Cannabinoids: New Promising Agents in the Treatment of Neurological Diseases


Abstract
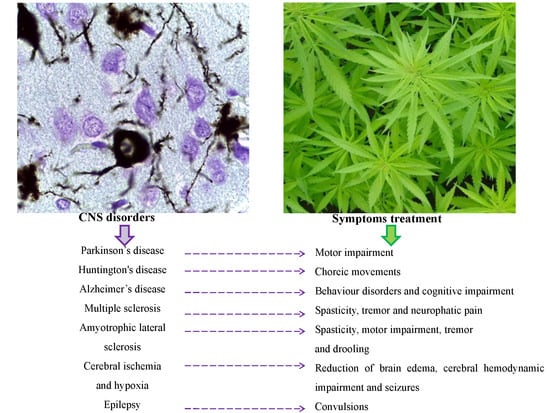
:
1. Introduction
2. Current Cannabinoid-Based Drugs
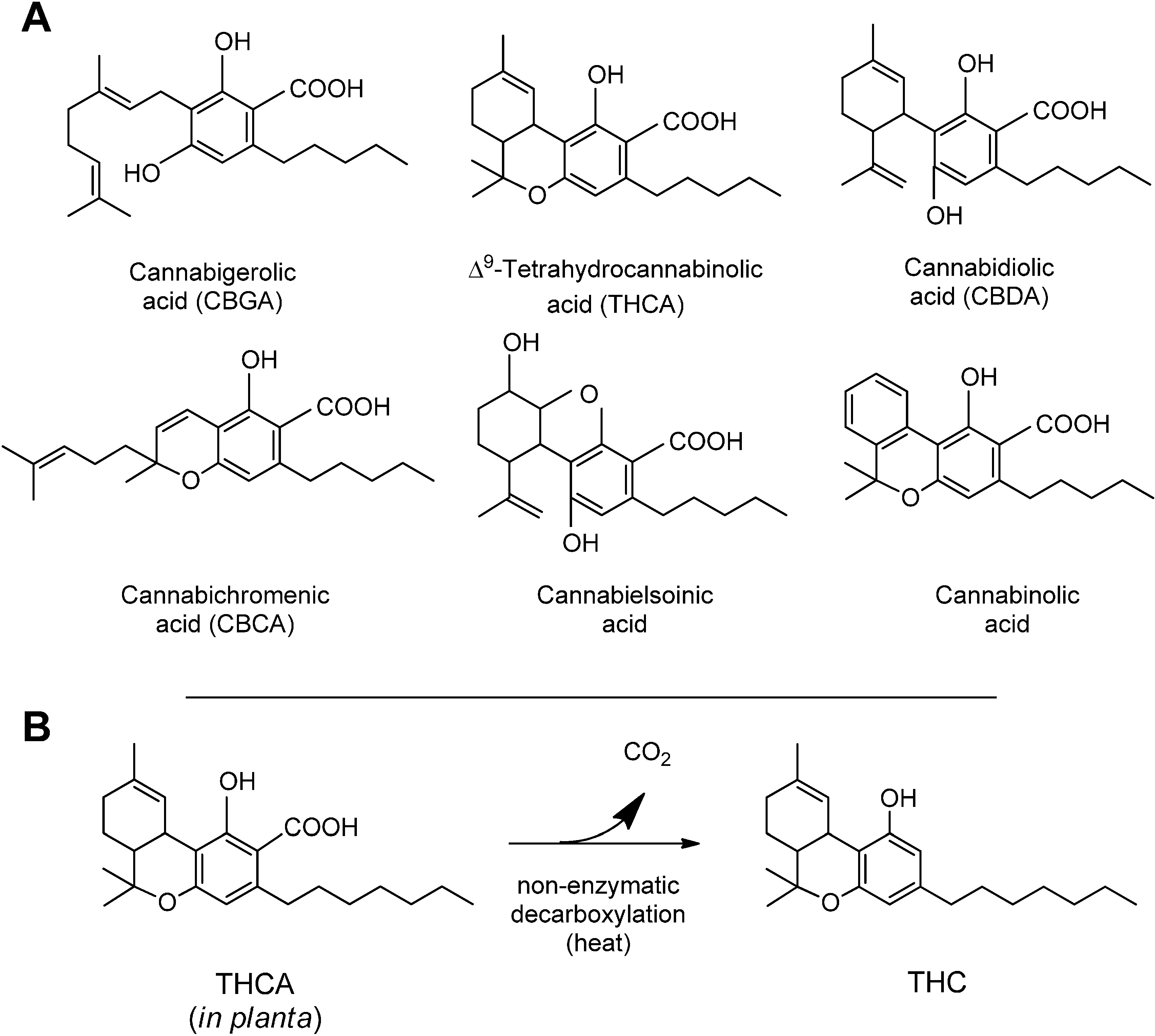
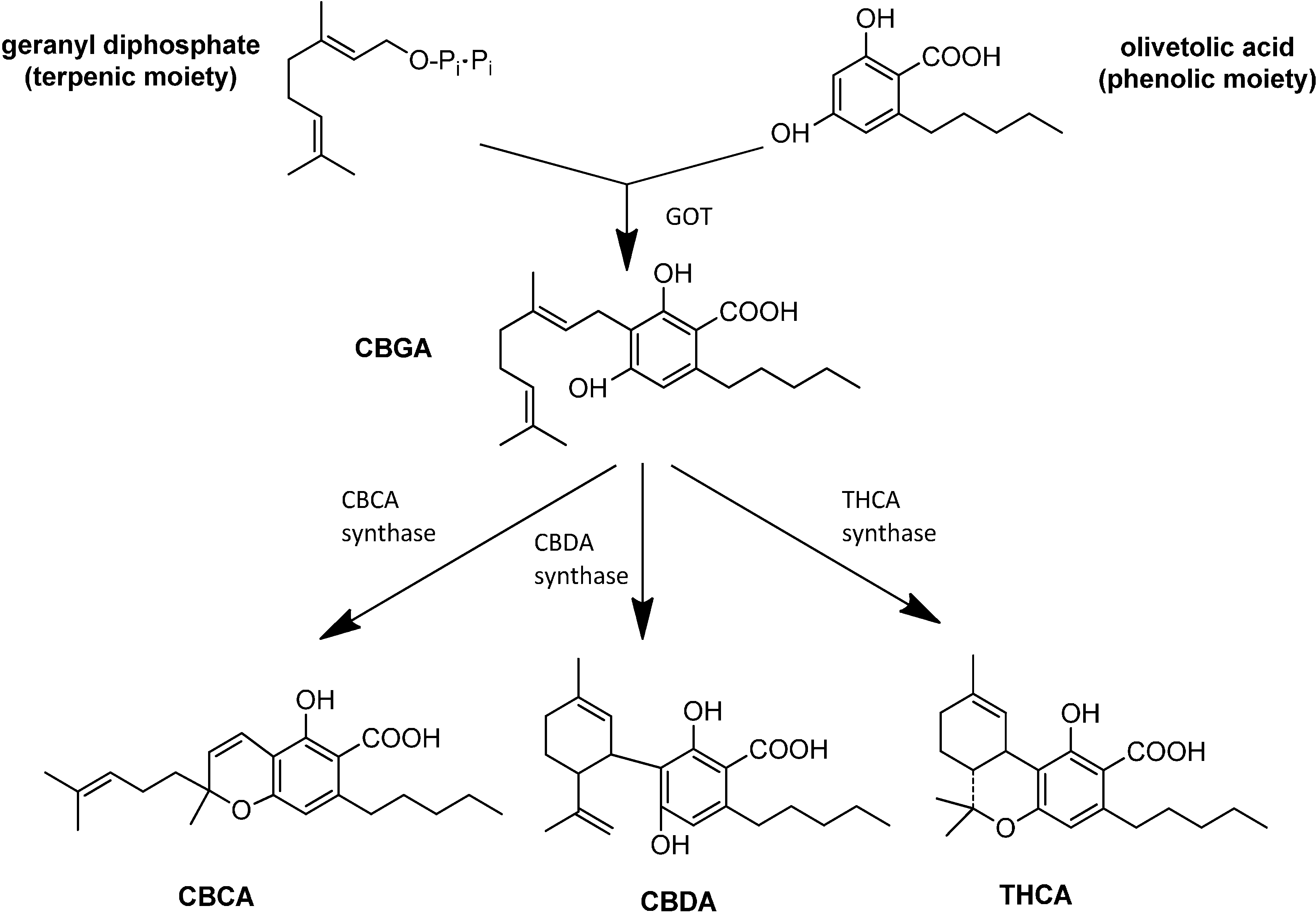
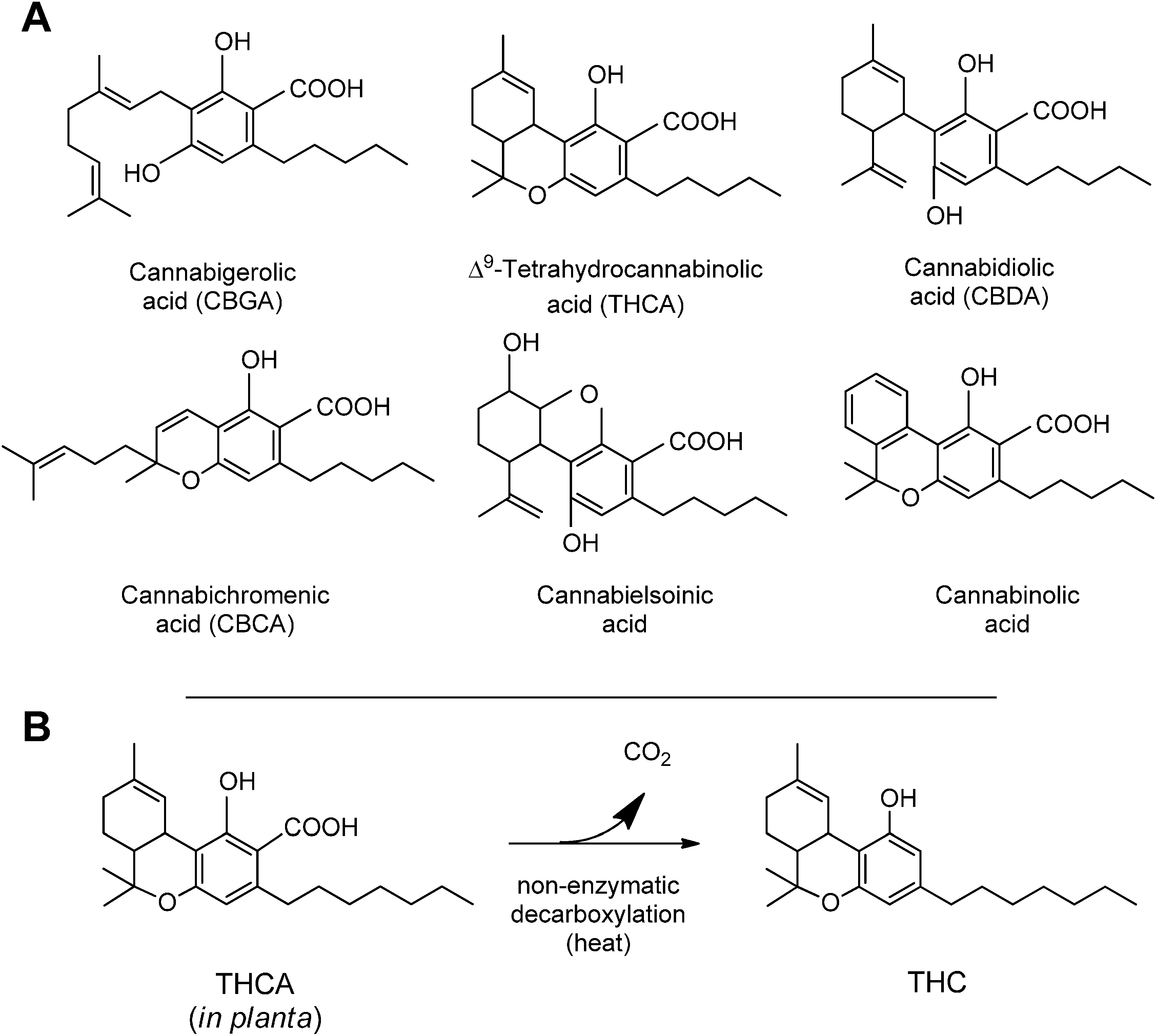
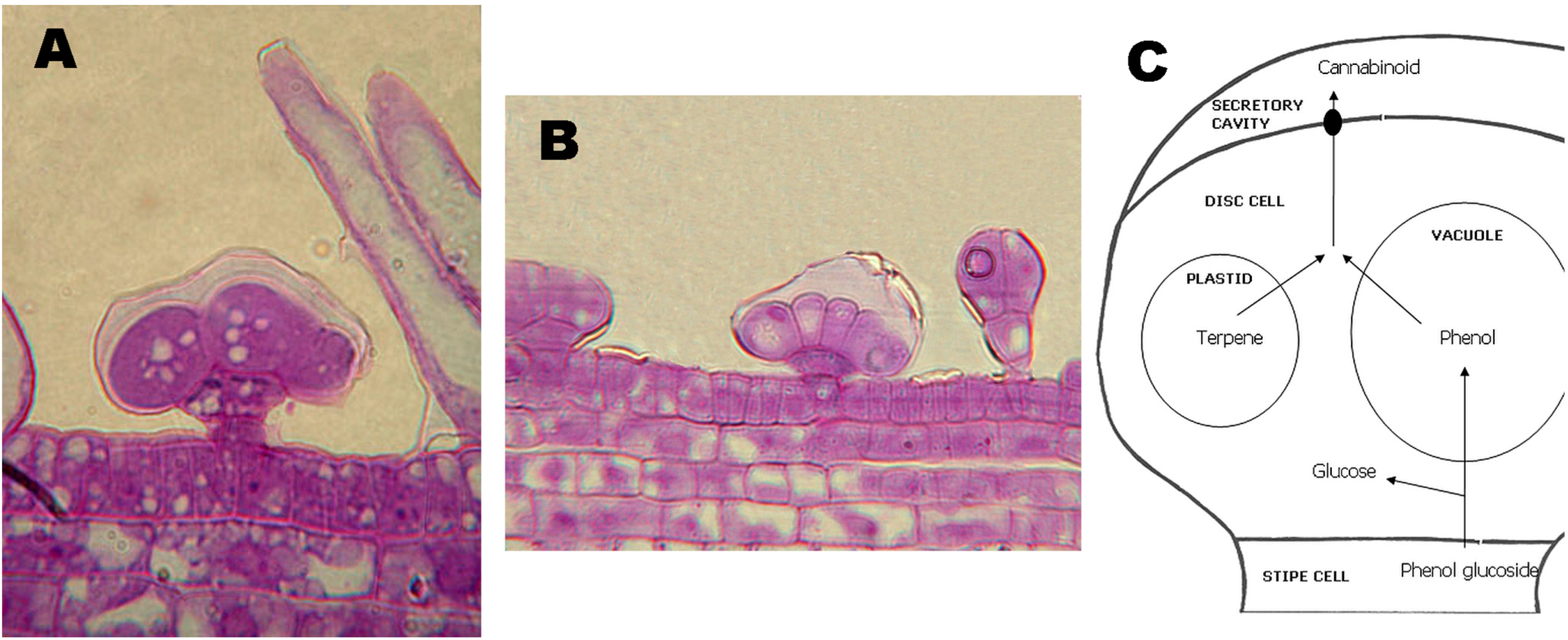
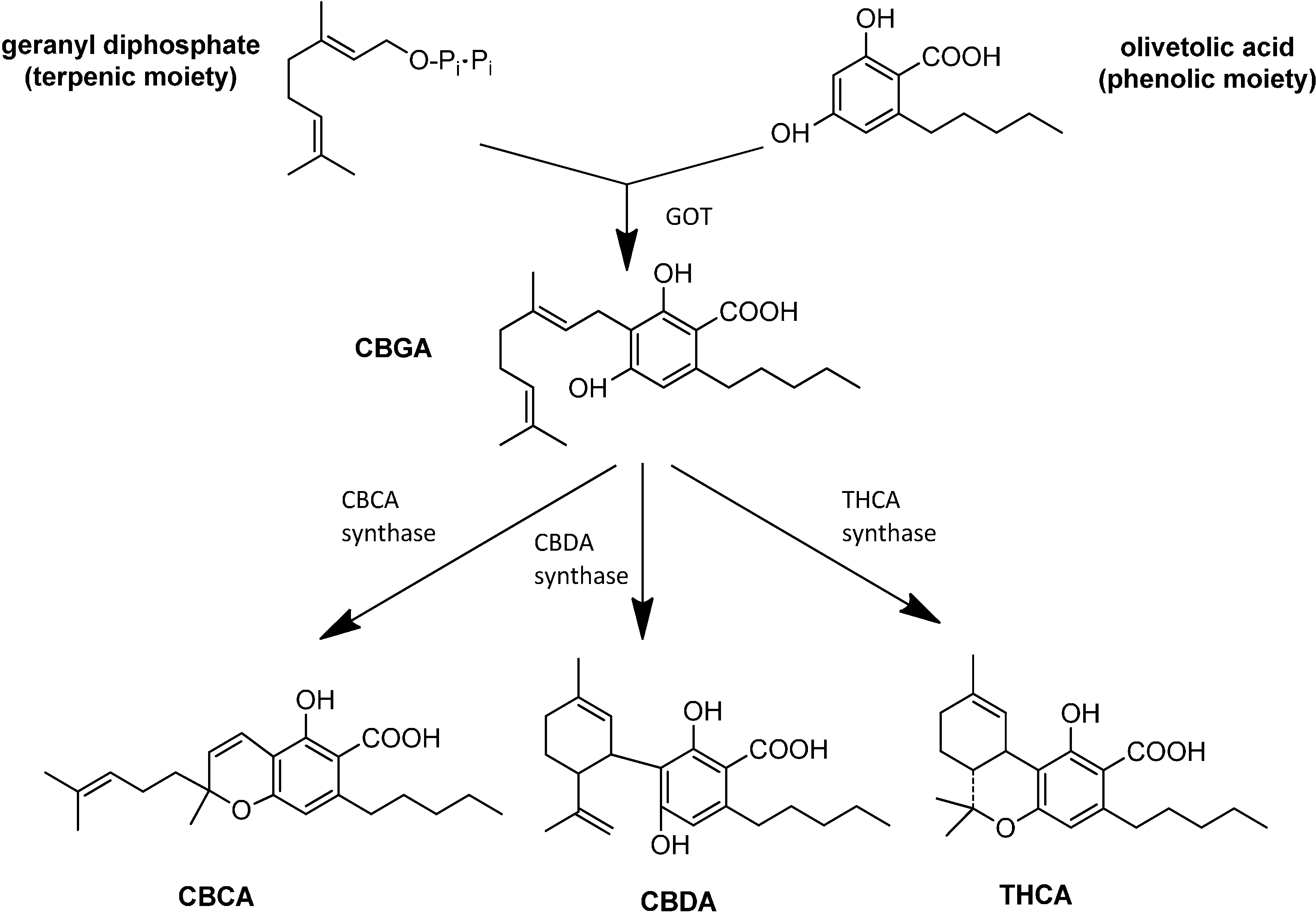
3. Synthesis and Production of Phytocannabinoids

4. Cannabinoid Receptors
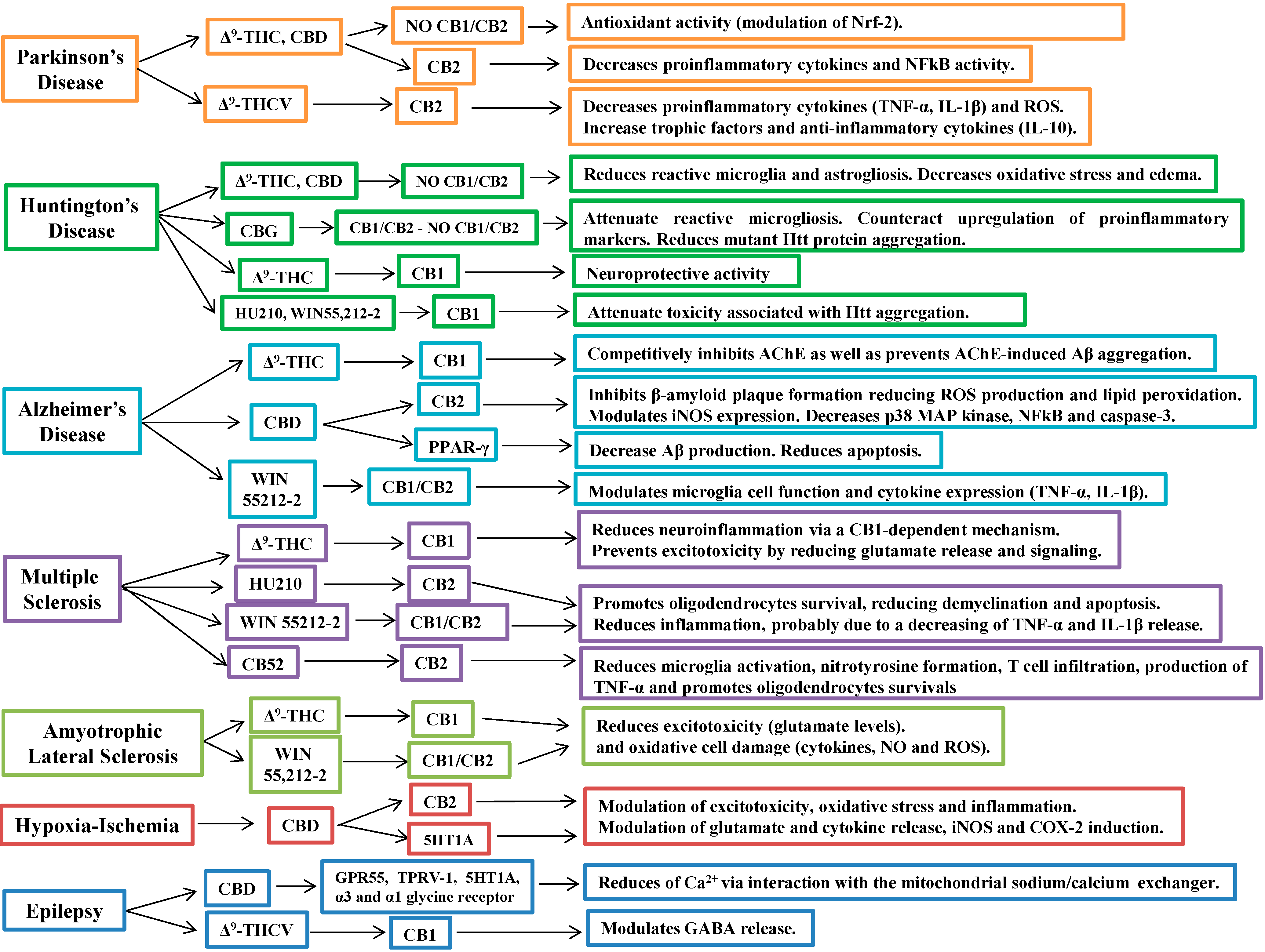
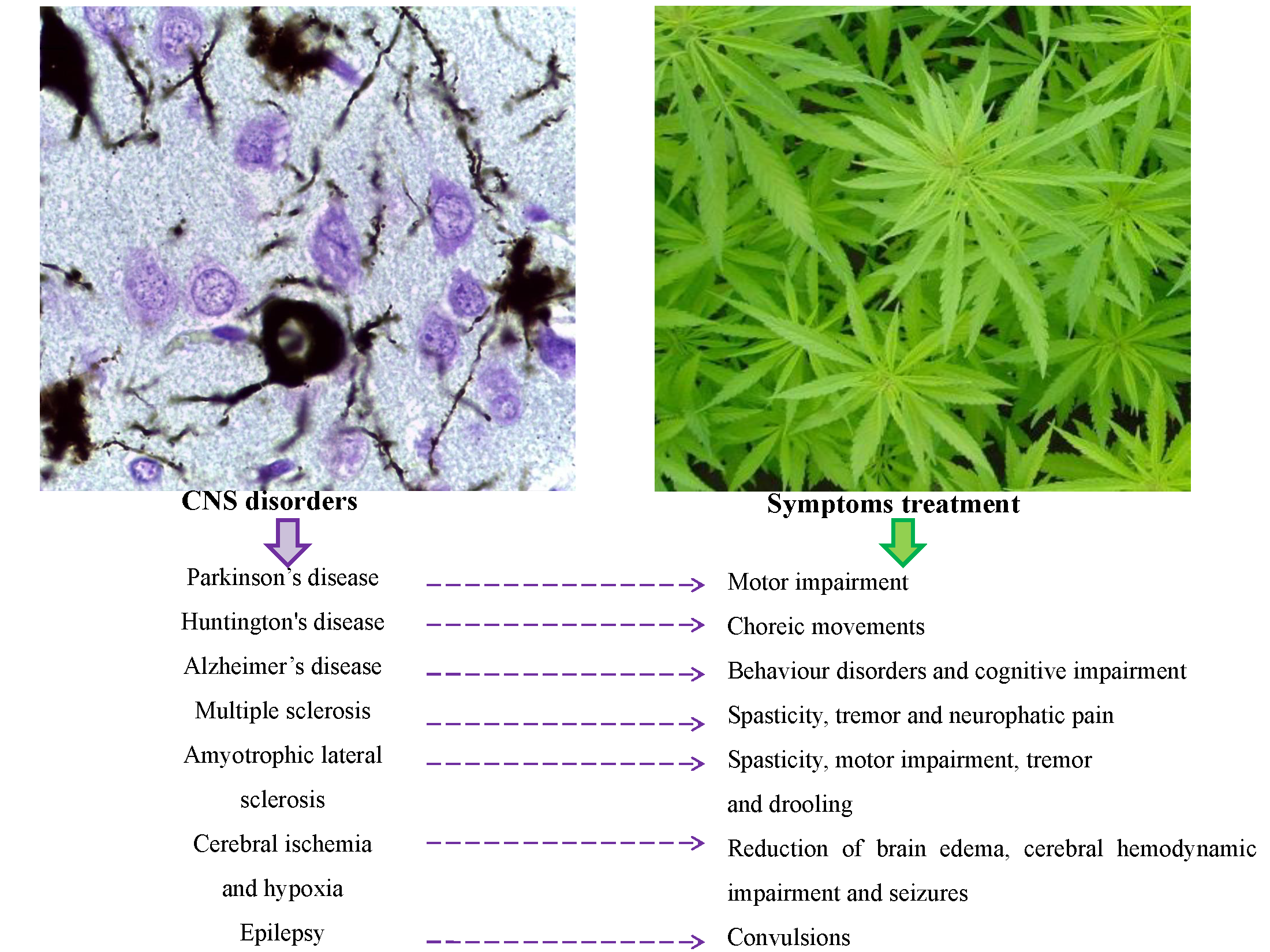
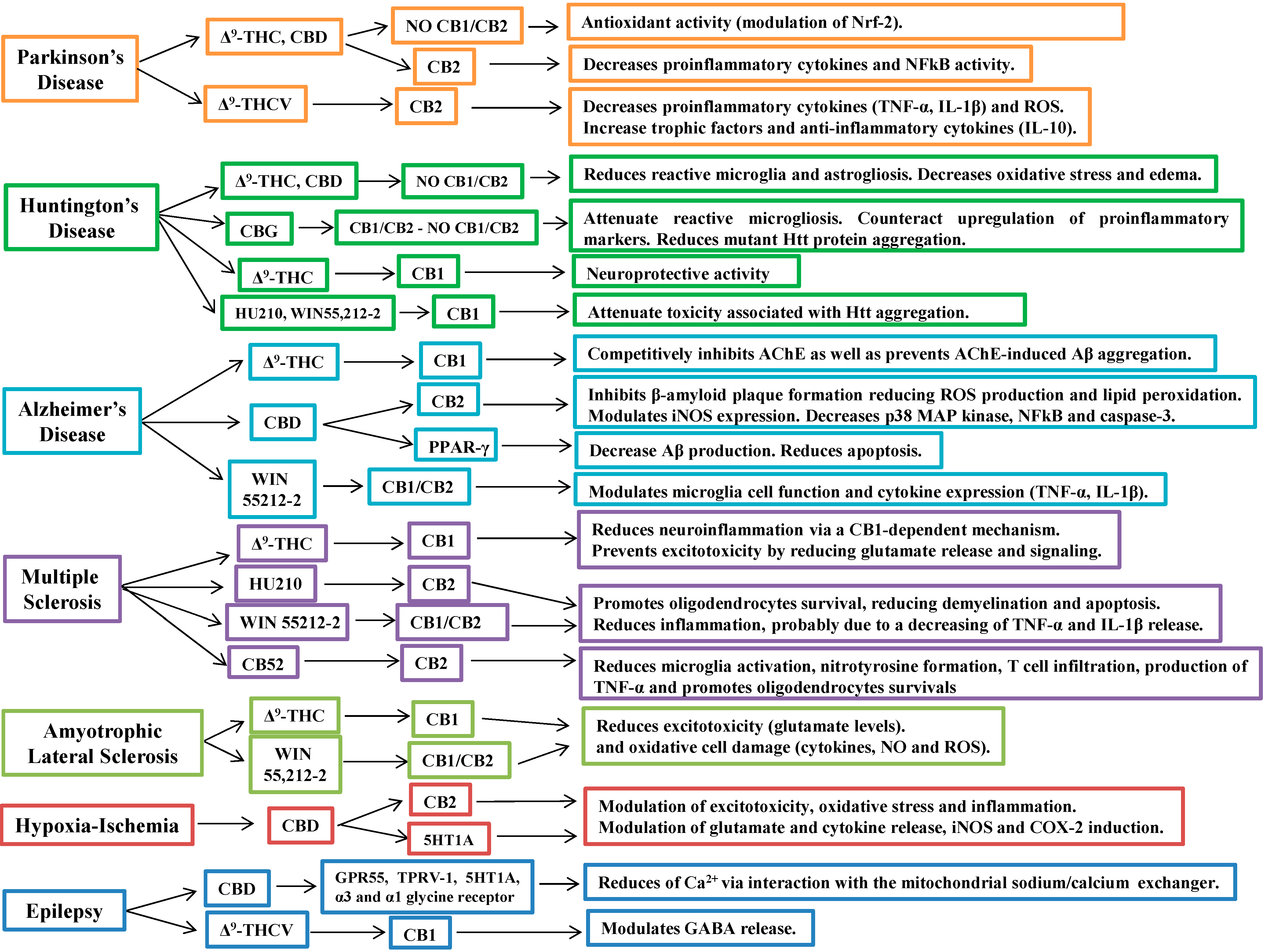
5. Cannabinoids in the Treatment of Neurodegenerative Diseases
5.1. Cannabinoids in Parkinson’s Disease
5.2. Cannabinoids in Huntington’s Disease
5.3. Cannabinoids in Alzheimer’s Disease
5.4. Cannabinoids in Multiple Sclerosis
5.5. Cannabinoids in Amyotrophic Lateral Sclerosis
5.6. Cannabinoids in Cerebral Ischemia and Hypoxia
6. Other Therapeutic Applications of Cannabinoids

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Therapeutic Cannabinoids | Therapeutic Targets | Ref. |
|---|---|---|---|
| PD | Δ9-THC | Tremor | Lastres-Becker et al. [81] |
| CBD | Dystonia and discinesia | Lastres-Becker et al. [81] | |
| WIN 55,212-2 + SR141716A (RIMONABANT) | Akinesia | Maneuf et al. [80] | |
| Δ9-THCV | Diskinesia | Garcia et al. [84] | |
| HD | Δ9-THC | Hyperkinesia and choreic movements | Chiarlone et al. [102] |
| CBG | Hyperkinesia | Valdeolivas et al. [104] | |
| Δ9-THC+ CBD (SATIVEX®) | Hyperkinesia and choreic movements | Sagredo et al. [106] | |
| HU210 and WIN55,212-2 | Hyperkinesia | Scotter et al. [108] | |
| AD | Δ9-THC | Behavior disorders and motor impairment | Eubanks et al. [117] |
| CBD | Learning behavior | Esposito et al. [119]; Martin-Moreno et al. [120] | |
| WIN 55,212-2 | Cognitive impairment | Martin-Moreno et al [120] | |
| Δ9-THC + CBD | Memory and learning impairment | Aso et al. [122] | |
| SYNTHETIC Δ9-THC (Dronabinol) | Nocturnal motor activity, agitation and anorexia | Walther et al. [123] | |
| MS | Δ9-THC | Spasticity | Lyman et al. [126] |
| HU210 and WIN 55,212-2 | Tremor and spasticity | Molina-Holgado et al. [127]; Cabral et al. [128]; Arevalo-Martin et al. [130] | |
| JWH-133 | Tremor and spasticity | Baker et al. [134]; Buccellato et al. [135] | |
| CB52 | Motor impairment | Ribeiro et al. [131] | |
| SYNTHETIC Δ9-THC (NABILONE) | Neuropathic pain | Turcotte et al. [144] | |
| Δ9-THC+ CBD (SATIVEX®) | Spasticity, neuropathic pain and bladder dysfunction | Vaney et al. [14]; Wilkinson et al. [15]; Freeman et al. [145] | |
| ALS | Δ9-THC | Motor impairment and spasticity | Raman et al. [149] |
| WIN 55,212-2 | Tremor and motor impairment | Bilsland et al. [150] | |
| AM1241 | Tremor and motor impairment | Kim et al. [153] | |
| Δ9-THC + CBD (SATIVEX®) | Motor impairment | Moreno-Martet et al. [154] | |
| Cerebral Ischemia and Hypoxia | CBD | Reduction of brain edema, cerebral hemodynamic impairment and seizures | Alvarez et al. [162]; Pazos et al. [166,168] |
| Epilepsy | CBD | Convulsions | Jones et al. [172] |
| CBDV | Convulsions | Scutt et al. [176]; de Petrocellis et al. [177] | |
| Δ9-THCV | Convulsions | Dennis et al. [180]; Ma et al. [181] |
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sakamoto, K.; Akiyama, Y.; Fukui, K.; Kamada, H.; Satoh, S. Characterization, genome sizes and morphology of sex chromosomes in hemp (Cannabis sativa L.). Cytologia 1998, 63, 459–464. [Google Scholar] [CrossRef]
- Mandolino, G. Marker assisted selection and genomics of industrial plants. In Improvement of Crop Plants for Industrial End Uses; Ranalli, P., Ed.; Springer: Dordrecht, The Netherlands, 2007; pp. 59–82. [Google Scholar]
- Forapani, S.; Carboni, A.; Paoletti, C.; Moliterni, V.M.C.; Ranalli, P.; Mandolino, G. Comparison of Hemp Varieties Using Random Amplified Polymorphic DNA Markers. Crop. Sci. 2001, 41, 1682–1689. [Google Scholar] [CrossRef]
- Baker, D.; Pryce, G.; Croxford, J.L.; Brown, P.; Pertwee, R.G.; Makriyannis, A.; Khanolkar, A.; Layward, L.; Fezza, F.; Bisogno, T.; et al. Endocannabinoids control spasticity in a multiple sclerosis model. FASEB J. 2001, 15, 300–302. [Google Scholar] [PubMed]
- Russo, E.B.; Jiang, H.E.; Li, X.; Sutton, A.; Carboni, A.; del Bianco, F.; Mandolino, G.; Potter, D.J.; Zhao, Y.X.; Bera, S.; et al. Phytochemical and genetic analyses of ancient cannabis from Central Asia. J. Exp. Bot. 2008, 59, 4171–4182. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B. History of cannabis and its preparations in saga, science, and sobriquet. Chem. Biodivers. 2007, 4, 1614–1648. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.; Guy, G.W. A tale of two cannabinoids: The therapeutic rationale for combining tetrahydrocannabinol and cannabidiol. Med. Hypotheses 2006, 66, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Gaoni, Y.A. Total Synthesis of Dl-δ-1-Tetrahydrocannabinol, the Active Constituent of Hashish. J. Am. Chem. Soc. 1965, 87, 3273–3275. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Diemel, L.T.; Pryce, G.; Baker, D. Cannabinoids and neuroprotection in CNS inflammatory disease. J. Neurol. Sci. 2005, 233, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Orgado, J.M.; Fernandez-Ruiz, J.; Romero, J. The endocannabinoid system in neuropathological states. Int. Rev. Psychiatry 2009, 21, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, N.E. Cannabinoids in the treatment of chemotherapy-induced nausea and vomiting: beyond prevention of acute emesis. J. Support. Oncol. 2007, 5, 1–19. [Google Scholar] [PubMed]
- Lutge, E.E.; Gray, A.; Siegfried, N. The medical use of cannabis for reducing morbidity and mortality in patients with HIV/AIDS. Cochrane Database Syst. Rev. 2013, 4. Available online: http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD005175.pub3/pdf/standard (accessed on 10 November 2014).
- Sutton, I.R.; Daeninck, P. Cannabinoids in the management of intractable chemotherapy-induced nausea and vomiting and cancer-related pain. J. Support Oncol. 2006, 4, 531–535. [Google Scholar] [PubMed]
- Vaney, C.; Heinzel-Gutenbrunner, M.; Jobin, P.; Tschopp, F.; Gattlen, B.; Hagen, U.; Schnelle, M.; Reif, M. Efficacy, safety and tolerability of an orally administered cannabis extract in the treatment of spasticity in patients with multiple sclerosis: A randomized, double-blind, placebo-controlled, crossover study. Mult. Scler. 2004, 10, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, J.D.; Whalley, B.J.; Baker, D.; Pryce, G.; Constanti, A.; Gibbons, S.; Williamson, E.M. Medicinal cannabis: Is Δ9-tetrahydrocannabinol necessary for all its effects? J. Pharm. Pharmacol. 2003, 55, 1687–1694. [Google Scholar] [CrossRef] [PubMed]
- Brenneisen, R. Chemistry and analysis of phytocannabinoids and other Cannabis constituents. In Marijuana and the Cannabinoids; ElSohly, M.A., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 17–49. [Google Scholar]
- Happyana, N.; Agnolet, S.; Muntendam, R.; van Dam, A.; Schneider, B.; Kayser, O. Analysis of cannabinoids in laser-microdissected trichomes of medicinal Cannabis sativa using LCMS and cryogenic NMR. Phytochemistry 2013, 87, 51–59. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, R.A.; Wijsbeek, J.; Breimer, D.D.; Vree, T.B.; van Ginneken, C.A.; van Rossum, J.M. Cannabinoids with a propyl side chain in cannabis: Occurrence and chromatographic behavior. Science 1972, 175, 778–779. [Google Scholar] [CrossRef] [PubMed]
- De Meijer, E.P.; Bagatta, M.; Carboni, A.; Crucitti, P.; Moliterni, V.M.; Ranalli, P.; Mandolino, G. The inheritance of chemical phenotype in Cannabis sativa L. Genetics 2003, 163, 335–346. [Google Scholar]
- Mandolino, G.; Carboni, A. Potential of marker assisted selection in hemp genetic improvement. Euphytica 2004, 140, 107–120. [Google Scholar] [CrossRef]
- Mahlberg, P.G.; Kim, E.S. Accumulation of cannabinoids in glandular trichomes of Cannabis (Cannabaceae). J. Ind. Hemp. 2004, 9, 15–36. [Google Scholar]
- Basile, A. Understanding the Regulating Mechanisms behind Cannabinoid Biosynthesis. Ph.D. Thesis, Scuola Superiore Sant’Anna, Pisa, Italy, May 2014. [Google Scholar]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Komatsu, K.; Taura, F.; Shoyama, Y. Purification and characterization of cannabichromenic acid synthase from Cannabis sativa. Phytochemistry 1998, 49, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Shoyama, Y.; Tamada, T.; Kurihara, K.; Takeuchi, A.; Taura, F.; Arai, S.; Blaber, M.; Shoyama, Y.; Morimoto, S.; Kuroki, R. Structure and function of 1-tetrahydrocannabinolic acid (THCA) synthase, the enzyme controlling the psychoactivity of Cannabis sativa. J. Mol. Biol. 2012, 423, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Shire, D.; Carillon, C.; Kaghad, M.; Calandra, B.; Rinaldi-Carmona, M.; le Fur, G.; Caput, D.; Ferrara, P. An amino-terminal variant of the central cannabinoid receptor resulting from alternative splicing. J. Biol. Chem. 1995, 270, 3726–3731. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Vu, H.K.; Larsson, N.; Groblewski, T.; Hjorth, S.; Elebring, T.; Sjogren, S.; Greasley, P.J. Identification and characterisation of a novel splice variant of the human CB1 receptor. FEBS Lett. 2005, 579, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Biegon, A.; Kerman, I.A. Autoradiographic study of pre- and postnatal distribution of cannabinoid receptors in human brain. NeuroImage 2001, 14, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Dragunow, M.; Faull, R.L. Cannabinoid receptors in the human brain: A detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 1997, 77, 299–318. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C.; Rahman, R.M.; Nair, S.M.; Sutherland, B.A.; Glass, M.; Appleton, I. Cerebral hypoxia-ischemia and middle cerebral artery occlusion induce expression of the cannabinoid CB2 receptor in the brain. Neurosci. Lett. 2007, 412, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Ross, R.A. Cannabinoid receptors and their ligands. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 101–121. [Google Scholar] [CrossRef]
- Maccarrone, M.; Finazzi-Agro, A. Endocannabinoids and their actions. Vitam. Horm. 2002, 65, 225–255. [Google Scholar] [PubMed]
- Mackie, K.; Devane, W.A.; Hille, B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol. Pharmacol. 1993, 44, 498–503. [Google Scholar] [PubMed]
- Buckley, N.E. The peripheral cannabinoid receptor knockout mice: An update. Br. J. Pharmacol. 2008, 153, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Valverde, O.; Karsak, M.; Zimmer, A. Analysis of the endocannabinoid system by using CB1 cannabinoid receptor knockout mice. Handb. Exp. Pharmacol. 2005, 117–145. [Google Scholar]
- Chuang, H.H.; Prescott, E.D.; Kong, H.; Shields, S.; Jordt, S.E.; Basbaum, A.I.; Chao, M.V.; Julius, D. Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 2001, 411, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Leffler, A.; Malmberg, A.B.; Martin, W.J.; Trafton, J.; Petersen-Zeitz, K.R.; Koltzenburg, M.; Basbaum, A.I.; Julius, D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 2000, 288, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Cristino, L.; de Petrocellis, L.; Pryce, G.; Baker, D.; Guglielmotti, V.; di Marzo, V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neuroscience 2006, 139, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Toth, Z.E.; Cortright, D.N.; Arzubi, M.K.; Krause, J.E.; Elde, R.; Guo, A.; Blumberg, P.M.; Szallasi, A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci. USA 2000, 97, 3655–3660. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J. Novel cannabinoid receptors. Br. J. Pharmacol. 2007, 152, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB(1) and CB(2). Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Henstridge, C.M.; Balenga, N.A.; Schroder, R.; Kargl, J.K.; Platzer, W.; Martini, L.; Arthur, S.; Penman, J.; Whistler, J.L.; Kostenis, E.; et al. GPR55 ligands promote receptor coupling to multiple signalling pathways. Br. J. Pharmacol. 2010, 160, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Kimura, S.; Toshida, T.; Ota, R.; Yamashita, A.; Sugiura, T. Lysophosphatidylinositol induces rapid phosphorylation of p38 mitogen-activated protein kinase and activating transcription factor 2 in HEK293 cells expressing GPR55 and IM-9 lymphoblastoid cells. J. Biochem. 2010, 147, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Buhl, A.M.; Johnson, N.L.; Dhanasekaran, N.; Johnson, G.L. G alpha 12 and G alpha 13 stimulate Rho-dependent stress fiber formation and focal adhesion assembly. J. Biol. Chem. 1995, 270, 24631–24634. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Hu, S.S.; Rimmerman, N.; Juknat, A.; Vogel, Z.; Walker, J.M.; Bradshaw, H.B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Gantz, I.; Muraoka, A.; Yang, Y.K.; Samuelson, L.C.; Zimmerman, E.M.; Cook, H.; Yamada, T. Cloning and chromosomal localization of a gene (GPR18) encoding a novel seven transmembrane receptor highly expressed in spleen and testis. Genomics 1997, 42, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Hoglund, P.J.; Gloriam, D.E.; Lagerstrom, M.C.; Schioth, H.B. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett. 2003, 554, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.M.; Sharp, T. A review of central 5-HT receptors and their function. Neuropharmacology 1999, 38, 1083–1152. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, J.S.; Aghajanian, G.K. (−)-Propranolol blocks the inhibition of serotonergic dorsal raphe cell firing by 5-HT1A selective agonists. Eur. J. Pharmacol. 1986, 128, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Araneda, R.; Andrade, R. 5-Hydroxytryptamine2 and 5-hydroxytryptamine 1A receptors mediate opposing responses on membrane excitability in rat association cortex. Neuroscience 1991, 40, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Moreira, F.A.; Gomes, F.V.; del Bel, E.A.; Guimaraes, F.S. Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Philos. Trans R. Soc. Lond. B Biol. Sci. 2012, 367, 3364–3378. [Google Scholar] [CrossRef] [PubMed]
- Kunos, G.; Osei-Hyiaman, D.; Batkai, S.; Sharkey, K.A.; Makriyannis, A. Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol. Sci. 2009, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Borrelli, F.; Capasso, R.; di Marzo, V.; Mechoulam, R. Non-psychotropic plant cannabinoids: New therapeutic opportunities from an ancient herb. Trends Pharmacol. Sci. 2009, 30, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Jean-Gilles, L.; Gran, B.; Constantinescu, C.S. Interaction between cytokines, cannabinoids and the nervous system. Immunobiology 2010, 215, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; de Filippis, D.; Maiuri, M.C.; de Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-κB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Mukhopadhyay, P.; Batkai, S.; Hasko, G.; Liaudet, L.; Drel, V.R.; Obrosova, I.G.; Pacher, P. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H610–H619. [Google Scholar] [CrossRef]
- Fagan, S.G.; Campbell, V.A. The influence of cannabinoids on generic traits of neurodegeneration. Br. J. Pharmacol. 2014, 171, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialog. Clin. Neurosci. 2004, 6, 259–280. [Google Scholar]
- Tabrez, S.; Jabir, N.R.; Shakil, S.; Greig, N.H.; Alam, Q.; Abuzenadah, A.M.; Damanhouri, G.A.; Kamal, M.A. A synopsis on the role of tyrosine hydroxylase in Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2012, 11, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.D., Jr.; Chang, M.L.; Klaidman, L. Parkinson’s disease—Redox mechanisms. Curr. Med. Chem. 2001, 8, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Brotchie, J.M. CB1 cannabinoid receptor signalling in Parkinson’s disease. Curr. Opin. Pharmacol. 2003, 3, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Gubellini, P.; Picconi, B.; Bari, M.; Battista, N.; Calabresi, P.; Centonze, D.; Bernardi, G.; Finazzi-Agro, A.; Maccarrone, M. Experimental parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. J. Neurosci. 2002, 22, 6900–6907. [Google Scholar] [PubMed]
- Pisani, A.; Fezza, F.; Galati, S.; Battista, N.; Napolitano, S.; Finazzi-Agro, A.; Bernardi, G.; Brusa, L.; Pierantozzi, M.; Stanzione, P.; Maccarrone, M. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Ann. Neurol. 2005, 57, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Pisani, V.; Moschella, V.; Bari, M.; Fezza, F.; Galati, S.; Bernardi, G.; Stanzione, P.; Pisani, A.; Maccarrone, M. Dynamic changes of anandamide in the cerebrospinal fluid of Parkinson’s disease patients. Mov. Disord. 2010, 25, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.J.; Mash, D.C.; Jenner, P. Expression of cannabinoid CB1 receptor mRNA in basal ganglia of normal and parkinsonian human brain. J. Neural. Transm. 2003, 110, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Pisani, V.; Madeo, G.; Tassone, A.; Sciamanna, G.; Maccarrone, M.; Stanzione, P.; Pisani, A. Homeostatic changes of the endocannabinoid system in Parkinson’s disease. Mov. Disord. 2011, 26, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.J.; Darrow, D.P.; Meier, K.T.; Robinson, J.; Schiehser, D.M.; Glahn, D.C.; Nadasdy, Z. Changes in GABA and glutamate concentrations during memory tasks in patients with Parkinson’s disease undergoing DBS surgery. Front. Hum. Neurosci. 2014, 8. [Google Scholar] [CrossRef]
- David, H.N. Towards a reconceptualization of striatal interactions between glutamatergic and dopaminergic neurotransmission and their contribution to the production of movements. Curr. Neuropharm. 2009, 7, 132–141. [Google Scholar] [CrossRef]
- Johnson, K.A.; Conn, P.J.; Niswender, C.M. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Koutsilieri, E.; Riederer, P. Excitotoxicity and new antiglutamatergic strategies in Parkinson’s disease and Alzheimer’s disease. Parkinsonism Relat. Disord. 2007, 13, S329–S331. [Google Scholar] [CrossRef] [PubMed]
- Marjama-Lyons, J.; Koller, W. Tremor-predominant Parkinson’s disease. Approaches to treatment. Drugs Aging 2000, 16, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Maneuf, Y.P.; Crossman, A.R.; Brotchie, J.M. The cannabinoid receptor agonist WIN 55,212–2 reduces D2, but not D1, dopamine receptor-mediated alleviation of akinesia in the reserpine-treated rat model of Parkinson’s disease. Exp. Neurol. 1997, 148, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Molina-Holgado, F.; Ramos, J.A.; Mechoulam, R.; Fernandez-Ruiz, J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: Relevance to Parkinson’s disease. Neurobiol. Dis. 2005, 19, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ruiz, J.; Moreno-Martet, M.; Rodriguez-Cueto, C.; Palomo-Garo, C.; Gomez-Canas, M.; Valdeolivas, S.; Guaza, C.; Romero, J.; Guzman, M.; Mechoulam, R.; et al. Prospects for cannabinoid therapies in basal ganglia disorders. Br. J. Pharmacol. 2011, 163, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arencibia, M.; Garcia, C.; Fernandez-Ruiz, J. Cannabinoids and Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Palomo-Garo, C.; Garcia-Arencibia, M.; Ramos, J.; Pertwee, R.; Fernandez-Ruiz, J. Symptom-relieving and neuroprotective effects of the phytocannabinoid Δ9-THCV in animal models of Parkinson’s disease. Br. J. Pharmacol 2011, 163, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Consroe, P. Brain cannabinoid systems as targets for the therapy of neurological disorders. Neurobiol Dis. 1998, 5, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Muller-Vahl, K.R.; Kolbe, H.; Schneider, U.; Emrich, H.M. Cannabis in movement disorders. Forsch Komplement. 1999, 6, 23–27. [Google Scholar] [CrossRef]
- Brotchie, J.M. Adjuncts to dopamine replacement: A pragmatic approach to reducing the problem of dyskinesia in Parkinson’s disease. Mov. Disord. 1998, 13, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Meschler, J.P.; Conley, T.J.; Howlett, A.C. Cannabinoid and dopamine interaction in rodent brain: Effects on locomotor activity. Pharmacol. Biochem. Behav. 2000, 67, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Sieradzan, K.A.; Fox, S.H.; Hill, M.; Dick, J.P.; Crossman, A.R.; Brotchie, J.M. Cannabinoids reduce levodopa-induced dyskinesia in Parkinson’s disease: A pilot study. Neurology 2001, 57, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.B.; Bain, P.G.; Teare, L.; Liu, X.; Joint, C.; Wroath, C.; Parkin, S.G.; Fox, P.; Wright, D.; Hobart, J.; et al. Cannabis for dyskinesia in Parkinson disease: A randomized double-blind crossover study. Neurology 2004, 63, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef] [PubMed]
- Francelle, L.; Galvan, L.; Brouillet, E. Possible involvement of self-defense mechanisms in the preferential vulnerability of the striatum in Huntington’s disease. Front. Cell Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Huntington’s disease: Pathogenesis to animal models. Pharmacol. Rep. 2010, 62, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, N.R.; Paulsen, J.; Braff, D.L.; Butters, N.; Geyer, M.A.; Swenson, M.R. Impaired prepulse inhibition of acoustic and tactile startle response in patients with Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 1995, 58, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; Pazos, M.R.; Valdeolivas, S.; Fernandez-Ruiz, J. Cannabinoids: Novel medicines for the treatment of Huntington’s disease. Recent Pat. CNS Drug. Discov. 2012, 7, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Groves, P.M. A theory of the functional organization of the neostriatum and the neostriatal control of voluntary movement. Brain Res. 1983, 286, 9–32. [Google Scholar]
- Penney, J.B., Jr.; Young, A.B. Speculations on the functional anatomy of basal ganglia disorders. Annu. Rev. Neurosci. 1983, 6, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Aguado, T.; Pazos, M.R.; Julien, B.; Carrasco, C.; Resel, E.; Sagredo, O.; Benito, C.; Romero, J.; Azcoitia, I.; et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain 2009, 132, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; Gonzalez, S.; Aroyo, I.; Pazos, M.R.; Benito, C.; Lastres-Becker, I.; Romero, J.P.; Tolon, R.M.; Mechoulam, R.; Brouillet, E.; et al. Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: Relevance for Huntington’s disease. Glia 2009, 57, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Chiarlone, A.; Bellocchio, L.; Blazquez, C.; Resel, E.; Soria-Gomez, E.; Cannich, A.; Ferrero, J.J.; Sagredo, O.; Benito, C.; Romero, J.; et al. A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc. Natl. Acad. Sci. USA 2014, 111, 8257–8262. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Valdeolivas, S.; Navarrete, C.; Cantarero, I.; Bellido, M.L.; Munoz, E.; Sagredo, O. Neuroprotective Properties of Cannabigerol in Huntington’s Disease: Studies in R6/2 Mice and 3-Nitropropionate-lesioned Mice. Neurotherapeutics 2014, 1–14. [Google Scholar] [CrossRef]
- Valdeolivas, S.; Satta, V.; Pertwee, R.G.; Fernandez-Ruiz, J.; Sagredo, O. Sativex-like combination of phytocannabinoids is neuroprotective in malonate-lesioned rats, an inflammatory model of Huntington’s disease: Role of CB1 and CB2 receptors. ACS Chem. Neurosci. 2012, 3, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Sagredo, O.; Pazos, M.R.; Satta, V.; Ramos, J.A.; Pertwee, R.G.; Fernandez-Ruiz, J. Neuroprotective effects of phytocannabinoid-based medicines in experimental models of Huntington’s disease. J. Neurosci. Res. 2011, 89, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ruiz, J.; Romero, J.; Velasco, G.; Tolon, R.M.; Ramos, J.A.; Guzman, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Goodfellow, C.E.; Graham, E.S.; Dragunow, M.; Glass, M. Neuroprotective potential of CB1 receptor agonists in an in vitro model of Huntington’s disease. Br. J. Pharmacol. 2010, 160, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Consroe, P.; Laguna, J.; Allender, J.; Snider, S.; Stern, L.; Sandyk, R.; Kennedy, K.; Schram, K. Controlled clinical trial of cannabidiol in Huntington’s disease. Pharmacol. Biochem. Behav. 1991, 40, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.; Rickards, H. Nabilone could treat chorea and irritability in Huntington’s disease. J. Neuropsychiatry Clin. Neurosci. 2006, 18, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Muller-Vahl, K.R.; Schneider, U.; Emrich, H.M. Nabilone increases choreatic movements in Huntington’s disease. Mov. Disord. 1999, 14, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.; Mitchell, I.; Patel, S.; Ives, N.; Rickards, H. A pilot study using nabilone for symptomatic treatment in Huntington’s disease. Mov. Disord. 2009, 24, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Mi, K.; Johnson, G.V. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2006, 3, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Tolon, R.M.; Pazos, M.R.; Nunez, E.; Castillo, A.I.; Romero, J. Cannabinoid CB2 receptors in human brain inflammation. Br. J. Pharmacol. 2008, 153, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Eubanks, L.M.; Rogers, C.J.; Beuscher, A.E., IV; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K. A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Scuderi, C.; Savani, C.; Steardo, L., Jr.; de Filippis, D.; Cottone, P.; Iuvone, T.; Cuomo, V.; Steardo, L. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1β and iNOS expression. Br. J. Pharmacol. 2007, 151, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Martin-Moreno, A.M.; Reigada, D.; Ramirez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharm. 2011, 79, 964–973. [Google Scholar] [CrossRef]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARgamma involvement. Phytother. Res. 2014, 28, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Aso, E.; Sanchez-Pla, A.; Vegas-Lozano, E.; Maldonado, R.; Ferrer, I. Cannabis-Based Medicine Reduces Multiple Pathological Processes in AbetaPP/PS1 Mice. J. Alzheimers Dis. 2014. Available online: http://scibite.com/site/library/2014_8/1/0/25125475.html (accessed on 10 November 2014).
- Walther, S.; Mahlberg, R.; Eichmann, U.; Kunz, D. Δ9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology 2006, 185, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Siffrin, V.; Brandt, A.U.; Herz, J.; Zipp, F. New insights into adaptive immunity in chronic neuroinflammation. Adv. Immunol. 2007, 96, 1–40. [Google Scholar] [PubMed]
- Pryce, G.; Baker, D. Control of spasticity in a multiple sclerosis model is mediated by CB1, not CB2, cannabinoid receptors. Br. J. Pharmacol. 2007, 150, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Lyman, W.D.; Sonett, J.R.; Brosnan, C.F.; Elkin, R.; Bornstein, M.B. Delta 9-tetrahydrocannabinol: A novel treatment for experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1989, 23, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Molina-Holgado, E.; Vela, J.M.; Arevalo-Martin, A.; Almazan, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids promote oligodendrocyte progenitor survival: Involvement of cannabinoid receptors and phosphatidylinositol-3 kinase/Akt signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [PubMed]
- Cabral, G.A.; Harmon, K.N.; Carlisle, S.J. Cannabinoid-mediated inhibition of inducible nitric oxide production by rat microglial cells: Evidence for CB1 receptor participation. Adv. Exp. Med. Biol. 2001, 493, 207–214. [Google Scholar] [PubMed]
- Klein, T.W.; Lane, B.; Newton, C.A.; Friedman, H. The cannabinoid system and cytokine network. Proc. Soc. Exp. Biol. Med. 2000, 225, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Arevalo-Martin, A.; Vela, J.M.; Molina-Holgado, E.; Borrell, J.; Guaza, C. Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J. Neurosci. 2003, 23, 2511–2516. [Google Scholar] [PubMed]
- Ribeiro, R.; Yu, F.; Wen, J.; Vana, A.; Zhang, Y. Therapeutic potential of a novel cannabinoid agent CB52 in the mouse model of experimental autoimmune encephalomyelitis. Neuroscience 2013, 254, 427–442. [Google Scholar] [CrossRef] [PubMed]
- Cabranes, A.; Venderova, K.; de Lago, E.; Fezza, F.; Sanchez, A.; Mestre, L.; Valenti, M.; Garcia-Merino, A.; Ramos, J.A.; di Marzo, V.; et al. Decreased endocannabinoid levels in the brain and beneficial effects of agents activating cannabinoid and/or vanilloid receptors in a rat model of multiple sclerosis. Neurobiol. Dis. 2005, 20, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Cabranes, A.; Pryce, G.; Baker, D.; Fernandez-Ruiz, J. Changes in CB1 receptors in motor-related brain structures of chronic relapsing experimental allergic encephalomyelitis mice. Brain Res. 2006, 1107, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.; Pryce, G.; Croxford, J.L.; Brown, P.; Pertwee, R.G.; Huffman, J.W.; Layward, L. Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature 2000, 404, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Buccellato, E.; Carretta, D.; Utan, A.; Cavina, C.; Speroni, E.; Grassi, G.; Candeletti, S.; Romualdi, P. Acute and chronic cannabinoid extracts administration affects motor function in a CREAE model of multiple sclerosis. J. Ethnopharmacol. 2011, 133, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Bahr, B.A.; Karanian, D.A.; Makanji, S.S.; Makriyannis, A. Targeting the endocannabinoid system in treating brain disorders. Expert Opin. Investig. Drugs 2006, 15, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Bari, M.; Rossi, S.; Prosperetti, C.; Furlan, R.; Fezza, F.; de Chiara, V.; Battistini, L.; Bernardi, G.; Bernardini, S.; et al. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain 2007, 130, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Jean-Gilles, L.; Feng, S.; Tench, C.R.; Chapman, V.; Kendall, D.A.; Barrett, D.A.; Constantinescu, C.S. Plasma endocannabinoid levels in multiple sclerosis. J. Neurol. Sci. 2009, 287, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.B.; Crone, C.; Hultborn, H. The spinal pathophysiology of spasticity—From a basic science point of view. Acta Physiol. 2007, 189, 171–180. [Google Scholar] [CrossRef]
- Paisley, S.; Beard, S.; Hunn, A.; Wight, J. Clinical effectiveness of oral treatments for spasticity in multiple sclerosis: A systematic review. Mult. Scler. 2002, 8, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, J.; Fox, P.; Sanders, H.; Wright, D.; Vickery, J.; Nunn, A.; Thompson, A.; Group, U.M.R. Cannabinoids for treatment of spasticity and other symptoms related to multiple sclerosis (CAMS study): Multicentre randomised placebo-controlled trial. Lancet 2003, 362, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, J.P.; Sanders, H.P.; Wright, D.E.; Vickery, P.J.; Ingram, W.M.; Reilly, S.M.; Nunn, A.J.; Teare, L.J.; Fox, P.J.; Thompson, A.J. Cannabinoids in multiple sclerosis (CAMS) study: Safety and efficacy data for 12 months follow up. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1664–1669. [Google Scholar] [CrossRef] [PubMed]
- Zajicek, J.; Ball, S.; Wright, D.; Vickery, J.; Nunn, A.; Miller, D.; Gomez Cano, M.; McManus, D.; Mallik, S.; Hobart, J. Cupid investigator group. Effect of dronabinol on progression in progressive multiple sclerosis (CUPID): A randomised, placebo-controlled trial. Lancet Neurol. 2013, 12, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, D.; Doupe, M.; Torabi, M.; Gomori, A.; Ethans, K.; Esfahani, F.; Galloway, K.; Namaka, M. Nabilone as an Adjunctive to Gabapentin for Multiple Sclerosis-Induced Neuropathic Pain: A Randomized Controlled Trial. Pain Med. 2014. [Google Scholar] [CrossRef]
- Freeman, R.M.; Adekanmi, O.; Waterfield, M.R.; Waterfield, A.E.; Wright, D.; Zajicek, J. The effect of cannabis on urge incontinence in patients with multiple sclerosis: A multicentre, randomised placebo-controlled trial (CAMS-LUTS). Int. Urogynecol. J. Pelvic Floor Dysfunct. 2006, 17, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, S.J.; Witherden, A.S.; Hafezparast, M.; Martin, J.E.; Fisher, E.M. Mice, the motor system, and human motor neuron pathology. Mamm. Genome 2000, 11, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Meyer, T.; Riepe, M.W. The role of excitotoxicity in ALS—What is the evidence? J. Neurol. 2000, 247, I7–I16. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, W. Oxidative stress in amyotrophic lateral sclerosis. J. Neurol. 2000, 247, I1–I6. [Google Scholar] [CrossRef] [PubMed]
- Raman, C.; McAllister, S.D.; Rizvi, G.; Patel, S.G.; Moore, D.H.; Abood, M.E. Amyotrophic lateral sclerosis: Delayed disease progression in mice by treatment with a cannabinoid. Amyotroph. Lateral Scler. Other Motor Neuron. Disord. 2004, 5, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Bilsland, L.G.; Dick, J.R.; Pryce, G.; Petrosino, S.; di Marzo, V.; Baker, D.; Greensmith, L. Increasing cannabinoid levels by pharmacological and genetic manipulation delay disease progression in SOD1 mice. FASEB J. 2006, 20, 1003–1005. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Hong, S.; Witting, A.; Moller, T.; Stella, N.; Kliot, M. Cannabinol delays symptom onset in SOD1 (G93A) transgenic mice without affecting survival. Amyotroph. Lateral Scler. Other Motor Neuron. Disord. 2005, 6, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, B.G.; Blazquez, C.; Gomez del Pulgar, T.; Guzman, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Moore, D.H.; Makriyannis, A.; Abood, M.E. AM1241, a cannabinoid CB2 receptor selective compound, delays disease progression in a mouse model of amyotrophic lateral sclerosis. Eur. J. Pharmacol. 2006, 542, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Martet, M.; Espejo-Porras, F.; Fernandez-Ruiz, J.; de Lago, E. Changes in endocannabinoid receptors and enzymes in the spinal cord of SOD1(G93A) transgenic mice and evaluation of a Sativex((R)) -like combination of phytocannabinoids: Interest for future therapies in amyotrophic lateral sclerosis. CNS Neurosci. Ther. 2014, 20, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6. [Google Scholar] [CrossRef]
- Carter, G.T.; Rosen, B.S. Marijuana in the management of amyotrophic lateral sclerosis. Am. J. Hosp. Palliat. Care 2001, 18, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Amtmann, D.; Weydt, P.; Johnson, K.L.; Jensen, M.P.; Carter, G.T. Survey of cannabis use in patients with amyotrophic lateral sclerosis. Am. J. Hosp. Palliat. Care 2004, 21, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; de Deyn, P.P. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg. 2009, 111, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Sahota, P.; Savitz, S.I. Investigational therapies for ischemic stroke: Neuroprotection and neurorecovery. Neurotherapeutics 2011, 8, 434–451. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Velayudhan, L.; van Diepen, E.; Marudkar, M.; Hands, O.; Suribhatla, S.; Prettyman, R.; Murray, J.; Baillon, S.; Bhattacharyya, S. Therapeutic potential of cannabinoids in neurodegenerative disorders: A selective review. Curr. Pharm. Des. 2014, 20, 2218–2230. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.J.; Lafuente, H.; Rey-Santano, M.C.; Mielgo, V.E.; Gastiasoro, E.; Rueda, M.; Pertwee, R.G.; Castillo, A.I.; Romero, J.; Martinez-Orgado, J. Neuroprotective effects of the nonpsychoactive cannabinoid cannabidiol in hypoxic-ischemic newborn piglets. Pediatr. Res. 2008, 64, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Mishima, K.; Hayakawa, K.; Abe, K.; Ikeda, T.; Egashira, N.; Iwasaki, K.; Fujiwara, M. Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism. Stroke 2005, 36, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Maas, A.I.; Murray, G.; Henney, H., III; Kassem, N.; Legrand, V.; Mangelus, M.; Muizelaar, J.P.; Stocchetti, N.; Knoller, N.; Pharmos, T.B.I.I. Efficacy and safety of dexanabinol in severe traumatic brain injury: Results of a phase III randomised, placebo-controlled, clinical trial. Lancet Neurol. 2006, 5, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.R.; Cinquina, V.; Gomez, A.; Layunta, R.; Santos, M.; Fernandez-Ruiz, J.; Martinez-Orgado, J. Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 2012, 63, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Tolon, M.R.; Fernandez-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martinez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of cannabidiol neuroprotection in hypoxic-ischemic newborn pigs: Role of 5HT(1A) and CB2 receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.S.; Lowenstein, D.H. Epilepsy. N. Engl. J. Med. 2003, 349, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Kwan, P.; Brodie, M.J. Emerging drugs for epilepsy. Expert Opin. Emerg. Drugs 2007, 12, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Compton, D.R.; Aceto, M.D.; Lowe, J.; Martin, B.R. In vivo characterization of a specific cannabinoid receptor antagonist (SR141716A): Inhibition of delta 9-tetrahydrocannabinol-induced responses and apparent agonist activity. J. Pharmacol. Exp. Ther. 1996, 277, 586–594. [Google Scholar] [PubMed]
- Jones, N.A.; Hill, A.J.; Smith, I.; Bevan, S.A.; Williams, C.M.; Whalley, B.J.; Stephens, G.J. Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Ther. 2010, 332, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Weston, S.E.; Jones, N.A.; Smith, I.; Bevan, S.A.; Williamson, E.M.; Stephens, G.J.; Williams, C.M.; Whalley, B.J. Δ9-Tetrahydrocannabivarin suppresses in vitro epileptiform and in vivo seizure activity in adult rats. Epilepsia 2010, 51, 1522–1532. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.J.; Mercier, M.S.; Hill, T.D.; Glyn, S.E.; Jones, N.A.; Yamasaki, Y.; Futamura, T.; Duncan, M.; Stott, C.G.; Stephens, G.J.; et al. Cannabidivarin is anticonvulsant in mouse and rat. Br. J. Pharmacol. 2012, 167, 1629–1642. [Google Scholar] [CrossRef] [PubMed]
- Ryan, D.; Drysdale, A.J.; Lafourcade, C.; Pertwee, R.G.; Platt, B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J. Neurosci. 2009, 29, 2053–2063. [Google Scholar] [CrossRef] [PubMed]
- Scutt, A.; Williamson, E.M. Cannabinoids stimulate fibroblastic colony formation by bone marrow cells indirectly via CB2 receptors. Calcif. Tissue Int. 2007, 80, 50–59. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allara, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Howell, F.; Williams, G.; Minassi, A.; Cascio, M.G.; Ligresti, A.; Matias, I.; Schiano-Moriello, A.; Paul, P.; Williams, E.J.; et al. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell. Biol. 2003, 163, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Pertwee, R.G. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr. Med. Chem. 2010, 17, 1360–1381. [Google Scholar] [CrossRef] [PubMed]
- Dennis, I.; Whalley, B.J.; Stephens, G.J. Effects of Delta9-tetrahydrocannabivarin on [35S]GTPgammaS binding in mouse brain cerebellum and piriform cortex membranes. Br. J. Pharmacol. 2008, 154, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.L.; Weston, S.E.; Whalley, B.J.; Stephens, G.J. The phytocannabinoid Δ9-tetrahydrocannabivarin modulates inhibitory neurotransmission in the cerebellum. Br. J. Pharmacol. 2008, 154, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Guimaraes, F.S. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology 2008, 199, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Russo, E.B.; Guy, G.W.; Robson, P.J. Cannabis, pain, and sleep: Lessons from therapeutic clinical trials of Sativex, a cannabis-based medicine. Chem. Biodivers. 2007, 4, 1729–1743. [Google Scholar] [CrossRef] [PubMed]
- Rahn, E.J.; Hohmann, A.G. Cannabinoids as pharmacotherapies for neuropathic pain: From the bench to the bedside. Neurotherapeutics 2009, 6, 713–737. [Google Scholar] [CrossRef] [PubMed]
- Borsook, D. Neurological diseases and pain. Brain 2012, 135, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Koltzenburg, M.; Scadding, J. Neuropathic pain. Curr. Opin. Neurol. 2001, 14, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Galuppo, M.; Giacoppo, S.; Bramanti, P.; Mazzon, E. Use of natural compounds in the management of diabetic peripheral neuropathy. Molecules 2014, 19, 2877–2895. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Trovato, A.E.; Comelli, F.; Giagnoni, G.; Colleoni, M. The non-psychoactive cannabis constituent cannabidiol is an orally effective therapeutic agent in rat chronic inflammatory and neuropathic pain. Eur. J. Pharmacol. 2007, 556, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Calignano, A.; la Rana, G.; Giuffrida, A.; Piomelli, D. Control of pain initiation by endogenous cannabinoids. Nature 1998, 394, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Farquhar-Smith, W.P.; Egertova, M.; Bradbury, E.J.; McMahon, S.B.; Rice, A.S.; Elphick, M.R. Cannabinoid CB(1) receptor expression in rat spinal cord. Mol. Cell. Neurosci. 2000, 15, 510–521. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacoppo, S.; Mandolino, G.; Galuppo, M.; Bramanti, P.; Mazzon, E. Cannabinoids: New Promising Agents in the Treatment of Neurological Diseases. Molecules 2014, 19, 18781-18816. https://doi.org/10.3390/molecules191118781
Giacoppo S, Mandolino G, Galuppo M, Bramanti P, Mazzon E. Cannabinoids: New Promising Agents in the Treatment of Neurological Diseases. Molecules. 2014; 19(11):18781-18816. https://doi.org/10.3390/molecules191118781
Chicago/Turabian StyleGiacoppo, Sabrina, Giuseppe Mandolino, Maria Galuppo, Placido Bramanti, and Emanuela Mazzon. 2014. "Cannabinoids: New Promising Agents in the Treatment of Neurological Diseases" Molecules 19, no. 11: 18781-18816. https://doi.org/10.3390/molecules191118781
APA StyleGiacoppo, S., Mandolino, G., Galuppo, M., Bramanti, P., & Mazzon, E. (2014). Cannabinoids: New Promising Agents in the Treatment of Neurological Diseases. Molecules, 19(11), 18781-18816. https://doi.org/10.3390/molecules191118781