Synthesis of Chiral Chalcone Derivatives Catalyzed by the Chiral Cinchona Alkaloid Squaramide

Abstract
:1. Introduction
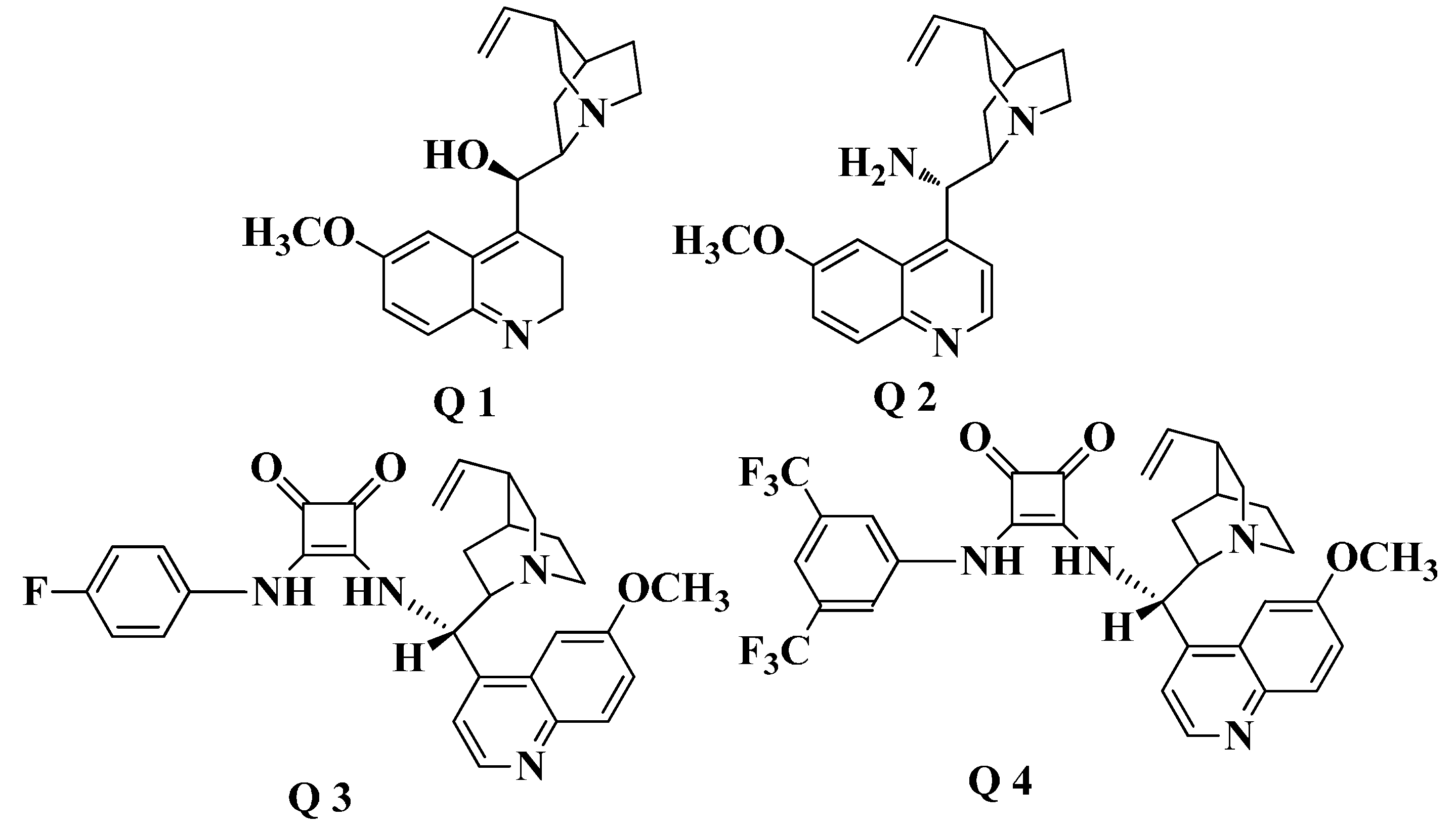
2. Results and Discussion
2.1. Chiral Catalysts Screening

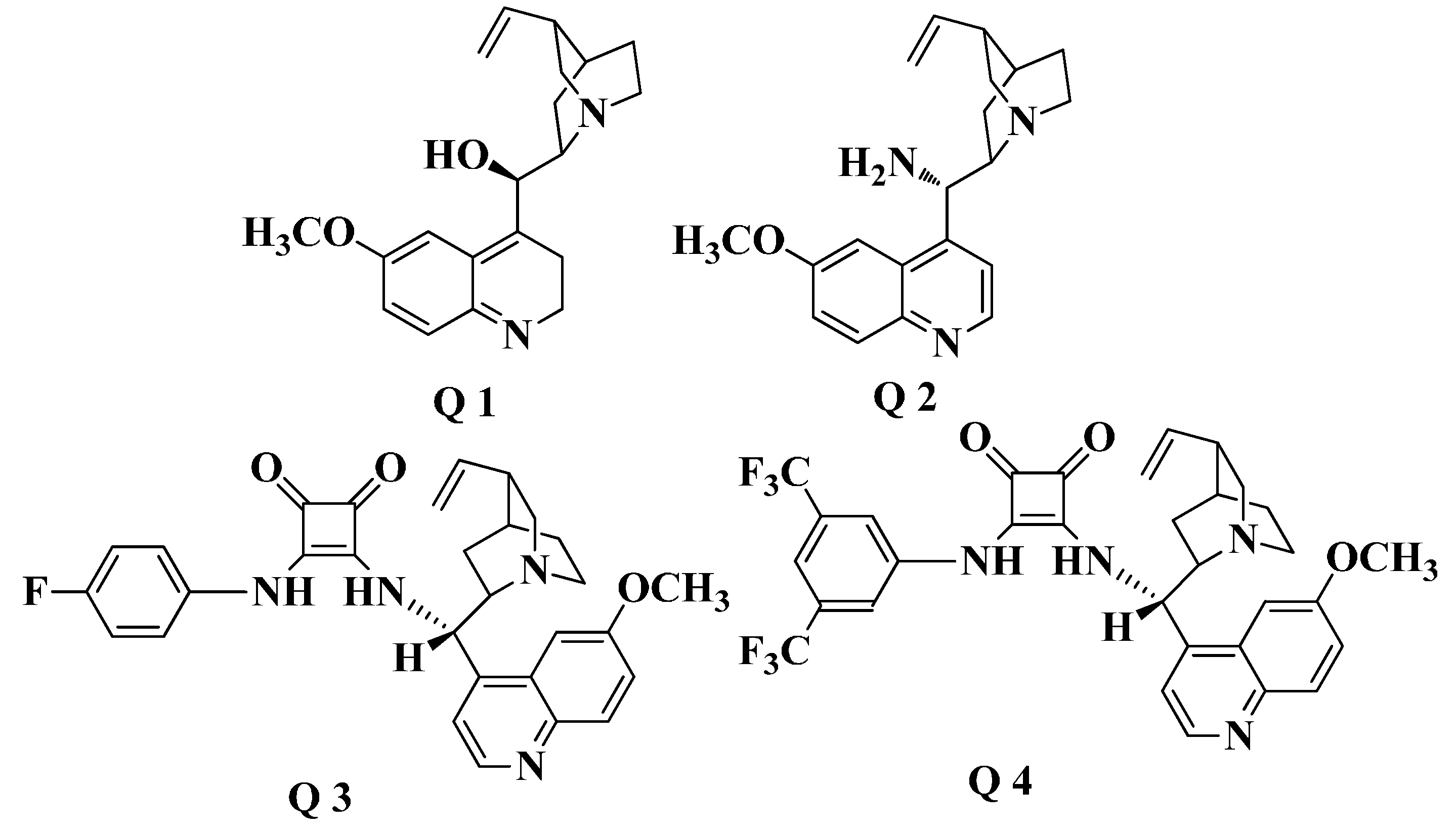{kind=link}
{kind=link}
| Entry | Catalyst | Solvent | Yield [%] [b] | ee [%] [c] |
|---|---|---|---|---|
| 1 | Q1 | CH2Cl2 | 20.2 | 60 |
| 2 | Q2 | CH2Cl2 | 35.0 | 45 |
| 3 | Q3 | CH2Cl2 | 37.2 | 70 |
| 4 | Q4 | CH2Cl2 | 46.0 | 96 |
| Entry | Temperature [°C] | Solvent | Yield [%] [b] | ee [%] [c] |
|---|---|---|---|---|
| 1 | rt | CH2Cl2 | 46.2 | 96 |
| 2 | rt | EtOH | 30.0 | 50 |
| 3 | rt | PhCH3 | 40.9 | 67 |
| 4 | 60 | PhCH3 | 60.2 | 55 |
| 5 | 0 | CH2Cl2 | 46.0 | 79 |
| 6 | 40 | CH2Cl2 | 50.2 | 85 |
| 7 | rt [d] | CH2Cl2 | 40.2 | 88 |
| 8 | rt [e] | CH2Cl2 | 55.2 | 97 |
2.2. Optimization Studies
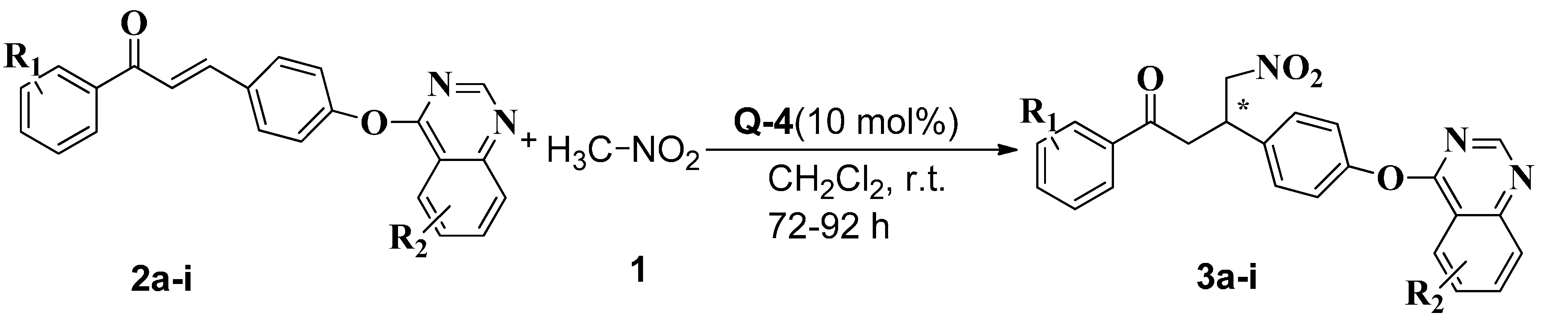
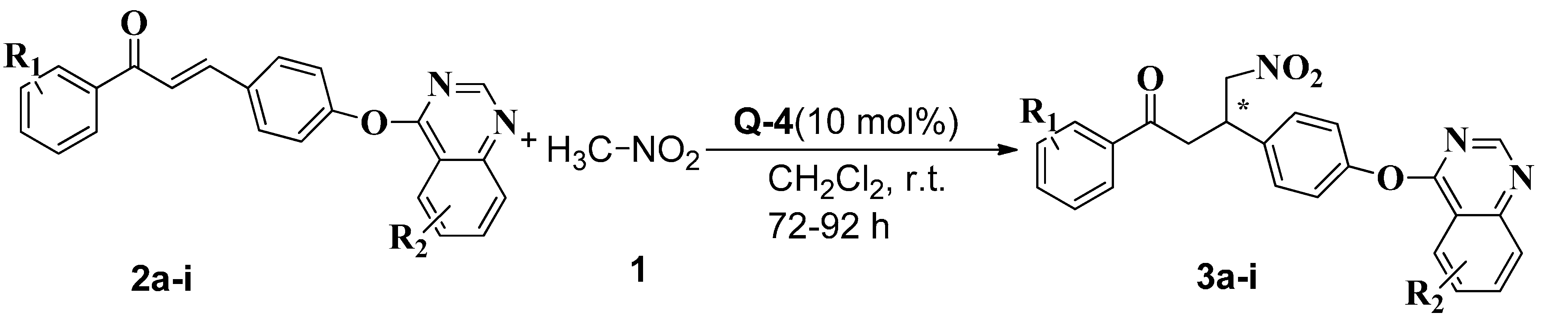
2.3. Synthesis of Chiral Chalcones Derivatives (−)-3a–i
| Compounds | R1 | R2 | Yield [%] [b] | ee [%] [c] |
|---|---|---|---|---|
| (−)-3a | H | 8-Me | 45.7 | 81.4 |
| (−)-3b | 2,4-diCl | 8-Me | 40.0 | 91.5 |
| (−)-3c | H | 6-Me | 40.0 | 86.0 |
| (−)-3d | 2,4-diCl | 6-Me | 38.0 | 91.8 |
| (−)-3e | 4-Cl | H | 35.0 | 99.0 |
| (−)-3f | 4-MeO | H | 42.2 | 92.0 |
| (−)-3g | 4-Cl | 6-Me | 46.1 | 95.9 |
| (−)-3h | 2-Cl | 6-Me | 40.3 | 96.0 |
| (−)-3i | 2-F | 6-Me | 42.5 | 92.5 |
3. Experimental Section
3.1. General Information
3.2. Preparation of Chiral Catalyst Q4
3.3. Preparation of Intermediates 2
3.4. Preparation of Title Chiral Compounds (−)-3a–3i
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, J.Z.; Cheng, C.C.; Shen, L.L.; Wang, Z.K.; Wu, S.B.; Li, W.L.; Chen, S.H.; Zhou, R.P.; Qiu, P.H. Synthetic chalcones with potent antioxidant ability on H2O2-induced apoptosis in PC12 cells. Int. J. Mol. Sci. 2014, 15, 18525–18539. [Google Scholar]
- Kumar, C.S.C.; Loh, W.S.; Ooi, C.W.; Quah, C.K.; Fun, H.K. Heteroarylchalcones: Design, synthesis, X-ray crystal structures and biological evaluation. Molecules 2013, 18, 12707–12724. [Google Scholar]
- Hamada, N.M.M.; Sharshira, E.M. Synthesis and antimicrobial evaluation of some heterocyclic chalcone derivatives. Molecules 2011, 16, 2304–2312. [Google Scholar]
- Nguyen, T.T.N.; Do, T.H.; Huynh, T.N.P.; Tran, C.D.T.; Thai, K.M. Synthesis and antibacterial activity of some heterocyclic chalcone analogues alone and in combination with antibiotics. Molecules 2012, 17, 6684–6696. [Google Scholar]
- Hassan, S.Y. Synthesis, antibacterial and antifungal activity of some new pyrazoline and pyrazole derivatives. Molecules 2013, 18, 2683–2711. [Google Scholar]
- Kang, J.E.; Cho, J.K.; Curtis-Long, M.J.; Ryu, H.W.; Kim, J.H.; Kim, H.J.; Yuk, H.J.; Kim, D.W.; Yuk, H.J.; Kim, D.W.; et al. Preparation of substituted pyridines and pyridazines with angiogenesis inhibiting activity for pharmaceutical use as antitumor agents. Molecules 2013, 18, 140–153. [Google Scholar]
- Solomon, V.R.; Lee, H. Anti-breast cancer activity of heteroarylchalcone derivatives. Biomed. Pharmacother. 2012, 66, 213–220. [Google Scholar]
- Kumar, D.; Kumar, N.M.; Akamatsu, K.; Kusaka, E.; Harada, H.; Ito, T. Synthesis and biological evaluation of indolylchalcones as antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 3916–3919. [Google Scholar]
- Domýngueza, J.N.; Charris, J.E.; Loboa, G.; de Domýnguezb, N.G.; Moreno, M.M.; Riggione, F.; Sanchez, E.; Olson, J.; Rosenthal, P.J. Synthesis of quinolinylchalcones and evaluation of their antimalarial activity. Eur. J. Med. Chem. 2001, 36, 555–560. [Google Scholar]
- Hayat, F.; Moseley, E.; Salahuddin, A.; Zyl, R.L.V.; Azam, A. Antiprotozoal activity of chloro-quinoline based chalcones. Eur. J. Med. Chem. 2011, 46, 1897–1905. [Google Scholar]
- Kotra, V.; Ganapathy, S.; Adapa, S.R. Synthesis of new quinolinylchalcones as anticancer and anti-inflammatory agents. Ind. J. Chem. 2010, 49B, 1109–1116. [Google Scholar]
- Rizvi, S.U.F.; Siddiqui, H.L.; Johns, M.; Detorio, M.; Schinazi, R.F. Anti-HIV-1 and cytotoxicity studies of piperidyl-thienylchalcones and their 2-pyrazoline derivatives. Med. Chem. Res. 2012, 21, 3741–3749. [Google Scholar]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A minireview. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar]
- Luo, J.; Jiang, C.H.; Wang, H.F.; Xu, L.W.; Lu, Y.X. Direct asymmetric Michael addition of phthalide derivatives to chalcones. Tetrahedron Lett. 2013, 54, 5261–5265. [Google Scholar]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14419. [Google Scholar]
- Yang, W.; Du, D.M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar]
- Herchl, R.; Waser, M. Asymmetric cyclopropanation of chalcones using chiral phase-transfer catalysts. Tetrahedron Lett. 2013, 54, 2472–2475. [Google Scholar]
- Liu, Y.; Sun, B.F.; Wang, B.M. Catalytic asymmetric conjugate addition of simple alkyl thiols to α,β-unsaturated N-acylated oxazolidin-2-oneswith bifunctional catalysts. J. Am. Chem. Soc. 2009, 131, 418–419. [Google Scholar]
- Ye, Z.; Malerich, P.; Viresh, H. Squaramide-catalyzed enantioselective of diphenylphosphite to nitroalkenes. Angew. Chem. Int. Ed. 2010, 122, 157–160. [Google Scholar]
- Xie, Y. Synthesis and Biological Activity of Chalcone Derivatives Containing Quinazoline Moiety. Master’s Thesis, Guizhou University, Guiyang, China, 20 June 2013. [Google Scholar]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, D.; Xie, Y.; Ding, Y.; Wu, J.; Hu, D. Synthesis of Chiral Chalcone Derivatives Catalyzed by the Chiral Cinchona Alkaloid Squaramide. Molecules 2014, 19, 19491-19500. https://doi.org/10.3390/molecules191219491
Xie D, Xie Y, Ding Y, Wu J, Hu D. Synthesis of Chiral Chalcone Derivatives Catalyzed by the Chiral Cinchona Alkaloid Squaramide. Molecules. 2014; 19(12):19491-19500. https://doi.org/10.3390/molecules191219491
Chicago/Turabian StyleXie, Dandan, Ying Xie, Yan Ding, Jian Wu, and Deyu Hu. 2014. "Synthesis of Chiral Chalcone Derivatives Catalyzed by the Chiral Cinchona Alkaloid Squaramide" Molecules 19, no. 12: 19491-19500. https://doi.org/10.3390/molecules191219491
APA StyleXie, D., Xie, Y., Ding, Y., Wu, J., & Hu, D. (2014). Synthesis of Chiral Chalcone Derivatives Catalyzed by the Chiral Cinchona Alkaloid Squaramide. Molecules, 19(12), 19491-19500. https://doi.org/10.3390/molecules191219491