3. Experimental
Full experimental details and spectroscopic characterization data is given is the proceeding section:
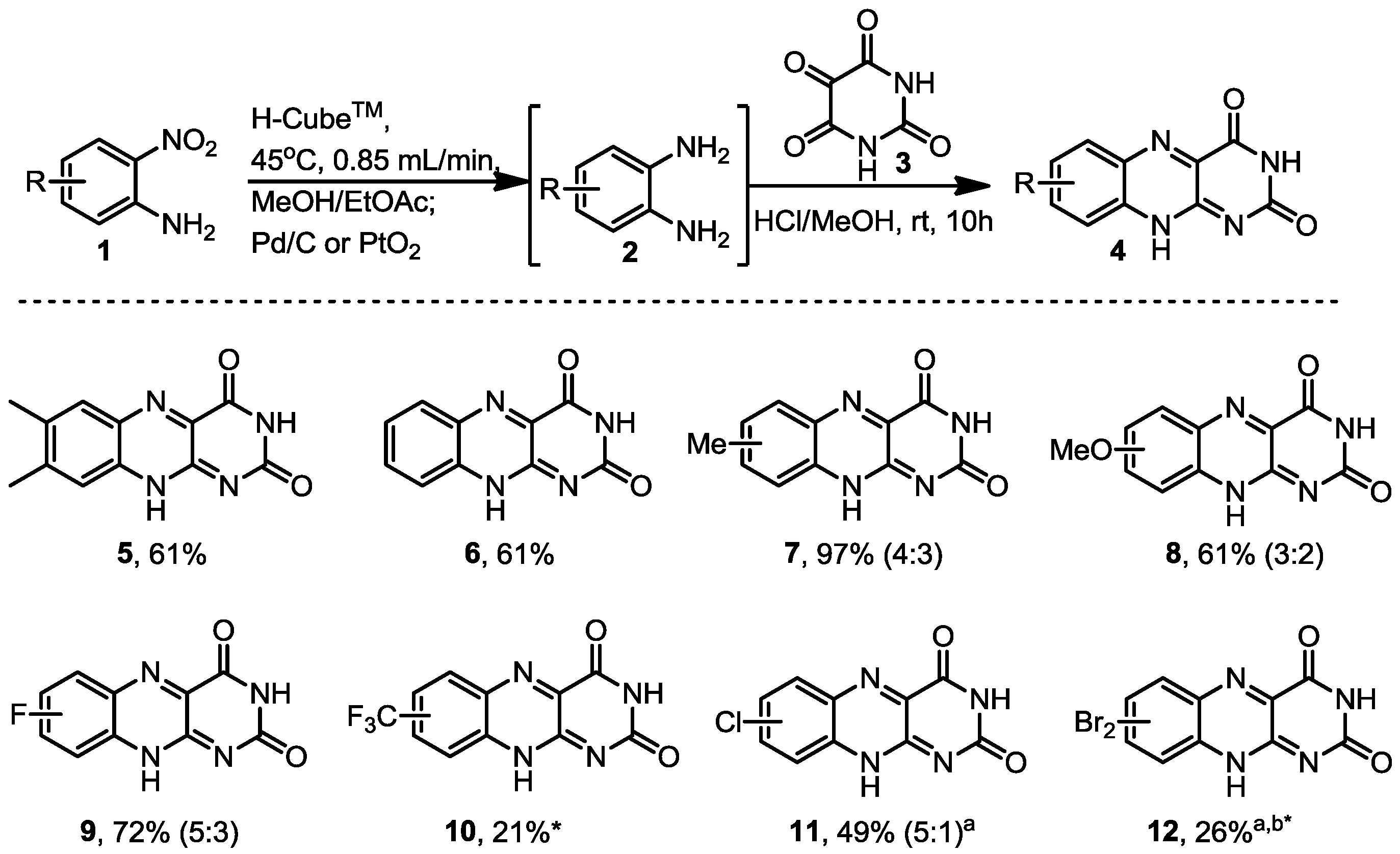
3.2. General Procedure for the Hydrogenation Reactions in Flow Using the H-Cube®
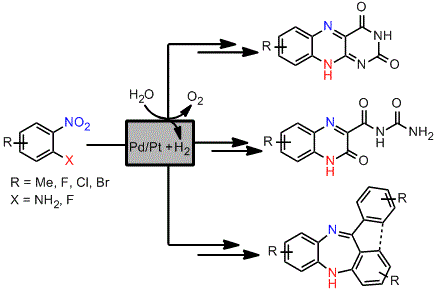
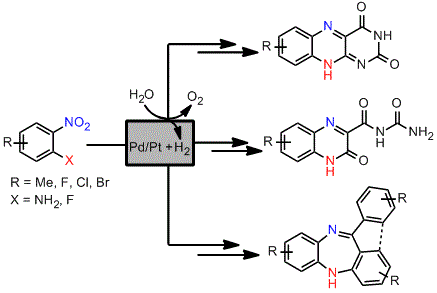
A solution of the nitroaniline (1 mmol, 0.1–0.25 M, MeOH/EtOAc, 1:1) was passed through the H-Cube®, which was equipped with a cartridge filled with the corresponding Pd/C or PtO2/C catalyst. Extra back pressure regulators were added to the H-Cube® set-up, with a pressure of 100 psi (6.9 bar) being applied before the solution entered the H-Cube® and a pressure of 250 psi (17.2 bar) applied to the exiting solution. The nitroanilines were pumped at different temperatures and flow rates depending on the substrate using full H2 mode. The exiting solutions were concentrated to determine the reaction conversion by 1H-NMR analysis and used subsequently due to the instability of the diamines. The catalyst cartridge was exchanged approximately every 8 runs.
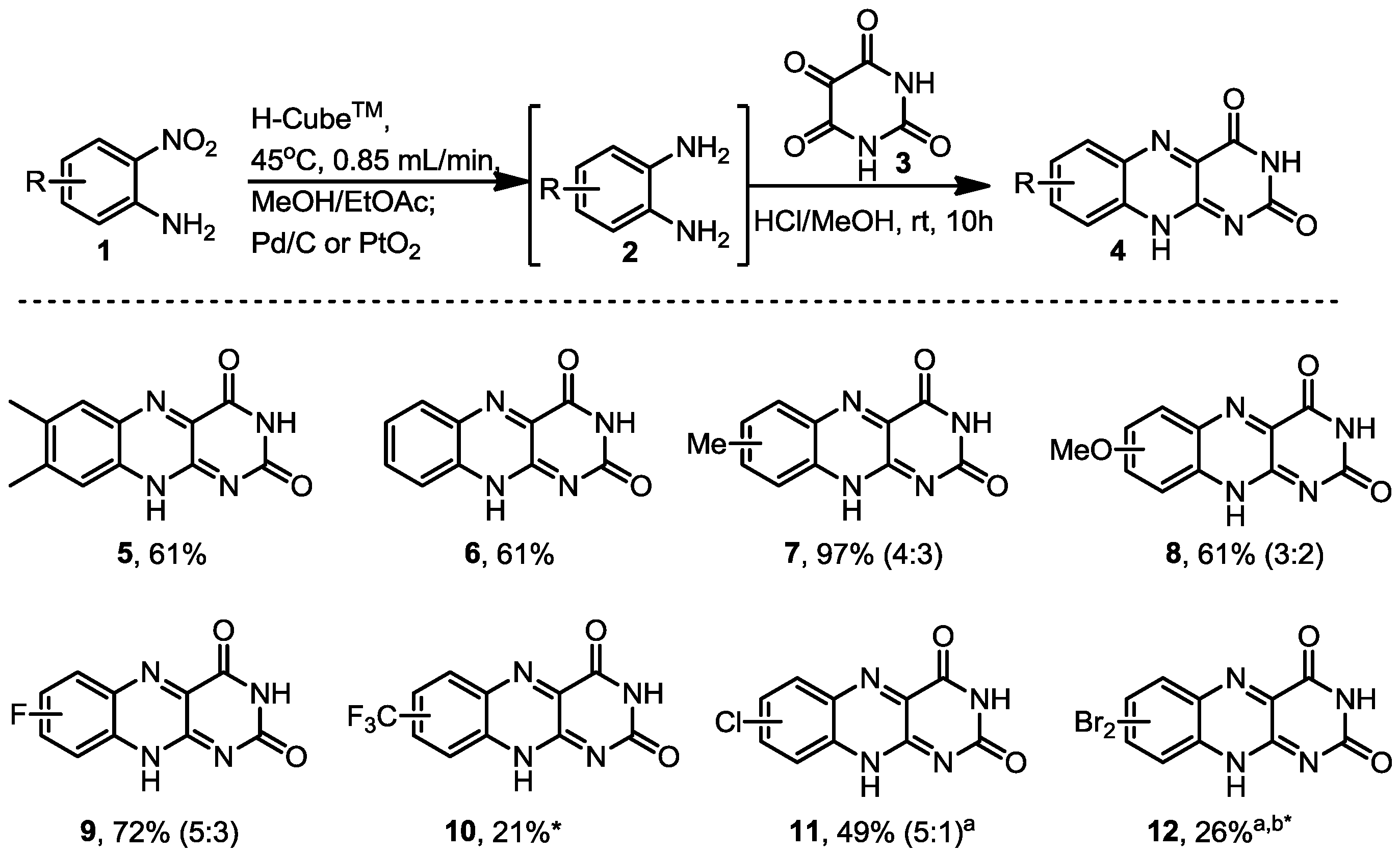
3.3. General Procedure for the Formation of the Riboflavine Analogues 5–12
The diamines generated by the hydrogenation process in flow were mixed with alloxane monohydrate (0.160 g, 1 mmol) and hydrogen chloride in methanol (1 mL of approx. 1.25 M concentration) and left to stir at room temperature overnight. The product was then filtered and the solid product was dried in vacuo.
7,8-Dimethylbenzo[g]pteridine-2,4(3H,10H)-dione (5). Prepared from 4,5-dimethyl-2-nitroaniline (0.166 g, 1 mmol) to give a yellow solid (0.147 g, 61% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 11.82 (1H, d, J = 1.8 Hz, NH), 11.65 (1H, s, NH), 7.90 (s, 1H), 7.69 (s, 1H), 3.15 (s, 6H, 2 × CH3); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 160.7 (C), 150.1 (C), 146.5 (C), 144.7 (C), 141.7 (C), 139.0 (C), 138.4 (C), 130.3 (C), 128.8 (CH), 125.9 (CH), 55.0 (CH3), 48.6 (CH3). IR (neat) ν/cm−1 = 3445.6 (w), 3187.7 (w), 3145.6 (w), 3073.2 (w), 2985.0 (w), 2951.7 (w), 2848.1 (w), 1734.3 (m), 1693.0 (s), 1629.8 (w), 1587.9 (m), 1578.0 (m), 1486.7 (w), 1441.5 (w), 1423.8 (w), 1386.7 (w), 1363.8 (m), 1349.3 (m), 1285.7 (s), 1221.3 (w), 1192.6 (w), 1143.9 (w), 1097.6 (w), 1035.0 (m), 1023.5 (w), 1001.9 (w), 911.5 (w), 882.9 (m), 816.2 (m), 797.9 (w), 770.5 (w), 754.5 (m), 736.2 (w), 683.9 (w), 661.9 (w). LC-MS: Rt = 4.57 min; HRMS (ESI): m/z calculated for C12H9N4O2 [M+H+]: 213.0731; found 241.0732.
Benzo[g]pteridine-2,4(3H,10H)-dione (6). Prepared from 2-nitroaniline (0.138 g, 1 mmol) to give a pale green solid (0.131 g, 61% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 11.94 (d, J = 1.6 Hz, 1H, NH), 11.75 (s, 1H, NH), 8.16 (d, J = 8.2 Hz, 1H), 7.92 (m, 2H, 2 and 3), 7.77 (m, 1H); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 160.5 (C), 150.2 (C), 146.9 (C), 142.7 (C), 139.3 (C), 133.4 (CH), 131.8 (C), 130.2 (CH), 128.5 (CH), 127.0 (CH). IR (neat) ν/cm−1: 3173.4 (w, br), 3084.4 (w, br), 28.43.1 (w, br), 1813.8 (w), 1786.4 (w), 1733.6 (m), 1687.9 (s), 1618.5 (w), 1582.9 (m), 1505.4 (w), 1485.5 (w), 1447.4 (m), 1390.4 (m), 1364.0 (m), 1334.4 (m), 1316.1 (m), 1271.4 (s), 1248.8 (m), 1212.6 (s), 1153.6 (w), 1144.9 (w), 1034.9 (w), 1015.0 (w), 989.7 (w), 915.6 (w), 866.9 (w), 811.8 (w), 763.6 (s, br), 705.1 (m), 677.8 (m). LC-MS: Rt 1.82 min; HRMS (ESI): m/z calculated for C10H5N4O2 [M+H+]: 213.0418; found 213.0411.
7-Methylbenzo[g]pteridine-2,4(3H,10H)-dione and 8-methylbenzo[g]pteridine-2,4(3H,10H)-dione (7). Prepared from 4-methyl-2-nitroaniline (0.152 g, 1 mmol) to give a yellow solid, with two regioisomers found in a 4:3 ratio (0.212 g, 96% yield). 1H-NMR gave broad peaks and not all quaternary centres were observed in the 13C-NMR. 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 12.99 (s, 1H, NH), 11.15 (s, 1H, NH), 7.68 (d, br, J = 18 Hz, 2H), 7.51 (d, J = 9 Hz, 1H), 7.27 (dd, J = 6.5 Hz, 20.3 Hz, 2H), 7.15 (s, 1H), 2.50 (m, 3H), 2.49 (m, 3H). To separate broad peaks: 1H-NMR (400 MHz, d6-DMSO + 3 drops TFA): δ/ppm = 8.03 (1H from major regioisomer, d, J = 8.5 Hz, H3), 7.96 (1H from minor regioisomer, s, H1), 7.71 (1H from minor regioisomer, dd, J = 8.4 Hz, 1.9 Hz, H2), 7.48 (1H from major regioisomer, dd, J = 8.9 Hz, 1.3 Hz, H2), 7.41 (1H from minor regioisomer, d, J = 8.4 Hz, H3), 7.27 (1H from major regioisomer, s, H1) 2.62 (3H from major regioisomer, s, CH3), 2.56 (3H from minor regioisomer, s, CH3); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 163.7 (C), 154.5 (C), 153.2 (C), 143.7 (C), 134.1 (CH), 132.7 (C), 131.4 (C), 130.5 (C), 129.8 (CH), 129.5 (CH), 129.0 (C), 125.9 (CH), 115.5 (CH), 115.3 (CH), 21.6 (CH3), 20.4 (CH3). IR (neat) ν/cm−1: 3395.5 (w), 3134.2 (w, br), 1724.7 (s), 1693.2 (s), 1656.2 (m), 1626.1 (m), 1583.3 (m), 1533.1 (w), 1509.2 (m), 1465.3 (w), 1371.3 (s), 1290.6 (w), 1278.8 (w), 1248.7 (w), 1199.4 (w), 1185.1 (w), 1146.0 (w), 1116.6 (w), 1025.9 (w), 961.8 (w), 914.6 (w), 883.2 (w), 860.5 (w), 825.8 (s), 810.3 (s), 758.4 (w), 719.8 (w), 697.7 (w), 681.6 (w). LC-MS: Rt 3.49 min; HRMS (ESI): m/z calculated for C11H9N4O3 [M+H+]: 227.0574; found 227.0571.
7-Methylbenzo[g]pteridine-2,4(3H,10H)-dione and 8-methyl-benzo[g]pteridine-2,4(3H,10H)-dione (7). Prepared from 5-methyl-2-nitroaniline (0.152 g, 1 mmol) to give a yellow solid, with two regioisomers found in a 4:3 ratio (0.140 g, 61% yield). This was indicated by 1H-NMR to be identical to tne above compounds 7.
7-Methoxybenzo[g]pteridine-2,4(3H,10H)-dione and 8-methoxybenzo[g]pteridine-2,4(3H,10H)-dione (8). Prepared from 4-methoxy-2-nitroaniline (0.206 g, 1 mmol) to give an orange solid, with two regioisomers found in a 3:2 ratio (0.1043 g, 43% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 11.86 (1H from minor regioisomer, d, J = 1.7 Hz, NH), 11.82 (1H from major regioisomer, d, J = 1.7 Hz, NH), 11.69 (1H from major regioisomer, s, NH), 11.66 (1H from minor regioisomer, s, NH), 8.02 (1H from minor regioisomer, d, J = 9.2 Hz, H3), 7.83 (1H from major isomer, d, J = 9.2 Hz, H3), 7.59 (1H from major regioisomer, dd, J = 9.2 Hz, 2.7 Hz, H2), 7.54 (1H from major regioisomer, d, J = 2.8 Hz, H1), 7.40 (1H from minor regioisomer, dd, J = 9.3 Hz, 2.3 Hz, H2), 7.22 (1H from minor regioisomer, d, J = 2.7 Hz, H1), 3.97 (3H from minor regioisomer, s, CH3), 3.80 (3H from major regioisomer, s, CH3); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 163.25 (C), 160.7 (C), 160.6 (C), 159.1 (C), 150.3 (C), 150.1 (C), 147.2 (C), 145.5 (C), 145.1 (C), 145.1 (C), 140.8 (C), 138.8 (C), 135.6 (C), 131.5 (CH), 130.9 (C), 128.4 (C), 128.0 (CH), 126.9 (CH), 107.5 (CH), 104.9 (CH), 56.3 (CH3), 56.0 (CH3). IR (neat) ν/cm−1: 3258.9 (w), 3071.0 (w, br), 2851.2 (w), 1726.3 (m), 1694.1 (s), 1621.3 (w), 1556.8 (m), 1509.3 (w), 1458.9 (w), 1439.0 (m), 1399.2 (w), 1354.6 (s), 1340.8 (m), 1330.1(m), 1314.0 (w), 1233.5 (m), 1212.9 (s), 1158.8 (w), 1142.7 (w), 1120.4 (w), 1008.4 (m), 960.0 (w), 877.6 (w), 850.7 (m), 794.2 (m), 763.1 (m), 749.2 (m), 704.8 (w), 659.4 (w). LC-MS: Rt 3.42 min; HRMS (ESI): m/z calculated for C11H7N4O3 [M+H+]: 243.0524; found 243.0527.
7-Fluorobenzo[g]pteridine-2,4(3H,10H)-dione and 8-Fluorobenzo[g]pteridine-2,4(3H,10H)-dione (9). Prepared from 4-fluoro-2-nitroaniline (0.106 g, 1 mmol) to give a green solid with two regioisomers found in a 5:3 ratio (0.1678 g, 73% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 12.04 (1H from minor regioisomer, d, J = 1.8 Hz, NH), 11.98 (1H from major regioisomer, d, J = 1.8 Hz, NH), 11.81 (1H from each regioisomer, br s, 2 × NH), 8.27 (1H from minor regioisomer, dd, J = 9.2 Hz, 6.1 Hz, H3), 8.02–7.99 (2H from major regioisomer, m, H1 and H2), 7.91–7.87 (1H from major regioisomer, m, H3), 7.74–7.69 (2H from minor regioisomer, m, H1 and H2); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 164.4 (C from minor regioisomer, d, J = 250.0 Hz, C-F), 164.4 (C from major regioisomer, d, J = 37.5 Hz, C-F), 160.4 (C), 160.3 (C), 150.1 (C, major regioisomer), 150.0 (C, minor regioisomer), 147.5 (C), 146.7 (C, d, J = 2.1 Hz), 143.9 (C from minor regioisomer, d, J = 14.4 Hz), 140.0 (C from major regioisomer), 139.5 (C from major regioisomer, d, J = 13.1 Hz), 136.6 (C), 133.0 (CH from minor regioisomer, d, J = 11.2 Hz, C3), 132.5 (C from major regioisomer), 131.3 (C from minor regioisomer, d, J = 3.0 Hz), 129.3 (CH from major regioisomer, d, J = 9.9 Hz, C3), 123.5 (CH from major regioisomer, d, J = 18.5 Hz, C2), 118.8 (CH from minor regioisomer, d, J = 26.2 Hz, C2), 113.3 (CH from major regioisomer, d, J = 21.5 Hz, C1), 110.7 (CH from minor regioisomer, d, J = 22.0 Hz, C1). IR (neat) ν/cm−1: 3182.7 (w), 3086.6 (w, br), 2845.9 (w), 1733.7 (m), 1691.9 (s), 1626.7 (m), 1583.9 (m), 1571.6 (m), 1510.4 (m), 1482.0 (w), 1455.3 (m), 1398.4 (w), 1354.5 (m), 1334.9 (s), 1298.6 (w), 1278.3 (s), 1243.1 (w), 1213.1 (s), 1157.4 (w), 1139.3 (w), 1108.9 (w), 1035.9 (w), 975.4 (w), 856.9 (s), 835.3 (s), 808.0 (m), 767.4 (m), 753.1 (s), 703.3 (w), 684.0 (w), 663.0 (m). LC-MS: Rt 2.85 min (mass peak of 233), another weaker peak seen at 3.45 min; HRMS (ESI): m/z calculated for C10H4N4O2F, [M+H+]: 231.0324; found 231.0323.
7-(Trifluoromethyl)benzo[g]pteridine-2,4(3H,10H)-dione or 8-(trifluoromethyl)benzo[g]pteridine-2,4(3H,10H)-dione (10). Prepared from 2-nitro-5-(trifluoromethyl)-aniline (0.206 g, 1 mmol) to give a pale yellow solid, one regioisomer was cleanly isolated (0.059 g, 21% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 12.11 (br s, 1H, NH), 11.87 (br s, 1H, NH), 8.35 (d, J = 8.7 Hz, H3), 8.21 (d, J = 0.6 Hz, H1), 7.98 (dd, J = 8.7 Hz, 2.0 Hz, H2); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 160.1 (C), 150.1 (C), 148.0 (C), 141.7 (C), 140.2 (C), 134.4 (C) 132.3 (C-CF3) 132.0 (CH), 124.7 (CH), 124.0 (CF3), 123.4 (CH). IR (neat) ν/cm−1: 3456.0 (w), 3054.8 (w, br), 2898.1 (w), 2825.2 (w), 1733.4 (w), 1689.9 (s), 1631.4 (w), 1584.7 (w), 1507.7 (w), 1489.4 (w), 1454.8 (w), 1394.7 (m), 1367.9 (w), 1336.9 (w), 1318.3 (m), 1290.0 (s), 1266.5 (m), 1257.6 (m), 1244.0 (m), 1205.2 (m), 1166.9 (s), 1158.8 (s), 1136.1 (s), 1111.1 (m), 1086.1 (w), 1031.0 (w), 997.5 (m), 941.0 (w), 908.4 (w) 900.3 (w), 859.9 (s), 823.3 (w) 804.2 (m), 774.2 (w), 755.8 (w), 710.3 (s), 678.7 (w), 655.8 (w). LC-MS: Rt 3.85 min; HRMS (ESI): m/z calculated for C11H4F3N4O2 [M+H+]: 281.0292; found 281.0287.
8-Chlorobenzo[g]pteridine-2,4(3H,10H)-dione and 7-chloro-benzo[g]pteridine-2,4(3H,10H)-dione (11). Prepared from 4-chloro-2-nitroaniline (0.172 g, 1 mmol) to give a pale yellow solid, with two regioisomers found in a 5:1 ratio (0.121 g, 49% yield). 1H-NMR gave broad peaks. 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 13.09 (1H from major regioisomer, br s, NH), 12.61 (1H from minor regioisomer, br s, NH), 10.98 (1H from major regioisomer, br s, NH), 8.25 (1H from minor regioisomer, s), 8.17 (1H from minor regioisomer, d, J = 9.0 Hz), 7.97–7.73 (4H, three from major regioisomer, one from minor regioisomer, br m). To separate broad peaks: 1H-NMR (400 MHz, d6-DMSO + 3 drops TFA): δ/ppm = 8.23 (1H from minor regioisomer, d, J = 1.5Hz, H1), 8.15 (1H from minor regioisomer, d, J = 9.0 Hz, H3), 7.93 (1H from major regioisomer, dd, J = 9.0 Hz, 2.1 Hz, H2), 7.74 (1H from minor regioisomer, dd, J = 9.0 Hz, 2.3 Hz, H2), 7.81 (1H from major regioisomer, m, H3), 6.70 (1H from major regioisomer, m, H1); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 160.3 (C), 153.1 (C), 150.1 (C), 147.6 (C), 147.2 (C), 143.1 (C), 141.4 (C), 141.4 (C), 139.4 (C), 137.8 (C), 133.7 (CH), 132.8 (C), 132.3 (C), 132.0 (CH), 131.7 (C), 131.4 (C), 128.6 (CH), 125.7 (CH), 117.5 (CH), 115.1 (CH). IR (neat) ν/cm−1: 3149.8 (w), 3085.3 (w, br), 2852.0 (w), 1728.1 (m), 1693.9 (s), 1661.4 (m), 1610.7 (m), 1578.6 (m), 1534.2 (w), 1490.7 (m), 1457.6 (w), 1365.9 (m), 1354.6 (s), 1335.6 (m), 1292.9 (m), 1274.7 (m), 1237.1 (w), 1190.2 (w), 1173.0 (w), 1145.2 (w), 1160.2 (w), 1082.7 (w), 1029.3 (w), 938.3 (w), 912.9 (w), 876.5 (w), 826.9 (m), 807.5 (m), 751.1 (w), 738.5 (w), 682.0 (w), 666.8 (w). LC-MS: Rt 3.60 min; HRMS (ESI): m/z calculated for C10H4N4O2Cl [M+H+]: 247.0028; found 247.0039.
6,8-Dibromobenzo[g]pteridine-2,4(3H,10H)-dione or 7,9-dibromobenzo[g]pteridine-2,4(3H,10H)-dione (12). Prepared from 2,4-dibromo-6-nitroaniline (0.294 g, 1 mmol) to give a pale green solid, one regioisomer was cleanly isolated (0.096 g, 26% yield). 1H-NMR (500 Hz, d6-DMSO): δ/ppm = 12.21 (s, 1H, NH), 11.85 (s, 1H, NH), 8.45 (d, J = 2.1 Hz, 1H), 8.43 (d, J = 2.1 Hz, 1H); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 159.9 (C), 150.0 (C), 147.8 (C), 139.9 (C), 139.4 (C), 138.2 (CH), 133.6 (C), 131.9 (CH), 122.0 (C), 120.2 (C). IR (neat) ν/cm−1: 3491.0 (w), 3183.1 (w) 3072.6 (w), 2849.1 (w), 1704.0 (s), 1603.2 (m), 1575.7 (w), 1558.3 (w), 1497.1 (w), 1468.2 (m), 1445.5 (m), 1386.8 (m), 1347.8 (m), 1319.7 (w), 1286.5 (s), 1210.7 (w), 1181.7 (w), 1136.6 (w), 1076.6 (w), 1028.2 (m), 1010.7 (m), 949.6 (m), 869.9 (m), 833.0 (m), 814.7 (m), 750.9 (w), 732.6 (w), 710.1 (w), 670.6 (w). LC-MS: Rt 4.02 min; HRMS (ESI): m/z calculated for C10H3N4O2Br2 [M+H+]: 368.8642; found 368.8628.
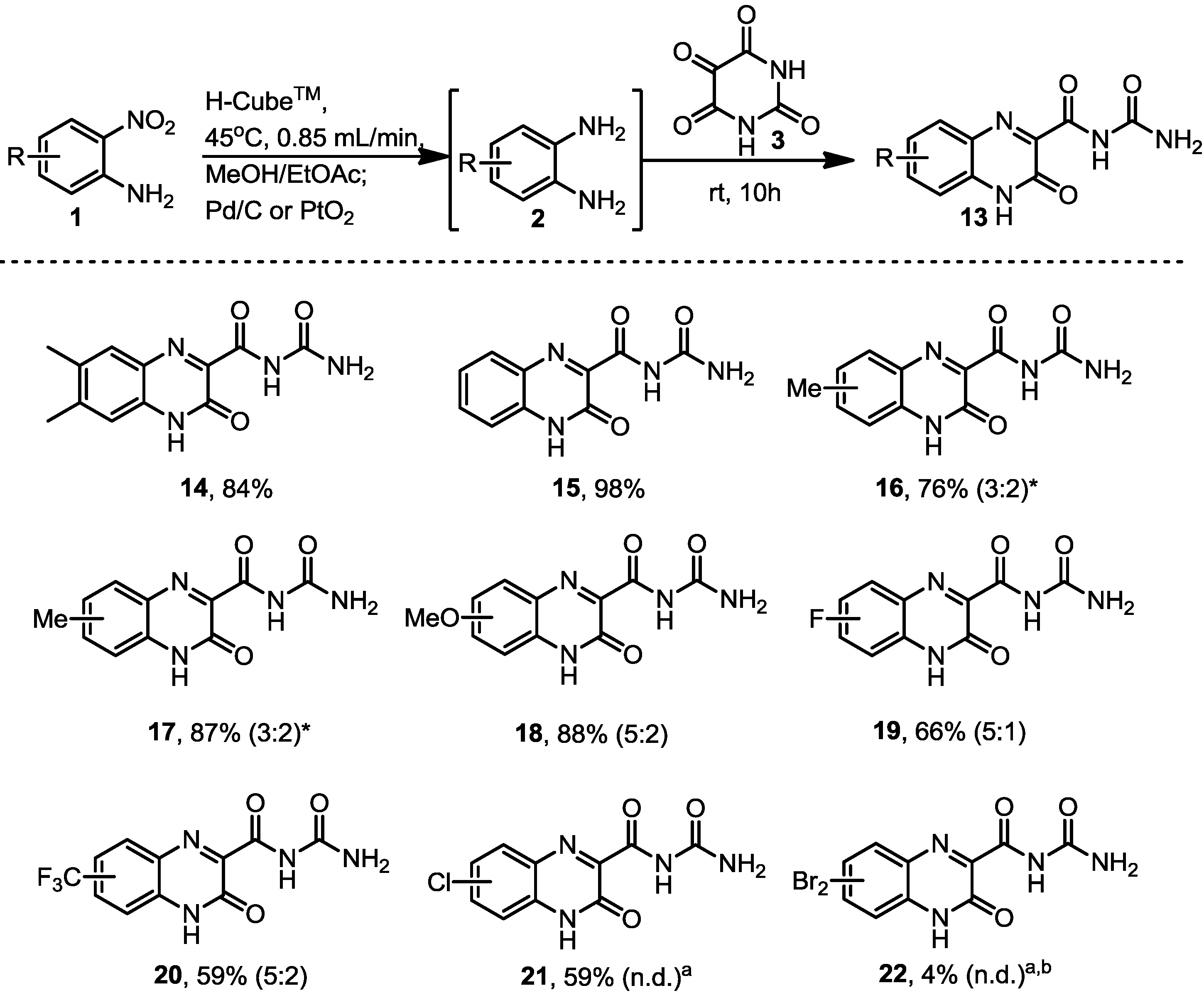
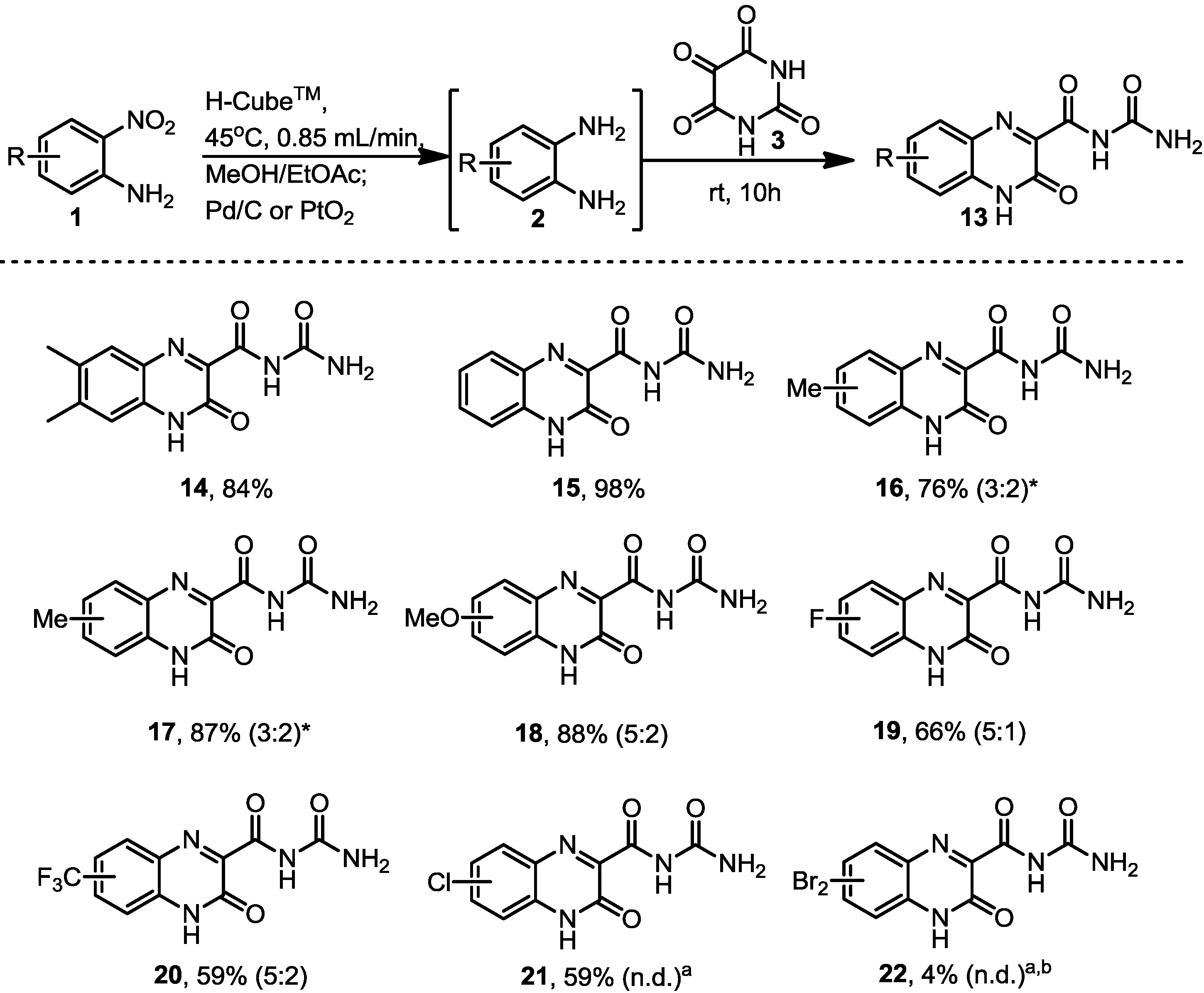
3.4. General Procedure for the Formation of the Dehydroquinoxalines Analogues 14–22
The diamines generated by the hydrogenation process in flow were mixed with alloxane monohydrate (0.160 g, 1 mmol) and left to stir at room temperature overnight. The product was then filtered and the solid product was dried in vacuo.
N-Carbamoyl-6,7-dimethyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide (14). Prepared from 4,5-dimethyl-2-nitroaniline (0.166 g, 1 mmol) to give a dull yellow solid (0.212 g, 84% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 12.97 (br s, 1H, NH), 11.21 (br s, 1H, NH), 7.74 (br s, 1H, NH from NH2), 7.69–7.65 (br m, 2H), 7.54 (br s, 1H, NH from NH2). To separate broad peaks: 1H-NMR (400 MHz, d6-DMSO + 3 drops TFA): δ/ppm = 6.55 (s, 1H), 6.45 (s, 1H), 5.72 (s, 2H from NH2); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 159.6 (C), 155.2 (C), 153.2 (C), 142.0 (C), 141.9 (C), 131.2 (C), 128.8 (C), 123.4 (C), 115.8 (CH), 115.6 (CH), 20.2 (CH3), 19.0 (CH3). IR (neat) ν/cm−1: 3377.1 (m), 3148.1 (w, br), 2823.3 (w, br), 1690.1 (s), 1645.9 (m), 1570.3 (s), 1487.5 (s), 1446.3 (s), 1390.6 (s), 1390.6 (s), 1367.5 (s), 1331.5 (m), 1286.8 (w), 1253.5 (m), 1196.5 (m), 1167.9 (s), 1111.9 (m), 1032.3 (w), 1011.4 (w), 967.3 (w), 919.9 (w), 860.8 (w), 820.5 (w), 801.8 (s), 752.8 (w), 741.6 (w), 683.7 (m). LC-MS: Rt 3.69 min; HRMS (ESI): m/z calculated for C12H11N4O3 [M+H+]: 259.0837; found 259.0841.
N-Carbamoyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide (15). Prepared from 2-nitroaniline (0.138 g, 1 mmol) to give a light green solid (0.228 g, 98% yield). 1H-NMR gave broad peaks. 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 13.03 (s, 1H, NH), 11.09 (s, 1H, NH), 7.92–7.37 (m br, 6H, NH2 + 4CH). To separate broad peaks: 1H-NMR (400 MHz, d6-DMSO + 3 drops TFA): δ/ppm = 8.97 (br s, 1H, NH2), 7.37 (br s, 1H, NH2), 6.83 (m, 2H), 6.68 (m, 2H); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 163.8 (C), 154.1 (C), 153.2 (C), 149.3 (C), 132.6 (CH), 131.3 (C), 129.7 (CH), 124.3 (CH), 124.0 (C), 115.8 (CH). IR (neat) ν/cm−1: 3366.5 (w), 3152.2 (w, br), 2969.0, (w), 2813.8 (w, br), 1718.3 (m), 1685.2 (s), 1643.2 (m), 1609.6 (w), 1571.0 (m), 1495.8 (m), 1433.8 (m), 1391.0 (s), 1360.1 (m), 1336.6 (w), 1293.6 (w), 1270.1 (w), 1249.6 (w), 1232.8 (w), 1183.2 (w), 1146.8 (m), 1100.7 (m), 1049.0 (w), 969.8 (w), 929.1 (w), 912.1 (m), 796.1 (m), 764.1 (s), 723.9 (w), 680.4 (w). LC-MS: Rt 2.63 min; HRMS (ESI): m/z calculated for C10H7N4O3 [M+H+]: 213.0524; found 231.0527.
N-Carbamoyl-7-methyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide and N-carbamoyl-8-methyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide (16). Prepared from 4-methyl-2-nitroaniline (0.152 g, 1 mmol) to give a bright yellow solid, with two regioisomers in a 3:2 ratio (0.187 g, 76% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 11.86 (1H from minor regioisomer, d, J = 1.7 Hz, NH), 11.84 (1H from major regioisomer, d, J = 1.7 Hz, NH) 11.68 (1H from major regioisomer, s, NH), 11.69 (1H from minor regioisomer, s, NH) , 8.00 (1H from minor regioisomer, d, J = 8.6 Hz, H1) 7.89 (1H from major regioisomer, s, H3), 7.78 (1H from major regioisomer, d, J = 8.6 Hz, H1), 7.72 (1H from major isomer, dd, J = 8.6 and 1.9 Hz, H2), 7.66 (1H from minor regioisomer, s, H3), 7.57 (1H from minor regioisomer, dd, J = 8.6 and 1.9 Hz, H2), 2.54 (3H from minor regioisomer, s, CH3), 2.48 (3H from major regioisomer, m, CH3). 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 160.6 (C), 160.6 (C), 150.2 (C), 150.1 (C), 147.0 (C), 146.4 (C), 144.3 (C), 142.8 (C), 141.1 (C), 139.4 (C), 138.6 (C), 137.9 (C), 135.7 (CH), 131.3 (C), 130.8 (CH), 130.6 (C), 129.7 (CH), 128.7 (CH), 126.6 (CH), 125.7 (CH), 21.7 (CH3), 21.1 (CH3). IR (neat) ν/cm−1: 3171.9 (w), 3075.1 (w), 2990.5 (w),2852.3 (w), 1726.5 (m), 1701.1 (s), 1622.7 (w), 1581.9 (m), 1564.7 (w), 1511.5 (w), 1476.0 (w), 1446.6 (w), 1392.9 (w), 1354.4 (m), 1337.5 (m), 1307.9 (w), 1281.0 (s), 1249.4 (w), 1207.9 (w), 1151.2 (w), 1121.8 (w), 1027.5 (w), 982.2 (w), 948.9 (w), 907.4 (w), 874.6 (w), 830.1 9s), 810.0 (m), 760.8 (w), 704.4 (w), 682.0 (w), 661.8 (w). LC-MS: Rt 3.43 min; HRMS (ESI): m/z calculated for C11H9N4O3 [M+H+]: 245.0680; found 245.0681.
N-Carbamoyl-7-methyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide and N-carbamoyl-8-methyl-3-oxo-3,4-dihydro-quinoxaline-2-carboxamide (17). Prepared from 5-methyl-2-nitroaniline (0.152 g, 1 mmol) to give a bright yellow solid, with two regioisomers found in a 3:2 ratio (0.215 g, 87% yield). This was gave an identical 1H-NMR to 16.
N-Carbamoyl-7-methoxy-3-oxo-3,4-dihydroquinoxaline-2-carboxamide and N-carbamoyl-8-methoxy-3-oxo-3,4-dihydroquinoxaline-2-carboxamide (18). Prepared from 4-methoxy-2-nitroaniline (0.206 g, 1 mmol) to give an orange solid, with two regioisomers found in a 5:2 ratio (0.231 g, 88% yield). 1H-NMR gave broad peaks and not all quaternary centres were observed in the 13C-NMR. 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 13.03 (s, 1H, NH), 11.23 (s, 1H, NH), 7.83-7.30 (br m, 6H), 7.03 (d, J = 8.7 Hz, 2H, NH2), 6.81 (d, J = 2.9 Hz, 2H, NH2), 3.88 (s, 3H, CH3), 3.84 (s, 3H, CH3). To separate broad peaks: 1H-NMR (400 MHz, d6-DMSO + 3 drops TFA): δ/ppm = 7.79 (1H from minor regioisomer, d, J = 9.0 Hz, CH, 2 or 3), 7.58 (1H from major regioisomer, s, H1), 7.00 (1H from minor regioisomer, d, J = 9.5 Hz, H2 or H3), 6.81 (1H from minor regioisomer, d, J = 2.5 Hz, H1) 6.69 (1H from major regioisomer, d, J = 9.3 Hz, H2 or H3), 6.60 (1H from major regioisomer, d, J = 9.3 Hz, H2 or H3), 6.44 (2H, one from each regioisomer, d, J = 6.9 Hz, 2NH from the NH2), 6.24 (2H, one from each regioisomer, d, J = 6.6 Hz, 2NH from the NH2); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 162.9 (C), 153.2 (C), 153.2 (C), 134.89 (C), 131.6 (CH), 126.9 (C), 116.7 (CH), 114.1 (CH), 110.5 (CH), 103.7 (CH), 97.6 (CH), 56.0 (CH3), 55.8 (CH3). IR (neat) ν/cm−1: 3394.4 (w), 3315.5 (w), 3149.3 (w,br), 2976.0 (w), 1731.0 (s), 1697.5 (s), 1676.5 (w), 1648.2 (s), 1618.5 (s), 1583.6 (s), 1502.4 (s) 1487 (s), 1471.4 (m), 1455.1 (m), 1441.4 (w), 1381.8 (s), 1370.6 (s)., 1280.7 (w), 1249.7 (m), 1222.3 (s), 1184.2 (s), 1150.6 (m), 1110.4 (m), 1031.7 (m), 1012.8 (m), 969.4 (w), 914.1 (w), 839.5 (s), 822.0 (s), 801.6 (s), 751.8 (w), 682.0 (w). LC-MS: Rt 3.44 min; HRMS (ESI): m/z calculated for C11H9N4O4 [M+H+]: 261.0629; found 261.0634.
N-Carbamoyl-7-fluoro-3-oxo-3,4-dihydroquinoxaline-2-carboxamide and N-carbamoyl-8-fluoro-3-oxo-3,4-dihydro-quinoxaline-2-carboxamide (19). Prepared from 4-fluoro-2-nitroaniline (0.106 g, 1 mmol) to give a pale green solid, with two regioisomers found in a 5:1 ratio (0.165 g, 66% yield). 1H-NMR gave broad peaks and not all quaternary centres were observed in the 13C-NMR. 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 13.07 (1H from major regioisomer, br s, NH), 12.55 (1H from minor regioisomer, br s, NH), 11.03 (1H from major regiosiomer, br s, NH), 10.34 (1H from minor regioisomer, br s, NH), 7.94 (1H from major regioisomer, br s, NH from NH2) 7.76–7.57 (5H from minor regioisomer and 1H from major regioisomer, br m, 2NH from NH2, 4CH), 7.38 (1H from major regioisomer, br s, NH from NH2), 7.25 (1H from major regioisomer, s), 7.07 (1H from major regioisomer, d, J = 4.1 Hz); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 163.7 (C), 159.1 (C), 157.2 (C), 154.0 (C-F), 134.4 (C), 132.4 (CH), 131.5 (C), 129.6 (CH), 128.4 (C), 121.0 (CH), 117.4 (C), 114.5 (CH), 112.6 (CH), 101.6 (CH). IR (neat) ν/cm−1: 3380.2 (w), 3152.2 (w, br), 2732.4 (w,br) 1721.4 (s), 1698.5 (s), 1649.5 (s), 1596.5 (m), 1578.9 (m), 1504.1 (s), 1485.6 (s), 1403.6 (s), 1364.1 (s), 1284.1 (w), 1246.3 (m), 1201.8 (m), 1179.4 (m), 1144.7 (w), 1120.1 (w), 1099.3 (m), 978.9 (w), 939.0 (w), 912.9 (w), 847.0 (m), 823.2 (s), 805.3 (s), 752.8 (w), 698.9 (w), 680.7 (m). LC-MS: Rt 3.23 min; HRMS (ESI): m/z calculated for C10H6N4O3F [M+H+]: 249.0429; found 249.0421.
N-Carbamoyl-3-oxo-7(trifluoromethyl)-3,4-dihydro-quinoxaline-2-carboxamide and N-carbamoyl-3-oxo-8(trifluoromethyl)-3,4-dihydroquinoxaline-2-carboxamide (20). Prepared from 2-nitro-5-(trifluromethyl)-aniline (0.206 g, 1 mmol) to give a pale yellow solid, with two regioisomers in a 5:2 ratio (0.178 g, 59% yield). 1H-NMR gave broad peaks and not all quaternary centres were observed in the 13C-NMR. 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 13.21 (1H from major regioisomer, s, NH), 12.78 (1H from minor regioisomer, br d, J = 40.35 Hz, NH), 10.95 (1H from major regioisomer, s, NH), 10.39 (1H from minor regioisomer, br, s, NH), 8.23–7.51 (6H, three from each regioisomer, m), 6.96 (2H, one from each regioisomer, br s, 2NH from the NH2’s), 6.23 (2H, one from both compounds, br s, 2NH from the NH2’s). To separate broad peaks: 1H-NMR (400 MHz, d6-DMSO + 3 drops TFA): δ/ppm = 7.97 (1H from minor regioisomer, s, NH from NH2), 7.92 (1H from minor regioisomer, s, NH from NH2), 7.76 (1H from minor regioisomer, s, H1), 7.62 (1H from minor regioisomer, s, H2 or H3), 7.51 (1H from minor regioisomer, d, J = 8.4 Hz, H2 or H3), 7.17 (1H from major regioisomer, d, J = 8.3, H2 or H3), 7.07 (1H from major regioisomer, s, H1), 7.01 (1H from major regioisomer, d, J = 8.3 Hz, NH from NH2), 6.96 (1H from major regioisomer, d, J = 8.3 Hz, NH from NH2), 6.81 (1H from major regioisomer, J = 8.3 Hz, H2 or H3); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 166.5 (CF3), 163.8 (C), 153.0 (C-CF3), 135.4 (C), 133.0 (C), 130.9 (CH), 130.4 (C), 128.4 (CH), 126.9 (CH), 125.1 (C), 124.7 (C, minor regioisomer), 122.8 (C, minor regioisomer), 122.5 (C, minor regioisomer), 120.1 (CH), 117.2 (CH), 113.0 (CH). IR (neat) ν/cm−1: 3520.9 (w), 3411.4 (w), 3243.7 (w), 3159.1 (w), 1736.8 (m), 1692.7 (s), 1674.6 (s), 1662.0 (s), 1625.9 (m), 1568.0 (w), 1506.1 (w), 1466.7 (w). 1412.1 (w), 1376.5 (m), 1320.3 (m), 1285.7 (w), 1260.6 (w), 1242.3 (w), 1189.4 (w), 1166.3 (w), 1148.9 (s), 1135.7 (s), 1112.7 (m), 1091.4 (m), 1064.7 (m), 957.1 (w),901.4 (m), 847.0 (m), 832.5 (m), 820.7 (w), 779.8 (w), 738.9 (w), 687.3 (m), 655.3 (m). LC-MS: Rt 3.80 min; HRMS (ESI): m/z calculated for C11H4F3N4O2 [M+H+]: 281.0292; found 281.0287.
N-Carbamoyl-6-chloro-3-oxo-3,4-dihydroquinoxaline-2-carboxamid and N-carbamoyl-7-chloro-3-oxo-3,4-dihydro-quinoxaline-2-carboxamide (21). Prepared from 4-chloro-2-nitroaniline (0.172 g, 1 mmol) to give a pale yellow solid, with two regioisomers in an unknown ratio due to overlapping 1H signals (0.157 g, 59% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 12.02 (s, 2H, 2NH), 11.79 (s, 2H, 2NH), 8.17 (d, J = 9.0 Hz, 2xH3), 7.97 (d, J = 2.3 Hz, 2 × H1), 7.93 (4H, m, 2H, 2NH2), 7.78 (dd, J = 2.3 Hz, 9.0 Hz, 2 × H2); 13C-NMR (125 MHz, d6-DMSO): δ/ppm = 160.3 (br, 2C), 150.1 (br, C), 147.6 (C), 147.2 (C), 143.1 (C), 141.4 (C), 139.4 (C), 137.9 (C), 137.8 (C), 133.7 (CH), 132.8 (C), 132.5 (C), 132.3 (C), 132.0 (CH), 129.1 (CH), 128.8 (CH), 128.6 (CH), 125.7 (CH). IR (neat) ν/cm−1: 3457.6 (w, br), 3184.9 (w), 3068.7 (w, br), 2850.5 (w), 1727.7 (m), 1697.9 (s), 1614.5 (m), 1578.8 (m), 1562.0 (w), 1489.7 9(w), 1449.0 (w), 1388.9 (m), 1354.7 (m), 1335.8 (m), 1292.5 (m), 1275.0 (m), 1245.9 (w), 1196.3 (w), 1145.6 (w), 1112.4 (w), 1071.0 (w), 1029.8 (m), 939.2 (w), 876.7 (w), 847.5 (s), 806.6 (m), 751.5 (m), 738.1 (m), 692.8 (w), 666.2 (w). LC-MS: Rt 3.60 min; HRMS (ESI): m/z calculated for C10H4N4O2Cl [M+H+]: 266.0207; found 266.0211.
6,8-Dibromo-N-carbamoyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide or 7,9-dibromo-N-carbamoyl-3-oxo-3,4-dihydroquinoxaline-2-carboxamide (22). Prepared from 2,4-dibromo-6-nitroaniline (0.294 g, 1 mmol) to give a mixture of a green solid and a liquid component in approximately a 1:2 ratio. Upon purification one regioisomer was cleanly isolated (estimated yield 8%). HRMS (ESI): m/z calculated for C10H5N4O3Br2 [M+H+]: 386.8734; found 386.8716. 1H-NMR (400 MHz, d6-DMSO): δ/ppm = 11.20 (s, 1H, NH), 11.04 (s, 1H, NH), 8.16 (d, J = 2.6 Hz, 1H), 8.05 (d, J = 2.2 Hz, 1H).
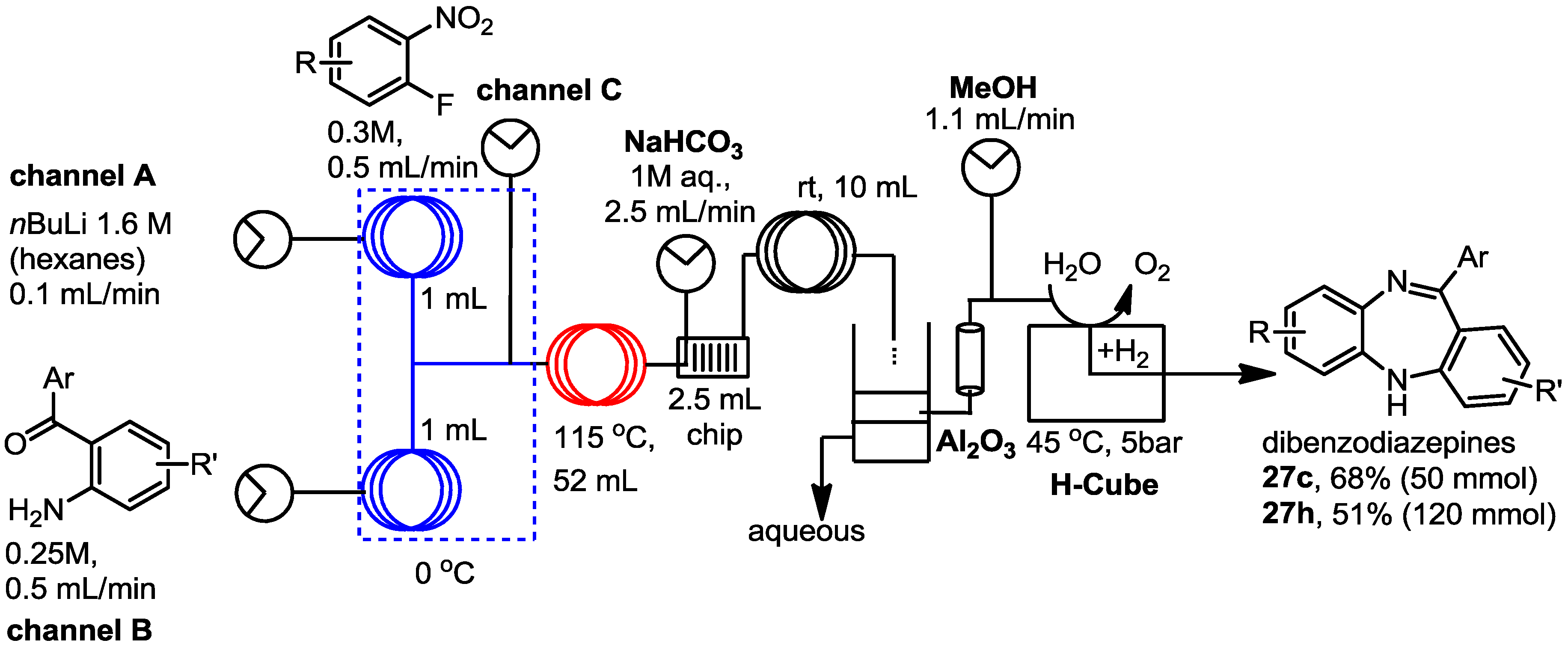
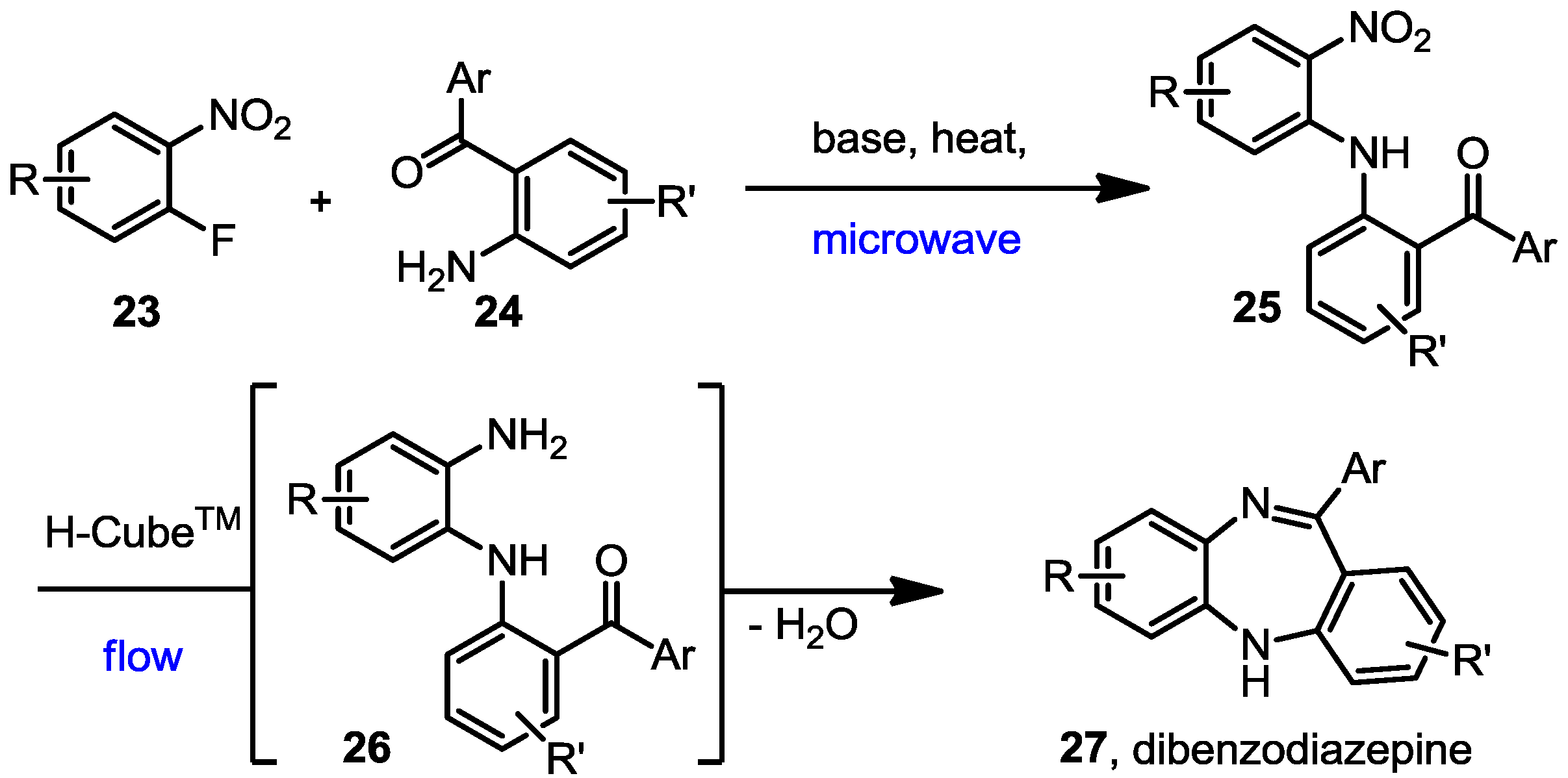
3.5. General Procedure for the Formation of 1,5-Dibenzodiazepines 27a–k
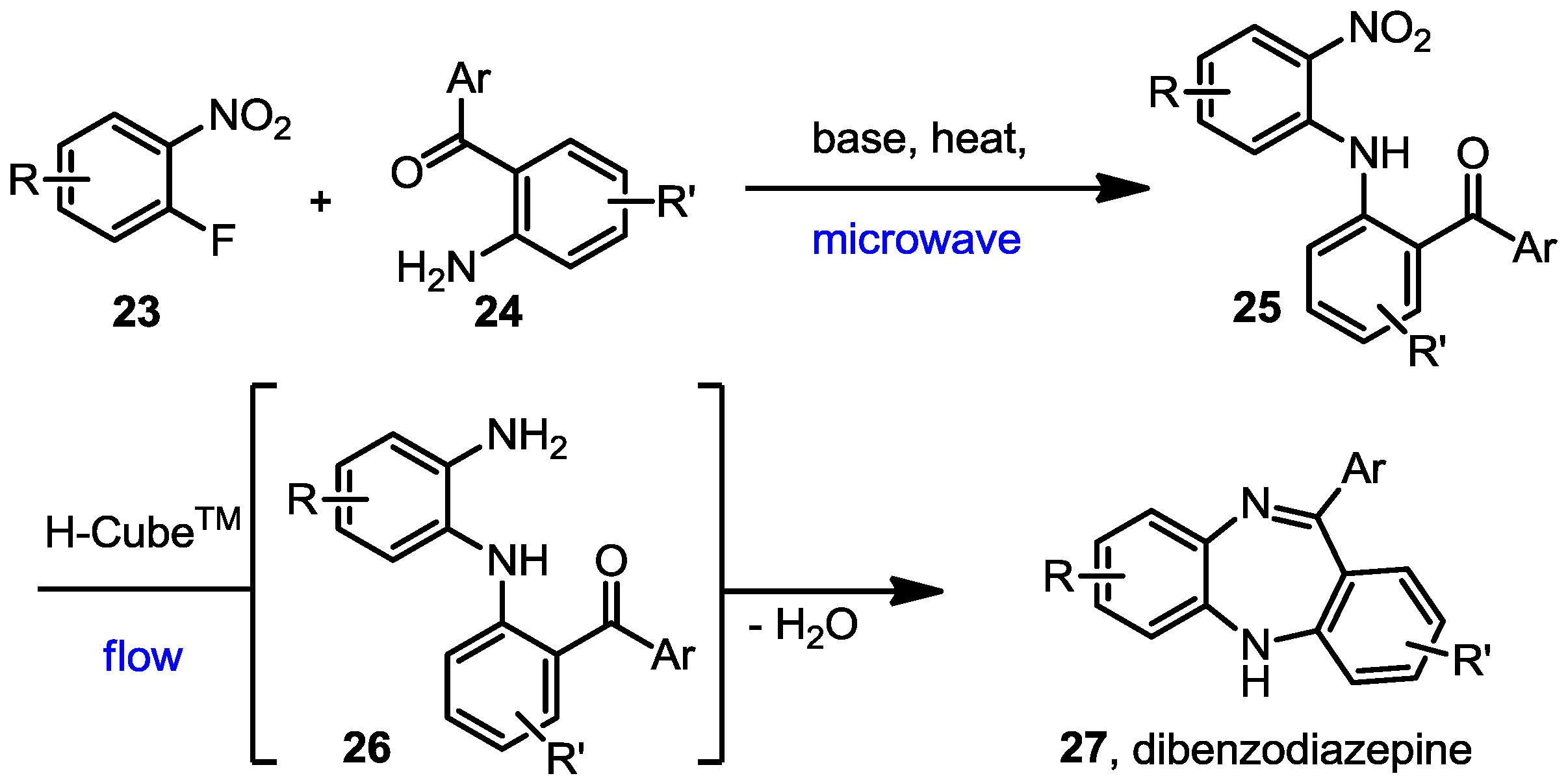
A mixture of the fluoro nitrobenzene derivative and the corresponding amino benzophenone/fluorenone (1:1) was dissolved in dry THF in a microwave vial. Lithium bis(trimethylsilyl)amide (LiHMDS, 1.0 M solution in THF) was added dropwise and the mixture heated to 100 °C for 30 to 90 min using a Biotage Initiator microwave instrument. The reaction was quenched by addition of water and the mixture extracted with EtOAc. The combined organic layers were dried over anhydrous Na2SO4 and the solvent evaporated under vacuum. Most of the intermediate products (25) were not further purified but directly hydrogenated in flow, although some of them were subjected to column chromatography and fully characterised to confirm the structure and complete data is shown below.
(2-((5-Chloro-2-nitrophenyl)amino)phenyl)(phenyl)-methanone. 1H-NMR (400 MHz, CDCl3): δ/ppm = 10.98 (s, 1H, NH), 8.09 (d, J = 8.8 Hz, 1H), 7.81 (d, J = 1.1 Hz, 1H), 7.79 (d, J = 1.5 Hz, 1H), 7.62–7.56 (m, 4H), 7.47–7.44 (m, 3H), 7.20 (dd, J = 7.6 Hz, 1.5 Hz, 1H), 6.80 (dd, J = 9.0 Hz, 2.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ 196.7 (C=O), 141.7 (C), 141.3 (C), 139.7 (C), 138.1 (C), 134.2 (C), 133.3 (CH), 133.3 (2 × CH), 130.5 (2 × CH), 129.8 (C), 128.8 (2 × CH), 128.5 (CH), 123.8 (CH), 122.5 (CH), 119.7 (CH), 117.1 (CH). IR (neat) ν/cm−1: 3301.0 (w), 3062.6 (w), 1643 (w), 1608.2 (m), 1596.2 (m), 1562.6 (s), 1486.6 (s), 1449.3 (m), 1409.4 (w), 1312.5 (m), 1335.1 (w), 1297.1 (m), 1248.8 (s), 1211.5 (m), 1180.2 (w), 1164.3 (w), 1103.8 (w), 1070.6 (w), 921.6 (m), 841.4 (w), 749.9 (m), 700.8 (m). HRMS (m/z) calculated for C19H14O3N2Cl, (M-H), 353.0687; found 353.0678
(5-Chloro-2-((5-chloro-2-nitrophenyl)amino)phenyl)-(phenyl)methanone. 1H-NMR (400 MHz, CDCl3): δ/ppm = 10.82 (s, 1H, NH), 8.11 (d, J = 9.2 Hz, 1H), 7.82 (d, J = 1.5 Hz, 1H), 7.80 (d, J = 1.4 Hz, 1H), 7.65–7.58 (m, 2H), 7.56 (s, 1H), 7.54 (d, J = 2.0 Hz, 1H), 7.53–7.48 (m, 2H), 7.40 (d, J = 2.0 Hz, 1H), 6.80 (dd, J = 9.2 Hz, 2.2 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ 195.3 (C=O), 141.8 (C), 140.9 (C), 138.2 (C), 137.1 (C), 134.1 (C), 133.4 (CH), 132.8 (CH), 132.2 (CH), 130.9 (C), 130.1 (2 x CH), 128.7 (C), 128.6 (2 × CH), 128.2 (CH), 123.6 (CH), 119.7 (CH), 116.6 (CH). IR (neat) ν/cm−1: 2673.4 (w), 2319.2 (w), 1646.9 (w), 1607.9 (m), 1568.3 (m), 1488.2 (s), 1395.9 (w), 1336.5 (w), 1294.2 (w), 1259.7 (m), 1240.5 (m), 1163.3 (w), 1104.2 (w), 930.4 (w), 826.6 (w), 752.2 (w). HRMS (m/z) calculated for C19H13O3N2Cl2, (M-H), 387.0298; found 387.0289.
(5-Chloro-2-((5-chloro-2-nitrophenyl)amino)phenyl)(3-chlorophenyl)methanone. 1H-NMR (400 MHz, CDCl3): δ/ppm = 11.33 (s, 1H, NH), 8.14 (d, J = 9.1 Hz, 1H), 7.57–7.40 (m, 8H), 6.94 (dd, J = 9.1 Hz, 2.2 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ 195.7 (C=O), 140.2 (C), 140.1 (C), 138.3 (C), 135.8 (C), 134.6 (CH), 133.5 (CH), 132.4 (CH), 131.8 (C), 130.7 (CH), 130.0 (CH), 128.5 (CH), 128.5 (C), 128.2 (C), 127.4 (CH), 122.2 (CH), 121.1 (CH), 118.3 (CH). IR (neat) ν/cm−1: 2738.9 (w), 1648.0 (w), 1607.8 (m), 1570.8 (m), 1488.2 (s), 1397.9 (w), 1336.5 (w), 1293.2 (s), 1247.5 (m), 1072.3 (w), 951.9 (w), 929.7 (w), 828.3 (w), 748.9 (w), 699.8 (w). HRMS (m/z) calculated for C19H11O3N2Cl3Na, 442.9727; found 442.9724.
5-Chloro-2-((5-chloro-2-nitrophenyl)amino)phenyl)(2-fluoro-phenyl)methanone. 1H-NMR (400 MHz, CDCl3): δ/ppm = 11.09 (s, 1H, NH), 8.11 (d, J = 9.2 Hz, 1H), 7.64–7.48 (m, 5H), 7.44 (d, J = 2.2 Hz, 1H), 7.28 (td, J = 7.7 Hz, 1.1 Hz, 1H), 7.13 (app t, 1H), 6.88 (dd, J = 9.0 Hz, 2.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3): δ 192.7 (C=O), 160.1 (d, J = 253.6 Hz, C-F), 141.7 (C), 140.4 (C), 138.6 (C), 134.8 (C), 134.3 (d, J = 8.4 Hz, CH), 133.7 (CH), 132.5 (d, J = 2.3 Hz, CH), 131.0 (d, J = 1.9 Hz, CH), 130.2 (C), 128.5 (CH), 128.1 (C), 126.4 (d, J = 13.6 Hz, C), 124.7 (d, J = 3.6 Hz, CH), 122.6 (CH), 120.2 (CH), 117.4 (CH), 116.5 (d, J = 21.6 Hz, CH). IR (neat) ν/cm−1: 2338.9 (w), 1648.9 (w), 1608.6 (m), 1570.1 (m), 1488.6 (s), 1453.1 (w), 1398.5 (w), 1336.3 (w), 1308.8 (w), 1259.7 (m), 1243.7 (m), 1213.9 (w), 1072.1 (w), 931.2 (w), 753.12 (m). HRMS (m/z) calculated for C19H12O3N2Cl2F (M+H), 405.0204; found 405.0197.
The nitro phenylamino derivatives 25 were hydrogenated in flow at 60 °C using a flow rate of 0.5 mL/min, following the general procedure described before. The generated diamines were collected over MgSO4 and stirred for a further 1–2 h at room temperature to enhance the cyclisation process. After filtration to remove the drying agent, catalytic formic acid was added to the remaining solution and the mixture was stirred at room temperature overnight. The solvent was then removed under vacuum and the final benzodiazepines were purified by column chromatography and fully characterised by NMR.
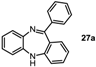
11-Phenyl-5H-dibenzo[b,e][1,4]diazepine (27a). Prepared from 1-fluoronitrobenzene and 2-aminobenzophenone (175 mg, 65% yield). 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.76 (d, J = 7.1 Hz, 2H), 7.48–7.42 (m, 3H), 7.36 (dd, J = 7.7 Hz, 1.0 Hz, 1H), 7.29 (td, J = 7.6 Hz, 1.1 Hz, 1H), 7.08 (td, J = 7.5 Hz, 1.0 Hz, 1H), 7.04 (m, 2H), 6.94 (t, J = 7.5 Hz, 1H), 6.77 (d, J = 7.9 Hz, 1H), 6.70 (dd, J = 7.6 Hz, 0.8 Hz, 1H), 5.03 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 169.6 (C), 154.5 (C), 142.7 (C), 141.4 (C), 140.9 (C), 132.2 (CH), 132.0 (CH), 130.0 (CH), 129.6 (2xCH), 128.7 (CH), 128.0 (2 × CH), 127.6 (C), 126.9 (CH), 124.2 (CH), 122.4 (CH), 119.8 (2 × CH); IR (neat) cm−1: 3354.0 (w), 3273.2 (w), 3051.8 (w), 2850.2 (w), 1601.9 (w, N=C), 1494.8 (w), 1460.2 (m), 1401.3 (w), 1317.0 (w), 1283.1 (w), 1234.6 (w), 1151.7 (w), 1108.2 (w), 958.5 (w), 944.3 (w), 853.1 (w), 802.7 (w), 756.4 (s), 694.7 (s), 657.7 (m); LC-MS: tr = 4.68, m/z = 271.32 (C19H14N2)+.
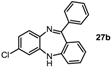
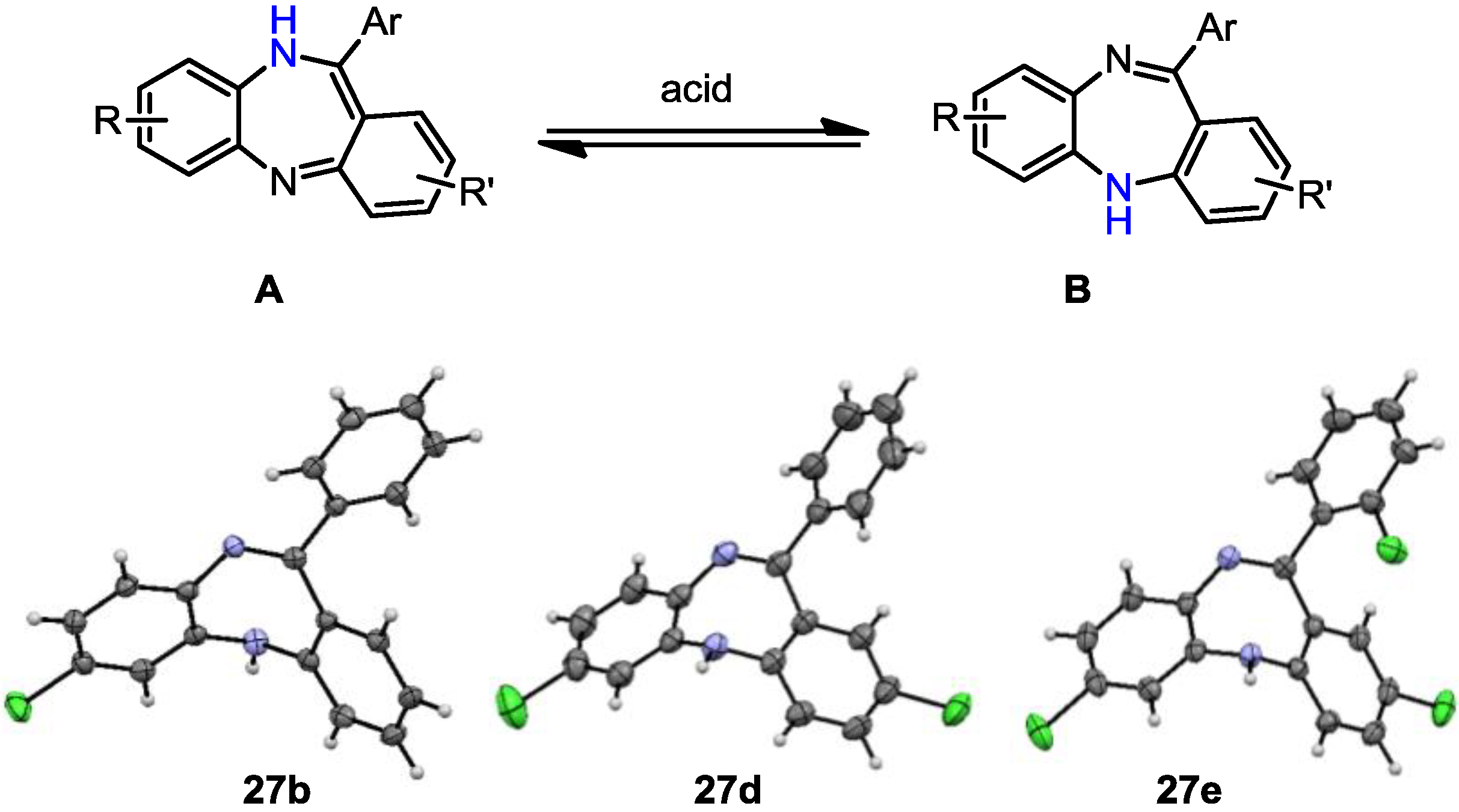
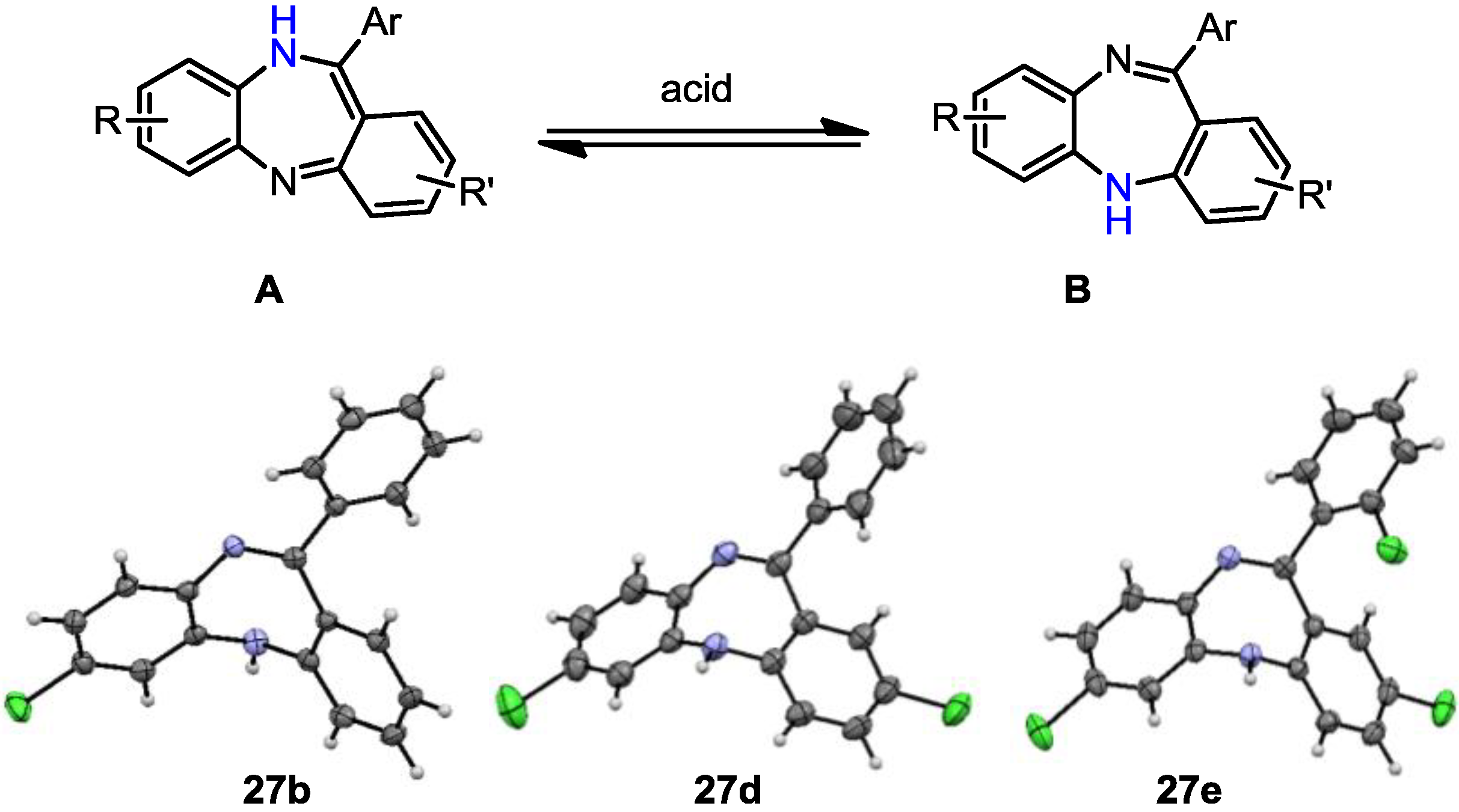
7-Chloro-11-phenyl-5H-dibenzo[b,e][1,4]diazepine (27b). Prepared from 4-chloro-2-fluoro-1-nitrobenzene and 2-aminobenzophenone (201 mg, 66% yield). 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.70 (d, J = 7.1 Hz, 2H), 7.47–7.40 (m, 3H), 7.32 (t, J = 7.0 Hz, 1H), 7.22 (d, J = 8.3 Hz, 1H), 7.02 (d, J = 8.0 Hz, 2H), 6.97 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 7.8 Hz, 1H), 6.72 (d, J = 1.8 Hz, 1H), 4.97 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 169.8 (C), 153.6 (C), 143.4 (C), 141.0 (C), 139.5 (C), 132.3 (C), 132.2 (CH), 132.1 (CH), 130.2 (CH), 129.6 (CH), 129.5 (2 × CH), 128.0 (2xCH), 127.5 (C), 124.2 (CH), 122.8 (CH), 119.9 (CH), 119.7 (CH); IR (neat) cm−1: 3359.8 (w, NH), 3053.7 (w), 1607.9 (s, N=C), 1573.1 (w), 1455.8 (s), 1433.3 (w), 1317.6 (w), 1285.6 (m), 1250.0 (w), 1117.5 (w), 1089.7 (w), 957.6 (w), 915.0 (m), 855.7 (m), 815.9 (s), 778.8 (s), 742.5 (s), 722.6 (m), 692.8 (s), 681.2 (s), 666.1 (m); LC-MS: tr = 5.13, m/z = 304.90 (C19H13ClN2)+. The structure was unambiguously confirmed by X-ray crystallography and deposited at the Cambridge Crystallographic Data Centre with the unique reference number CCDC 867823; Formula: C19H13ClN2, unit cell parameters: a = 16.4748(5) Å, b = 5.6296(2) Å, c = 16.0146(7) Å, α = 90°, β = 102.528(2)°, γ = 90°, space group: P21/c.
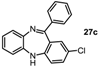
2-Chloro-11-phenyl-5H-dibenzo[b,e][1,4]diazepine (27c). Prepared from 1-fluoronitrobenzene and 2-amino-4-chlorobenzophenone (110 mg, 72% yield). 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.71 (d, J = 7.1 Hz, 2H), 7.49-7.42 (m, 3H), 7.32 (dd, J = 7.6 Hz, 1.4 Hz, 1H), 7.27–7.25 (m, 1H), 7.09–7.03 (m, 2H), 6.99 (d, J = 2.3 Hz, 1H), 6.74 (d, J = 8.5 Hz, 1H), 6.71 (dd, J = 7.5 Hz, 1.3 Hz, 1H), 5.02 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 153.0 (C), 142.2 (C), 140.5 (C), 140.4 (C), 131.8 (CH), 131.6 (CH), 130.2 (CH), 129.5 (2 × CH), 128.8 (CH), 128.2 2 × (CH), 127.9 (C), 127.8 (C), 127.7 (C), 127.2 (CH), 124.5 (CH), 121.1 (CH), 119.8 (CH); IR (neat) cm−1: 3269.5 (w, NH), 3053.0 (w), 1610.6 (w, N=C), 1569.3 (w), 1489.0 (w), 1461.7 (w), 1421.2 (w), 1318.9 (w), 1280.2 (w), 1234.7 (w), 1120.4 (w), 964.9 (w), 823.2 (m), 806.3 (m), 760.2 (s), 738.6 (s), 698.1 (s), 667.8 (w); LC-MS: tr = 5.09, m/z = 305.03 (C19H13ClN2)+.
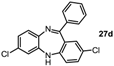
2,7-Dichloro-11-phenyl-5H-dibenzo[b,e][1,4]diazepine (27d). Prepared from 4-chloro-2-fluoro-1-nitrobenzene and 2-amino-4-chlorobenzophenone (99 mg, 59% yield). 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.69 (d, J = 7.2 Hz, 2H), 7.49-7.40 (m, 3H), 7.27 (m, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.04 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 6.99 (d, J = 2.3 Hz, 1H), 6.73–6.71 (m, 2H), 4.99 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 168.2 (C), 152.0 (C), 142.9 (C), 140.3 (C), 139.2 (C), 132.4 (C), 131.9 (CH), 131.6 (CH), 130.5 (CH), 129.7 (CH), 129.6 (2 × CH), 129.4 (C), 128.9 (C), 128.3 (2 × CH), 124.5 (CH), 121.2 (CH), 119.8 (CH); IR (neat) cm−1: 3359.6 (w), 3259.7 (w), 3054.2 (w), 1608.7 (m, N=C), 1573.9 (w), 1454.7 (m), 1316.9 (m), 1287.3 (w), 1249.8(w), 1117.0 (w), 1087.6 (w), 957.9 (w), 915.2 (m), 858.1 (m), 816.5 (m), 778.8 (m), 742.8 (m), 691.9 (s); LC-MS: tr = 5.35, m/z = 339.22 (C19H12Cl2N2)+. The structure was unambiguously confirmed by X-ray crystallography and deposited at the Cambridge Crystallographic Data Centre with the unique reference number CCDC 867824; Formula: C19H12Cl2N2, unit cell parameters: a = 14.2135(5) Å, b = 9.2512(4) Å, c = 25.0981(10) Å, α = 90°, β = 103.764(2)°, γ = 90°, space group: C2/c.
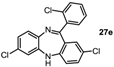
2,7-Dichloro-11-(2-chlorophenyl)-5H-dibenzo[b,e][1,4]-diazepine (27e). Prepared from 4-chloro-2-fluoro-1-nitrobenzene and 2-amino-2’,4-dichloro-benzophenone (131 mg, 70% yield). 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.54 (m, 1H), 7.40–7.36 (m, 3H), 7.18 (dd, J = 8.5 Hz, 2.4 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 6.98 (dd, J = 8.4 Hz, 2.2 Hz, 1H), 6.70 (d, J = 2.1 Hz, 1H), 6.65–6.63 (m, 2H), 5.47 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 167.7 (C), 151.5 (C), 143.6 (C), 140.0 (C), 138.4 (C), 133.6 (C), 132.6 (C), 132.2 (CH), 130.8 (CH), 130.6 (CH), 130.3 (CH), 130.2 (CH), 130.1 (CH), 129.7 (C), 128.4 (C), 127.0 (CH), 124.3 (CH), 120.9 (CH), 120.0 (CH); IR (neat) cm−1: 3257.3 (w, NH), 3051.1 (w), 2919.2 (w), 2850.0 (w), 1605.9 (m, N=C), 1489.2 (w), 1446.1 (m), 1434.2 (m), 1390.9 (w), 1315.9 (m), 1258.5 (w), 1236.7 (w), 1204.1 (w), 1155.6 (w), 1123.8 (w), 1085, 2 (w), 1057.6 (w), 968.8 (w), 942.8(w), 916.4(m), 880.9 (m), 857.7 (s), 822.1 (s), 747.5 (s), 727.9 (s); LC-MS: tr = 5.26, m/z = 374.94 (C19H11Cl3N2)+. The structure was unambiguously confirmed by X-ray crystallography. and deposited at the Cambridge Crystallographic Data Centre with the unique reference number CCDC 867822; Formula: C19H11Cl3N2, unit cell parameters: a = 15.5464(3) Å, b = 9.3738(2) Å, c = 24.0677(5) Å, α = 90°, β = 107.942(1)°, γ = 90°, space group: C2/c.
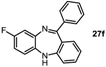
8-Fluoro-11-phenyl-5H-dibenzo[b,e][1,4]diazepine (27f). Prepared from 1,4-difluoro-2-nitrobenzene and 2-aminobenzophenone (192 mg, 67% yield). 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.71 (d, J = 7.3 Hz, 2H), 7.48–7.40 (m, 3H), 7.32 (td, J = 7.6 Hz, 1.2 Hz, 1H), 7.04 (d, J = 9.2 Hz, 2H), 6.96 (t, J = 7.5 Hz, 1H), 6.79 (d, J = 7.9 Hz, 1H), 6.75 (td, J = 8.2 Hz, 2.8 Hz 1H), 6.66–6.63 (m, 1H), 4.95 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 170.6 (C), 159.8 (d, J = 240.7 Hz, C-F), 154.5 (C), 142.1 (d, J = 10.6 Hz, C), 140.9 (C), 138.7 (d, J = 2.5 Hz, C), 132.2 (d, J = 9.2 Hz, CH), 130.3 (CH), 129.7 (2 × CH), 128.0 (2 × CH), 127.5 (C), 122.6 (CH), 120.2 (d, J = 9.1 Hz, CH), 119.7 (2 × CH), 114.7 (d, J = 23.4 Hz, CH), 113.0 (d, J = 22.9 Hz, CH); IR (neat) cm−1: 3350.8 (w), 3061.9 (w), 1600.9 (m, N=C), 1570.4 (w), 1497.7 (m), 1461.6 (s), 1389.2 (w), 1318.2 (w), 1285.9 (w), 1257.3 (m), 1169.8 (w), 1135.0 (w), 1099.1 (w), 970.8 (m), 906.5 (w), 869.2 (w), 807.7 (m), 750.1 (s), 721.8 (s), 694.3 (s), 660.3 (m); LC-MS: tr = 4.99, m/z = 288.92 (C19H13FN2)+.
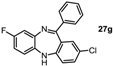
2-Chloro-8-fluoro-11-phenyl-5H-dibenzo[b,e][1,4]diazepine (27g). Prepared from 1,4-difluoro-2-nitrobenzene and 2-amino-4-chlorobenzophenone (81 mg, 50% yield) 1H-NMR (600 MHz, CDCl3): δ/ppm = 7.70 (d, J = 7.4 Hz, 2H), 7.50-7.42 (m, 3H), 7.28 (dd, J = 8.5 Hz, 2.3 Hz, 1H), 7.03 (dd, J = 9.5 Hz, 2.8 Hz, 1H), 7.00 (d, J = 2.3 Hz, 1H), 6.77–6.73 (m, 2H), 6.64 (m, 1H), 4.93 (s, 1H); 13C-NMR (150 MHz, CDCl3): δ 169.0, 159.9 (d, J = 241.4 Hz, C-F), 153.0, 141.8 (d, J = 10.8 Hz), 140.2, 138.2 (d, J = 2.6 Hz), 131.9, 131.6, 130.6, 129.5, 128.8, 128.3, 128.1, 121.1, 120.2 (d, J = 9.0 Hz), 114.8 (d, J = 23.5 Hz), 113.3 (d, J = 22.9 Hz); IR (neat) cm−1: 3272.0 (w, NH), 3064.0 (w), 1598.2 (w), 15.72.3 (w), 1491.6 (w), 1463.7 (s), 1318.2 (w), 1258.0 (s), 1206.4 (w), 1138.9 (w), 1119.2 (m), 1100.9 (w), 976.8 (w), 954.8 (w), 909.0 (w), 868.1 (m), 820.2 (m), 774.6 (w), 755.5 (m), 721.9 (s), 692.3(s); LC-MS: tr = 5.15, m/z = 322.84 (C19H12ClFN2)+.
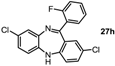
2,7-Dichloro-11-(2-fluorophenyl)-5H-dibenzo[b,e][1,4]diazepine (27h). Prepared from 4-chloro-2-fluoro-1-nitrobenzene and 2-amino-4-chloro-2’-fluoro-benzophenone (105 mg, 33% yield). 1H-NMR (400 MHz, CDCl3): δ/ppm = 7.67 (td, J = 7.6 Hz, 1.9 Hz, 1H), 7.46–7.40 (m, 1H), 7.25 (dd, J = 7.5 Hz, 1.0 Hz, 1H), 7.22–7.19 (m, 1H), 7.15 (d, J = 8.5 Hz, 1H), 7.09–7.04 (m, 1H), 6.98 (dd, J = 8.6 Hz, 2.0 Hz, 1H), 6.83 (d, J = 2.4 Hz, 1H), 6.82–6.80 (m, 1H), 5.83 (s, 1H); 13C-NMR (100 MHz, CDCl3): δ 164.9 (C), 160.5 (d, J = 250.0 Hz, C-F), 151.1 (C), 143.6 (C), 138.7 (C), 133.2 (C), 132.1 (CH), 131.7 (d, J = 8.3 Hz, CH), 131.3 (d, J = 2.5 Hz, CH), 130.1 (CH), 130.1 (d, J = 1.7 Hz, C), 130.0 (C), 129.0 (d, J = 12.4 Hz, C), 128.4 (C), 124.3 (d, J = 3.3 Hz, CH), 124.2 (CH), 121.2 (CH), 120.0 (CH), 116.2 (d, J = 17.5 Hz, CH); IR (neat) cm−1: 3235.0 (w), 2949.9 (w), 2663.3 (w), 1610.7 (m), 1482.9 (m), 1454.4 (s), 1389.8 (w), 1316.2 (w), 1290.5 (w), 1237.0 (w), 1161.9 (w), 1089.1 (w), 1035.0 (w), 919.2 (w), 838.7 (w), 756.5 (w); HRMS (m/z) calculated for C19H12N2Cl2F (M+H) 357.0356; found 357.0349.
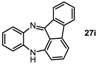
13H-Benzo[b]fluoreno[1,9-ef][1,4]diazepine (27i). Prepared from 1-fluoronitrobenzene and 2-aminofluorenone (120 mg, 51% yield). 1H-NMR (400 MHz, CDCl3): δ/ppm = 7.55 (d, J = 7.5 Hz, 1H), 7.28 (m, 2H), 7.20 (m, 1H), 6.85 (dd, J = 7.7 Hz, 1.5 Hz, 1H), 6.70 (t, J = 7.7 Hz, 1H), 6.66 (td, J = 7.7 Hz, 1.5 Hz, 1H), 6.45 (d, J = 7.4 Hz, 1H), 6.52 (dt, J = 7.4 Hz, 1.5 Hz, 1H), 5.82 (dd, J = 8.0 Hz, 1.5 Hz, 1H), 5.61 (d, J = 7.8 Hz, 1H), 4.41 (s, 1H, NH); 13C-NMR (100 MHz, CDCl3): δ 165.1 (C), 147.3 (C), 144.5 (C), 142.0 (C), 141.0 (C), 138.5 (C), 137.7 (C), 136.0 (CH), 135.4 (CH), 131.5 (CH), 130.9 (CH), 128.4 (CH), 123.3 (CH), 122.8 (CH), 121.8 (C), 120.2 (CH), 119.2 (CH), 114.3 (CH), 111.9 (CH). IR (neat) cm−1: 3400.1 (w), 3061.8 (w), 2925 (w), 1645.6 (w), 1623.4 (m), 1597.0 (m), 1466.0 (s), 1454.0 (m), 1434.5 (w), 1412.5 (w), 1321.8 (m), 1157.0 (w), 1058.7 (w), 958.5 (w), 779.1 (w), 747.5 (s). HRMS (m/z) calculated for C19H13N2 (M+H), 269.1073; found 269.1068.
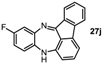
10-Fluoro-13H-benzo[b]fluoreno[1,9-ef][1,4]diazepine (27j). Prepared from 1,4-difluoro-2-nitrobenzene and 2-aminofluorenone (105 mg, 37% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 7.42 (dd, J = 7.2 Hz, 1.0 Hz, 1H), 7.37 (d, J = 1.0 Hz, 1H), 7.34 (m, 1H), 7.20 (td, J = 7.5 Hz, 1.0 Hz, 1H), 6.99 (m, 1H, NH), 6.70 (td, J = 7.5 Hz, 1.7 Hz, 1H), 6.53 (m, 1H), 6.46 (dd, J = 7.2 Hz, 0.7 Hz, 1H), 6.35 (dd, J = 9.8 Hz, 3.0 Hz, 1H), 6.02 (dd, J = 8.9 Hz, 5.8 Hz, 1H), 5.73 (dd, J = 7.5 Hz, 0.7 Hz, 1H); 13C-NMR (125 MHz, d6-DMSO): δ 166.0 (C), 157.6 (d, 295.2, C-F), 148.7 (C), 143.5 (C), 141.4 (C), 138.8 (d, J = 9.1 Hz, C), 138.7 (d, J = 3.3 Hz, C), 136.2 (CH), 136.0 (C), 131.9 (CH), 122.1 (CH), 120.8 (d, J = 28.4 Hz, CH), 120.4 (C), 120.4 (CH), 119.9 (d, J = 10.1 Hz, CH), 116.8 (d, J = 27.1 Hz, CH), 114.7 (C), 111.1 (CH). IR (neat) cm−1: 3390.0 (w, NH), 3060.0 (w), 2327.6 (w), 1650.0 (w), 1597.8 (s), 1468.1 (s), 1455.1 (m), 1421.7 (m), 1320.0 (w), 1266.3 (m), 1210.5 (w), 1160.3 (w), 974.7 (w), 957 (w), 878.5 (w), 749.4 (m). HRMS (m/z) calculated for C19H12N2F (M+H), 287.0979; found 287.0972.
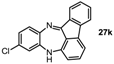
11-Chloro-13H-benzo[b]fluoreno[1,9-ef][1,4]diazepine (27k). Prepared from 4-chloro-2-fluoro-1-nitrobenzene and 2-aminofluorenone (105 mg, 42% yield). 1H-NMR (500 MHz, d6-DMSO): δ/ppm = 7.43 (dd, J = 7.1 Hz, 1.0 Hz, 1H), 7.37 (d, J = 1.4 Hz, 1H), 7.34 (dd, J = 7.8 Hz, 1.0 Hz, 1H), 7.21 (td, J = 7.5 Hz, 1.0 Hz, 1H), 7.18 (m, 1H, NH), 6.73 (t, J = 8.0 Hz, 1H), 6.56 (d, J = 8.1 Hz, 1H), 6.52 (d, J = 7.5 Hz, 1H), 6.41 (dd, J = 8.5 Hz, 2.4 Hz, 1H), 6.09 (d, J = 2.4 Hz, 1H), 5.73 (dd, J = 8.2 Hz, 0.7 Hz, 1H); 13C-NMR (125 MHz, d6-DMSO): δ 164.2 (C), 147.4 (C), 143.4 (C), 1471.3 (C), 136.6 (CH), 136.5 (C), 136.0 (C), 135.9 (CH), 134.8 (C), 131.8 (CH), 128.3 (CH), 122.0 (CH), 121.4 (CH), 120.8 (C), 120.5 (CH), 118.2 (CH), 114.7 (CH), 111.81 (CH). IR (neat) cm−1: 3395.1 (w, NH), 2926.3 (m), 2851.7 (m), 2308.1 (w), 1720.5 (w), 1650.0 (w), 1627.2 (w), 1598.5 (m), 1575.6 (m), 1464.8 (s), 1422.2 (m), 1314.2 (w), 1104.5 (w), 966.6 (w), 806.7 (w), 778.7 (w), 750.4 (m). HRMS (m/z) calculated for C19H12N2Cl (M+H), 303.0684; found 303.0675.
Crystallographic data for compounds
27b,
27d and
27e have been deposited with the accession numbers CCDC 867822, 867823 and 867824contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via
http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail:
deposit@ccdc.cam.ac.uk)


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}