ROS-Dependent Antiproliferative Effect of Brassinin Derivative Homobrassinin in Human Colorectal Cancer Caco2 Cells

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
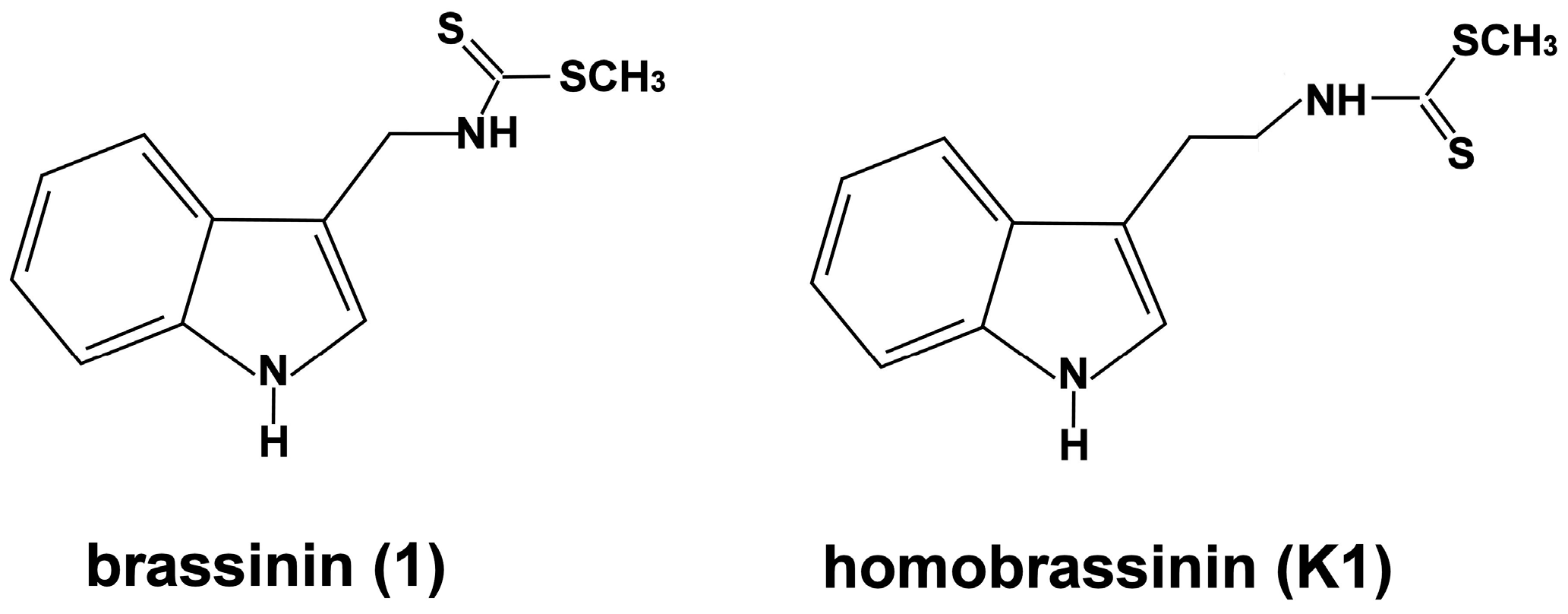
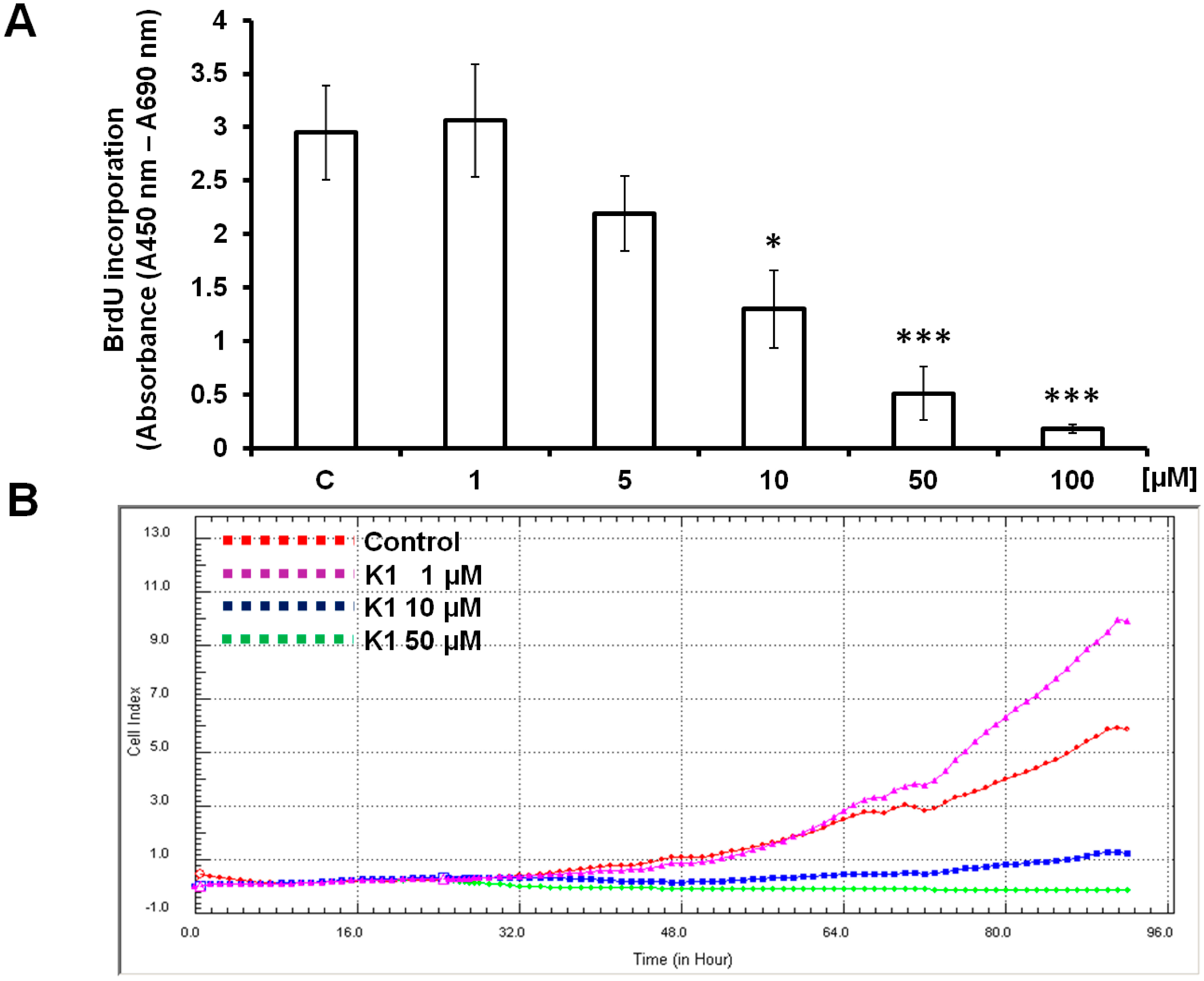
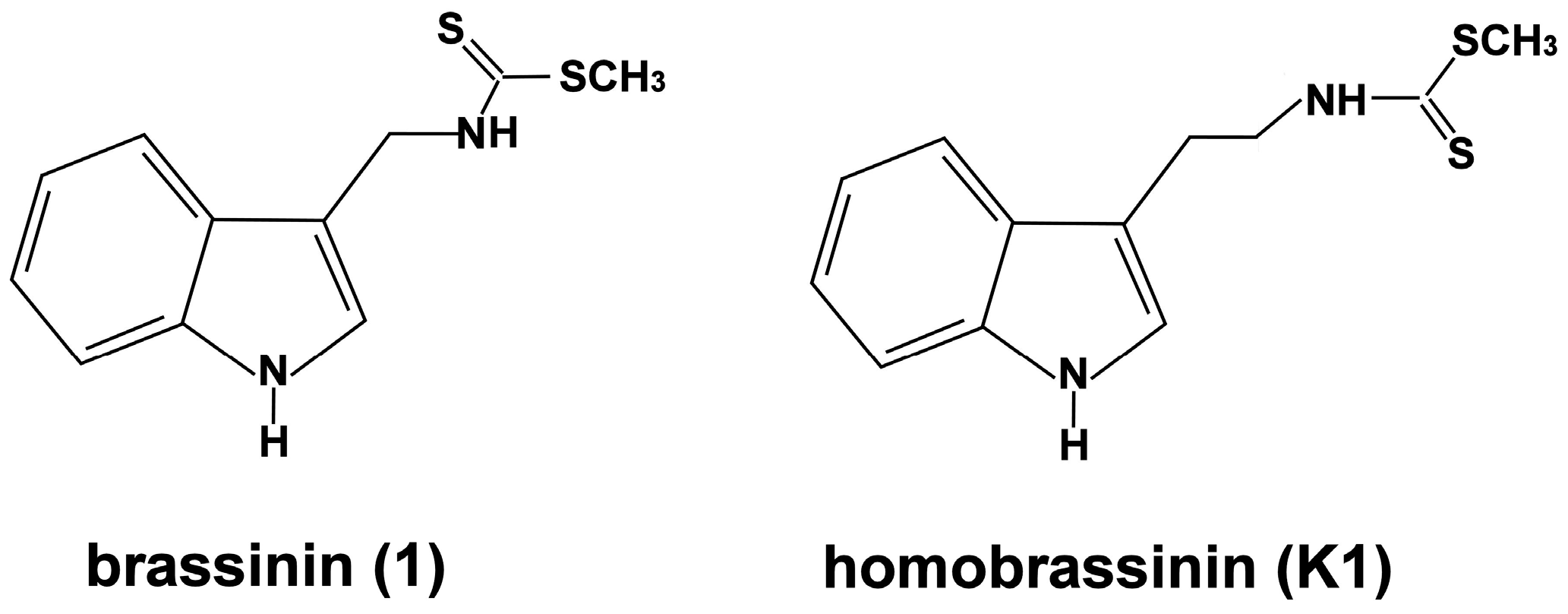
2.1. Effect of Brassinin and Its Derivatives on Cell Proliferation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cancer Cell Lines | |||||||
|---|---|---|---|---|---|---|---|---|
| Jurkat | Caco2 | HepG2 | HCT-116 | A549 | HeLa | MCF-7 | MDA-MB-231 | |
| 1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| K1 | 28.2 ± 1.2 | 8.0 ± 0.6 | 21.3 ± 2.3 | 27.3 ± 1.8 | 33.4 ± 0.8 | 26.1 ± 2.4 | 35.0 ± 2.1 | 22.8 ± 1.4 |
| K10 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| K124 | 30.0 ± 0.5 | >100 | >100 | 30.7 ± 2.9 | >100 | 34.0 ± 0.7 | 92.0 ± 2.7 | >100 |
| K49 | 39.6 ± 1.8 | 89.4 ± 1.3 | 37.4 ± 3.7 | 76.0 ± 2.5 | 22.8 ± 0.5 | 45.6 ± 1.4 | 62.0 ± 3.4 | >100 |
| K170 | 44.3 ± 3.2 | >100 | >100 | 53.7 ± 2.1 | 96.5 ± 4.1 | >100 | >100 | >100 |
| 47 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
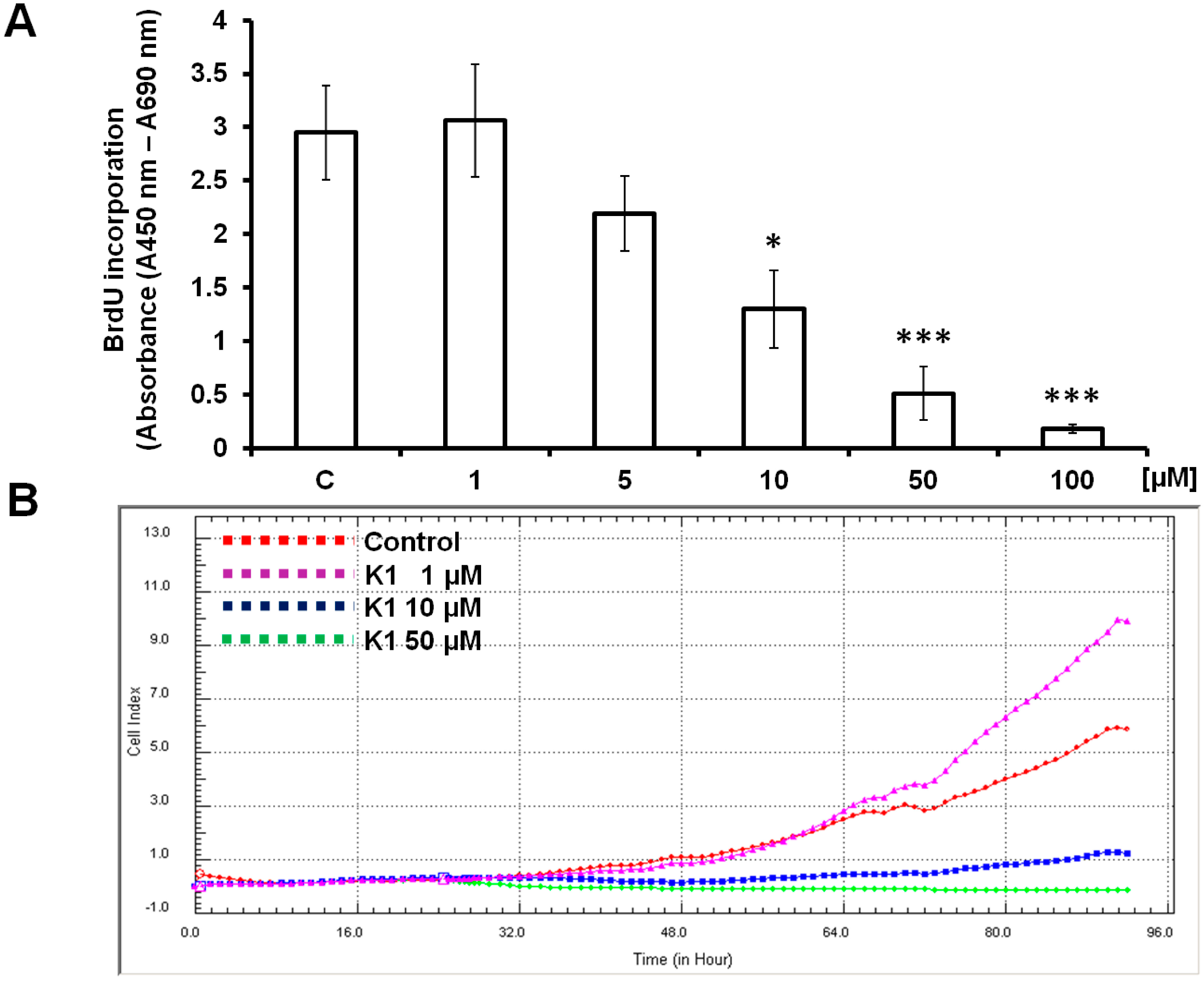
2.2. K1 Blocks Cell Cycle at G2/M Transition
| Treatment | Time (h) | Sub-G1 | G0/G1 | S | G2/M |
|---|---|---|---|---|---|
| Control | 0.29 ± 0.15 | 40.81 ± 3.29 | 18.25 ± 3.90 | 36.64 ± 2.29 | |
| K1 | 24 | 1.49 ± 0.37 | 21.34 ± 3.13 ** | 6.69 ± 1.56 * | 70.48 ± 2.28 *** |
| K1 | 48 | 3.36 ± 1.67 * | 30.89 ± 2.81 * | 9.58 ± 2.43 | 56.20 ± 2.58 ** |
| K1 | 72 | 5.58 ± 1.16 * | 30.17 ± 1.65 * | 11.47 ± 1.65 | 52.79 ± 2.77 ** |
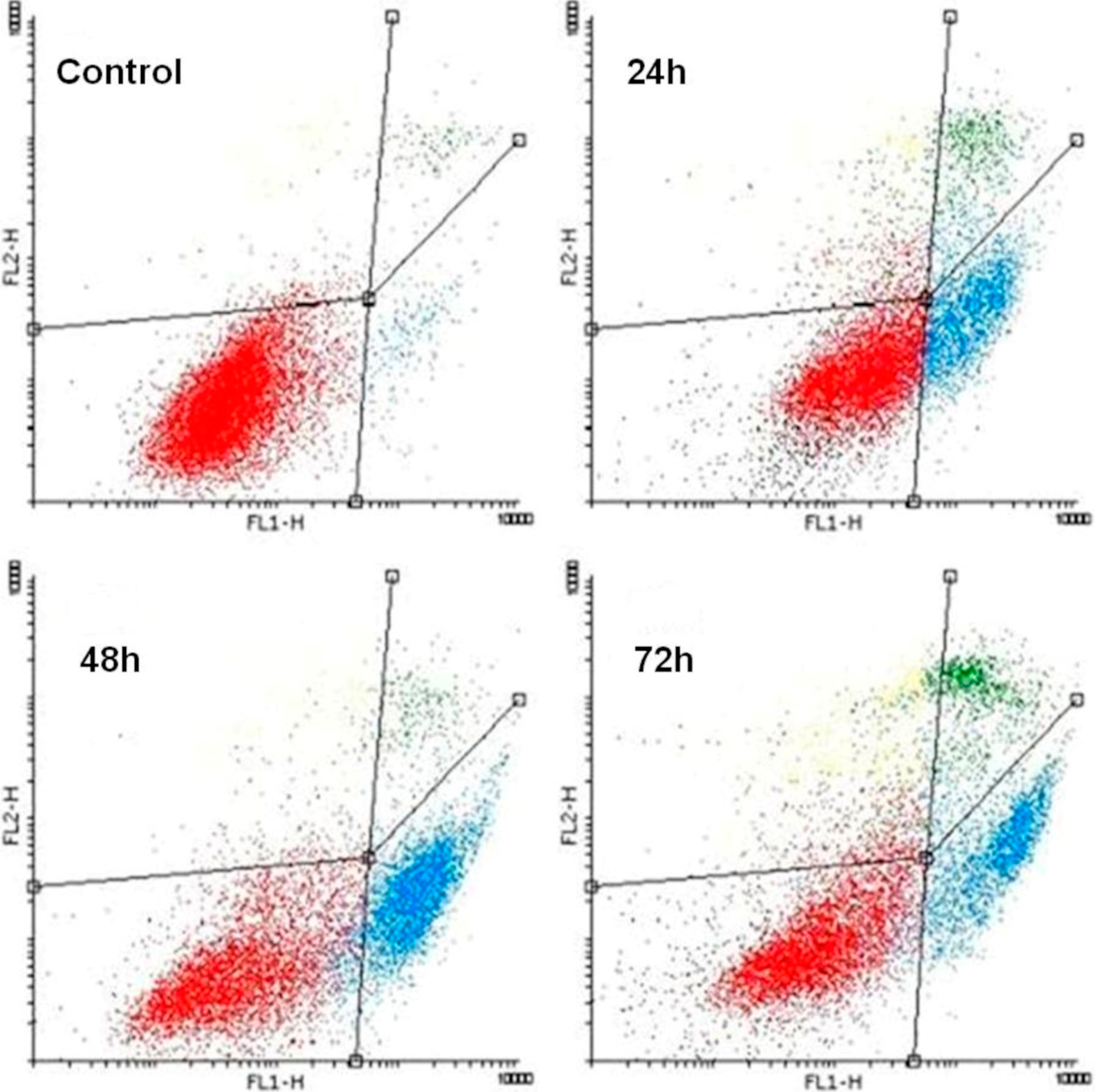
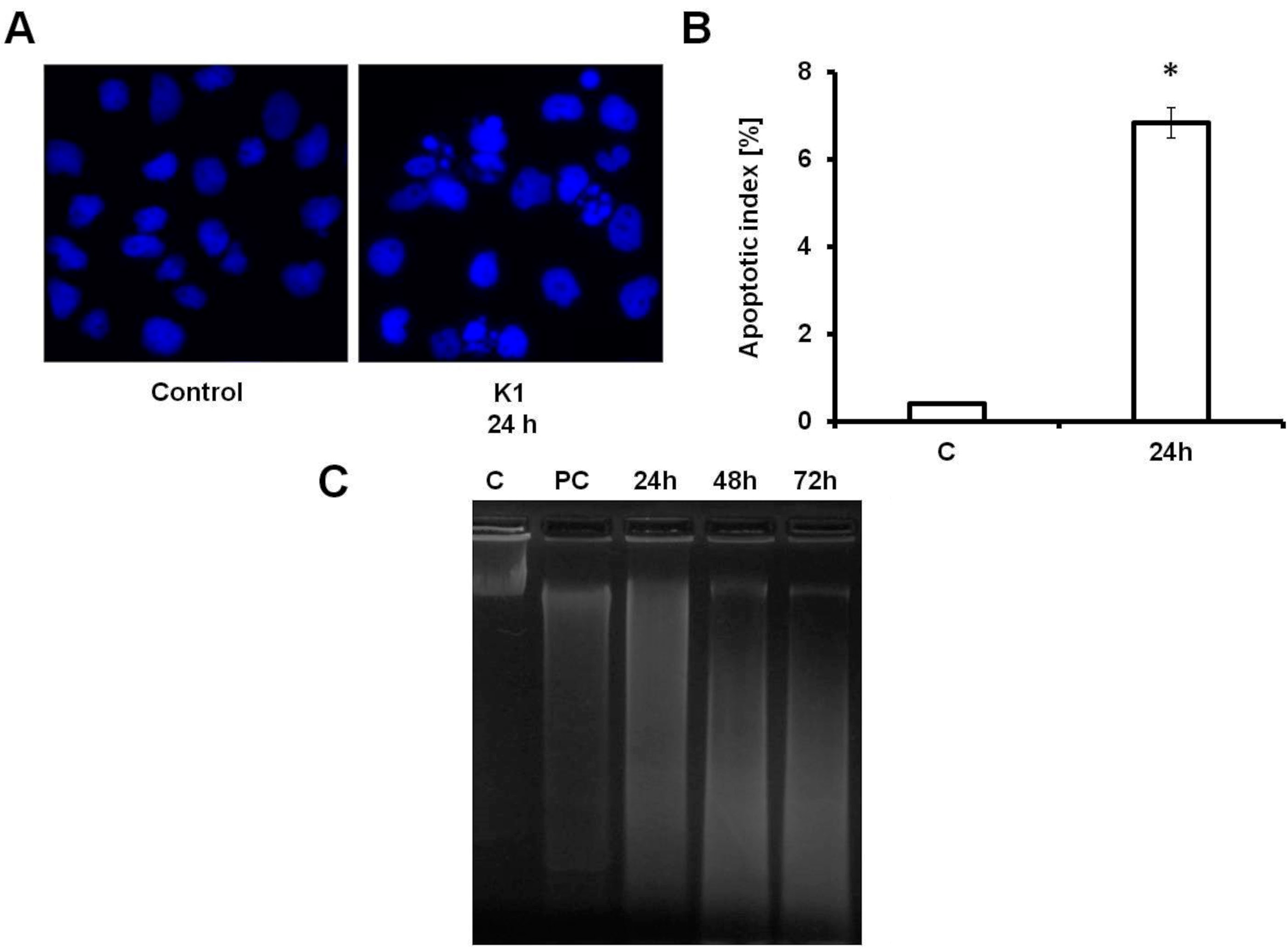
2.3. K1 Induces Apoptotic Cell Death
| Treatment | Time (h) | An−/PI− | An+/PI− | An+/PI+ |
|---|---|---|---|---|
| Control | 94.07 ± 1.32 | 3.17 ± 0.60 | 2.05 ± 0.50 | |
| K1 | 24 | 67.81 ± 1.18 ** | 18.54 ± 1.63 ** | 13.75 ± 2.43 * |
| K1 | 48 | 49.92 ± 3.20 *** | 38.55 ± 2.47 *** | 11.81 ± 2.24 * |
| K1 | 72 | 42.36 ± 3.65 *** | 37.07 ± 2.66 *** | 20.85 ± 3.69 ** |
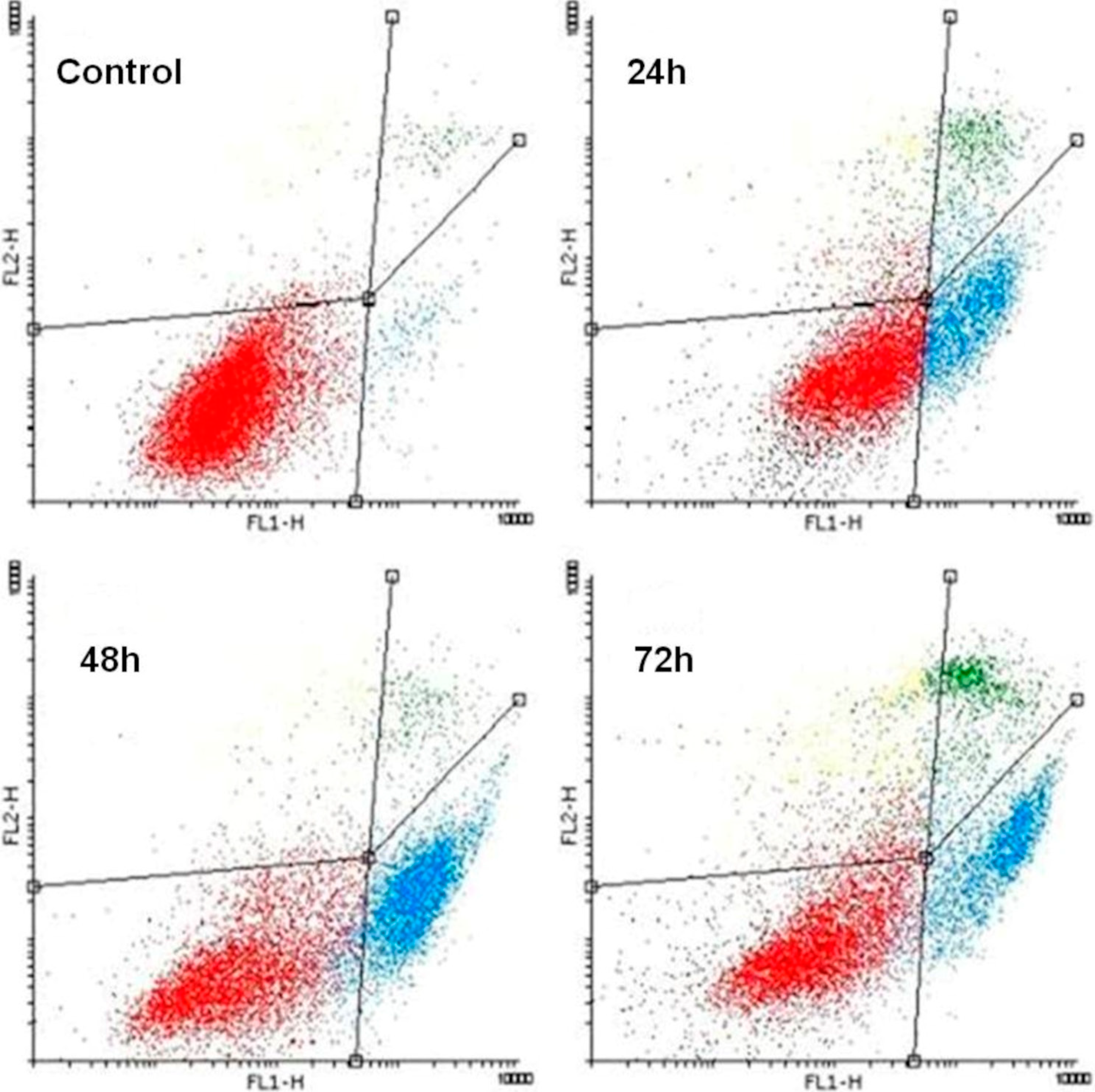
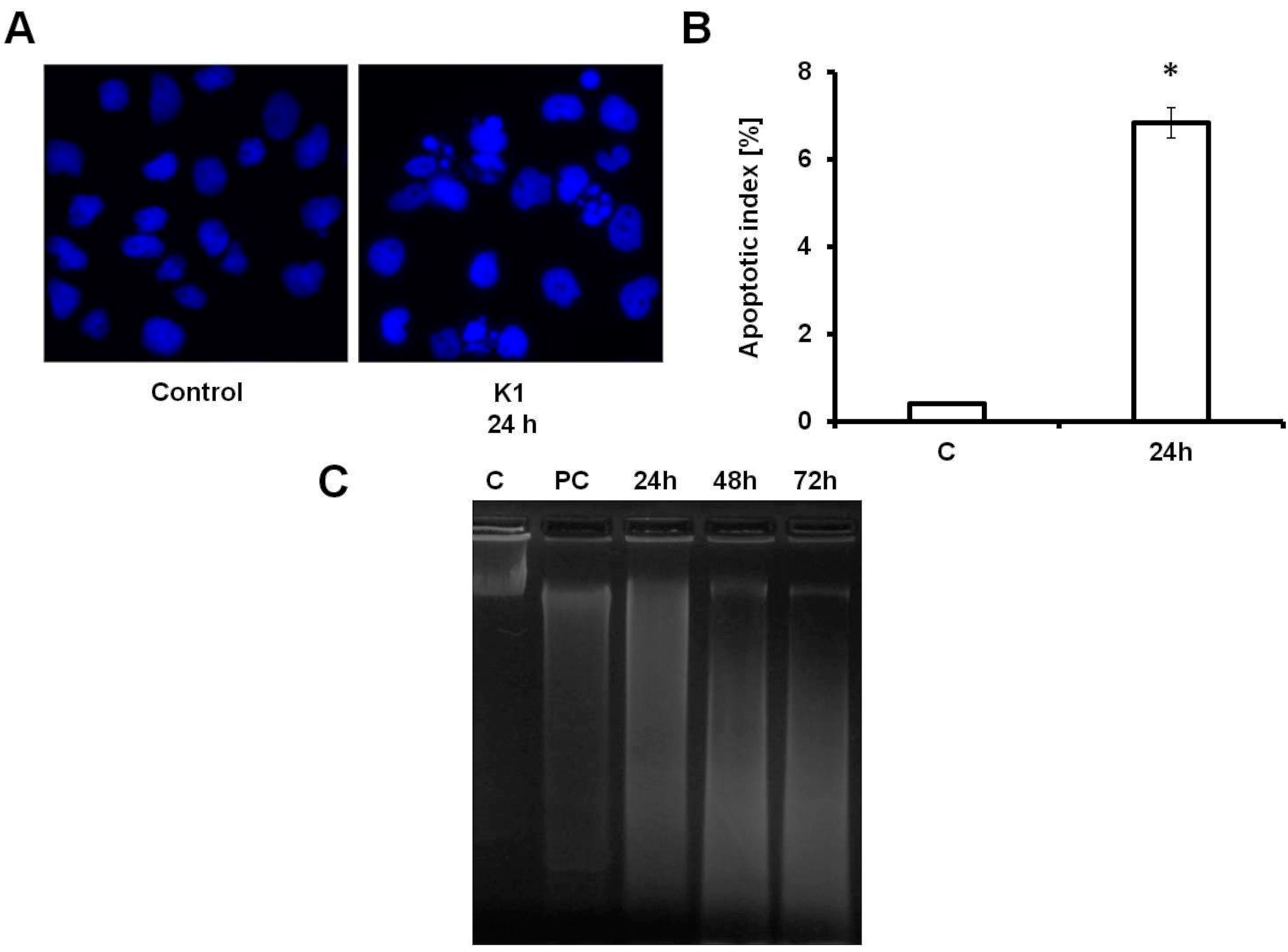
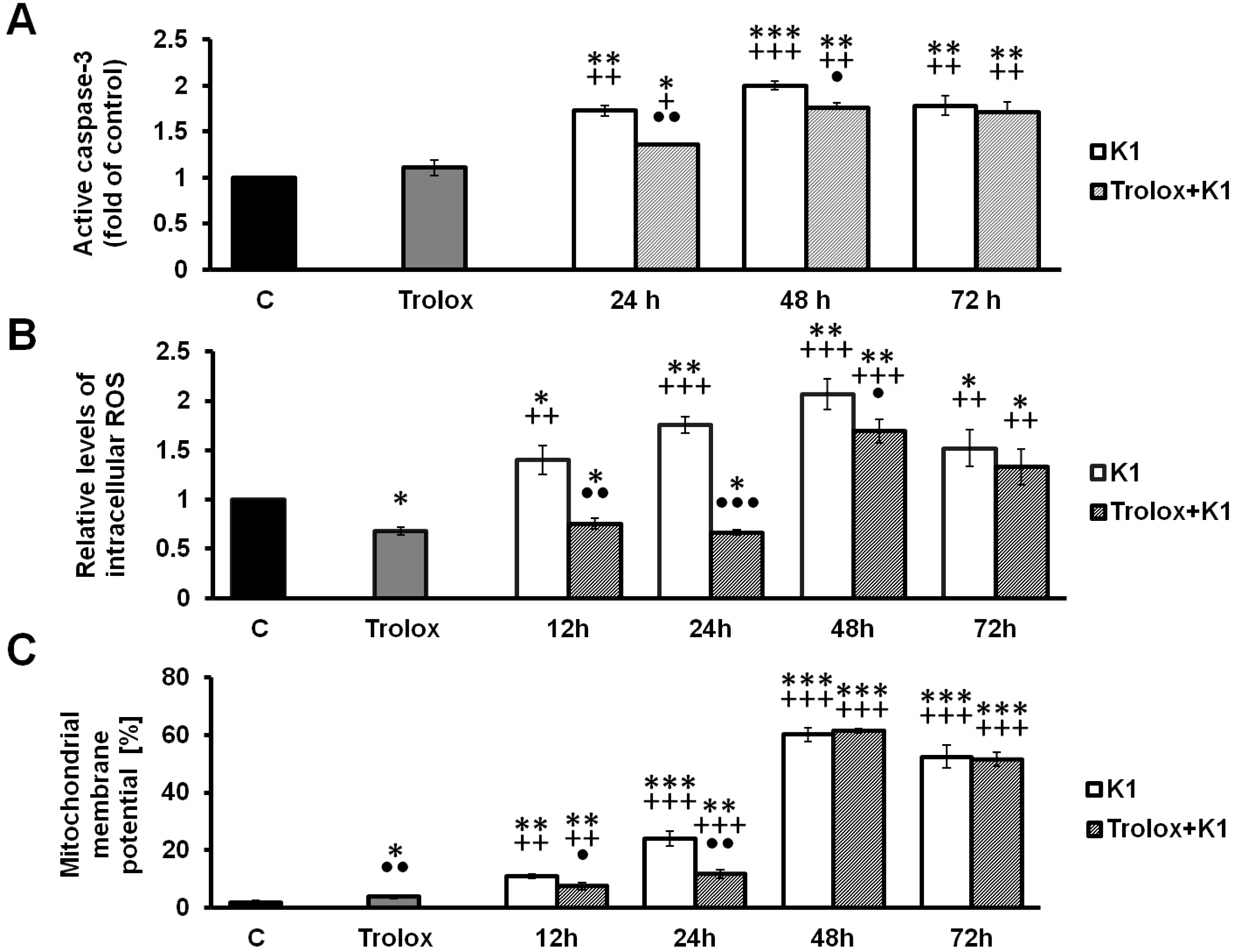
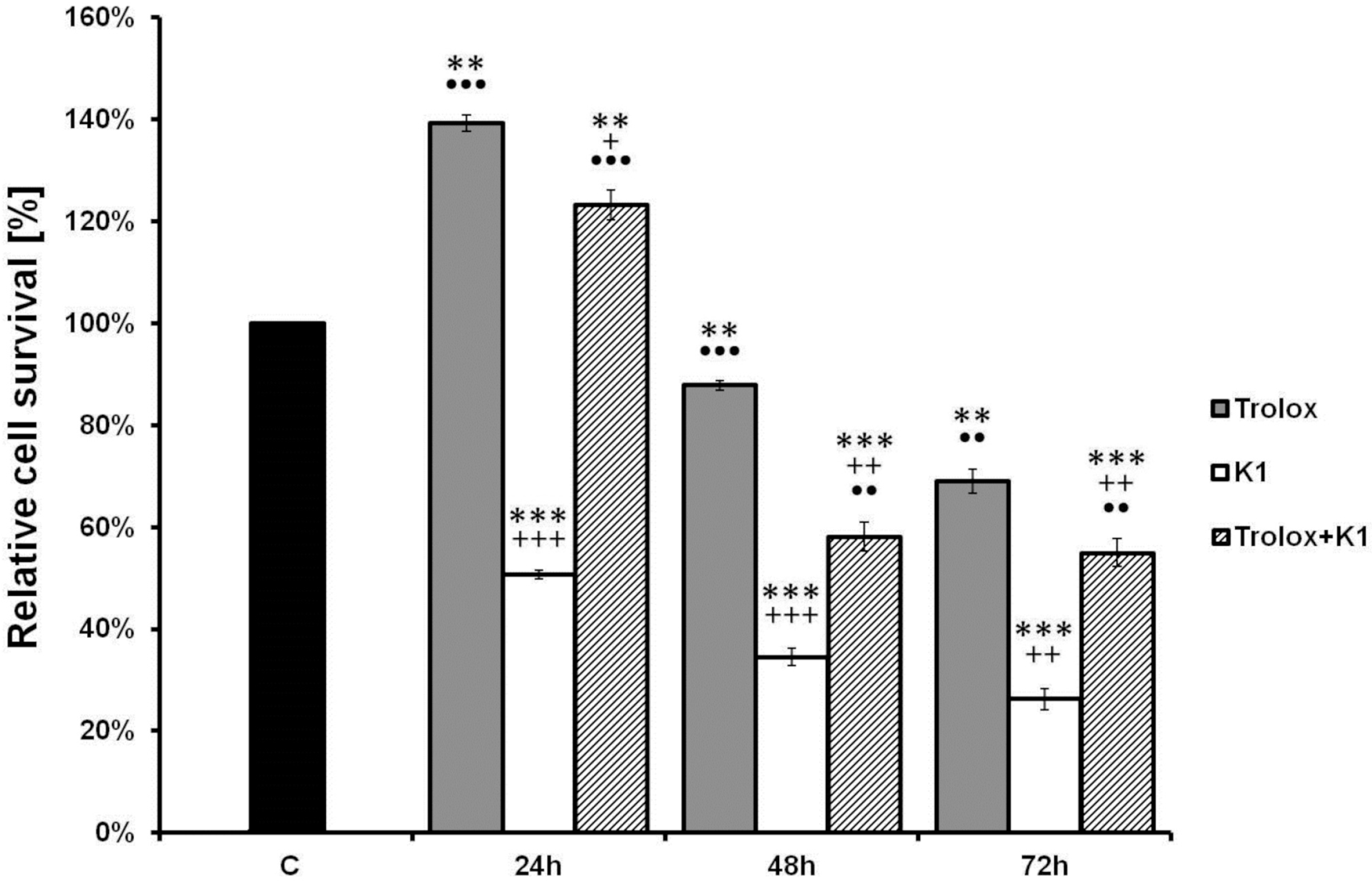
2.4. Effect of K1 on ROS Formation
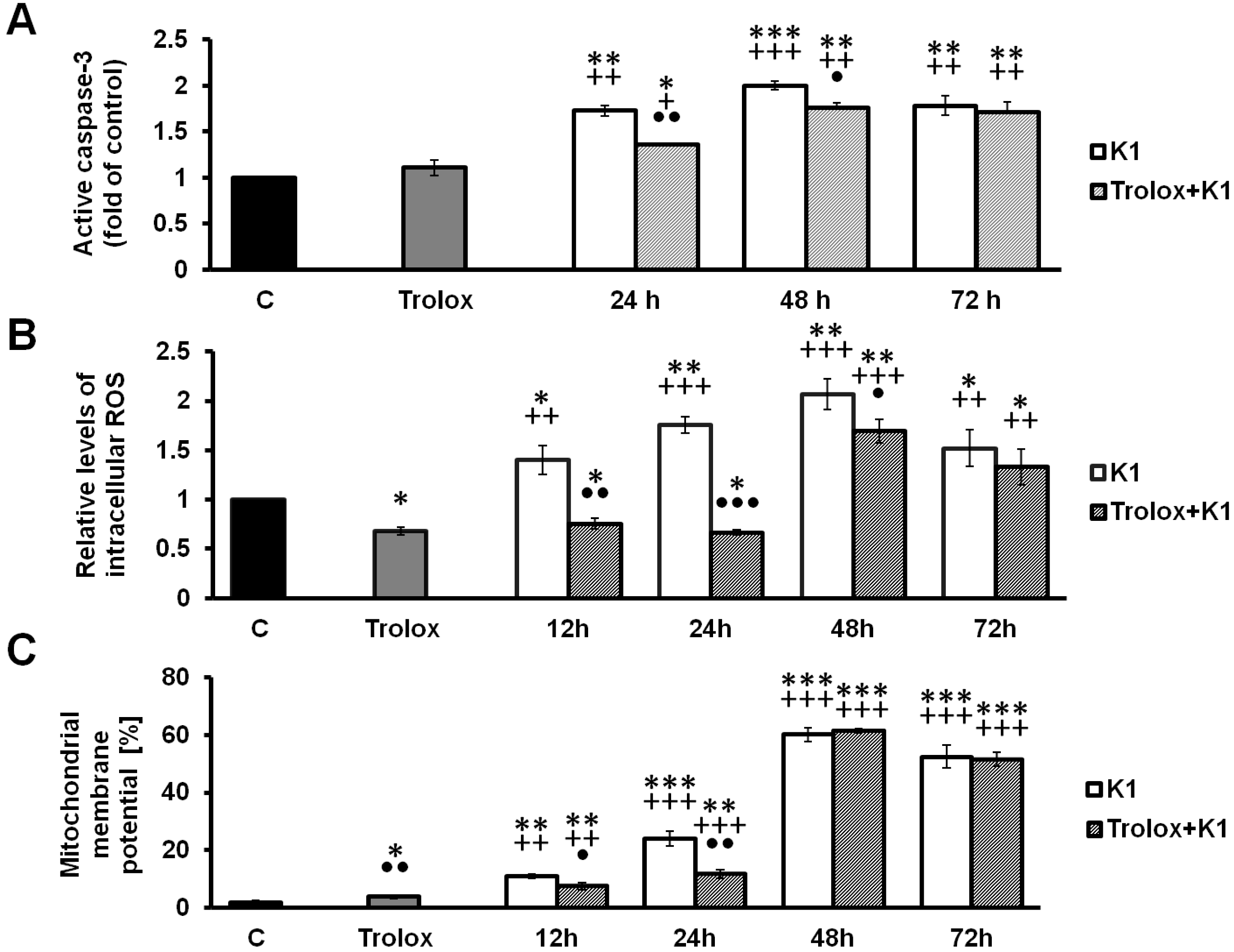
2.5. K1-Induced Mitochondrial Dysfunction
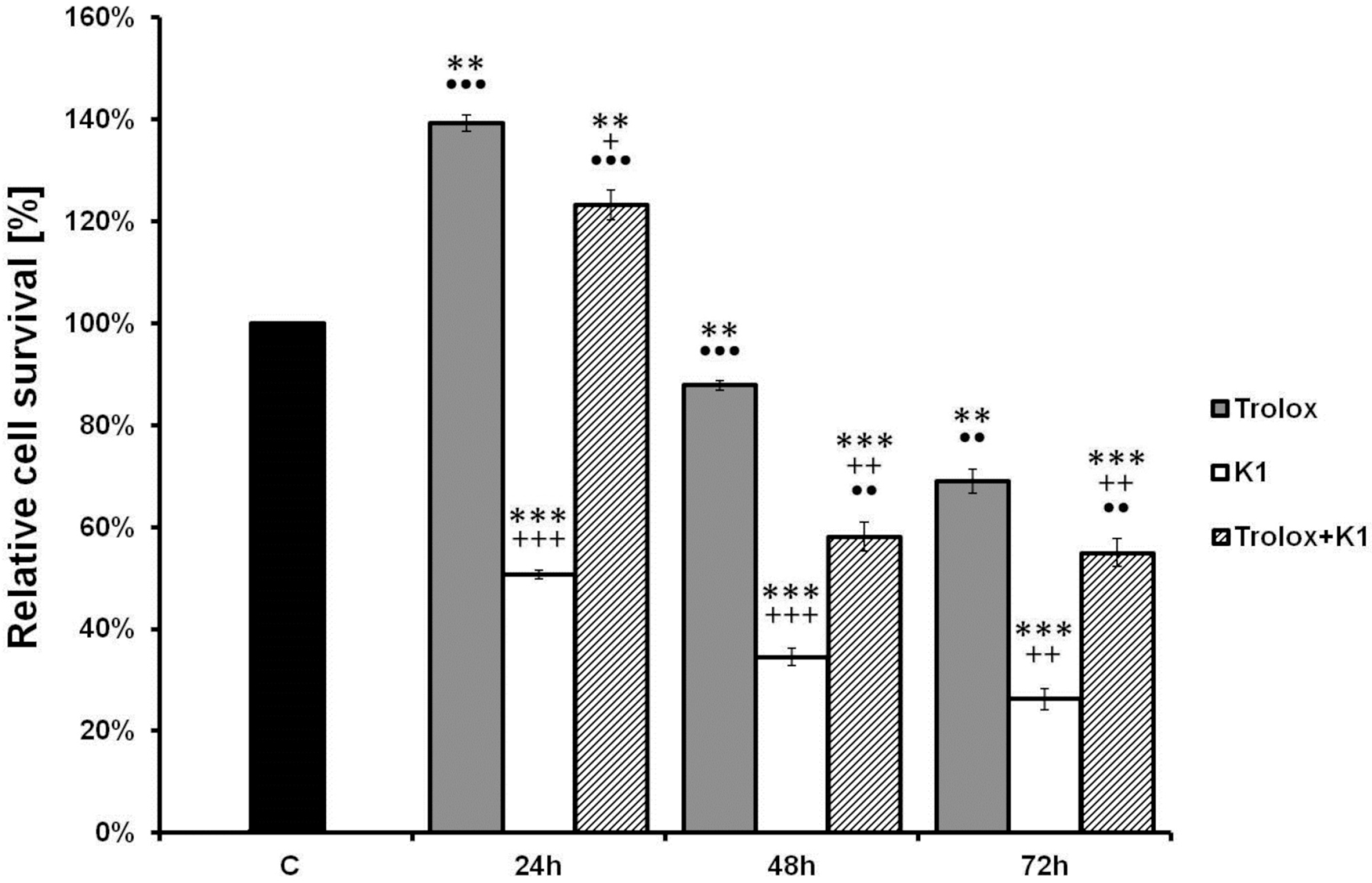
2.6. Activation of Caspase-3 by K1
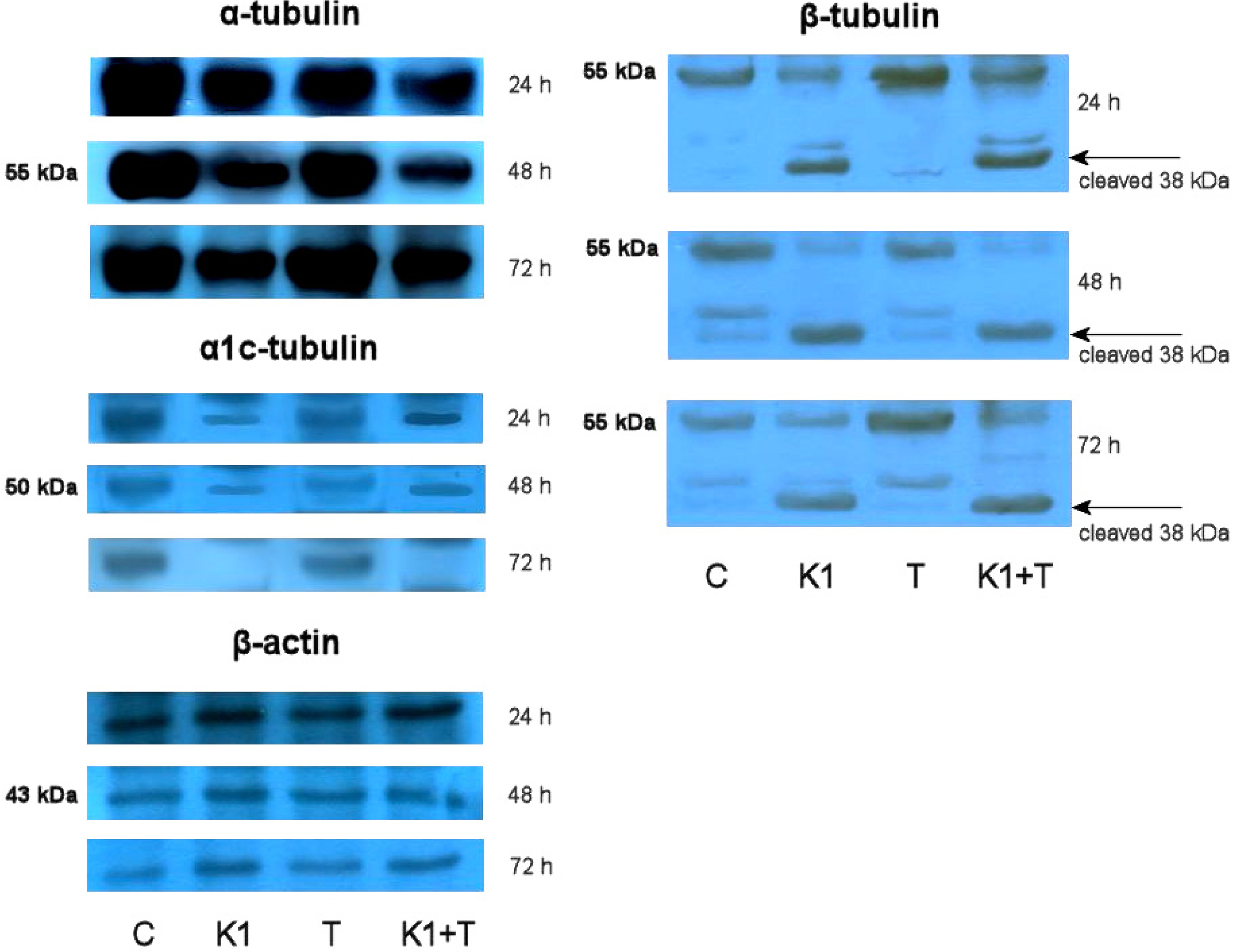
2.7. Effect of K1 on Gene Expression of Tubulins
| Genes | Normalized Ratio | ||
|---|---|---|---|
| 24 h | 48 h | 72 h | |
| α-tubulin | 5.59 | 1.68 | 3.35 |
| α1-tubulin | 1.89 | 0.65 | 1.47 |
| β5-tubulin | 0.29 | 0.99 | 0.48 |
2.8. Effect of K1 on Cytoskeletal Tubulins Assembly
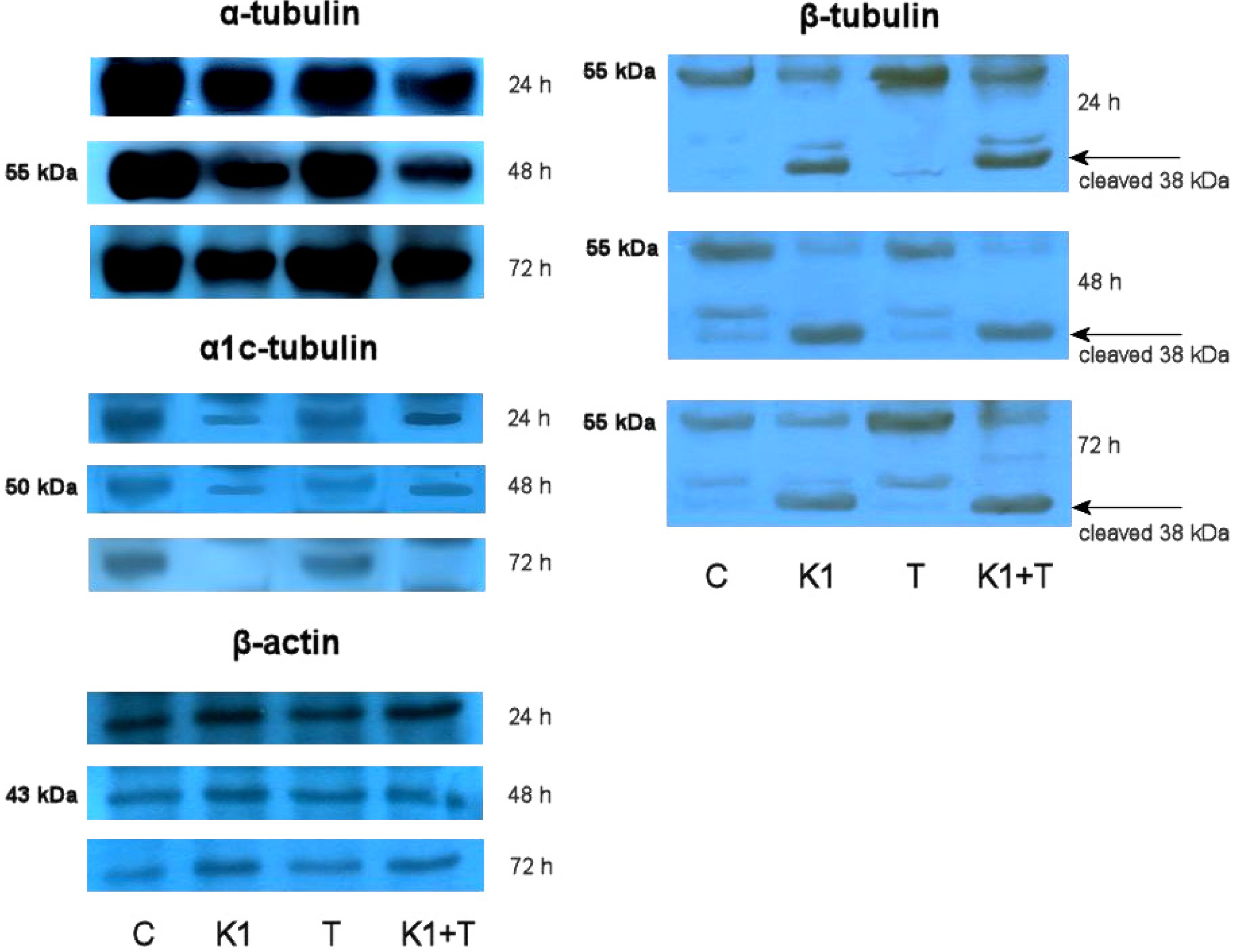
2.9. Discussion
3. Experimental Section
3.1. Test Compounds
3.2. Cell Culture
3.3. Growth Inhibition Assay
3.4. 5-Bromo-2'-Deoxyuridine (BrdU) Cell Proliferation Assay
3.5. xCELLigence Cell Analysis System
3.6. Experimental Design for Flow Cytometry Analysis
3.7. Analysis of Cell Cycle
3.8. Annexin V-FITC Labelling
3.9. Measurement of ROS
3.10. Detection of MMP
3.11. Detection of Active Caspase 3
3.12. DNA Fragmentation Assay
3.13. DAPI Staining
3.14. RNA Isolation and cDNA Synthesis
3.15. Quantitative Real-Time PCR
3.16. Western Blot Analysis
3.17. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Han, B.; Li, X.; Yu, T. Cruciferous vegetables consumption and the risk of ovarian cancer: A meta-analysis of observational studies. Diagn. Pathol. 2014, 9, 1–7. [Google Scholar] [CrossRef]
- Tse, G.; Eslick, G.D. Cruciferous vegetables and risk of colorectal neoplasms: A systematic review and meta-analysis. Nutr. Cancer 2014, 66, 128–139. [Google Scholar] [CrossRef]
- Liu, B.; Mao, Q.; Wang, X.; Zhou, F.; Luo, J.; Wang, C.; Lin, Y.; Zheng, X.; Xie, L. Cruciferous vegetables consumption and risk of renal cell carcinoma: A meta-analysis. Nutr. Cancer 2013, 65, 668–676. [Google Scholar] [CrossRef]
- Aras, U.; Gandhi, Y.A.; Masso-Welch, P.A.; Morris, M.E. Chemopreventive and anti-angiogenic effects of dietary phenethyl isothiocyanate in an N-methyl nitrosourea-induced breast cancer animal model. Biopharm. Drug Dispos. 2013, 34, 98–106. [Google Scholar] [CrossRef]
- Abdull Razis, A.F.; Noor, N.M. Cruciferous vegetables: Dietary phytochemicals for cancer prevention. Asian Pac. J. Cancer Prev. 2013, 14, 1565–1570. [Google Scholar] [CrossRef]
- Izutani, Y.; Yogosawa, S.; Sowa, Y.; Sakai, T. Brassinin induces G1 phase arrest through increase of p21 and p27 by inhibition of the phosphatidylinositol 3-kinase signalling pathway in human colon cancer cells. Int. J. Oncol 2012, 40, 816–824. [Google Scholar]
- Kim, S.M.; Park, J.H.; Kim, K.D.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H. Brassinin induces apoptosis in PC-3 human prostate cancer cells through the suppression of PI3K/Akt/mTOR/S6K1 signalling cascades. Phytother. Res. 2014, 28, 423–431. [Google Scholar] [CrossRef]
- Smith, B.; Randle, D.; Mezencev, R.; Thomas, L.; Hinton, C.; Odero-Marah, V. Camalexin-induced apoptosis in prostate cancer cells involves alterations of expression and activity of lysosomal protease cathepsind. Molecules 2014, 19, 3988–4005. [Google Scholar] [CrossRef]
- Pedras, M.S.; Yaya, E.E.; Glawischnig, E. The phytoalexins from cultivated and wild crucifers: Chemistry and biology. Nat. Prod. Rep. 2011, 28, 1381–1405. [Google Scholar] [CrossRef]
- Jeandet, P.; Clement, C.; Courot, E.; Cordelier, S. Modulation of phytoalexin biosynthesis in engineered plants for disease resistance. Int. J. Mol. Sci. 2013, 14, 14136–14170. [Google Scholar] [CrossRef]
- Borriello, A.; Bencivenga, D.; Caldarelli, I.; Tramontano, A.; Borgia, A.; Zappia, V.; Della Ragione, F. Resveratrol: From basic studies to bedside. Cancer Treat. Res. 2014, 159, 167–184. [Google Scholar] [CrossRef]
- Kim, H.J.; Lim, J.S.; Kim, W.K.; Kim, J.S. Soyabean glyceollins: Biological effects and relevance to human health. Proc. Nutr. Soc. 2012, 71, 166–174. [Google Scholar] [CrossRef]
- Moody, C.J.; Roffey, J.R.; Stephens, M.A.; Stratford, I.J. Synthesis and cytotoxic activity of indolyl thiazoles. Anticancer Drugs 1997, 8, 489–499. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, B.; Zang, W.; Wang, X.; Liu, Z.; Li, W.; Jia, J. Resveratrol inhibits the growth of gastric cancer by inducing G1 phase arrest and senescence in a Sirt1-dependent manner. PLoS One 2013, 8, e70627. [Google Scholar]
- Romagnolo, D.F.; Davis, C.D.; Milner, J.A. Phytoalexins in cancer prevention. Front. Biosci. 2012, 17, 2035–2058. [Google Scholar] [CrossRef]
- Yin, H.T.; Tian, Q.Z.; Guan, L.; Zhou, Y.; Huang, X.E.; Zhang, H. In vitro and in vivo evaluation of the antitumor efficiency of resveratrol against lung cancer. Asian Pac. J. Cancer Prev. 2013, 14, 1703–1706. [Google Scholar] [CrossRef]
- Mezencev, R.; Mojzis, J.; Pilatova, M.; Kutschy, P. Antiproliferative and cancer chemopreventive activity of phytoalexins: Focus on indole phytoalexins from crucifers. Neoplasma 2003, 50, 239–245. [Google Scholar]
- Curillova, Z.; Kutschy, P.; Solcaniova, E.; Pilatova, M.; Mojzis, J.; Kovacik, V. Synthesis and antiproliferative activity of 1-methoxy-, 1-(α-d-ribofuranosyl)- and 1-(β-d-ribofuranosyl)brassenin B. ARKIVOC 2008, 8, 85–104. [Google Scholar]
- Mezencev, R.; Kutschy, P.; Salayova, A.; Curillova, Z.; Mojzis, J.; Pilatova, M.; McDonald, J. Anticancer properties of 2-piperidyl analogues of the natural indole phytoalexin 1-methoxyspirobrassinol. Chemotherapy 2008, 54, 372–378. [Google Scholar] [CrossRef]
- Kutschy, P.; Salayova, A.; Curillova, Z.; Kozar, T.; Mezencev, R.; Mojzis, J.; Pilatova, M.; Balentova, E.; Pazdera, P.; Sabol, M.; et al. 2-(Substituted phenyl)amino analogs of 1-methoxyspirobrassinol methyl ether: synthesis and anticancer activity. Bioorg. Med. Chem. 2009, 17, 3698–3712. [Google Scholar] [CrossRef]
- Pilatova, M.; Ivanova, L.; Kutschy, P.; Varinska, L.; Saxunova, L.; Repovska, M.; Sarissky, M.; Seliga, R.; Mirossay, L.; Mojzis, J. In vitro toxicity of camalexin derivatives in human cancer and non-cancer cells. Toxicol. In Vitro 2013, 27, 939–944. [Google Scholar] [CrossRef]
- Takasugi, M.; Katsui, N.; Shirata, A. Isolation of 3 novel sulfur-containing phytoalexins from the Chinese-cabbage brassica-campestris L ssp pekinensis (cruciferae). J. Chem Soc. Chem Commun. 1986, 14, 1077–1078. [Google Scholar] [CrossRef]
- Mehta, R.G.; Liu, J.; Constantinou, A.; Thomas, C.F.; Hawthorne, M.; You, M.; Gerhuser, C.; Pezzuto, J.M.; Moon, R.C.; Moriarty, R.M. Cancer chemopreventive activity of brassinin, a phytoalexin from cabbage. Carcinogenesis 1995, 16, 399–404. [Google Scholar] [CrossRef]
- Csomos, P.; Zupko, I.; Rethy, B.; Fodor, L.; Falkay, G.; Bernath, G. Isobrassinin and its analogues: Novel types of antiproliferative agents. Bioorg. Med. Chem. Lett. 2006, 16, 6273–6276. [Google Scholar] [CrossRef]
- Pilatova, M.; Sarissky, M.; Kutschy, P.; Mirossay, A.; Mezencev, R.; Curillova, Z.; Suchy, M.; Monde, K.; Mirossay, L.; Mojzis, J. Cruciferous phytoalexins: Antiproliferative effects in T-Jurkat leukemic cells. Leuk. Res. 2005, 29, 415–421. [Google Scholar] [CrossRef]
- Monde, K.; Taniguchi, T.; Miura, N.; Kutschy, P.; Curillova, Z.; Pilatova, M.; Mojzis, J. Chiral cruciferous phytoalexins: Preparation, absolute configuration, and biological activity. Bioorg. Med. Chem. 2005, 13, 5206–5212. [Google Scholar] [CrossRef]
- Kutschy, P.; Sykora, A.; Curillova, Z.; Repovska, M.; Pilatova, M.; Mojzis, J.; Mezencev, R.; Pazdera, P.; Hromjakova, T. Glyoxyl analogs of indole phytoalexins: Synthesis and anticancer activity. Collect. Czech. Chem. C 2010, 75, 887–903. [Google Scholar] [CrossRef]
- Budovska, M.; Pilatova, M.; Varinska, L.; Mojzis, J.; Mezencev, R. The synthesis and anticancer activity of analogs of the indole phytoalexins brassinin, 1-methoxyspirobrassinol methyl ether and cyclobrassinin. Bioorg. Med. Chem. 2013, 21, 6623–6633. [Google Scholar] [CrossRef]
- Ocenas, P.; Tomasova, L.; Kutschy, P.; Pazdera, P.; Mojzis, J.; Pilatova, M. Spirocyclisation of phytoalexin 1-methoxybrassinin in the presence of Grignard reagents. Chem. Pap. 2013, 67, 631–642. [Google Scholar] [CrossRef]
- Chripkova, M.; Drutovic, D.; Pilatova, M.; Mikes, J.; Budovska, M.; Vaskova, J.; Brogginy, M.; Mirossay, L.; Mojzis, J. Brassinin and its derivatives as potential anticancer agents. Toxicol. In Vitro 2014, 28, 907–915. [Google Scholar]
- Banerjee, T.; Duhadaway, J.B.; Gaspari, P.; Sutanto-Ward, E.; Munn, D.H.; Mellor, A.L.; Malachowski, W.P.; Prendergast, G.C.; Muller, A.J. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene 2008, 27, 2851–2857. [Google Scholar] [CrossRef]
- Patel, P.B.; Thakkar, V.R. l-Carvone induces p53, caspase 3 mediated apoptosis and inhibits the migration of breast cancer cell lines. Nutr. Cancer 2014, 66, 453–462. [Google Scholar] [CrossRef]
- Zhao, B.; Li, X. Altholactone induces reactive oxygen species-mediated apoptosis in bladder cancer T24 cells through mitochondrial dysfunction, MAPK-p38 activation and Akt suppression. Oncol. Rep. 2014, 31, 2769–2775. [Google Scholar]
- Mezencev, R.; Updegrove, T.; Kutschy, P.; Repovska, M.; McDonald, J.F. Camalexin induces apoptosis in T-leukemia Jurkat cells by increased concentration of reactive oxygen species and activation of caspase-8 and caspase-9. J. Nat. Med. 2011, 65, 488–499. [Google Scholar] [CrossRef]
- Smith, B.A.; Neal, C.L.; Chetram, M.; Vo, B.; Mezencev, R.; Hinton, C.; Odero-Marah, V.A. The phytoalexin camalexin mediates cytotoxicity towards aggressive prostate cancer cells via reactive oxygen species. J. Nat. Med. 2013, 67, 607–618. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- Long, X.; Fan, M.; Bigsby, R.M.; Nephew, K.P. Apigenin inhibits antiestrogen-resistant breast cancer cell growth through estrogen receptor-alpha-dependent and estrogen receptor-alpha-independent mechanisms. Mol. Cancer Ther. 2008, 7, 2096–2108. [Google Scholar] [CrossRef]
- ElAttar, T.M.; Virji, A.S. Modulating effect of resveratrol and quercetin on oral cancer cell growth and proliferation. Anticancer Drugs 1999, 10, 187–193. [Google Scholar] [CrossRef]
- Baron-Menguy, C.; Bocquet, A.; Guihot, A.L.; Chappard, D.; Amiot, M.J.; Andriantsitohaina, R.; Loufrani, L.; Henrion, D. Effects of red wine polyphenols on postischemic neovascularization model in rats: Low doses are proangiogenic, high doses anti-angiogenic. FASEB J. 2007, 21, 3511–3521. [Google Scholar] [CrossRef]
- Choi, E.J.; Kim, G.H. Antiproliferative activity of daidzein and genistein may be related to ERalpha/c-erbB-2 expression in human breast cancer cells. Mol. Med. Rep. 2013, 7, 781–784. [Google Scholar]
- Downing, K.H.; Nogales, E. Tubulin and microtubule structure. Curr. Opin. Cell. Biol. 1998, 10, 16–22. [Google Scholar] [CrossRef]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Kang, J.; Pervaiz, S. Mitochondria: Redox metabolism and dysfunction. Biochem. Res. Int. 2012, 2012, 896751. [Google Scholar]
- Green, D.R. Apoptotic pathways: Ten minutes to dead. Cell 2005, 121, 671–674. [Google Scholar] [CrossRef]
- Erejuwa, O.O.; Sulaiman, S.A.; Wahab, M.S. Effects of honey and its mechanisms of action on the development and progression of cancer. Molecules 2014, 19, 2497–2522. [Google Scholar] [CrossRef]
- Villena, J.; Madrid, A.; Montenegro, I.; Werner, E.; Cuellar, M.; Espinoza, L. Diterpenylhydroquinones from natural ent-labdanes induce apoptosis through decreased mitochondrial membrane potential. Molecules 2013, 18, 5348–5359. [Google Scholar] [CrossRef]
- Yang, T.; Li, M.H.; Liu, J.; Huang, N.; Li, N.; Liu, S.N.; Liu, Y.; Zhang, T.; Zou, Q.; Li, H. Benzimidazole derivative, BMT-1, induces apoptosis in multiple myeloma cells via a mitochondrial-mediated pathway involving H+/K+-ATPase inhibition. Oncol. Rep. 2014. [Google Scholar] [CrossRef]
- Thangam, R.; Senthilkumar, D.; Suresh, V.; Sathuvan, M.; Sivasubramanian, S.; Pazhanichamy, K.; Gorlagunta, P.K.; Kannan, S.; Gunasekaran, P.; Rengasamy, R.; et al. Induction of ROS-dependent mitochondria-mediated intrinsic apoptosis in MDA-MB-231 cells by glycoprotein from codium decorticatum. J. Agric. Food Chem. 2014, 62, 3410–3421. [Google Scholar] [CrossRef]
- Duan, D.; Zhang, B.; Yao, J.; Liu, Y.; Fang, J. Shikonin targets cytosolic thioredoxin reductase to induce ROS-mediated apoptosis in human promyelocytic leukemia HL-60 cells. Free Radic. Biol. Med. 2014, 70, 182–193. [Google Scholar] [CrossRef]
- Gaspari, P.; Banerjee, T.; Malachowski, W.P.; Muller, A.J.; Prendergast, G.C.; DuHadaway, J.; Bennett, S.; Donovan, A.M. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J. Med. Chem. 2006, 49, 684–692. [Google Scholar] [CrossRef]
- Kutschy, P.; Sabol, M.; Maruskova, R.; Curillova, Z.; Dzurilla, M.; Geci, I.; Alfoldi, J.; Kovacik, V. A linear synthesis of 1-(beta-d-glucopyranosyl)brassinin, -brassenin A, -brassenin B and 9-(beta-d-glucopyranosyl)-cyclobrassinin. Collect. Czech. Chem. Commun. 2004, 69, 850–866. [Google Scholar] [CrossRef]
- Budovska, M.; Kutschy, P.; Kozar, T.; Gondova, T.; Petrovaj, J. Synthesis of spiroindoline phytoalexin (S)-(−)-spirobrassinin and its unnatural (R)-(+)-enantiomer. Tetrahedron 2013, 69, 1092–1104. [Google Scholar] [CrossRef]
- Ivanova, L.; Varinska, L.; Pilatova, M.; Gal, P.; Solar, P.; Perjesi, P.; Smetana, K., Jr.; Ostro, A.; Mojzis, J. Cyclic chalcone analogue KRP6 as a potent modulator of cell proliferation: An in vitro study in HUVECs. Mol. Biol. Rep. 2013, 40, 4571–4580. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Limame, R.; Wouters, A.; Pauwels, B.; Fransen, E.; Peeters, M.; Lardon, F.; de Wever, O.; Pauwels, P. Comparative analysis of dynamic cell viability, migration and invasion assessments by novel real-time technology and classic endpoint assays. PLoS One 2012, 7, e46536. [Google Scholar] [CrossRef]
- Ke, N.; Wang, X.; Xu, X.; Abassi, Y.A. The xCELLigence system for real-time and label-free monitoring of cell viability. Methods Mol. Biol. 2011, 740, 33–43. [Google Scholar] [CrossRef]
- Sample Availability: Sample of the compound K1 is available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kello, M.; Drutovic, D.; Chripkova, M.; Pilatova, M.; Budovska, M.; Kulikova, L.; Urdzik, P.; Mojzis, J. ROS-Dependent Antiproliferative Effect of Brassinin Derivative Homobrassinin in Human Colorectal Cancer Caco2 Cells. Molecules 2014, 19, 10877-10897. https://doi.org/10.3390/molecules190810877
Kello M, Drutovic D, Chripkova M, Pilatova M, Budovska M, Kulikova L, Urdzik P, Mojzis J. ROS-Dependent Antiproliferative Effect of Brassinin Derivative Homobrassinin in Human Colorectal Cancer Caco2 Cells. Molecules. 2014; 19(8):10877-10897. https://doi.org/10.3390/molecules190810877
Chicago/Turabian StyleKello, Martin, David Drutovic, Martina Chripkova, Martina Pilatova, Mariana Budovska, Lucia Kulikova, Peter Urdzik, and Jan Mojzis. 2014. "ROS-Dependent Antiproliferative Effect of Brassinin Derivative Homobrassinin in Human Colorectal Cancer Caco2 Cells" Molecules 19, no. 8: 10877-10897. https://doi.org/10.3390/molecules190810877
APA StyleKello, M., Drutovic, D., Chripkova, M., Pilatova, M., Budovska, M., Kulikova, L., Urdzik, P., & Mojzis, J. (2014). ROS-Dependent Antiproliferative Effect of Brassinin Derivative Homobrassinin in Human Colorectal Cancer Caco2 Cells. Molecules, 19(8), 10877-10897. https://doi.org/10.3390/molecules190810877