Tungsten Trioxide as a Visible Light Photocatalyst for Volatile Organic Carbon Removal


Abstract
:1. Introduction
2. Results and Discussion
2.1. Effect of Sulphate Anions and pH on WO3 Characteristics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Precursor Solution | Molar Ratio  | Final pH | Morphology | Crystalline Phase (Chemical Formula) | Surface Area (m2/g) | Band-Gap (eV) |
|---|---|---|---|---|---|---|---|
| W0 | 20 mL 0.1 M H2WO4 | - | 1.61 | slabs with columnar clusters | monoclinic (WO3) + orthorhombic (WO3·⅓H2O) | n.a. | n.a. |
| W0.3NaS | 20 mL 0.1 M H2WO4 + SO42− (from Na2SO4) | 0.3 | 1.3 | columnar crystals | monoclinic (WO3) + orthorhombic (WO3·⅓H2O) | n.a. | n.a. |
| W7.6NaS | 20 mL 0.1 M H2WO4 + SO42− (from Na2SO4) | 7.6 | 1.15 | nanobundles | hexagonal (WO3) | 44.4 | 2.68 |
| W0.3HS | 20 mL 0.1 M H2WO4 + H2SO4 | 0.3 | <0.3 | nanocubes | monoclinic (WO3) + orthorhombic (WO3·⅓H2O) | n.a. | n.a. |
| W7.6HS | 20 mL 0.1 M H2WO4 + H2SO4 | 7.6 | <0.3 | nanocubes | monoclinic (WO3) + orthorhombic (WO3·⅓H2O) | 7.0 | 2.75 |
| WSA | Commercial WO3 (Sigma Aldrich) | - | - | nanoparticles | monoclinic (WO3) | 8.3 | 2.61 |
2.2. Pt/WO3 Characteristics

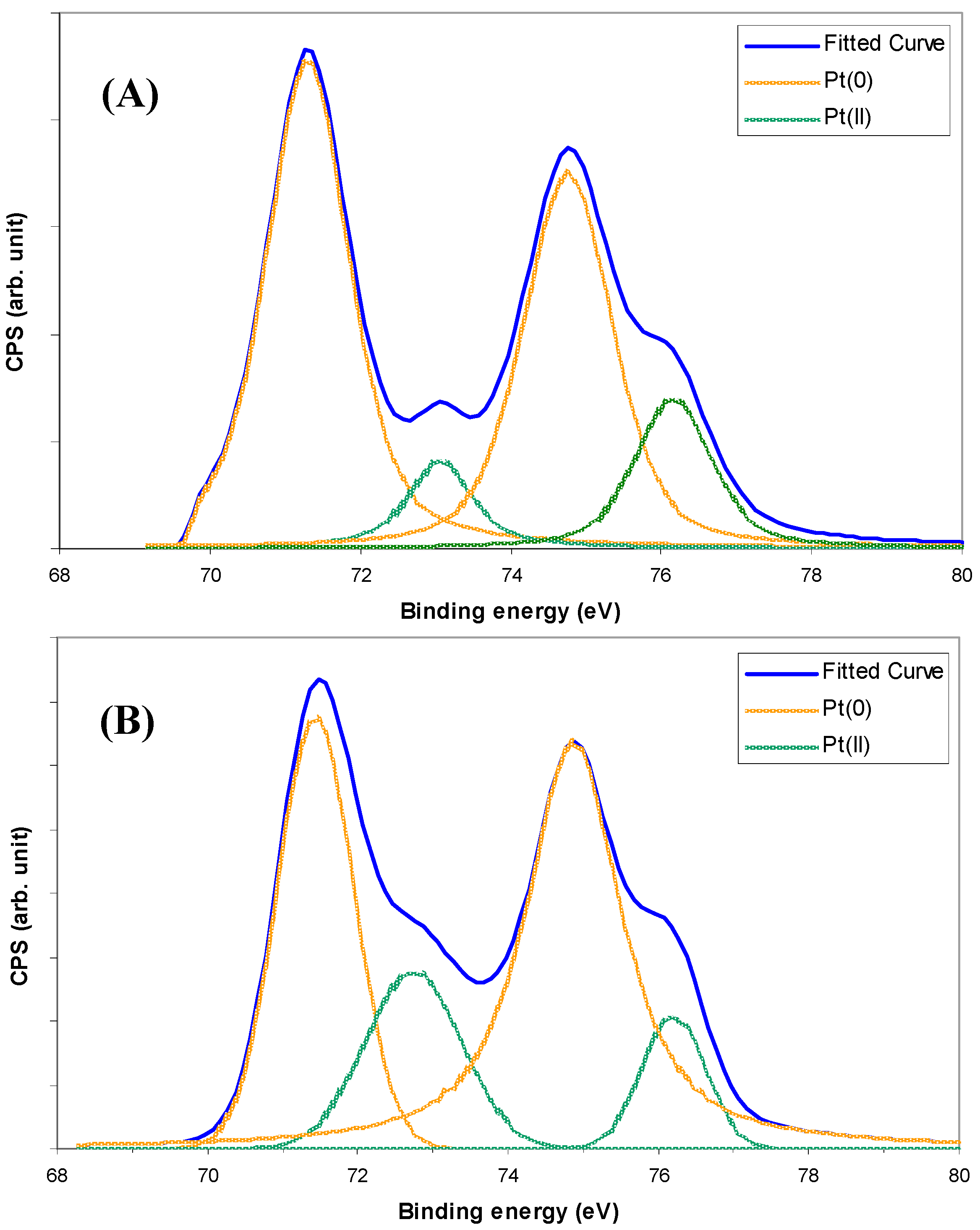
2.3. Ethylene Photodegradation
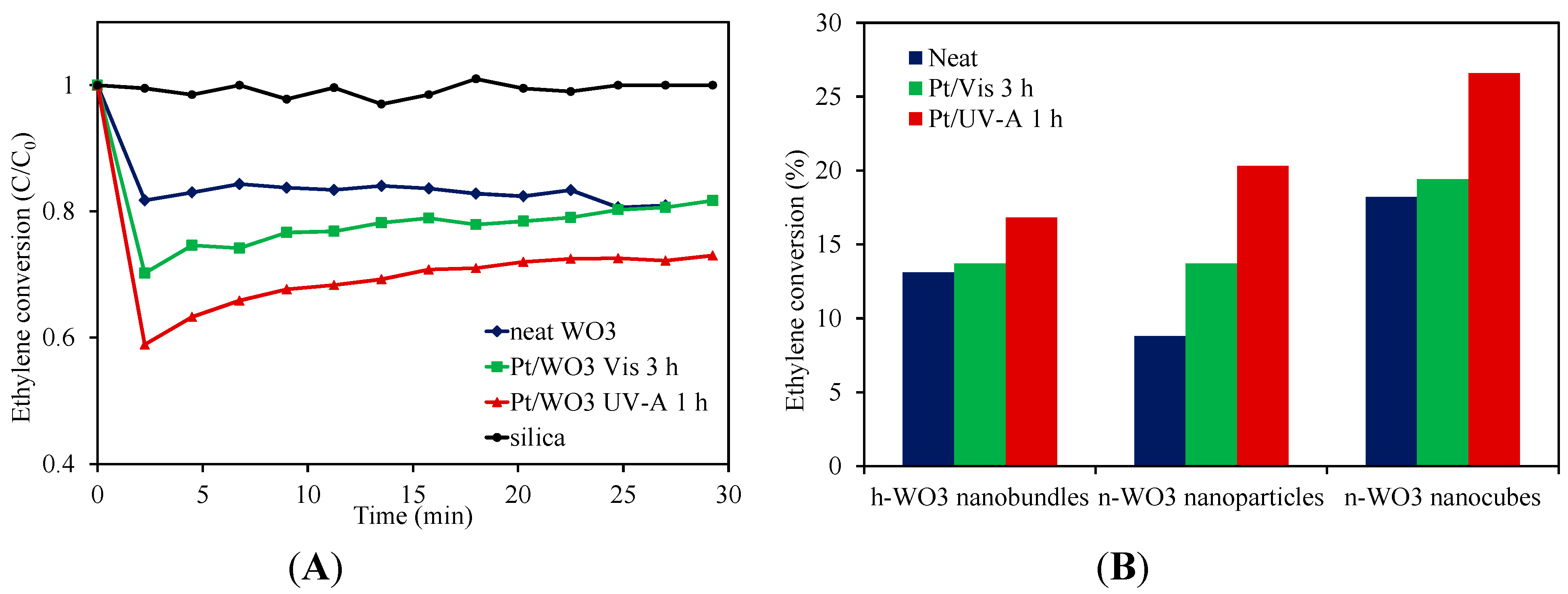
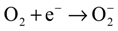
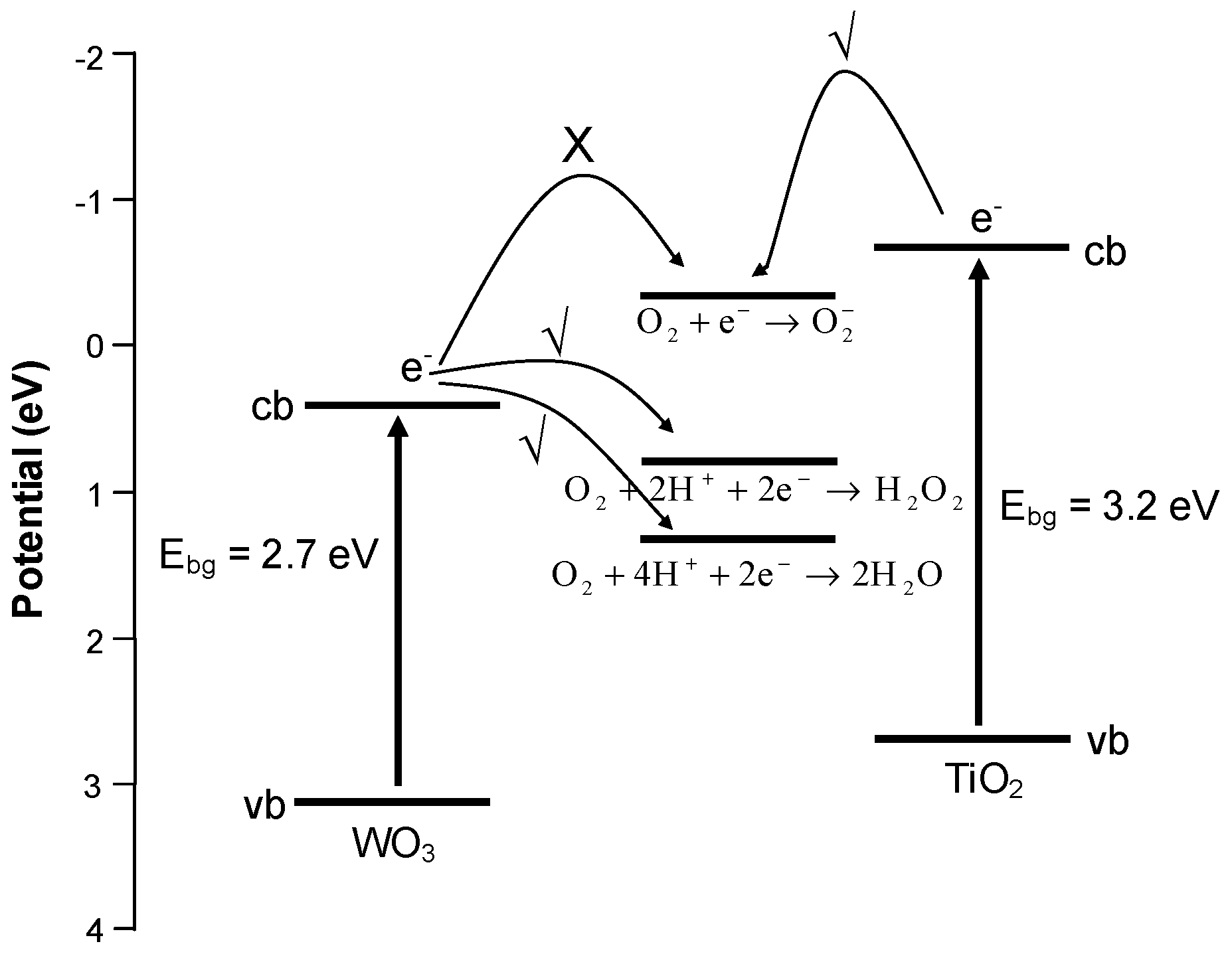
3. Experimental Section
3.1. Materials
3.2. Hydrothermal Synthesis of WO3 Nanostructures
3.3. Photodeposition of Platinum on WO3 Nanostructured Supports
3.4. Photocatalytic Oxidation of Ethylene
3.5. Characterisation
4. Conclusions
Supplementary Materials
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kotzias, D. Indoor air and human exposure assessment-needs and approaches. Exp. Toxicol. Pathol. 2005, 57, 5–7. [Google Scholar] [CrossRef]
- Kostiainen, R. Volatile organic compounds in the indoor air of normal and sick houses. Atmos. Environ. 1995, 29, 693–702. [Google Scholar] [CrossRef]
- Jo, W.K.; Park, K.H. Heterogeneous photocatalysis of aromatic and chlorinated volatile organic compounds (vocs) for non-occupational indoor air application. Chemosphere 2005, 57, 555–565. [Google Scholar] [CrossRef]
- Mo, J.; Zhang, Y.; Xu, Q.; Lamson, J.J.; Zhao, R. Photocatalytic purification of volatile organic compounds in indoor air: A literature review. Atmos. Environ. 2009, 43, 2229–2246. [Google Scholar] [CrossRef]
- Wang, S.; Ang, H.M.; Tade, M.O. Volatile organic compounds in indoor environment and photocatalytic oxidation: State of the art. Environ. Int. 2007, 33, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Qi, L. Template-free fabrication of hierarchically flower-like tungsten trioxide assemblies with enhanced visible-light-driven photocatalytic activity. J. Hazard. Mater. 2009, 169, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.P.; Yang, B.Q.; Zhang, H.X.; Feng, P.X. Tungsten oxide nanorods array and nanobundle prepared by using chemical vapor deposition technique. Nanoscale Res.Lett. 2007b, 2, 405–409. [Google Scholar]
- Ponzoni, A.; Comini, E.; Ferroni, M.; Sberveglieri, G. Nanostructured WO3 deposited by modified thermal evaporation for gas-sensing applications. Thin Solid Films 2005, 490, 81–85. [Google Scholar] [CrossRef]
- Baeck, S.H.; Choi, K.S.; Jaramillo, T.F.; Stucky, G.D.; McFarland, E.W. Enhancement of photocatalytic and electrochromic properties of electrochemically fabricated mesoporous WO3 thin films. Adv. Mater. 2003, 15, 1269–1273. [Google Scholar] [CrossRef]
- Arutanti, O.; Ogi, T.; Nandiyanto, A.B.D.; Iskandar, F.; Okuyama, K. Controllable crystallite and particle sizes of WO3 particles prepared by a spray-pyrolysis method and their photocatalytic activity. Am. Inst. Chem. Eng. J. 2014, 60, 41–49. [Google Scholar] [CrossRef]
- Satishkumar, B.C.; Govindaraj, A.; Nath, M.; Rao, C.N.R. Synthesis of metal oxide nanorods using carbon nanotubes as templates. J. Mater. Chem. 2000, 10, 2115–2119. [Google Scholar] [CrossRef]
- Badilescu, S.; Ashrit, P.V. Study of sol-gel prepared nanostructured WO3 thin films and composites for electrochromic applications. Solid State Ion. 2003, 158, 187–197. [Google Scholar] [CrossRef]
- Song, X.C.; Zheng, Y.F.; Yang, E.; Wang, Y. Large-scale hydrothermal synthesis of WO3 nanowires in the presence of K2SO4. Mater. Lett. 2007, 61, 3904–3908. [Google Scholar] [CrossRef]
- Salmaoui, S.; Sediri, F.; Gharbi, N. Characterization of h-WO3 nanorods synthesized by hydrothermal process. Polyhedron 2010, 29, 1771–1775. [Google Scholar] [CrossRef]
- Gu, Z.; Zhai, T.; Gao, B.; Sheng, X.; Wang, Y.; Fu, H.; Ma, Y.; Yao, J. Controllable assembly of WO3 nanorods/nanowires into hierarchical nanostructures. J. Phys. Chem. B 2006, 110, 23829–23836. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Nataraj, D.; Mangalaraj, D.; Djaoued, Y.; Robichaud, J.; Khyzhun, O.Y. Controlled growth of WO3 nanostructures with three different morphologies and their structural, optical, and photodecomposition studies. Nanoscale Res. Lett. 2009, 4, 1335–1342. [Google Scholar] [PubMed]
- Huang, K.; Pan, Q.; Yang, F.; Ni, S.; Wei, X.; He, D. Controllable synthesis of hexagonal WO3 nanostructures and their application in lithium batteries. J. Phys. D: Appl. Phys. 2008, 41, 155417. [Google Scholar]
- Xu, Z.; Tabata, I.; Hirogaki, K.; Hisada, K.; Wang, T.; Wang, S.; Hori, T. Preparation of platinum-loaded cubic tungsten oxide: A highly efficient visible light-driven photocatalyst. Mater. Lett. 2011, 65, 1252–1256. [Google Scholar] [CrossRef]
- Amano, F.; Ishinaga, E.; Yamakata, A. Effect of particle size on the photocatalytic activity of WO3 particles for water oxidation. J. Phys. Chem. C 2013, 117, 22584–22590. [Google Scholar] [CrossRef]
- Denny, F.; Scott, J.; Chiang, K.; Teoh, W.Y.; Amal, R. Insight towards the role of platinum in the photocatalytic mineralisation of organic compounds. J. Mol. Catal. A: Chem. 2007, 263, 93–102. [Google Scholar] [CrossRef]
- Abe, R.; Takami, H.; Murakami, N.; Ohtani, B. Pristine simple oxides as visible light driven photocatalysts: Highly efficient decomposition of organic compounds over platinum-loaded tungsten oxide. J. Am. Chem. Soc. 2008, 130, 7780–7781. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, A.; Palmisano, L.; Marc, G.; Venezia, A.M. Influence of platinum on catalytic activity of polycrystalline WO3 employed for phenol photodegradation in aqueous suspension. Sol. Energy Mater. Sol. Cells 1998, 51, 203–219. [Google Scholar] [CrossRef]
- Zhang, G.; Guan, W.; Shen, H.; Zhang, X.; Fan, W.; Lu, C.; Bai, H.; Xiao, L.; Gu, W.; Shi, W. Organic additives-free hydrothermal synthesis and visible-light-driven photodegradation of tetracyline of WO3 nanosheets. Ind. Eng. Chem. Res. 2014, 53, 5443–5450. [Google Scholar] [CrossRef]
- Bamwenda, G.R.; Arakawa, H. The visible light induced photocatalytic activity of tungsten trioxide powders. Appl. Catal. A: Gen. 2001, 210, 181–191. [Google Scholar] [CrossRef]
- Murata, A.; Oka, N.; Nakamura, S.; Shigesato, Y. Visible-light active photocatalytic WO3 films loaded with Pt nanoparticles deposited by sputtering. J. Nanosci. Nanotechnol. 2012, 12, 5082–5086. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.G.; Sakai, G.; Shimanoe, K.; Miura, N.; Yamazoe, N. Preparation of aqueous sols of tungsten oxide dihydrate from sodium tungstate by an ion-exchange method. Sens. Actuator B-Chem. 2002, 87, 63–72. [Google Scholar] [CrossRef]
- Livage, J.; Guzman, G. Aqueous precursors for electrochromic tungsten oxide hydrates. Solid State Ion. 1996, 84, 205–211. [Google Scholar] [CrossRef]
- Gu, Z.; Li, H.; Zhai, T.; Yang, W.; Xia, Y.; Ma, Y.; Yao, J. Large-scale synthesis of single-crystal hexagonal tungsten trioxide nanowires and electrochemical lithium intercalation into the nanocrystals. J. Solid State Chem. 2007, 180, 98–105. [Google Scholar] [CrossRef]
- Wang, H.L.; Ma, X.D.; Qian, X.F.; Yin, J.; Zhu, Z.K. Selective synthesis of CdWO4 short nanorods and nanofibers and their self-assembly. J. Solid State Chem. 2004, 177, 4588–4596. [Google Scholar] [CrossRef]
- Gu, Z.; Ma, Y.; Yang, W.; Zhang, G.; Yao, J. Self-assembly of highly oriented one-dimensional h-WO3 nanostructures. Chem. Commun. 2005, 28, 3597–3599. [Google Scholar] [CrossRef]
- Reis, K.P.; Ramanan, A.; Whittingham, M.S. Hydrothermal synthesis of sodium tungstates. Chem. Mater. 1990, 2, 219–221. [Google Scholar] [CrossRef]
- Wang, J.; Khoo, E.; Lee, P.S.; Ma, J. Controlled synthesis of WO3 nanorods and their electrochromic properties in H2SO4 electrolyte. J. Phys. Chem. C 2009, 113, 9655–9658. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 88th Ed. ed; CRC Press: Boca Raton, FL, USA, 2007–2008; pp. 8-20–8-29. [Google Scholar]
- Lee, J.; Choi, W. Photocatalytic reactivity of surface platinized TiO2: Substrate specificity and the effect of Pt oxidation state. J. Phys. Chem. B 2005, 109, 7399–7406. [Google Scholar] [CrossRef] [PubMed]
- Vorontsov, A.; Savinov, E.; Zhensheng, J. Influence of the form of photodeposited platinum on titania upon its photocatalytic activity in CO and acetone oxidation. J. Photochem. Photobiol. A: Chem. 1999, 125, 113–117. [Google Scholar] [CrossRef]
- Kato, H.; Asakura, K.; Kudo, A. Highly efficient water splitting into H2 and O2 over lanthanum-doped NaTaO3 photocatalysts with high crystallinity and surface nanostructure. J. Am. Chem. Soc. 2003, 125, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.P.; Liu, G.; Yin, L.; Cheng, H.-M. Crystal facet-dependent photocatalytic oxidation and reduction reactivity of monoclinic WO3 for solar energy conversion. J. Mater. Chem. 2012, 22, 6746–6751. [Google Scholar] [CrossRef]
- Wang, C.M.; Heller, A.; Gerischer, H. Palladium catalysis of O2 reduction by electrons accumulated on TiO2 particles during photoassisted oxidation of organic compounds. J. Am. Chem. Soc. 1992, 114, 5230–5234. [Google Scholar] [CrossRef]
- Lee, S.L.; Scott, J.; Chiang, K.; Amal, R. Nanosized metal deposits on titanium dioxide for augmenting gas-phase toluene photooxidation. J. Nanopart. Res. 2009, 11, 209–219. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wicaksana, Y.; Liu, S.; Scott, J.; Amal, R. Tungsten Trioxide as a Visible Light Photocatalyst for Volatile Organic Carbon Removal. Molecules 2014, 19, 17747-17762. https://doi.org/10.3390/molecules191117747
Wicaksana Y, Liu S, Scott J, Amal R. Tungsten Trioxide as a Visible Light Photocatalyst for Volatile Organic Carbon Removal. Molecules. 2014; 19(11):17747-17762. https://doi.org/10.3390/molecules191117747
Chicago/Turabian StyleWicaksana, Yossy, Sanly Liu, Jason Scott, and Rose Amal. 2014. "Tungsten Trioxide as a Visible Light Photocatalyst for Volatile Organic Carbon Removal" Molecules 19, no. 11: 17747-17762. https://doi.org/10.3390/molecules191117747
APA StyleWicaksana, Y., Liu, S., Scott, J., & Amal, R. (2014). Tungsten Trioxide as a Visible Light Photocatalyst for Volatile Organic Carbon Removal. Molecules, 19(11), 17747-17762. https://doi.org/10.3390/molecules191117747