Radiolabeling Strategies for Tumor-Targeting Proteinaceous Drugs

Abstract
:1. Introduction
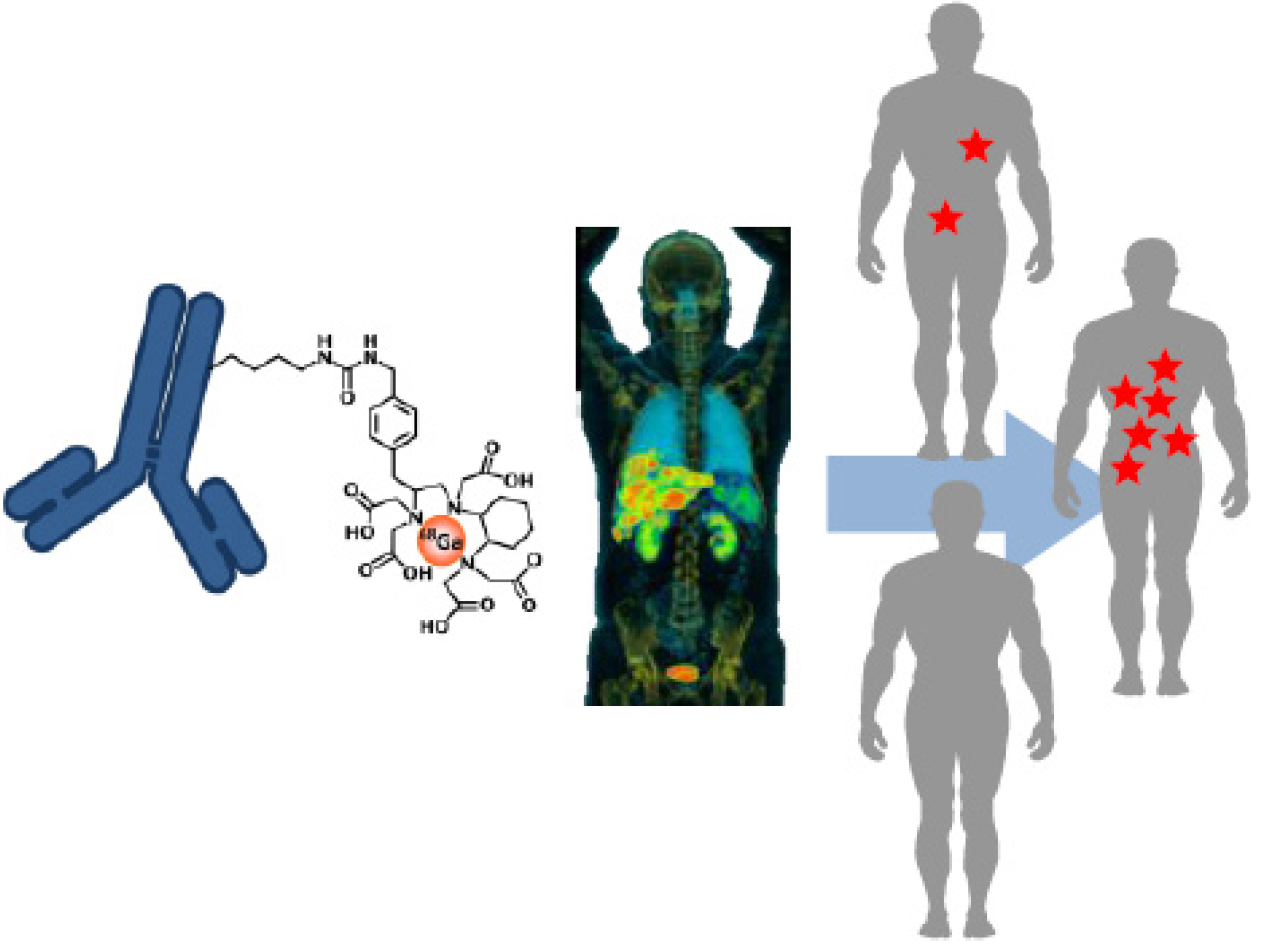
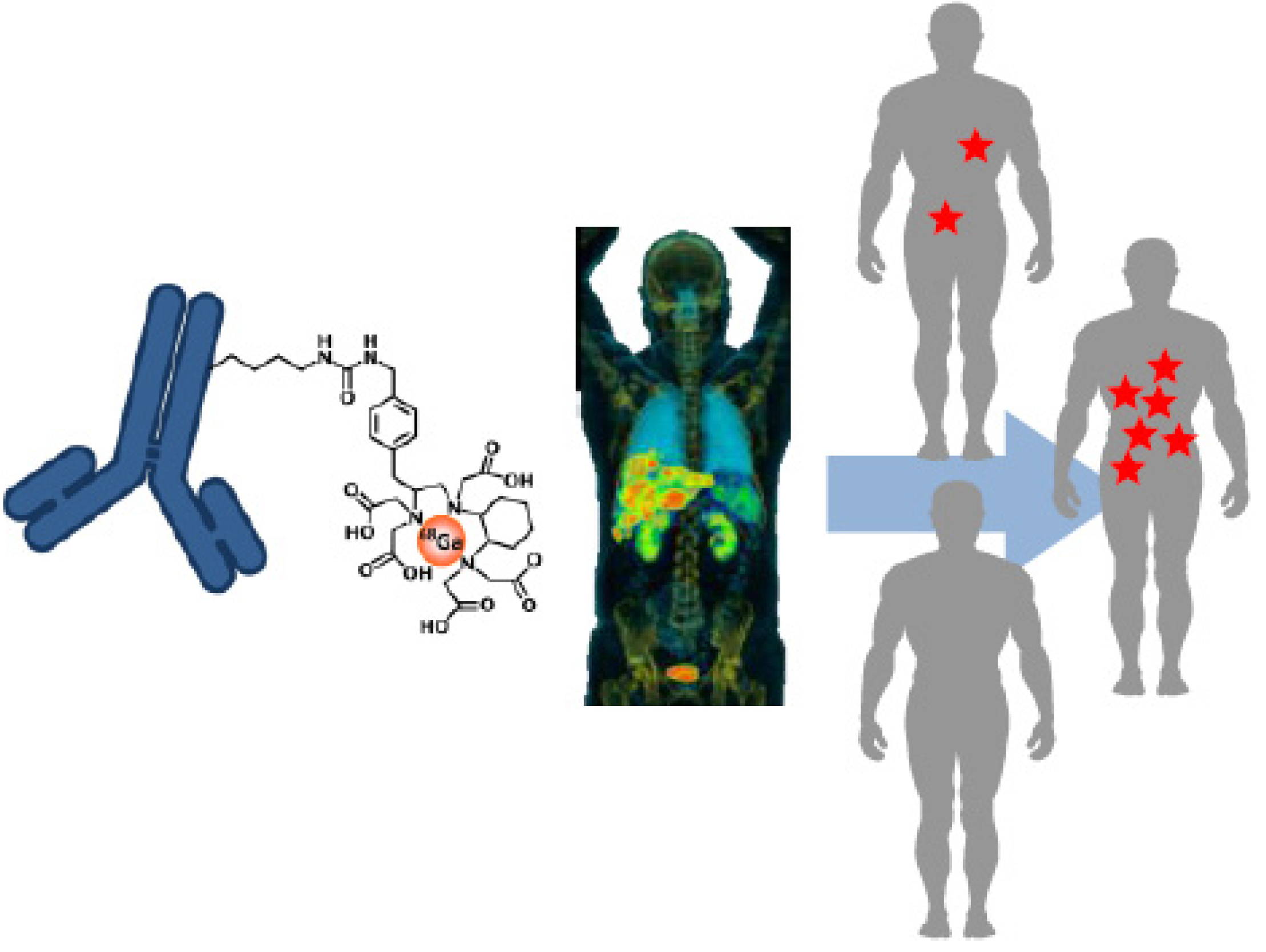
2. Molecular Imaging

{kind=link}
{kind=link}
{kind=link}
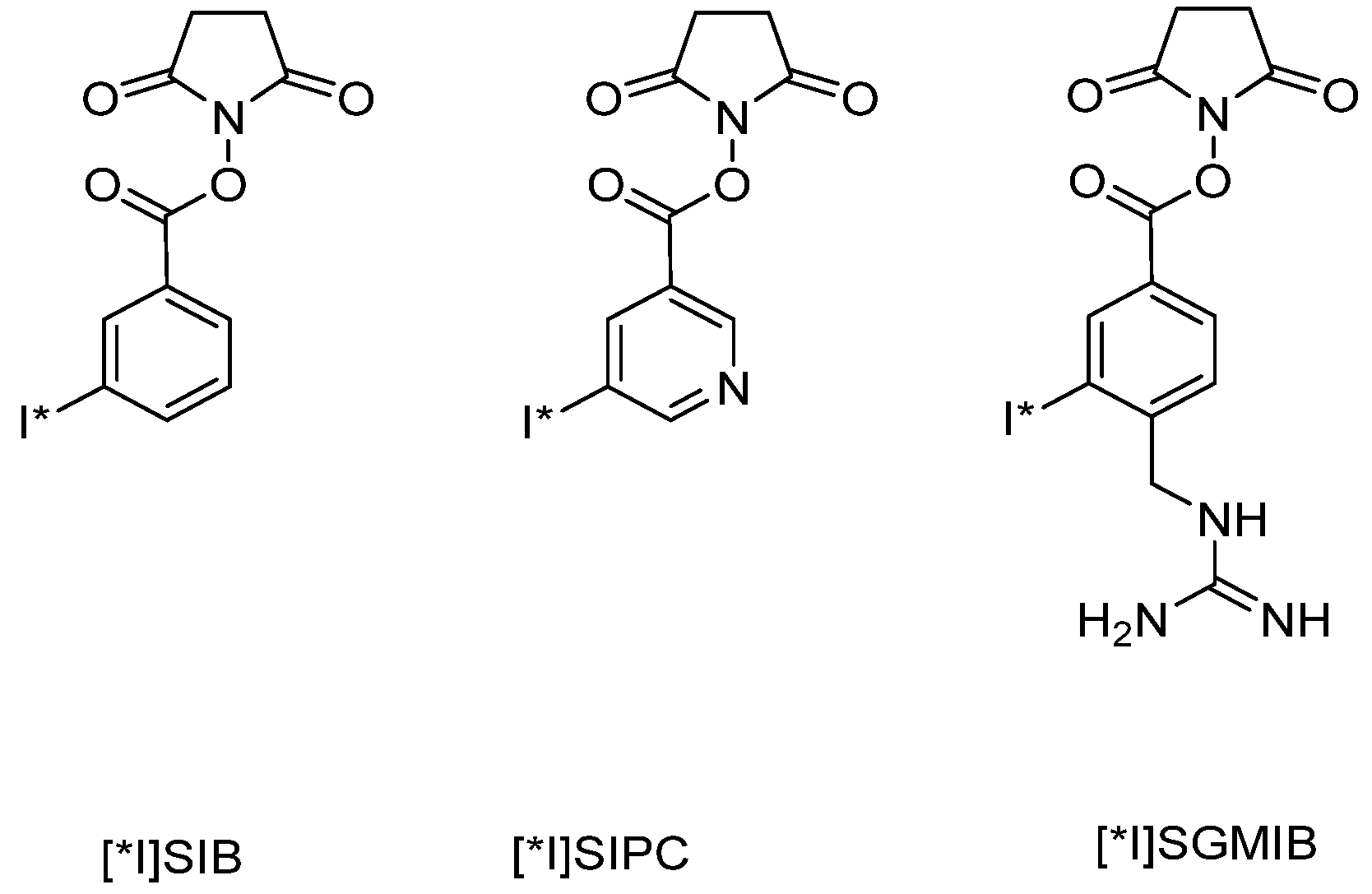{kind=link}
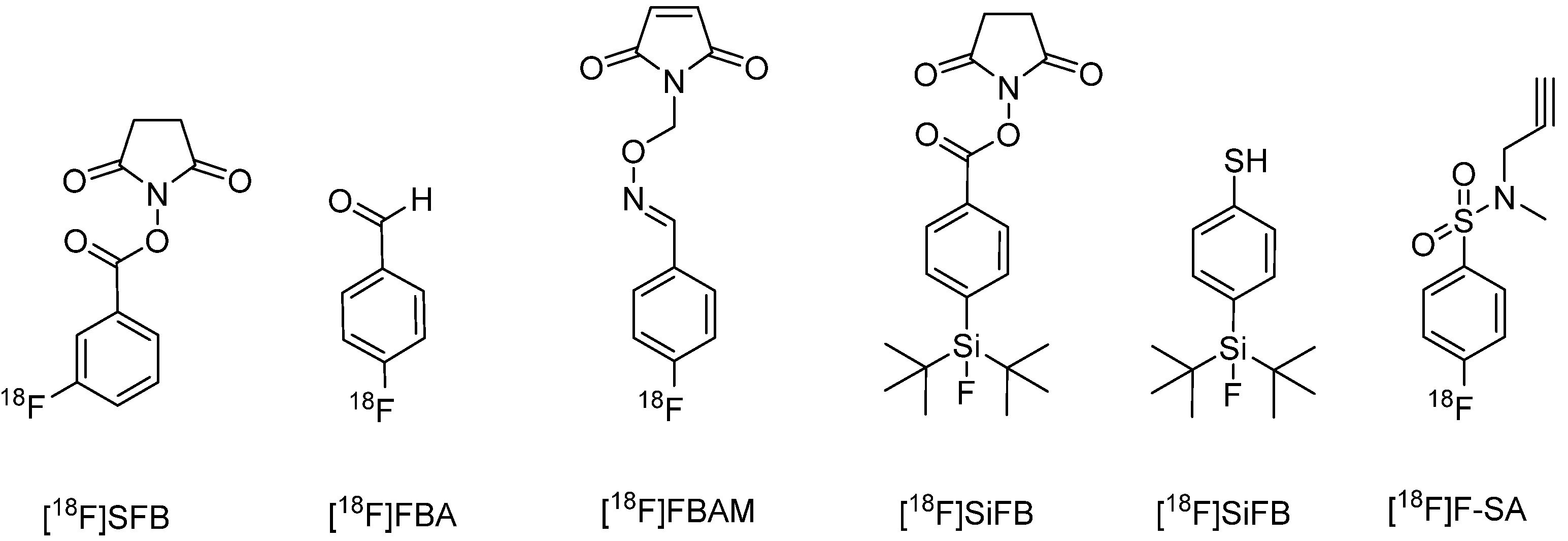{kind=link}
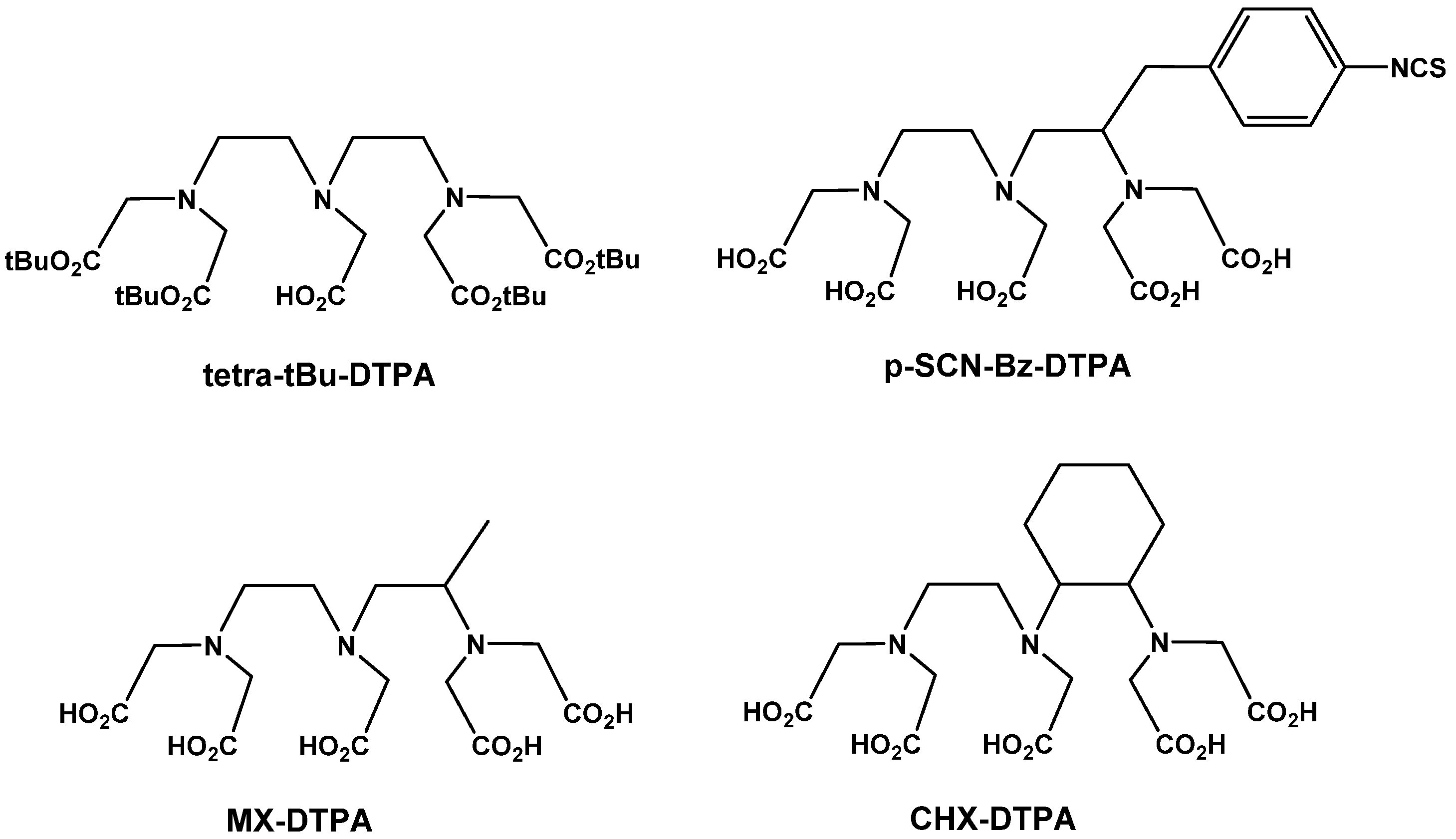{kind=link}
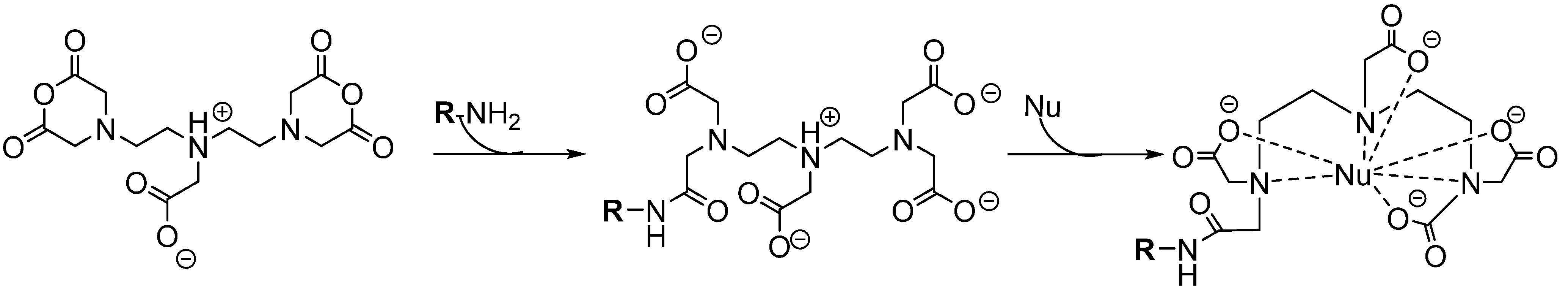{kind=link}
{kind=link}
| Radionuclide | Mode of Decay | Half-life | Emax (mean) |
|---|---|---|---|
| 18F | β+ | 109.8 min | 0.63 MeV |
| 68Ga | β+ | 68 min | 1.90 MeV |
| 99mTc | IC | 6.02 h | 0.14 MeV |
| 111In | EC | 2.8 d | 0.24 MeV |
| 123I | EC | 13.2 h | 0.16 MeV |
| Isotope | Half-life | Application |
|---|---|---|
| 123I | 13.2 h | SPECT imaging |
| 124I | 4.2 days | PET imaging |
| 125I | 59.4 days | Small animal imaging & in vivo studies |
| 131I | 8.0 days | SPECT imaging & therapy |
3. Radiotherapy
| Name | Protein | Antibody Form | Radionuclide attached | Target | Application | Clinical Trial | Method of Attachment |
|---|---|---|---|---|---|---|---|
| Rituximab | Anti-CD20 mAB | Chimeric | N/A | CD20 | B-cell non-Hodgkin’s lymphoma | FDA approved | N/A |
| Trastuzumab | Anti-HER2 mAB | Humanized | N/A | HER2 | HER2 positive breast cancer | FDA approved | N/A |
| Pertuzumab | Anti-HER2 mAB | Humanized | N/A | HER2 | HER2 Positive breast cancer | FDA approved | N/A |
| Avastin® (bevacizumab) | Anti-VEGF mAB | Humanized | N/A | VEGF-A | Angiogenesis inhibitor | FDA approved | N/A |
| Zevalin® (ibritumomab) | Anti-CD20 mAB | mu IgG1 | 90Y | CD20 | B-cell non-Hodgkin’s lymphoma | FDA approved | MX-DTPA |
| Bexxar® (tositumomab) | Anti-CD20 mAB | mu IgG2a | 131I | CD20 | B-cell non-Hodgkin's lymphoma | FDA approved | Iodination |
| Epratuzumab | Anti-CD22 mAB | Humanized | 90Y | CD22 | B-cell non-Hodgkin’s lymphoma | Phase I/II | DOTA |
| Veltuzumab | Anti-CD20 mAB | Humanized | N/A | CD20 | B-cell non-Hodgkin’s lymphoma | Phase I/II | N/A |
| Clivatuzumab | Anti-MUC1 | Humanized | 90Y | MUC1 | Pancreatic cancer | Phase I/II | DOTA |
4. Radionuclides
| Radionuclide | Mode of Decay | Half-life | Eav | Mean Tissue Range |
|---|---|---|---|---|
| 90Y | β | 2.7 d | 2.27 MeV | 2.76 mm |
| 131I | β,γ | 8.0 d | 0.61 MeV | 0.40 mm |
| 177Lu | β, γ | 6.7 d | 0.50 MeV | 0.28 mm |
| 225Ac | α, β | 10.0 d | 6.83 MeV | 0.04–0.1 mm |
| 213Bi | α | 47.5 min | 8.32 MeV | 0.04–0.1 mm |
| 212Bi | α | 1.0 h | 6.21 MeV | 0.04–0.1 mm |
| 211At | α | 7.2 h | 6.79 MeV | 0.04–0.1 mm |
| 212Pb | β | 10.6 h | 0.57 MeV | 0.6 mm |
5. Proteins as Site-Specific Drugs
5.1. Design of Molecular Imaging Agents Based on Proteins
5.2. Protein-Based Carrier System
6. Radiolabeling Strategies of Proteins
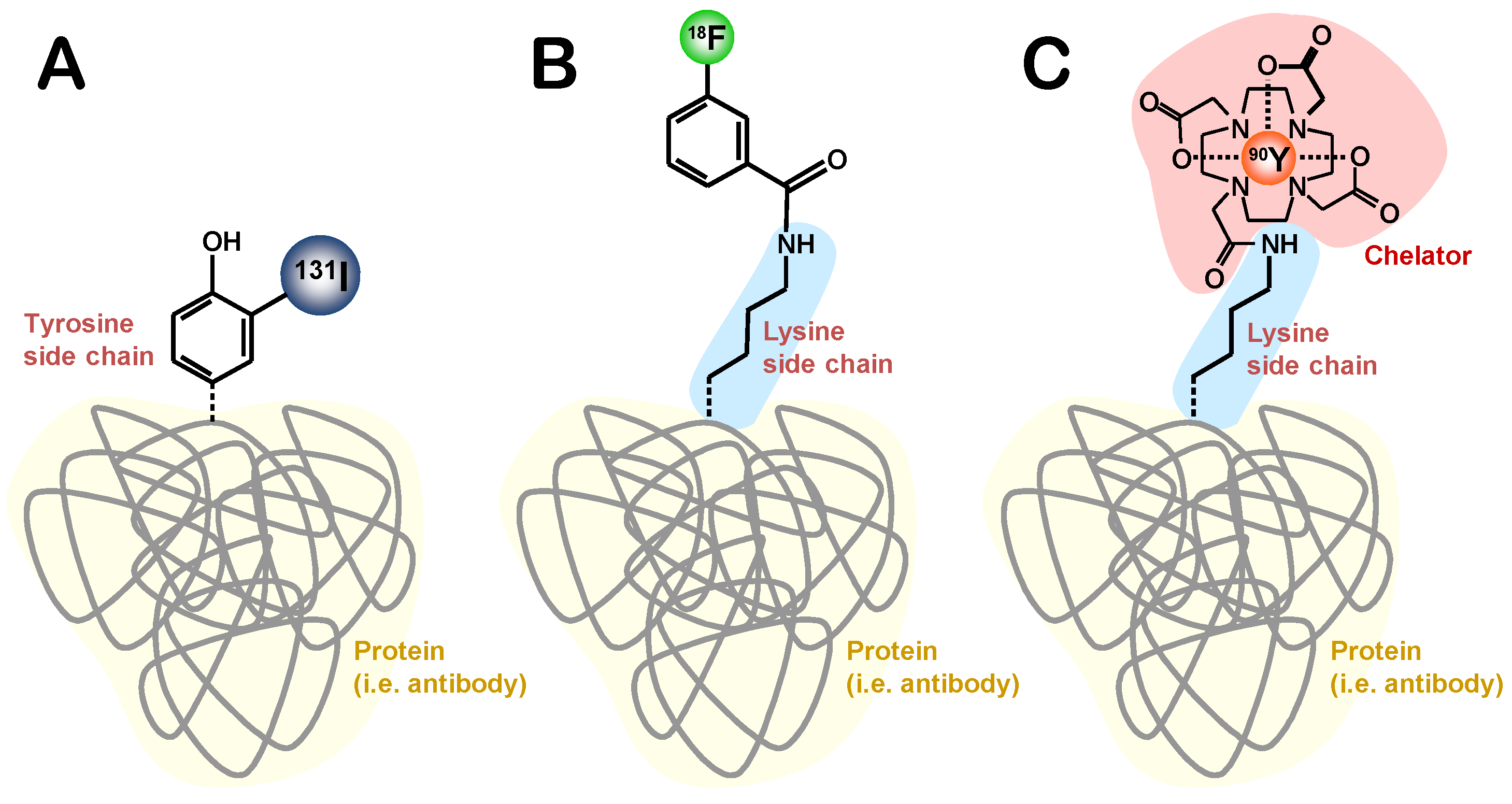
6.1. Methods of Radiolabelling
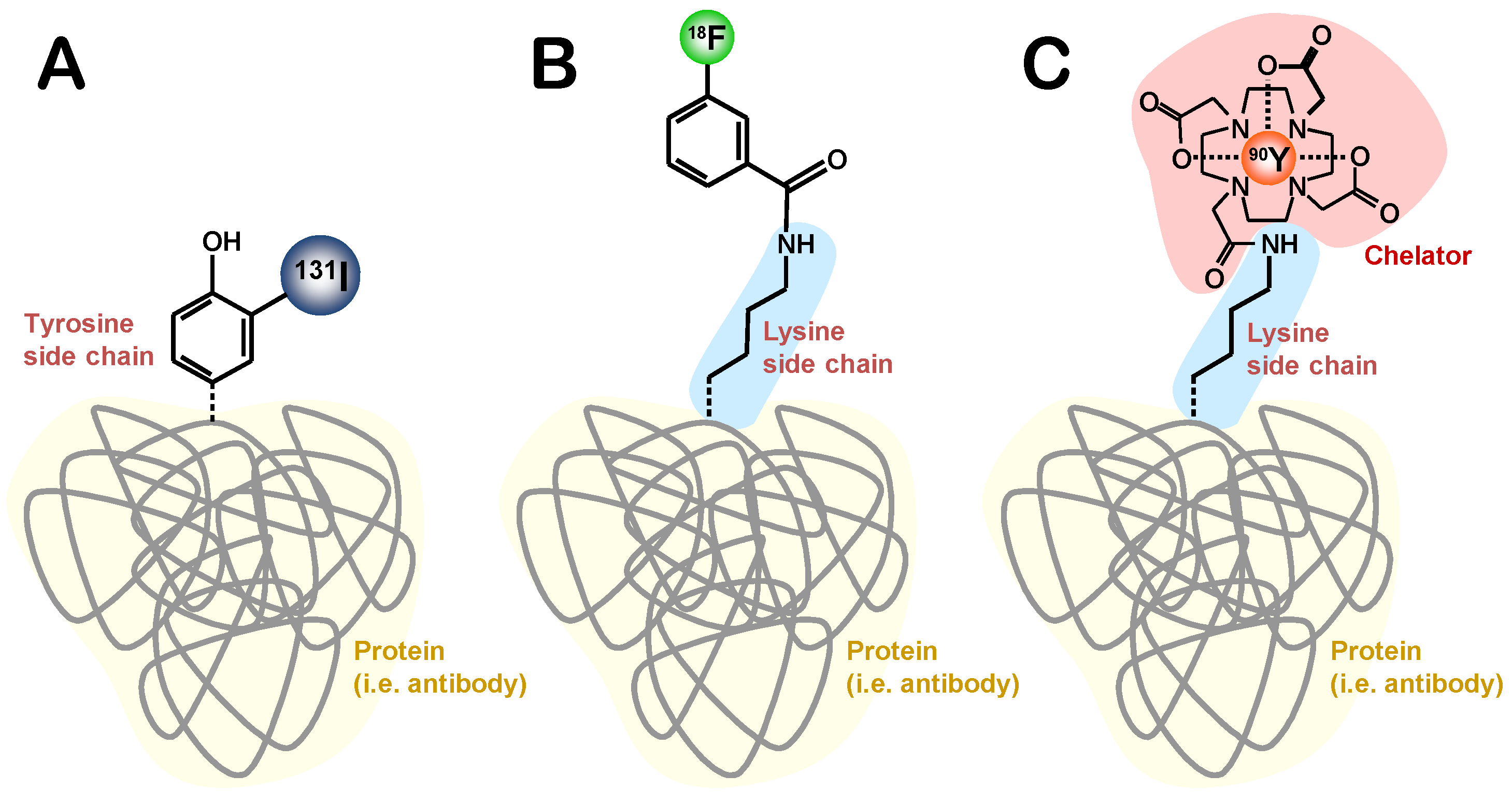
6.1.1 Radiohalogenation
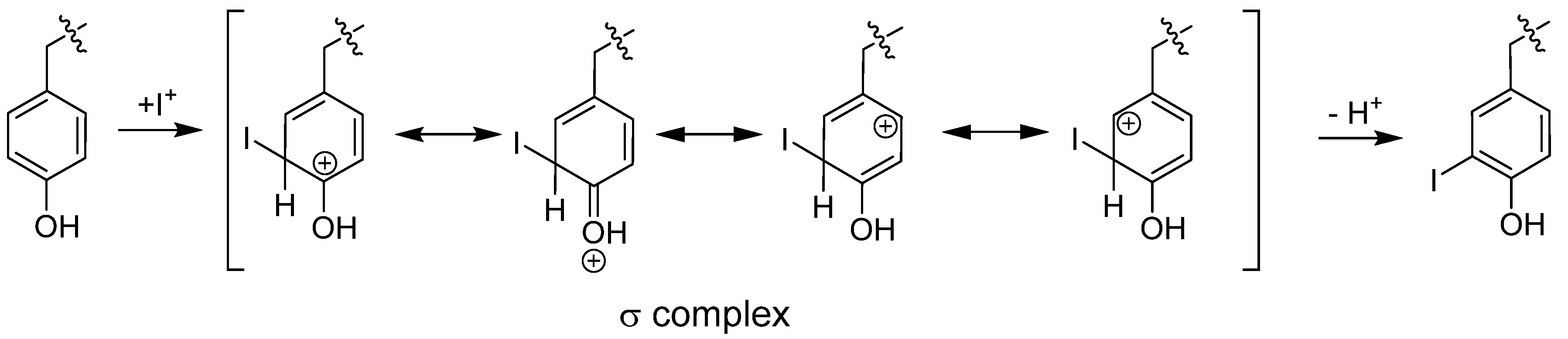
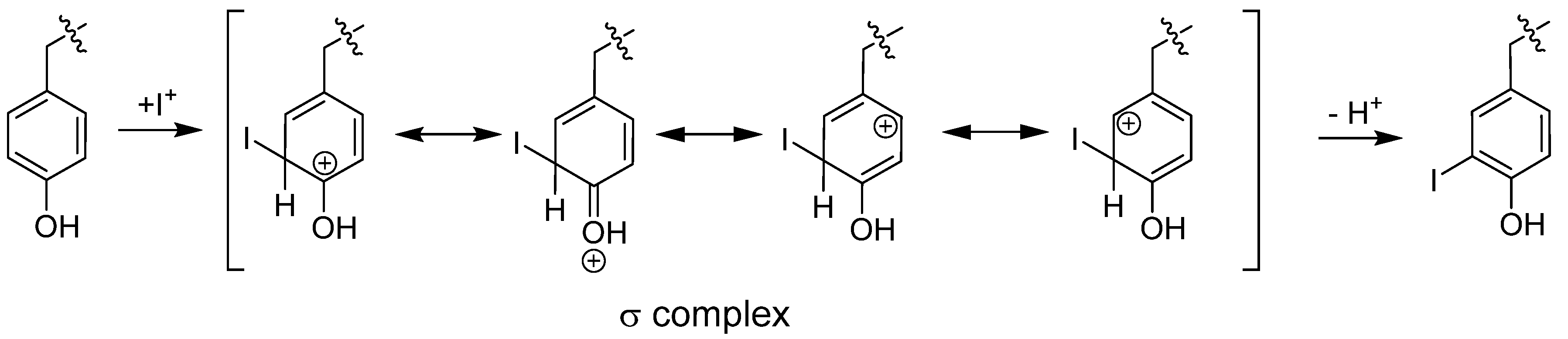
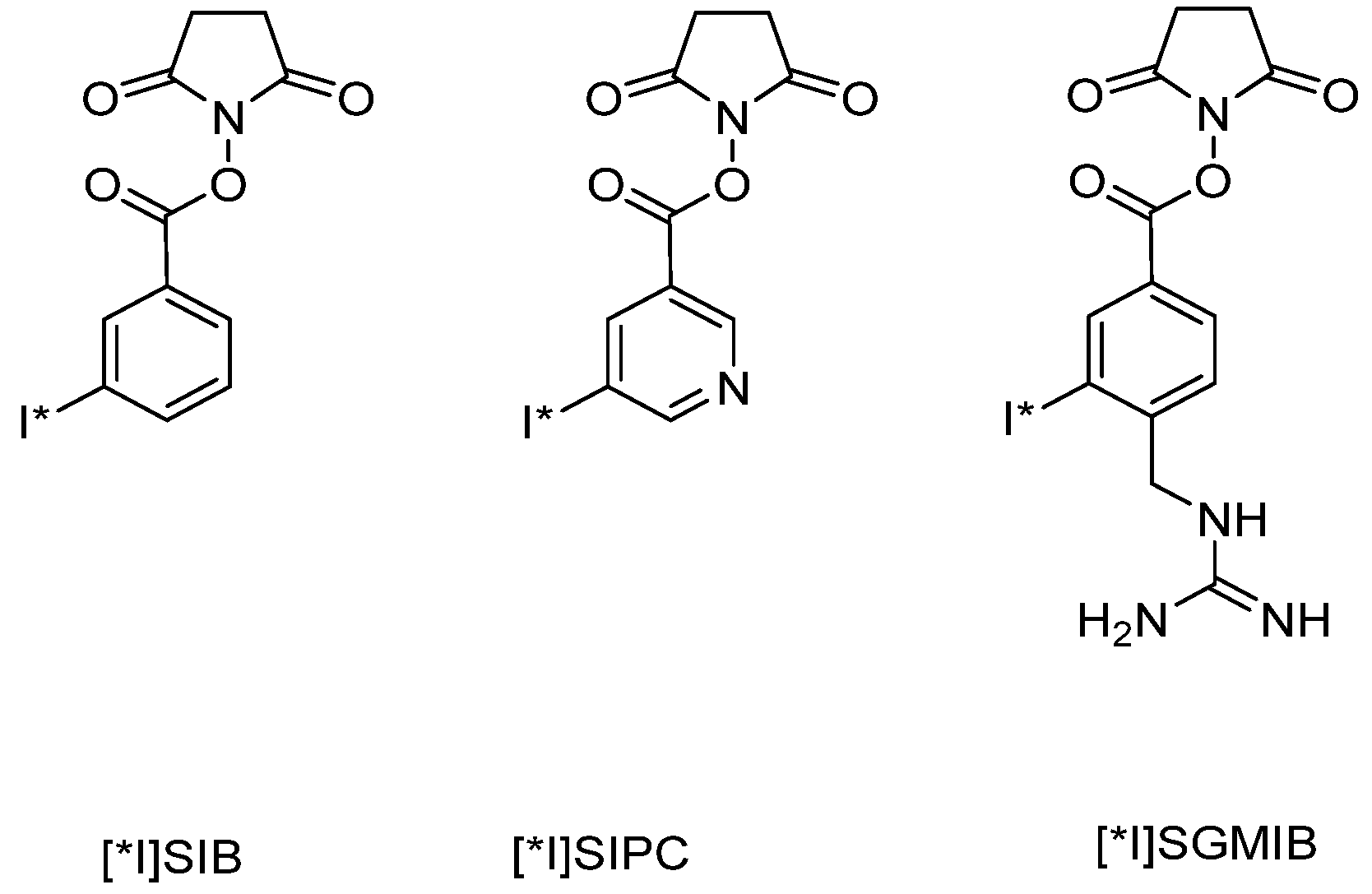
6.1.1.1. Radioiodination of Proteins
6.1.1.2. Protein-Labeling with 18F
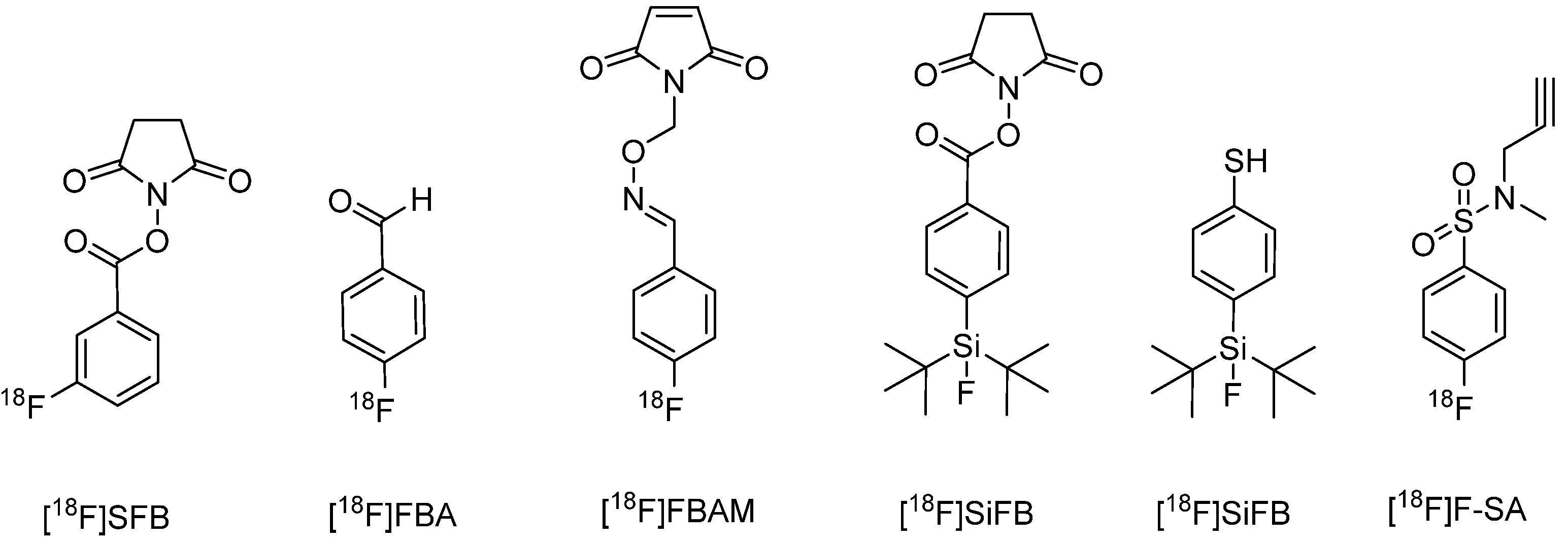
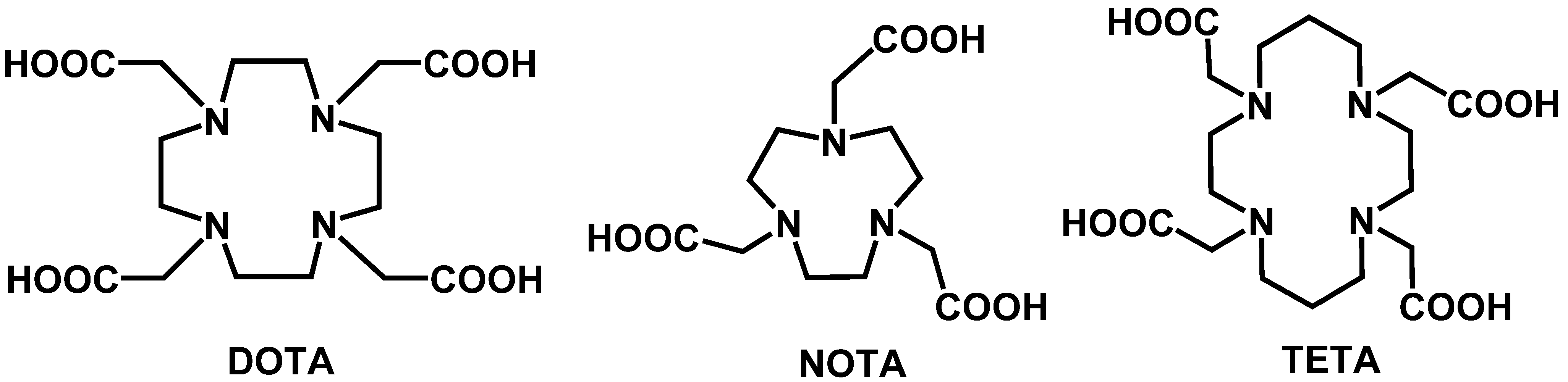
6.1.2. Chelation
6.1.2.1. Acyclic Chelators
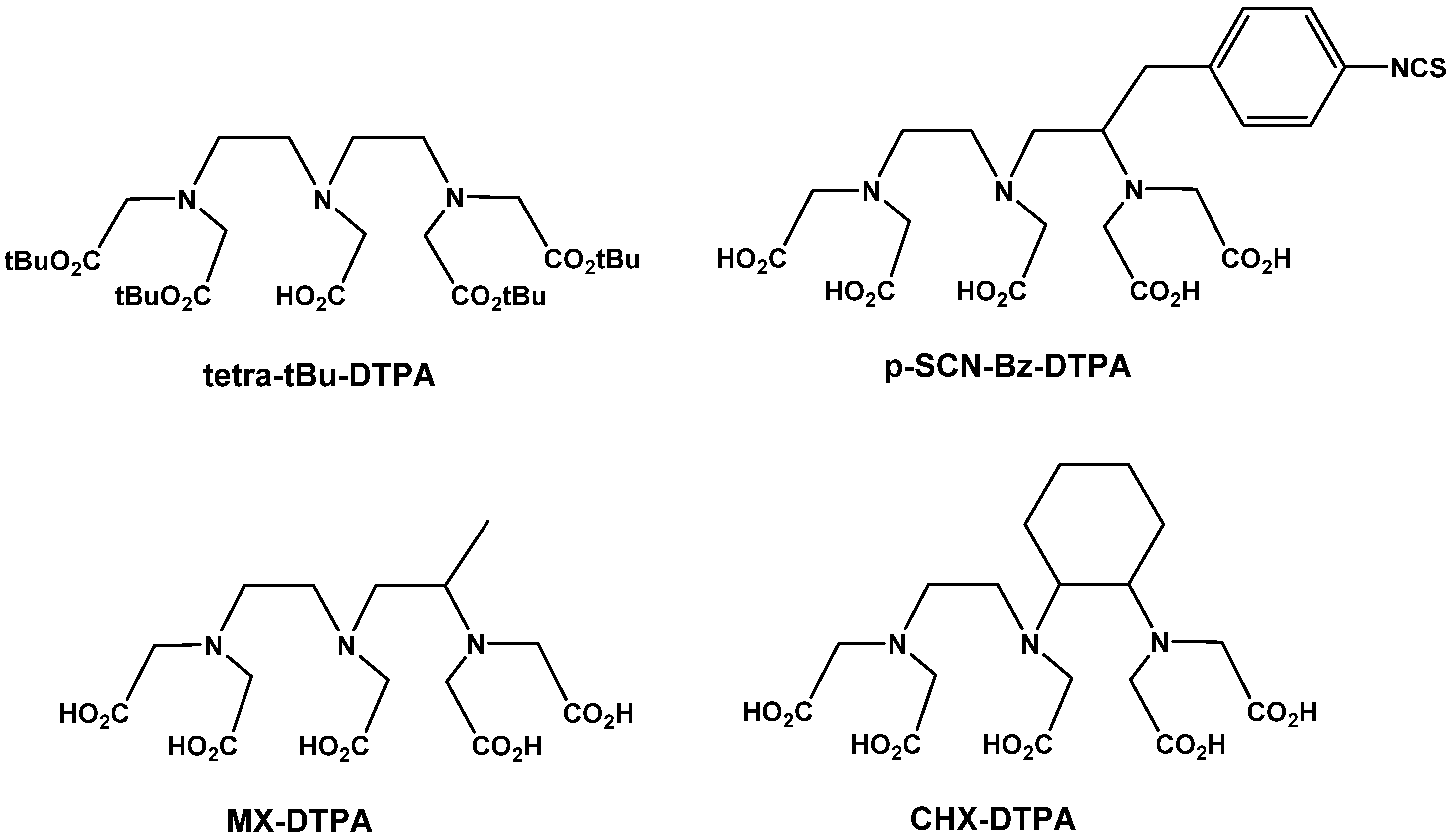
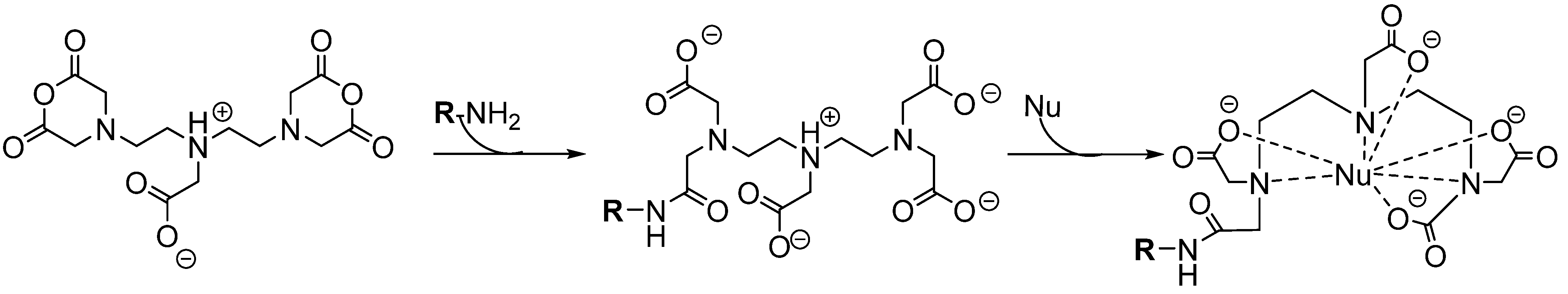
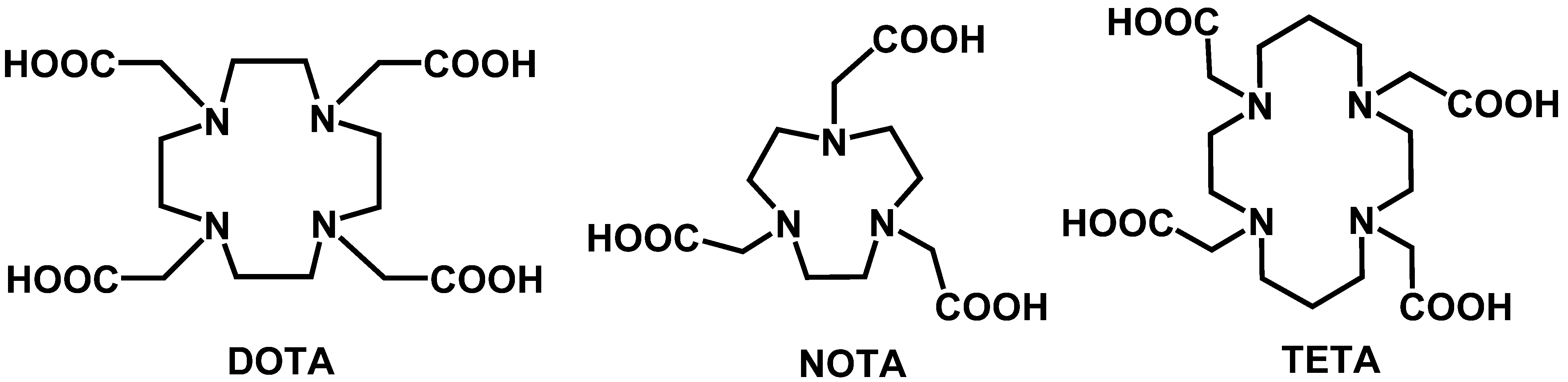
6.1.2.2. Macrocyclic Chelators
7. Metallic Nuclide Labeled Pharmaceuticals
7.1. 99mTc-Labeled Antibody Radiopharmaceuticals
7.2. Metallic Radionuclides Other than Tc
8. In-Vivo Pretargeting Strategies
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mankoff, D.A.; Link, J.M.; Linden, H.M.; Sundararajan, L.; Krohn, K.A. Tumor receptor imaging. J. Nucl. Med. 2008, 49 Suppl 2, 149S–163S. [Google Scholar]
- Heston, T.F.; Wahl, R.L. Molecular imaging in thyroid cancer. Cancer Imaging 2010, 10, 1–7. [Google Scholar]
- Selzner, M.; Hany, T.F.; Wildbrett, P.; McCormack, L.; Kadry, Z.; Clavien, P.-A. Does the novel PET/CT imaging modality impact on the treatment of patients with metastatic colorectal cancer of the liver? Ann. Surg. 2004, 240, 1027–1036. [Google Scholar] [CrossRef]
- Frangioni, J.V. New technologies for human cancer imaging. J. Clin Oncol. 2008, 26, 4012–4021. [Google Scholar]
- Belhocine, T.; Steinmetz, N.; Hustinx, R.; Bartsch, P.; Jerusalem, G.; Seidel, L.; Rigo, P.; Green, A. Increased uptake of the apoptosis-imaging agent 99mTc recombinant human annexin V in human tumors after one course of chemotherapy as a predictor of tumor. Clin. Cancer Res. 2002, 8, 2766–2774. [Google Scholar]
- Schrevens, L.; Lorent, N.; Dooms, C.; Vansteenkiste, J. The role of PET scan in diagnosis, staging, and management of non-small cell lung cancer. Oncologist 2004, 9, 633–643. [Google Scholar] [CrossRef]
- Daniels, S.; Tohid, S.F.M.; Velanguparackel, W.; Westwell, A.D. The role and future potential of fluorinated biomarkers in positron emission tomography. Expert Opin. Drug Discov. 2010, 5, 291–304. [Google Scholar] [CrossRef]
- Li, Z.; Conti, P.S. Radiopharmaceutical chemistry for positron emission tomography. Adv. Drug Delivery Rev. 2010, 62, 1031–1051. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Conti, M. State of the art and challenges of time-of-flight PET. Phys. Med. 2009, 25, 1–11. [Google Scholar] [CrossRef]
- Wängler, C.; Moldenhauer, G.; Eisenhut, M.; Haberkorn, U.; Mier, W. Antibody-dendrimer conjugates: the number, not the size of the dendrimers, determines the immunoreactivity. Bioconjug. Chem. 2008, 19, 813–820. [Google Scholar] [CrossRef]
- Wängler, C.; Buchmann, I.; Eisenhut, M.; Haberkorn, U.; Mier, W. Radiolabeled peptides and proteins in cancer therapy. Protein Pept. Lett. 2007, 14, 273–279. [Google Scholar] [CrossRef]
- Boswell, C.A.; McQuade, P.; Weisman, G.R.; Wong, E.H.; Anderson, C.J. Optimization of labeling and metabolite analysis of copper-64-labeled azamacrocyclic chelators by radio-LC-MS. Nucl. Med. Biol. 2005, 32, 29–38. [Google Scholar] [CrossRef]
- Chamarthy, M. Radioimmunotherapy of non-Hodgkin’s lymphoma: From the ‘magic bullets’ to ‘radioactive magic bullets’. Yale J. Biol. Med. 2011, 84, 391–407. [Google Scholar]
- Rao, A.V.; Akabani, G.; Rizzieri, D.A. Radioimmunotherapy for Non-Hodgkin’s Lymphoma. Clin. Med. Res. 2005, 3, 157–165. [Google Scholar] [CrossRef]
- Frampas, E.; Rousseau, C.; Bodet-Milin, C.; Barbet, J.; Chatal, J.-F.; Kraeber-Bodéré, F. Improvement of radioimmunotherapy using pretargeting. Front. Oncol. 2013, 3, 159. [Google Scholar]
- Sharkey, R.M.; Goldenberg, D.M. Cancer radioimmunotherapy. Future Oncol. 2012, 3, 349–370. [Google Scholar]
- Oriuchi, N.; Higuchi, T.; Hanaoka, H.; Iida, Y.; Endo, K. Current status of cancer therapy with radiolabeled monoclonal antibody. Ann. Nucl. Med. 2005, 19, 355–365. [Google Scholar] [CrossRef]
- Yuan, J.; Yeasky, T.M.; Rhee, M.C.; Glazer, P.M. Frequent T:A-->G:C transversions in X-irradiated mouse cells. Carcinogenesis 1995, 16, 83–88. [Google Scholar] [CrossRef]
- Snyder, A.R. Review of radiation-induced bystander effects. Hum. Exp. Toxicol. 2004, 23, 87–89. [Google Scholar] [CrossRef]
- Sofou, S. Radionuclide carriers for targeting of cancer. Int. J. Nanomedicine 2008, 3, 181–199. [Google Scholar] [CrossRef]
- Kassis, A. Therapeutic radionuclides: Biophysical and radiobiologic principles. Semin. Nucl. Med. 2008, 38, 358–366. [Google Scholar] [CrossRef]
- Zoller, F.; Eisenhut, M.; Haberkorn, U.; Mier, W. Endoradiotherapy in cancer treatment--basic concepts and future trends. Eur. J. Pharmacol. 2009, 625, 55–62. [Google Scholar] [CrossRef]
- Couturier, O.; Supiot, S.; Degraef-Mougin, M.; Faivre-Chauvet, A.; Carlier, T.; Chatal, J.-F.; Davodeau, F.; Cherel, M. Cancer radioimmunotherapy with alpha-emitting nuclides. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 601–614. [Google Scholar] [CrossRef]
- Miao, Y.; Owen, N.; Fisher, D. Therapeutic efficacy of a 188RE-Labeled alpha-melanocyte-stimulating hormone peptide analog in murine and human melanoma-bearing mouse models. J. Nucl. Med. 2005, 121–129. [Google Scholar]
- Volkert, W.A.; Hoffman, T.J. Therapeutic radiopharmaceuticals. Chem. Rev. 1999, 99, 2269–2292. [Google Scholar] [CrossRef]
- McGrath, N. A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ. 2010, 87, 1348–1349. [Google Scholar] [CrossRef]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keth, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar]
- Le, X.-F.; Pruefer, F.; Bast, R.C. HER2-Targeting Antibodies Modulate the Cyclin-Dependent Kinase inhibitor p27Kip1 via multiple signaling pathways. Cell. Cycle 2005, 4, 87–95. [Google Scholar] [CrossRef]
- Cardinale, D.; Colombo, A.; Torrisi, R.; Sandri, M.T.; Civelli, M.; Salvatici, M.; Lamantia, G.; Colombo, N.; Cortinovis, S.; Dessanai, M.A.; et al. Trastuzumab-induced cardiotoxicity: clinical and prognostic implications of troponin I evaluation. J. Clin. Oncol. 2010, 28, 3910–3916. [Google Scholar] [CrossRef]
- Kinders, R.; Parchment, R.; Ji, J.; Kummar, S.; Murgo, A.; Gutierrez, M. Phase 0 clinical trials in cancer drug development: From FDA guidance to clinical practice. Mol. Interv. 2007, 7, 325–334. [Google Scholar] [CrossRef]
- Wong, J.Y.; Raubitschek, A.; Yamauchi, D.; Williams, L.E.; Wu, A.M.; Yazaki, P.; Shively, J.E.; Colcher, D.; Somlo, G. A pretherapy biodistribution and dosimetry study of indium-111-radiolabeled trastuzumab in patients with human epidermal growth factor receptor 2-overexpressing breast cancer. Cancer Biother. Radiopharm. 2010, 25, 387–394. [Google Scholar] [CrossRef]
- Chames, P.; van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Brit. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Barbet, J.; Bardiès, M.; Bourgeois, M.; Chérel, M.; Davodeau, F.; Faivre-Chauvet, A.; Gestin, J.-F.; Kraeber-Bodéré, F. Radiolabeled antibodies for cancer imaging and therapy. Methods Mol. Biol. 2012, 907, 681–697. [Google Scholar] [CrossRef]
- Chakrabarti, M.C.; Le, N.; Paik, C.H.; de Graff, W.G.; Carrasquillo, J.A. Prevention of radiolysis of monoclonal antibody during labeling. J. Nucl. Med. 1996, 37, 1384–1388. [Google Scholar]
- Booy, E.P.; Johar, D.; Maddika, S.; Pirzada, H.; Sahib, M.M.; Gehrke, I.; Loewen, S.; Louis, S.F.; Kadkhoda, K.; Mowat, M.; et al. Monoclonal and bispecific antibodies as novel therapeutics. Arch. Immunol. Ther. Exp. 2006, 54, 85–101. [Google Scholar] [CrossRef]
- Beck, A.; Wurch, T.; Bailly, C.; Corvaia, N. Strategies and challenges for the next generation of therapeutic antibodies. Nat. Rev. Immunol. 2010, 10, 345–352. [Google Scholar] [CrossRef]
- Blagosklonny, M. Analysis of FDA approved anticancer drugs reveals the future of cancer therapy. Cell. Cycle 2004, 1035–1042. [Google Scholar]
- Grunberg, J.; Jeger, S.; Sarko, D.; Dennler, P.; Zimmermann, K.; Mier, W.; Schibli, R. DOTA-functionalized polylysine: A high number of DOTA chelates positively influences the biodistribution of enzymatic conjugated anti-tumor antibody chCE7agl. PLoS One 2013, 8, e60350. [Google Scholar]
- Jeger, S.; Zimmermann, K.; Blanc, A.; Grunberg, J.; Honer, M.; Hunziker, P.; Struthers, H.; Schibli, R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew. Chem. Int. Ed. Engl. 2010, 49, 9995–9997. [Google Scholar] [CrossRef]
- Milenic, D.; Brechbiel, M. Targeting of Radio-Isotopes for Cancer Therapy. Cancer Biol. Ther. 2004, 361–370. [Google Scholar]
- Nikula, T.K.; Bocchia, M.; Curcio, M.J.; Sgouros, G.; Ma, Y.; Finn, R.D.; Scheinberg, D.A. Impact of the high tyrosine fraction in complementarity determining regions: Measured and predicted effects of radioiodination on IgG immunoreactivity. Mol. Immunol. 1995, 32, 865–872. [Google Scholar] [CrossRef]
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef]
- McLaughlin, P.; Grillo-López, A.J.; Link, B.K.; Levy, R.; Czuczman, M.S.; Williams, M.E.; Heyman, M.R.; Bence-Bruckler, I.; White, C.A.; Cabanillas, F.; et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J. Clin. Oncol. 1998, 16, 2825–2833. [Google Scholar]
- Chang, C.-H.; Rossi, E.; Wang, Y.; Cardillo, T.; Goldenberg, D. The development of bispecific hexavalent antibodies as a novel class of DOCK-AND-LOCKTM (DNLTM) complexes. Antibodies 2013, 2, 353–370. [Google Scholar] [CrossRef]
- Rossi, E.A.; Chang, C.-H.; Losman, M.J.; Sharkey, R.M.; Karacay, H.; McBride, W.; Cardillo, T.M.; Hansen, H.J.; Qu, Z.; Horak, I.D.; et al. Pretargeting of carcinoembryonic antigen-expressing cancers with a trivalent bispecific fusion protein produced in myeloma cells. Clin. Cancer Res. 2005, 11, 7122–7129. [Google Scholar] [CrossRef]
- Gomes, C.M.; Abrunhosa, A.J.; Ramos, P.; Pauwels, E.K.J. Molecular imaging with SPECT as a tool for drug development. Adv. Drug Delivery Rev. 2011, 63, 547–554. [Google Scholar] [CrossRef]
- Phan, H.T.T.; Jager, P.L.; Paans, A.M.J.; Plukker, J.T.M.; Sturkenboom, M.G.G.; Sluiter, W.J.; Wolffenbuttel, B.H.R.; Dierckx, R.A.J.O.; Links, T.P. The diagnostic value of 124I-PET in patients with differentiated thyroid cancer. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 958–965. [Google Scholar] [CrossRef]
- Verel, I.; Visser, G.W.M.; Boerman, O.C.; van Eerd, J.E.M.; Finn, R.; Boellaard, R.; Vosjan, M.J.W.D.; Stigter-van Walsum, M.; Snow, G.B.; van Dongen, G.A.M.S. Long-lived positron emitters zirconium-89 and iodine-124 for scouting of therapeutic radioimmunoconjugates with PET. Cancer Biother. Radiopharm. 2003, 18, 655–661. [Google Scholar] [CrossRef]
- Juweid, M.E. Radioimmunotherapy with 131I-rituximab: What we know and what we don’t know. Cancer Biother. Radiopharm. 2003, 18, 489–495. [Google Scholar] [CrossRef]
- Tolmachev, V. Choice of Radionuclides and Radiolabelling Techniques; Springer: Dordrecht, The Netherlands, 2008. [Google Scholar]
- Wilbur, D.S. Radiohalogenation of proteins: an overview of radionuclides, labeling methods, and reagents for conjugate labeling. Bioconjug. Chem. 1992, 3, 433–470. [Google Scholar] [CrossRef]
- Amartey, J.K.; Esguerra, C.; Al-Otaibi, B.; Al-Jammaz, I.; Al-Qahtani, M.; Parhar, R.S. Prosthetic radioiodination of interleukin-8 ([(123/131)I]-IL-8): Biological behavior in a mouse infection model. Appl. Radiat. Isotopes 2005, 62, 39–47. [Google Scholar] [CrossRef]
- Dillman, R.O. Radiolabeled Anti-CD20 Monoclonal Antibodies for the Treatment of B-Cell Lymphoma. J. Clin. Oncol. 2002, 20, 3545–3557. [Google Scholar] [CrossRef]
- Garg, P.K.; Garg, S.; Zalutsky, M.R. Fluorine-18 labeling of monoclonal antibodies and fragments with preservation of immunoreactivity. Bioconjug. Chem. 1991, 2, 44–49. [Google Scholar] [CrossRef]
- Zalutsky, M.R. Current status of therapy of solid tumors: brain tumor therapy. J. Nucl. Med. 2005, 46, 151S–156S. [Google Scholar]
- Yang, Y.; Liu, N.; Zan, L.; Liao, J.; Jin, J. Radioiodination of insulin using N-succinimidyl 5-(tributylstannyl)-3-pyridinecarboxylate (SPC) as a bi-functional linker: Synthesis and biodistribution in mice. J. Radioanal. Nucl. Chem. 2006, 268, 205–210. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Affleck, D.J.; Li, J.; Welsh, P.; Zalutsky, M.R. A polar substituent-containing acylation agent for the radioiodination of internalizing monoclonal antibodies: N-succinimidyl 4-guanidinomethyl-3-[131I]iodobenzoate ([131I]SGMIB). Bioconjug. Chem. 2001, 12, 428–438. [Google Scholar] [CrossRef]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthesis of 11C, 18F, 15O, and 13N radiolabels for positron emission tomography. Angew. Chem. Int. Ed. Engl. 2008, 47, 8998–9033. [Google Scholar] [CrossRef]
- Jamous, M.; Haberkorn, U.; Mier, W. Synthesis of peptide radiopharmaceuticals for the therapy and diagnosis of tumor diseases. Molecules 2013, 18, 3379–3409. [Google Scholar] [CrossRef]
- Ramenda, T.; Steinbach, J.; Wuest, F. 4-[18F]Fluoro-N-methyl-N-(propyl-2-yn-1-yl)benzene-sulfonamide ([18F]F-SA): A versatile building block for labeling of peptides, proteins and oligonucleotides with fluorine-18 via Cu(I)-mediated click chemistry. Amino Acids 2013, 44, 1167–1180. [Google Scholar] [CrossRef]
- Hou, S.; Phung, D.L.; Lin, W.-Y.; Wang, M.-W.; Liu, K.; Shen, C.K.-F. Microwave-assisted one-pot synthesis of N-succinimidyl-4[ 18F]fluorobenzoate ([18F]SFB). JoVE 2011, 1, 4–9. [Google Scholar]
- Kostikov, A.P.; Chin, J.; Orchowski, K.; Schirrmacher, E.; Niedermoser, S.; Jurkschat, K.; Iovkova-Berends, L.; Wängler, C.; Wängler, B.; Schirrmacher, R. Synthesis of [(18)F]SiFB: A prosthetic group for direct protein radiolabeling for application in positron emission tomography. Nat. Protoc. 2012, 7, 1956–1963. [Google Scholar] [CrossRef]
- Berndt, M.; Pietzsch, J.; Wuest, F. Labeling of low-density lipoproteins using the 18F-labeled thiol-reactive reagent N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl]maleimide. Nucl. Med. Biol. 2007, 34, 5–15. [Google Scholar] [CrossRef]
- Flavell, R.R.; Kothari, P.; Bar-Dagan, M.; Synan, M.; Vallabhajosula, S.; Friedman, J.M.; Muir, T.W.; Ceccarini, G. Site-specific (18)F-labeling of the protein hormone leptin using a general two-step ligation procedure. J. Am. Chem. Soc. 2008, 130, 9106–9112. [Google Scholar] [CrossRef]
- Wängler, B.; Kostikov, A.P.; Niedermoser, S.; Chin, J.; Orchowski, K.; Schirrmacher, E.; Iovkova-Berends, L.; Jurkschat, K.; Wängler, C.; Schirrmacher, R. Protein labeling with the labeling precursor [(18)F]SiFA-SH for positron emission tomography. Nat. Protoc. 2012, 7, 1964–1969. [Google Scholar] [CrossRef]
- Chang, Y.S.; Jeong, J.M.; Lee, Y.-S.; Kim, H.W.; Rai, G.B.; Lee, S.J.; Lee, D.S.; Chung, J.-K.; Lee, M.C. Preparation of 18F-human serum albumin: a simple and efficient protein labeling method with 18F using a hydrazone-formation method. Bioconjug. Chem. 2005, 16, 1329–1333. [Google Scholar] [CrossRef]
- Ranadive, G.N.; Rosenzweig, H.S.; Epperly, M.W.; Seskey, T.; Bloomer, W.D. A new method of technetium-99m labeling of monoclonal antibodies through sugar residues. A study with TAG-72 specific CC-49 antibody. Nucl. Med. Biol. 1993, 20, 719–726. [Google Scholar] [CrossRef]
- Rhodes, B.A. Direct labeling of proteins with 99mTc. Int. J. Rad. Appl. Instrum. B 1991, 18, 667–676. [Google Scholar] [CrossRef]
- Bartholomä, M.D.; Gottumukkala, V.; Zhang, S.; Baker, A.; Dunning, P.; Fahey, F.H.; Treves, S.T.; Packard, A.B. Effect of the prosthetic group on the pharmacologic properties of 18F-labeled rhodamine B, a potential myocardial perfusion agent for positron emission tomography (PET). J. Med. Chem. 2012, 55, 11004–11012. [Google Scholar] [CrossRef]
- Pruszyński, M.; Bilewicz, A.; Zalutsky, M. Preparation of Rh[16aneS4-diol]211At complexes as potential precursors for astatine radiopharmaceuticals. Part I: Synthesis. Bioconjug. Chem. 2008, 19, 958–965. [Google Scholar] [CrossRef]
- Liu, S.; Edwards, D. Bifunctional chelators for therapeutic lanthanide radiopharmaceuticals. Bioconjug. Chem. 2001, 12, 7–34. [Google Scholar]
- Heppeler, A.; Froidevaux, S.; Mäcke, H.R.; Jermann, E.; Béhé, M.; Powell, P.; Hennig, M. Radiometal-labelled macrocyclic chelator-derivatised somatostatin analogue with superb tumour-targeting properties and potential for receptor-mediated internal radiotherapy. Chem-Eur. J. 1999, 5, 1974–1981. [Google Scholar] [CrossRef]
- Liu, G.; Hnatowich, D. Labeling biomolecules with radiorhenium—A review of the bifunctional chelators. Anti-Cancer Agents Med. Chem. 2007, 7, 367–377. [Google Scholar]
- Wu, C.; Kobayashi, H.; Sun, B.; Yoo, T. Stereochemical influence on the stability of radio-metal complexes in vivo. Synthesis and Evaluation of the Four Stereoisomers of 2-(p-Nitrobenzyl)-trans--CyDTPA. Bioorg. Med. Chem. 1997, 5, 1925–1934. [Google Scholar] [CrossRef]
- Brechbiel, M. Bifunctional Chelates for Metal Nuclides. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 166–173. [Google Scholar]
- Brechbiel, M.; Gansow, O. Synthesis of 1-(p-isothiocyanatobenzyl) derivatives of DTPA and EDTA. Antibody labeling and tumor-imaging studies. Inorg. Chem. 1986, 20892, 1511–1513. [Google Scholar]
- Arano, Y.; Akizawa, H. Conventional and high-yield synthesis of DTPA-Conjugated peptides: Application of a monoreactive DTPA to DTPA-D-Phe-Ocreotide synthesis. Bioconjug. Chem. 1997, 1802, 442–446. [Google Scholar] [CrossRef]
- McMurry, T.J.; Pippin, C.G.; Wu, C.; Deal, K.A.; Brechbiel, M.W.; Mirzadeh, S.; Gansow, O.A. Physical parameters and biological stability of yttrium(III) diethylenetriaminepentaacetic acid derivative conjugates. J. Med. Chem. 1998, 41, 3546–3549. [Google Scholar] [CrossRef]
- Liu, S. Bifunctional coupling agents for radiolabeling of biomolecules and target-specific delivery of metallic radionuclides. Adv. Drug Delivery Rev. 2008, 60, 1347–1370. [Google Scholar] [CrossRef]
- Brom, M.; Joosten, L.; Oyen, W.J.; Gotthardt, M.; Boerman, O.C. Improved labelling of DTPA- and DOTA-conjugated peptides and antibodies with 111In in HEPES and MES buffer. EJNMMI Res. 2012, 2, 4. [Google Scholar] [CrossRef]
- Hnatowich, D.J.; Virzi, F.; Doherty, P.W. DTPA-coupled antibodies labeled with yttrium-90. J. Nucl. Med. 1985, 26, 503–509. [Google Scholar]
- Meares, C.F.; McCall, M.J.; Reardan, D.T.; Goodwin, D.A.; Diamanti, C.I.; McTigue, M. Conjugation of antibodies with bifunctional chelating agents: isothiocyanate and bromoacetamide reagents, methods of analysis, and subsequent addition of metal ions. Anal. Biochem. 1984, 142, 68–78. [Google Scholar] [CrossRef]
- Dillman, R.O. Radioimmunotherapy of B-cell lymphoma with radiolabelled anti-CD20 monoclonal antibodies. Clin. Exp. Med. 2006, 6, 1–12. [Google Scholar] [CrossRef]
- Perk, L.; Visser, G. 89Zr as a PET Surrogate Radioisotope for Scouting Biodistribution of the Therapeutic Radiometals 90Y and 177Lu in Tumor-Bearing Nude Mice After Coupling to the Internalizing Antibody Cetuximab. J. Nucl. Med 2005, 46, 1898–1906. [Google Scholar]
- Hnatowich, D.J.; Layne, W.; Childs, R.; Lanteigne, D.; Davis, M. Radioactive Labeling of Antibody: A simple and Efficient Method. Science 1983, 220, 613–615. [Google Scholar]
- Frullano, L.; Caravan, P. Strategies for the preparation of bifunctional gadolinium(III) Chelators. Curr. Org. Synth. 2011, 8, 535–565. [Google Scholar] [CrossRef]
- Arano, Y.; Uezono, T.; Akizawa, H.; Ono, M.; Wakisaka, K.; Nakayama, M.; Sakahara, H.; Konishi, J.; Yokoyama, A. Reassessment of diethylenetriaminepentaacetic acid (DTPA) as a chelating agent for indium-111 labeling of polypeptides using a newly synthesized monoreactive DTPA derivative. J. Med. Chem. 1996, 39, 3451–3460. [Google Scholar] [CrossRef]
- Visser, G.W.; Gerretsen, M.; Herscheid, J.D.; Snow, G.B.; van Dongen, G. Labeling of monoclonal antibodies with rhenium-186 using the MAG3 chelate for radioimmunotherapy of cancer: a technical protocol. J. Nucl. Med. 1993, 34, 1953–1963. [Google Scholar]
- Fichna, J.; Janecka, A. Synthesis of target-specific radiolabeled peptides for diagnostic imaging. Bioconjug. Chem. 2003, 14, 3–17. [Google Scholar] [CrossRef]
- Roselli, M.; Schlom, J.; Gansow, O.A.; Brechbiel, M.W.; Mirzadeh, S.; Pippin, C.G.; Milenic, D.E.; Colcher, D. Comparative biodistribution studies of DTPA-derivative bifunctional chelates for radiometal labeled monoclonal antibodies. Int. J. Radiat. Appl. Instrum. B Nucl. Med. Biol. 1991, 18, 389–394. [Google Scholar] [CrossRef]
- Hosken, G.; Allan, C. Structure of the copper (II) complex of a highly preorganised tetradentate ligand based on bispidine (3,7-diazabicyclo-[3.3.1]nonane). Dalton Trans. 1995, 22, 3705–3708. [Google Scholar] [CrossRef]
- Camera, L.; Kinuya, S.; Garmestani, K.; Wu, C.; Brechbiel, M.W.; Pai, L.H.; McMurry, T.J.; Gansow, O.A.; Pastan, I.; Paik, C.H. Evaluation of the serum stability and in vivo biodistribution of CHX-DTPA and other ligands for yttrium labeling of monoclonal antibodies. J. Nucl. Med. 1994, 35, 882–889. [Google Scholar]
- Kobayashi, H.; Wu, C.; Yoo, T.M.; Sun, B.F.; Drumm, D.; Pastan, I.; Paik, C.H.; Gansow, O.A.; Carrasquillo, J.A.; Brechbiel, M.W. Evaluation of the in vivo biodistribution of yttrium-labeled isomers of CHX-DTPA-conjugated monoclonal antibodies. J. Nucl. Med. 1998, 39, 829–836. [Google Scholar]
- Tolmachev, V.; Xu, H.; Wållberg, H.; Ahlgren, S.; Hjertman, M.; Sjöberg, A.; Sandström, M.; Abrahmsén, L.; Brechbiel, M.W. Evaluation of a maleimido derivative of CH-A" DTPA for site-specific labeling of Affibody molecules. Bioconjug. Chem. 2009, 19, 1579–1587. [Google Scholar]
- Lewis, M.R.; Kao, J.Y.; Anderson, A.L.; Shively, J.E.; Raubitschek, A. An improved method for conjugating monoclonal antibodies with N-hydroxysulfosuccinimidyl DOTA. Bioconjug. Chem. 2001, 12, 320–324. [Google Scholar] [CrossRef]
- Magerstädt, M.; Gansow, O.A.; Brechbiel, M.W.; Colcher, D.; Baltzer, L.; Knop, R.H.; Girton, M.E.; Naegele, M. Gd(DOTA): An alternative to Gd(DTPA) as a T1,2 relaxation agent for NMR imaging or spectroscopy. Magn. Reson. Med. 1986, 3, 808–812. [Google Scholar] [CrossRef]
- Wang, X.; Jin, T.; Comblin, V. A kinetic investigation of the lanthanide dota chelates. Stability and rates of formation and of dissociation of a macrocyclic gadolinium(111) polyaza polycarboxylic mri contrast agent. Inorg. Chem. 1992, 31, 1095–1099. [Google Scholar] [CrossRef]
- Lewis, M.R.; Shively, J.E. Maleimidocysteineamido-DOTA derivatives: new reagents for radiometal chelate conjugation to antibody sulfhydryl groups undergo pH-dependent cleavage reactions. Bioconjug. Chem. 1998, 9, 72–86. [Google Scholar] [CrossRef]
- Jalilian, A. Preparation Quality Control and Biodistribution Studies of two [111In]-Rituximab Immunoconjugates. Sci. Pharm. 2008, 76, 151–170. [Google Scholar] [CrossRef]
- Sano, K.; Okada, M.; Hisada, H.; Shimokawa, K.; Saji, H.; Maeda, M.; Mukai, T. In vivo evaluation of a radiogallium-labeled bifunctional radiopharmaceutical, Ga-DOTA-MN2, for hypoxic tumor imaging. Biol. Pharm. Bull. 2013, 36, 602–608. [Google Scholar] [CrossRef]
- Panwar, P.; Iznaga-Escobar, N. Radiolabeling and biological evaluation of dota-ph-al derivative conjugated to anti-EGFR antibody ior egf/r3 for targeted tumor imaging and therapy. Cancer Biol. Ther. 2005, 854–860. [Google Scholar] [CrossRef]
- Wong, J.Y.; Chu, D.Z.; Williams, L.E.; Liu, A.; Zhan, J.; Yamauchi, D.M.; Wilczynski, S.; Wu, A.M.; Yazaki, P.J.; Shively, J.E.; et al. A phase I trial of (90)Y-DOTA-anti-CEA chimeric T84.66 (cT84.66) radioimmunotherapy in patients with metastatic CEA-producing malignancies. Cancer Biother. Radiopharm. 2006, 21, 88–100. [Google Scholar] [CrossRef]
- Bass, L.a.; Wang, M.; Welch, M.J.; Anderson, C.J. In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug. Chem. 2000, 11, 527–532. [Google Scholar] [CrossRef]
- Blower, P.J.; Lewis, J.S.; Zweit, J. Copper radionuclides and radiopharmaceuticals in nuclear medicine. Nucl. Med. Biol. 1996, 23, 957–980. [Google Scholar] [CrossRef]
- Dearling, J.L.J.; Voss, S.D.; Dunning, P.; Snay, E.; Fahey, F.; Smith, S.V.; Huston, J.S.; Meares, C.F.; Treves, S.T.; Packard, A.B. Imaging cancer using PET--the effect of the bifunctional chelator on the biodistribution of a (64)Cu-labeled antibody. Nucl. Med. Biol. 2011, 38, 29–38. [Google Scholar] [CrossRef]
- Zhang, Y.; Hong, H.; Engle, J.W.; Bean, J.; Yang, Y.; Leigh, B.R.; Barnhart, T.E.; Cai, W. Positron emission tomography imaging of CD105 expression with a 64Cu-labeled monoclonal antibody: NOTA is superior to DOTA. PLoS One 2011, 6, e28005. [Google Scholar]
- Lewis, M.R.; Boswell, C.a.; Laforest, R.; Buettner, T.L.; Ye, D.; Connett, J.M.; Anderson, C.J. Conjugation of monoclonal antibodies with TETA using activated esters: biological comparison of 64Cu-TETA-1A3 with 64Cu-BAT-2IT-1A3. Cancer Biother. Radiopharm. 2001, 16, 483–494. [Google Scholar] [CrossRef]
- Cole, W.C.; DeNardo, S.J.; Meares, C.F.; McCall, M.J.; DeNardo, G.L.; Epstein, A.L.; O’Brien, H.A.; Moi, M.K. Comparative serum stability of radiochelates for antibody radiopharmaceuticals. J. Nucl. Med. 1987, 28, 83–90. [Google Scholar]
- Anderson, C.; Dehdashti, F. 64Cu-TETA-Octreotide as a PET Imaging Agent for Patients with Neuroendocrine Tumors. J. Nucl. Med. 2001, 213–221. [Google Scholar]
- Clarke, M.; Podbielski, L. Medical Diagnostic Imaging With Complexes of 99mTC. Coord. Chem. Rev. 1987, 78, 253–331. [Google Scholar] [CrossRef]
- Steffens, M.G.; Oosterwijk, E.; Kranenborg, M.H.; Manders, J.M.; Debruyne, F.M.; Corstens, F.H.; Boerman, O.C. In vivo and in vitro characterizations of three 99mTc-labeled monoclonal antibody G250 preparations. J. Nucl. Med. 1999, 40, 829–836. [Google Scholar]
- Oosterwijk-Wakka, J.C.; Boerman, O.C.; Mulders, P.F.A.; Oosterwijk, E. Application of monoclonal antibody G250 recognizing carbonic anhydrase IX in renal cell carcinoma. Int. J. Mol. Sci. 2013, 14, 11402–11423. [Google Scholar] [CrossRef]
- Van Gog, F.B.; Visser, G.W.; Gowrising, R.W.; Snow, G.B.; van Dongen, G.A. Synthesis and evaluation of 99mTc/99Tc-MAG3-biotin conjugates for antibody pretargeting strategies. Nucl. Med. Biol. 1998, 25, 611–619. [Google Scholar] [CrossRef]
- Lub-de Hooge, M.N.; Kosterink, J.G.W.; Perik, P.J.; Nijnuis, H.; Tran, L.; Bart, J.; Suurmeijer, A.J.H.; de Jong, S.; Jager, P.L.; de Vries, E.G.E. Preclinical characterisation of 111In-DTPA-trastuzumab. Brit. J. Pharmacol. 2004, 143, 99–106. [Google Scholar] [CrossRef]
- Todorovska, A.; Roovers, R.C.; Dolezal, O.; Kortt, A.A.; Hoogenboom, H.R.; Hudson, P.J. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J. Immunol. Meth. 2001, 248, 47–66. [Google Scholar] [CrossRef]
- Eder, M.; Knackmuss, S.; Le Gall, F.; Reusch, U.; Rybin, V.; Little, M.; Haberkorn, U.; Mier, W.; Eisenhut, M. 68Ga-labelled recombinant antibody variants for immuno-PET imaging of solid tumours. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 1397–1407. [Google Scholar] [CrossRef]
- Wong, J.Y.C.; Chu, D.Z.; Yamauchi, D.M.; Williams, L.E.; Liu, A.; Wilczynski, S.; Wu, A.M.; Shively, J.E.; Doroshow, J.H.; Raubitschek, A.A. A phase I radioimmunotherapy trial evaluating 90 yttrium-labeled anti-carcinoembryonic antigen (CEA) chimeric T84.66 in patients with metastatic CEA-producing malignancies. Clin. Cancer Res. 2000, 6, 38–55. [Google Scholar]
- Wong, J.; Shibata, S.; Williams, L.; Kwok, C. A phase I trial of 90Y-anti-carcinoembryonic antigen chimeric T84.66 radioimmunotherapy with 5-fluorouracil in patients with metastatic colorectal cancer. Clin. Cancer Res. 2003, 5842–5852. [Google Scholar]
- Crudo, J.L.; Edreira, M.M.; Obenaus, E.R.; Chinol, M.; Paganelli, G.; de Castiglia, S.G. Optimization of antibody labeling with rhenium-188 using a prelabeled MAG3 chelate. Int. J. Pharm. 2002, 248, 173–182. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M.; Paganelli, G.; Barbet, J.; Chatal, J.-F. Antibody pretargeting advances cancer radioimmunodetection and radioimmunotherapy. J. Clin. Oncol. 2006, 24, 823–834. [Google Scholar] [CrossRef]
- McBride, W.; Zanzonico, P. Bispecific Antibody Pretargeting PET (ImmunoPET) with an 124I-Labeled Hapten-peptide. J. Nucl. Med. 2006, 47, 1678–1688. [Google Scholar]
- Govindan, S.V.; Goldenberg, D.M. New antibody conjugates in cancer therapy. Scientific World Journal 2010, 10, 2070–2089. [Google Scholar] [CrossRef]
- Moro, M.; Pelagi, M.; Fulci, G.; Paganelli, G. Tumor cell targeting with antibody-avidin complexes and biotinylated tumor necrosis factor α. Cancer Res. 1997, 57, 1922–1928. [Google Scholar]
- Goodwin, D.A.; Meares, C.F.; McTigue, M.; Chaovapong, W.; Diamanti, C.I.; Ransone, C.H.; McCall, M.J. Pretargeted immunoscintigraphy: effect of hapten valency on murine tumor uptake. J. Nucl. Med. 1992, 33, 2006–2013. [Google Scholar]
- Le Doussal, J.M.; Martin, M.; Gautherot, E.; Delaage, M.; Barbet, J. In vitro and in vivo targeting of radiolabeled monovalent and divalent haptens with dual specificity monoclonal antibody conjugates: enhanced divalent hapten affinity for cell-bound antibody conjugate. J. Nucl. Med. 1989, 30, 1358–1366. [Google Scholar]
- Axworthy, D.B.; Reno, J.M.; Hylarides, M.D.; Mallett, R.W.; Theodore, L.J.; Gustavson, L.M.; Su, F.; Hobson, L.J.; Beaumier, P.L.; Fritzberg, A.R. Cure of human carcinoma xenografts by a single dose of pretargeted yttrium-90 with negligible toxicity. Proc. Natl. Acad. Sci. USA 2000, 97, 1802–1807. [Google Scholar] [CrossRef]
- Grana, C.; Chinol, M.; Robertson, C. Pretargeted adjuvant radioimmunotherapy with yttrium-90-biotin in malignant glioma patients: a pilot study. Brit. J. Cancer 2002, 207–212. [Google Scholar] [CrossRef]
- Grana, C.M.; Chinol, M.; Cicco, C.D.; Bartolomei, M.; Cremonesi, M.; Bodei, L.; Rocca, P.A.; Pacifici, M.; Tiberini, S.; Baio, S.M.; et al. Eleven-year experience with the avidin-biotin pretargeting system in glioblastoma: Toxicity, Efficacy and survival. Open Nucl. Med. J. 2012, 4, 14–20. [Google Scholar] [CrossRef]
- Moosmayer, D.; Berndorff, D.; Chang, C.-H.; Sharkey, R.M.; Rother, A.; Borkowski, S.; Rossi, E.A.; McBride, W.J.; Cardillo, T.M.; Goldenberg, D.M.; et al. Bispecific antibody pretargeting of tumor neovasculature for improved systemic radiotherapy of solid tumors. Clin. Cancer Res. 2006, 12, 5587–5595. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; White, B.J.; Affleck, D.J.; Zhao, X.G.; Welsh, P.C.; McDougald, D.; Choi, J.; Zalutsky, M.R. SIB-DOTA: A trifunctional prosthetic group potentially amenable for multi-modal labeling that enhances tumor uptake of internalizing monoclonal antibodies. Bioorg. Med. Chem. 2012, 20, 6929–6939. [Google Scholar] [CrossRef]
- Vaneycken, I.; Devoogdt, N.; van Gassen, N.; Vincke, C.; Xavier, C.; Wernery, U.; Muyldermans, S.; Lahoutte, T.; Caveliers, V. Preclinical screening of anti-HER2 nanobodies for molecular imaging of breast cancer. FASEB J. 2011, 25, 2433–2446. [Google Scholar] [CrossRef]
- Vaneycken, I.; D’Huyvetter, M.; Hernot, S.; de Vos, J.; Xavier, C.; Devoogdt, N.; Caveliers, V.; Lahoutte, T. Immuno-imaging using nanobodies. Curr. Opin. Biotechnol. 2011, 22, 877–881. [Google Scholar] [CrossRef]
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsen, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlen, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef]
- Glaser, M.; Iveson, P.; Hoppmann, S.; Indrevoll, B.; Wilson, A.; Arukwe, J.; Danikas, A.; Bhalla, R.; Hiscock, D. Three methods for 18F labeling of the HER2-binding affibody molecule ZHER2:2891 including preclinical assessment. J. Nucl. Med. 2013, 54, 1981–1988. [Google Scholar] [CrossRef]
- Morgenstern, A.; Bruchertseifer, F.; Apostolidis, C. Bismuth-213 and actinium-225—generator performance and evolving therapeutic applications of two generator-derived alpha-emitting radioisotopes. Curr. Radiopharm. 2012, 5, 221–227. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sugiura, G.; Kühn, H.; Sauter, M.; Haberkorn, U.; Mier, W. Radiolabeling Strategies for Tumor-Targeting Proteinaceous Drugs. Molecules 2014, 19, 2135-2165. https://doi.org/10.3390/molecules19022135
Sugiura G, Kühn H, Sauter M, Haberkorn U, Mier W. Radiolabeling Strategies for Tumor-Targeting Proteinaceous Drugs. Molecules. 2014; 19(2):2135-2165. https://doi.org/10.3390/molecules19022135
Chicago/Turabian StyleSugiura, Grant, Helen Kühn, Max Sauter, Uwe Haberkorn, and Walter Mier. 2014. "Radiolabeling Strategies for Tumor-Targeting Proteinaceous Drugs" Molecules 19, no. 2: 2135-2165. https://doi.org/10.3390/molecules19022135
APA StyleSugiura, G., Kühn, H., Sauter, M., Haberkorn, U., & Mier, W. (2014). Radiolabeling Strategies for Tumor-Targeting Proteinaceous Drugs. Molecules, 19(2), 2135-2165. https://doi.org/10.3390/molecules19022135