Isocorydine Derivatives and Their Anticancer Activities

Abstract
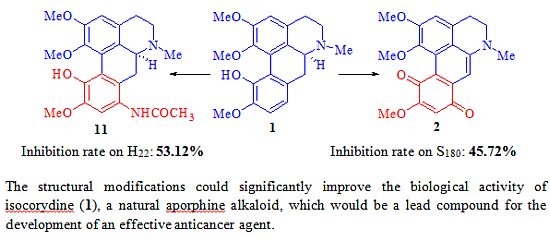
:
1. Introduction
2. Results and Discussion
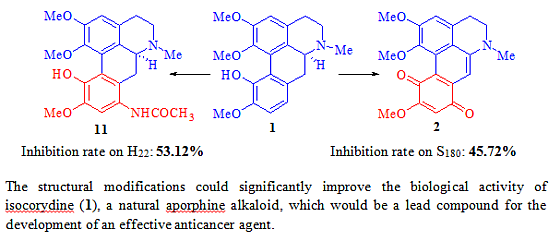
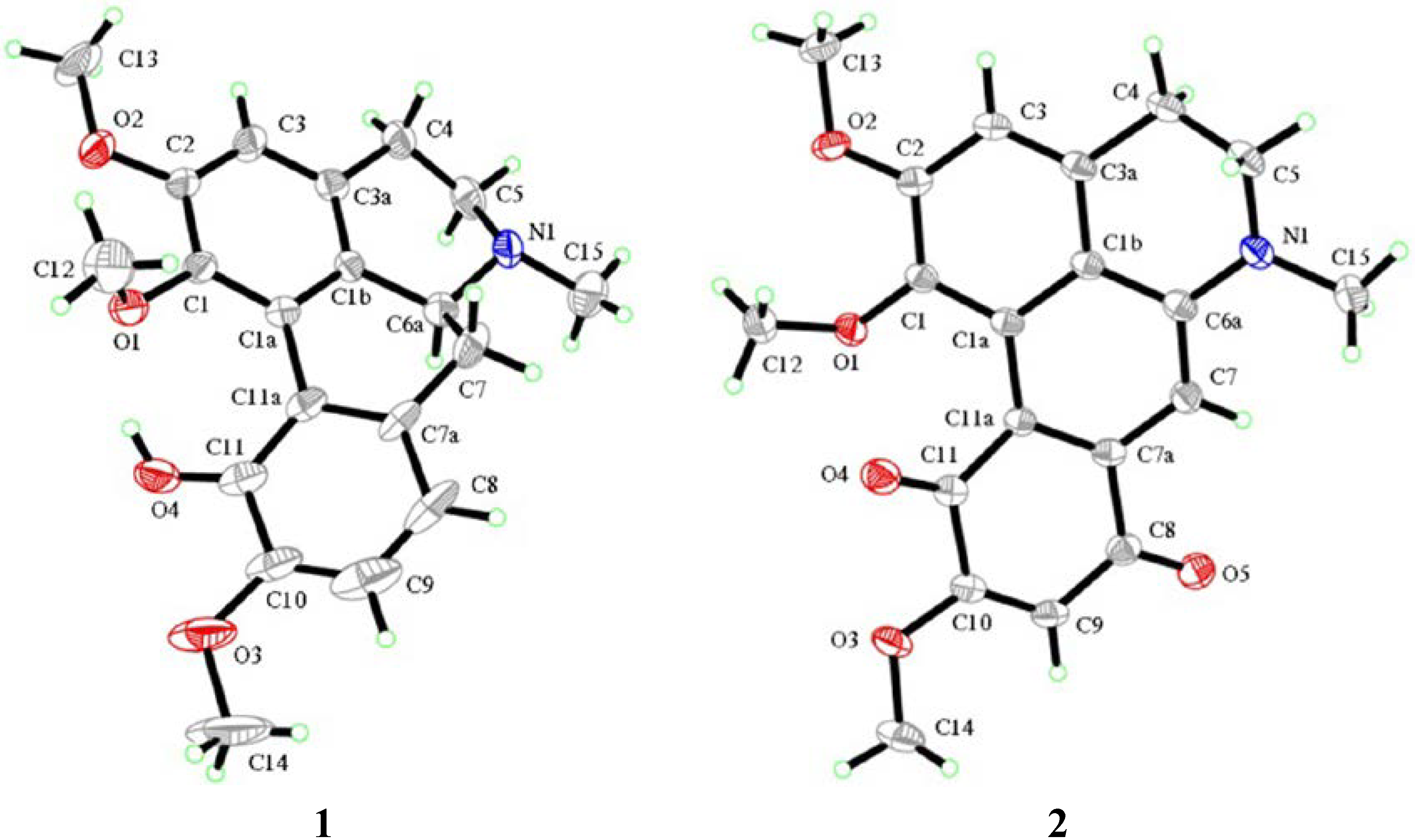
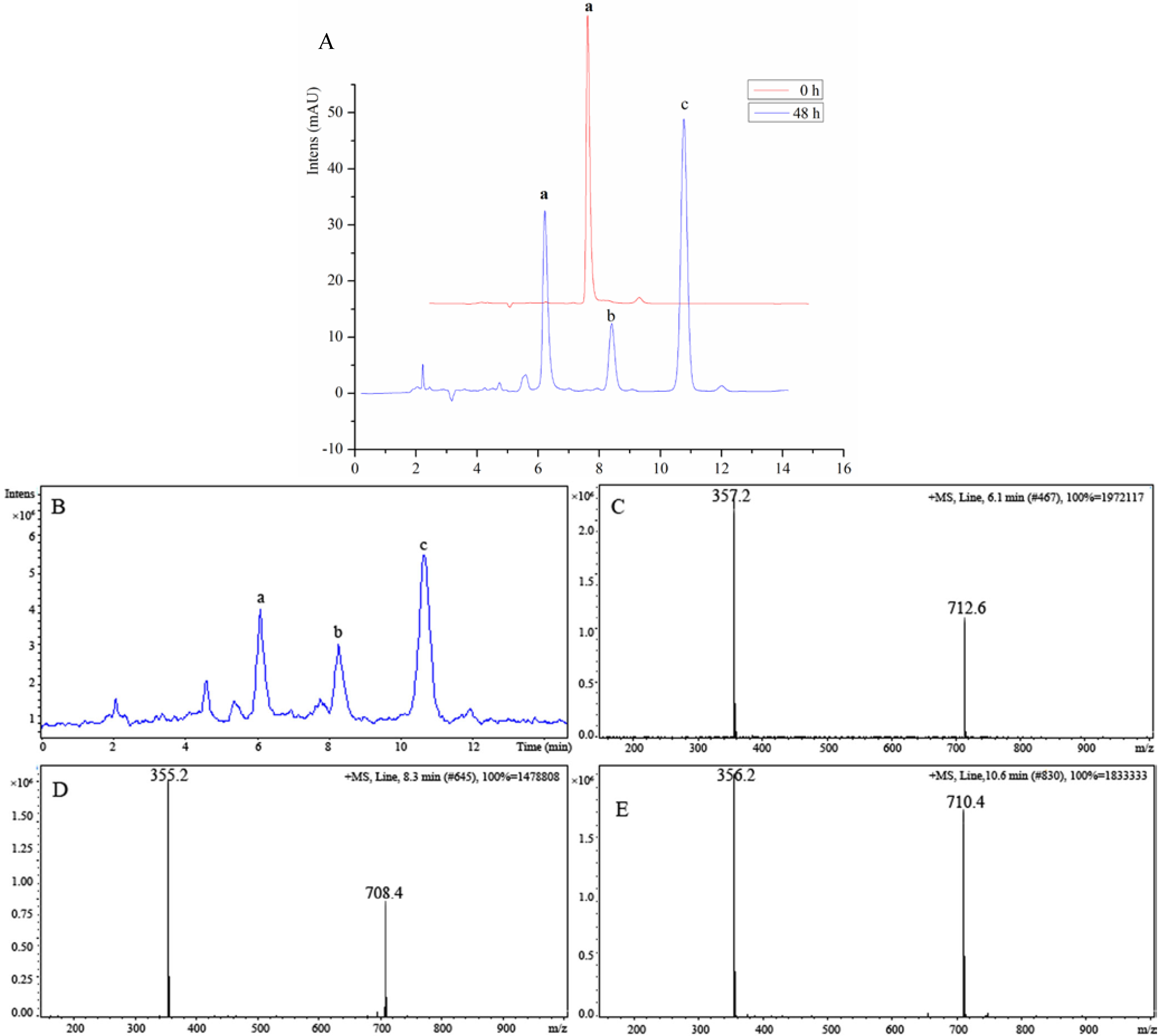
2.1. Chemistry
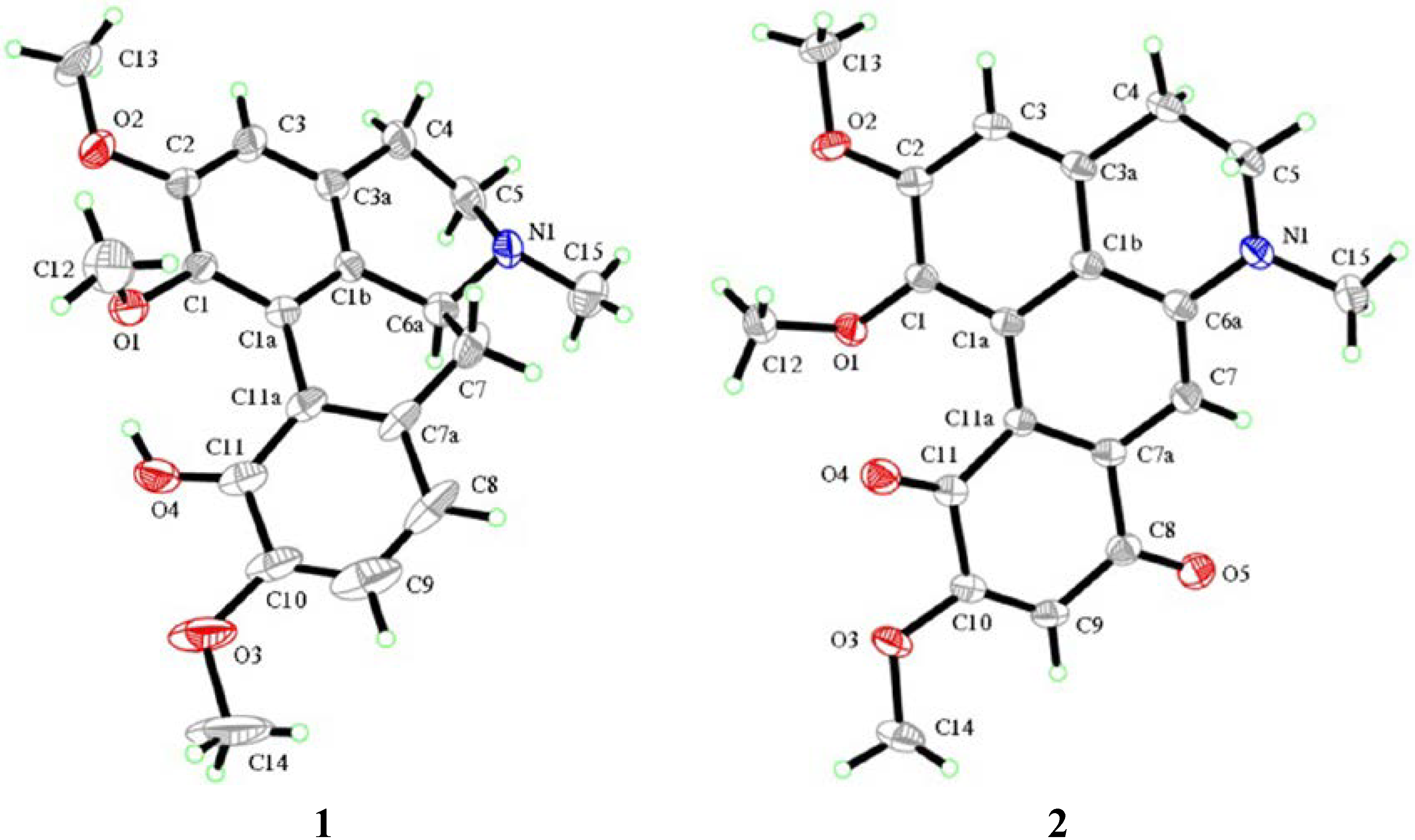
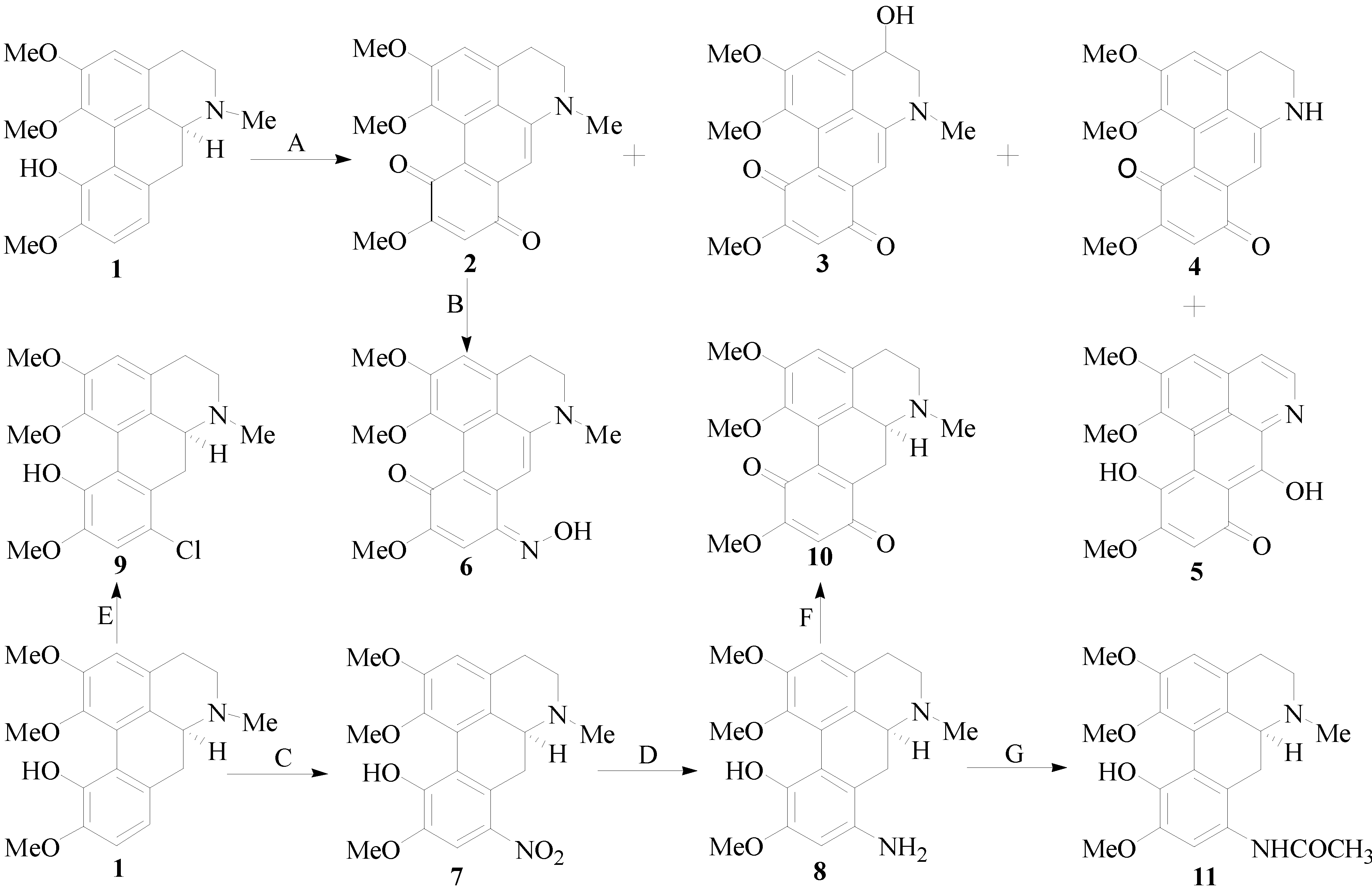
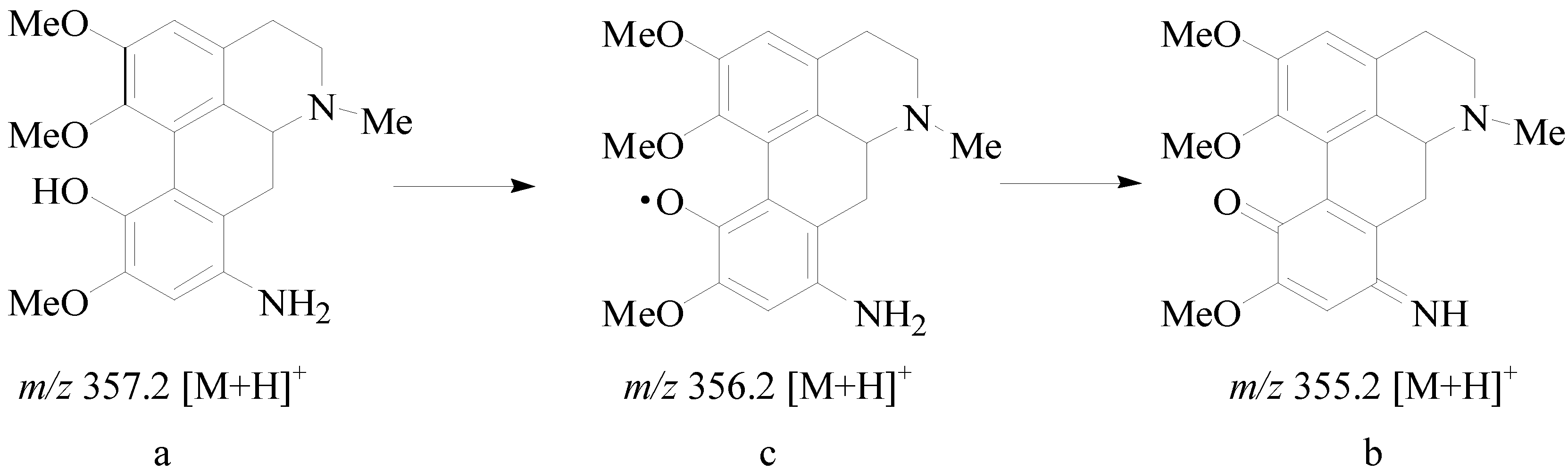
2.2. Biological Activity
2.2.1. In Vitro Anticancer Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
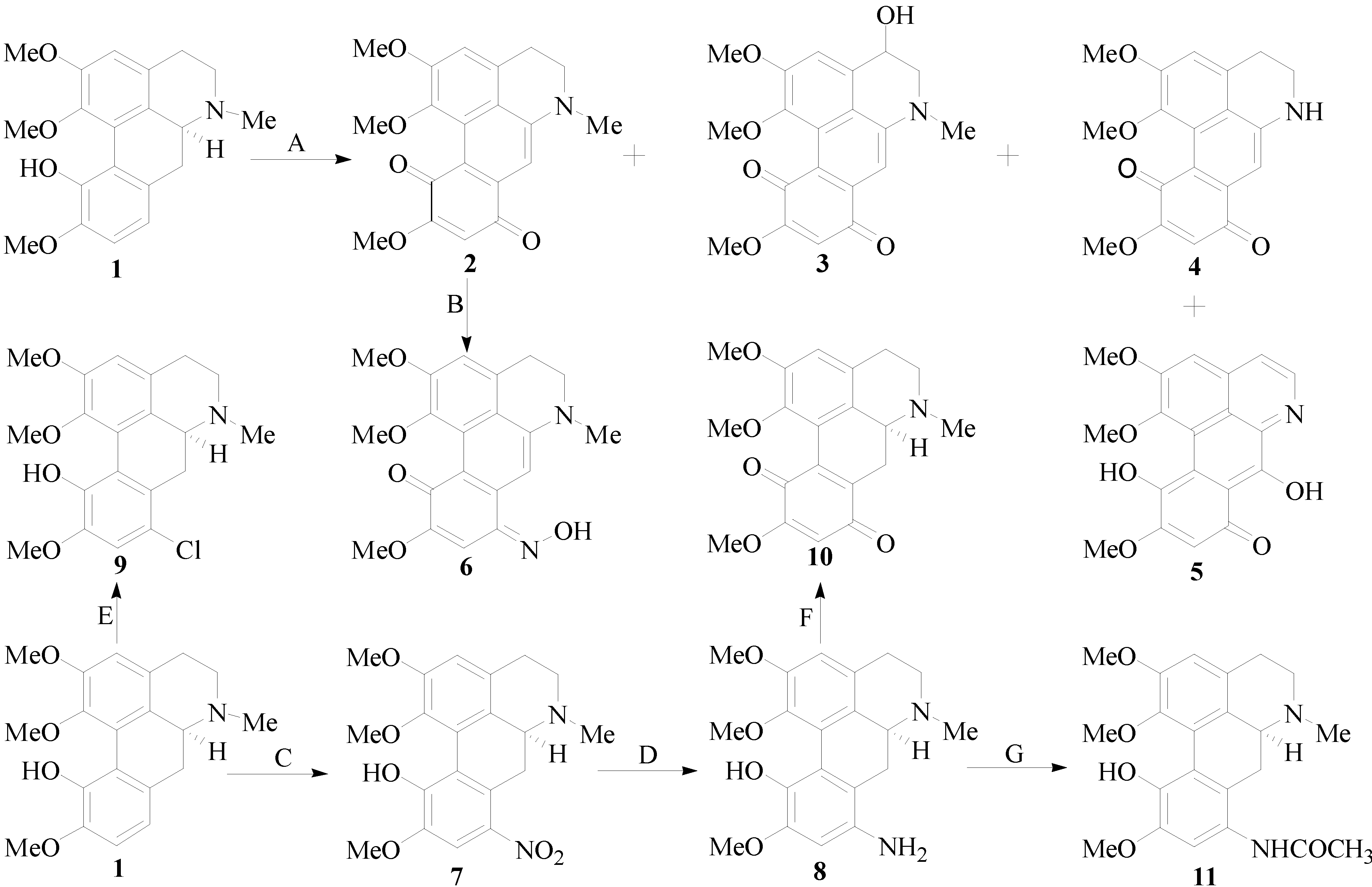{kind=link}
{kind=link}
| Compounds | Mean IC50 (μM) | ||
|---|---|---|---|
| HepG2 | A549 | SGC7901 | |
| 1 | >200.00 | >200.00 | >200.00 |
| 2 | 186.97 | 197.73 | >200.00 |
| 6 | 78.10 | 63.70 | 67.91 |
| 7 | >200.00 | >200.00 | >200.00 |
| 8 | 56.18 | 7.53 | 14.80 |
| 9 | >200.00 | >200.00 | >200.00 |
| 10 | 20.42 | 8.59 | 14.03 |
| 11 | >200.00 | >200.00 | >200.00 |
| Cisplatin | 0.67 | 1.02 | 0.88 |
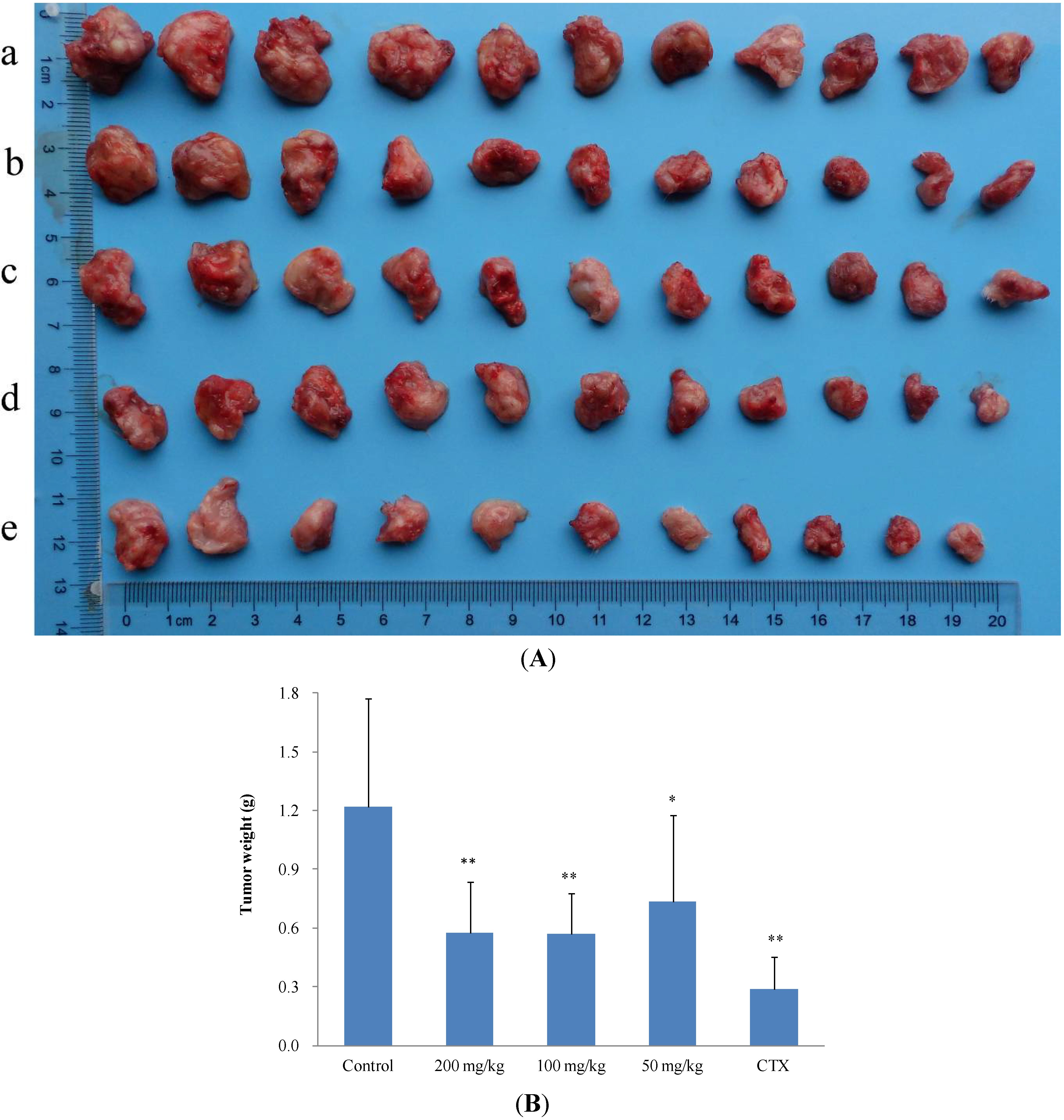
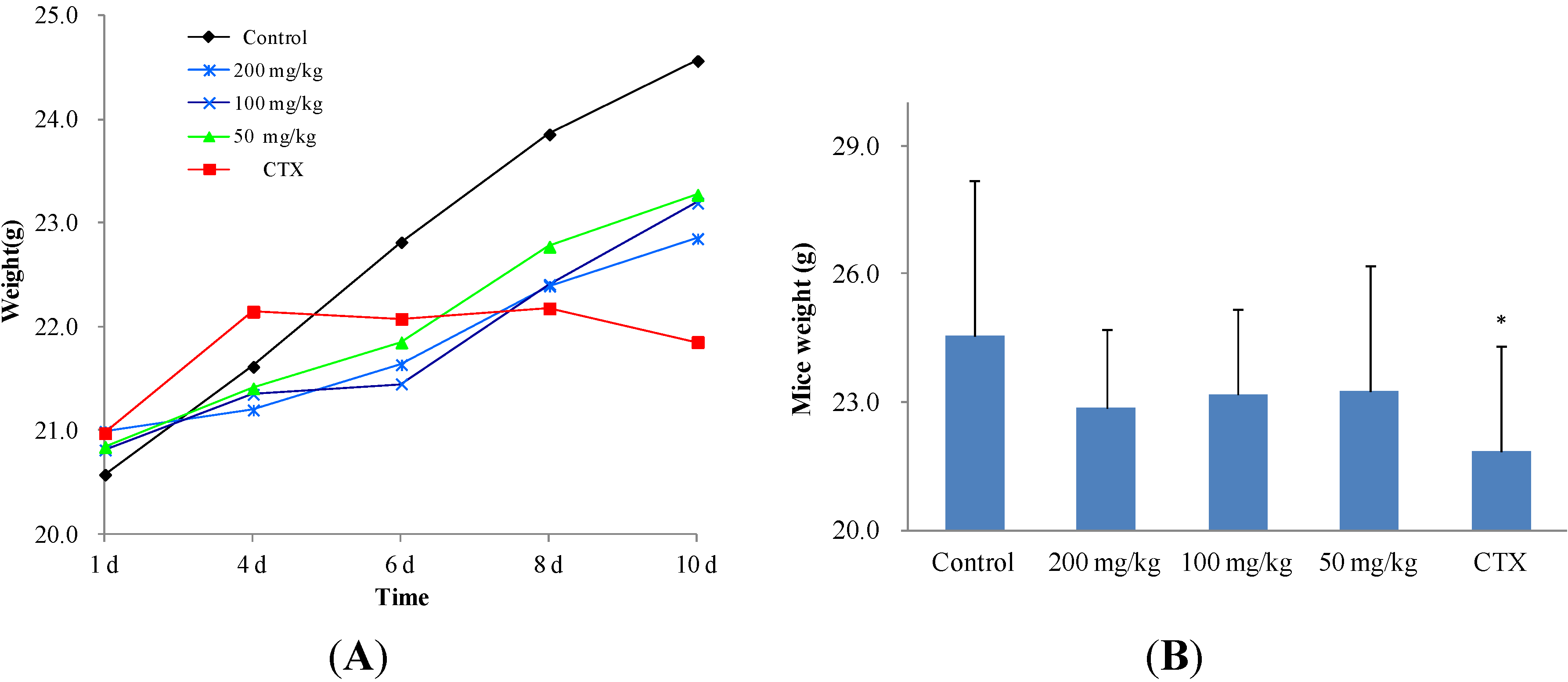
2.2.2. In Vivo Anticancer Activity
| Treatment group | Mice number | Dose (mg/kg/d) | Tumor weight (g, Mean ± SD) | Inhibition ratio (%) |
|---|---|---|---|---|
| Control | 11 | - | 1.2917 ± 0.0935 | - |
| 2 | 11 | 200 | 0.6431 ± 0.0197 ** | 50.21 |
| 2 | 11 | 100 | 0.7011 ± 0.0480 * | 45.72 |
| 2 | 11 | 50 | 0.9661 ± 0.0720 | 25.21 |
| CTX | 11 | 20 | 0.4645 ± 0.0152 ** | 64.04 |
| Treatment group | Mice number | Dose (mg/kg/d) | Tumor weight (g, Mean ± SD) | Inhibition ratio (%) |
|---|---|---|---|---|
| Control | 11 | - | 1.2189 ± 0.5520 | - |
| 11 | 11 | 200 | 0.5764 ± 0.2615 ** | 52.71 |
| 11 | 11 | 100 | 0.5714 ± 0.2087 ** | 53.12 |
| 11 | 11 | 50 | 0.7359 ± 0.4401 * | 39.63 |
| CTX | 11 | 20 | 0.2902 ± 0.1610 ** | 76.19 |
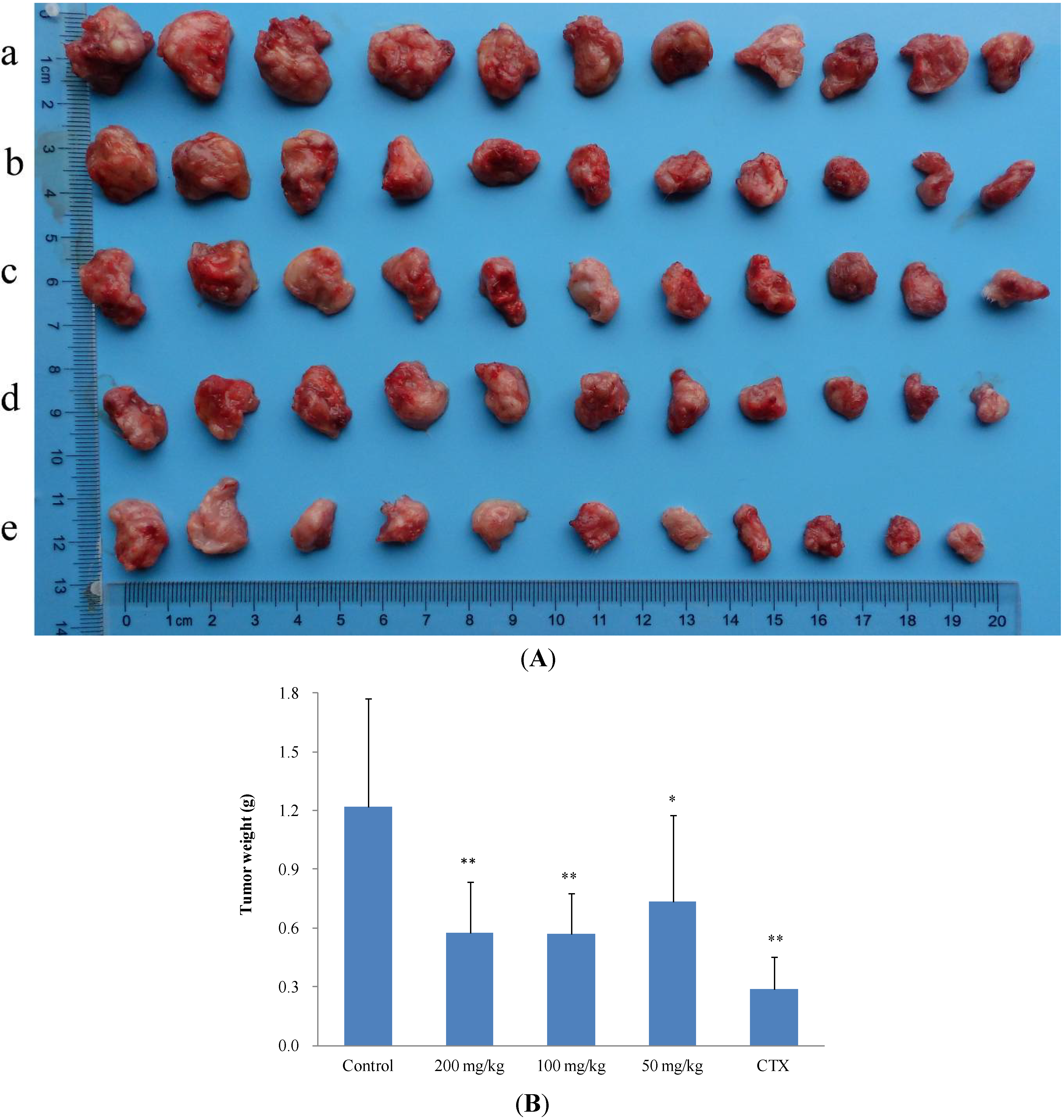
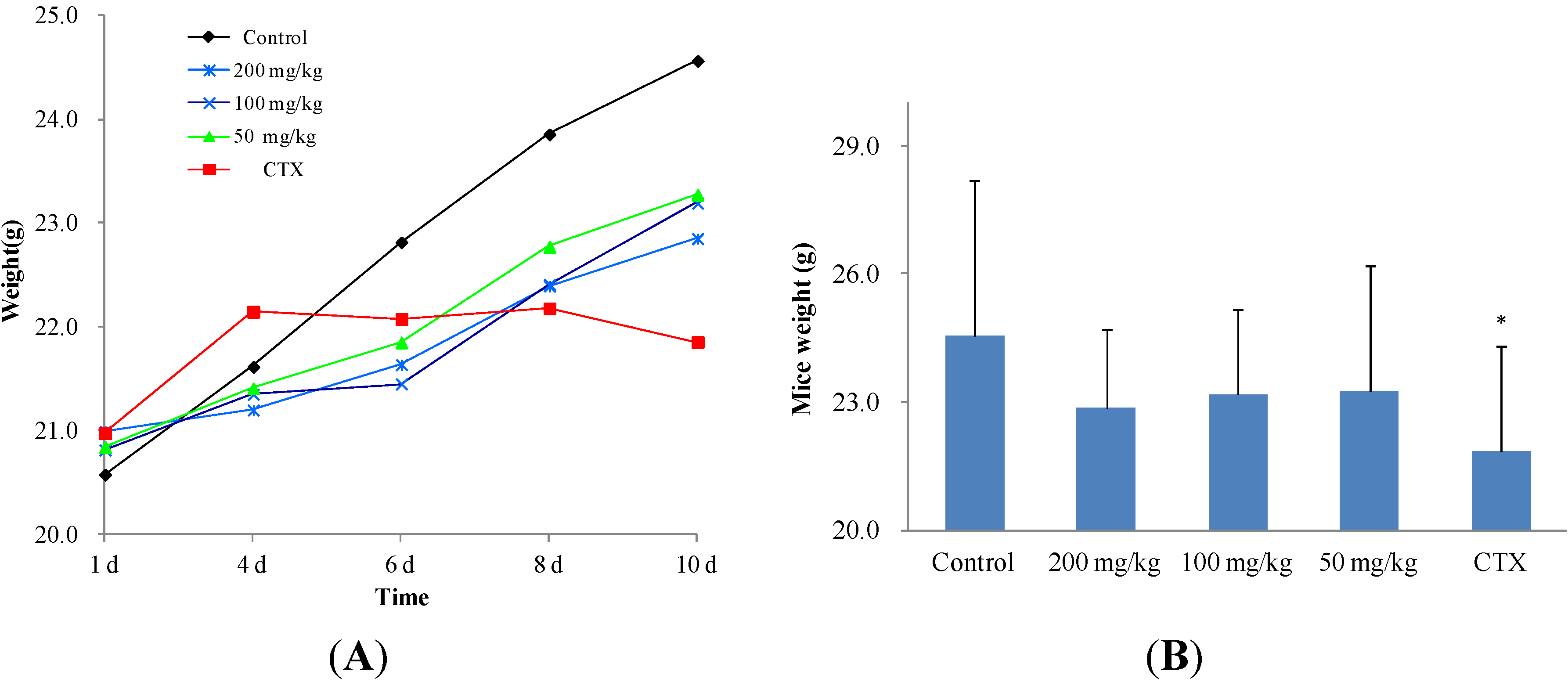
2.3. The Structure and Activity Relationships of ICD Alkaloid
3. Experimental
3.1. Chemistry
3.1.1. Materials
3.2. General Procedure for ICD Derivatives Preparations
3.2.1. Isolation of ICD (1)
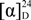 + 205.0 (c 0.1, MeOH); 1H-NMR (400 MHz, CD3COCD3) δ: 6.74 (1H, s, H-3), 3.36 (2H, m, H-4), 2.49 (2H, m, H-5), 2.61 (1H, dd, J = 16.4, 3.6 Hz, H-6a), 2.99 (2H, m, H-7), 7.00 (1H, d, J = 8.0 Hz, H-8), 7.14 (1H, d, J = 8.0 Hz, H-9), 3.04 (3H, s, N-CH3), 3.67 (3H, s, 1-OCH3), 3.88 (3H, s, 2-OCH3), 3.79 (3H, s, 10-OCH3); 13C-NMR (100 MHz, CD3COCD3) δ: 142.5 (C-1), 124.2 (C-1a), 126.2 (C-1b), 152.0 (C-2), 111.4 (C-3), 130.7 (C-3a), 35.9 (C-4), 52.7 (C-5), 63.0 (C-6a), 29.5 (C-7), 128.6 (C-7a), 119.4 (C-8), 112.1 (C-9), 149.1 (C-10), 144.1 (C-11), 124.3 (C-11a), 65.5 (1-OCH3), 55.4 (2-OCH3), 52.7 (10-OCH3), 43.4 (N-CH3). HRMS (ESI): m/z calcd. for C20H24NO4 [M + H]+ 342.1699, obsd. 342.1647.
+ 205.0 (c 0.1, MeOH); 1H-NMR (400 MHz, CD3COCD3) δ: 6.74 (1H, s, H-3), 3.36 (2H, m, H-4), 2.49 (2H, m, H-5), 2.61 (1H, dd, J = 16.4, 3.6 Hz, H-6a), 2.99 (2H, m, H-7), 7.00 (1H, d, J = 8.0 Hz, H-8), 7.14 (1H, d, J = 8.0 Hz, H-9), 3.04 (3H, s, N-CH3), 3.67 (3H, s, 1-OCH3), 3.88 (3H, s, 2-OCH3), 3.79 (3H, s, 10-OCH3); 13C-NMR (100 MHz, CD3COCD3) δ: 142.5 (C-1), 124.2 (C-1a), 126.2 (C-1b), 152.0 (C-2), 111.4 (C-3), 130.7 (C-3a), 35.9 (C-4), 52.7 (C-5), 63.0 (C-6a), 29.5 (C-7), 128.6 (C-7a), 119.4 (C-8), 112.1 (C-9), 149.1 (C-10), 144.1 (C-11), 124.3 (C-11a), 65.5 (1-OCH3), 55.4 (2-OCH3), 52.7 (10-OCH3), 43.4 (N-CH3). HRMS (ESI): m/z calcd. for C20H24NO4 [M + H]+ 342.1699, obsd. 342.1647.3.2.2. Synthesis of Isocorydione (2)
3.2.3. 4-Hydroxy-isocorydione (3)
3.2.4. N-demethyl-isocorydione (4)
3.2.5. 7-Hydroxy-N-demethyl-isocorydione (5)
3.2.6. 8-Hydroxylamine-isocorydione (6)
3.2.7. Synthesis of 8-Nitro-isocorydine (7)
+ 170.0 (c 0.1, MeOH); 1H-NMR (400 MHz, CDCl3): 6.76 (1H, s, H-3), 3.00 (1H, m, H-4), 2.40–2.48 (1H, J = 17.2, 2.8 Hz, H-4), 2.68–2.72 (2H, dd, J = 17.2, 2.8 Hz, H-5), 3.67 (1H, dd, J = 14.4, 3.2 Hz, H-6a), 3.00–3.21 (2H, m, H-7), 7.52 (1H, s, H-9), 3.46 (3H, s, N-CH3), 3.71 (3H, s, 1-OCH3), 3.96 (3H, s, 2-OCH3), 3.90 (3H, s, 10-OCH3); 13C-NMR (100 MHz, CDCl3): 142.3 (C-1), 128.0 (C-1a), 130.2 (C-1b), 151.4 (C-2), 112.1 (C-3), 127.5 (C-3a), 29.1 (C-4), 52.6 (C-5), 62.4 (C-6a), 31.0 (C-7), 121.3 (C-7a), 141.8 (C-8), 107.1 (C-9), 148.8 (C-10), 148.7 (C-11), 123.8 (C-11a), 61.6 (1-OCH3), 56.3 (2-OCH3), 55.9 (10-OCH3), 43.9 (N-CH3). HRMS (ESI): m/z calcd. for C20H23N2O6 [M + H]+ 387.1551, obsd. 387.1565.3.2.8. 8-Amino-isocorydine (8)
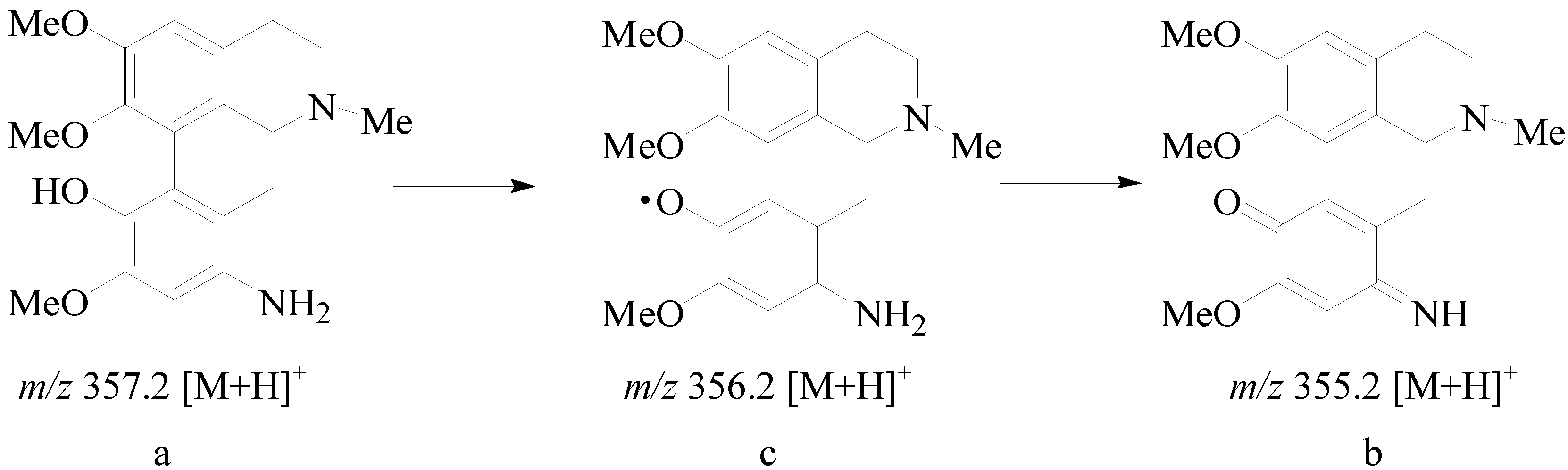
+ 170.0 (c 0.1, MeOH);1H-NMR (400 MHz, CDCl3): 6.60 (1H, s, H-3), 3.00 (2H, m, H-4), 2.43 (2H, m, H-5), 3.00 (1H, m, H-6a), 2.88 (1H, dd, J = 16.4, 2.0 Hz, H-7), 2.60 (1H, dd, J = 16.4, 2.0 Hz, H-7), 4.35 (2H, s, 8-NH2), 6.34 (1H, s, H-9), 2.51 (3H, s, N-CH3), 3.56 (3H, s, 1-OCH3), 3.79 (3H, s, 2-OCH3), 3.76 (3H, s, 10-OCH3); 13C-NMR (100 MHz, CDCl3): 142.2 (C-1), 125.9 (C-1a), 128.9 (C-1b), 151.2 (C-2), 113.0 (C-3), 127.6 (C-3a), 29.2 (C-4), 52.2 (C-5), 62.2 (C-6a), 36.6 (C-7), 120.6 (C-7a), 135.4 (C-8), 102.0 (C-9), 149.2 (C-10), 136.9 (C-11), 110.7 (C-11a), 62.8 (1-OCH3), 55.8 (2-OCH3), 55.4 (10-OCH3), 43.9 (N-CH3). HRMS (ESI): m/z calcd. for C20H25N2O4 [M + H]+ 357.1809, obsd. 357.1816.3.2.9. 8-Chloro-isocorydine (9)
+ 175.0 (c 0.1, MeOH) 1H-NMR (400 MHz, CDCl3): 6.74 (1H, s, H-3), 2.31–2.31 (2H, m, H-4), 2.91–2.95 (2H, m, H-5), 3.06 (1H, m, H-6a), 2.67–2.69 (2H, m, H-7), 7.50 (1H, s, H-9), 2.38 (3H, s, N-CH3), 3.60 (3H, s, 1-OCH3), 3.85 (3H, s, 2-OCH3), 3.79 (3H, s, 10-OCH3); 13C-NMR (100 MHz, CDCl3): 142.1 (C-1), 128.5 (C-1a), 130.1 (C-1b), 151.2 (C-2), 112.0 (C-3), 127.4 (C-3a), 29.6 (C-4), 52.5 (C-5), 62.3 (C-6a), 30.9 (C-7), 121.2 (C-7a), 141.7 (C-8), 107.0 (C-9), 148.3 (C-10), 148.7 (C-11),123.7 (C-11a), 61.5 (1-OCH3), 56.3 (2-OCH3), 55.9 (10-OCH3), 43.9 (N-CH3). HRMS (ESI): m/z calcd. for C20H22NO4Cl [M]+ 375.1237, obsd. 375.2060.3.2.10. 6a,7-Dihydrogen-isocorydione (10)
3.2.11. 8-Acetamino-isocorydine (11)
+ 172.0 (c 0.1, MeOH); 1H-NMR (400 MHz, CD3Cl): 7.53 (1H, s, H-3), 3.00 (2H, m, H-4), 2.43 (2H, m, H-5), 3.00 (1H, m, H-6a), 2.88 (1H, dd, J = 16.4, 2.0 Hz, H-7), 2.60 (1H, dd, J = 16.4, 2.0 Hz, H-7), 6.73 (1H, s, H-9), 2.48 (3H, s, N-CH3), 3.43 (3H, s, 1-OCH3), 3.90 (3H, s, 2-OCH3), 3.86 (3H, s, 10-OCH3); 2.22 (3H, s, COCH3). 13C-NMR (100 MHz, CD3Cl): 146.3 (C-1), 128.0 (C-1a), 128.5 (C-1b), 151.9 (C-2), 113.2 (C-3), 126.4 (C-3a), 29.4 (C-4), 52.7 (C-5), 61.8 (C-6a), 31.0 (C-7), 122.3 (C-7a), 141.4 (C-8), 107.1 (C-9), 150.9 (C-10), 146.1 (C-11),123.8 (C-11a), 61.3 (1-OCH3), 56.1 (2-OCH3), 55.5 (10-OCH3), 43.6 (N-CH3), 21.1 (COCH3). HRMS (ESI): m/z calcd. for C22H27N2O5 [M + H]+ 399.1914, obsd. 399.1911.3.3. Biological Assay
3.3.1. Materials
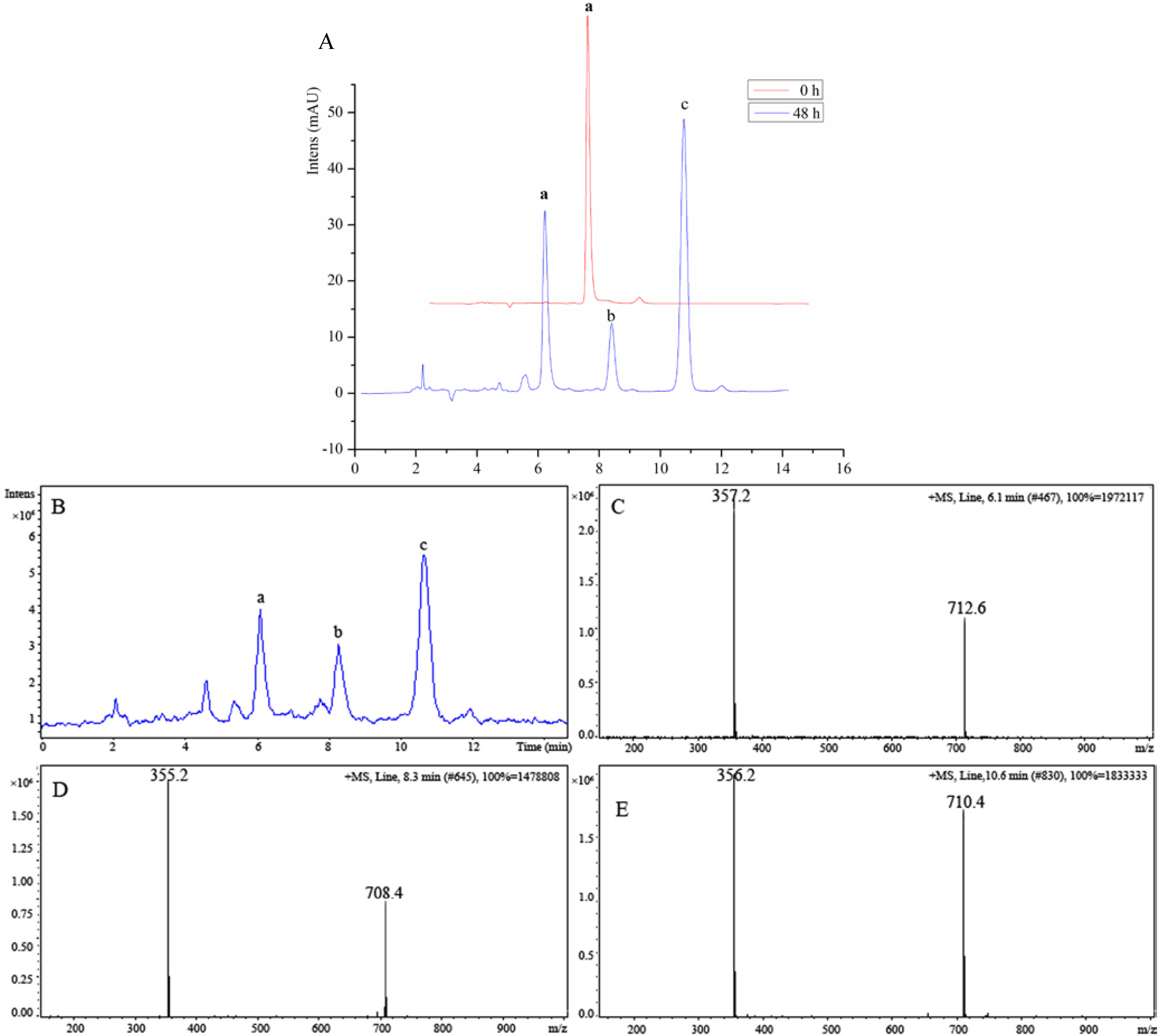
3.3.2. Anticancer Activity in Vitro
3.3.3. Anticancer Activity in Vivo
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, Y.J.; Liu, J.X.; Di, D.L.; Li, M.; Fen, Y. Structural and mechanistic bases of the anticancer activity of natural aporphinoid alkaloids. Curr. Top. Med. Chem. 2013, 13, 2116–2126. [Google Scholar]
- Stévigny, C.; Bailly, C.; Quetin-Leclercq, J. Cytotoxic and antitumor potentialities of aporphinoid alkaloids. Curr. Med. Chem. Anticancer Agents 2005, 5, 173–182. [Google Scholar]
- Gerhardt, D.; Horn, A.; Gaelzer, M.; Frozza, R.; Delgado-Canedo, A.; Pelegrini, A.; Henriques, A.; Lenz, G.; Salbego, C. Boldine: A potential new antiproliferative drug against glioma cell lines. Investig. New Drugs 2009, 27, 517–525. [Google Scholar]
- Zhang, A.; Zhang, Y.; Branfman, A.; Baldessarini, R.; Neumeyer, J. Advances in development of dopaminergic aporphinoids. J. Med. Chem. 2007, 50, 171–181. [Google Scholar]
- Ponnala, S.; Chaudhary, S.; Gonzalez-sarrias, A.; Seeram, N.; Harding, W. Cytotoxicity of aporphines in human colon cancer cell lines HCT-116 and Caco-2: An SAR study. Bioorg. Med. Chem. Lett. 2011, 21, 4462–4464. [Google Scholar]
- Guo, C.C.; Yu, C.H.; Li, L.; Wang, Y.Q.; Wang, S.J.; Wang, W.H.; Hu, H.H.; Xu, S.Y.; Yu, L.S.; Jiang, H.D.; et al. Rapid determination of isocorydine in rat plasma and tissues using liquid chromatography—Tandem mass spectrometry and its applications to pharmacokinetics and tissue distribution. Xenobiotica 2012, 42, 466–476. [Google Scholar] [CrossRef]
- Dang, Y.; Gong, H.F.; Liu, J.X.; Yu, S.J. Alkaloid from Dicranostigma leptopodum (Maxim) Fedde. Chin. Chem. Lett. 2009, 20, 1218–1220. [Google Scholar]
- Liu, D.H.; Zhang, T.C.; Liu, J.X.; Di, D.L.; Dang, Y. Chemical constituents of alkaloids from Dicranostigma leptopodum. Chin. Tradit. Herb Drugs 2011, 42, 1505–1508. [Google Scholar]
- Sun, H.F.; Hou, H.L.; Lu, P.; Zhang, L.X.; Zhao, F.Y.; Ge, C.; Wang, T.P.; Yao, M.; Li, J.J. Isocorydine inhibits cell proliferation in hepatocellular carcinoma cell lines by inducing G2/M cell cycle arrest and apoptosis. PLoS One 2012, 7, e36808. [Google Scholar]
- Lu, P.; Sun, H.F.; Zhang, L.X.; Hou, H.L; Zhang, L.; Zhao, F.Y.; Ge, C.; Yao, M.; Wang, T.P.; Li, J.J. Isocorydine targets the drug-resistant cellular side population through PDCD4-related apoptosis in hepatocellular carcinoma. Mol. Med. 2012, 18, 1136–1146. [Google Scholar]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef]
- Ferreira, M.L.R.; Pascoli, I.C.; Nascimento, I.R.; Zukerman-Schpector, J.; Lopes, L.M.X. Aporphine and bisaporphine alkaloids from Aristolochia lagesiana var. intermedia. Photochemistry 2010, 71, 569–478. [Google Scholar]
- Zhang, T.C.; Ye, H.L.; Liu, J.X.; Di, D.L. Study on semi-synthetic transforming technology for the natural product of isocorydione. Acta Pharm. Sin. 2011, 46, 1471–1475. [Google Scholar]
- Chia, Y.C.; Chang, F.R.; Wu, Y.C. Two p-Quinonoid aporphine alkaloids from Fissistigma balansae. J. Nat. Prod. 1998, 61, 1430–1432. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar]
- Wu, Z.Q.; Patel, A.; Dave, R.; Yuan, X.D. Development of acetaminophin proline prodrug. Bioorg. Med. Chem. Lett. 2010, 20, 3851–3854. [Google Scholar]
- Woo, S.H.; Sun, N.J.; Cassady, J.M.; Snapka, R.M. Topoisomerase II inhibition by aporphine alkaloids. Biochem. Pharmacol. 1999, 57, 1141–1145. [Google Scholar]
- Ribár, B.; Lazar, D.; Gašić, O.; Kanyó, I.; Engel, P. Structure of isocorydine. Acta Cryst. 1992, C48, 945–947. [Google Scholar]
- Sample Availability: All products reported in this paper are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhong, M.; Liu, Y.; Liu, J.; Di, D.; Xu, M.; Yang, Y.; Li, W.; Chen, Y.; Liu, J. Isocorydine Derivatives and Their Anticancer Activities. Molecules 2014, 19, 12099-12115. https://doi.org/10.3390/molecules190812099
Zhong M, Liu Y, Liu J, Di D, Xu M, Yang Y, Li W, Chen Y, Liu J. Isocorydine Derivatives and Their Anticancer Activities. Molecules. 2014; 19(8):12099-12115. https://doi.org/10.3390/molecules190812099
Chicago/Turabian StyleZhong, Mei, Yanjuan Liu, Junxi Liu, Duolong Di, Mengrou Xu, Yaya Yang, Wenguang Li, Yali Chen, and Jinxia Liu. 2014. "Isocorydine Derivatives and Their Anticancer Activities" Molecules 19, no. 8: 12099-12115. https://doi.org/10.3390/molecules190812099
APA StyleZhong, M., Liu, Y., Liu, J., Di, D., Xu, M., Yang, Y., Li, W., Chen, Y., & Liu, J. (2014). Isocorydine Derivatives and Their Anticancer Activities. Molecules, 19(8), 12099-12115. https://doi.org/10.3390/molecules190812099