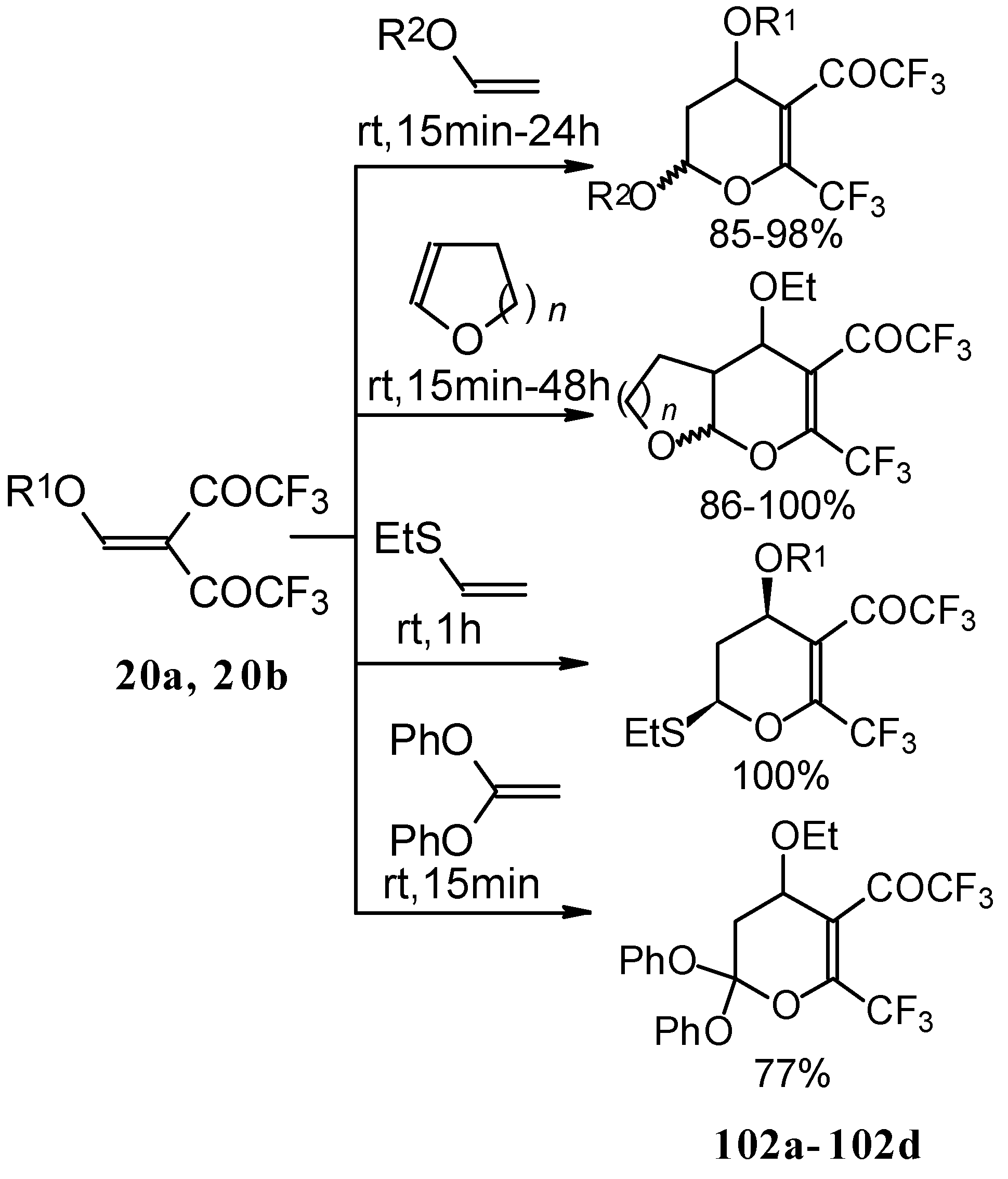
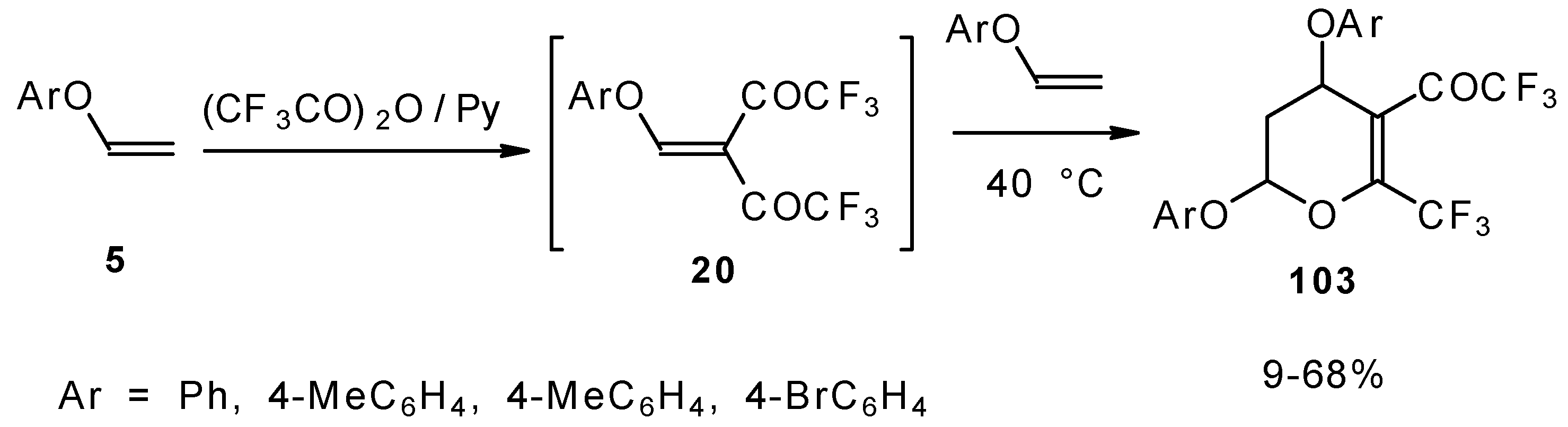
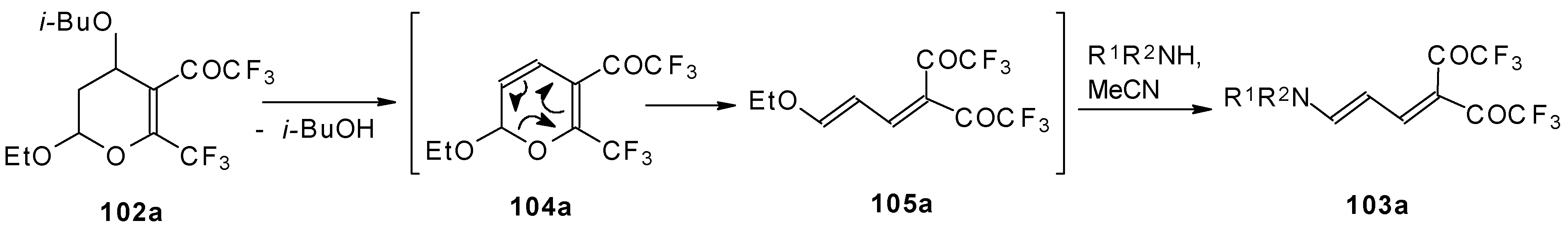
Scheme 58). Thus, the corresponding 2-trifluoromethyl substituted dihydropyrans were prepared in a one-pot reaction from aryl vinyl ethers in good yield.
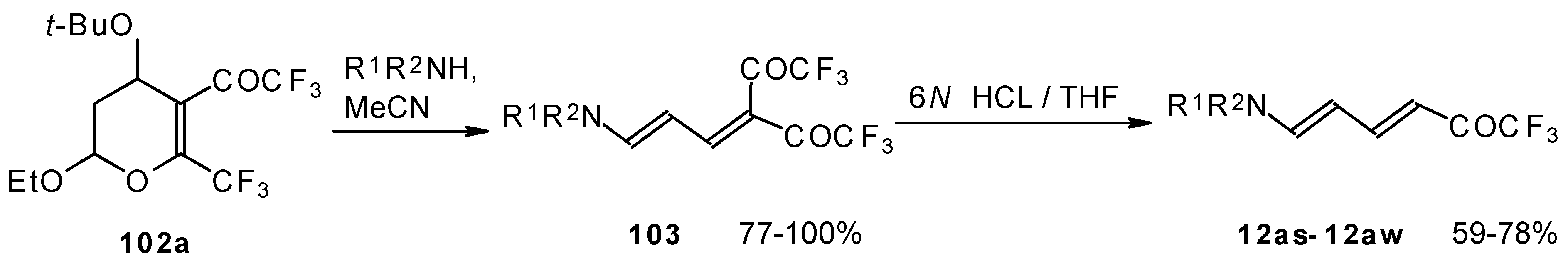
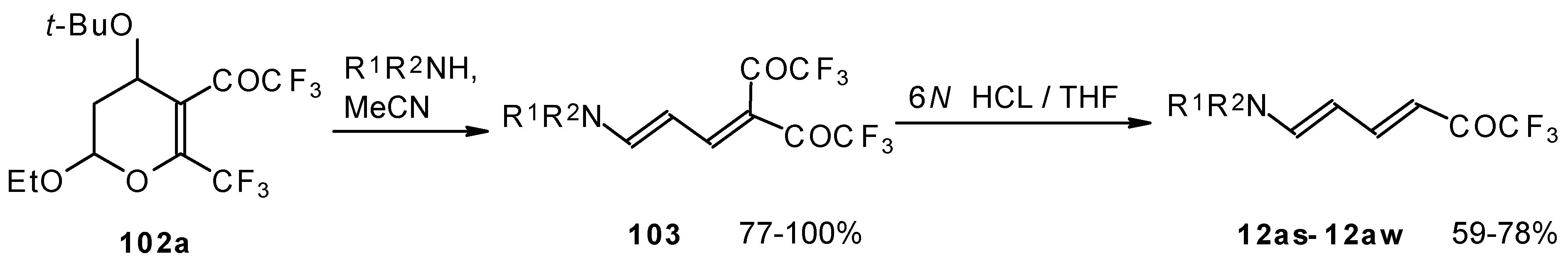
Cleavage of the corresponding dihydropyrans with dialkylamines opens up the route to 1-amino-4,4-bis(trifluoroacetyl)-1,3-butadienes (
Scheme 59). These ketones can be converted to the 1-amino-4-trifluoroacetyl-1,3-butadienes in the presence of HCl in good yields [
100].
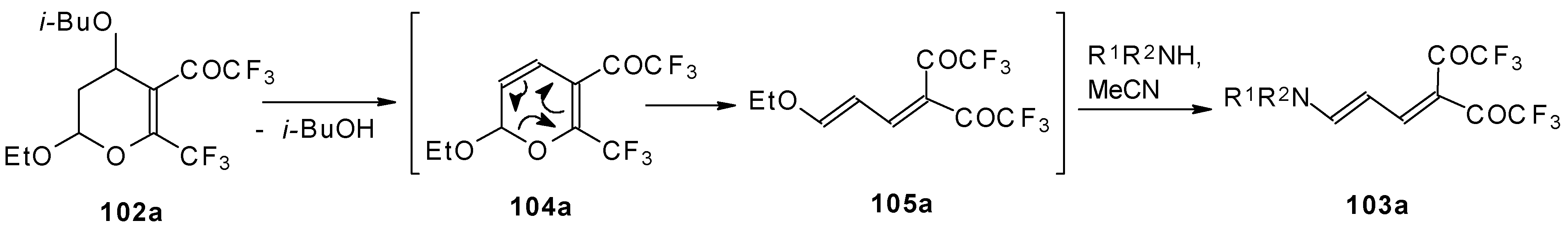
A suggested reaction mechanism involves elimination of
i-butanol, electrocyclic ring opening and nucleophilic O-N exchange. This transformation takes place only for CF
3 containing pyrans (
Scheme 60). It was found that the same reaction for the corresponding non-fluorinated compound did not proceed.
The same authors found it possible to prepare 1-amino-4,4-bis(trifluoroacetyl)-1,3-butadiene by trifluoroacylation of 1-amino-4-trifluoroacetyl-1,3-butadiene. This reaction proceeds quantitatively at room temperature (
Scheme 61).
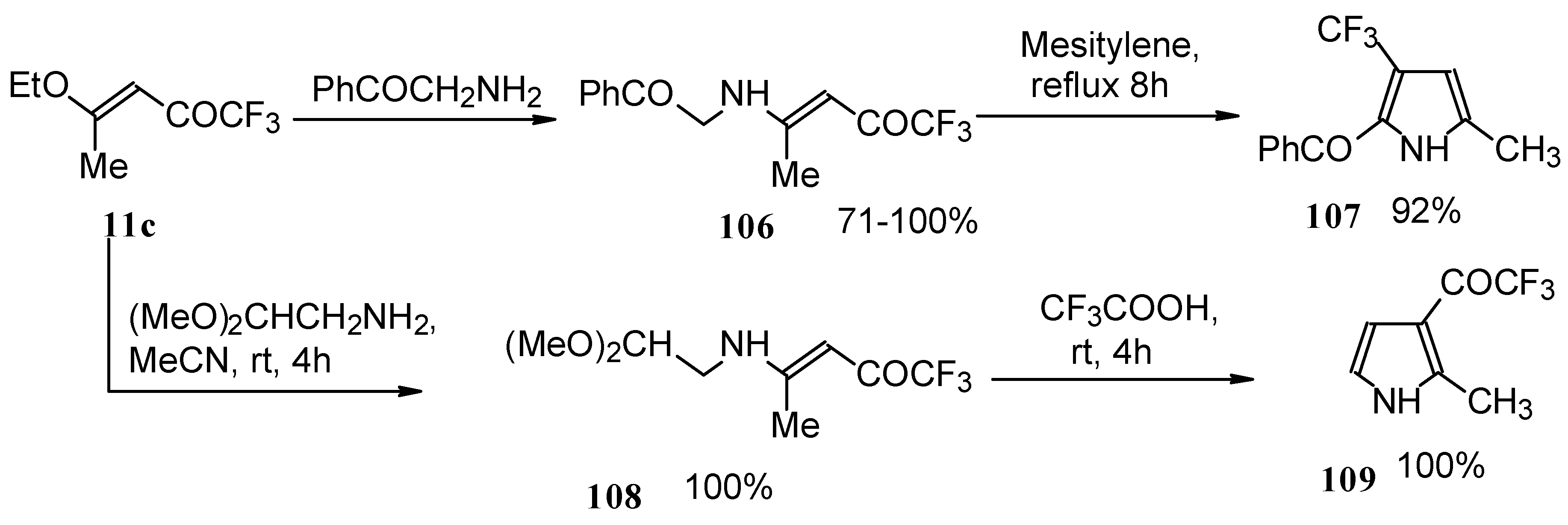
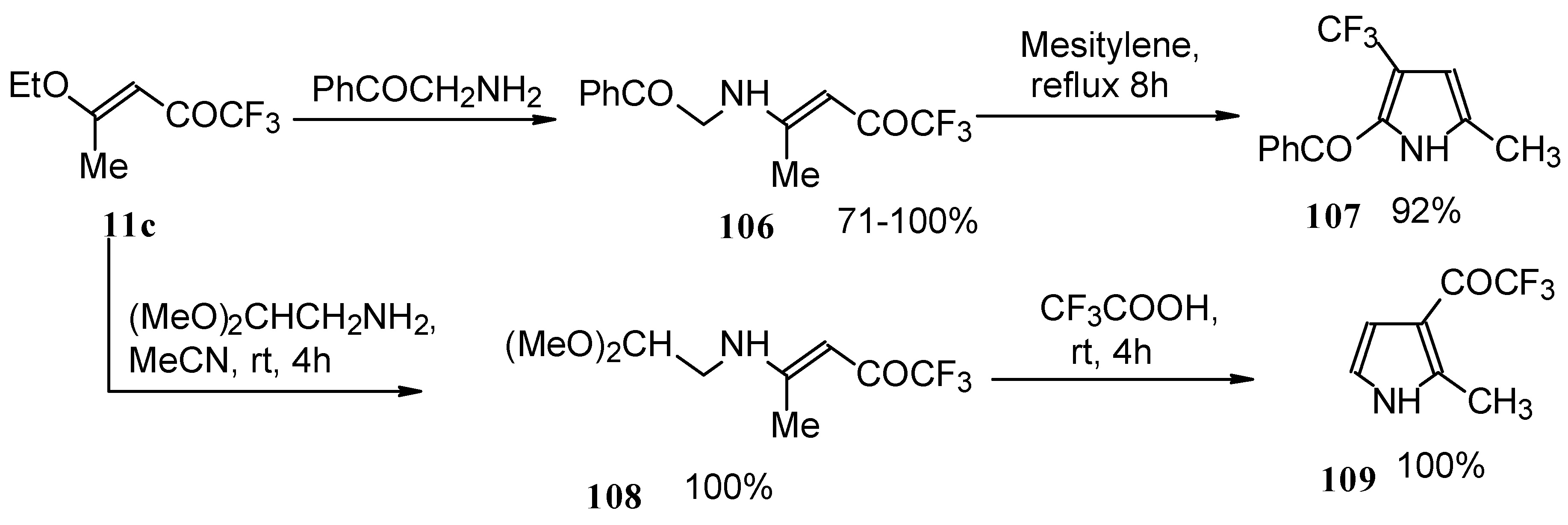
Trifluromethyl-containing pyrroles were prepared in excellent yields by reaction of trifluoroacetylated vinyl ethers with α-amino ketones or acetals of aminoaldehydes with subsequent cyclization of the adducts by heating in mesitylene or trifluoroacetic acid (
Scheme 62) [
101,
102].
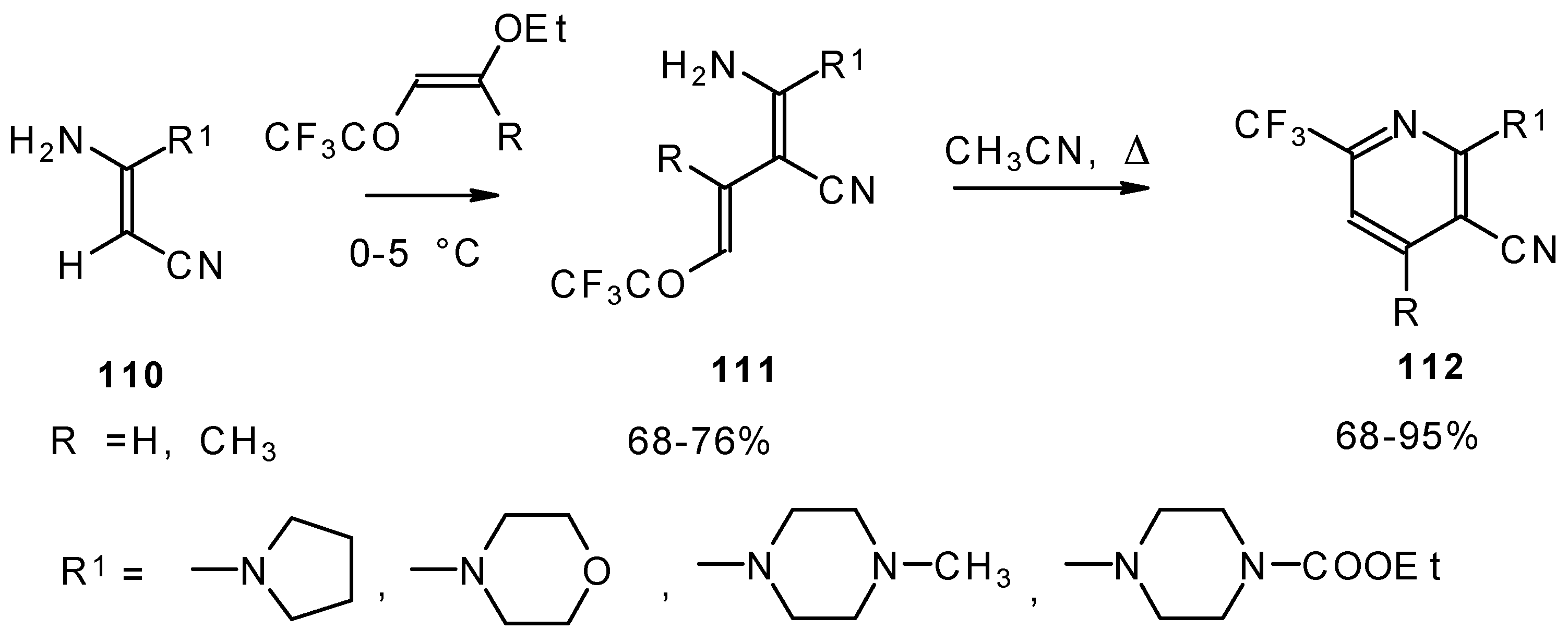
CF
3-Substituted pyridines were prepared by the reaction of enaminonitriles with trifluoroacylated vinyl ethers. Acyclic intermediates were isolated in the case of 4-ethoxy-1,1,1-trifluoro-3-buten-2-one (
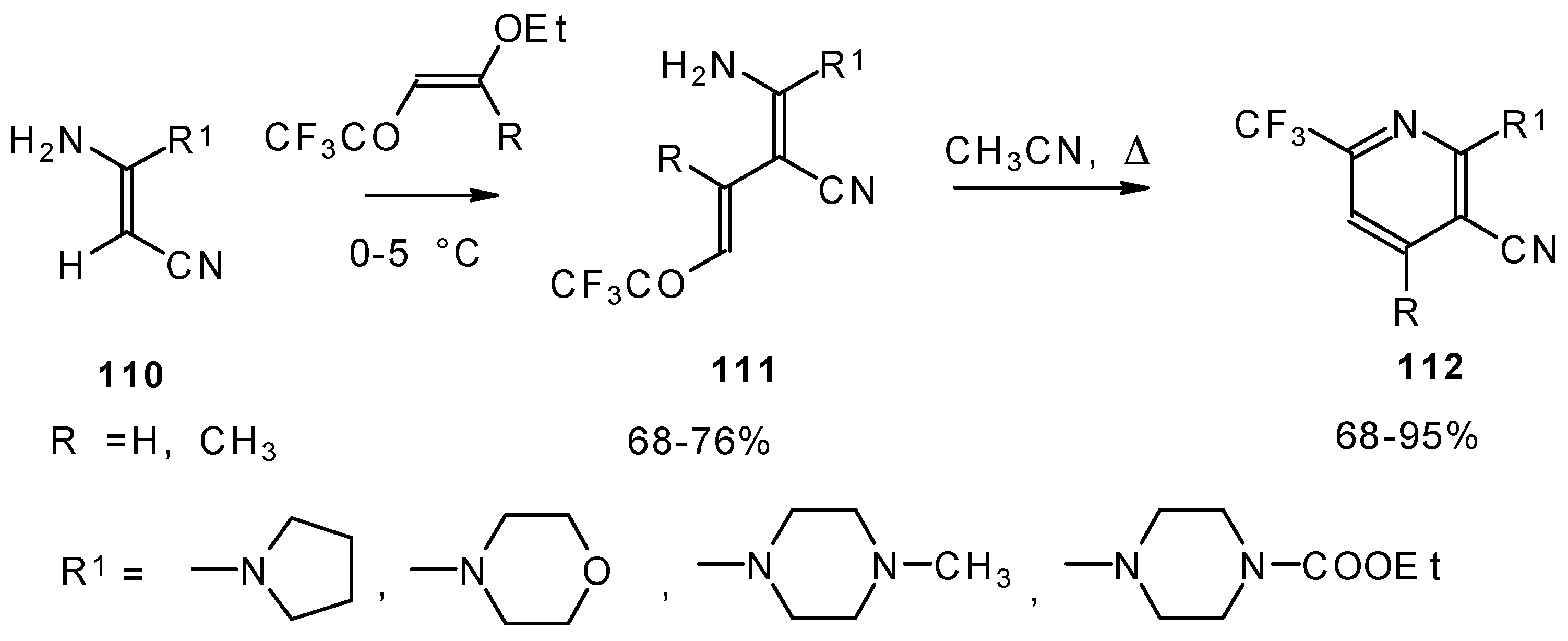
Scheme 63) [
103]. The raising of the reaction temperature leads to heterocyclization to the substituted CF
3 pyridines.
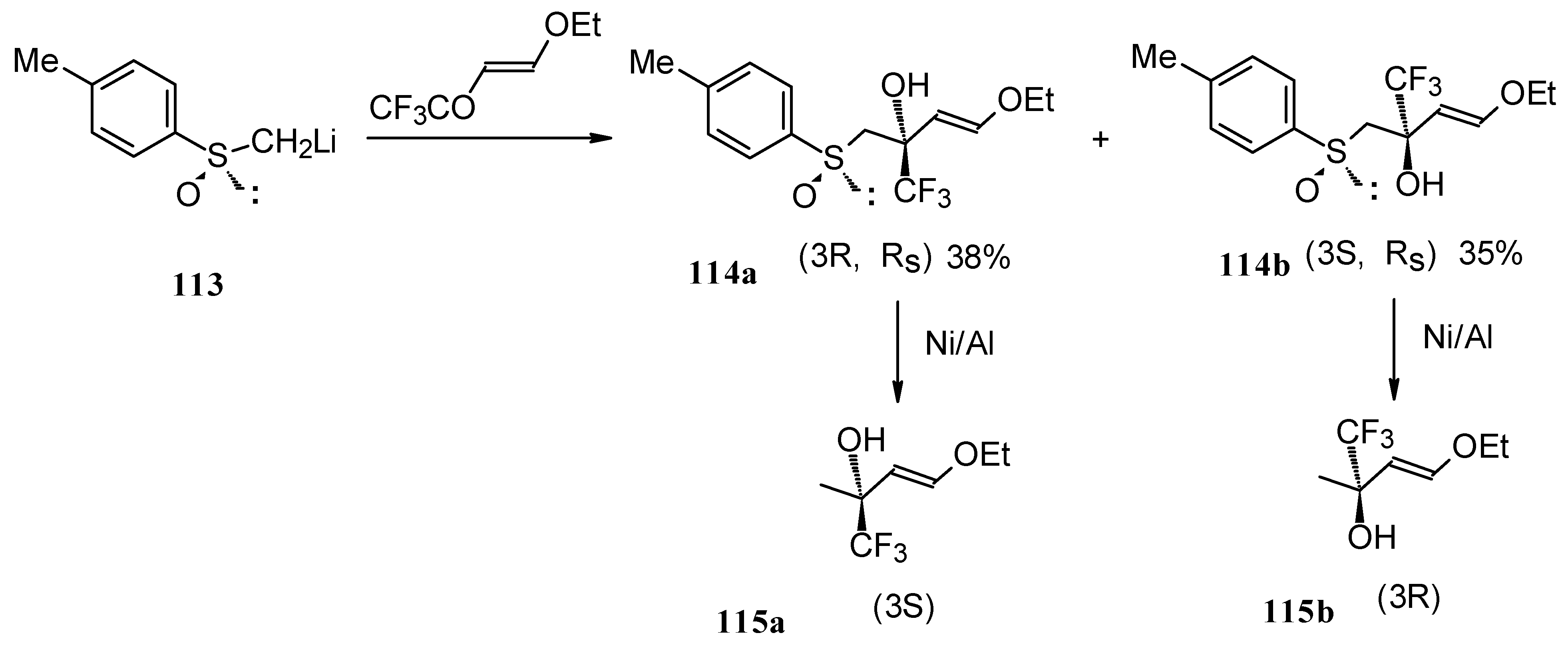
The reaction of 4-ethoxy-1,1,1-trifluoro-3-buten-2-one with α-sulfinyl carbanions was investigated (
Scheme 64) [
104].
The reaction with the lithium salt of (R)-methyl
p-tolylsulfoxide proceeds in THF at -60 °C totally regioselectively, affording only a tertiary alcohol, through a 1,2-addition (
Scheme 64). Stereoselectivity of addition was poor, diastereoisomeric alcohols were formed in a 1:1 ratio. The diastereoisomers could be separated by column chromatography. Subsequent desulphurization with Raney Ni resulted in enantiomerically pure CF
3 substituted alcohols [
104].
These transformations proceed cleanly under mild conditions in contrast to non-fluorinated enones of similar structure which give a complex mixture of products of both 1,2- and 1,4-addition [
104].
Another type of trifluoromethyl containing ketone used for the preparation of a CF3 substituted heterocycles is a ketone without a heteroatom substituent at β-position. In contrast to the above mentioned reactions this ketone gives rise to hydrogenated heterocycles.
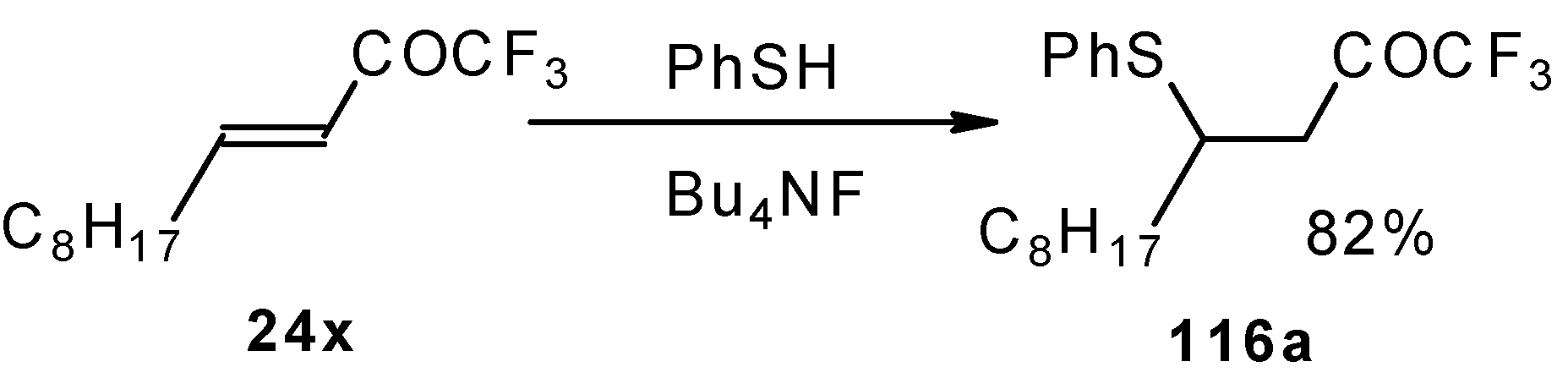
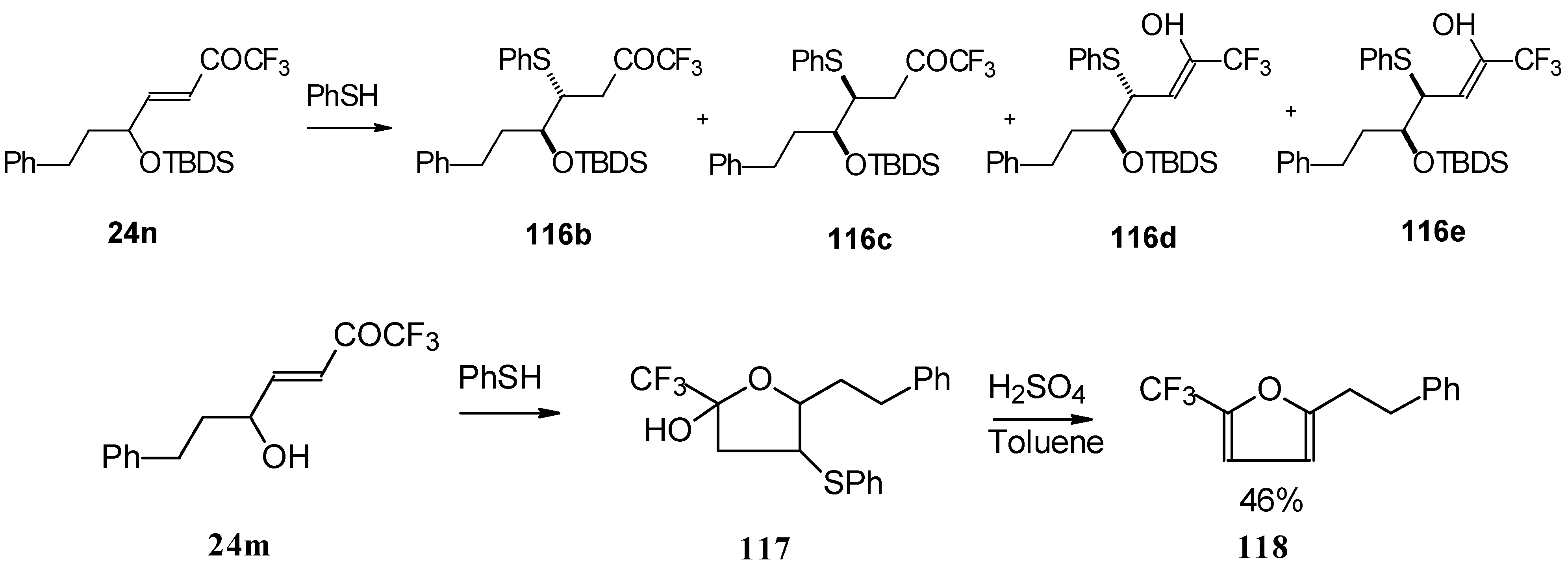
The thiol addition reaction to unsaturated CF
3 ketones was investigated (
Scheme 65 and
Scheme 66). The reaction proceeds as a 1,4-addition to give the corresponding keto sulphides [
50].
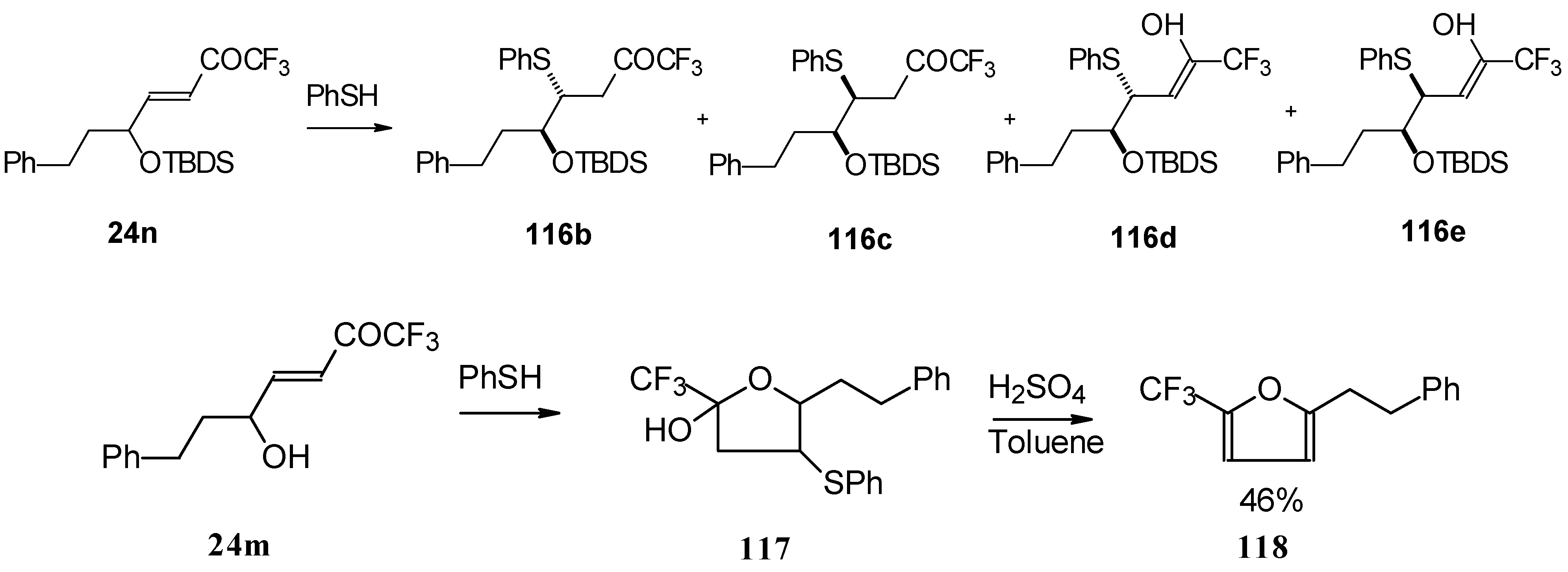
In the case of γ-silyloxy CF
3 enone a diastereomeric mixture of keto sulphides and enol sulphides in a 1/1 ratio was formed. In the case of γ-hydroxy CF
3 enone the corresponding tetrahydrofuran was isolated which is easily converted to the furan (
Scheme 66). Also the addition of glutathione was investigated [
50].
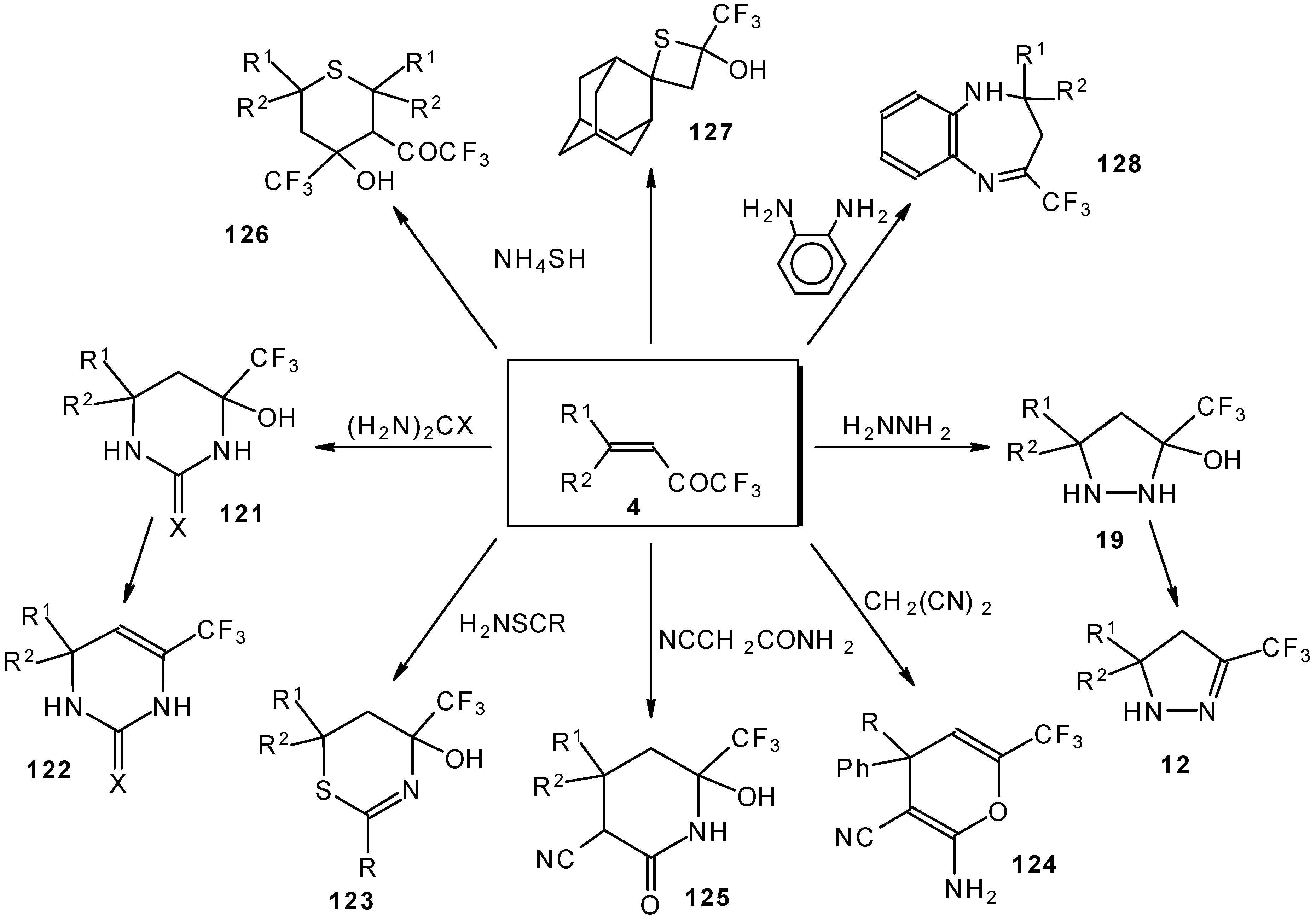
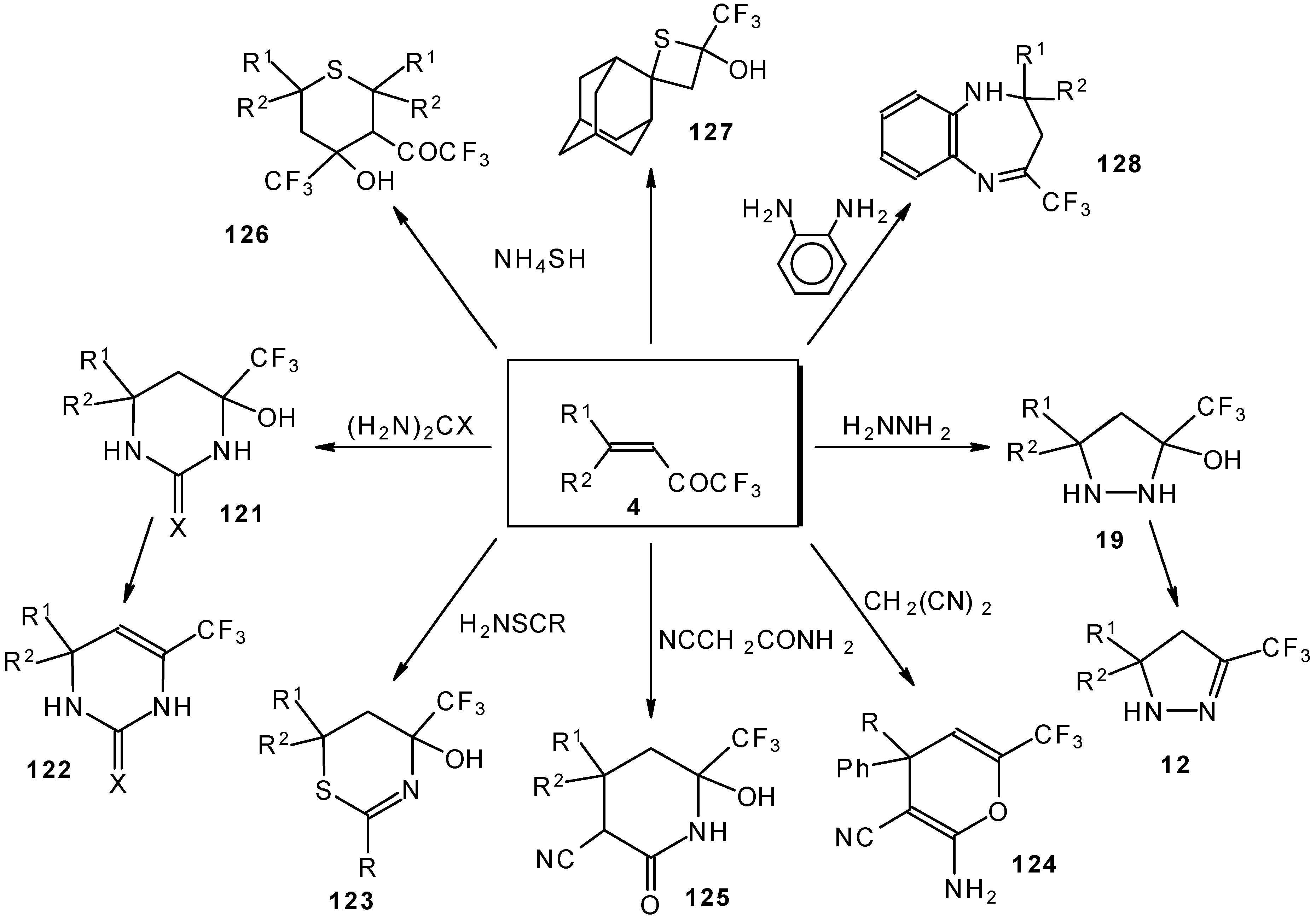
It was found that the reactions of some trifluoromethyl ketones of this type proceed easily with various nucleophiles. The CF
3 enones studied for the preparation of heterocycles have other substituents at the double bond and their reactivity is rather different (
Scheme 67).
Heterocyclic derivatives of various structures, including one with a heterocyclic spiro fragment and a small ring, are obtained.
These reactions open up the possibility of a one-step synthesis of CF
3-containing derivatives of pyrazolidines, pyrazolines, isoxazoles, thiazines, thiopyrans, thietane, pyrimidines, pyridines, pyrans and benzodiazepines (
Scheme 68).
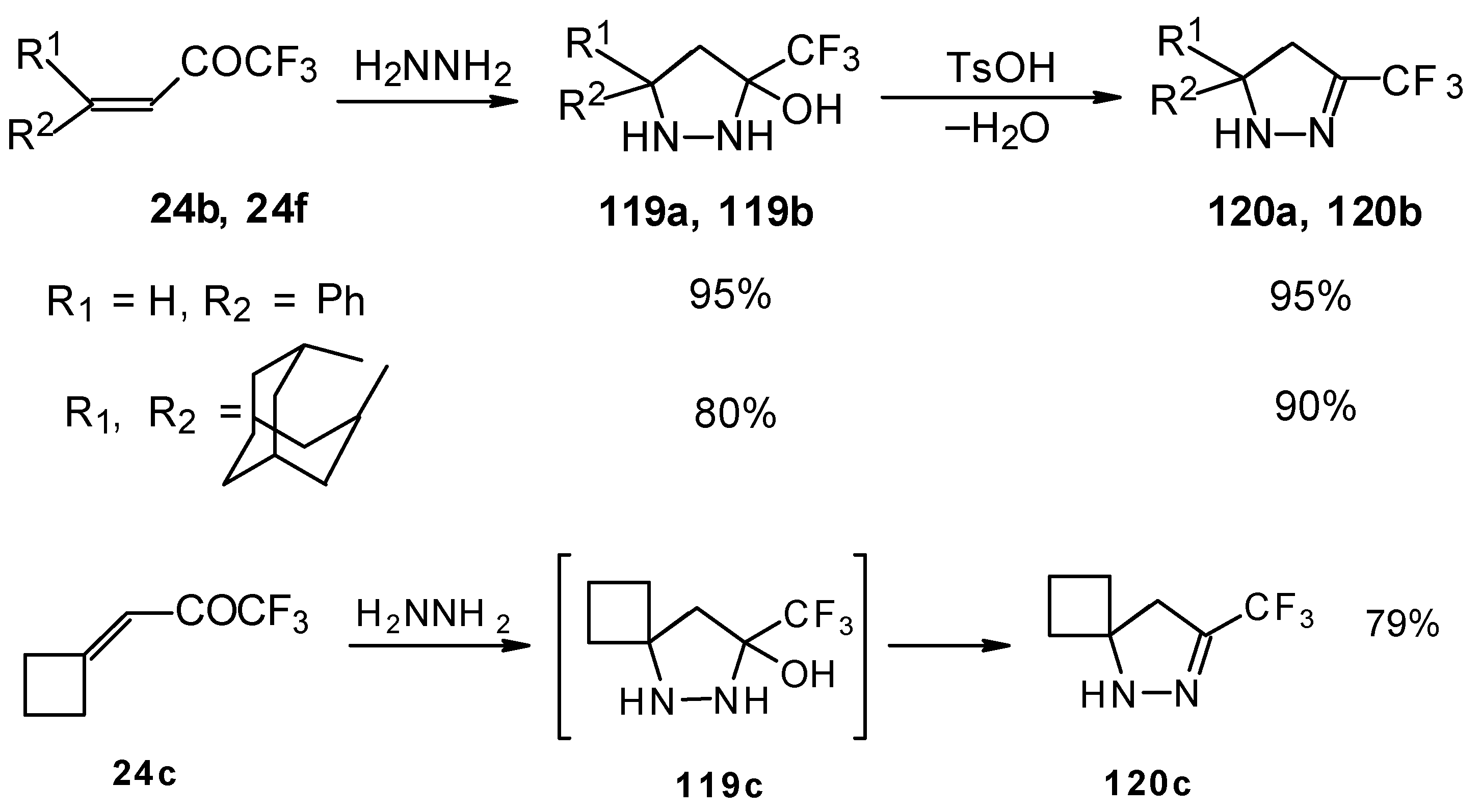
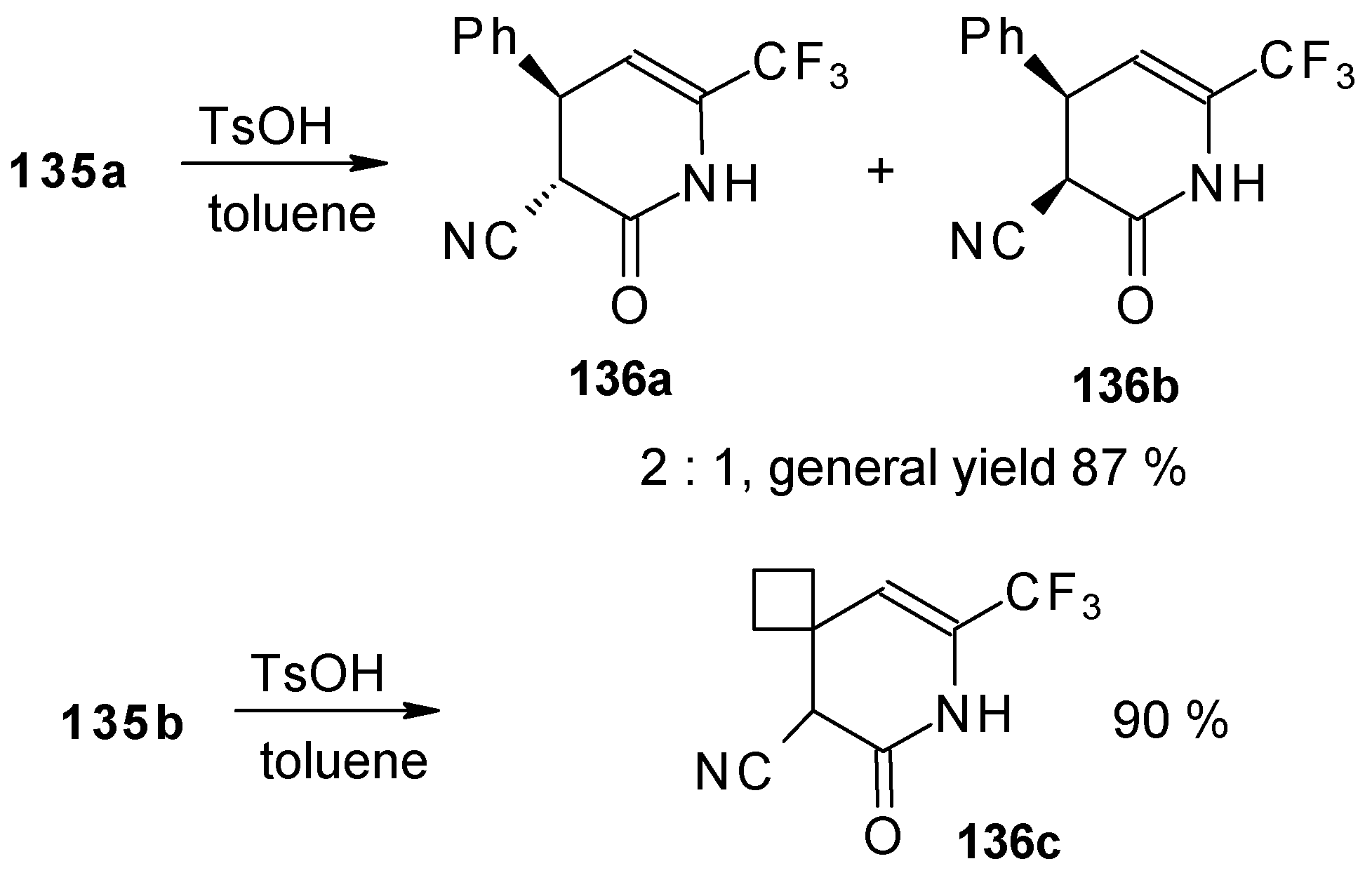
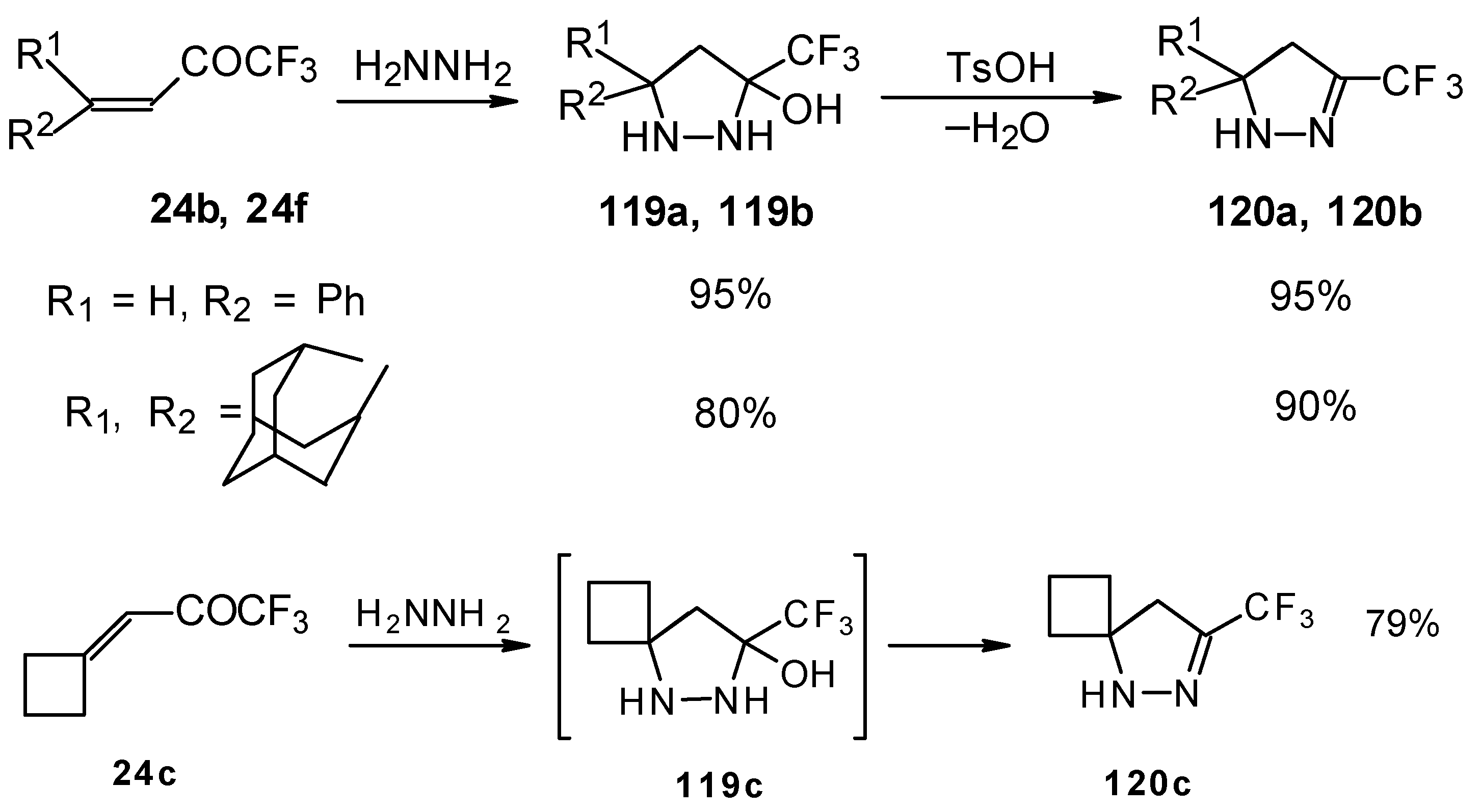
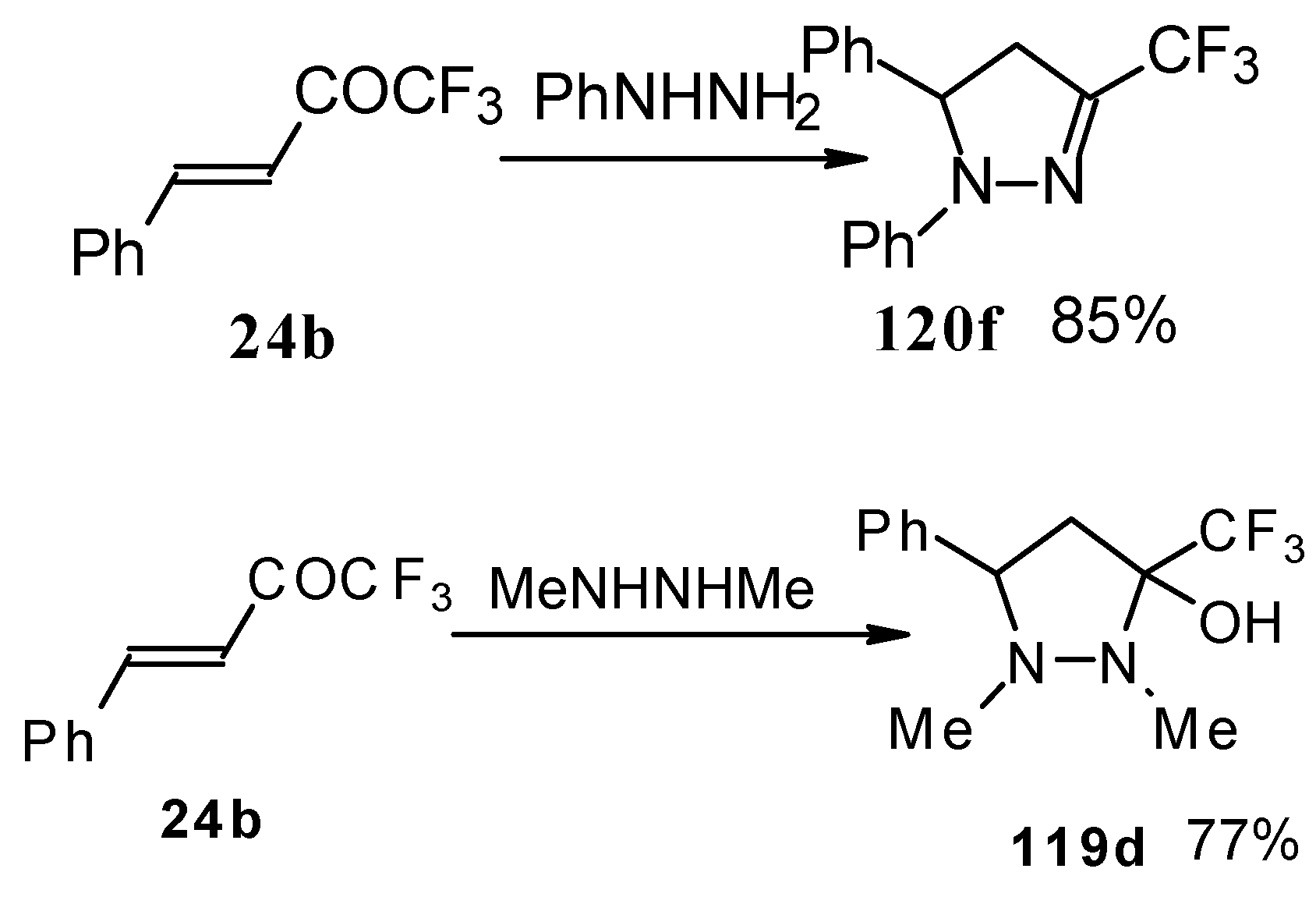
The reaction of these ketones with different hydrazines proceeds readily under mild conditions (
Scheme 69) [
105]. In the case of the hydrazine the reaction results in the formation of the corresponding pyrazolidines, in contrast to non-fluorinated ketones which react with hydrazine to give the corresponding pyrazolines. The pyrazolidines obtained contain a rather stable aminoalcohol fragment due to a electron withdrawing action of the CF
3 group, however it was found possible to transform them to pyrazolines by heating a toluene solution with a catalytic amount of
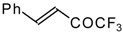
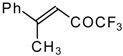
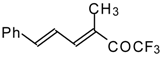
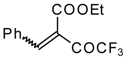
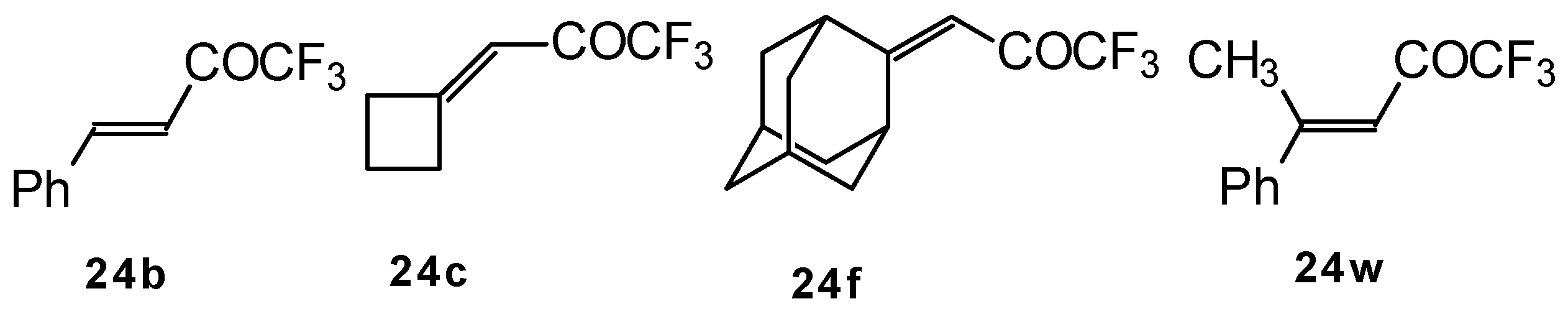
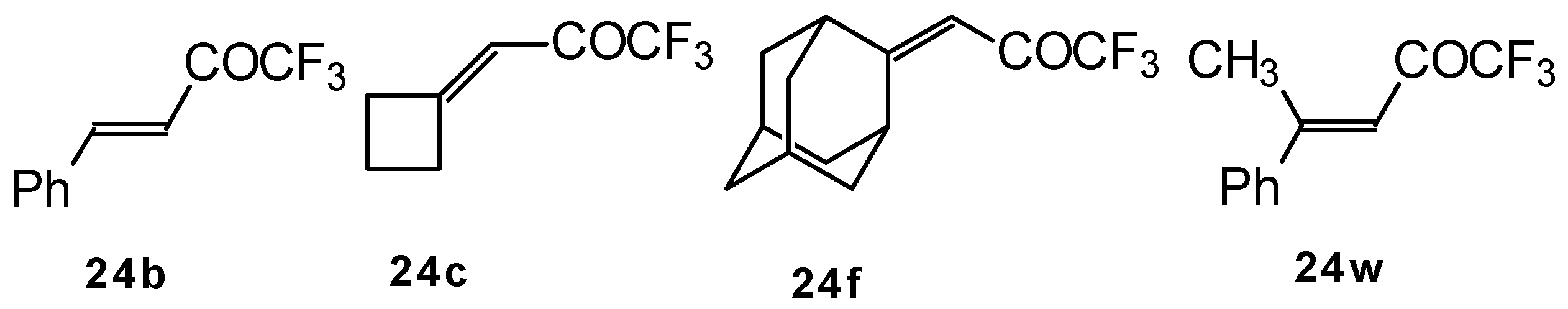
p-toluenesulfonic acid. In some cases the elimination of water proceeds spontaneously, for example in the case of the pyrazolidine derivative of the ketone containing a cyclobutane fragment. In the case of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b the pyrazolidines obtained eliminated water spontaneously, but this transformation required
ca. two months [
105].
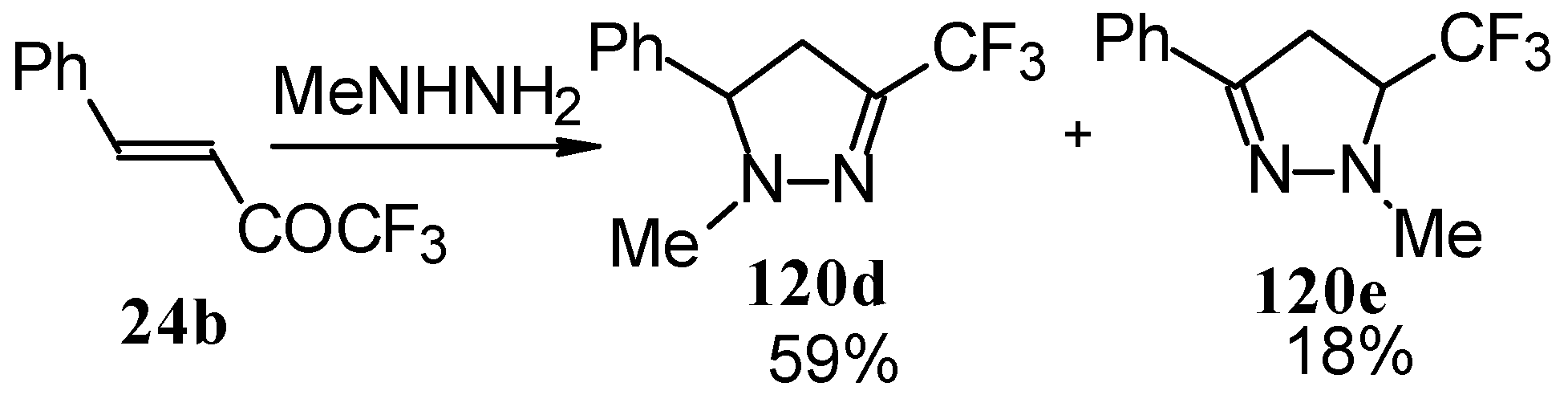
The reaction of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b with different substituted hydrazines was studied. The heterocyclization can proceed by two routes. The first route is a primary Michael addition of a nucleophile to an enone with subsequent cyclization, and the second route is a primary addition of a nucleophile at the carbonyl group with subsequent cyclization. However, it seems possible that both routes may take place depending on the reaction conditions and the structure of the hydrazine [
105].
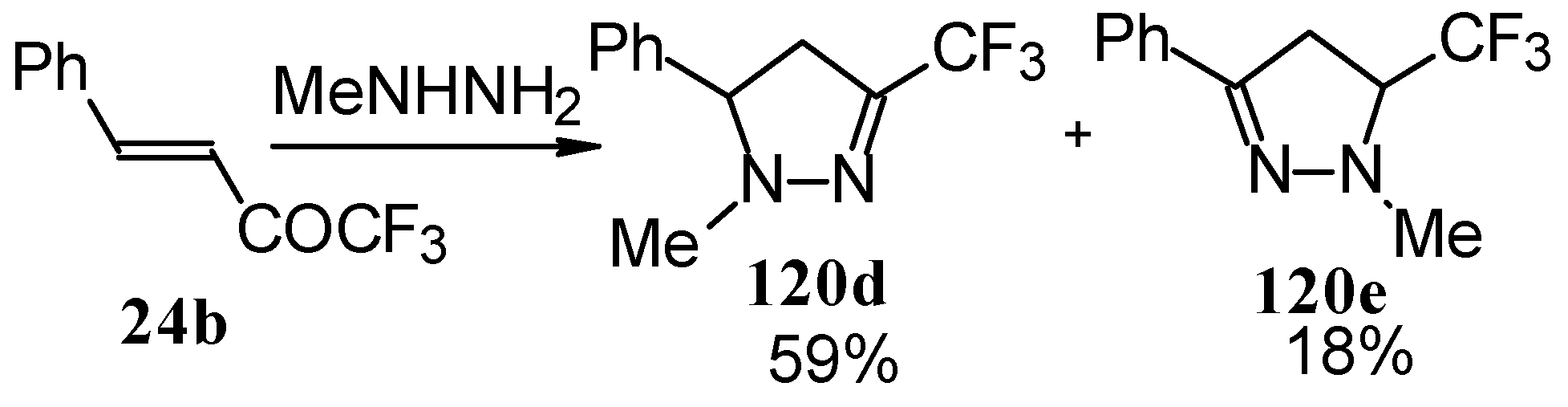
In the case of methyl hydrazine two possible regioisomers of the pyrazoline were isolated in a ~3:1 ratio (
Scheme 70). It is possible that this regiochemistry observed is due to both the similar nucleophilicity of the two nucleophilic centers of methylhydrazine and the possibility of attack of the nucleophile at both the double bond and the carbonyl group.
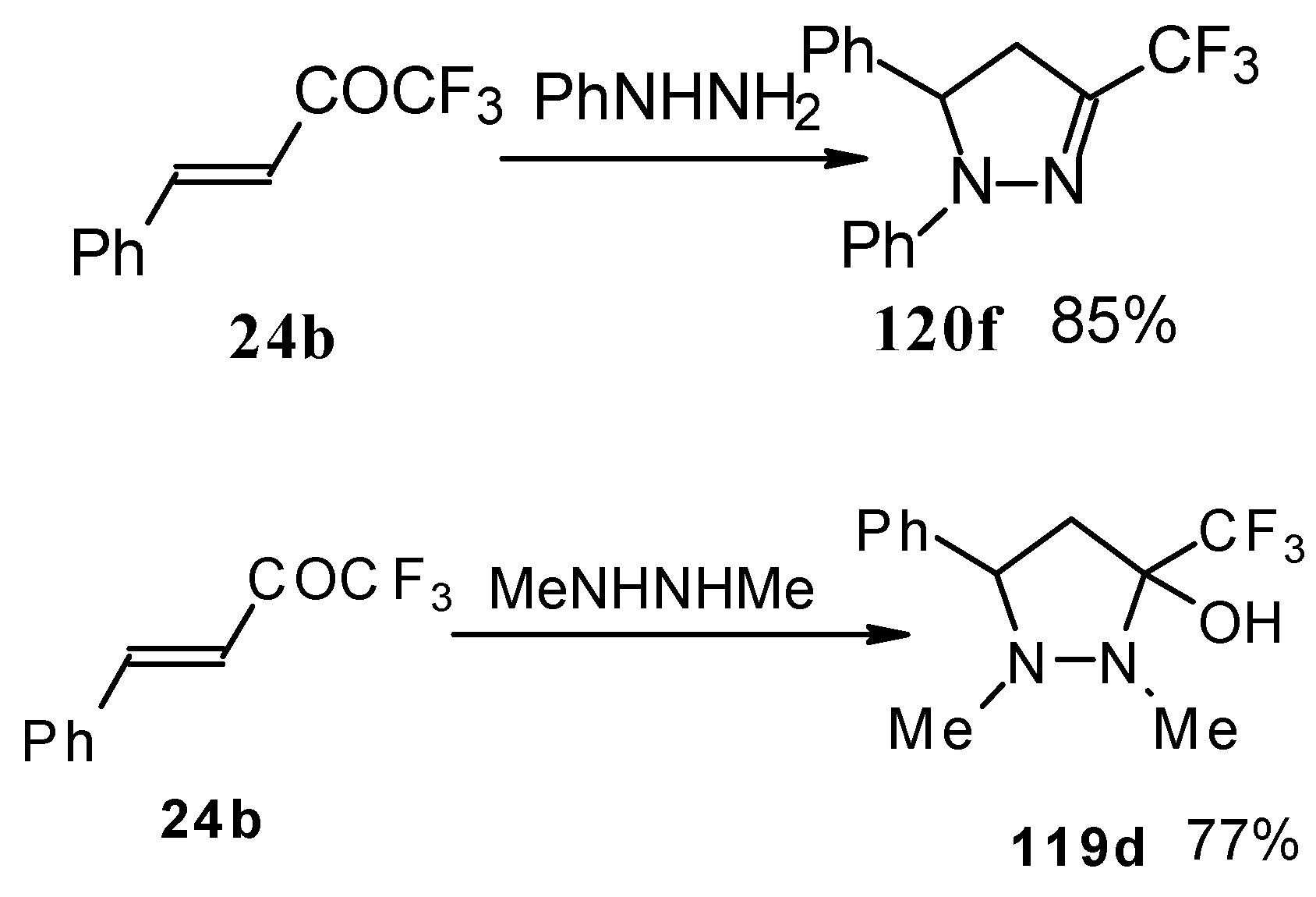
In the case of phenyl hydrazine, reaction leads to formation of the corresponding pirazoline and here the primary attack of the nucleophile takes place at the carbonyl group of the ketone (
Scheme71) [
105]. In the case of 1,2-dimethylhydrazine the corresponding pirazolidine was obtained in good yield.
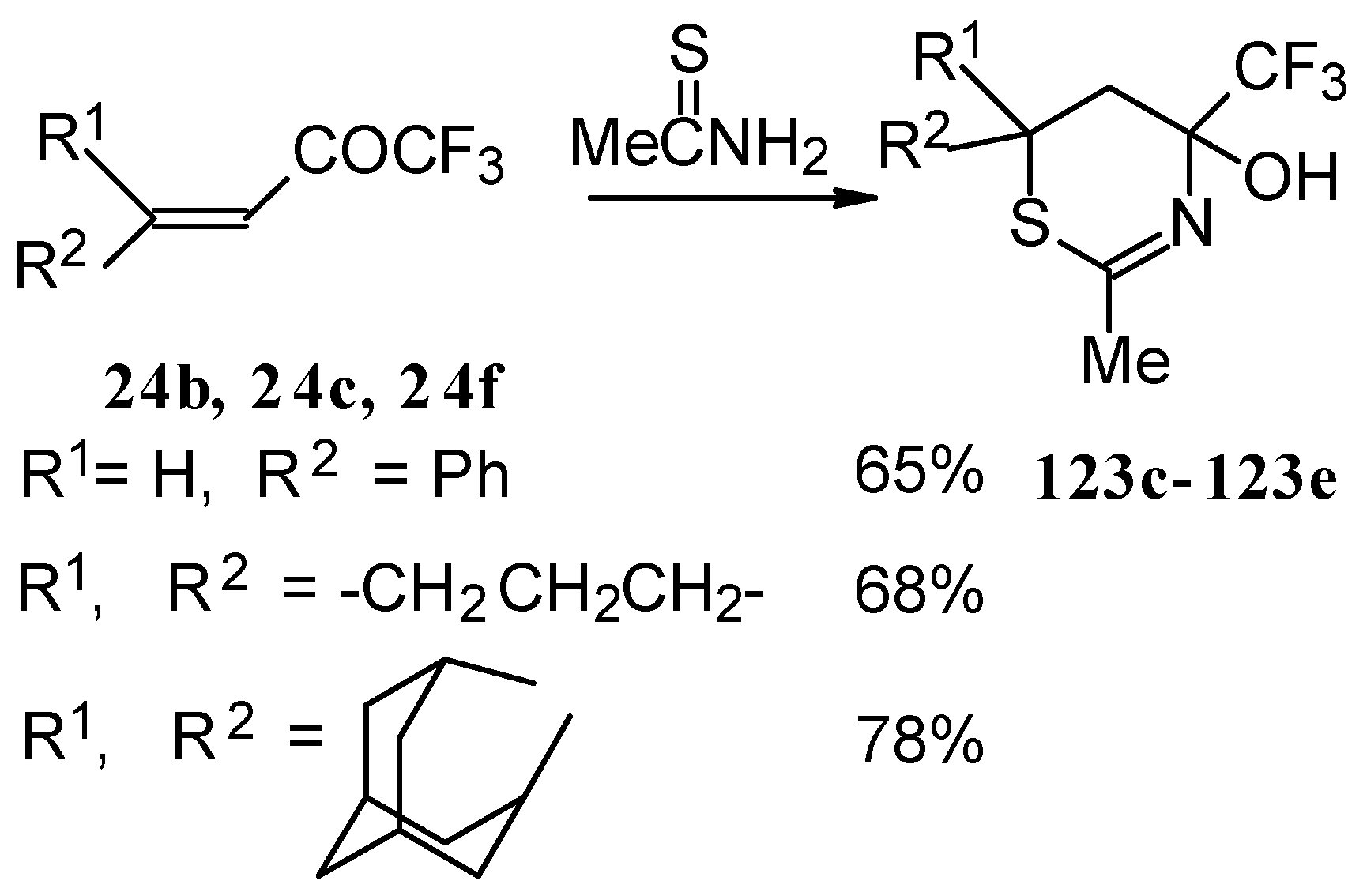
The reaction with various urea derivatives results in tetra- or dihydropyrimidines, bearing a CF
3 substituent, in excellent yields (
Scheme 72) [
106].
The reaction of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
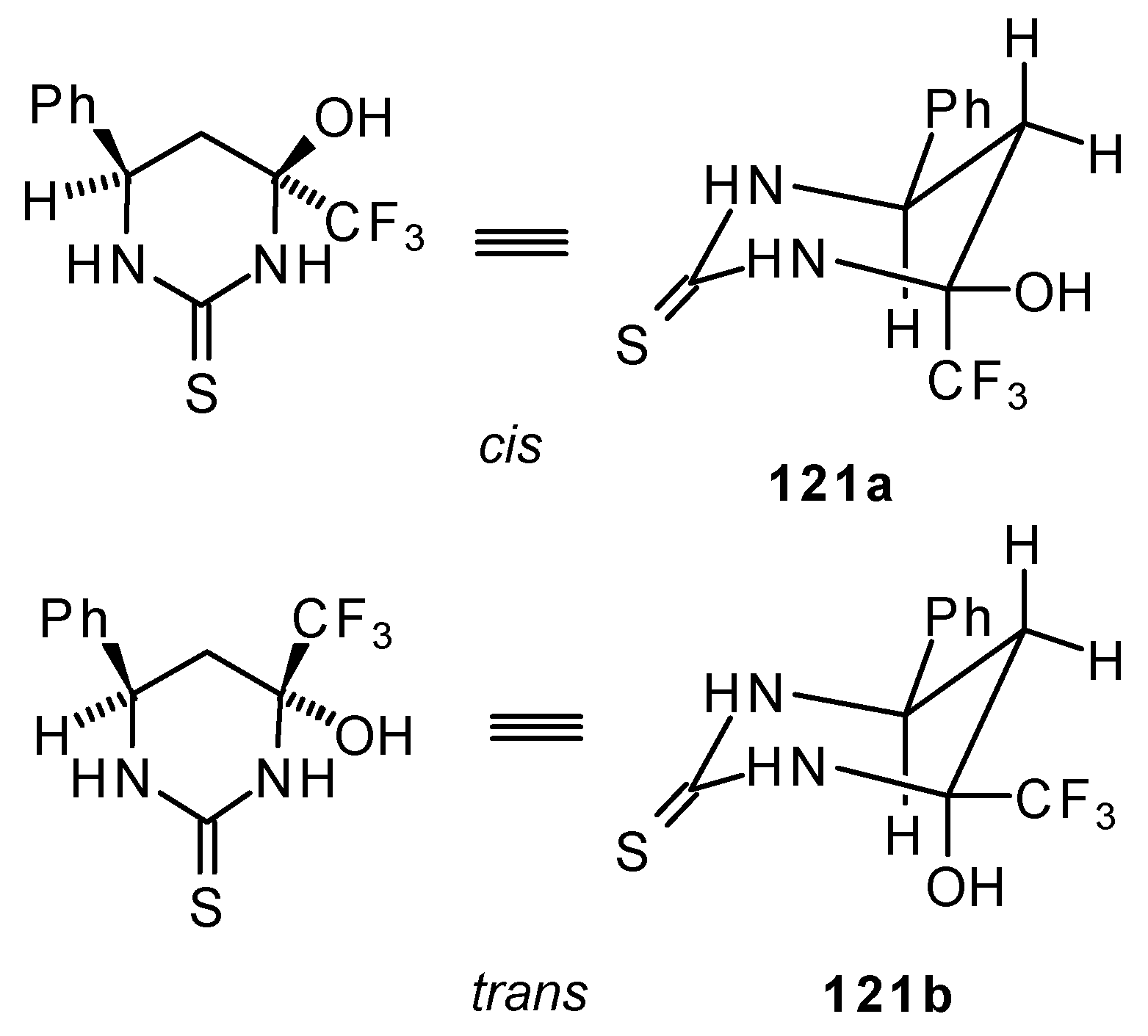
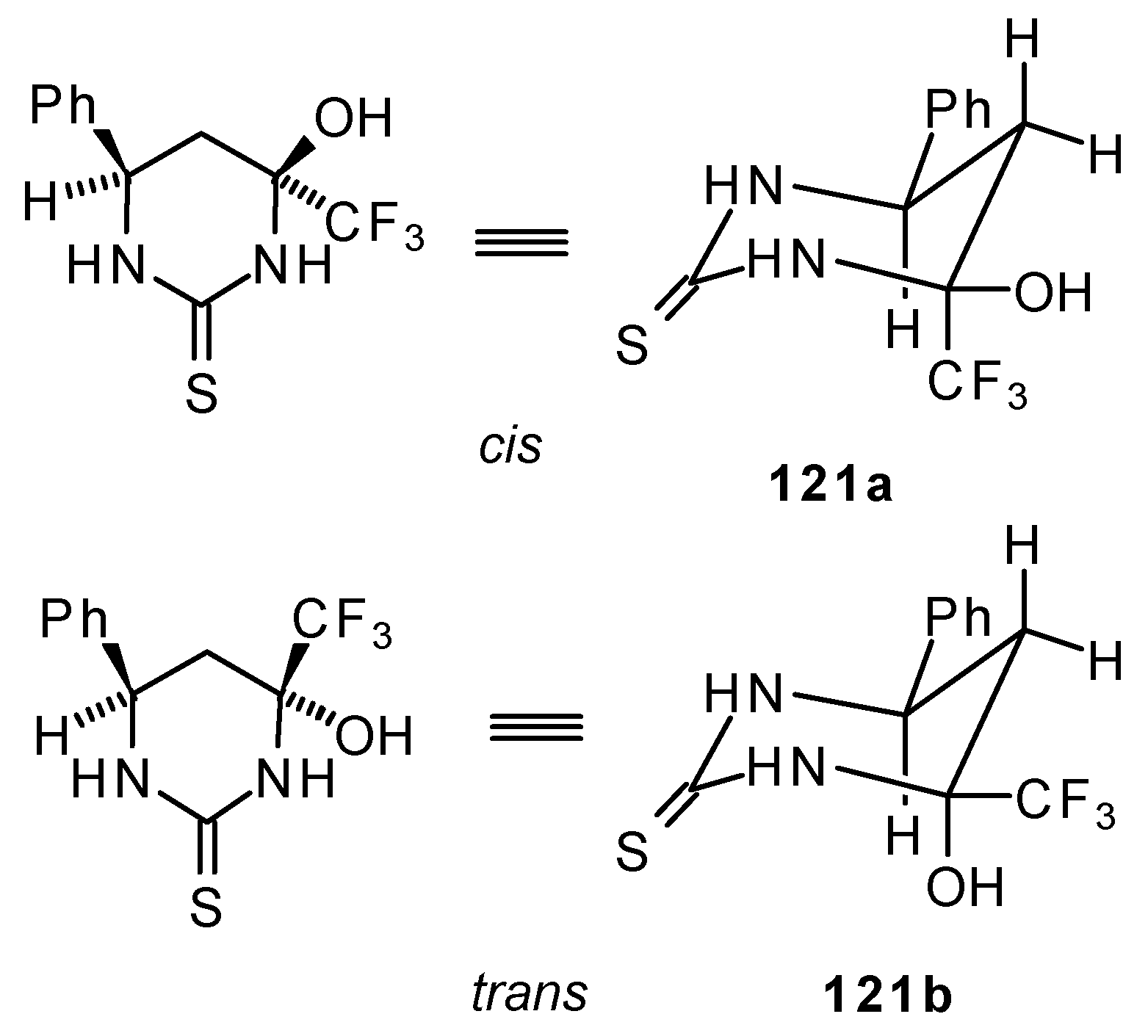
24b with urea derivatives could result in a mixture of two possible diastereoisomers of tetrahydropyrimidines with
cis and
tras arranged phenyl and CF
3 groups (
Scheme 73).
However the reaction with urea and thiourea proceeds stereoselectively to give preferably isomers having diequatorially arranged Ph and CF3 moieties. Only trace amounts of other diastereoisomers are observed. The configuration of the products was determined by NOE data and X-ray crystallography.
It is possible that the transition state for the cyclization of β-trifluoroacetylstyrene
24b to the corresponding pyrimidine has a chair conformation with the bulky groups (Ph and CF
3) equatorial. It is known that a trifluoromethyl moiety is almost double the bulk of a methyl group and does not fall much behind that of a
tert-butyl group. The energy required for the conformational promotion from the equatorial to the axial position for a trifluoromethyl group is more, than for an isopropyl group [
107].
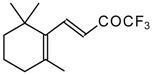
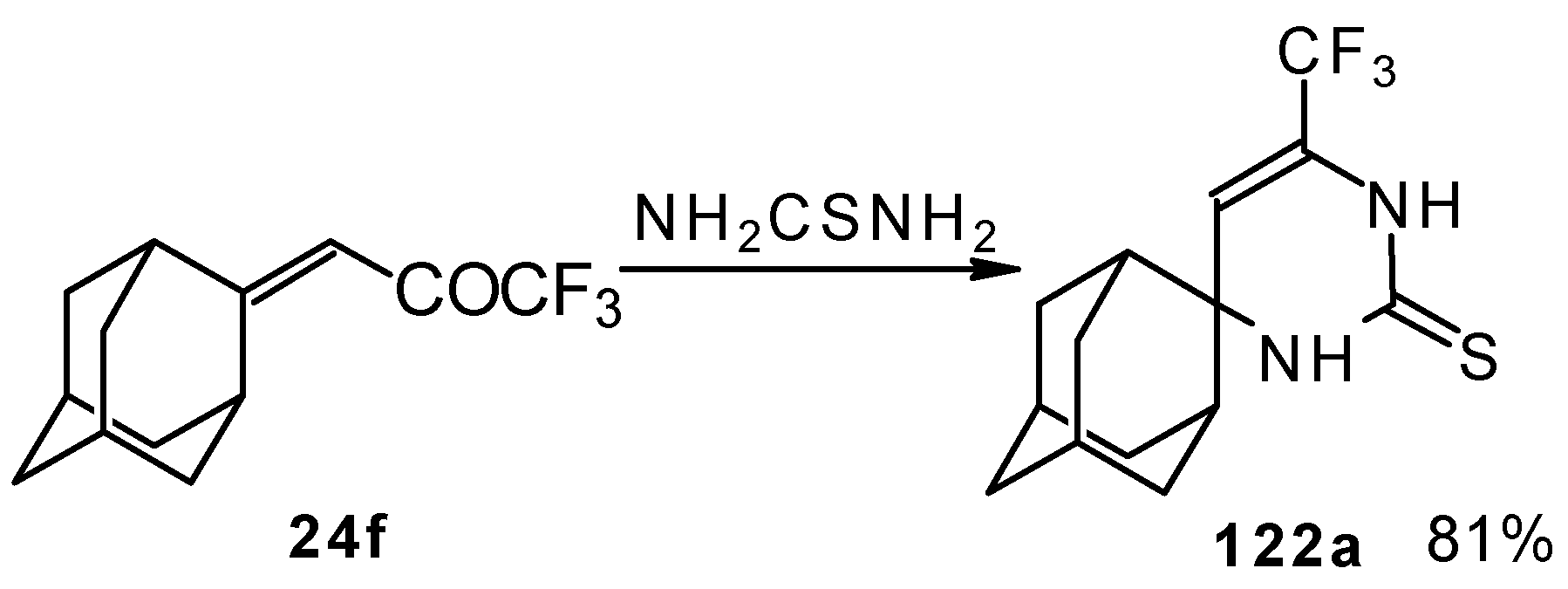
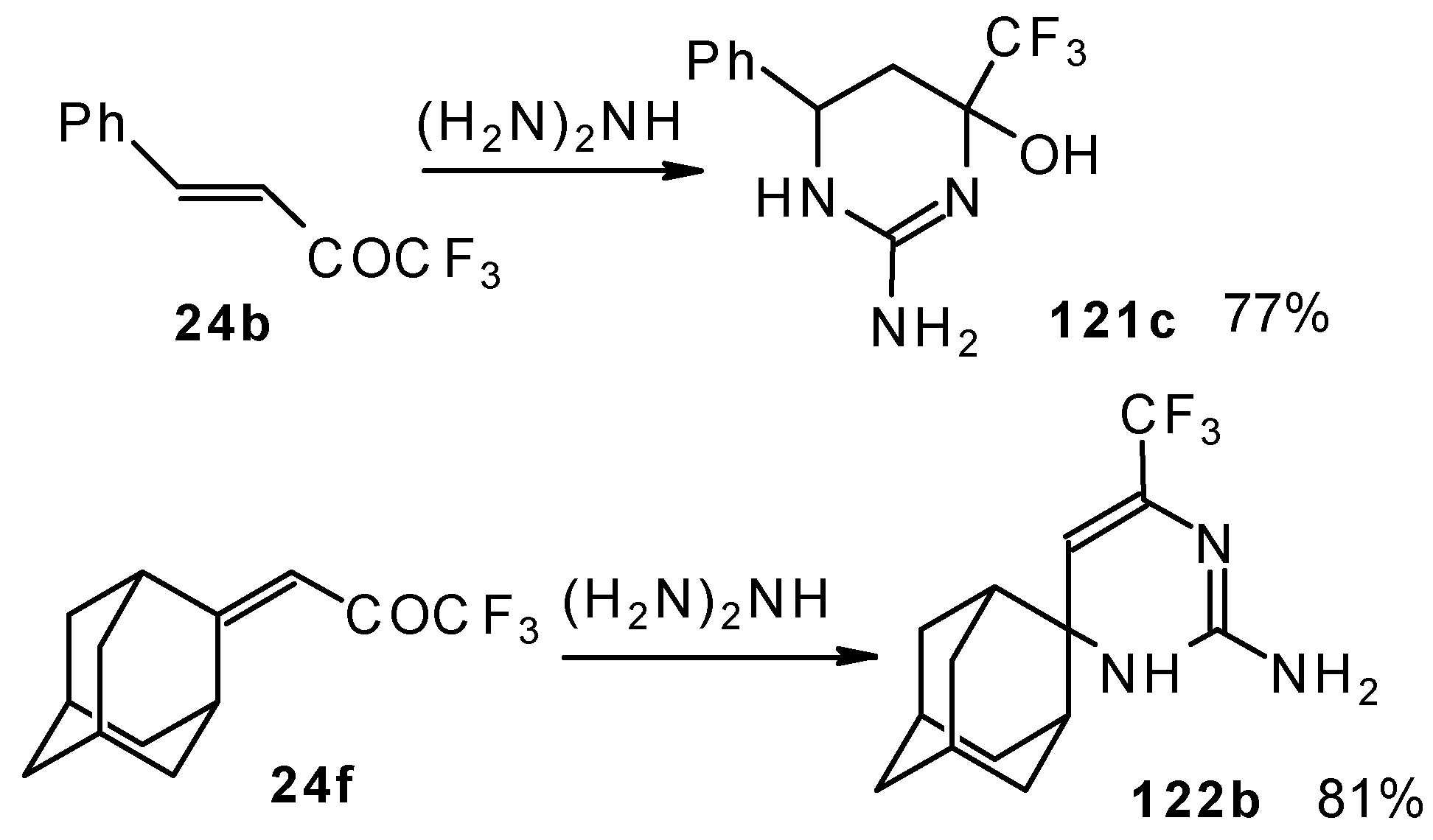
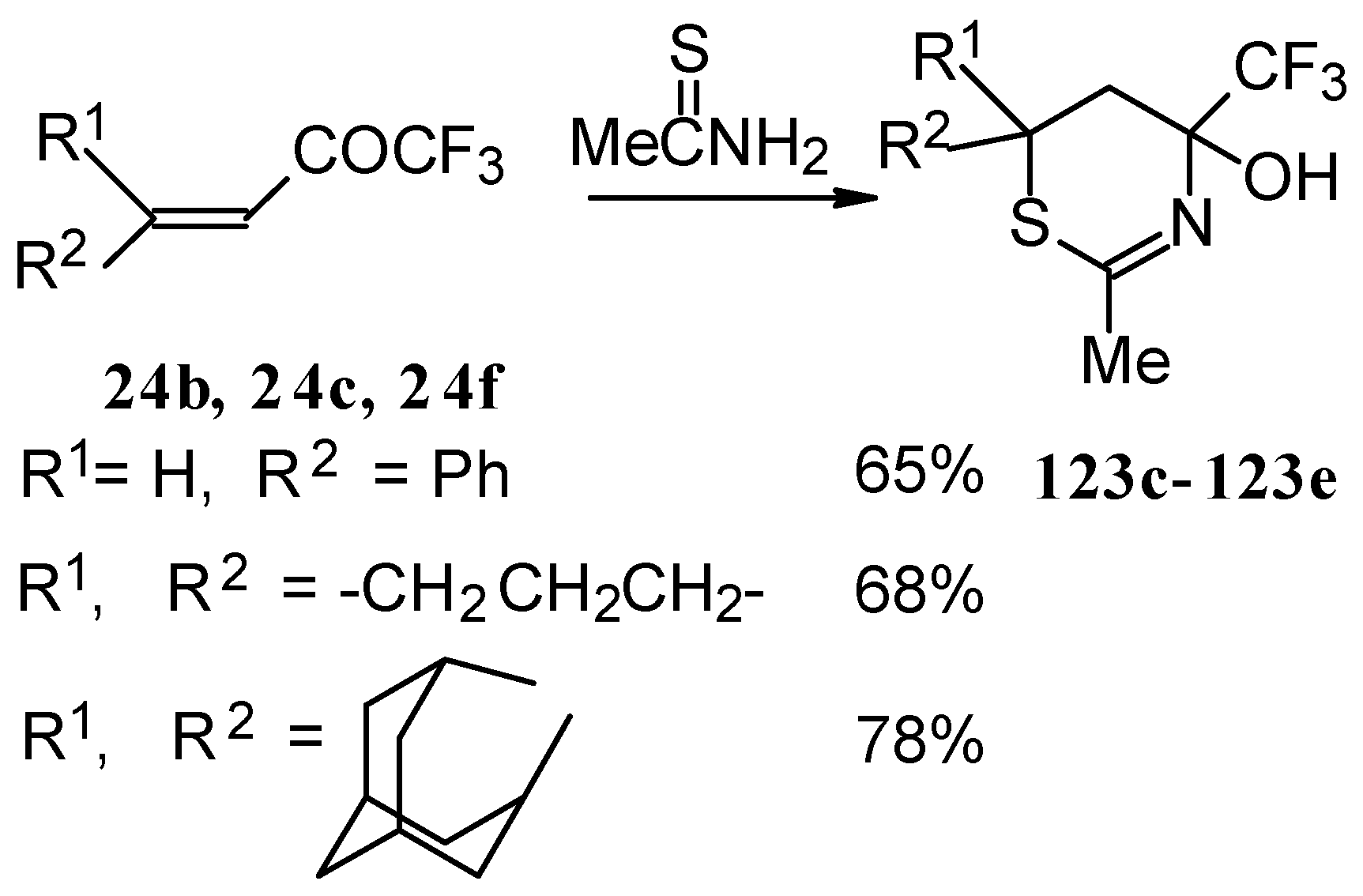
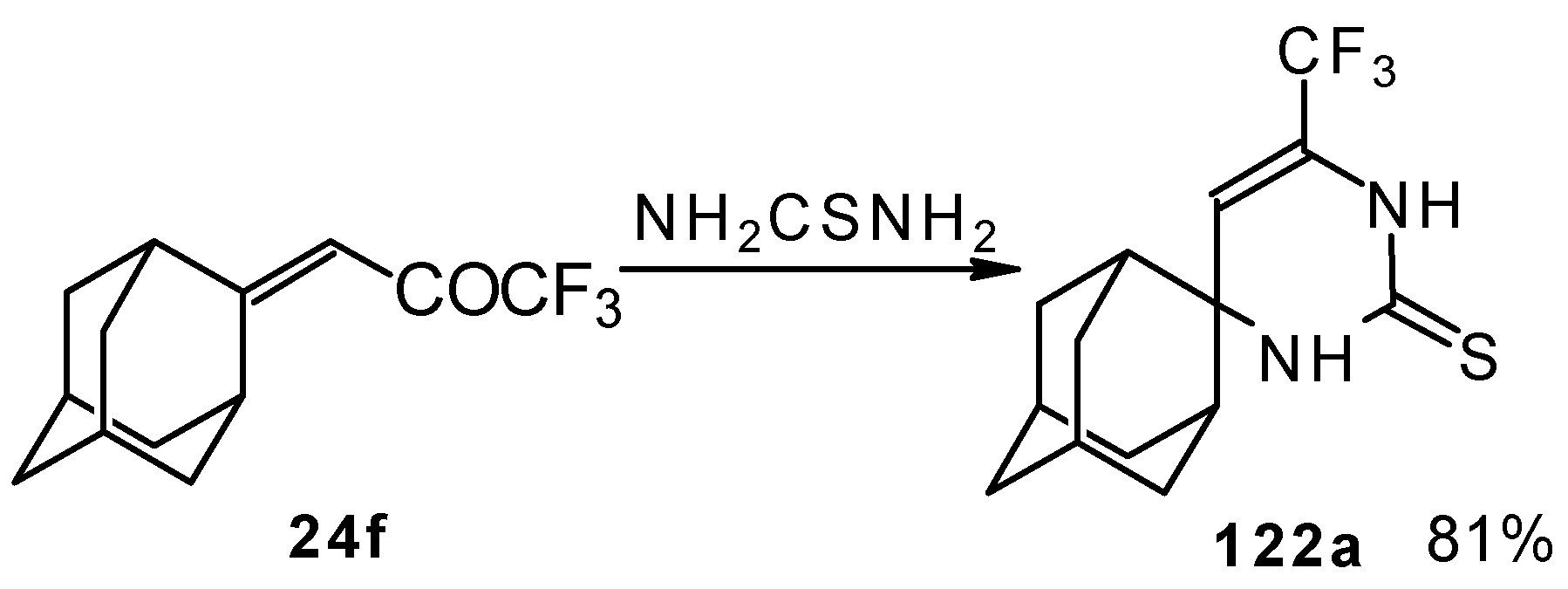
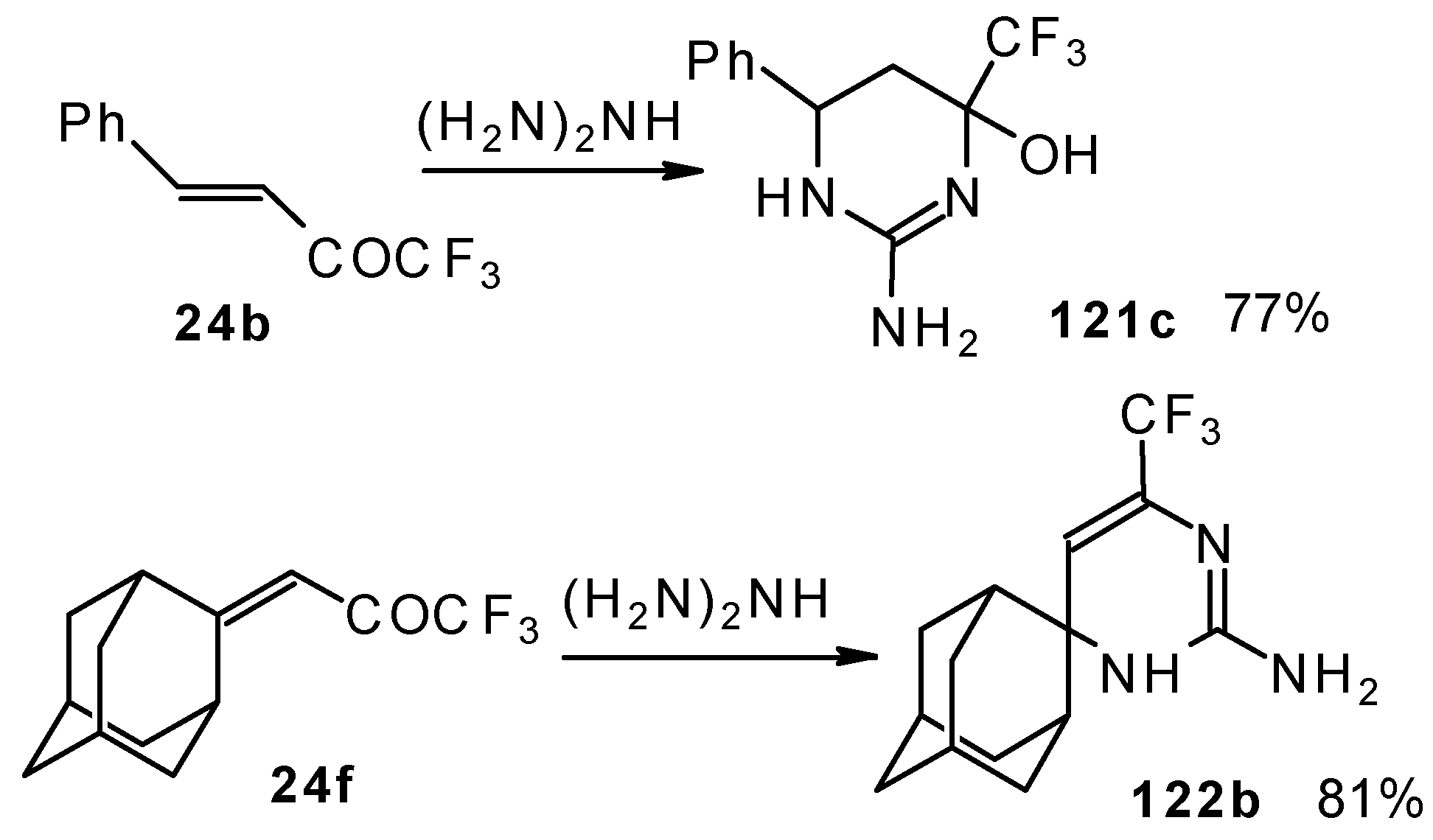
In the case of an adamantane containing CF
3 ketone
24f reaction with thiourea under the same conditions gives rise to the corresponding dihydropyrimidinethione (
Scheme 74).
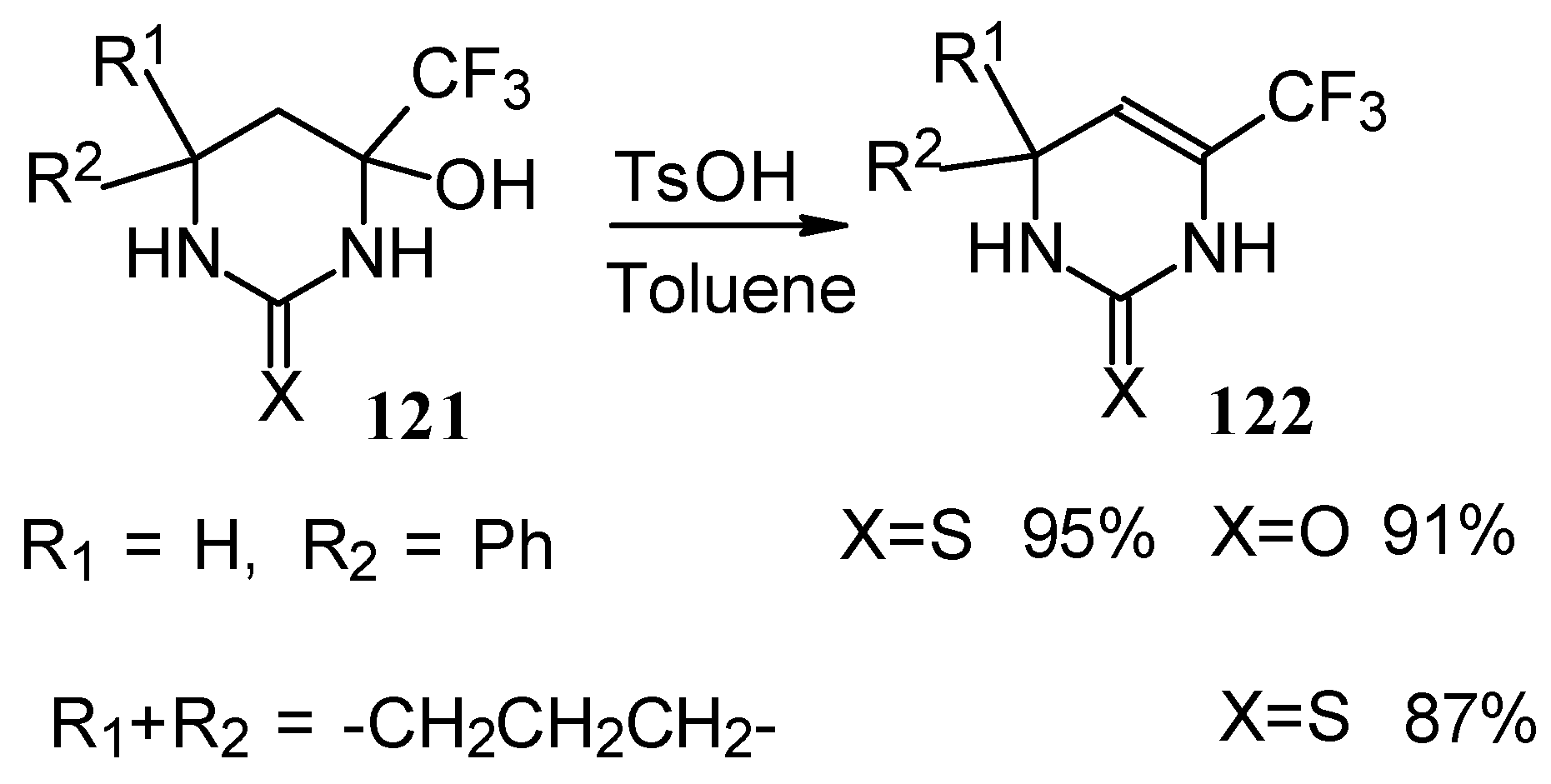
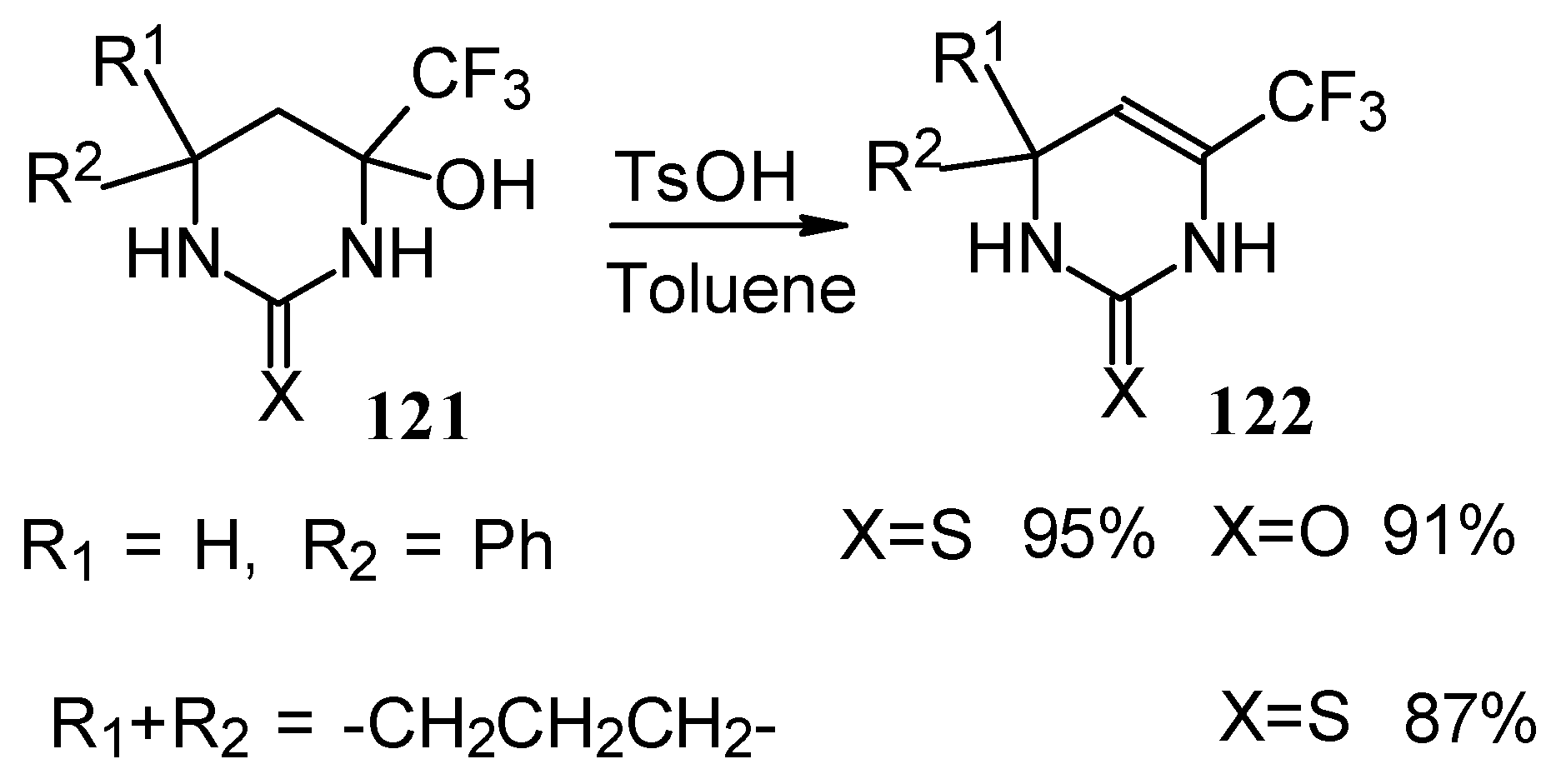
The possibility of water elimination from tetrahydropyrimidines to transform them to the corresponding dihydroderivatives has been investigated (
Scheme 75). This reaction proceeds smoothly when the compounds are heated in toluene with catalytic amounts of
p-toluenesulfonic acid.
Reactions with guanidine proceed analogously to give the corresponding CF
3-containing aminopyrimidines (
Scheme 76).
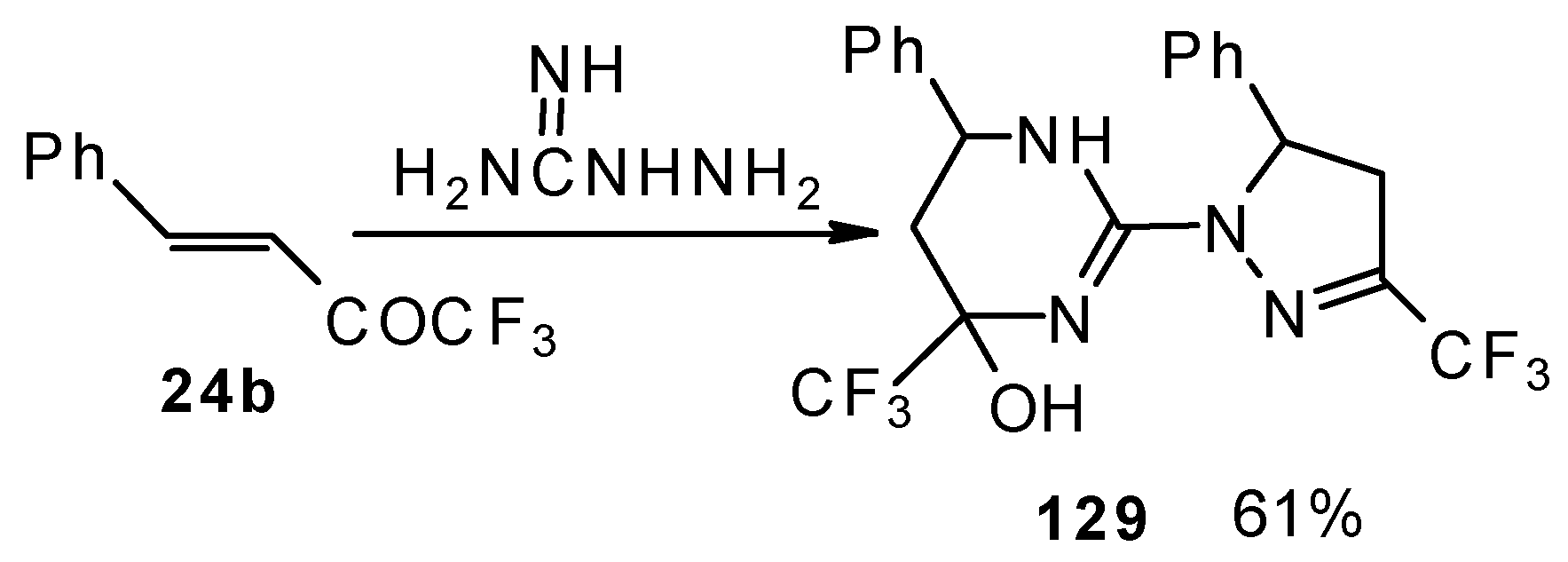
The reaction of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b with aminoguanidine proceeds unusually, to give the product having two heterocyclic fragments: tetrahydropyrimidine and pyrazoline (
Scheme 77). Tetrahydropyrimidines with a CF
3 group are more stable to elimination of water than the corresponding CF
3-containing pyrazolidines and this tendency is also realized in the case of this compound.
The formation of CF
3-containing dihydrothiazines [
108] by reaction of these ketones with thiourea under acidic conditions was also investigated(
Scheme 78). The reaction proceeds regiospecifically to give only one possible isomer of thiazine, the product of addition of sulphur to the double bond and nitrogen to the carbonyl group. We believe that this reactivity is according to the hard and soft acid and base principles [
109].
The reaction of ethylenic CF
3 ketones with thioacetamide proceeds in the similar way. All CF
3 substituted thiazines have stable aminoalcohol fragments and do not eliminate water under acidic conditions (
Scheme 79).
The reaction of CF
3-containing ketones with malononitrile under a variety of conditions was investigated (
Scheme 80,
Scheme 81,
Scheme 82,
Scheme 83 and
Scheme 84) [
110]. The reaction path is very sensitive to the conditions of the reaction and the structure of the starting ketone. 1,1,1-Trifluoro-4-phenyl-3-buten-2-one
24b reacts with malononitrile and a catalytic amount of pyrrolidine or triethylamine to form the corresponding pyran
124a (
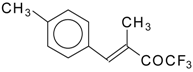
Scheme 80). The reaction of 1,1,1-trifluoro-4-methyl-4-phenyl-3-buten-2-one
24w with malononitrile under the same conditions proceeds analogously.
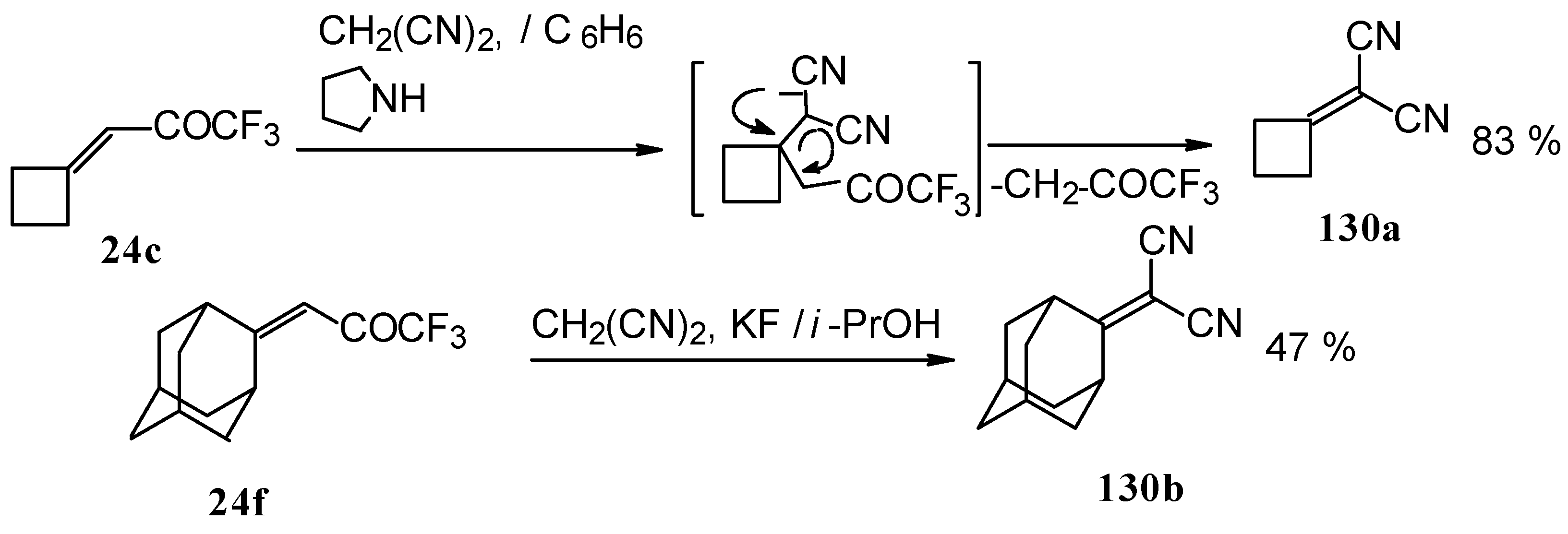
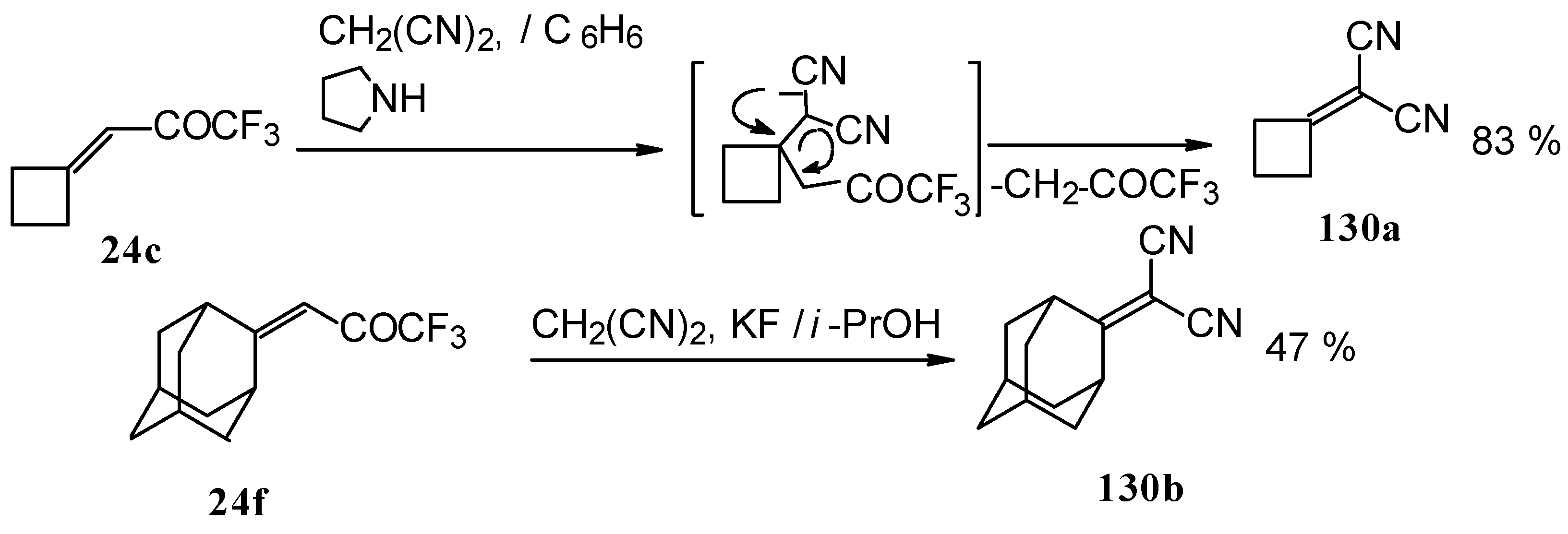
Ketones having an exocyclic double bond react very differently. Only the corresponding alkylidene dinitriles were obtained under the same conditions (
Scheme 81).
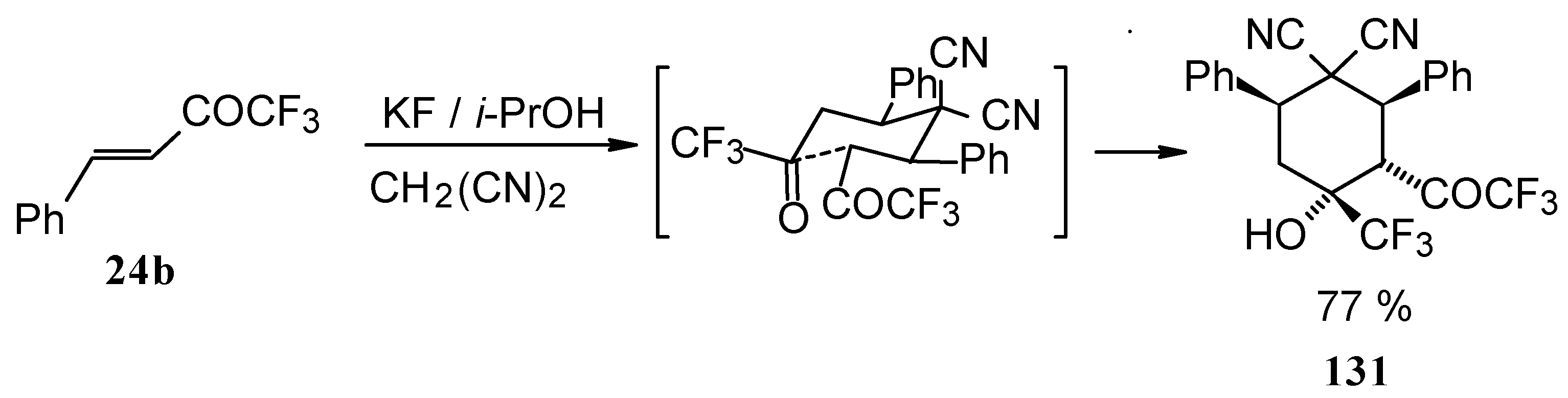
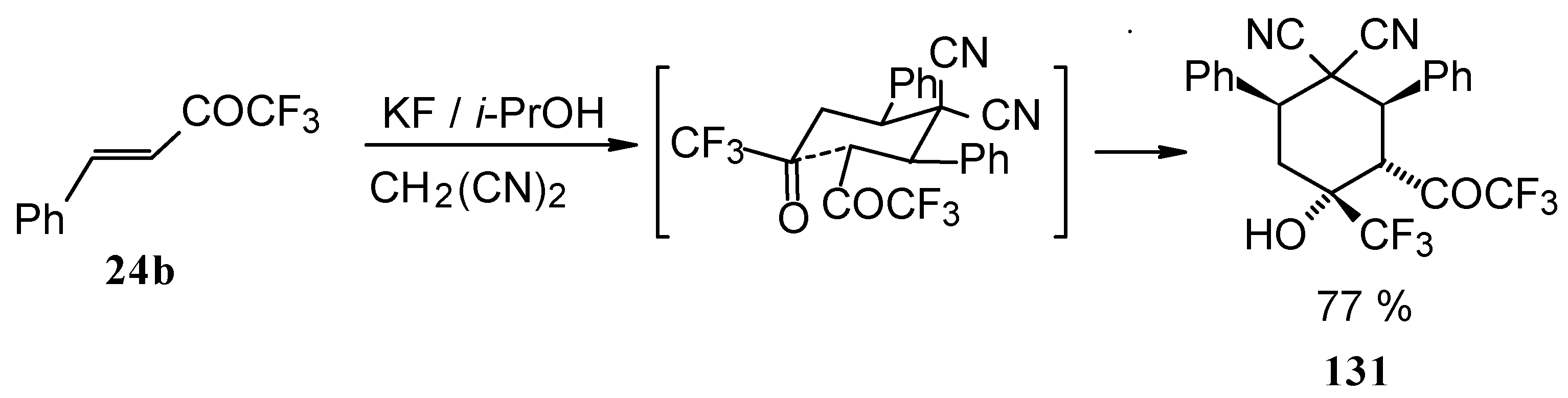
When the reaction of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b with malononitrile was carried out with potassium fluoride as a base the corresponding cyclohexanol - the product of Michael addition of malononitrile to the double bond of two molecules of this ketone with subsequent aldol condensation was obtained (
Scheme 82). The reaction proceeds stereospecifically to give only one isomer out of a possible eight.
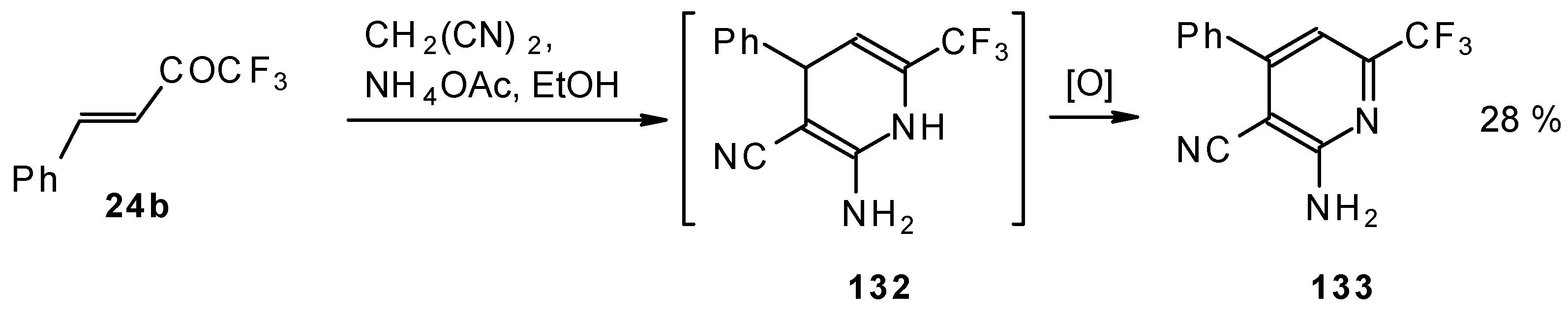
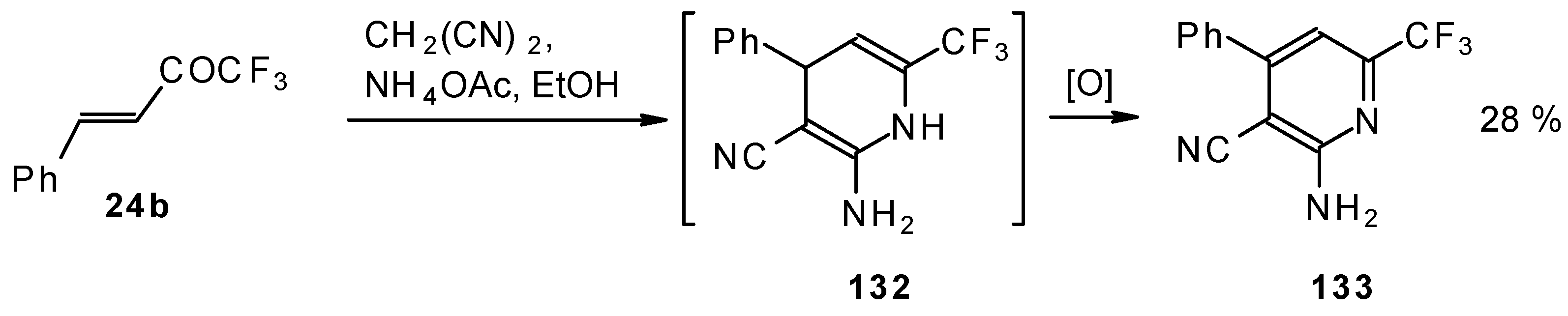
In the case of the reaction of malononitrile in the presence of ammonium acetate only in the case of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b was the corresponding pyridine isolated (
Scheme 83).
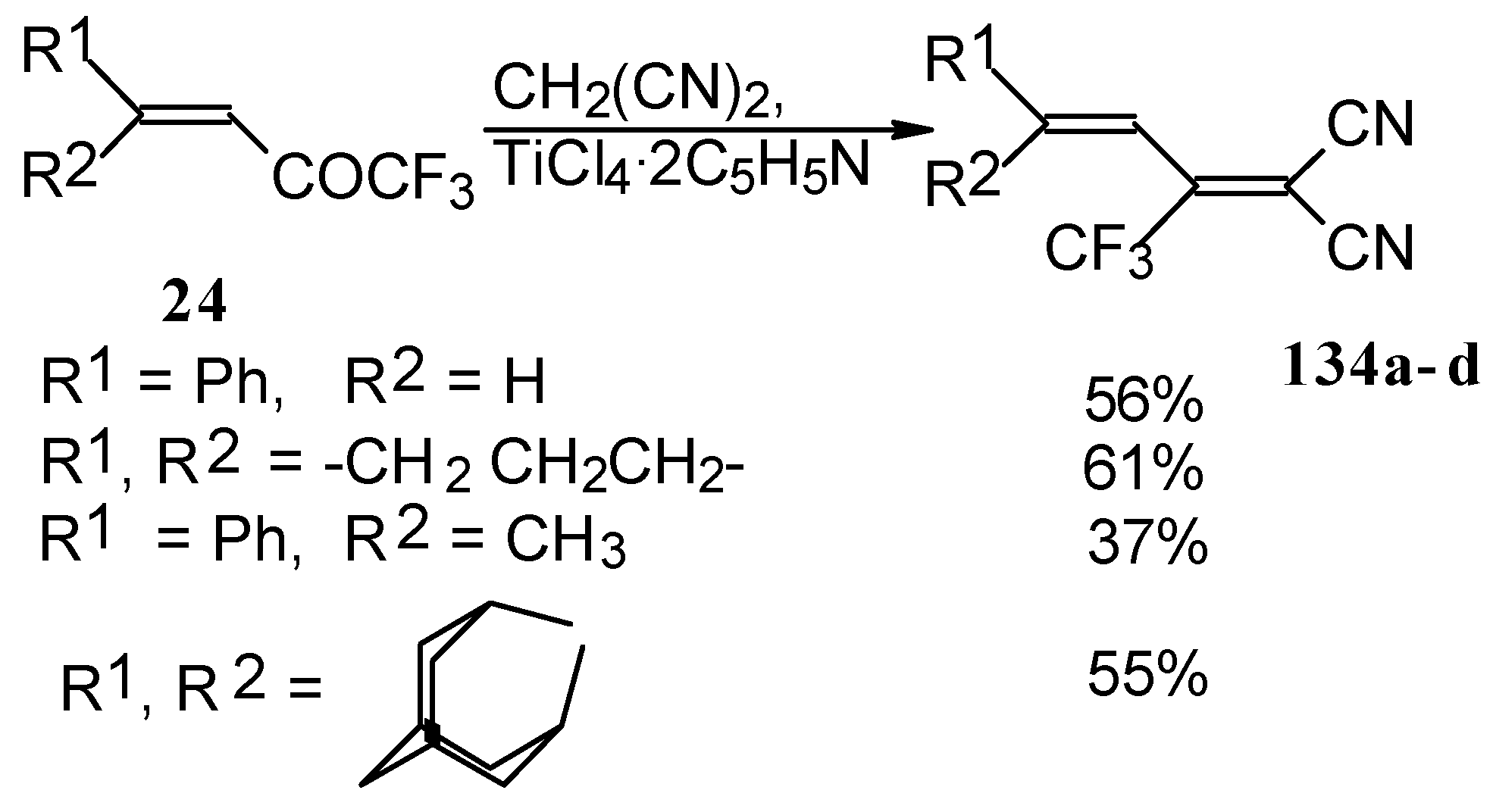
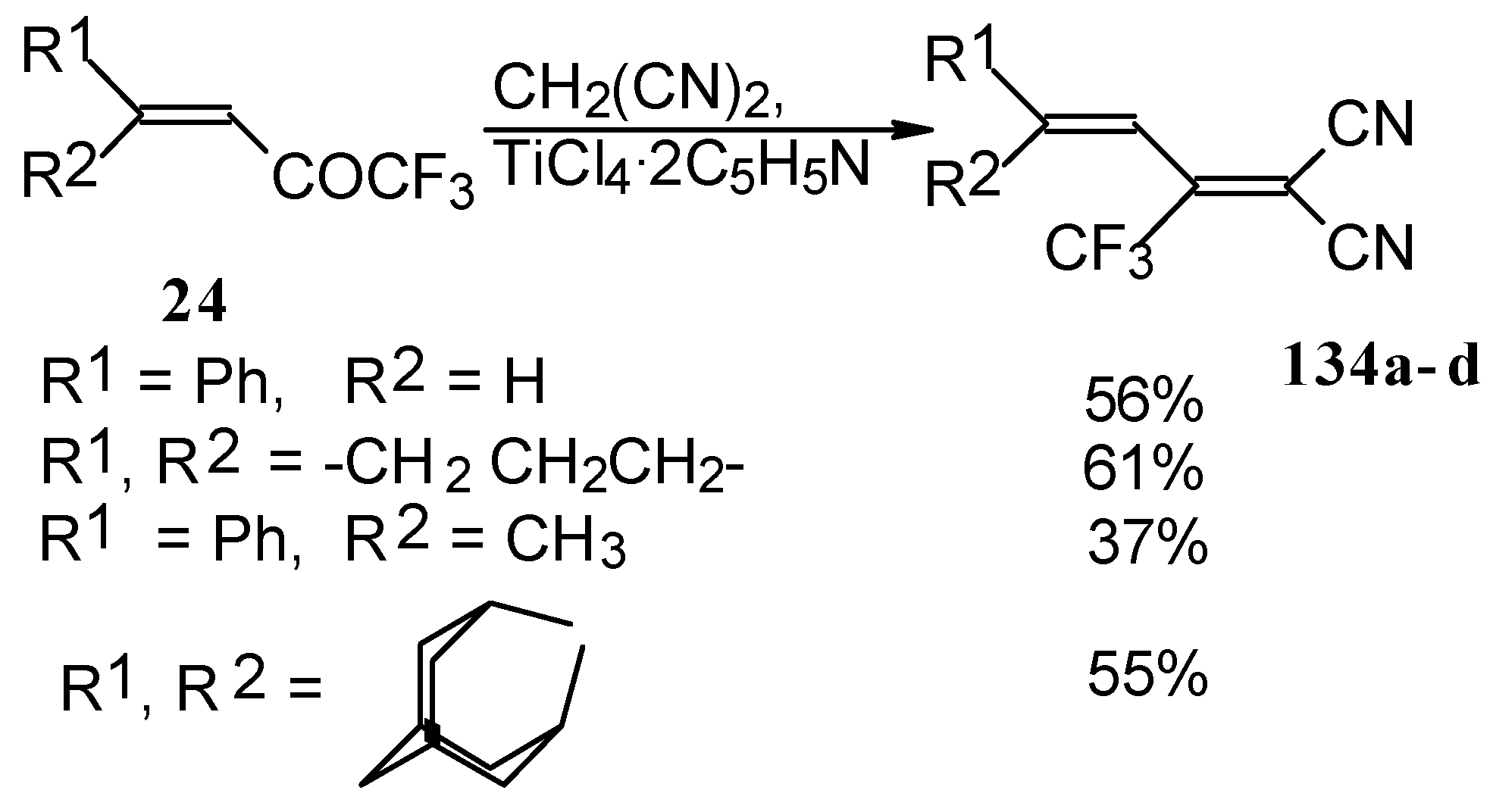
The reaction with malononitrile as a Knoevenagel condensation was also investigated (
Scheme 84). In the case of the use of the TiCl
4/2Py complex in dichloromethane the corresponding 1,1-dicyano-2-trifluoromethyl-1,3-butadienes were prepared in 37-61% yield [
110].
In the case of the reaction with cyanoacetamide with a catalytic amount of KF the corresponding tetrahydropyridines were isolated. This reaction proceeds stereospecifically (
Scheme 85).
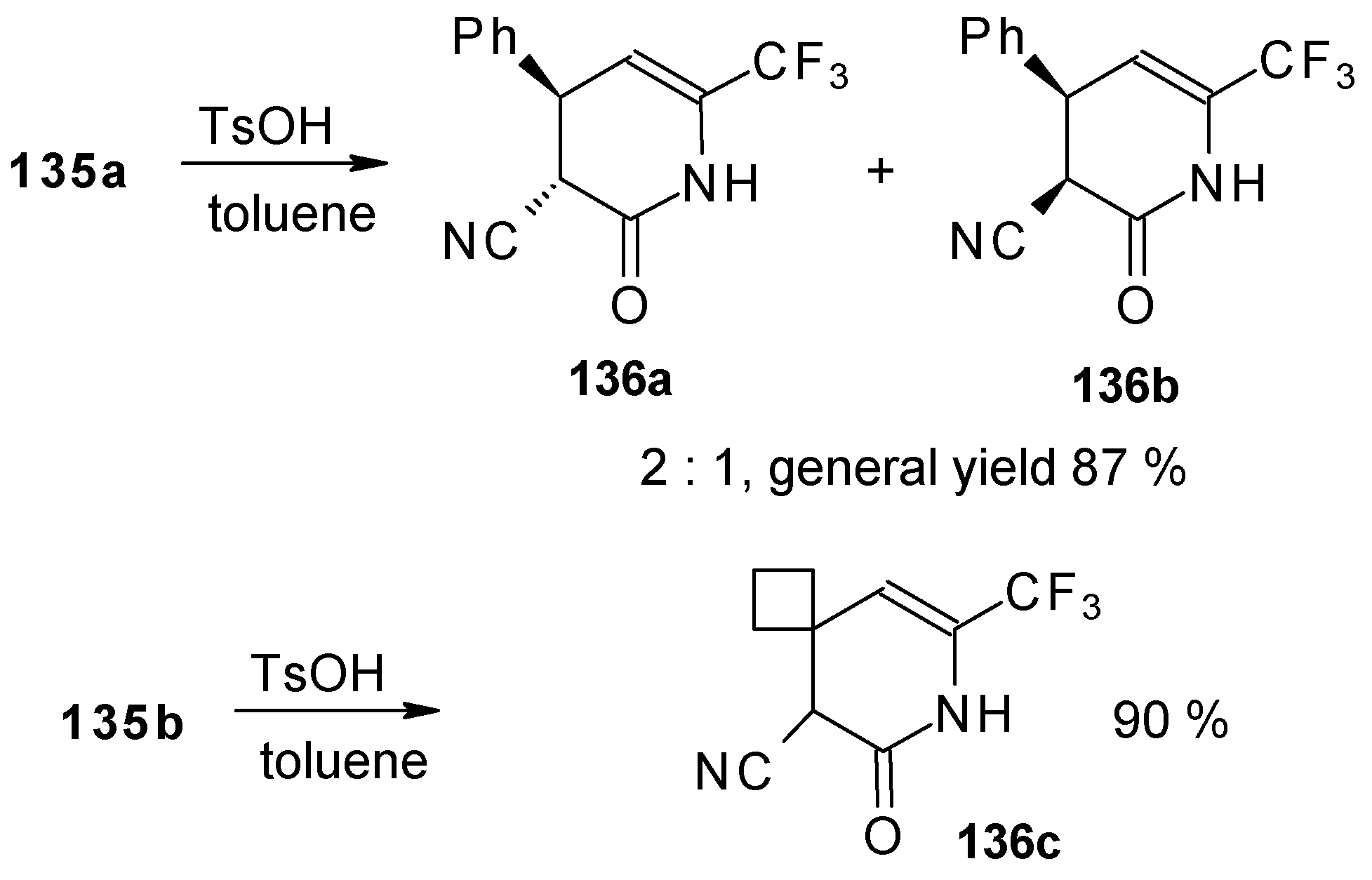
Reflux of a toluene solution of these piperidines with a catalytic amount of
p-toluenesulfonic acid results in elimination of water to give the corresponding dihydropyridines (
Scheme 86). In the case of the derivative of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b a mixture of two diastereoisomers is formed [
110].
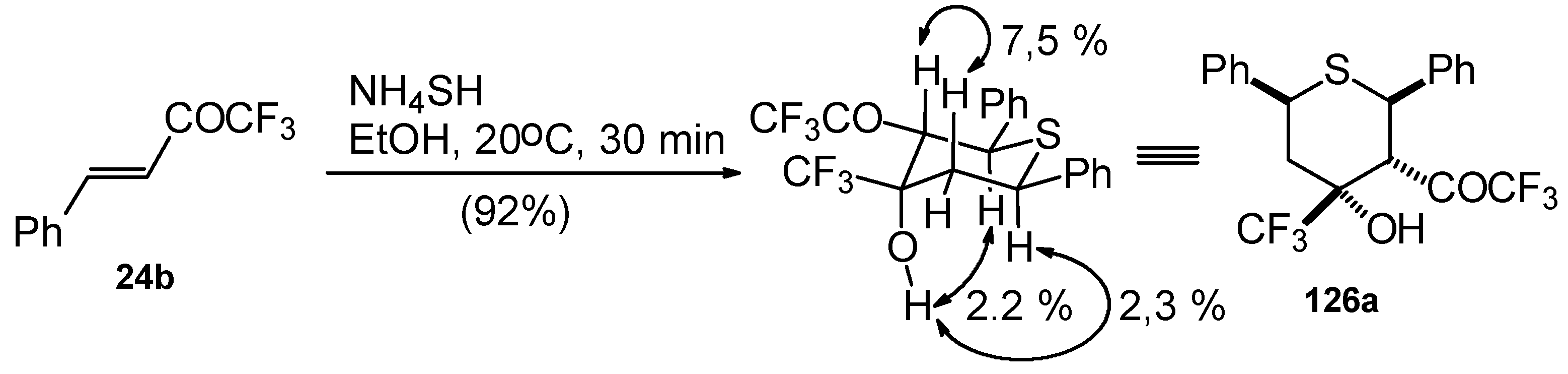
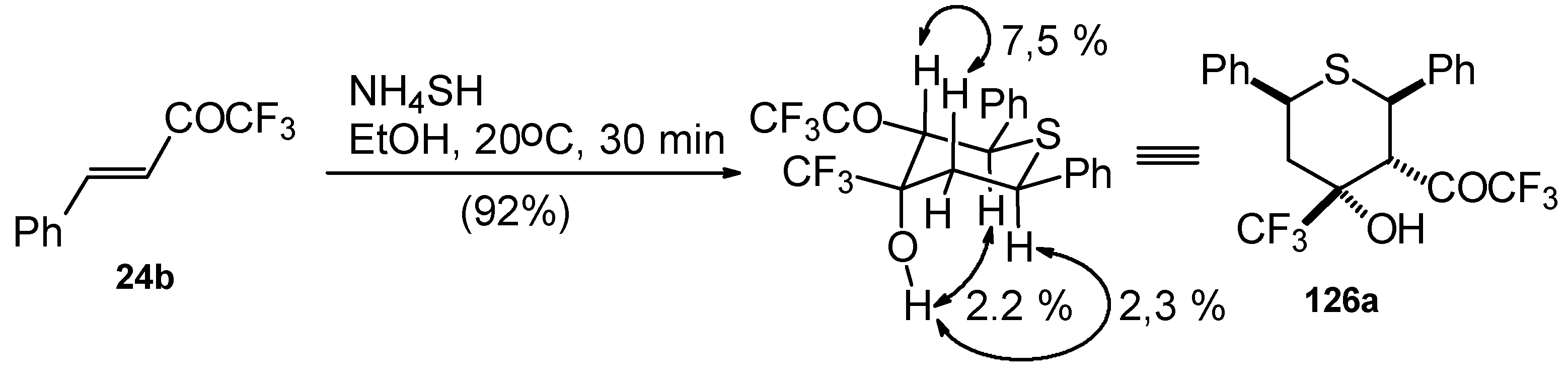
The reaction of CF
3 bearing enones with NH
4SH takes place under very mild conditions [
111]. The reaction of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b proceeds stereospecifically, giving only one diastereoisomer out of eight possible. The configuration of the obtained tetrahydrothiopyran was established by 2D
1H-
1H NOESY experiments. This compound has Ph, Ph, CF
3 and COCF
3 groups equatorially arranged, and an axial OH group (
Scheme 87).
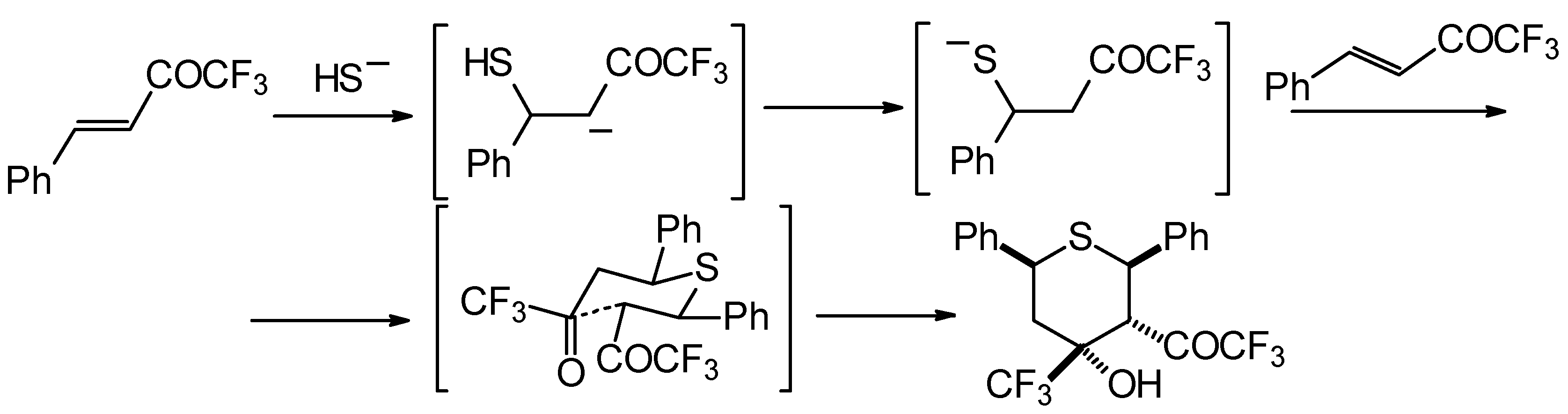
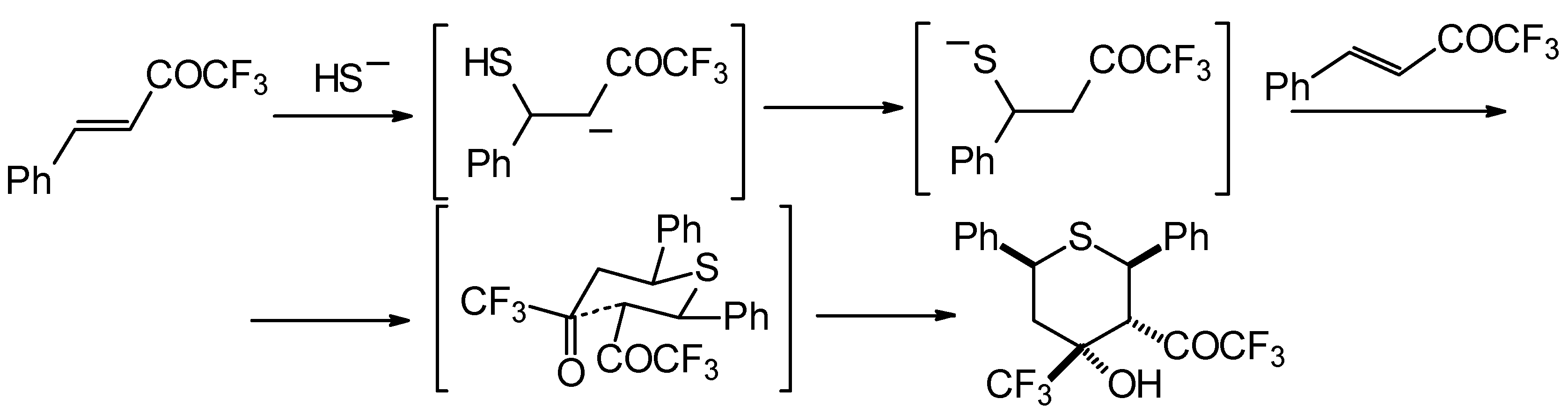
Formally the course of this reaction is the Michael addition of hydrosulphide anion to the double bonds of two molecules of 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b with subsequent aldol condensation. It was supposed that the transition state of cyclization has a chair conformation with all bulky groups (Ph, Ph, COCF
3, CF
3) equatorial (
Scheme 88).
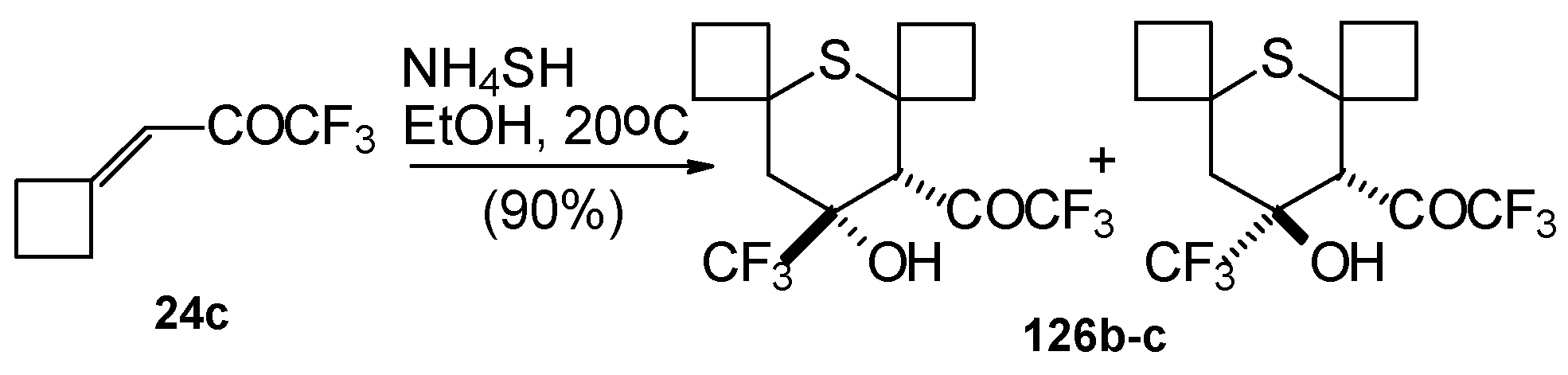
Reaction of NH
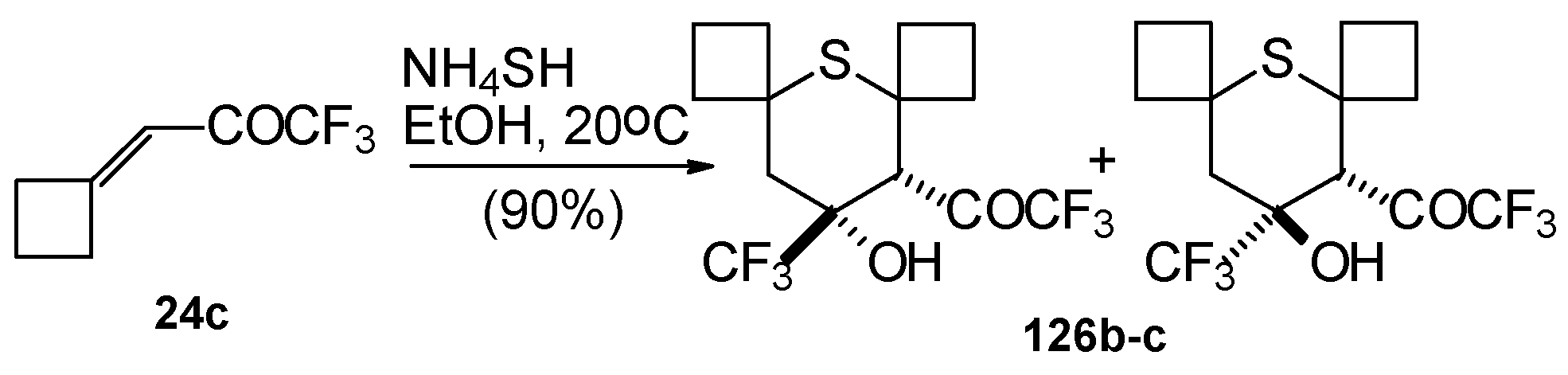
4SH with ketone derivative of methylenecyclobutane
24c leads to a 1/1 mixture of
cis- and
trans-diastereoisomers of the corresponding thiopyran (
Scheme 89). Formation of two diastereoisomers in the case of this ketone, as compared to one diastereoisomer with the reaction of ketone
24b, is explainable by steric differences in the substituents in these ketones (H and Ph) compared with
24c (cyclobutane ring).
A very unusual result was obtained in the reaction of ammonium hydrosulphide with a ketone containing an adamantane fragment
24f. The only product was the corresponding 4-membered trifluoromethyl-containing heterocyclic ring
127 (
Scheme 90). 2-Thietanols have not been described previously, apparently because the semithioacetal fragment is unstable in the 4-membered rings. We assume that this compound is stable due to both the trifluoromethyl group stabilizing the geminal HO-C-S fragments [
112] and the adamantane structure, which stabilizes spiro attached small rings [
113]. Formation of the corresponding tetrahydrothiopyrans as in the cases of other CF
3 ketones does not take place for this ketone, apparently due to steric reasons. An attempt to make the analogous thietanols from other CF
3 ketones at low temperature with a 20-fold excess of ammonium hydrosulphide was unsuccessful and only the corresponding tetrahydrothiopyrans were isolated.
The reaction 1,1,1-trifluoro-4-methyl-4-phenyl-3-buten-2-one
24w with ammonium hydrosulphide under similar conditions results in the corresponding mercaptan
137 (
Scheme 91)
. In spite of having a similar structure to 1,1,1-trifluoro-4-phenyl-3-buten-2-one
24b, this enone is much less reactive. It seems likely that the formation of the corresponding tetrahydrothiopyran does not take place due to 1,3-diaxial repulsions of the methyl groups. Apparently one of the reasons for cyclization in the case of adamantane containing ketones, is due to stabilization of thietanol by adamantane. In the case of ketone β-trifluoroacetyl methylstyrene this type of reaction does not occur [
111].
The oxidation of obtained thiopyrans and thietane by hydrogen peroxide in acetic acid was also investigated. The corresponding sulfones were obtained in good yield (
Scheme 92).
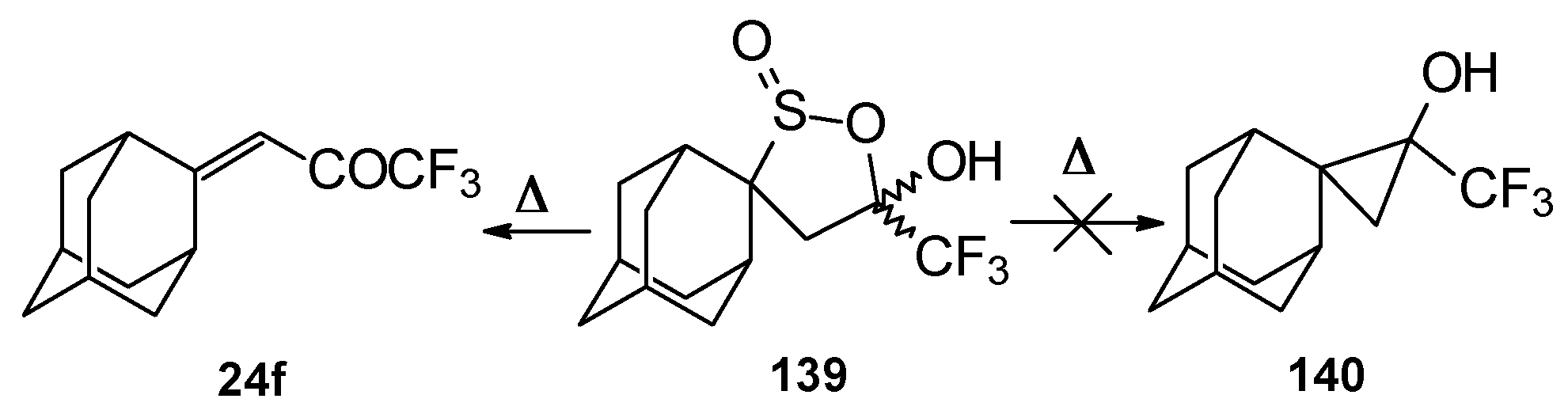
Oxidation of 2-thietanol proceeds unusually. The corresponding sultine
139 as a mixture of diastereoisomers (2.7/1) is formed (
Scheme 93). This is probably due to the labile semi-thioacetal fragment in sultine which facilitates the cleavage of the thietane ring. The structure of the
trans-diastereoisomer of sultine was confirmed by X-ray crystallography.
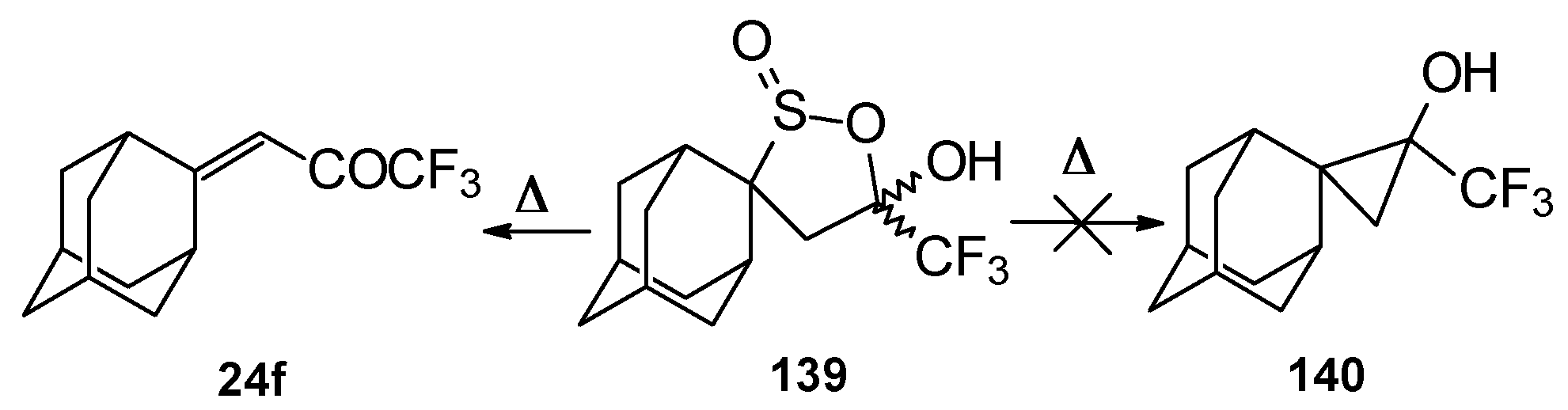
An attempt to thermally extrude sulphur dioxide from this sultine to make the corresponding cyclopropane was unsuccessful, only the starting ketone was obtained (
Scheme 94) [
111].
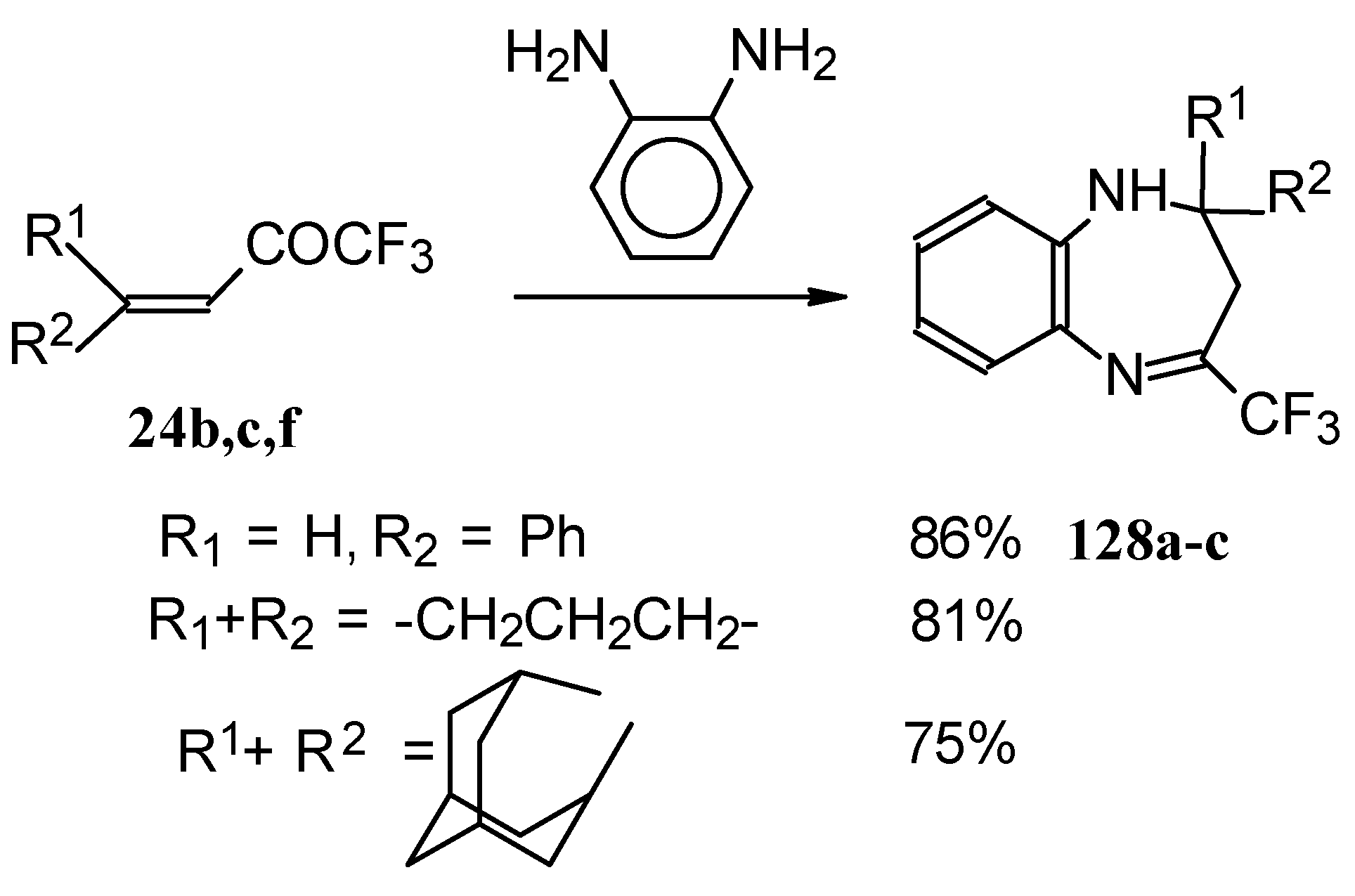
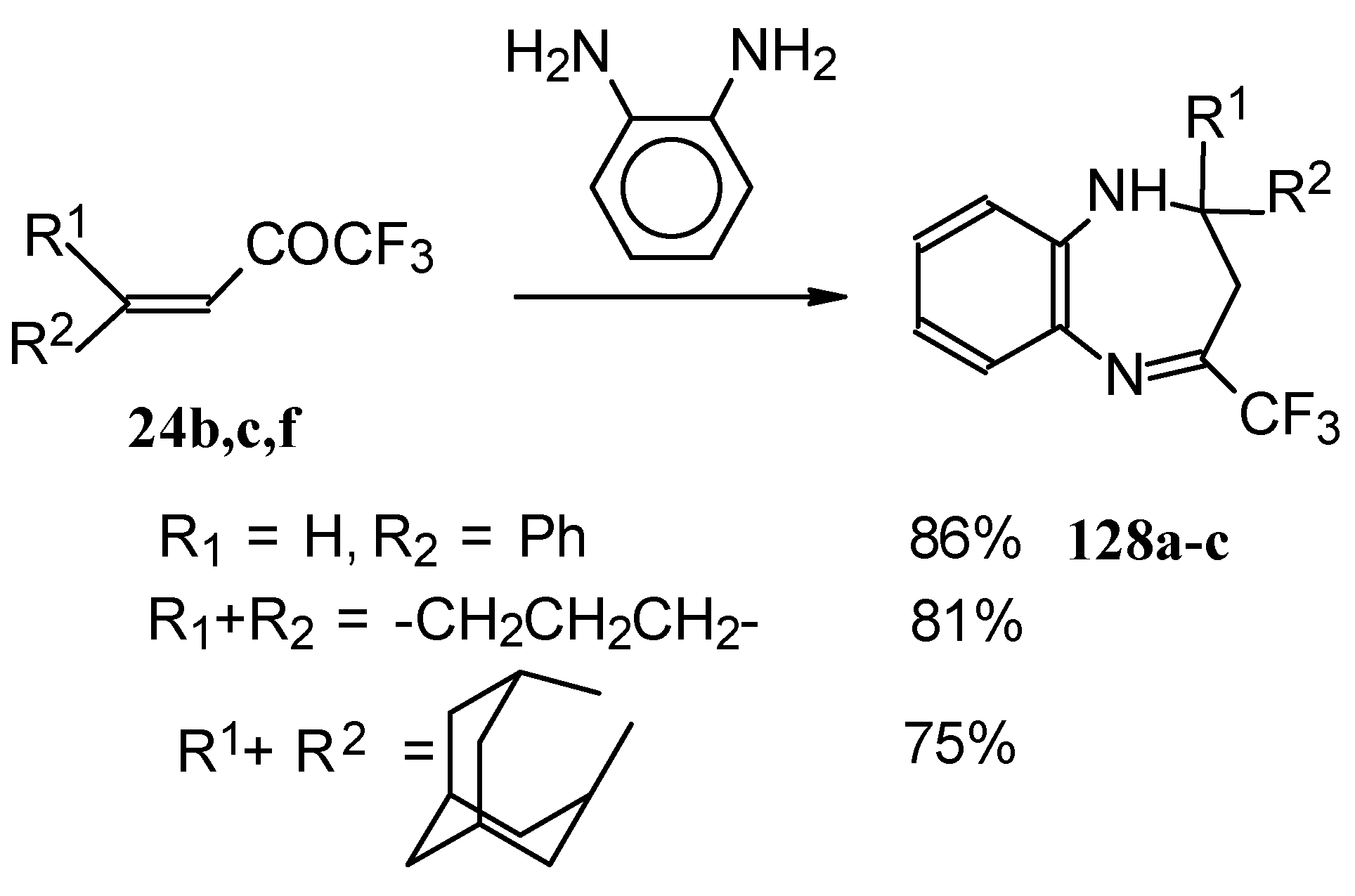
The reaction of CF
3 ethylenic ketones with
o-phenylenediamine was also investigated [
114]. As a result the corresponding 2,3-dihydro-1,5-benzodiazepines
128 were obtained (
Scheme 95). In this case elimination of water proceeds spontaneously to give conjugation of the aromatic ring and the CN double bond.
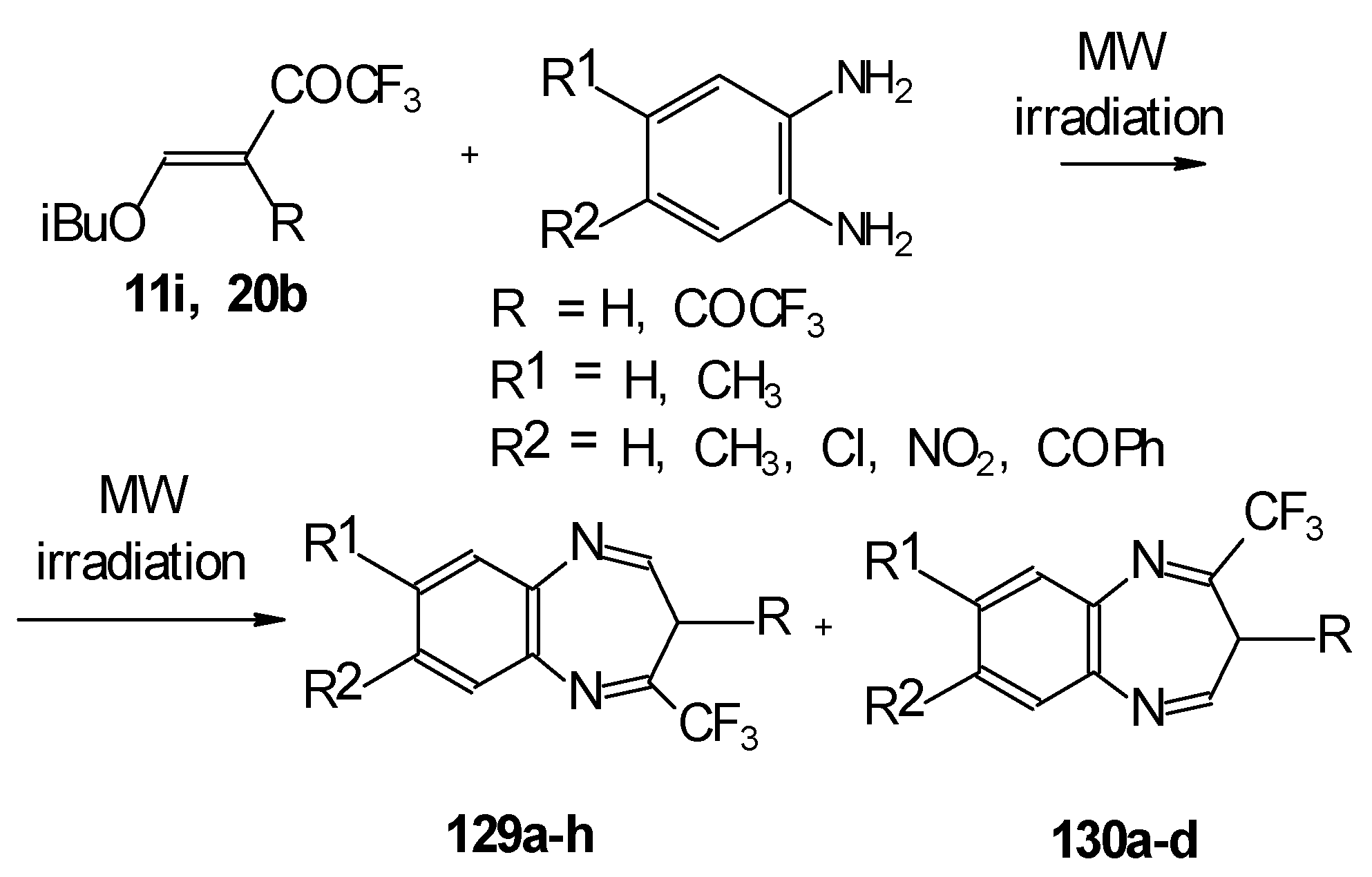
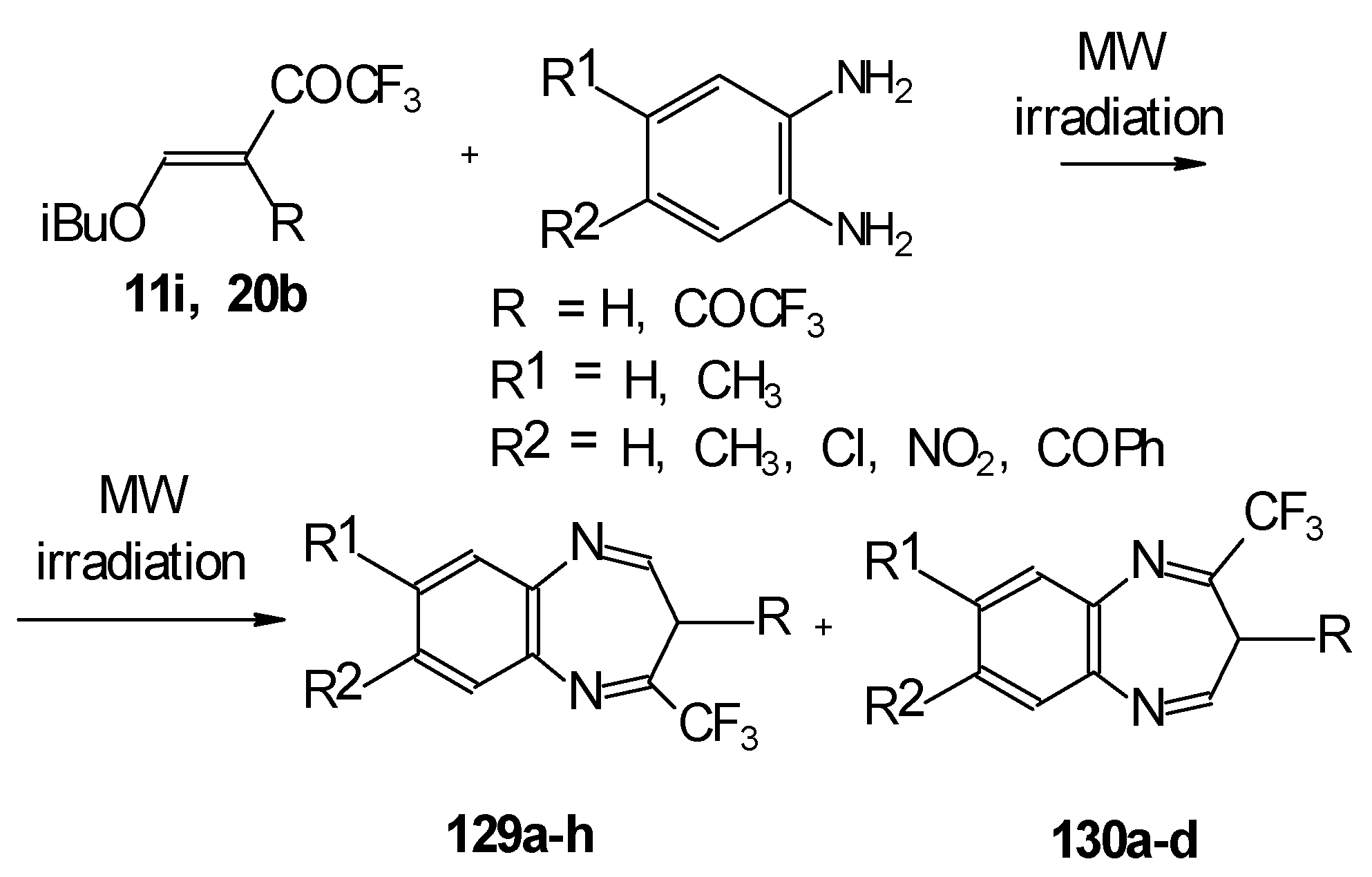
In the case of β-alkoxy substituted CF
3 ethylenic ketones the reaction with
o-phenylendiamines in refluxing xylene led to complex mixture of products. However, this reaction proceeds readily using microwave irradiation to give a 4-CF
3 and 2- CF
3 substituted (1H,5)aryldiazepines in 73-93% yield (
Scheme 96) [
115].
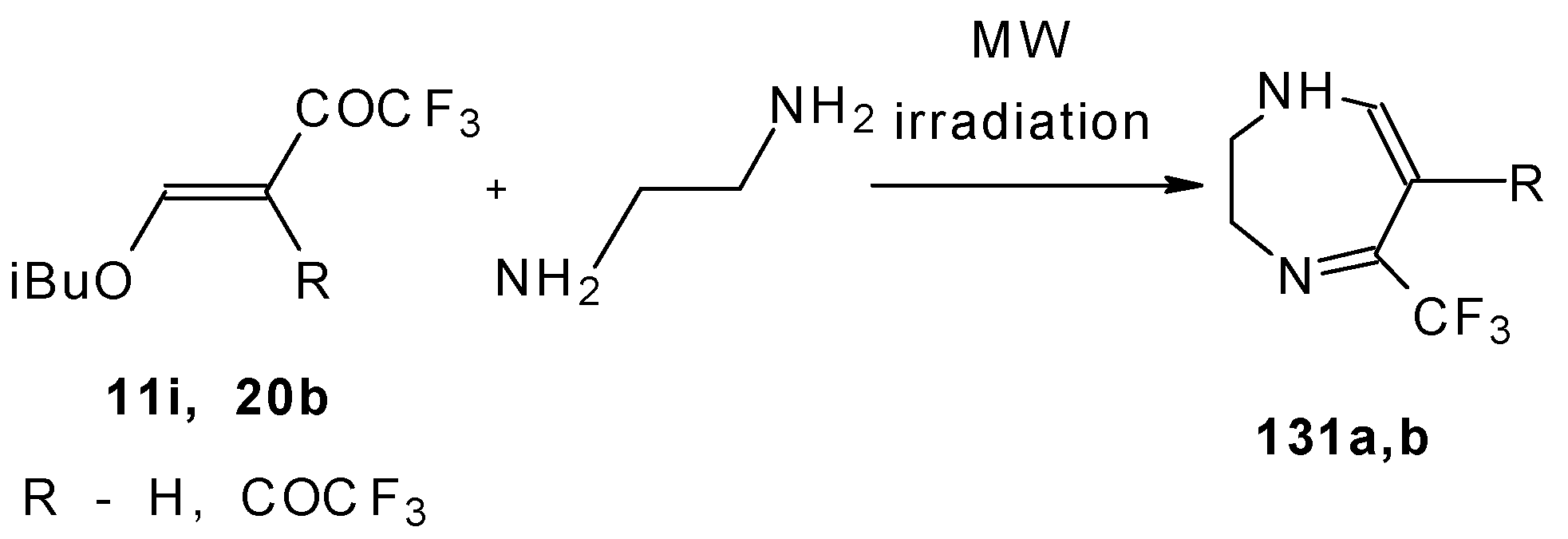
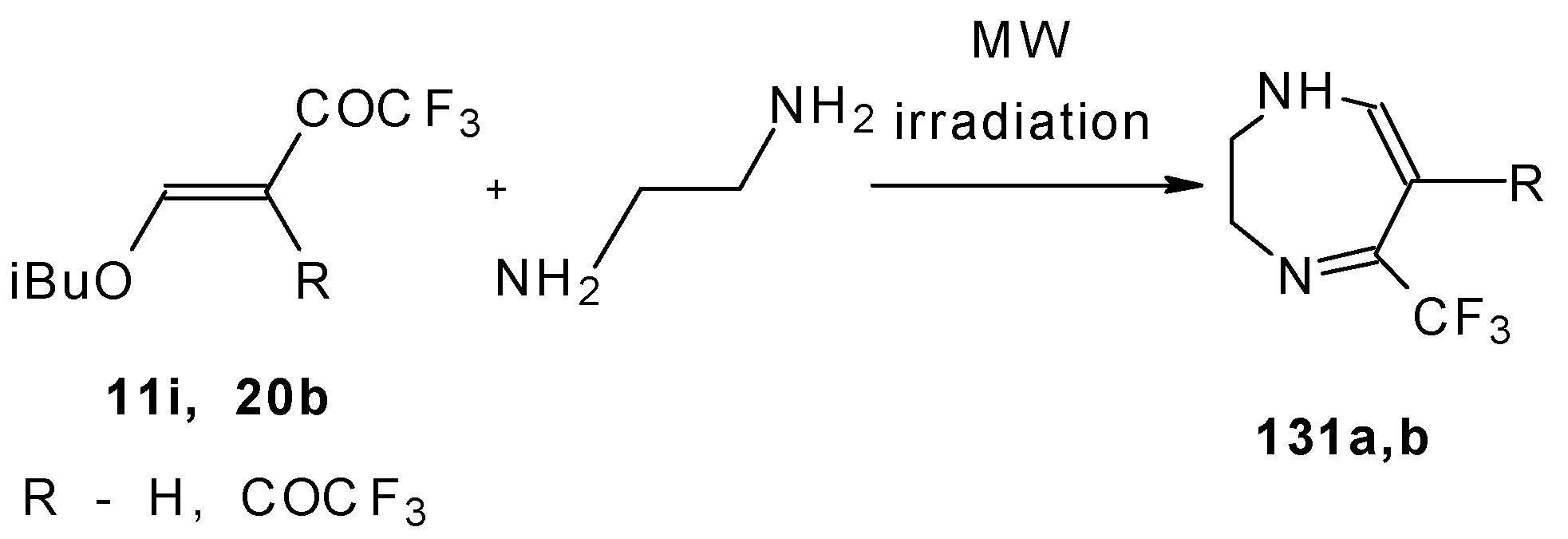
The reaction of ketones
11i and
20b with ethylendiamine proceeds analogiously (
Scheme 97) [
115].


{kind=link}
{kind=link}
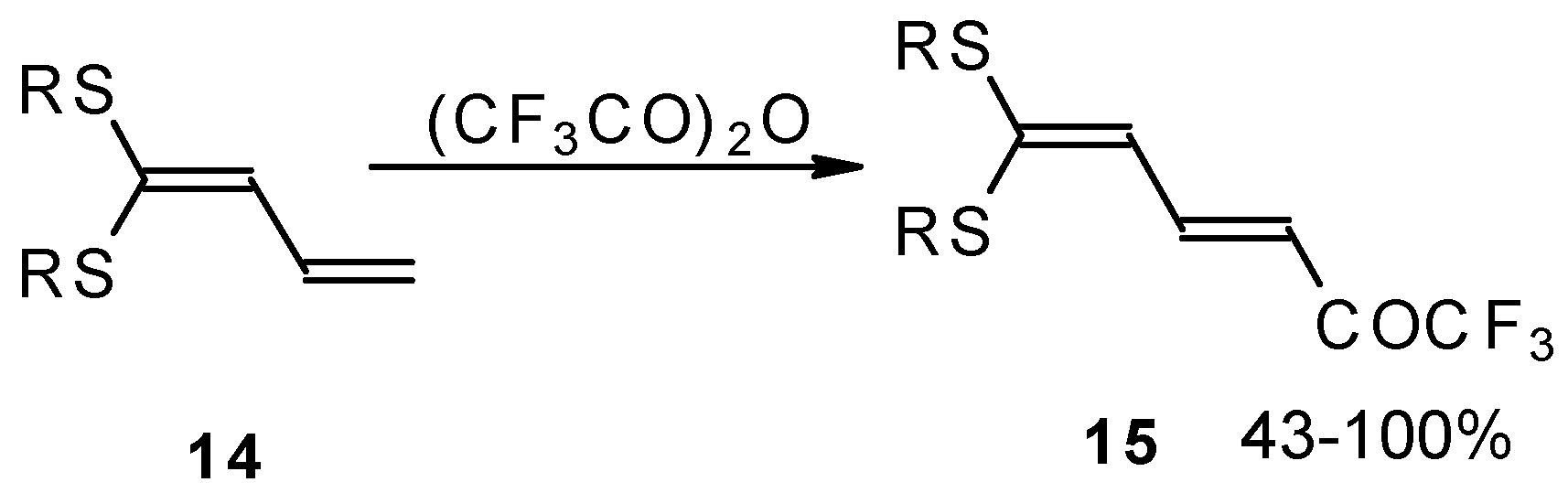{kind=link}
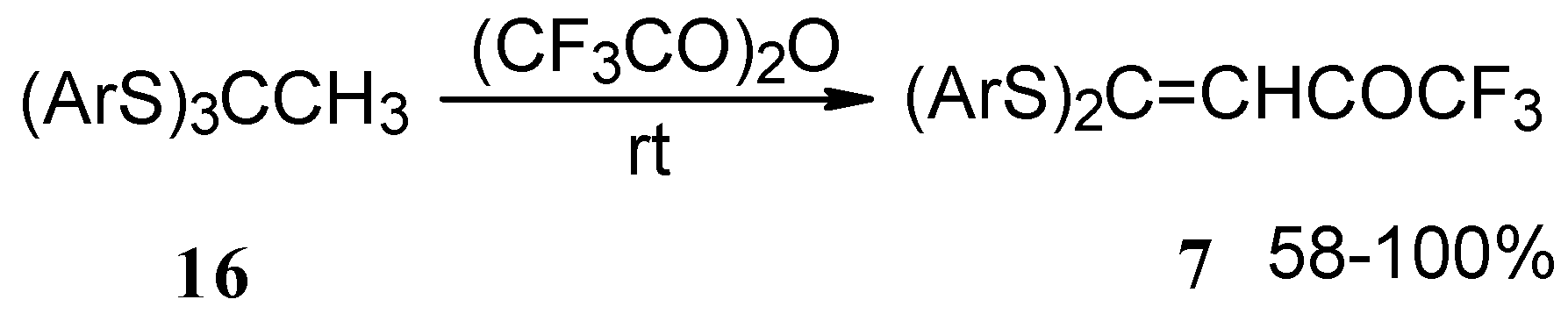{kind=link}
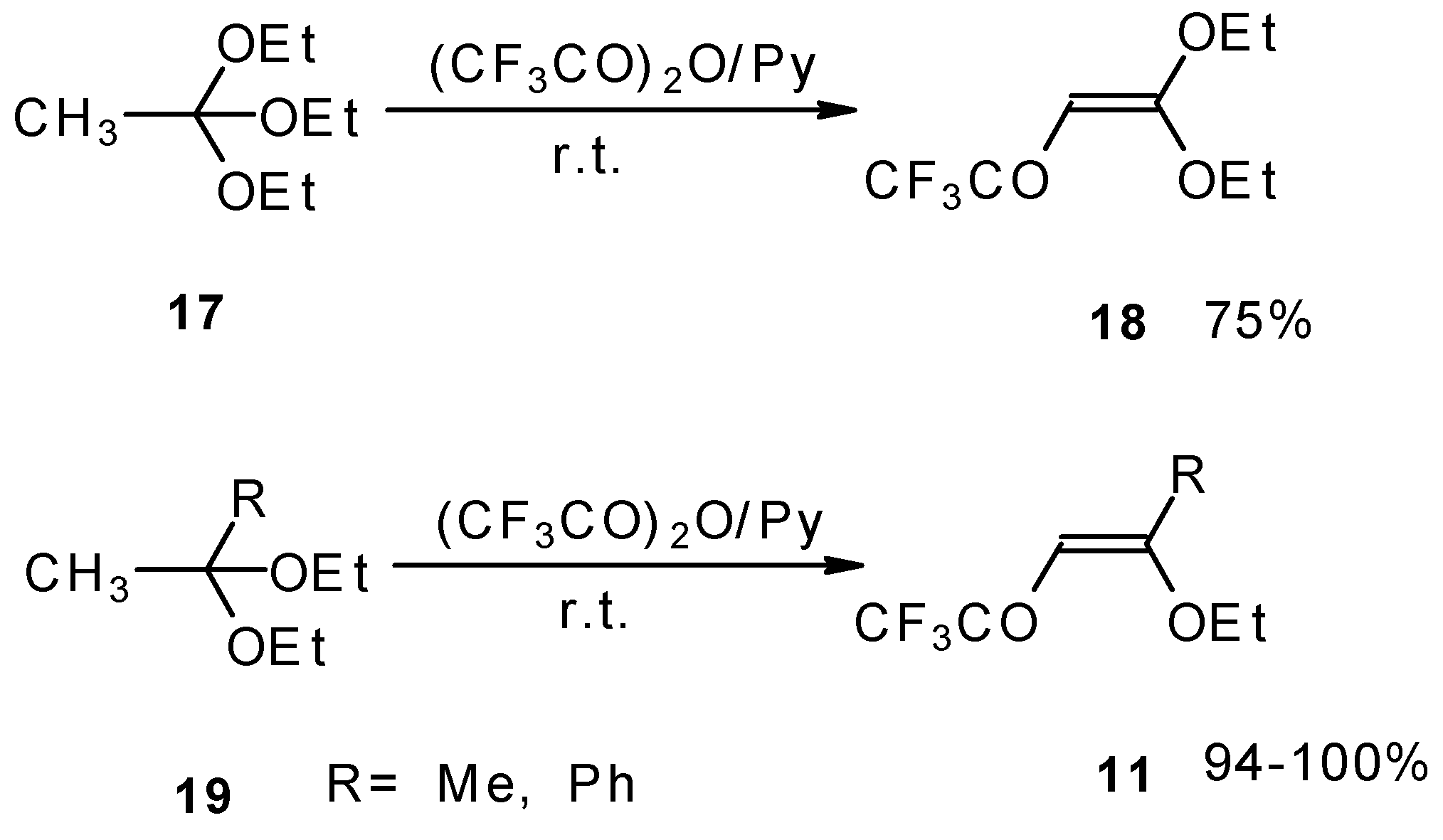{kind=link}
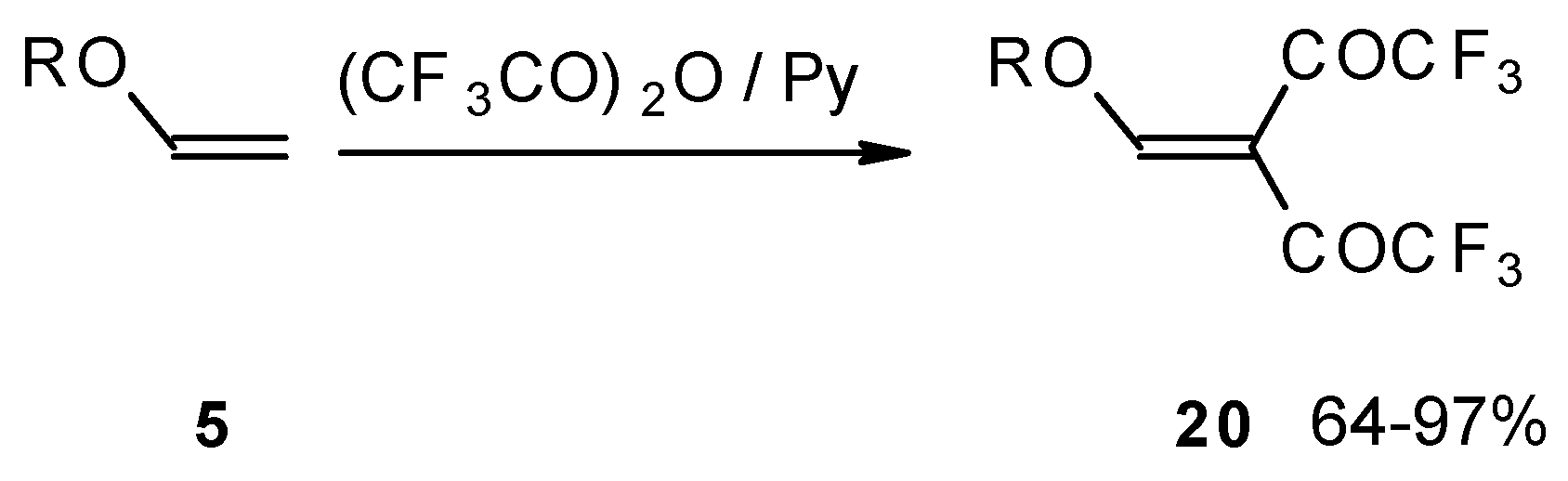{kind=link}
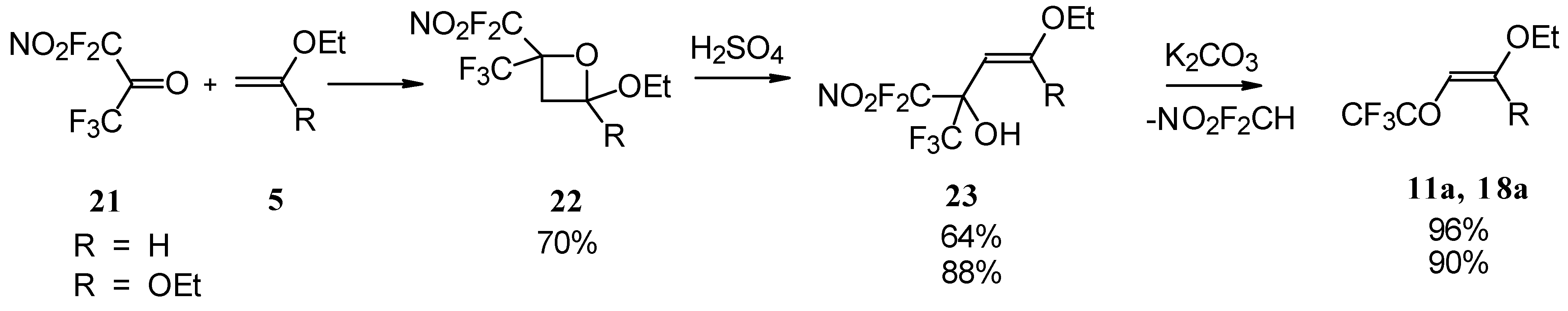{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
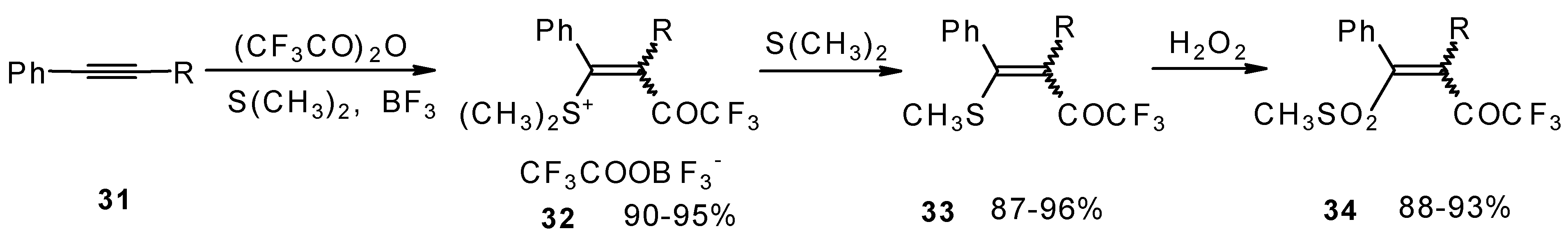{kind=link}
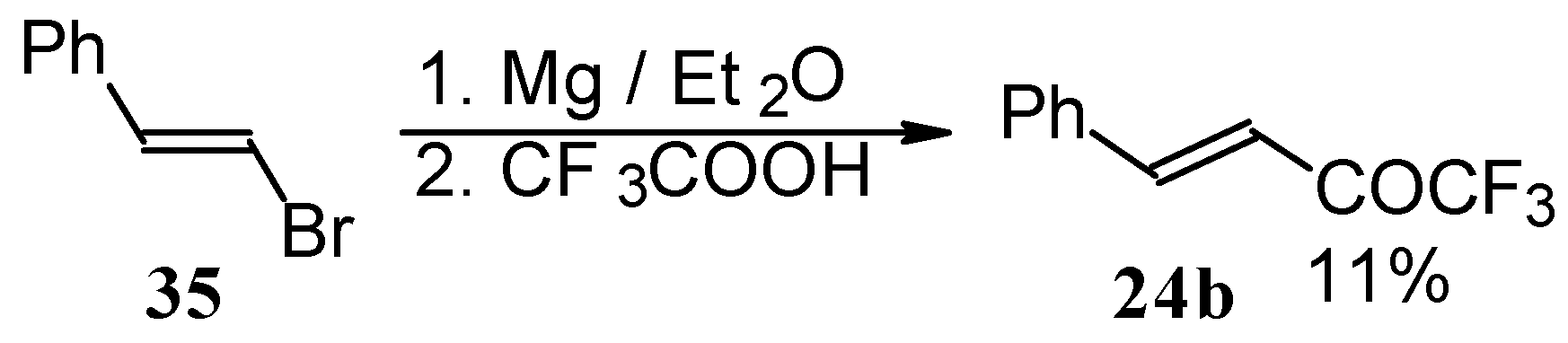{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
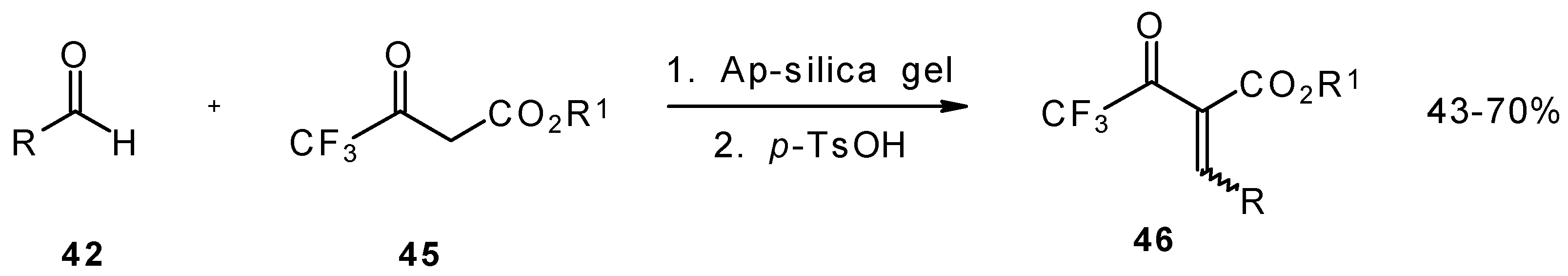{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
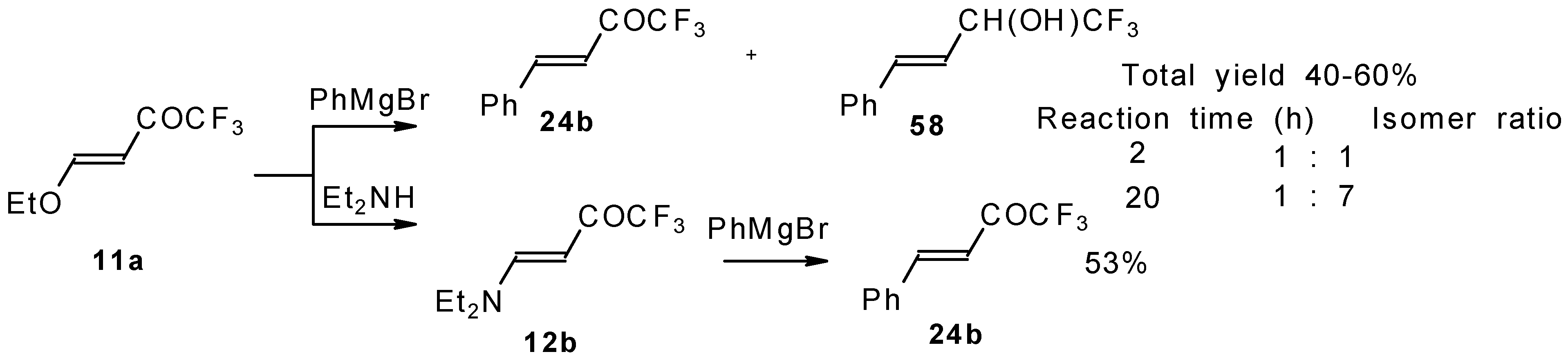{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
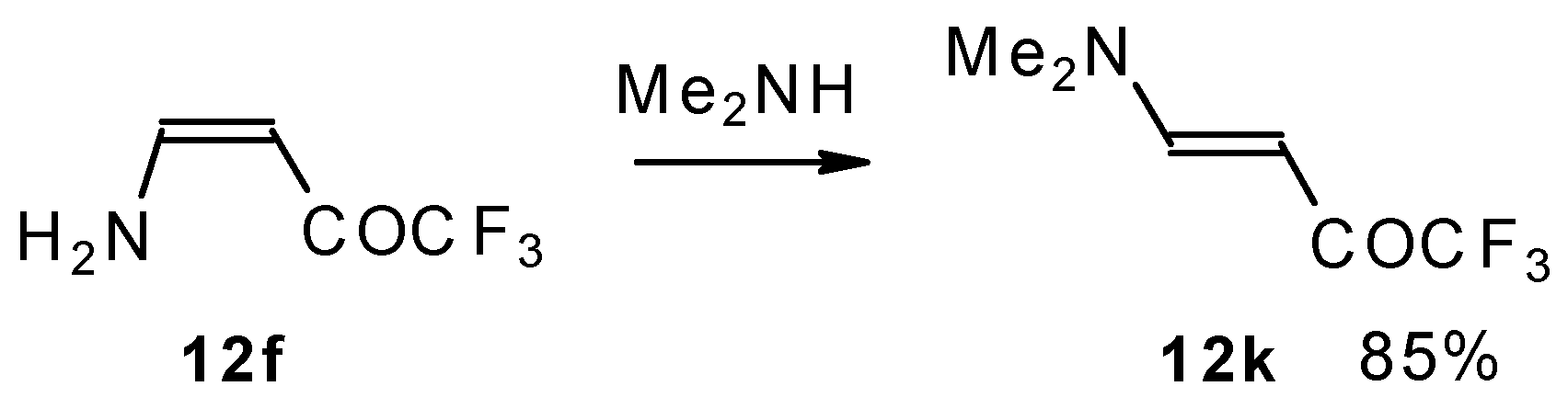{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
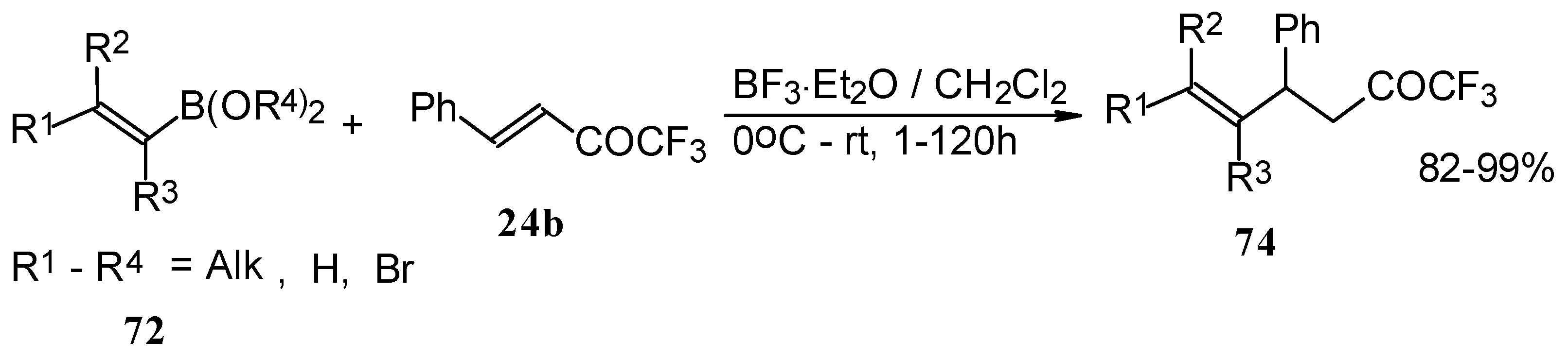{kind=link}
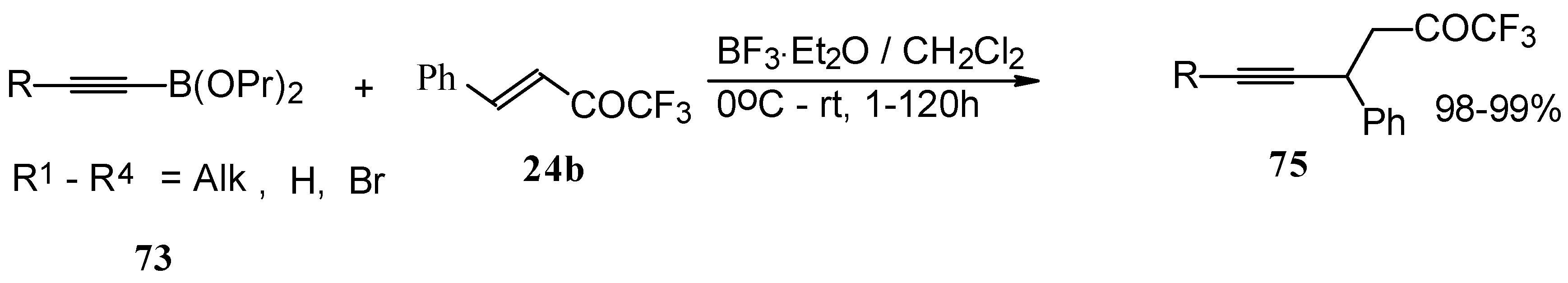{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
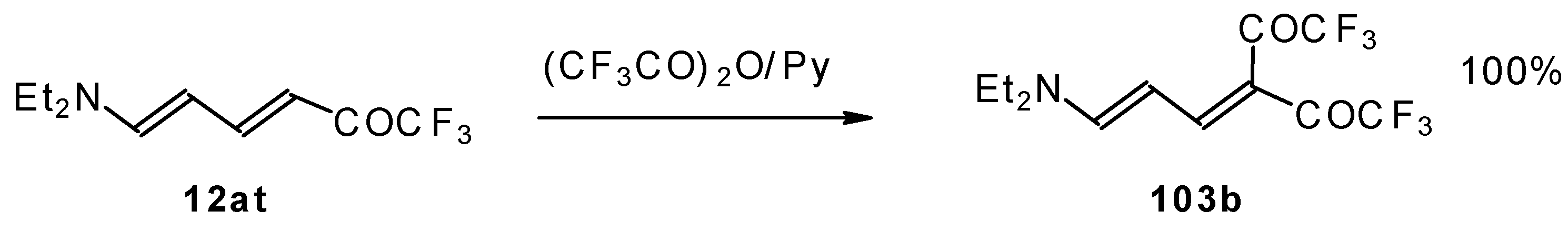{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
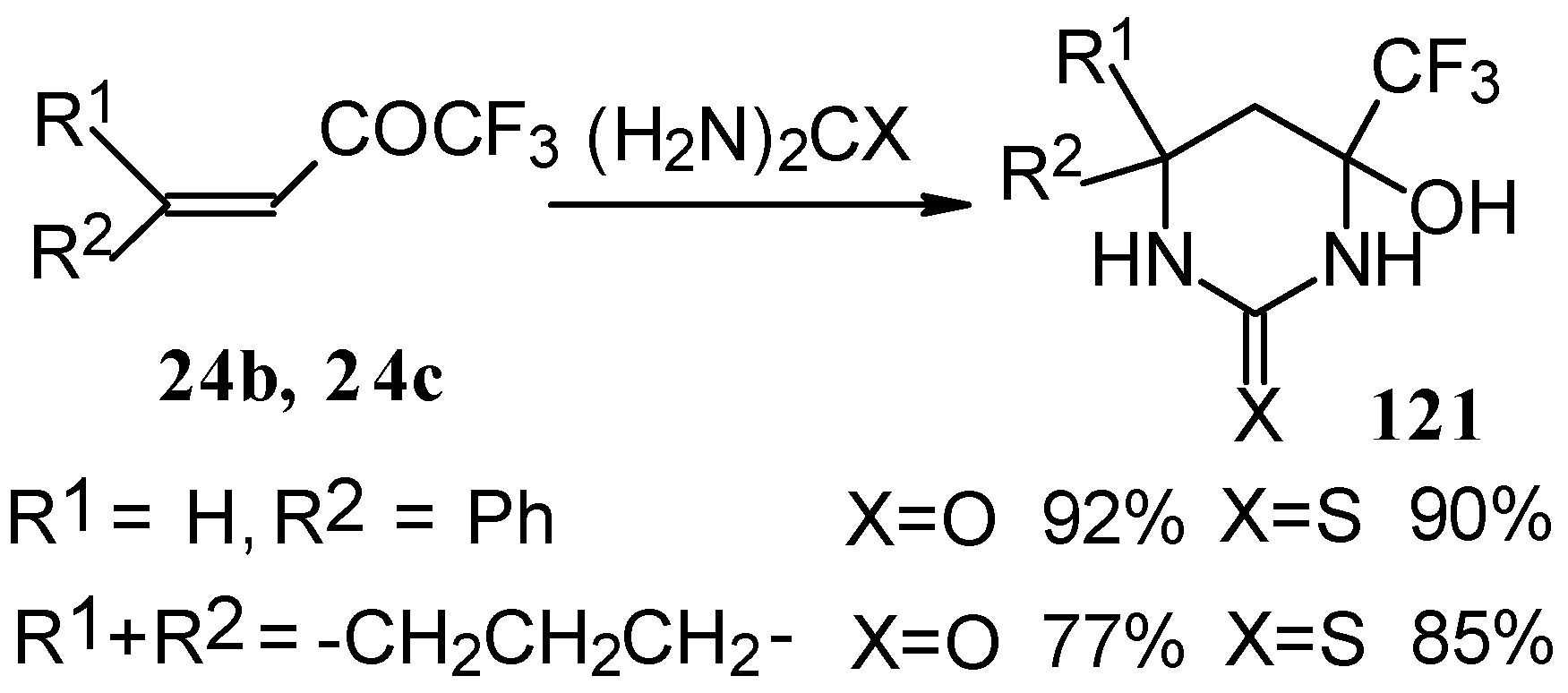{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
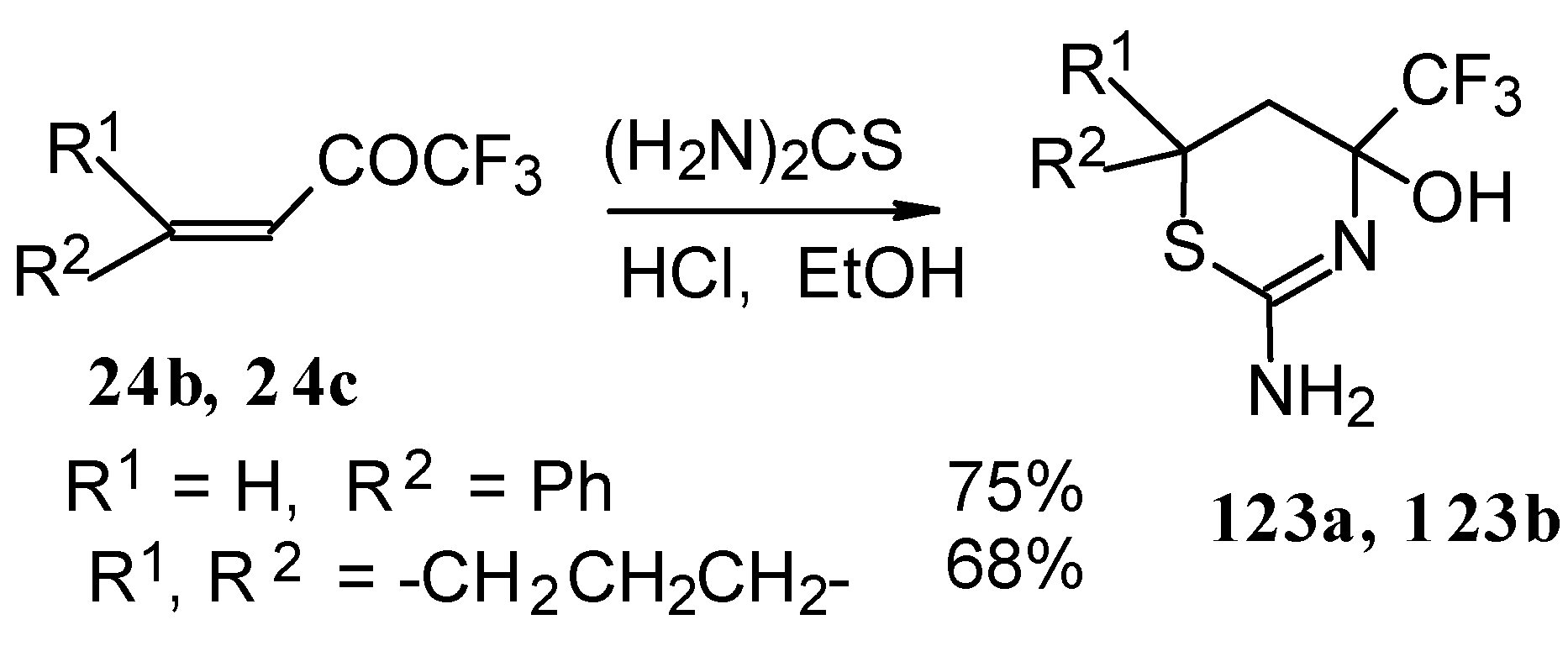{kind=link}
{kind=link}
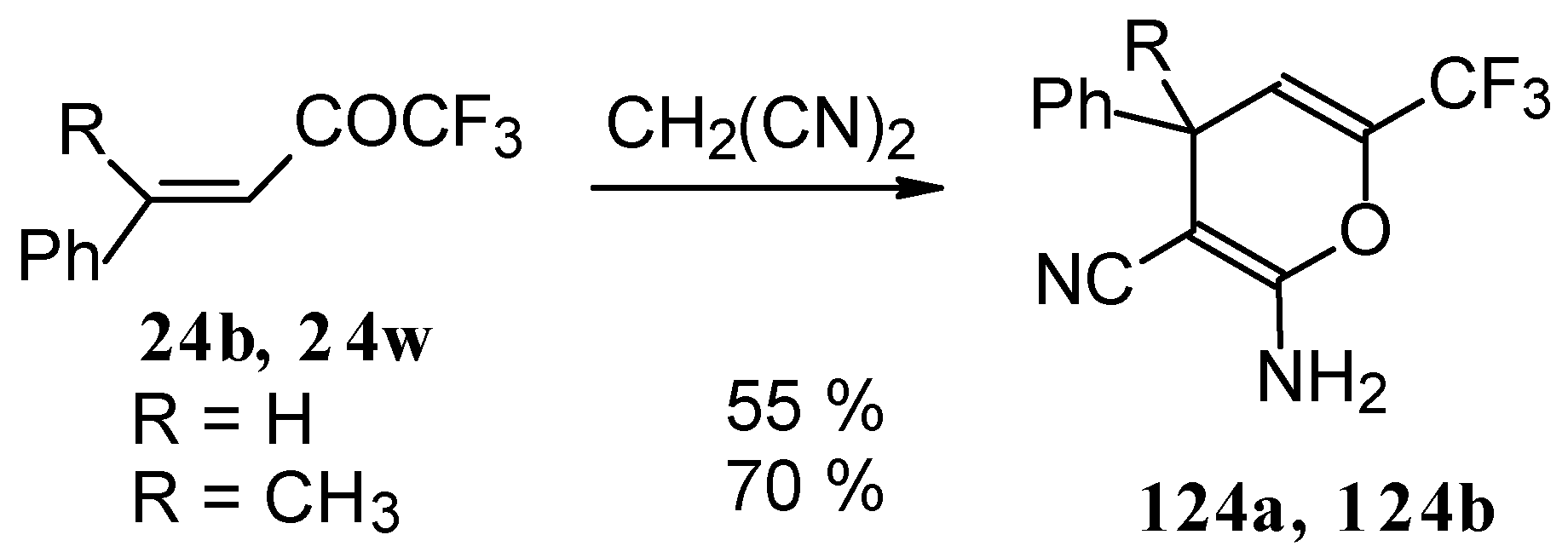{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
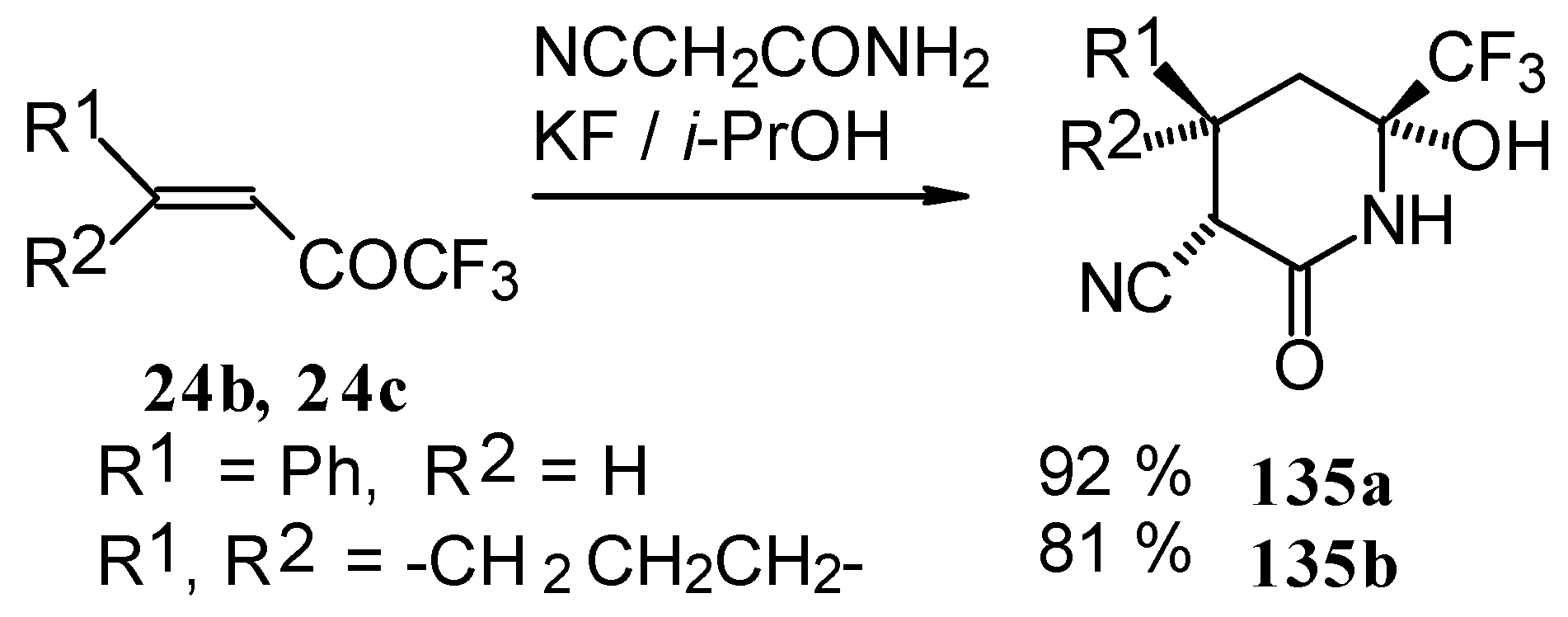{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
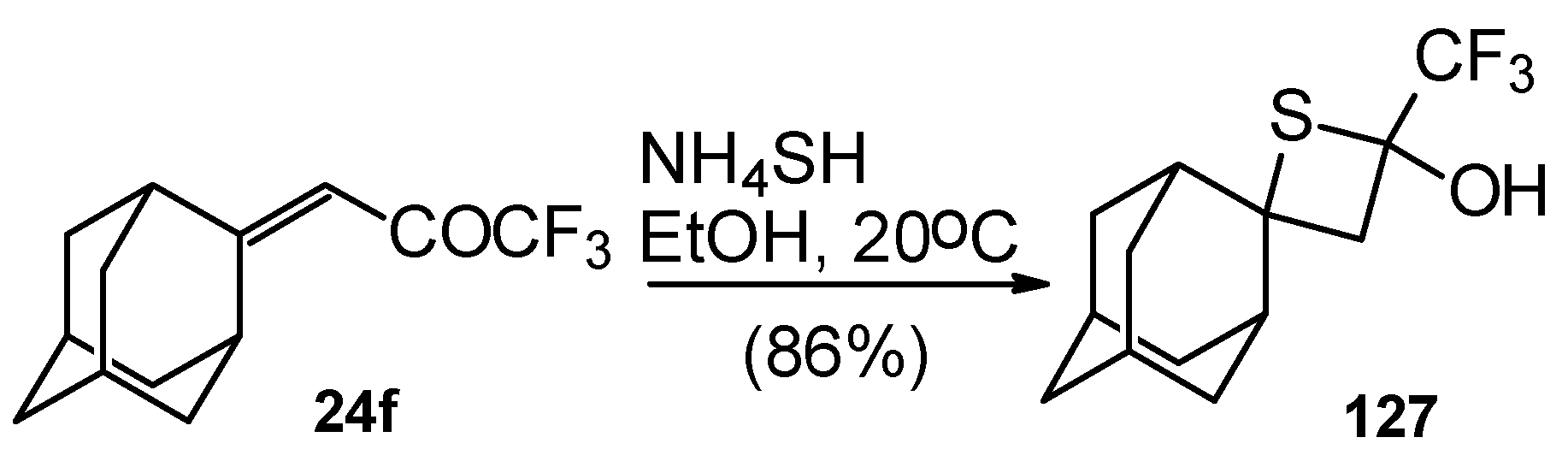{kind=link}
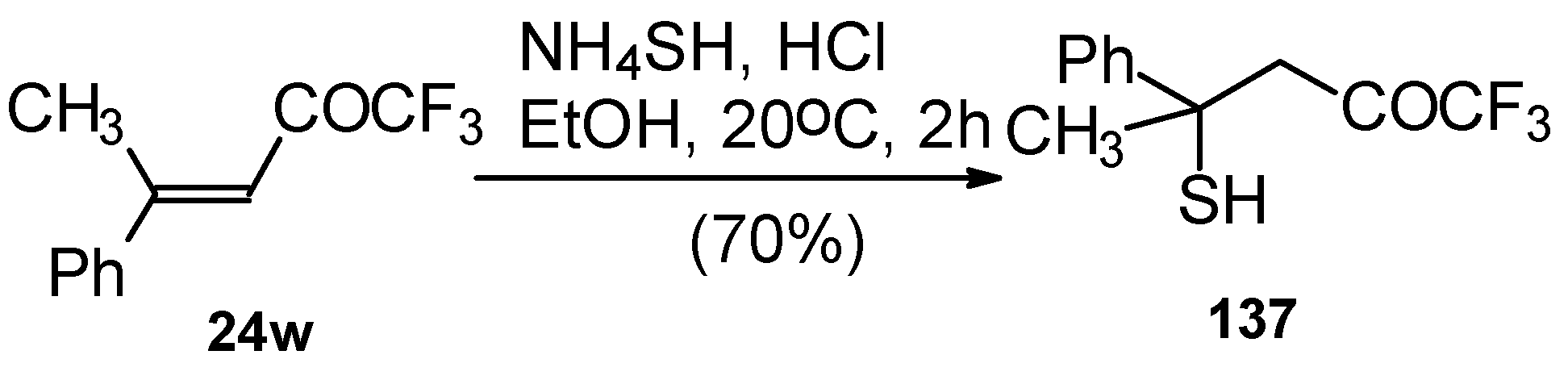{kind=link}
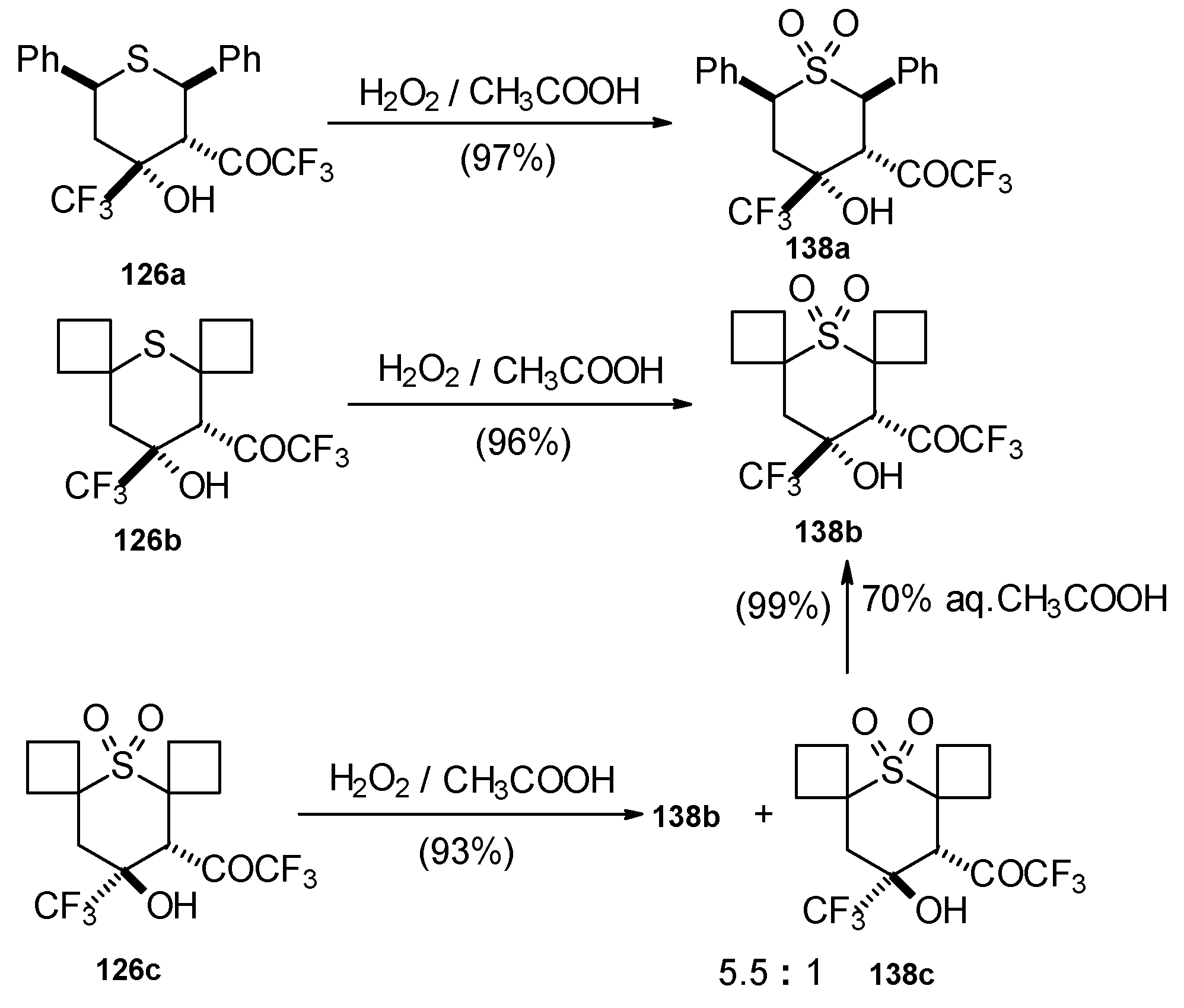{kind=link}
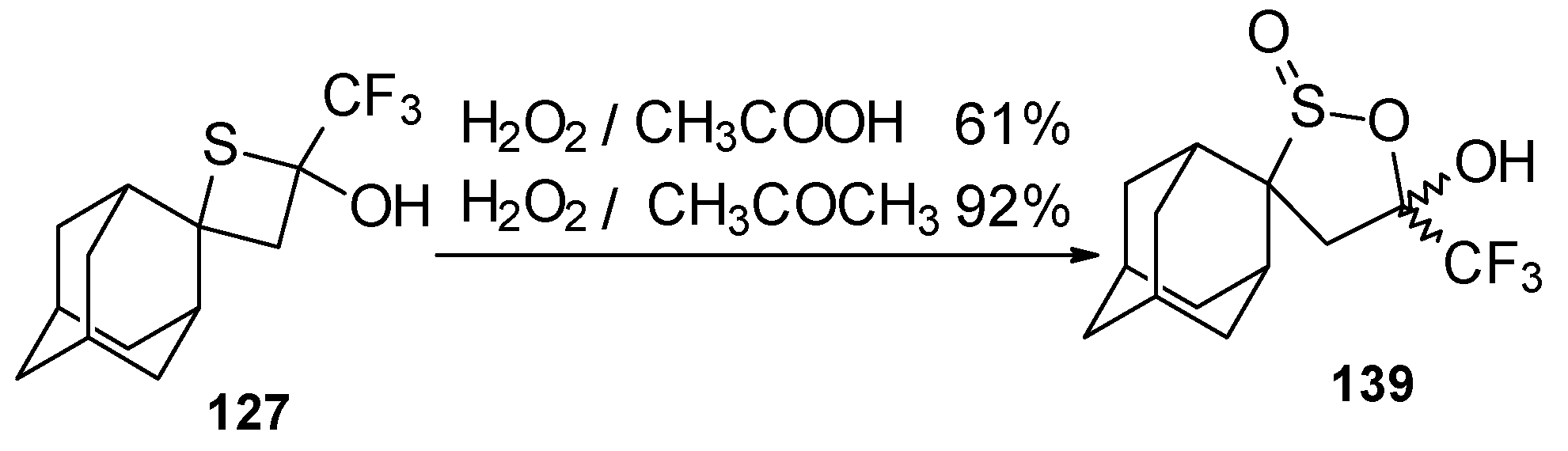{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}