Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design

Abstract
:1. Introduction

| Portion of Drugs | Family of Drug Target |
|---|---|
| 26.8% | Rhodopsin-like GPCRs |
| 13.0% | Nuclear receptors |
| 7.9% | Ligand-gated ion channels |
| 5.5% | Voltage-gated ion channels |
| 4.1% | Penicillin-binding protein |
| 3.0% | Myeloperoxidase-like |
| 2.7% | Sodium: neurotransmitter symporter family |
| 2.3% | Type II DNA topoisomerase |
| ≈35% | (other) |
2. Challenges in Protein-Ligand Docking
2.1. Scoring Methods
2.1.1. Force-Field-Based Potentials
2.1.2. Empirical Scoring Functions
2.1.3. Statistical Potentials
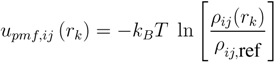
2.1.4. Summary
2.2. Sampling Methods
2.3. Recent Topics
2.3.1. Structural Water
2.3.2. Ligand Promiscuity
2.3.3. Accurate Models of the Protein Receptor
3. Protein-Ligand Docking Approaches
3.1. Screening for New Inhibitors
3.2. Hybrid Approaches for Drug Design
3.3. Mechanistic Studies Using Inverse Docking
4. Docking Benchmarks and Evaluation
4.1. Making Testable Predictions
4.2. Assuming Lack of Knowledge of the Native, Bound Conformation
4.3. Assessing Binding Mode Predictions Involving Symmetric Molecules
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Liljefors, T.; Krogsgaard-Larsen, P.; Madsen, U. Textbook of Drug Design and Discovery, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Khanna, I. Drug discovery in pharmaceutical industry: Productivity challenges and trends. Drug Discov. Today 2012, 17, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar]
- Schneider, G.; Böhm, H.J. Virtual screening and fast automated docking methods. Drug Discov. Today 2002, 7, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Scior, T.; Bender, A.; Tresadern, G.; Medina-Franco, J.L.; Martínez-Mayorga, K.; Langer, T.; Cuanalo-Contreras, K.; Agrafiotis, D.K. Recognizing pitfalls in virtual screening: A critical review. J. Chem. Inf. Model. 2012, 52, 867–881. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Martin, Y.C. The Information Content of 2D and 3D Structural Descriptors Relevant to Ligand-Receptor Binding. J. Chem. Inf. Comput. Sci. 1997, 37, 1–9. [Google Scholar]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Brooijmans, N.; Kuntz, I.D. Molecular recognition and docking algorithms. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 335–373. [Google Scholar] [PubMed]
- Leach, A.R.; Shoichet, B.K.; Peishoff, C.E. Prediction of protein-ligand interactions. Docking and scoring: Successes and gaps. J. Med. Chem. 2006, 49, 5851–5855. [Google Scholar]
- Huang, S.Y.; Zou, X. Advances and Challenges in Protein-Ligand Docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [PubMed]
- Hansch, C. The physicochemical approach to drug design and discovery (QSAR). Drug Dev. Res. 1981, 1, 267–309. [Google Scholar] [CrossRef]
- Ortiz, A.R.; Pisabarro, M.T.; Gago, F.; Wade, R.C. Prediction of drug binding affinities by comparative binding energy analysis. J. Med. Chem. 1995, 38, 2681–2691. [Google Scholar] [PubMed]
- Cereto-Massagué, A.; Ojeda, M.J.; Joosten, R.P.; Valls, C.; Mulero, M.; Salvado, M.J.; Arola-Arnal, A.; Arola, L.; Garcia-Vallvé, S.; Pujadas, G. The good,the bad and the dubious: VHELIBS,a validation helper for ligands and binding sites. J. Cheminform. 2013, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Hetényi, C.; van der Spoel, D. Toward prediction of functional protein pockets using blind docking and pocket search algorithms. Protein Sci. 2011, 20, 880–893. [Google Scholar] [CrossRef] [PubMed]
- DesJarlais, R.L.; Sheridan, R.P.; Seibel, G.L.; Dixon, J.S.; Kuntz, I.D.; Venkataraghavan, R. Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure. J. Med. Chem. 1988, 31, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Meng, E.C.; Shoichet, B.K. Structure-Based Molecular Design. Acc. Chem. Res. 1994, 27, 117–123. [Google Scholar] [CrossRef]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, D.T.; Lang, P.T.; Pegg, S.; Pettersen, E.; Kuntz, I.D.; Brooijmans, N.; Rizzo, R.C. Development and validation of a modular,extensible docking program: DOCK 5. J. Comput. Aided Mol. Des. 2006, 20, 601–619. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to model RNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Böhm, H.J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T.; Klebe, G. A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 1996, 261, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX incremental construction algorithm for protein-ligand docking. Proteins 1999, 37, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K. Glide: A new approach for rapid,accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid,accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function,efficient optimization,and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [PubMed]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function to predict protein-ligand interactions: I. Derivation of interaction potentials. J. Comput. Chem. 2006, 27, 1866–1875. [Google Scholar]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function to predict protein-ligand interactions: II. Validation of the scoring function. J. Comput. Chem. 2006, 27, 1876–1882. [Google Scholar]
- Huang, S.Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Proteins 2007, 66, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Efficient molecular docking of NMR structures: Application to HIV-1 protease. Protein Sci. 2007, 16, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein-ligand interactions. J. Chem. Inf. Model. 2010, 50, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.P.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Protein-ligand docking in the new millennium–a retrospective of 10 years in the field. Curr. Med. Chem. 2013, 20, 2296–2314. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Skolnick, J. FINDSITE(comb): A threading/structure-based,proteomic-scale virtual ligand screening approach. J. Chem. Inf. Model. 2013, 53, 230–240. [Google Scholar] [PubMed]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Zhou, H.X. Calculation of protein-ligand binding affinities. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 21–42. [Google Scholar] [PubMed]
- Huang, S.Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein-ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899–12908. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, O.; Estrada, T.P.; Doren, D.J.; Taufer, M.; Brooks, C.L., 3rd; Armen, R.S. Evaluation of several two-step scoring functions based on linear interaction energy, effective ligand size, and empirical pair potentials for prediction of protein-ligand binding geometry and free energy. J. Chem. Inf. Model. 2011, 51, 2047–2065. [Google Scholar]
- Nicolini, P.; Frezzato, D.; Gellini, C.; Bizzarri, M.; Chelli, R. Toward quantitative estimates of binding affinities for protein-ligand systems involving large inhibitor compounds: A steered molecular dynamics simulation route. J. Comput. Chem. 2013, 34, 1561–1576. [Google Scholar] [CrossRef] [PubMed]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins,Nucleic Acids,and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef]
- Jones, J.E. On the Determination of Molecular Fields. II. From the Equation of State of a Gas. Proc. R. Soc. Lond. A 1924, 106, 463–477. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Gilson, M.K.; Rashin, A.; Fine, R.; Honig, B. On the calculation of electrostatic interactions in proteins. J. Mol. Biol. 1985, 184, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.A.; Pickup, B.T.; Nicholls, A. A smooth permittivity function for Poisson–Boltzmann solvation methods. J. Comput. Chem. 2001, 22, 608–640. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Rocchia, W.; Sridharan, S.; Nicholls, A.; Alexov, E.; Chiabrera, A.; Honig, B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: Applications to the molecular systems and geometric objects. J. Comput. Chem. 2002, 23, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- Bashford, D.; Case, D.A. Generalized born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000, 51, 129–152. [Google Scholar] [PubMed]
- Hawkins, G.D.; Cramer, C.J.; Truhlar, D.G. Pairwise solute descreening of solute charges from a dielectric medium. Chem. Phys. Lett. 1995, 246, 122–129. [Google Scholar]
- Grycuk, T. Deficiency of the Coulomb-field approximation in the generalized Born model: An improved formula for Born radii evaluation. J. Chem. Phys. 2003, 119, 4817–4826. [Google Scholar]
- Feig, M.; Onufriev, A.; Lee, M.S.; Im, W.; Case, D.A.; Brooks, C.L., 3rd. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004, 25, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Zou, X. Electrostatics of ligand binding: Parametrization of the generalized Born model and comparison with the Poisson-Boltzmann approach. J. Phys. Chem. B 2006, 110, 9304–9313. [Google Scholar] [PubMed]
- Tjong, H.; Zhou, H.X. GBr(6): A parameterization-free,accurate,analytical generalized born method. J. Phys. Chem. B 2007, 111, 3055–3061. [Google Scholar] [PubMed]
- Srinivasan, J.; Miller, J.; Kollman, P.A.; Case, D.A. Continuum solvent studies of the stability of RNA hairpin loops and helices. J. Biomol. Struct. Dyn. 1998, 16, 671–682. [Google Scholar] [PubMed]
- Zou, X.; Yaxiong; Kuntz, I.D. Inclusion of Solvation in Ligand Binding Free Energy Calculations Using the Generalized-Born Model. J. Am. Chem. Soc. 1999, 121, 8033–8043. [Google Scholar] [CrossRef]
- Wang, J.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in Reproducing the Binding Free Energies to HIV-1 RT of TIBO Derivatives and Predicting the Binding Mode to HIV-1 RT of Efavirenz by Docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R. Free energy landscape of protein folding in water: Explicit vs. implicit solvent. Proteins 2003, 53, 148–161. [Google Scholar]
- Liu, H.Y.; Kuntz, I.D.; Zou, X. Pairwise GB/SA Scoring Function for Structure-based Drug Design. J. Phys. Chem. B 2004, 108, 5453–5462. [Google Scholar]
- Liu, H.Y.; Grinter, S.Z.; Zou, X. Multiscale generalized Born modeling of ligand binding energies for virtual database screening. J. Phys. Chem. B 2009, 113, 11793–11799. [Google Scholar] [PubMed]
- Purisima, E.O.; Hogues, H. Protein-ligand binding free energies from exhaustive docking. J. Phys. Chem. B 2012, 116, 6872–6879. [Google Scholar] [PubMed]
- Kollman, P. Free energy calculations: Applications to chemical and biochemical phenomena. Chem. Rev. 1993, 93, 2395–2417. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.J. Prediction of binding constants of protein ligands: A fast method for the prioritization of hits obtained from de novo design or 3D database search programs. J. Comput. Aided Mol. Des. 1998, 12, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lu, Y.; Wang, S. Comparative evaluation of 11 scoring functions for molecular docking. J. Med. Chem. 2003, 46, 2287–2303. [Google Scholar] [CrossRef] [PubMed]
- Temiz, N.A.; Trapp, A.; Prokopyev, O.A.; Camacho, C.J. Optimization of minimum set of protein-DNA interactions: A quasi exact solution with minimum over-fitting. Bioinformatics 2010, 26, 319–325. [Google Scholar] [PubMed]
- Wang, R.; Lai, L.; Wang, S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J. Comput. Aided Mol. Des. 2002, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Scheraga, H.A. Model of protein folding: Incorporation of a one-dimensional short-range (Ising) model. Proc. Natl. Acad. Sci. USA 1977, 74, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, S.; Jernigan, R.L. Estimation of effective interresidue contact energies from protein crystal structures: Quasi-chemical approximation. Macromolecules 1985, 18, 534–552. [Google Scholar]
- Thomas, P.D.; Dill, K.A. Statistical potentials extracted from protein structures: How accurate are they? J. Mol. Biol. 1996, 257, 457–469. [Google Scholar] [CrossRef]
- Thomas, P.D.; Dill, K.A. An iterative method for extracting energy-like quantities from protein structures. Proc. Natl. Acad. Sci. USA 1996, 93, 11628–11633. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Chapter 14—Mean-Force Scoring Functions for Protein–Ligand Binding. In Annual Reports in Computational Chemistry; Wheeler, R.A., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; Volume 6, pp. 280–296. [Google Scholar]
- Muegge, I.; Martin, Y.C.; Hajduk, P.J.; Fesik, S.W. Evaluation of PMF scoring in docking weak ligands to the FK506 binding protein. J. Med. Chem. 1999, 42, 2498–2503. [Google Scholar] [CrossRef] [PubMed]
- Sippl, M.J.; Ortner, M.; Jaritz, M.; Lackner, P.; Flöckner, H. Helmholtz free energies of atom pair interactions in proteins. Fold Des. 1996, 1, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, J. Knowledge-based energy functions for computational studies of proteins. In Computational Methods for Protein Structure Prediction and Modeling; Springer: New York, NY, USA, 2007; pp. 71–123. [Google Scholar]
- Munson, P.J.; Singh, R.K. Statistical significance of hierarchical multi-body potentials based on Delaunay tessellation and their application in sequence-structure alignment. Protein Sci. 1997, 6, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.T.; Leelananda, S.P.; Kloczkowski, A.; Jernigan, R.L. Combining statistical potentials with dynamics-based entropies improves selection from protein decoys and docking poses. J. Phys. Chem. B 2012, 116, 6725–6731. [Google Scholar] [PubMed]
- Jernigan, R.L.; Bahar, I. Structure-derived potentials and protein simulations. Curr. Opin. Struct. Biol. 1996, 6, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Skolnick, J. How do potentials derived from structural databases relate to “true” potentials? Protein Sci. 1998, 7, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Martin, Y.C. A general and fast scoring function for protein-ligand interactions: A simplified potential approach. J. Med. Chem. 1999, 42, 791–804. [Google Scholar] [PubMed]
- Zhou, H.; Zhou, Y. Distance-scaled,finite ideal-gas reference state improves structure-derived potentials of mean force for structure selection and stability prediction. Protein Sci. 2002, 11, 2714–2726. [Google Scholar] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins 2008, 72, 557–579. [Google Scholar] [CrossRef] [PubMed]
- Ravikant, D.V.S.; Elber, R. Energy design for protein-protein interactions. J. Chem. Phys. 2011, 135, 065102. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. A knowledge-based scoring function for protein-RNA interactions derived from a statistical mechanics-based iterative method. Nucleic Acids Res. 2014. [CrossRef]
- Sippl, M.J. Calculation of conformational ensembles from potentials of mean force. An approach to the knowledge-based prediction of local structures in globular proteins. J. Mol. Biol. 1990, 213, 859–883. [Google Scholar]
- Grinter, S.Z.; Zou, X. A Bayesian statistical approach of improving knowledge-based scoring functions for protein-ligand interactions. J. Comput. Chem. 2014, 35, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Zhu, Q.; Zhou, Y. A knowledge-based energy function for protein-ligand,protein-protein,and protein-DNA complexes. J. Med. Chem. 2005, 48, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Gohlke, H.; Hendlich, M.; Klebe, G. Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 2000, 295, 337–356. [Google Scholar] [CrossRef] [PubMed]
- Velec, H.F.G.; Gohlke, H.; Klebe, G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 2005, 48, 6296–6303. [Google Scholar] [CrossRef] [PubMed]
- DeWitte, R.; Shakhnovich, E. SMoG: De Novo Design Method Based on Simple,Fast,and Accurate Free Energy Estimates. 1. Methodology and Supporting Evidence. J. Am. Chem. Soc. 1996, 118, 11733–11744. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Advances and Challenges in Protein-Ligand Docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Corkery, J.J.; Murcko, M.A.; Walters, W.P. Consensus scoring: A method for obtaining improved hit rates from docking databases of three-dimensional structures into proteins. J. Med. Chem. 1999, 42, 5100–5109. [Google Scholar] [CrossRef] [PubMed]
- Terp, G.E.; Johansen, B.N.; Christensen, I.T.; Jørgensen, F.S. A new concept for multidimensional selection of ligand conformations (MultiSelect) and multidimensional scoring (MultiScore) of protein-ligand binding affinities. J. Med. Chem. 2001, 44, 2333–2343. [Google Scholar] [PubMed]
- Plewczynski, D.; Łazniewski, M.; von Grotthuss, M.; Rychlewski, L.; Ginalski, K. VoteDock: Consensus docking method for prediction of protein-ligand interactions. J. Comput. Chem. 2011, 32, 568–581. [Google Scholar]
- Erickson, J.A.; Jalaie, M.; Robertson, D.H.; Lewis, R.A.; Vieth, M. Lessons in molecular recognition: The effects of ligand and protein flexibility on molecular docking accuracy. J. Med. Chem. 2004, 47, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Kuntz, I.D.; Bodian, D.L. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R.; Kuntz, I.D. Conformational analysis of flexible ligands in macromolecular receptor sites. J. Comput. Chem. 1992, 13, 730–748. [Google Scholar] [CrossRef]
- Lorber, D.M.; Shoichet, B.K. Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 2005, 5, 739–749. [Google Scholar] [PubMed]
- Damm, K.L.; Carlson, H.A. Exploring experimental sources of multiple protein conformations in structure-based drug design. J. Am. Chem. Soc. 2007, 129, 8225–8235. [Google Scholar] [CrossRef] [PubMed]
- Bottegoni, G.; Kufareva, I.; Totrov, M.; Abagyan, R. Four-dimensional docking: A fast and accurate account of discrete receptor flexibility in ligand docking. J. Med. Chem. 2009, 52, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Apostolakis, J.; Plückthun, A.; Caflisch, A. Docking small ligands in flexible binding sites. J. Comput. Chem. 1998, 19, 21–37. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, V.; Marinelli, L.; Cosconati, S.; La Motta, C.; Sartini, S.; Mugnaini, L.; Da Settimo, F.; Novellino, E.; Parrinello, M. Sampling protein motion and solvent effect during ligand binding. Proc. Natl. Acad. Sci. USA 2012, 109, 1467–1472. [Google Scholar] [PubMed]
- Rarey, M.; Kramer, B.; Lengauer, T. The particle concept: Placing discrete water molecules during protein-ligand docking predictions. Proteins 1999, 34, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Sahai, M.A.; Biggin, P.C. Quantifying water-mediated protein-ligand interactions in a glutamate receptor: A DFT study. J. Phys. Chem. B 2011, 115, 7085–7096. [Google Scholar] [PubMed]
- Lie, M.A.; Thomsen, R.; Pedersen, C.N.S.; Schiøtt, B.; Christensen, M.H. Molecular docking with ligand attached water molecules. J. Chem. Inf. Model. 2011, 51, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, X.; Zhang, J.Z.H. Improving the scoring of protein-ligand binding affinity by including the effects of structural water and electronic polarization. J. Chem. Inf. Model. 2013, 53, 1306–1314. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Bradley, P.; Baker, D. Protein-protein docking with backbone flexibility. J. Mol. Biol. 2007, 373, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, G.; Meiler, J. Rosetta Ligand docking with flexible XML protocols. Methods Mol. Biol. 2012, 819, 143–155. [Google Scholar] [PubMed]
- Huggins, D.J.; Tidor, B. Systematic placement of structural water molecules for improved scoring of protein-ligand interactions. Protein Eng. Des. Sel. 2011, 24, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Taboureau, O.; Jørgensen, F.S. In silico predictions of hERG channel blockers in drug discovery: From ligand-based and target-based approaches to systems chemical biology. Comb. Chem. High Throughput Screen. 2011, 14, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, R.; Deeds, E.J.; Karanicolas, J. Structural properties of non-traditional drug targets present new challenges for virtual screening. J. Chem. Inf. Model. 2013, 53, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Nueno, V.I.; Ritchie, D.W. Using consensus-shape clustering to identify promiscuous ligands and protein targets and to choose the right query for shape-based virtual screening. J. Chem. Inf. Model. 2011, 51, 1233–1248. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Lin, X.; Guo, Z.; Huang, N. Identifying multiple-target ligands via computational chemogenomics approaches. Curr. Top. Med. Chem. 2012, 12, 1363–1375. [Google Scholar] [PubMed]
- Shrinivasan, M.; Skariyachan, S.; Aparna, V.; Kolte, V.R. Homology modelling of CB1 receptor and selection of potential inhibitor against Obesity. Bioinformation 2012, 8, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Skariyachan, S.; Mahajanakatti, A.B.; Sharma, N.; Karanth, S.; Rao, S.; Rajeswari, N. Structure based virtual screening of novel inhibitors against multidrug resistant superbugs. Bioinformation 2012, 8, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Skariyachan, S.; Prakash, N.; Bharadwaj, N. In silico exploration of novel phytoligands against probable drug target of Clostridium tetani. Interdiscip. Sci. 2012, 4, 273–281. [Google Scholar] [PubMed]
- Kar, R.K.; Ansari, M.Y.; Suryadevara, P.; Sahoo, B.R.; Sahoo, G.C.; Dikhit, M.R.; Das, P. Computational elucidation of structural basis for ligand binding with Leishmania donovani adenosine kinase. Biomed. Res. Int. 2013, 2013, 609289:1–609289:14. [Google Scholar]
- Tahir, R.A.; Sehgal, S.A.; Khattak, N.A.; Khan Khattak, J.Z.; Mir, A. Tumor necrosis factor receptor superfamily 10B (TNFRSF10B): An insight from structure modeling to virtual screening for designing drug against head and neck cancer. Theor. Biol. Med. Model. 2013, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Skariyachan, S.; Jayaprakash, N.; Bharadwaj, N.; Narayanappa, R. Exploring insights for virulent gene inhibition of multidrug resistant Salmonella typhi,Vibrio cholerae,and Staphylococcus areus by potential phytoligands via in silico screeningd. J. Biomol. Struct. Dyn. 2014, 32, 1379–1395. [Google Scholar] [CrossRef] [PubMed]
- Merlino, A.; Vieites, M.; Gambino, D.; Laura Coitiño, E. Homology modeling of T. cruzi and L. major NADH-dependent fumarate reductases: Ligand docking, molecular dynamics validation, and insights on their binding modes. J. Mol. Graph. Model. 2014, 48, 47–59. [Google Scholar]
- Orry, A.J.W.; Abagyan, R. Preparation and refinement of model protein-ligand complexes. Methods Mol. Biol. 2012, 857, 351–373. [Google Scholar] [PubMed]
- Combs, S.A.; Deluca, S.L.; Deluca, S.H.; Lemmon, G.H.; Nannemann, D.P.; Nguyen, E.D.; Willis, J.R.; Sheehan, J.H.; Meiler, J. Small-molecule ligand docking into comparative models with Rosetta. Nat. Protoc. 2013, 8, 1277–1298. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.W.; Meiler, J. Using RosettaLigand for small molecule docking into comparative models. PLoS One 2012, 7, e50769. [Google Scholar] [PubMed]
- Mahasenan, K.V.; Li, C. Novel inhibitor discovery through virtual screening against multiple protein conformations generated via ligand-directed modeling: A maternal embryonic leucine zipper kinase example. J. Chem. Inf. Model. 2012, 52, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminform. 2012, 4, P17. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Li, Y.; Cao, R.; Zhong, W.; Zheng, Z.; Wang, G.; Xiao, J.; Li, S. Novel substituted heteroaromatic piperazine and piperidine derivatives as inhibitors of human enterovirus 71 and coxsackievirus a16. Molecules 2013, 18, 5059–5071. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [PubMed]
- Ahmed, L.; Rasulev, B.; Turabekova, M.; Leszczynska, D.; Leszczynski, J. Receptor-and ligand-based study of fullerene analogues: comprehensive computational approach including quantum-chemical,QSAR and molecular docking simulations. Org. Biomol. Chem. 2013, 11, 5798–5808. [Google Scholar] [CrossRef] [PubMed]
- Ballante, F.; Caroli, A.; Wickersham, Richard B, r.; Ragno, R. Hsp90 Inhibitors, Part 1: Definition of 3-D QSAutogrid/R Models as a Tool for Virtual Screening. J. Chem. Inf. Model. 2014, 54, 956–969. [Google Scholar]
- Caroli, A.; Ballante, F.; Wickersham, R.B., 3rd; Corelli, F.; Ragno, R. Hsp90 Inhibitors, Part 2: Combining Ligand-Based and Structure-Based Approaches for Virtual Screening Application. J. Chem. Inf. Model. 2014, 54, 970–977. [Google Scholar]
- Alcaro, S.; Musetti, C.; Distinto, S.; Casatti, M.; Zagotto, G.; Artese, A.; Parrotta, L.; Moraca, F.; Costa, G.; Ortuso, F.; et al. Identification and characterization of new DNA G-quadruplex binders selected by a combination of ligand and structure-based virtual screening approaches. J. Med. Chem. 2013, 56, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Grinter, S.Z.; Liang, Y.; Huang, S.Y.; Hyder, S.M.; Zou, X. An inverse docking approach for identifying new potential anti-cancer targets. J. Mol. Graph. Model. 2011, 29, 795–799. [Google Scholar] [PubMed]
- Chen, Y.Z.; Zhi, D.G. Ligand-protein inverse docking and its potential use in the computer search of protein targets of a small molecule. Proteins 2001, 43, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.; Kellenberger, E.; Bret, G.; Müller, P.; Rognan, D. Recovering the true targets of specific ligands by virtual screening of the protein data bank. Proteins 2004, 54, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Li, H.; Zhang, H.; Liu, X.; Kang, L.; Luo, X.; Zhu, W.; Chen, K.; Wang, X.; Jiang, H. PDTD: A web-accessible protein database for drug target identification. BMC Bioinform. 2008, 9, 104. [Google Scholar] [CrossRef]
- Kumar, S.P.; Pandya, H.A.; Desai, V.H.; Jasrai, Y.T. Compound prioritization from inverse docking experiment using receptor-centric and ligand-centric methods: A case study on Plasmodium falciparum Fab enzymes. J. Mol. Recognit. 2014, 27, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Setzer, W.N. In-silico Leishmania target selectivity of antiparasitic terpenoids. Molecules 2013, 18, 7761–7847. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Fang, X.; Lu, Y.; Wang, S. The PDBbind database: Collection of binding affinities for protein-ligand complexes with known three-dimensional structures. J. Med. Chem. 2004, 47, 2977–2980. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Construction and test of ligand decoy sets using MDock: Community structure-activity resource benchmarks for binding mode prediction. J. Chem. Inf. Model. 2011, 51, 2107–2114. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, J.B.; Smith, R.D.; Yang, C.Y.; Ung, P.M.U.; Lexa, K.W.; Khazanov, N.A.; Stuckey, J.A.; Wang, S.; Carlson, H.A. CSAR Benchmark Exercise of 2010: Selection of the Protein-Ligand Complexes. J. Chem. Inf. Model. 2011, 51, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, K.Y.J. Computational fragment-based screening using RosettaLigand: The SAMPL3 challenge. J. Comput. Aided Mol. Des. 2012, 26, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, J.B., Jr.; Smith, R.D.; Damm-Ganamet, K.L.; Ahmed, A.; Esposito, E.X.; Delproposto, J.; Chinnaswamy, K.; Kang, Y.N.; Kubish, G.; Gestwicki, J.E.; et al. CSAR data set release 2012: Ligands,affinities,complexes,and docking decoys. J. Chem. Inf. Model. 2013, 53, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Skillman, A.G.; Geballe, M.T.; Nicholls, A. SAMPL2 challenge: Prediction of solvation energies and tautomer ratios. J. Comput. Aided Mol. Des. 2010, 24, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Grinter, S.Z.; Yan, C.; Huang, S.Y.; Jiang, L.; Zou, X. Automated large-scale file preparation,docking,and scoring: Evaluation of ITScore and STScore using the 2012 Community Structure-Activity Resource benchmark. J. Chem. Inf. Model. 2013, 53, 1905–1914. [Google Scholar] [CrossRef] [PubMed]
- Bolia, A.; Gerek, Z.N.; Ozkan, S.B. BP-Dock: A Flexible Docking Scheme for Exploring Protein-Ligand Interactions Based on Unbound Structures. J. Chem. Inf. Model. 2014, 54, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Ten Brink, T.; Victor Paul Raj, F.R.D.; Keil, M.; Exner, T.E. Are predefined decoy sets of ligand poses able to quantify scoring function accuracy? J. Comput. Aided Mol. Des. 2012, 26, 185–197. [Google Scholar] [CrossRef]
- Vajda, S.; Hall, D.R.; Kozakov, D. Sampling and scoring: A marriage made in heaven. Proteins 2013, 81, 1874–1884. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Rizzo, R.C. Implementation of the hungarian algorithm to account for ligand symmetry and similarity in structure-based design. J. Chem. Inf. Model. 2014, 54, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Head, M.S.; Given, J.A.; Gilson, M.K. “Mining Minima”: Direct Computation of Conformational Free Energy. J. Phys. Chem. A 1997, 101, 1609–1618. [Google Scholar]
- Ruvinsky, A.M. Role of binding entropy in the refinement of protein-ligand docking predictions: Analysis based on the use of 11 scoring functions. J. Comput. Chem. 2007, 28, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grinter, S.Z.; Zou, X. Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design. Molecules 2014, 19, 10150-10176. https://doi.org/10.3390/molecules190710150
Grinter SZ, Zou X. Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design. Molecules. 2014; 19(7):10150-10176. https://doi.org/10.3390/molecules190710150
Chicago/Turabian StyleGrinter, Sam Z., and Xiaoqin Zou. 2014. "Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design" Molecules 19, no. 7: 10150-10176. https://doi.org/10.3390/molecules190710150
APA StyleGrinter, S. Z., & Zou, X. (2014). Challenges, Applications, and Recent Advances of Protein-Ligand Docking in Structure-Based Drug Design. Molecules, 19(7), 10150-10176. https://doi.org/10.3390/molecules190710150