Synthetic Advances in Macrosphelides: Natural Anticancer Agents

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
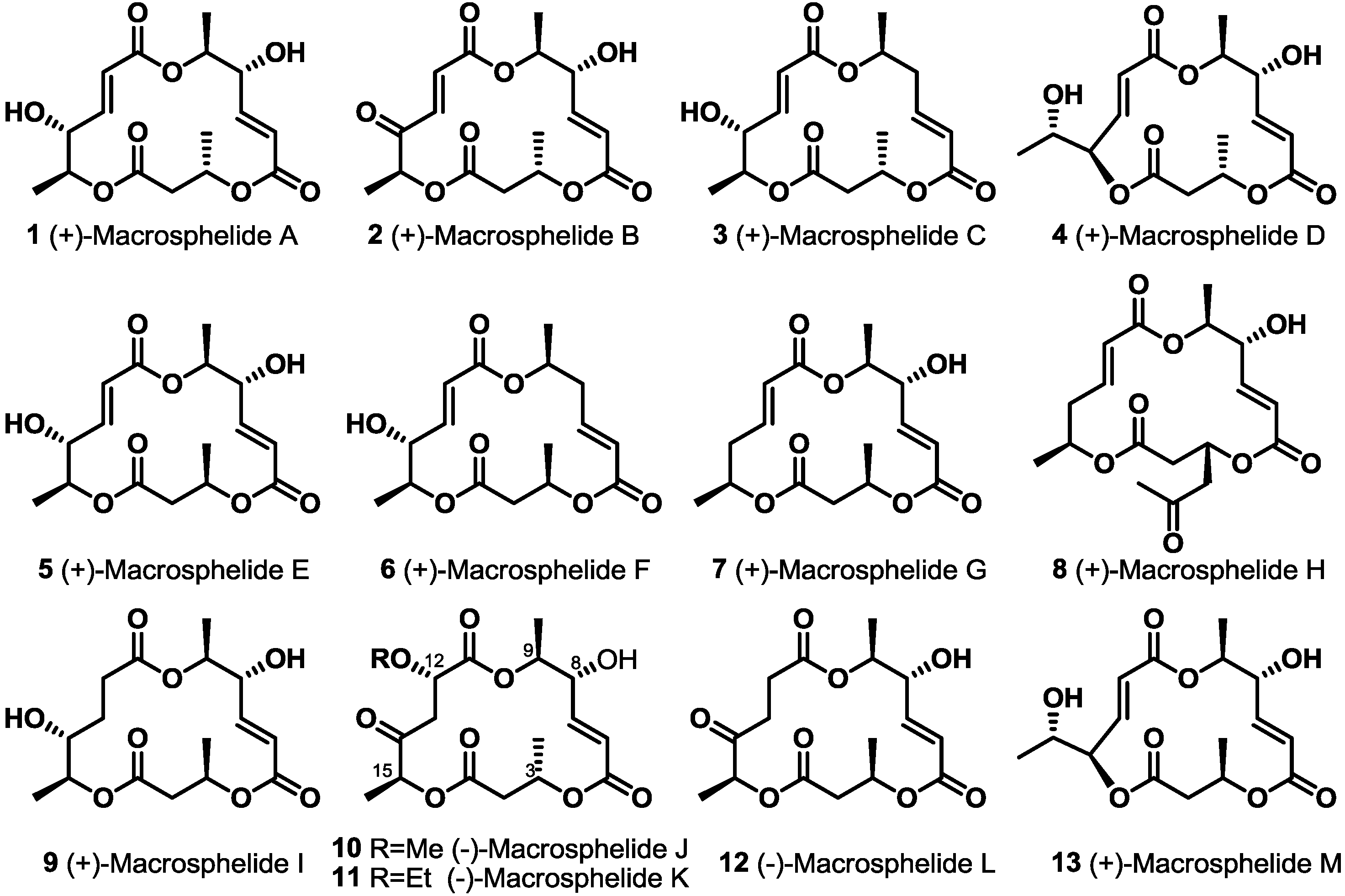
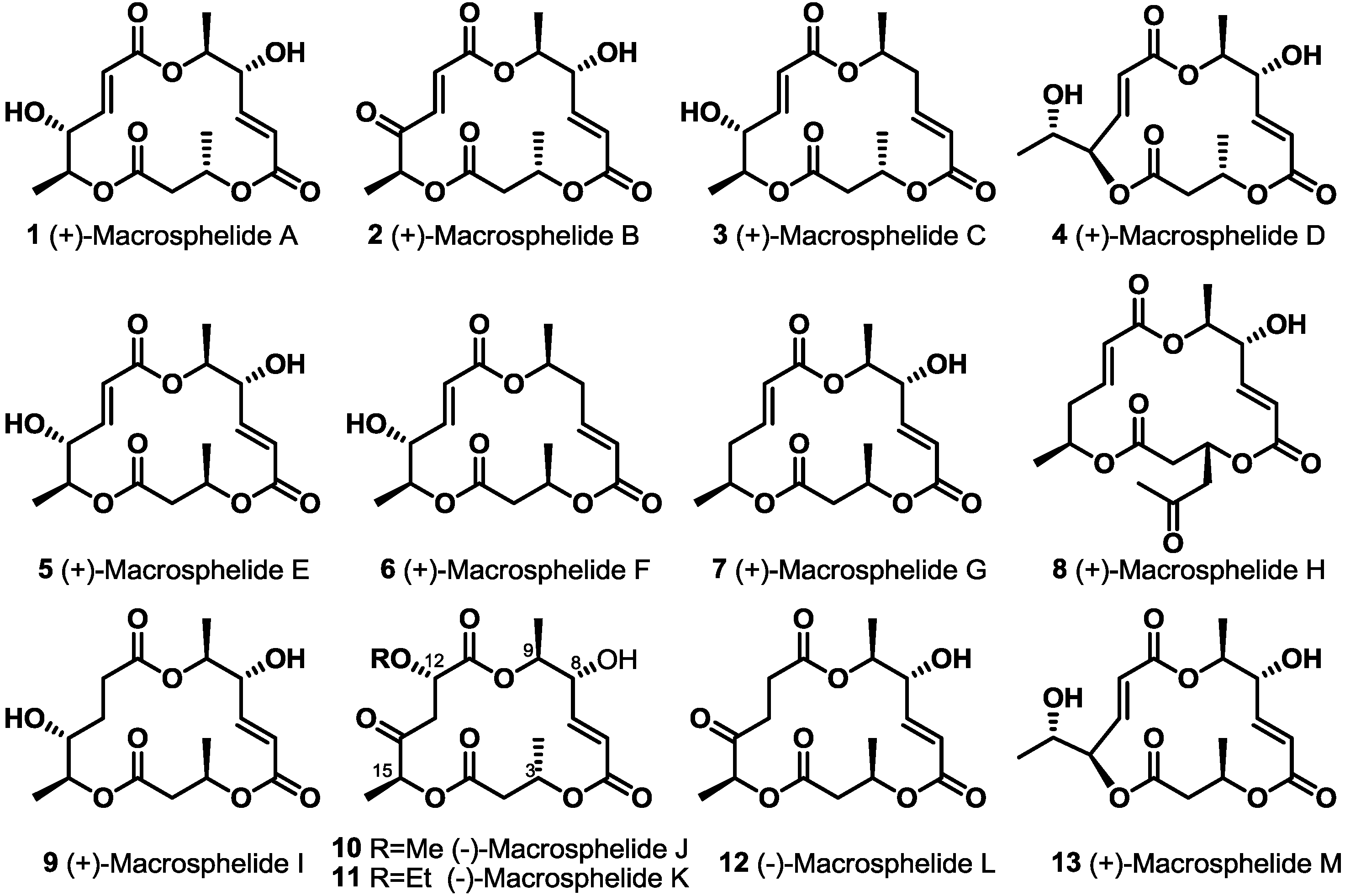
:1. Introduction
2. Results and Discussion
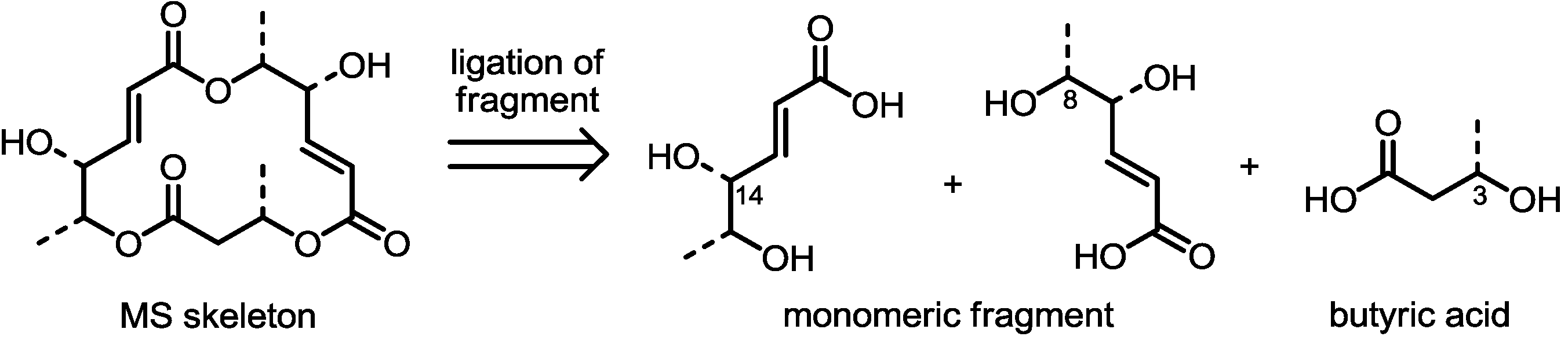
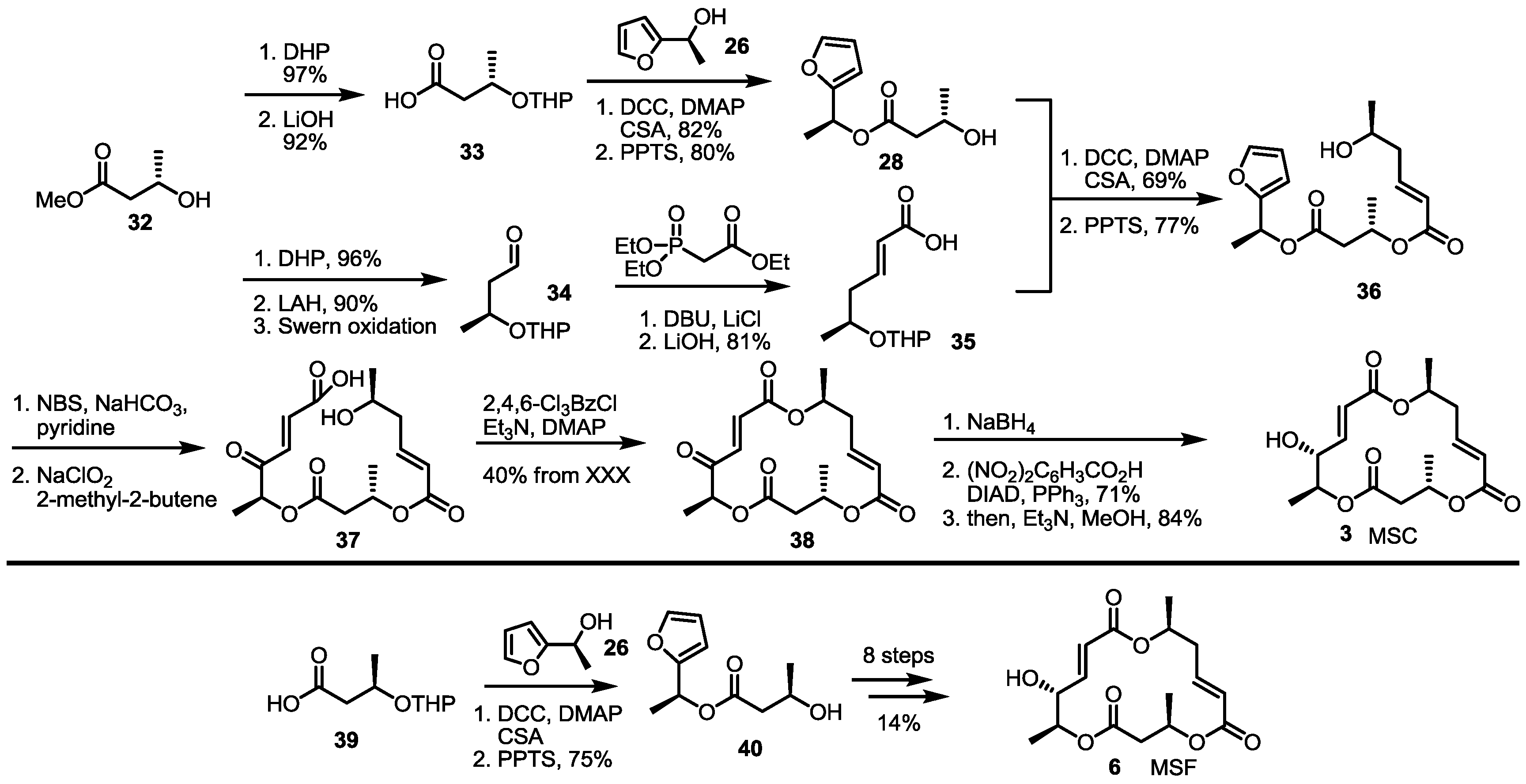
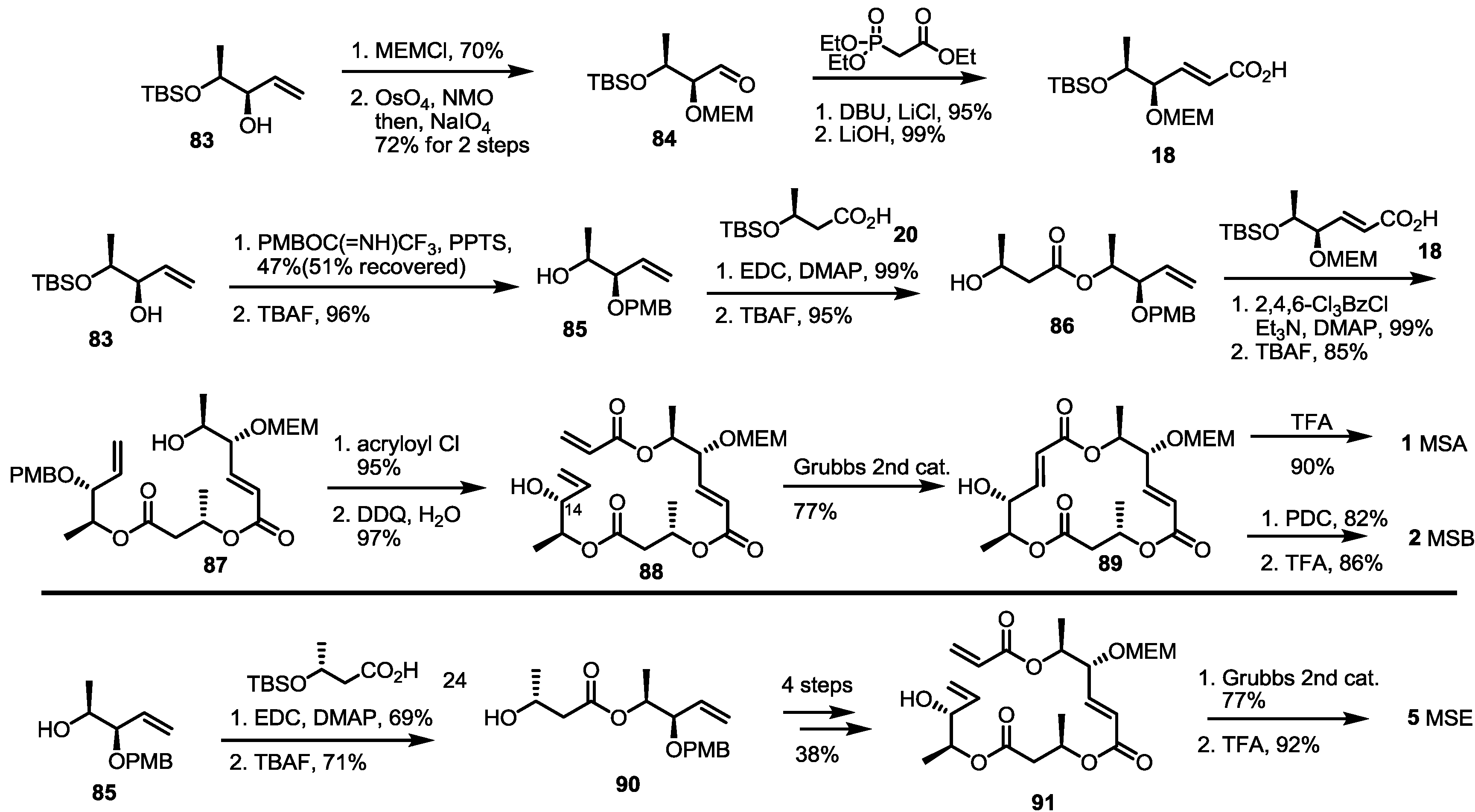
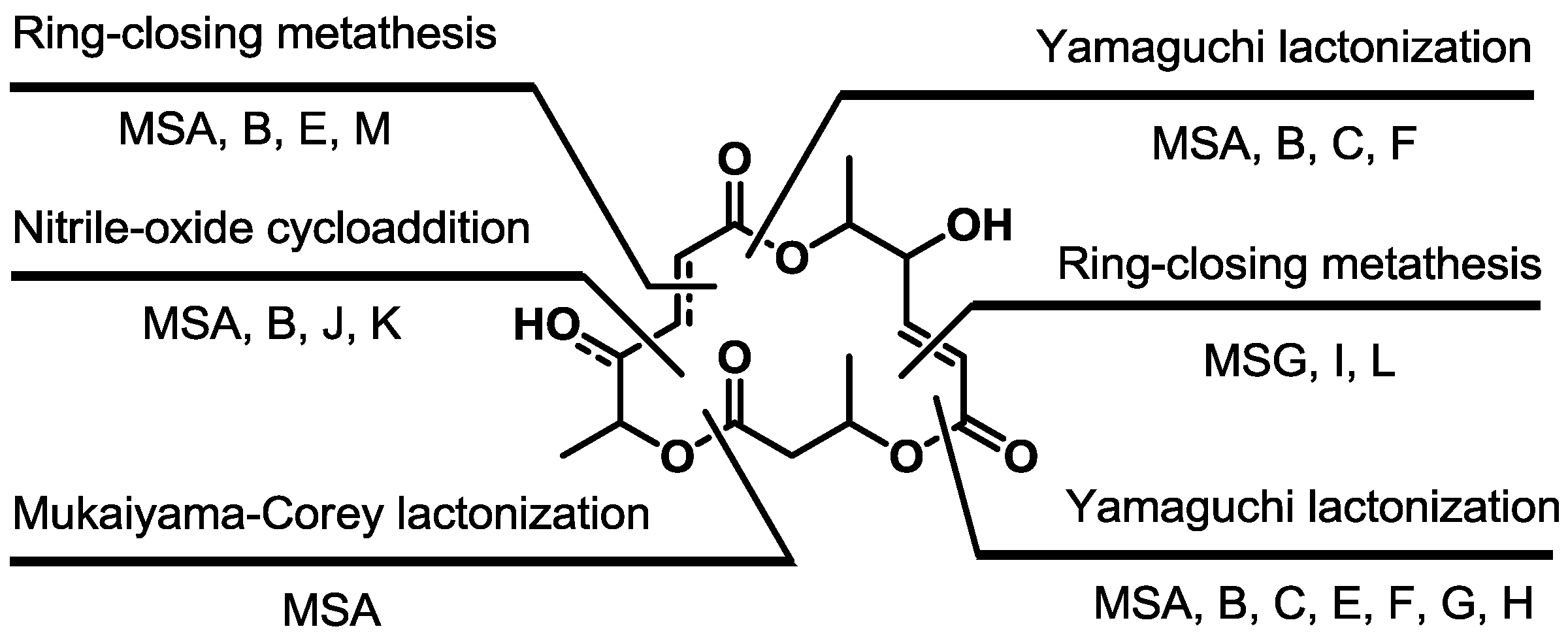
2.1. Synthetic Features of MS
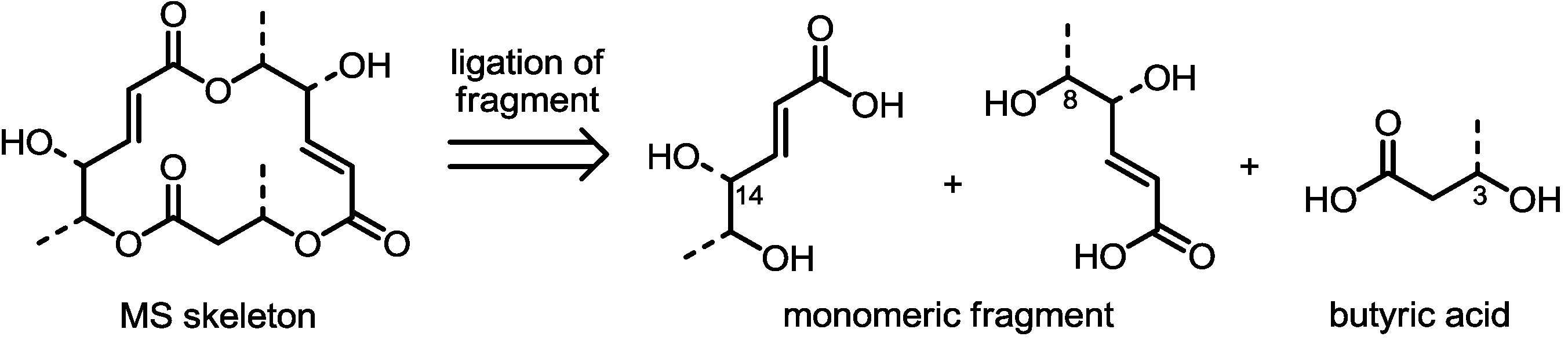
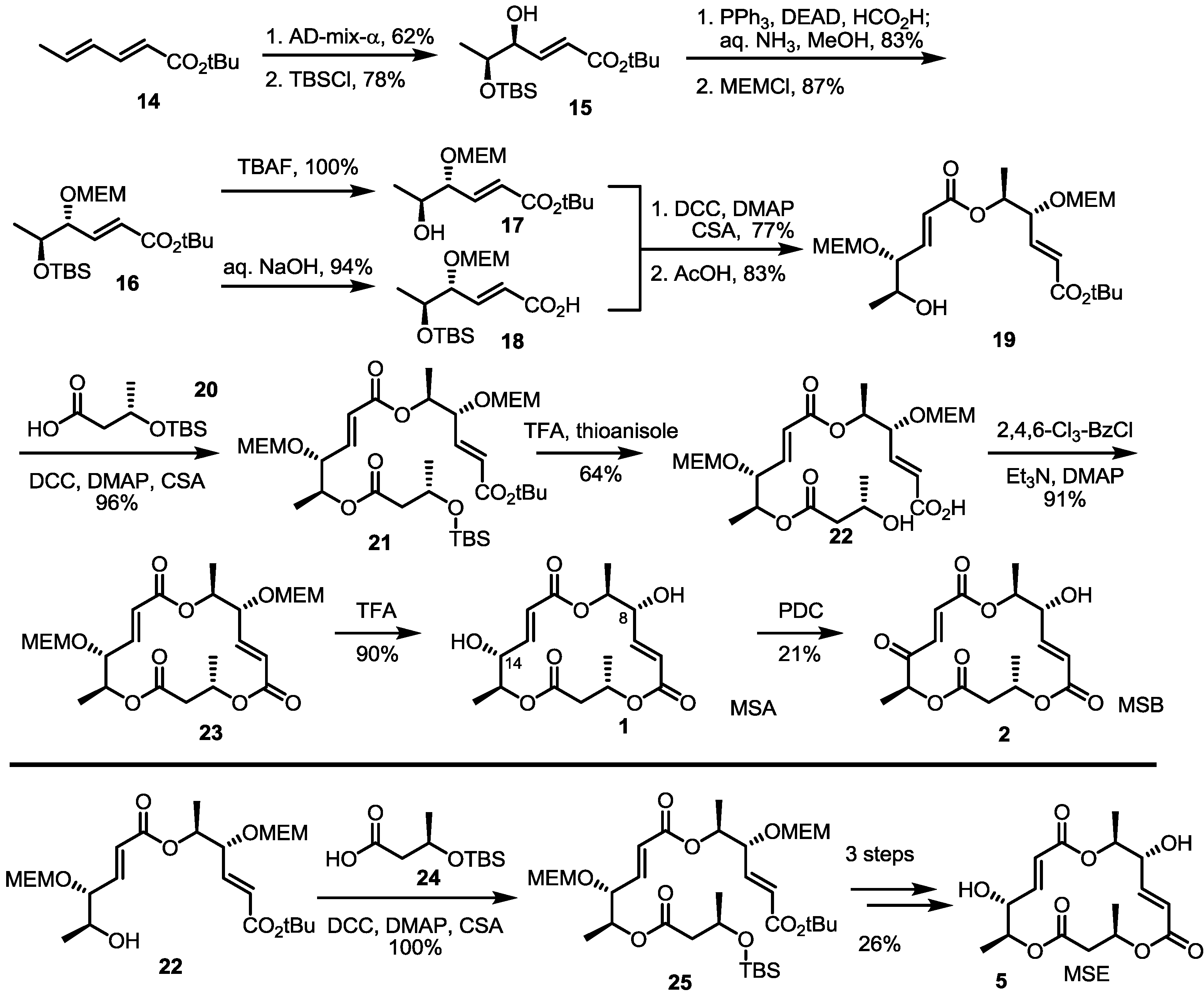
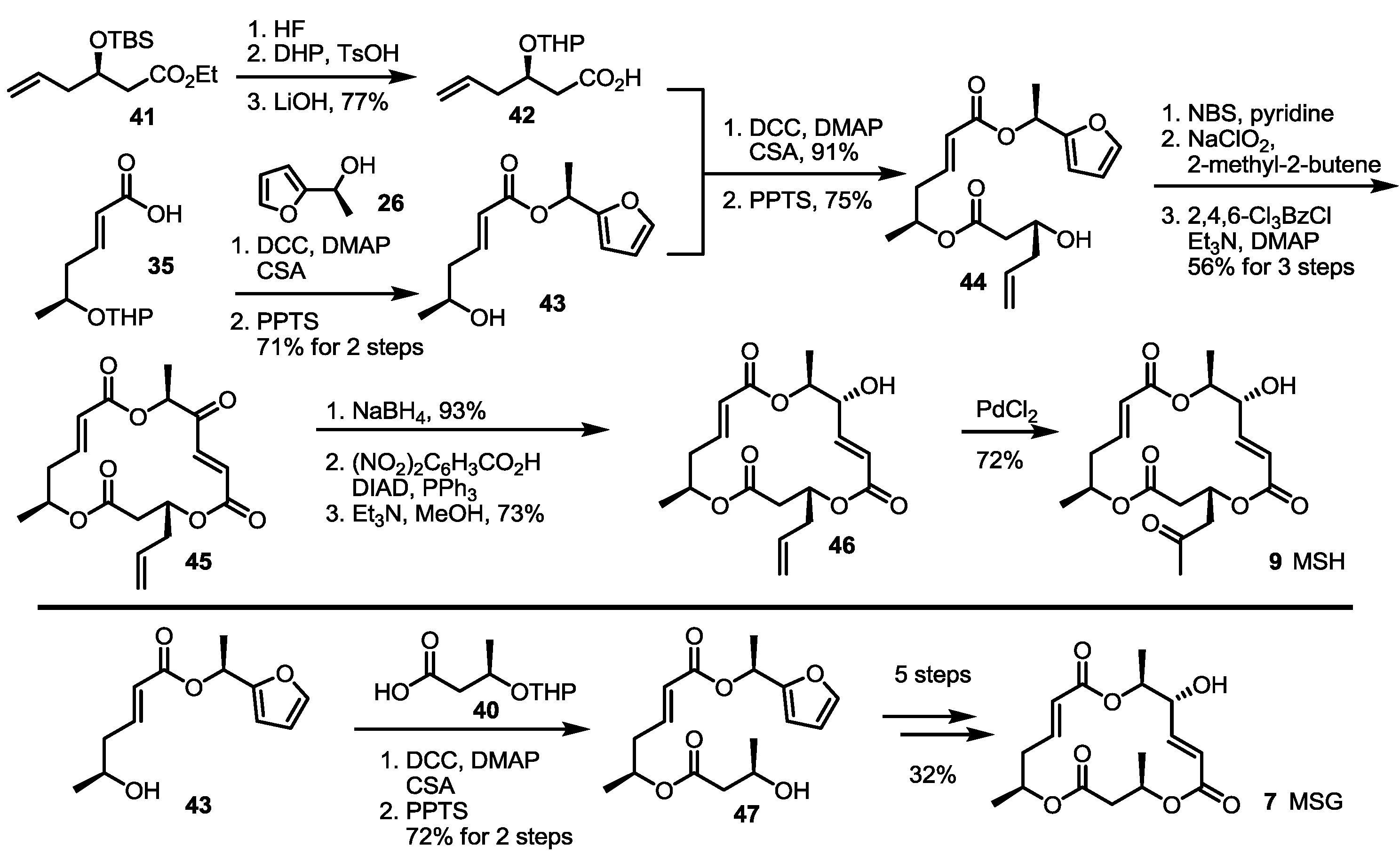
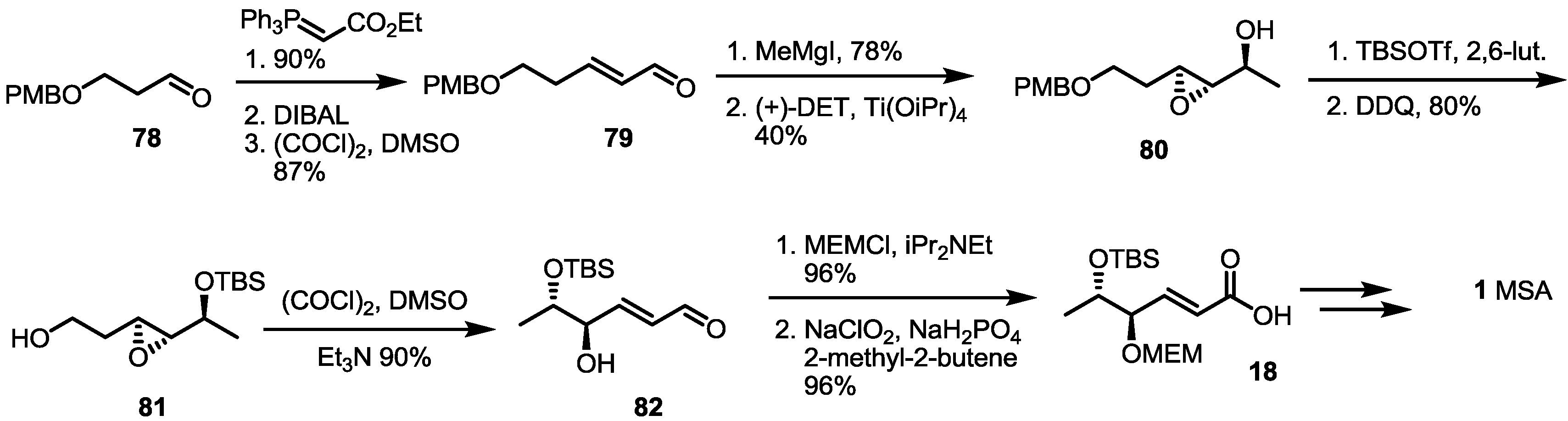
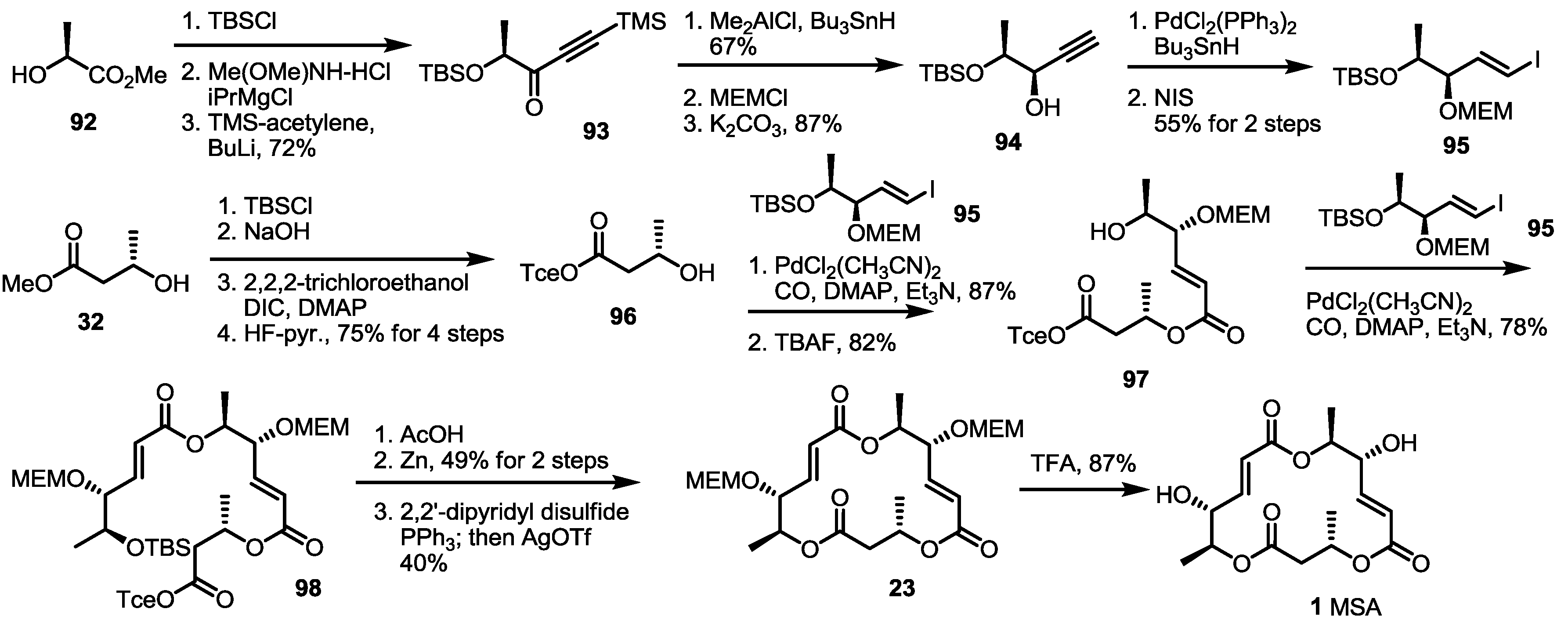
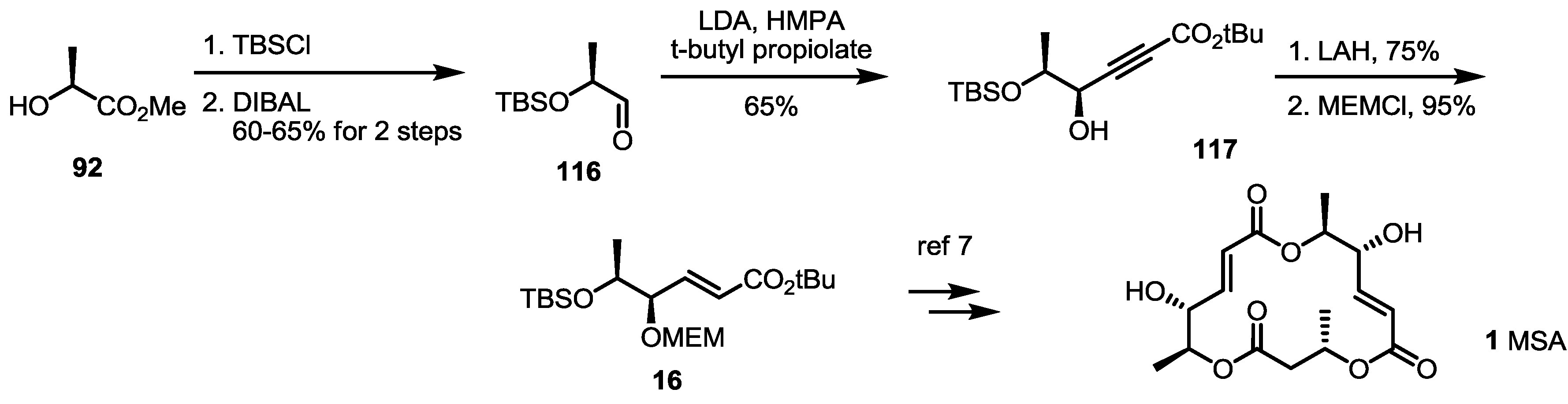
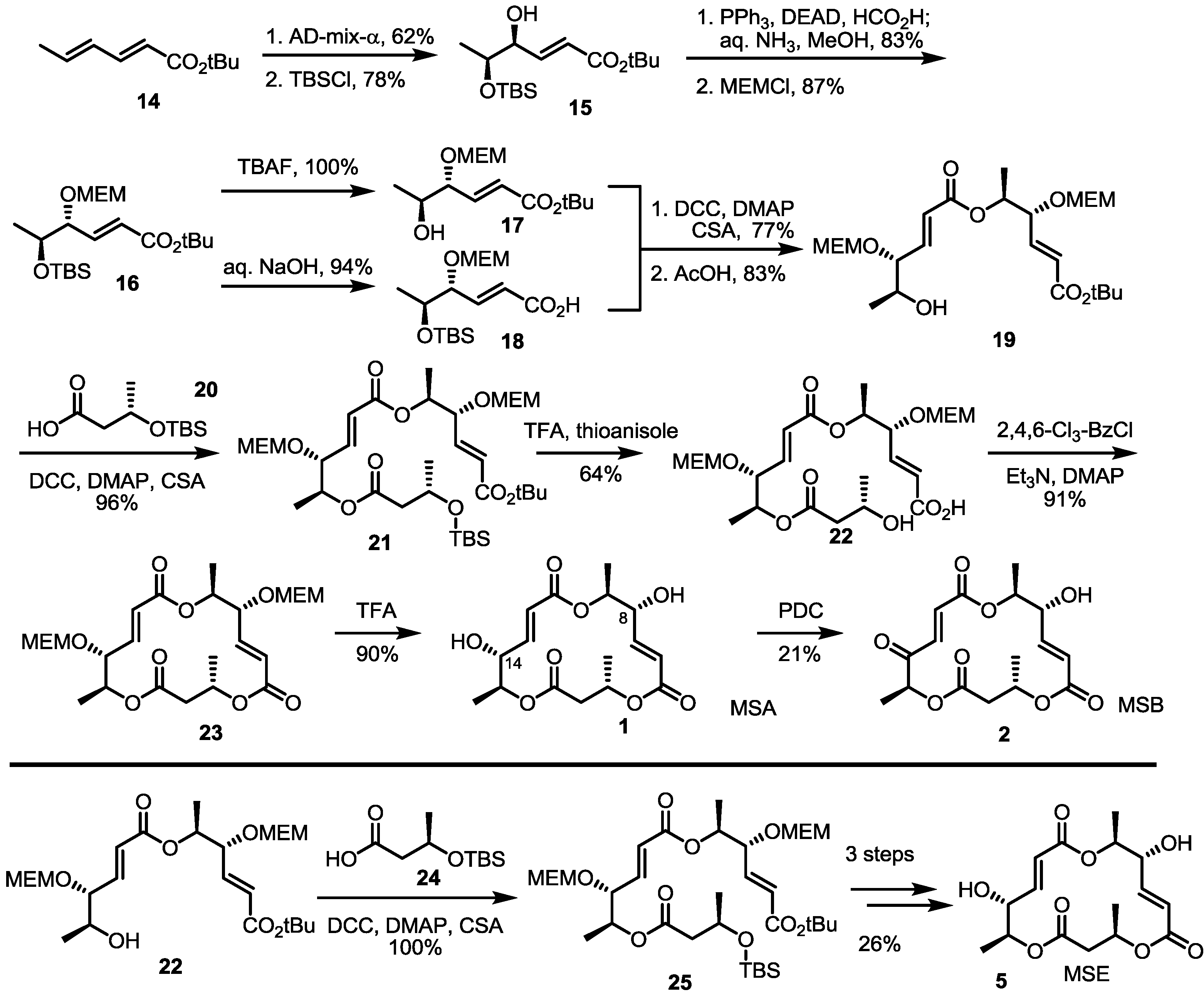
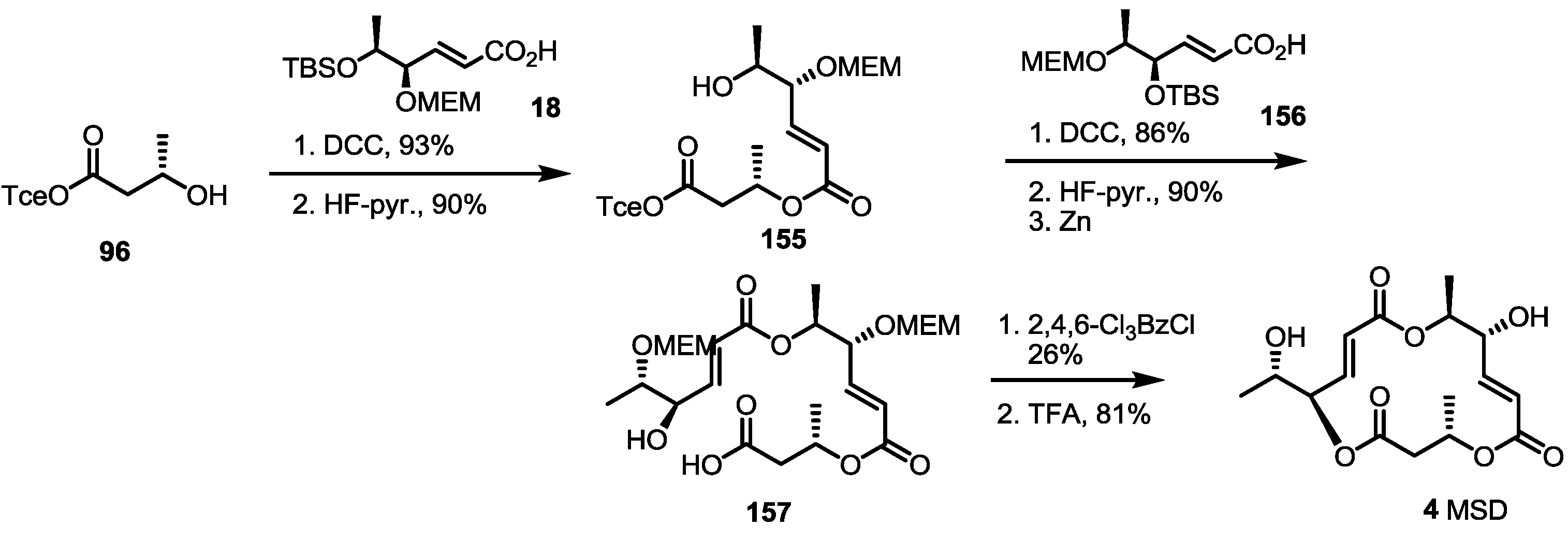
2.2. The First Total Synthesis of MSA and MSB
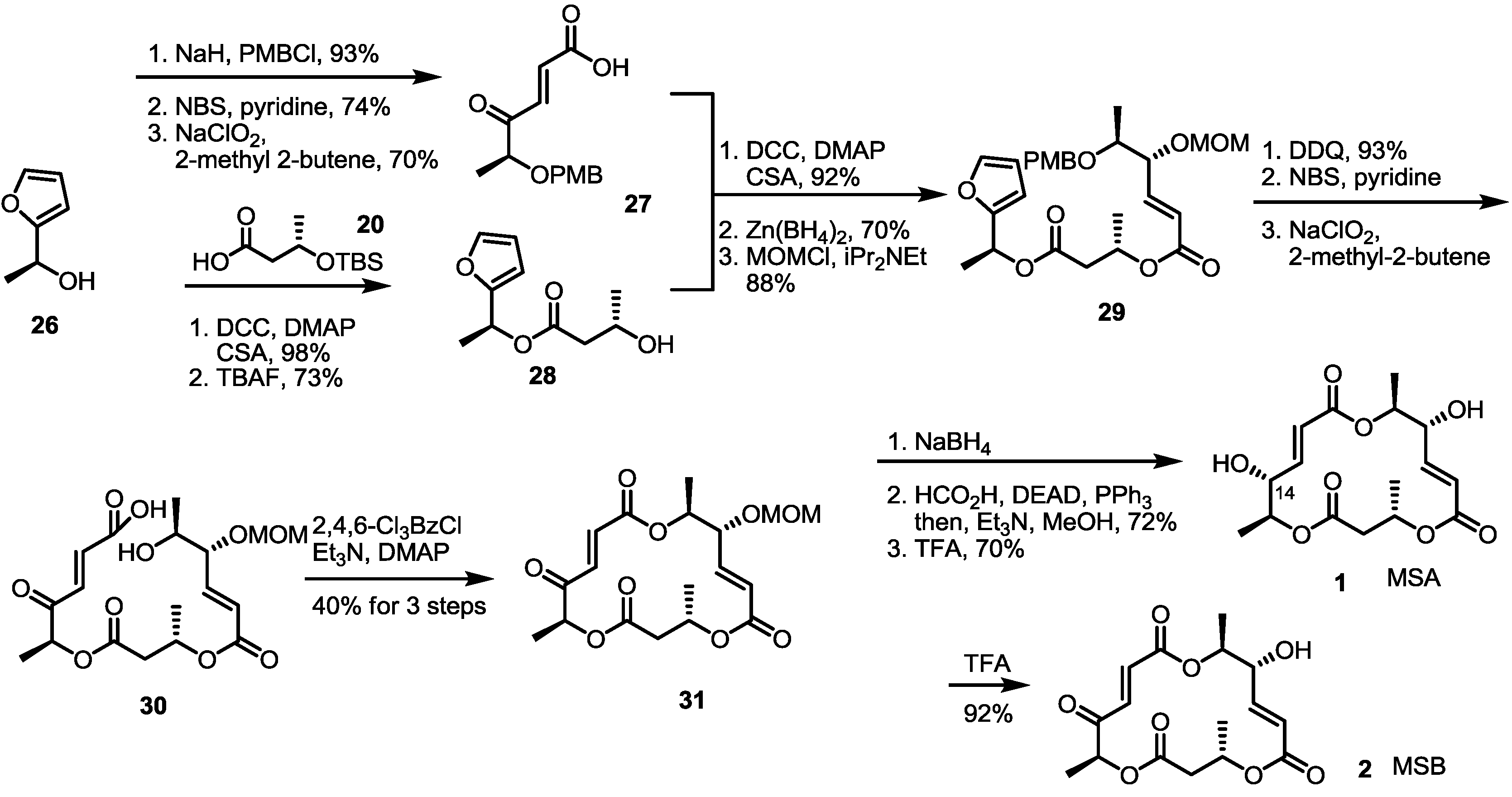
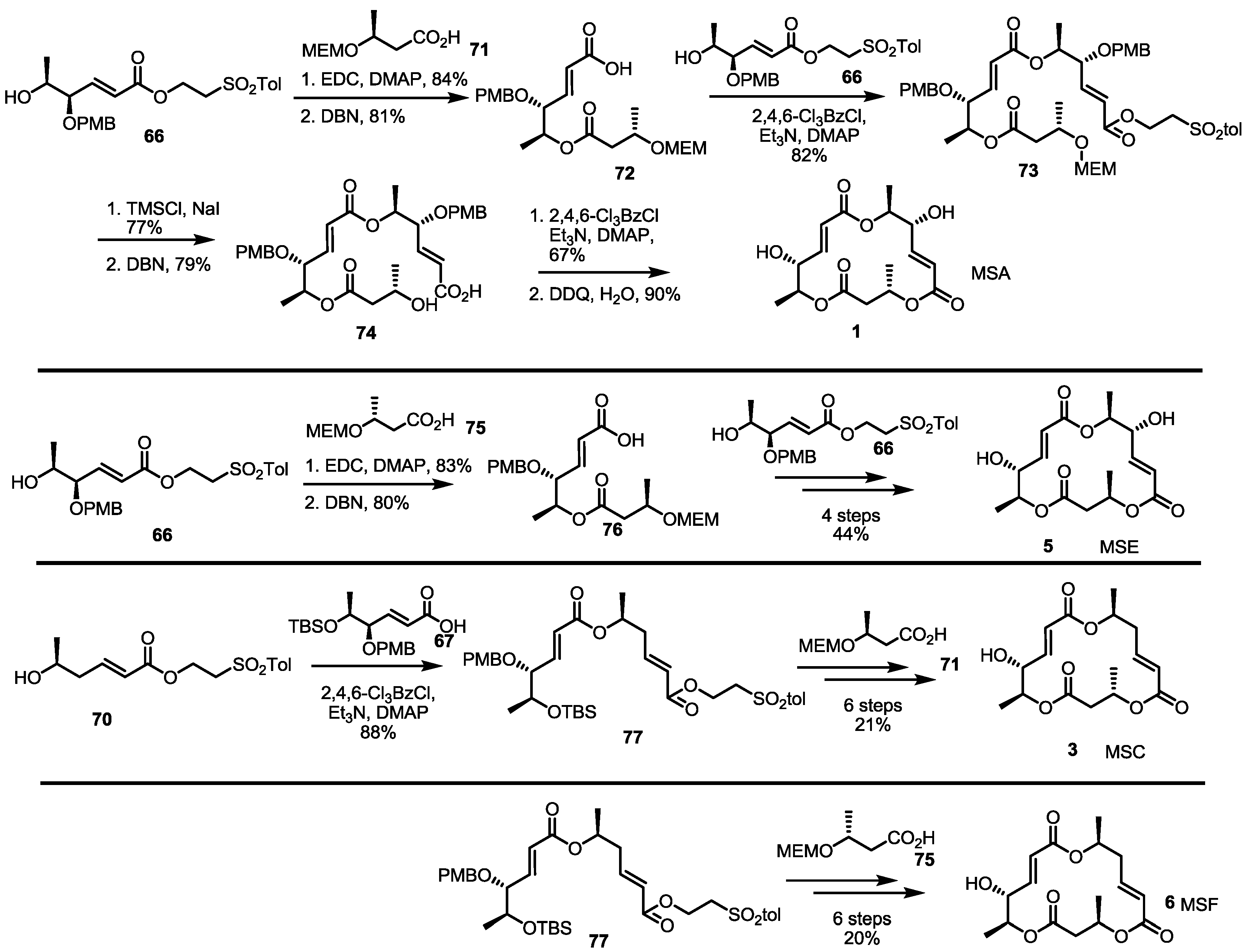
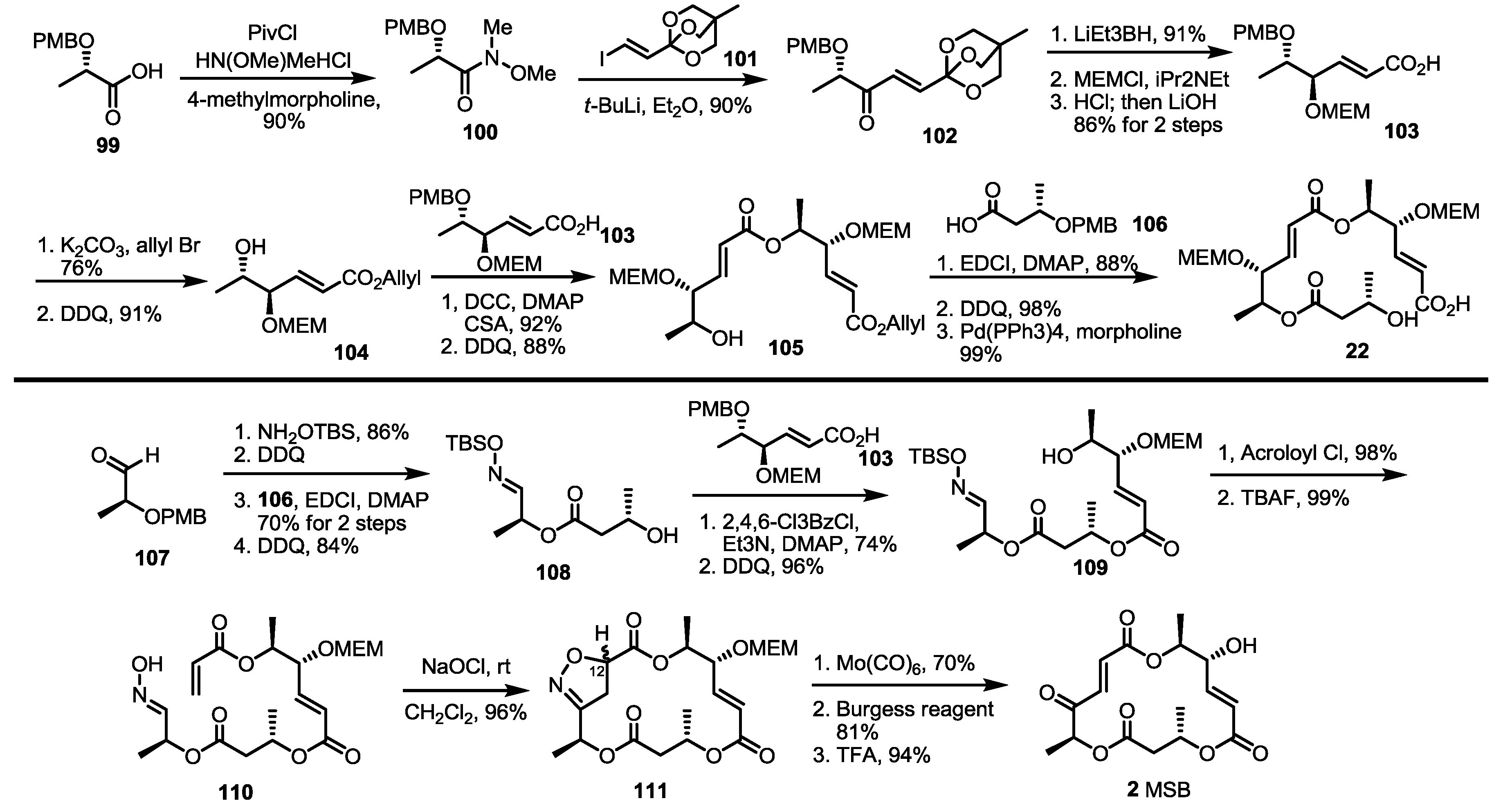
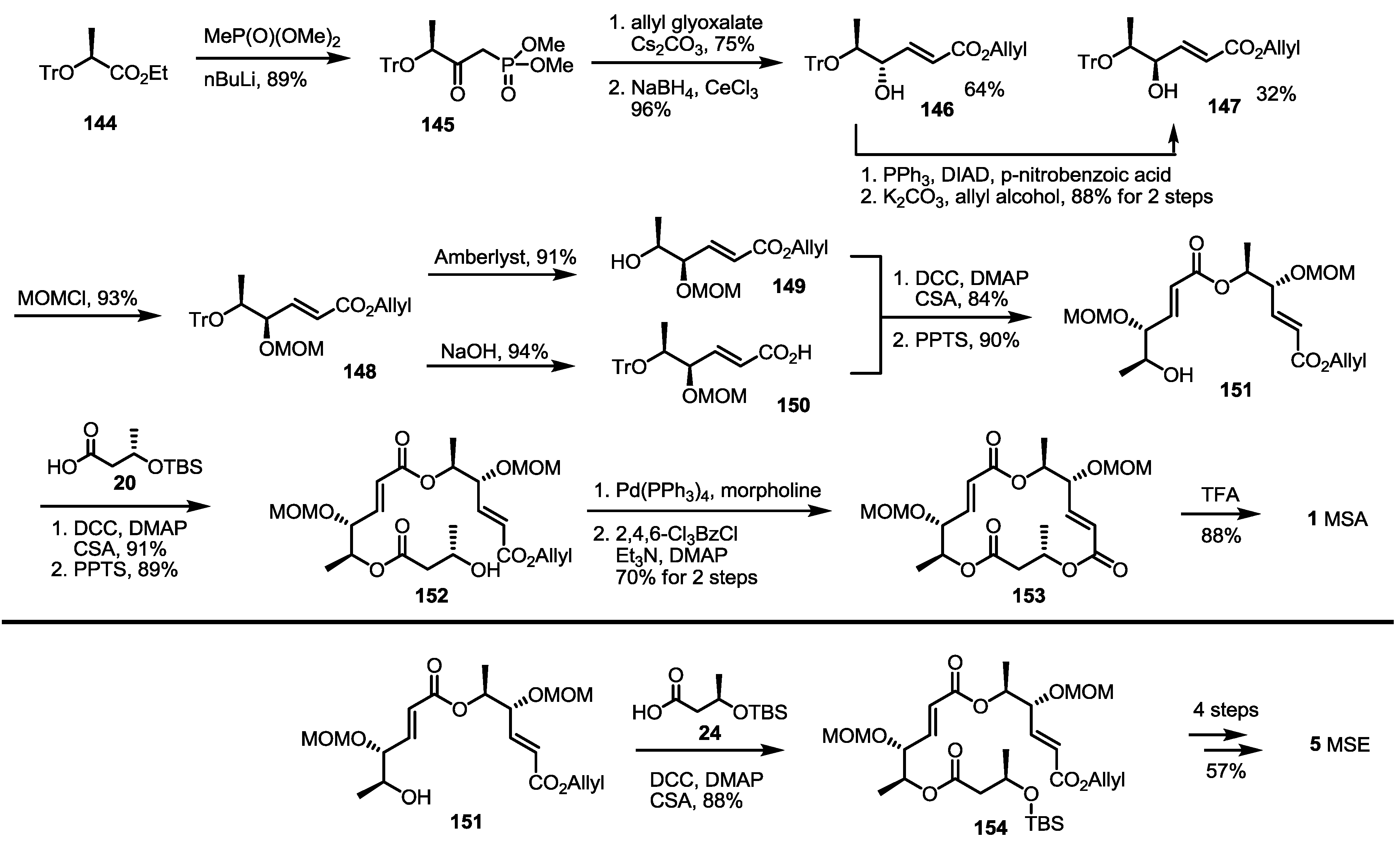
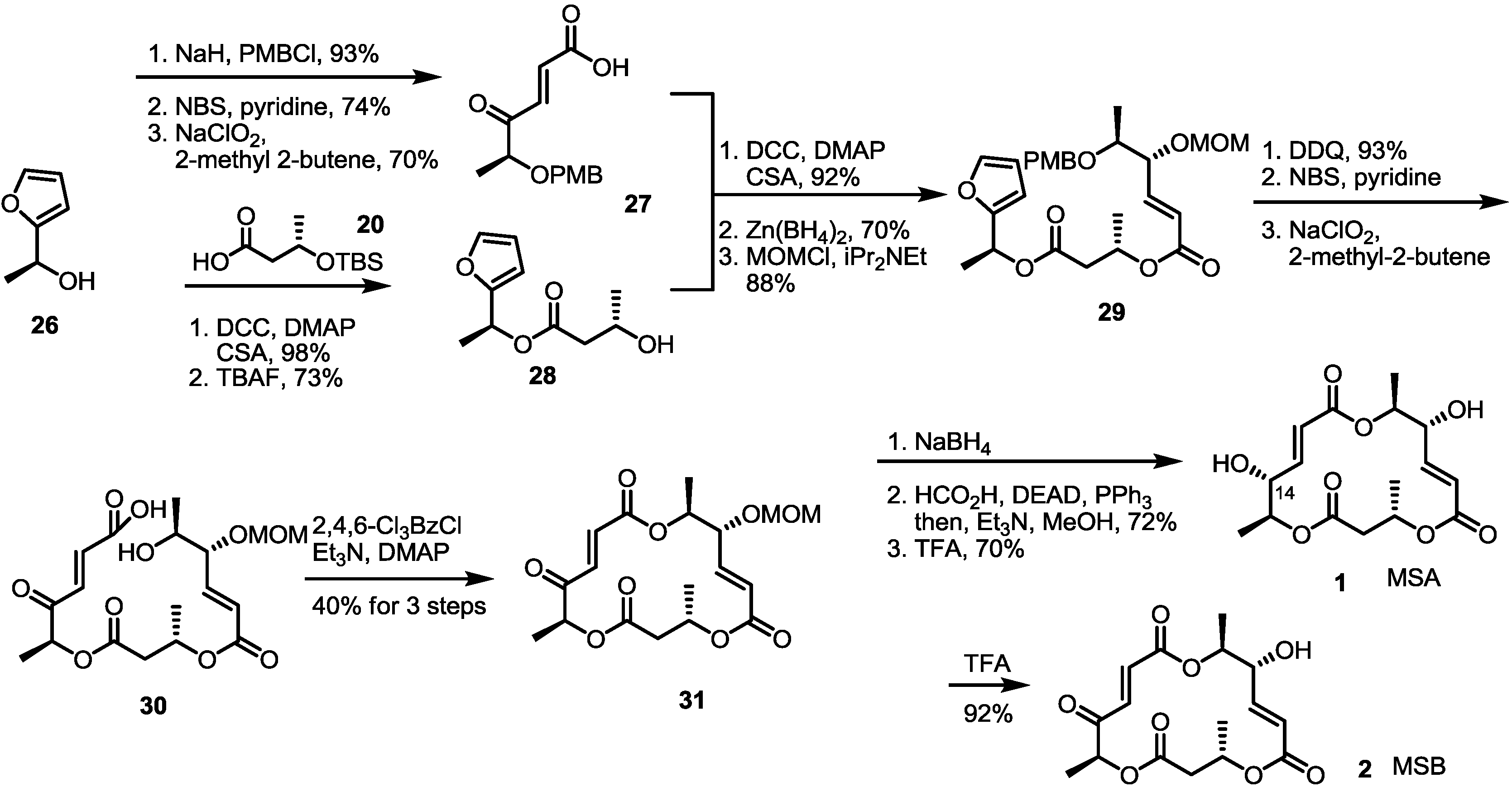
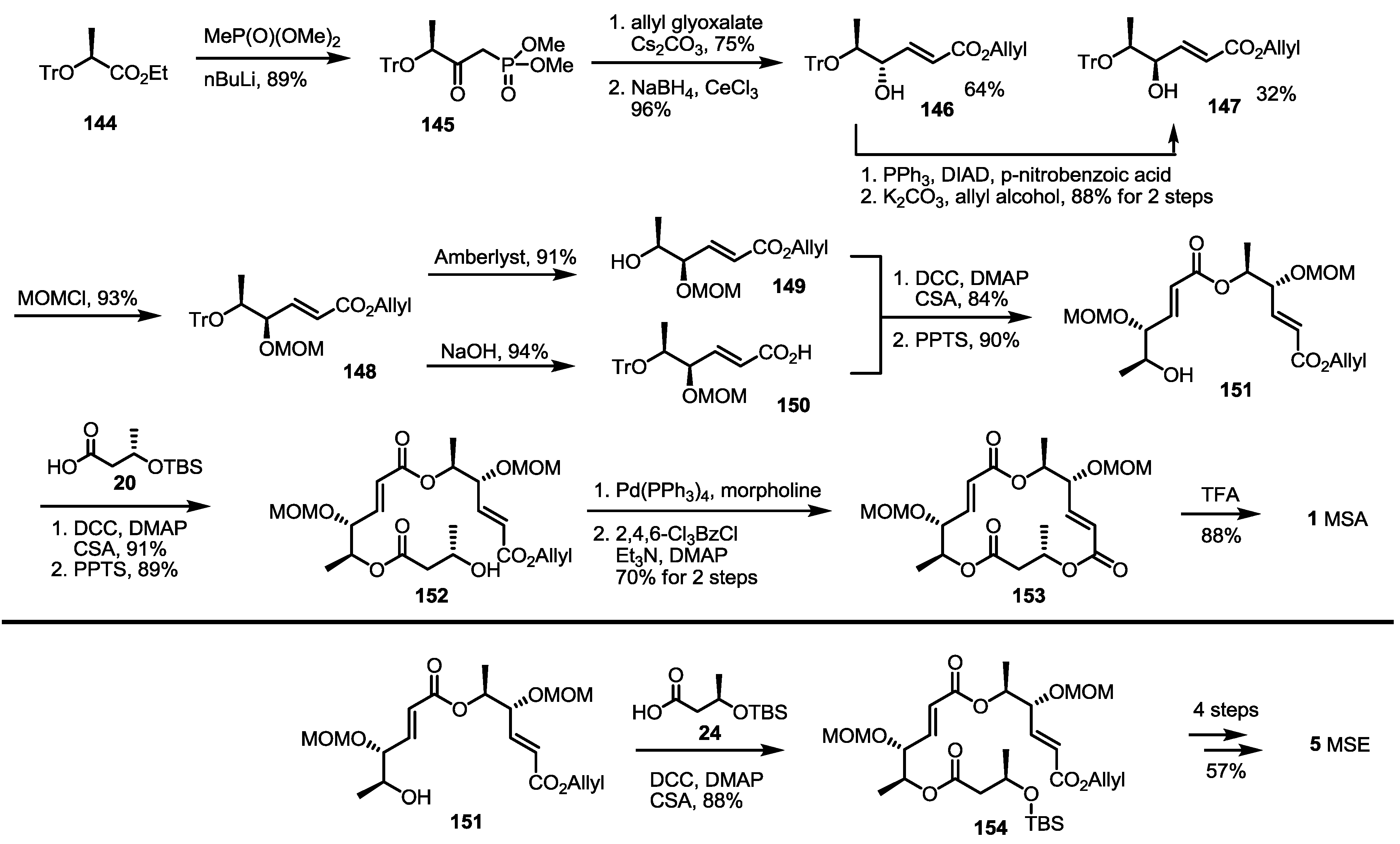
2.3. Synthesis of MSA and MSB by Furan Oxidation
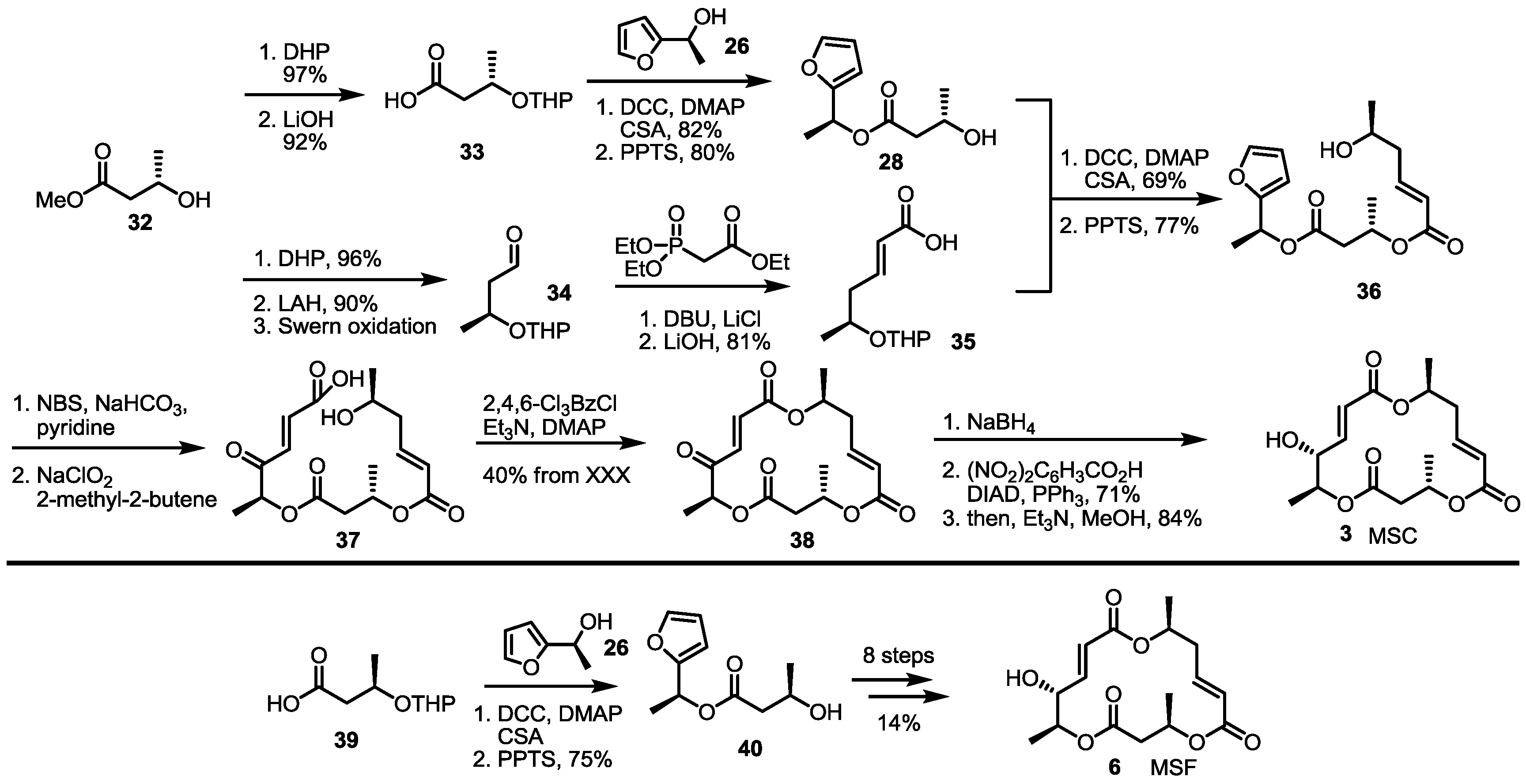
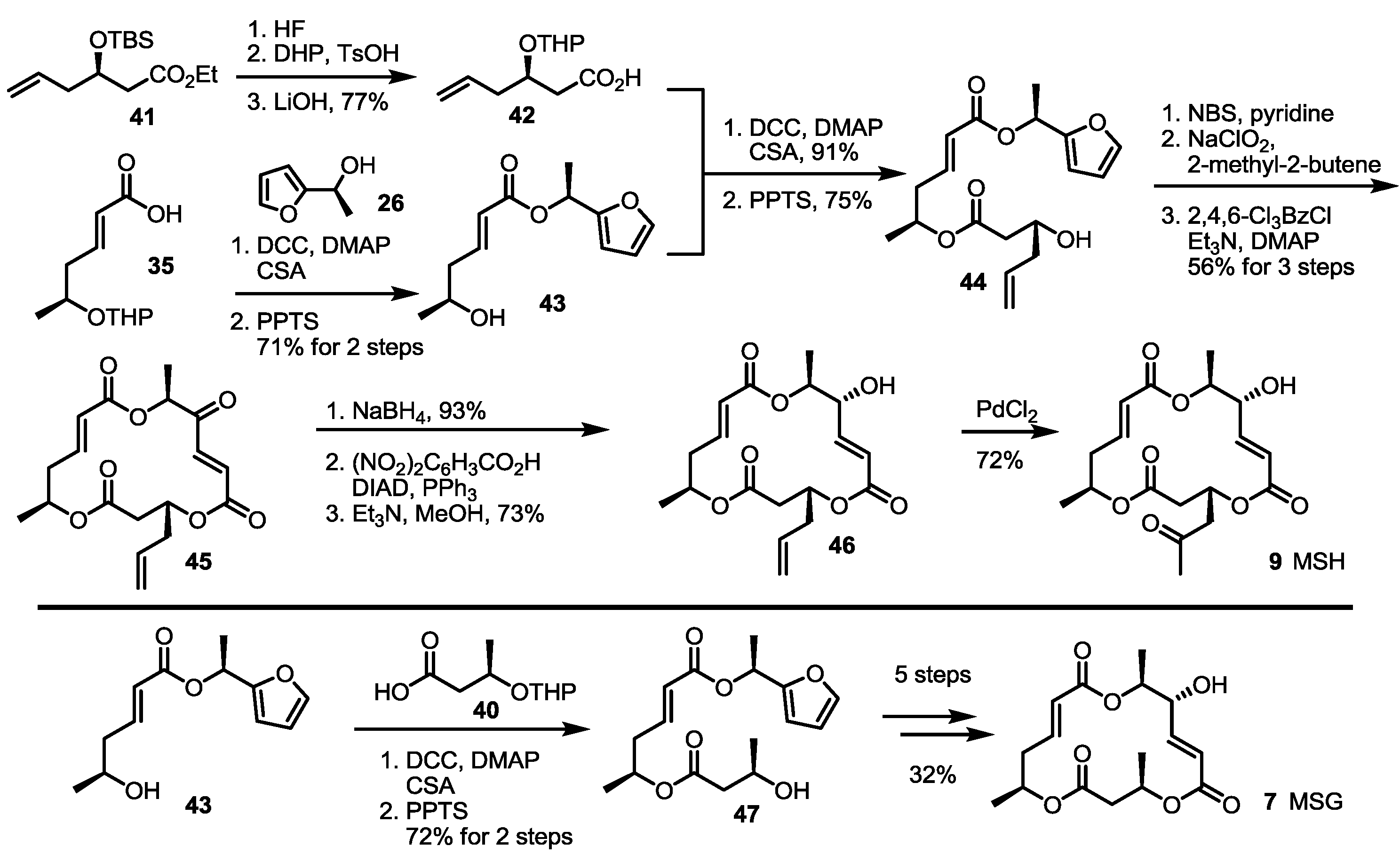
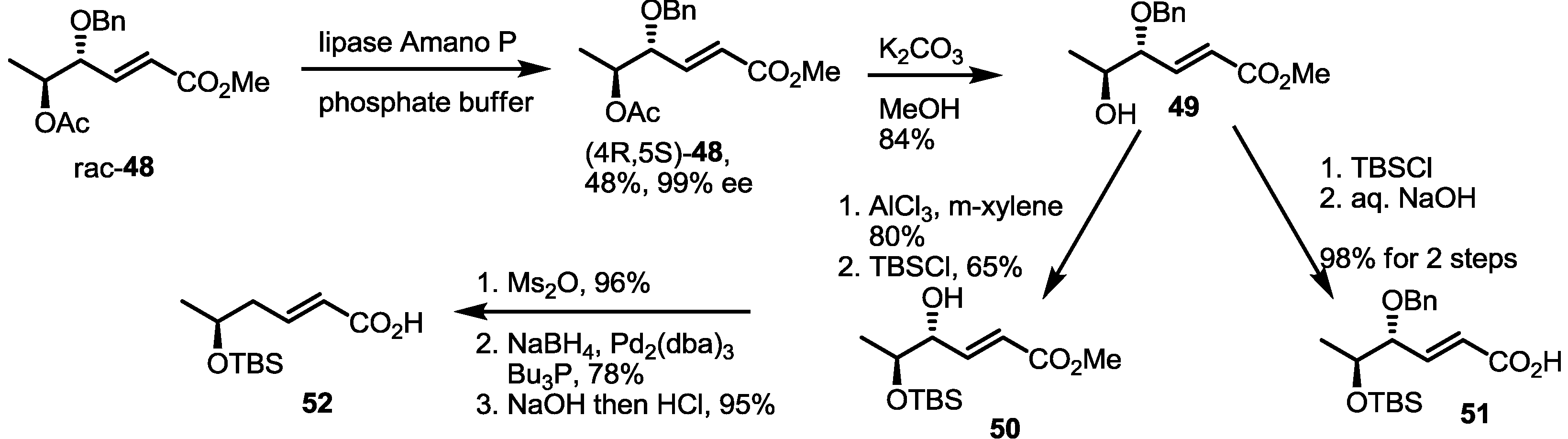
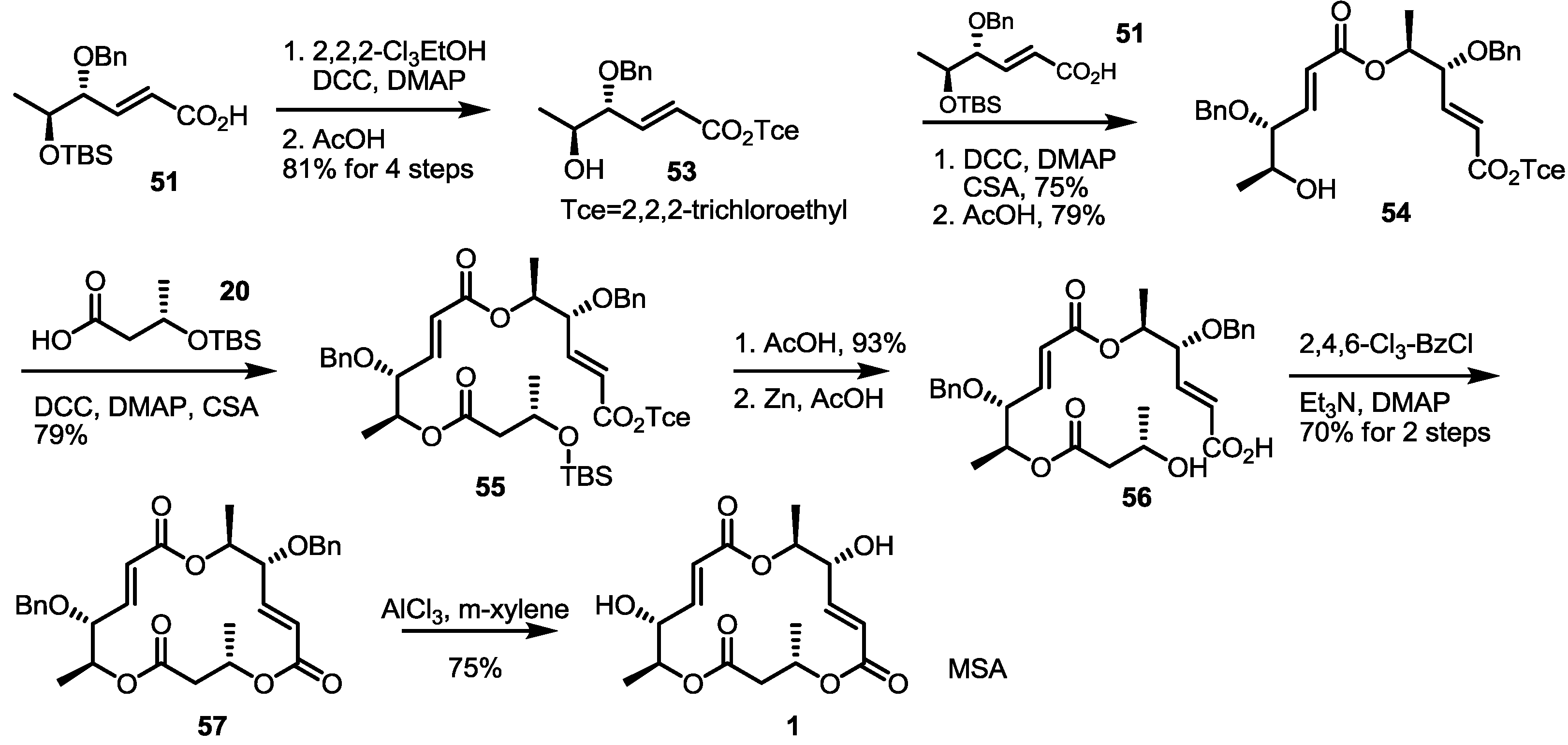
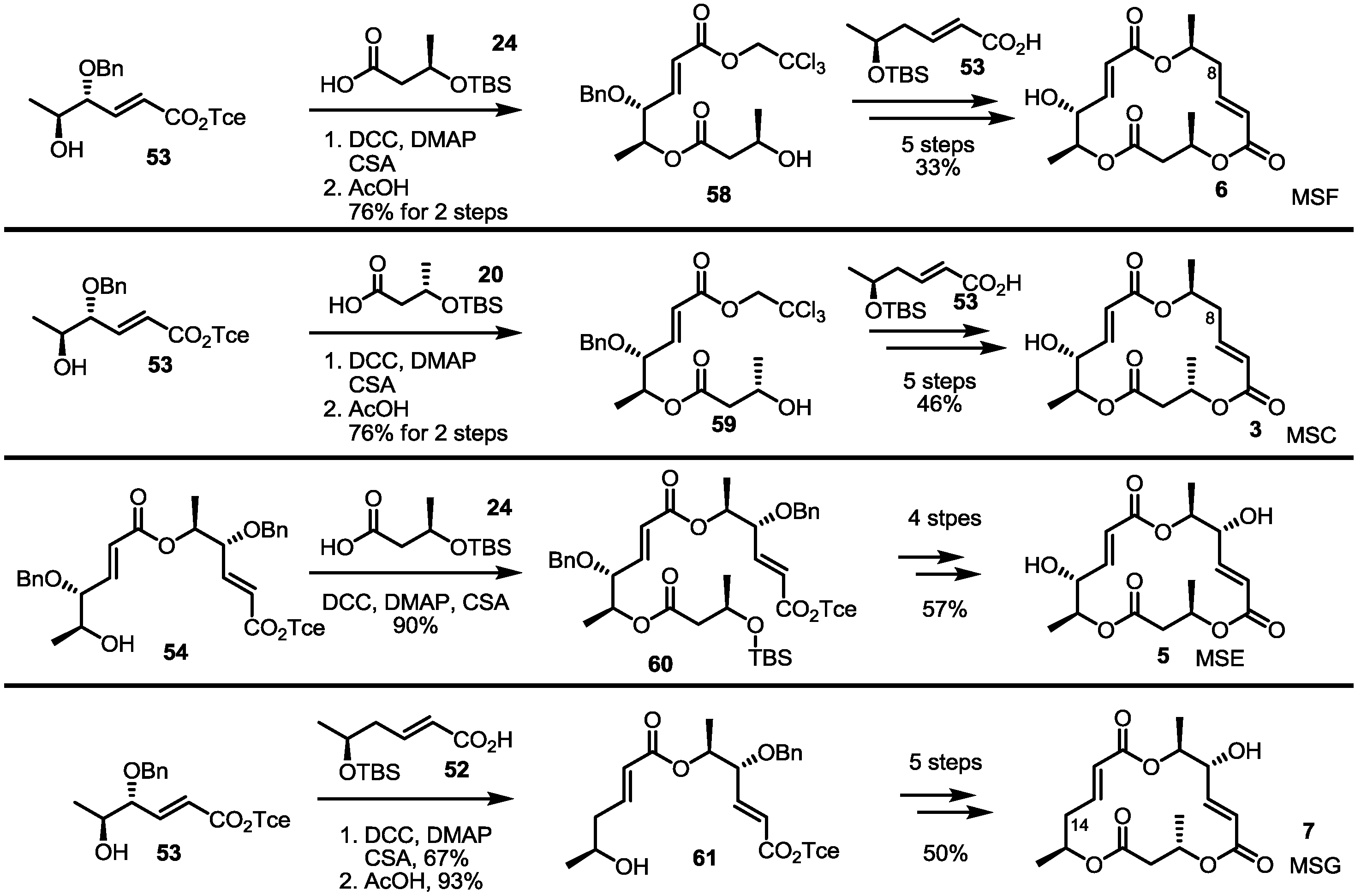
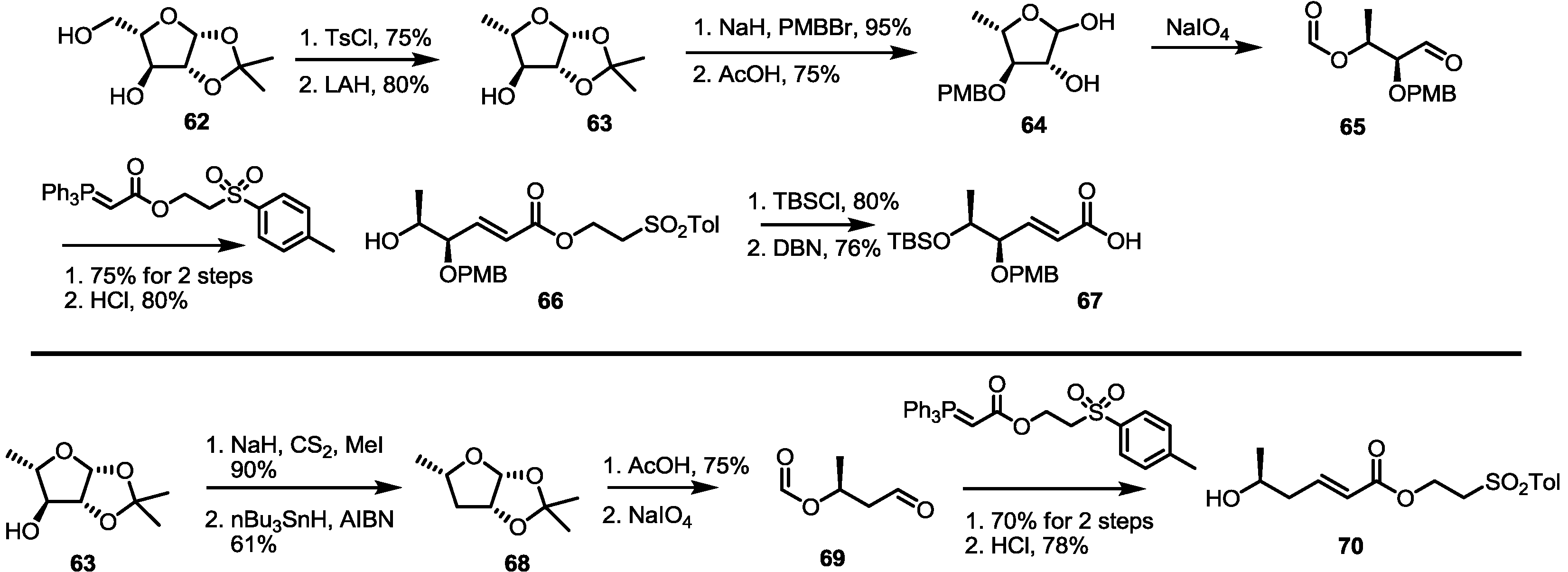
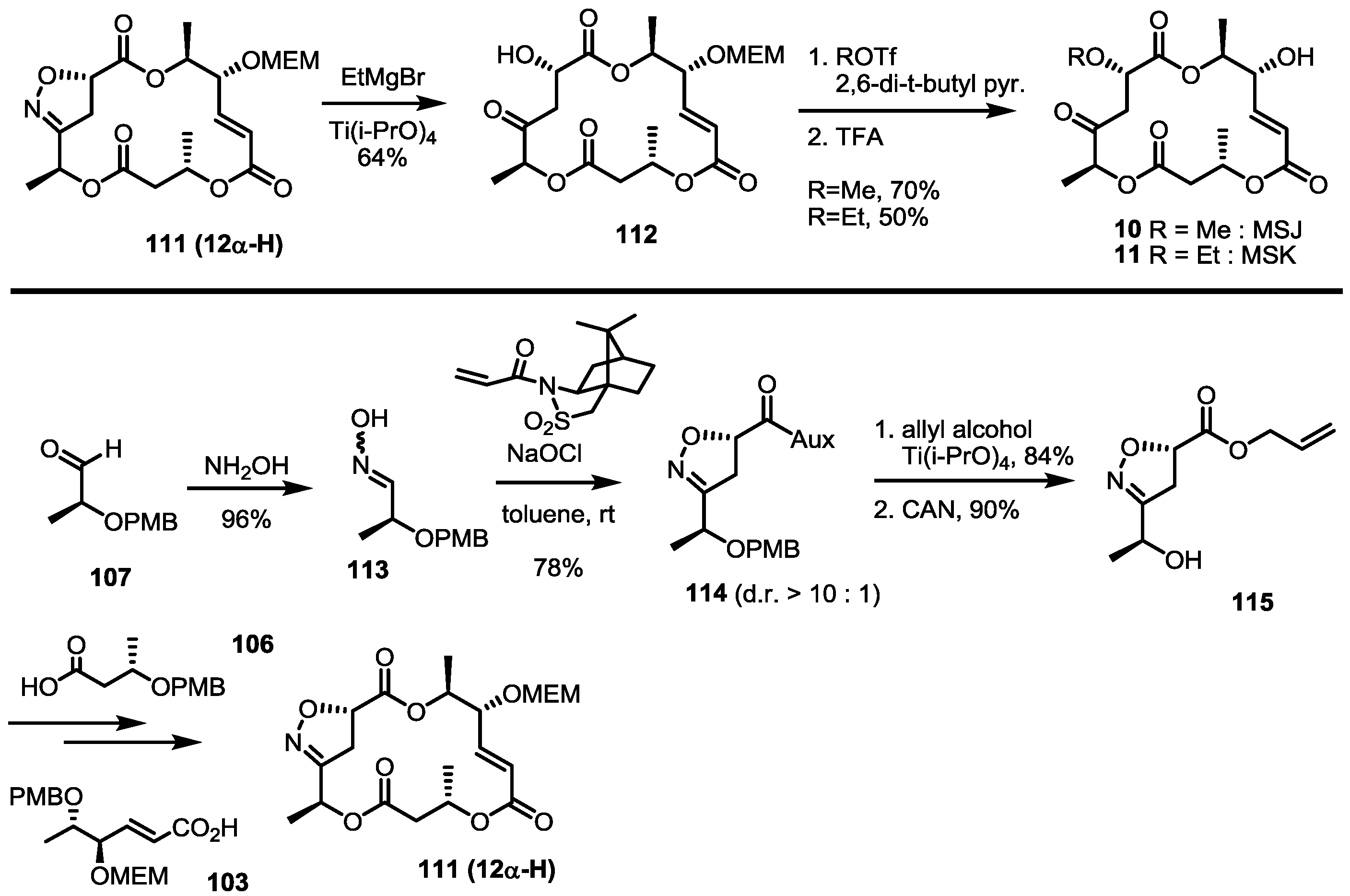
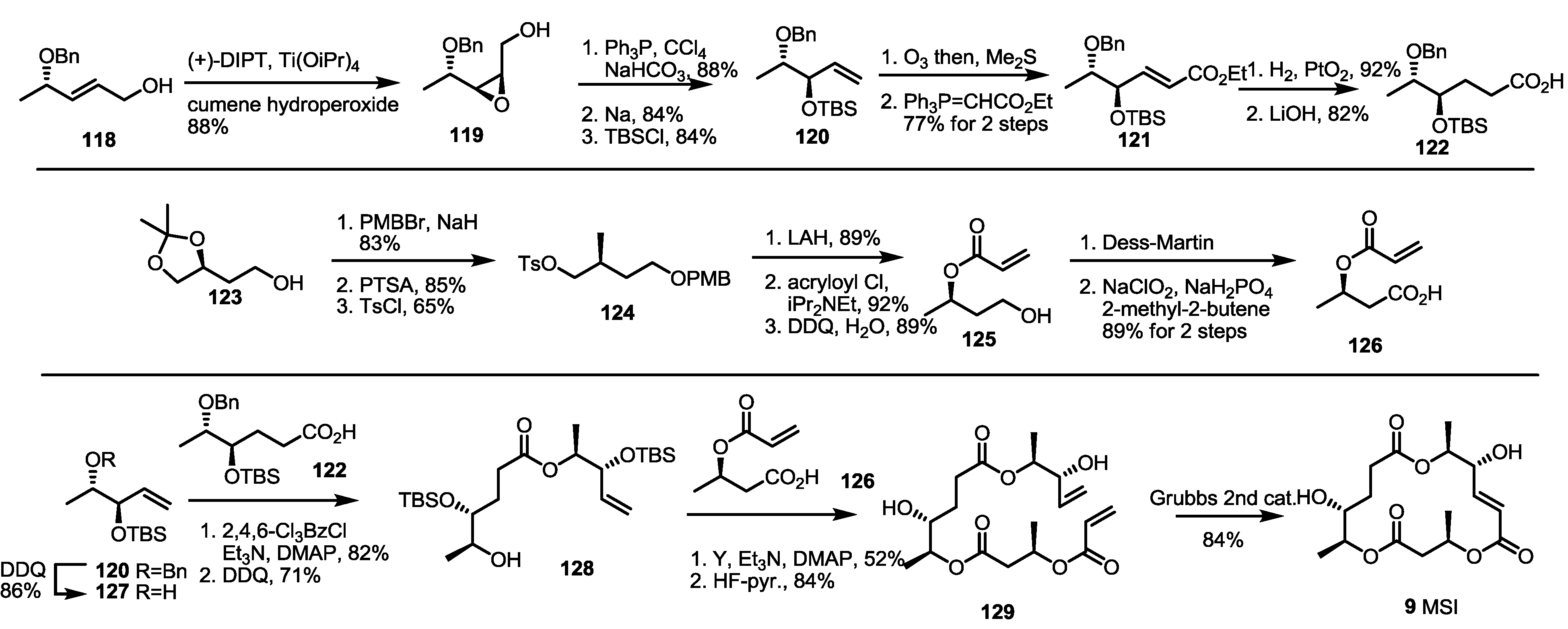
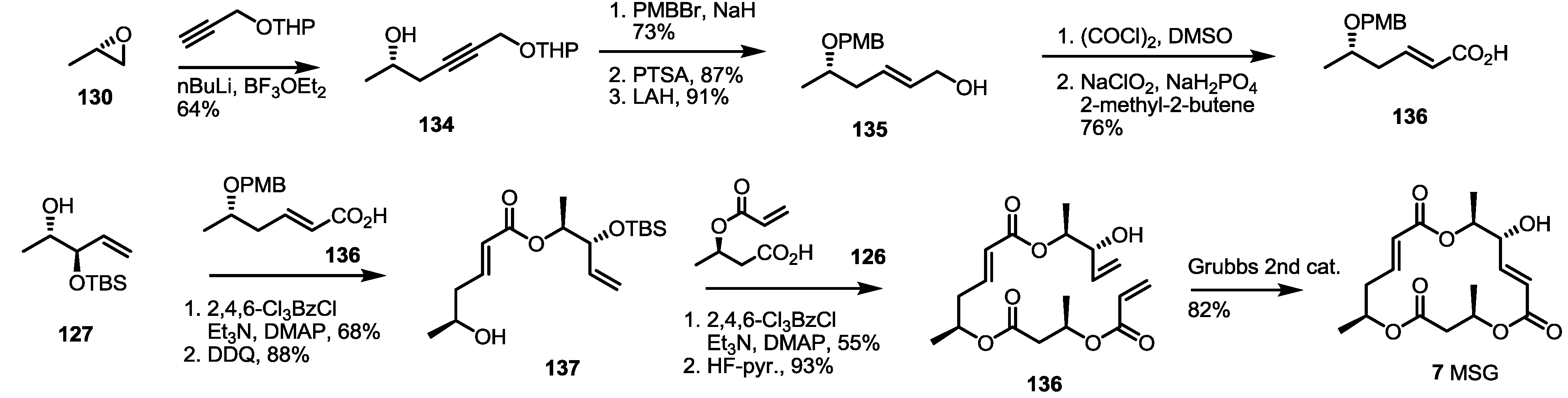
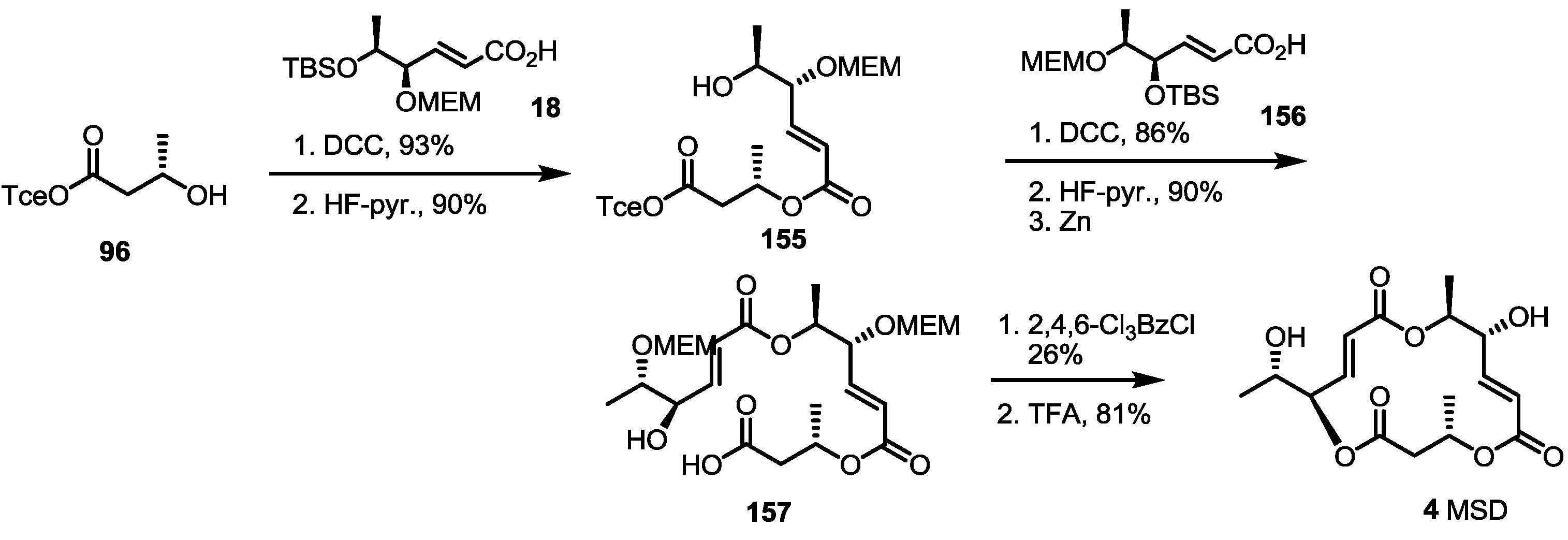
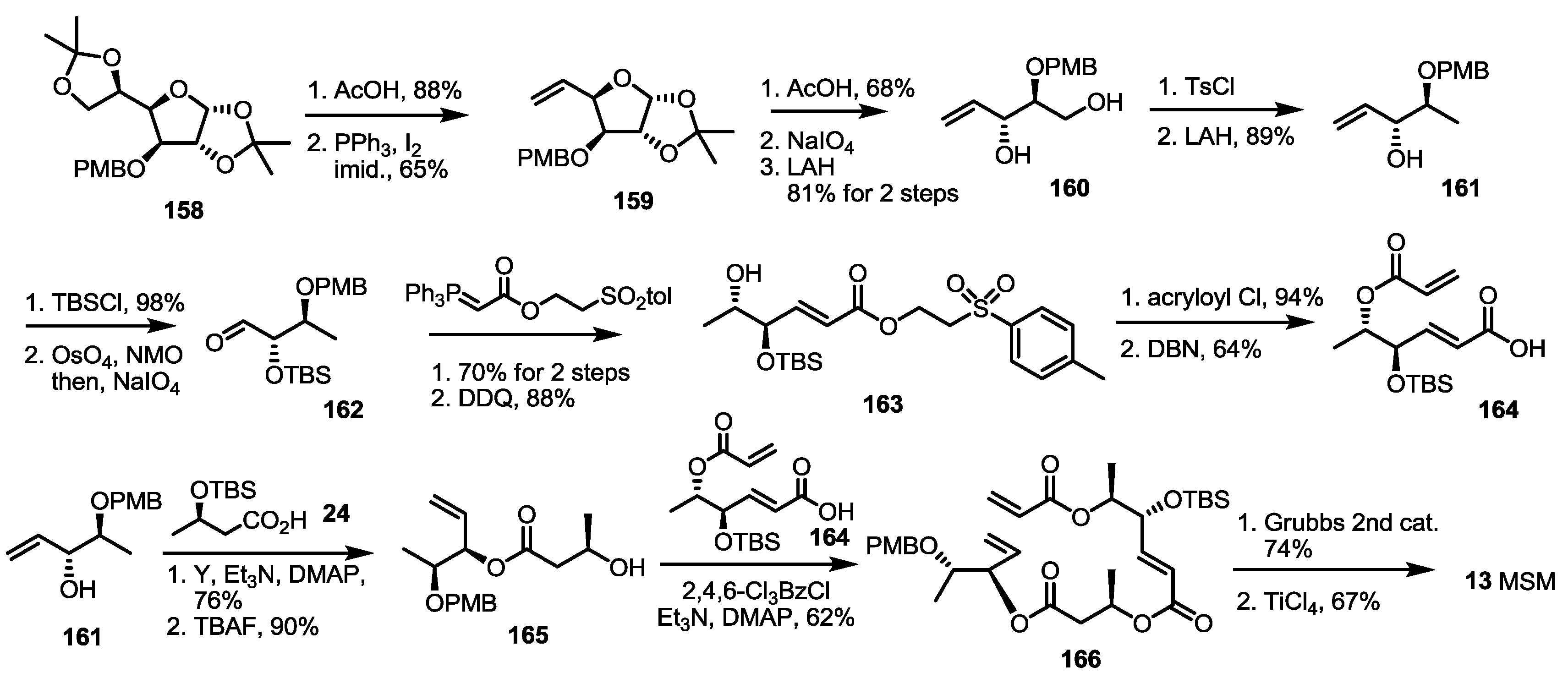
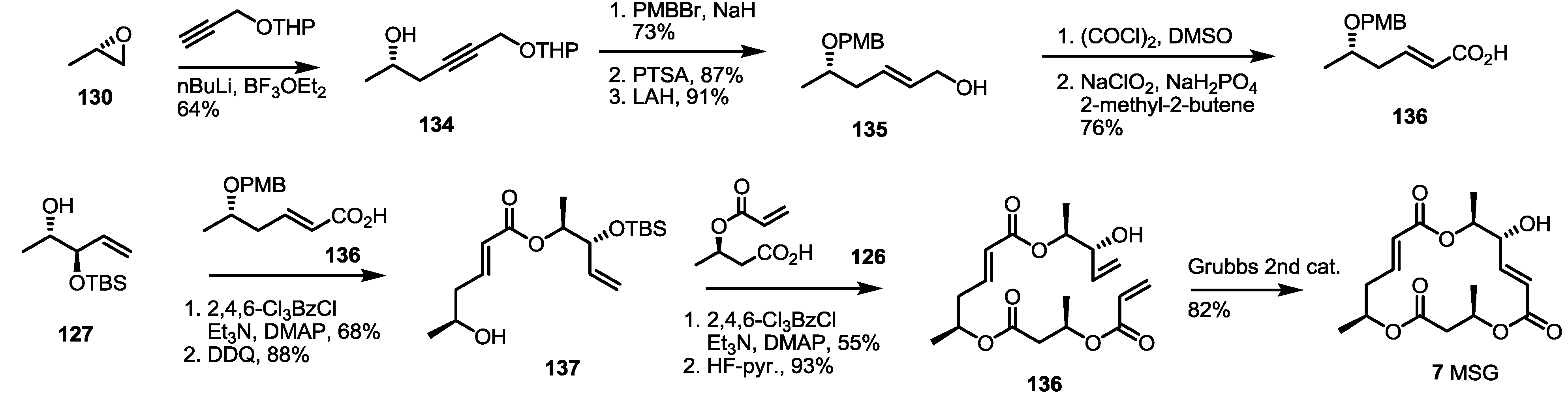
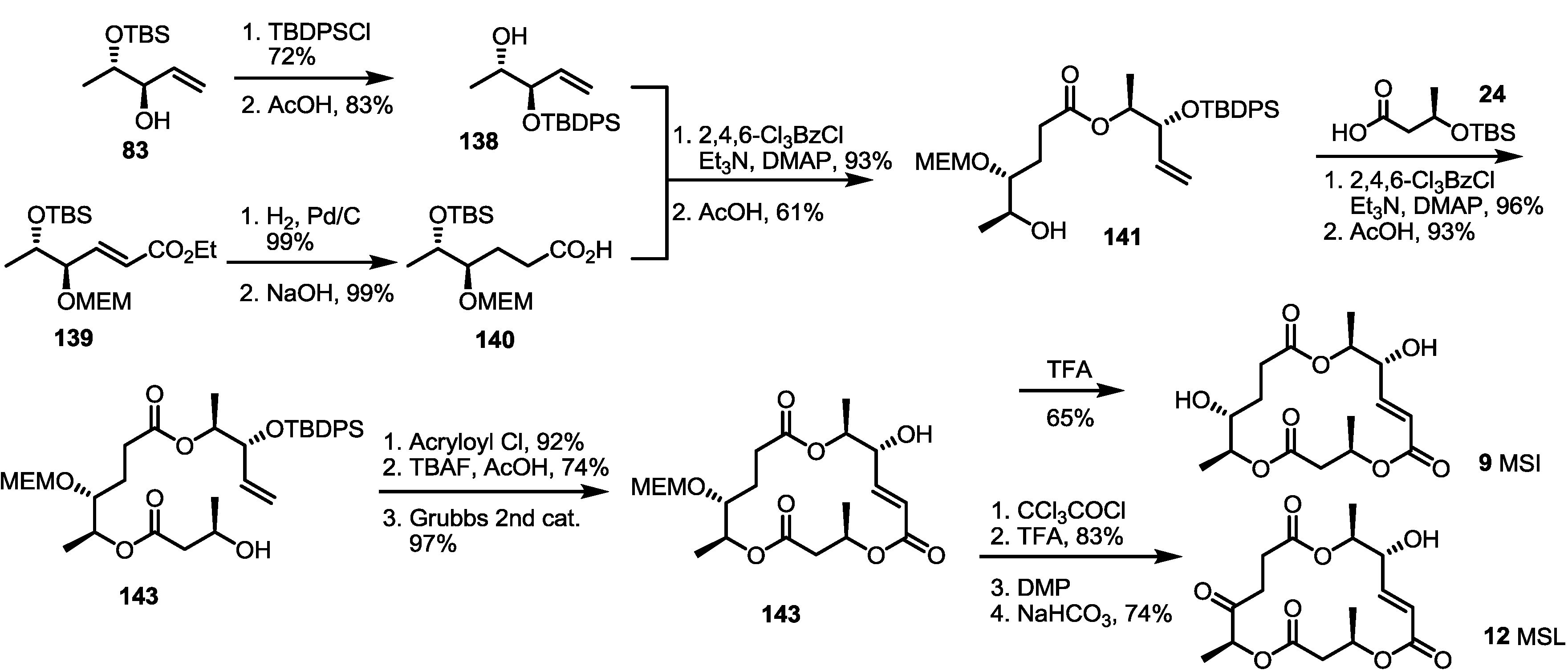
2.4. Synthesis of MS by Enzymatic Resolution
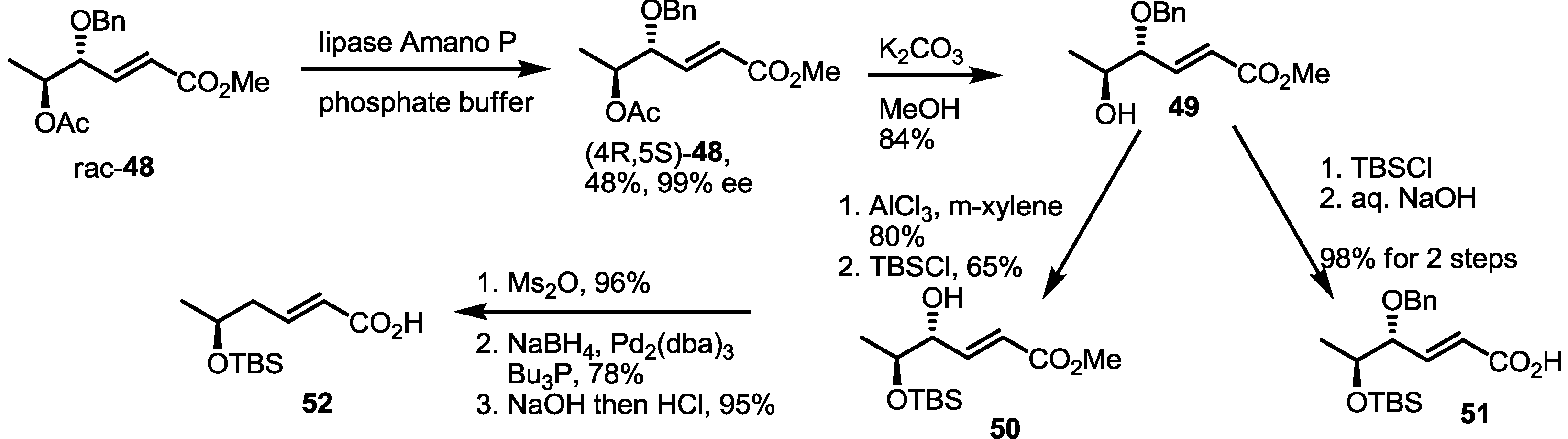
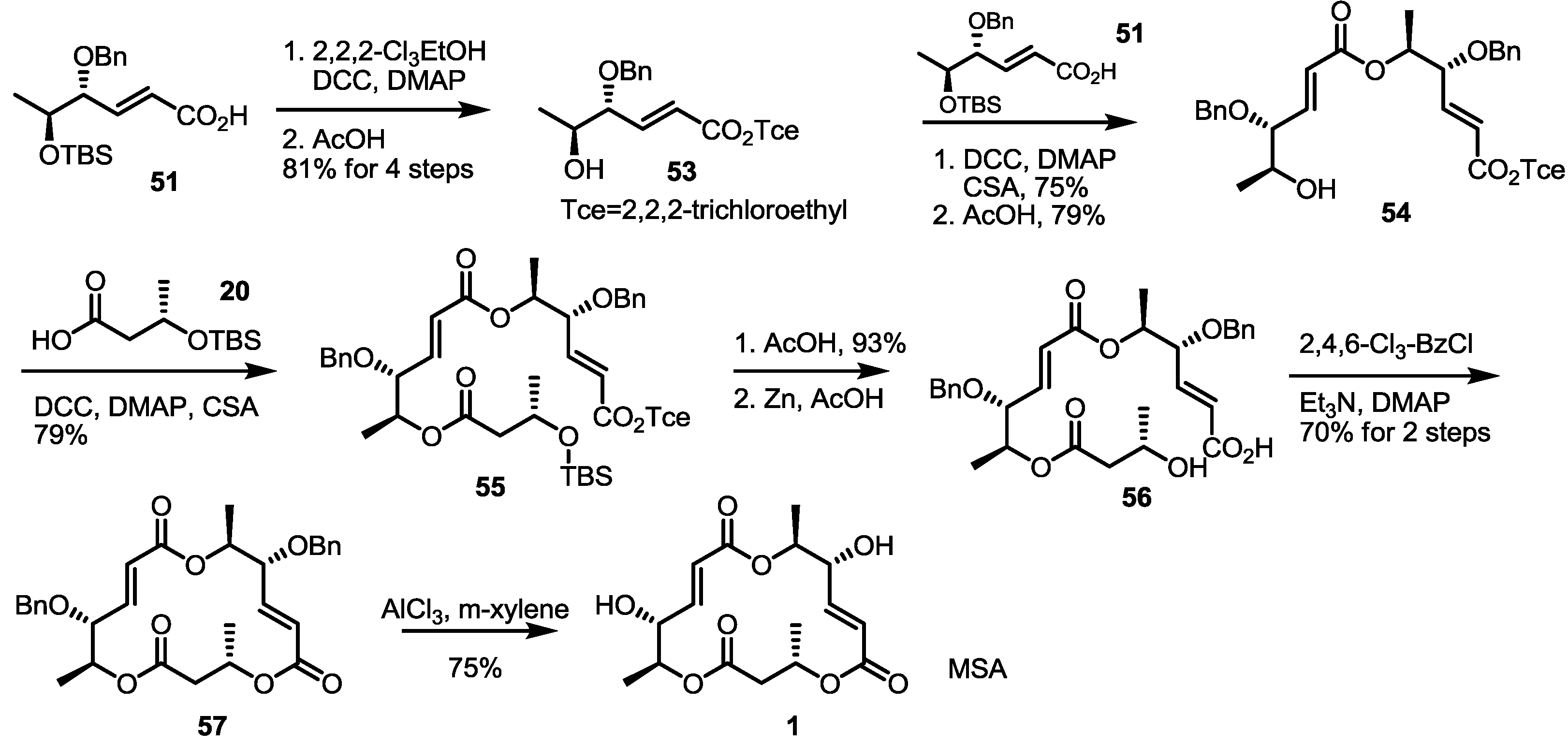
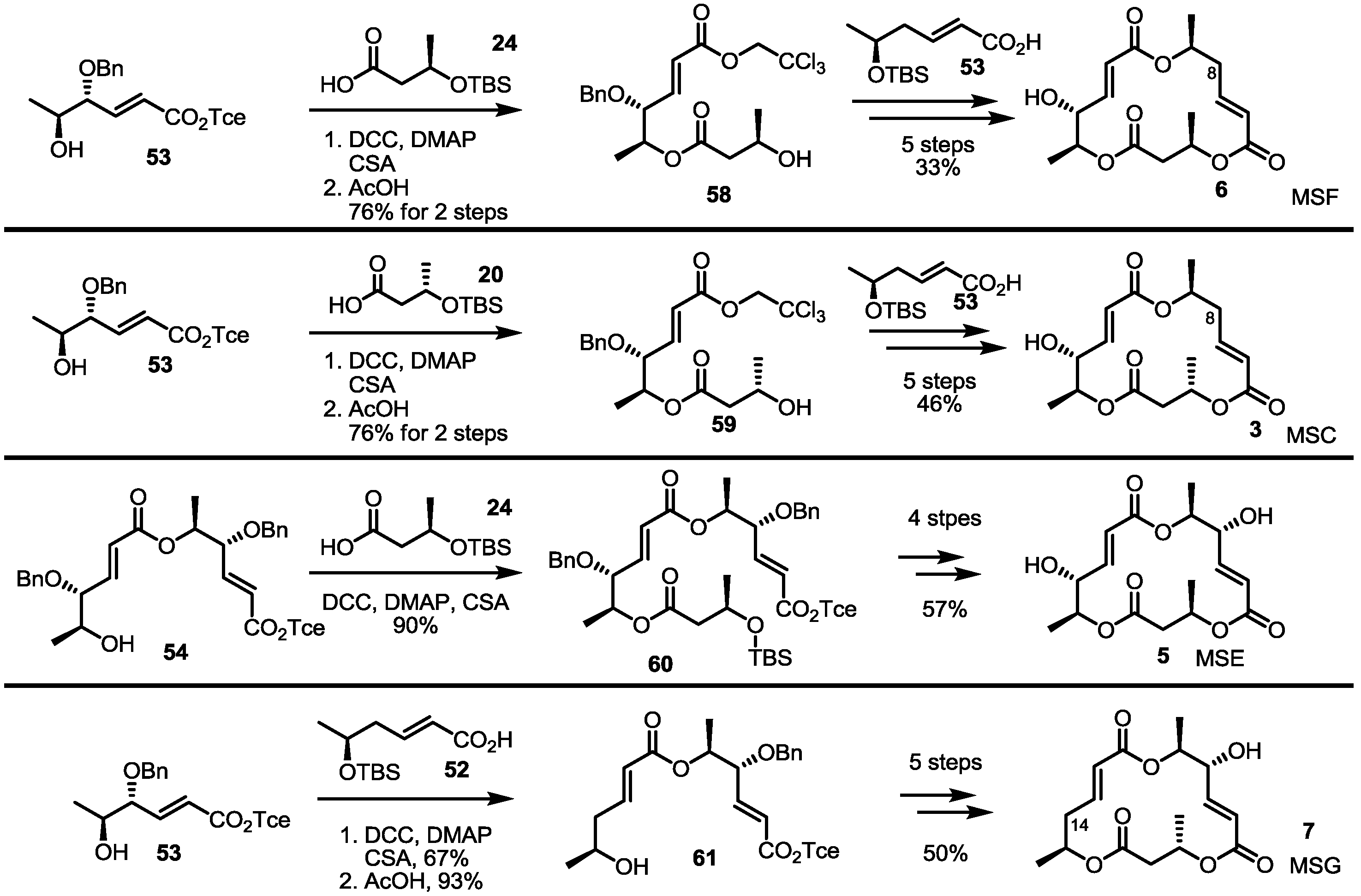
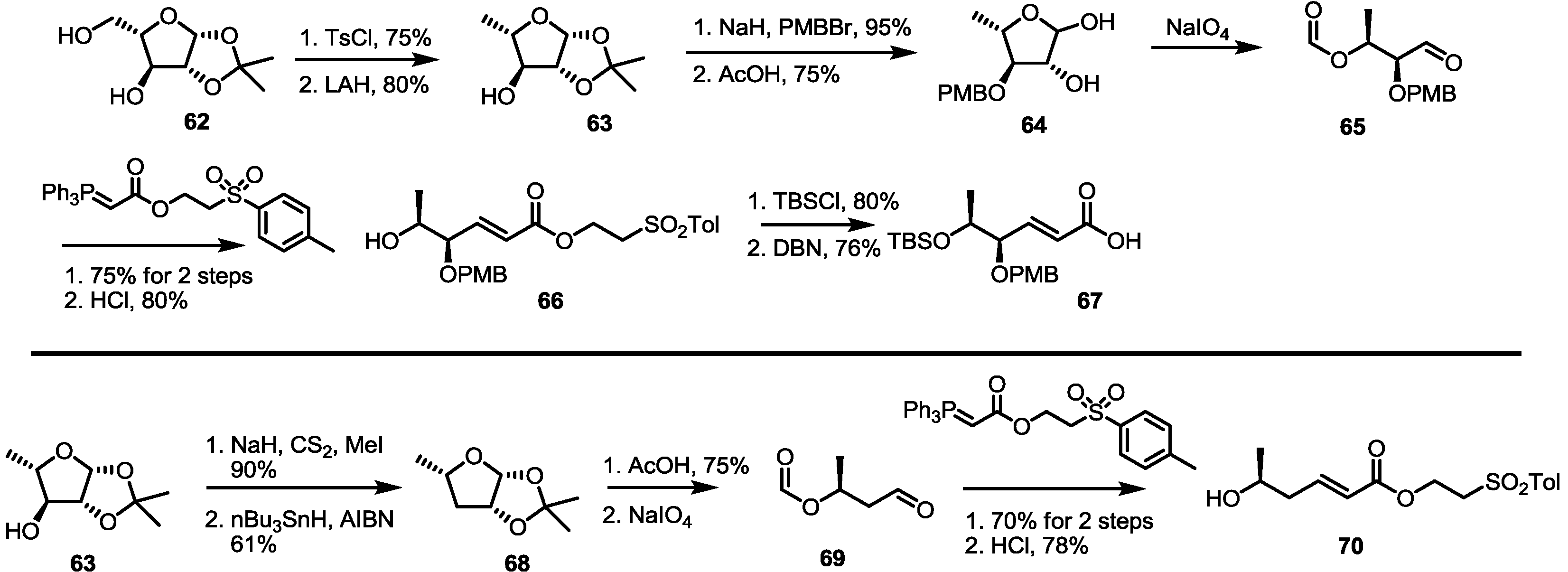
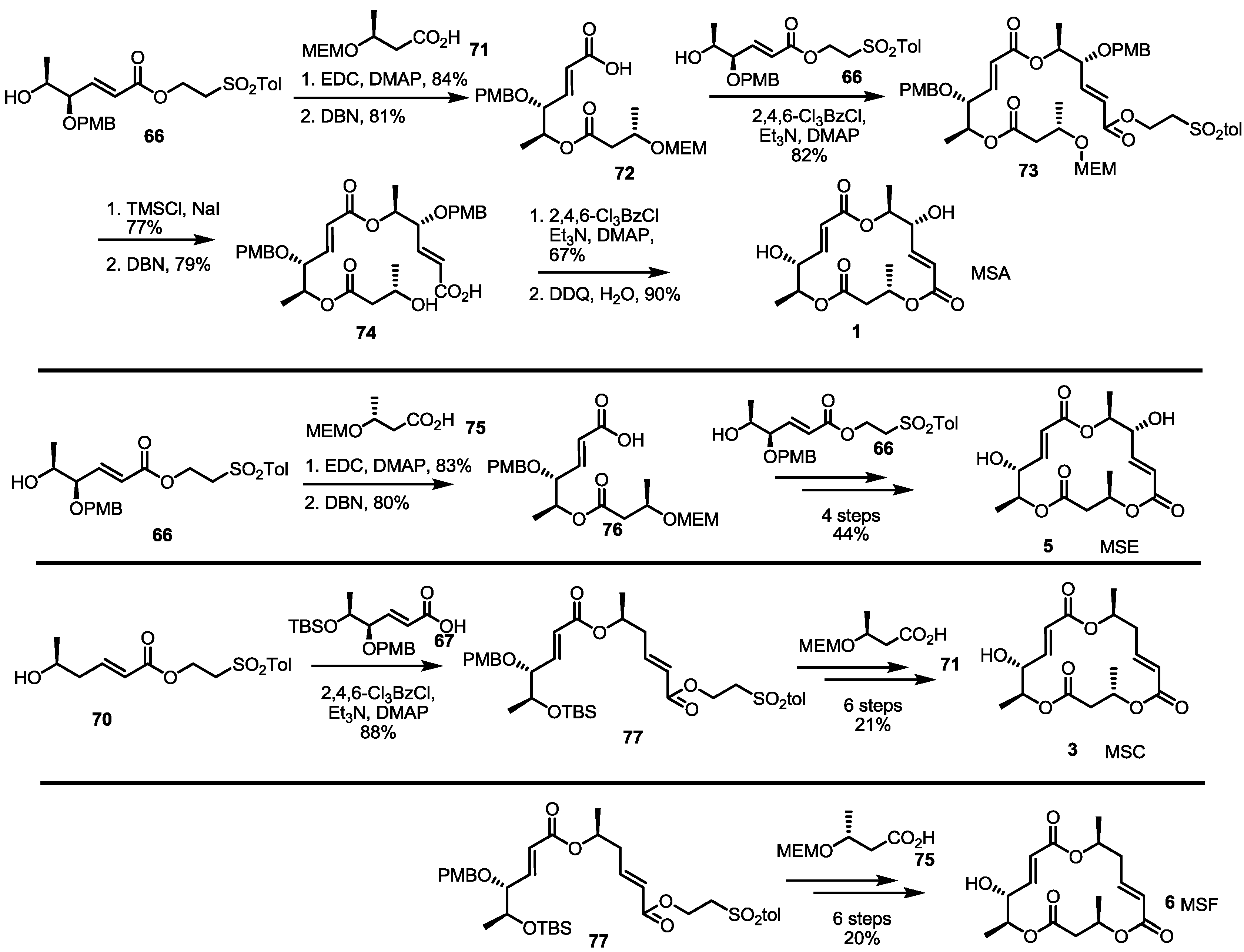
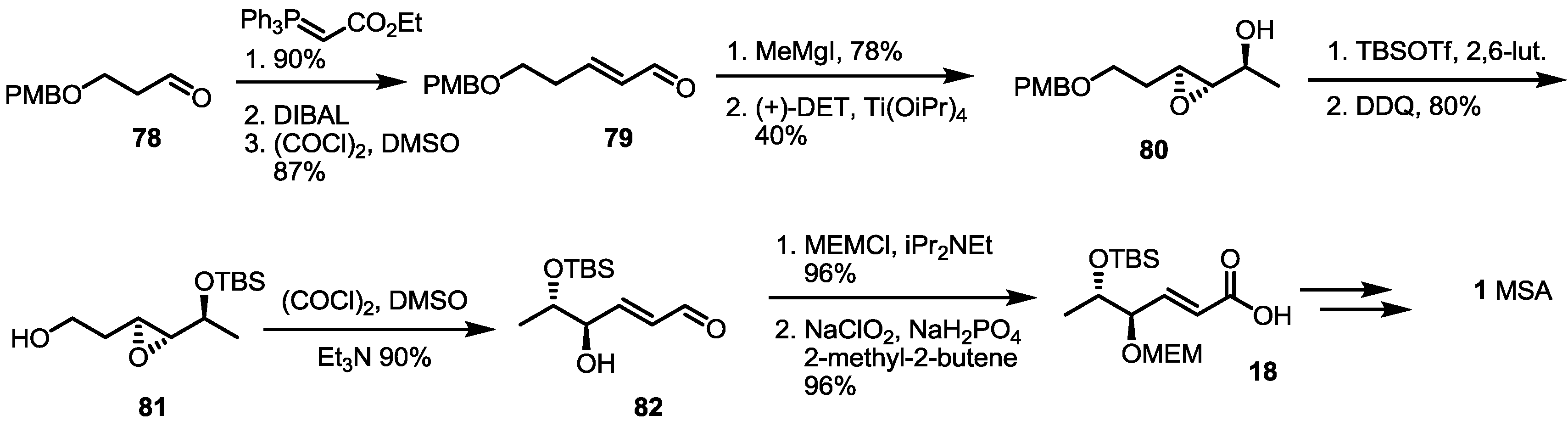
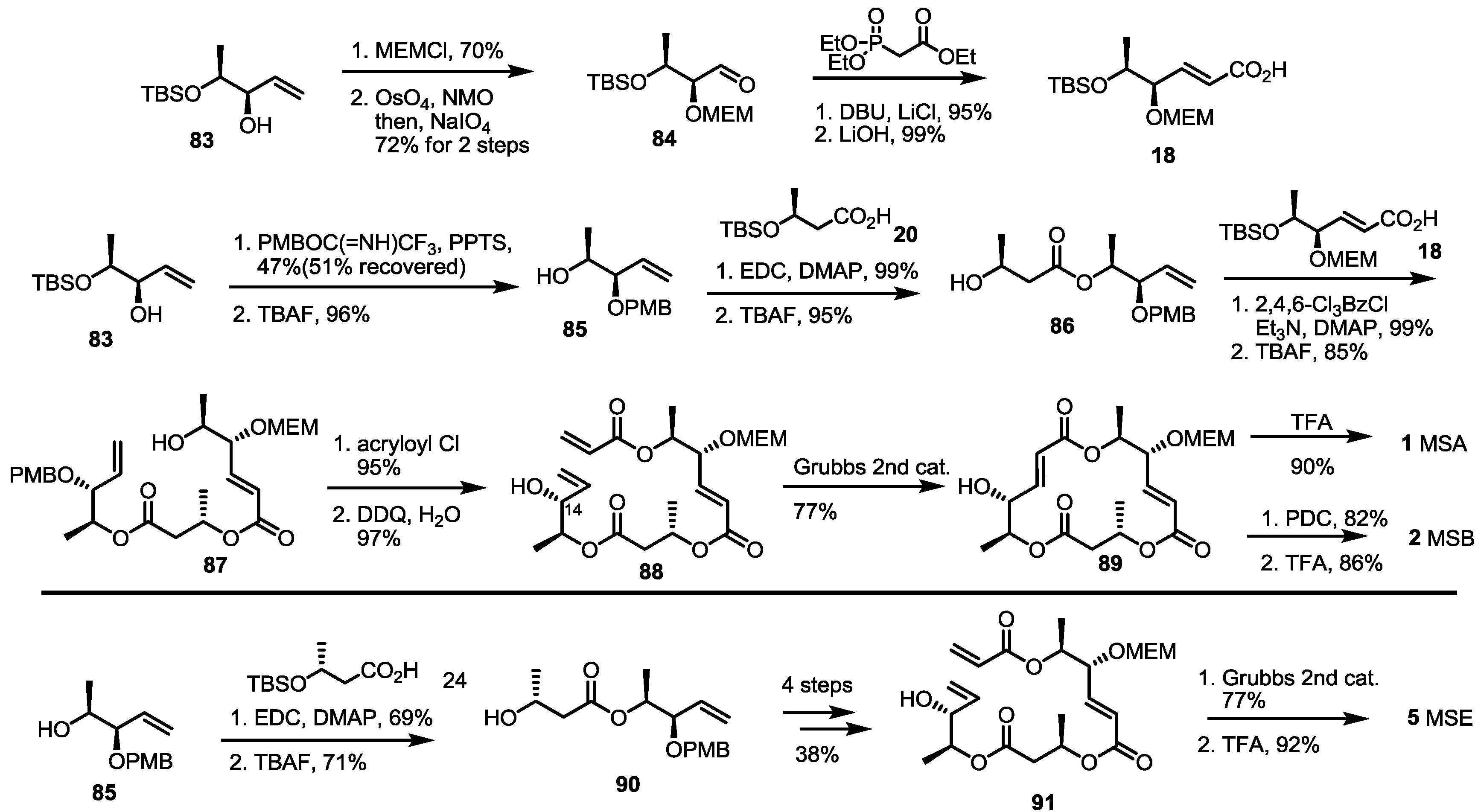
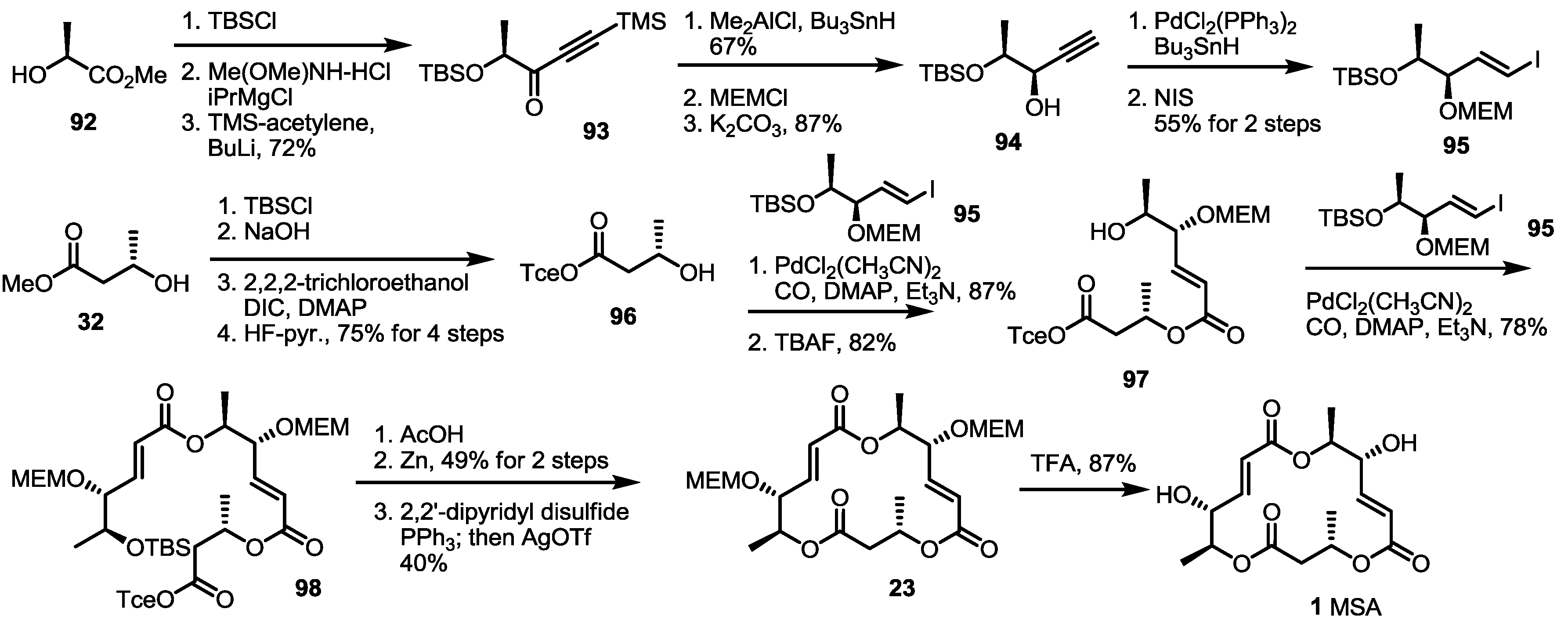
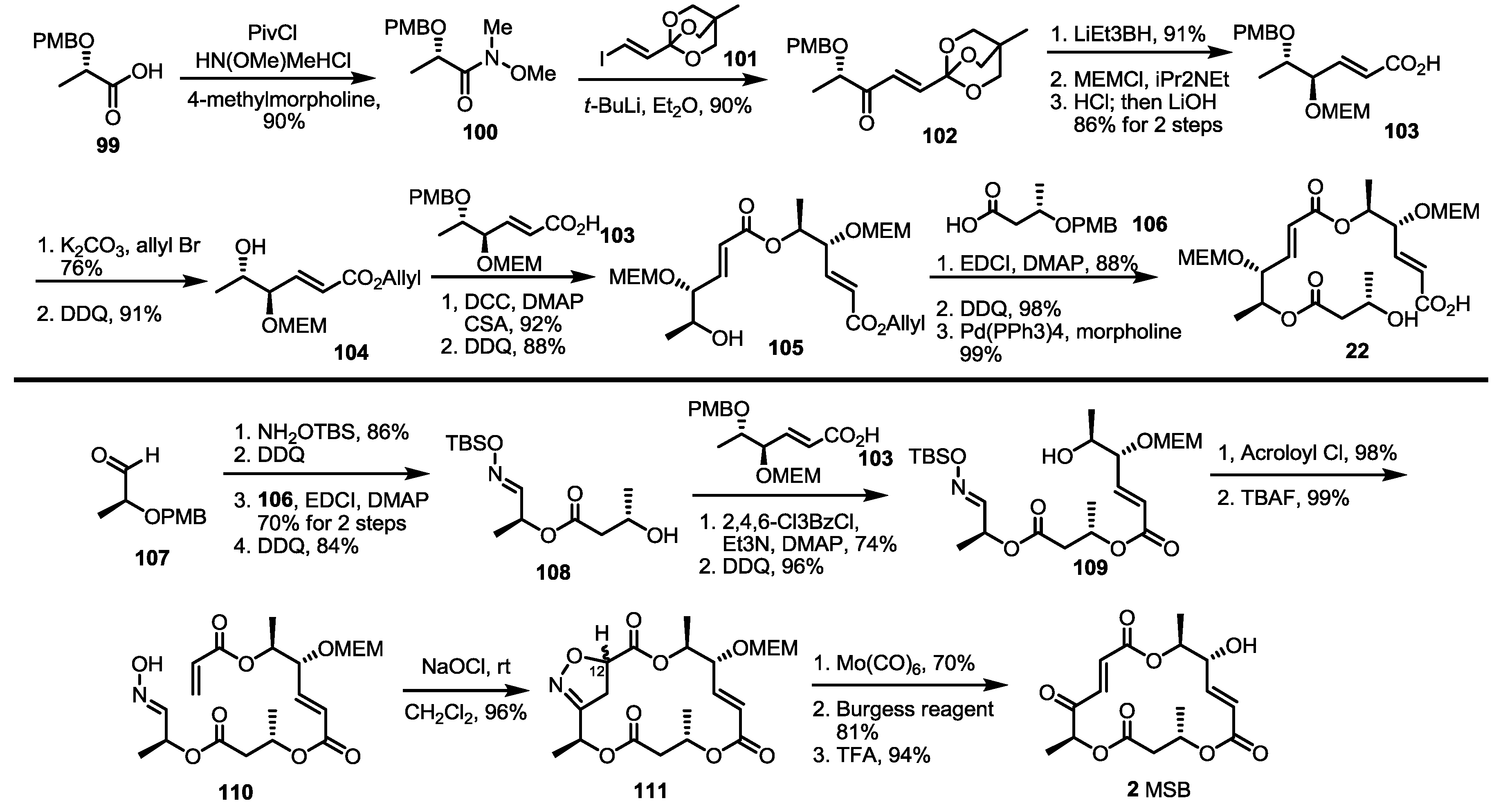

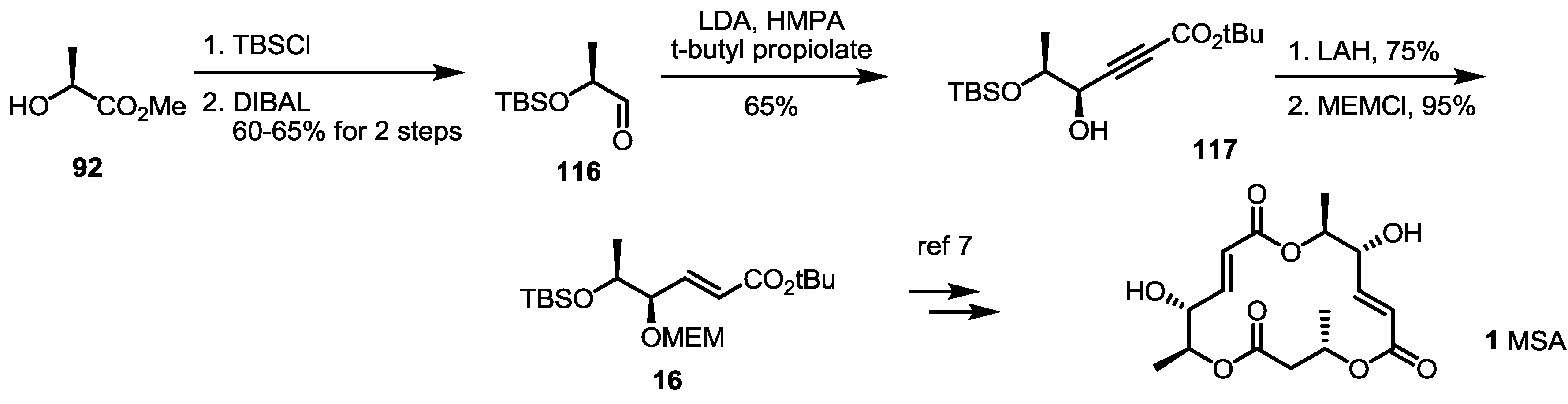
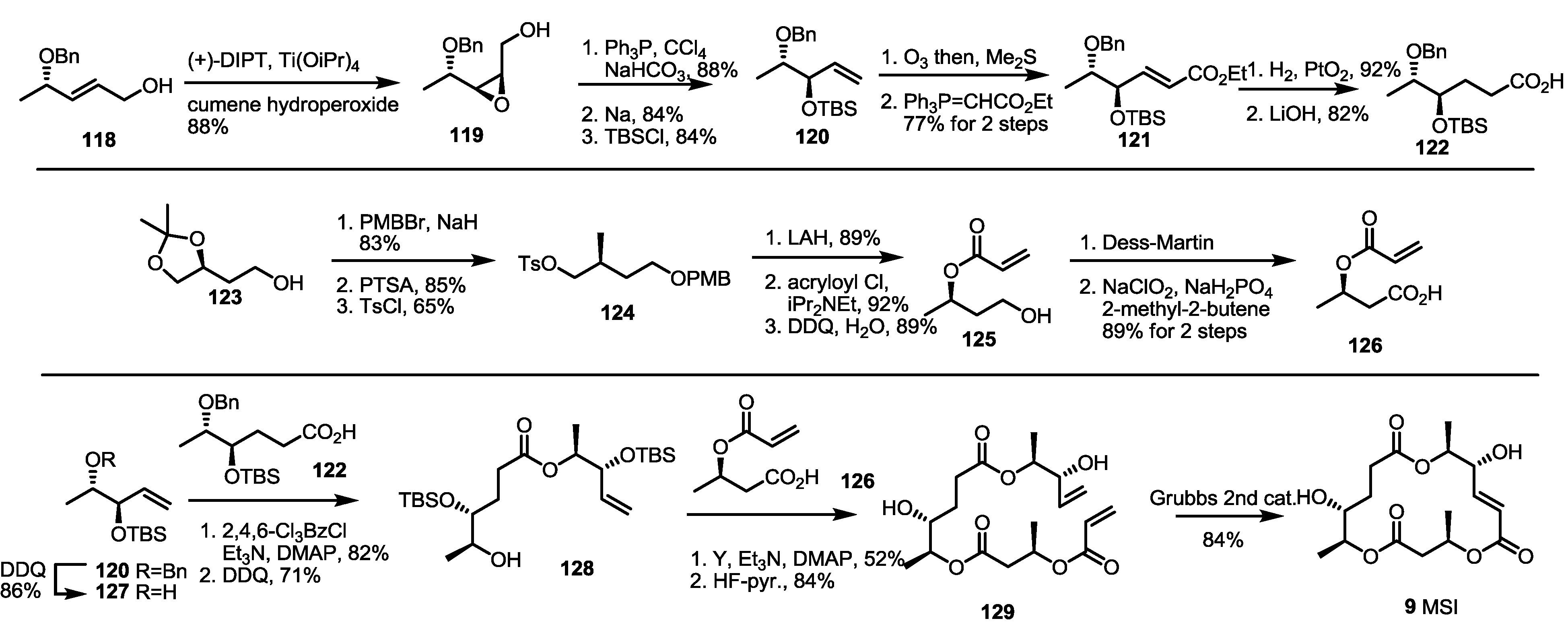
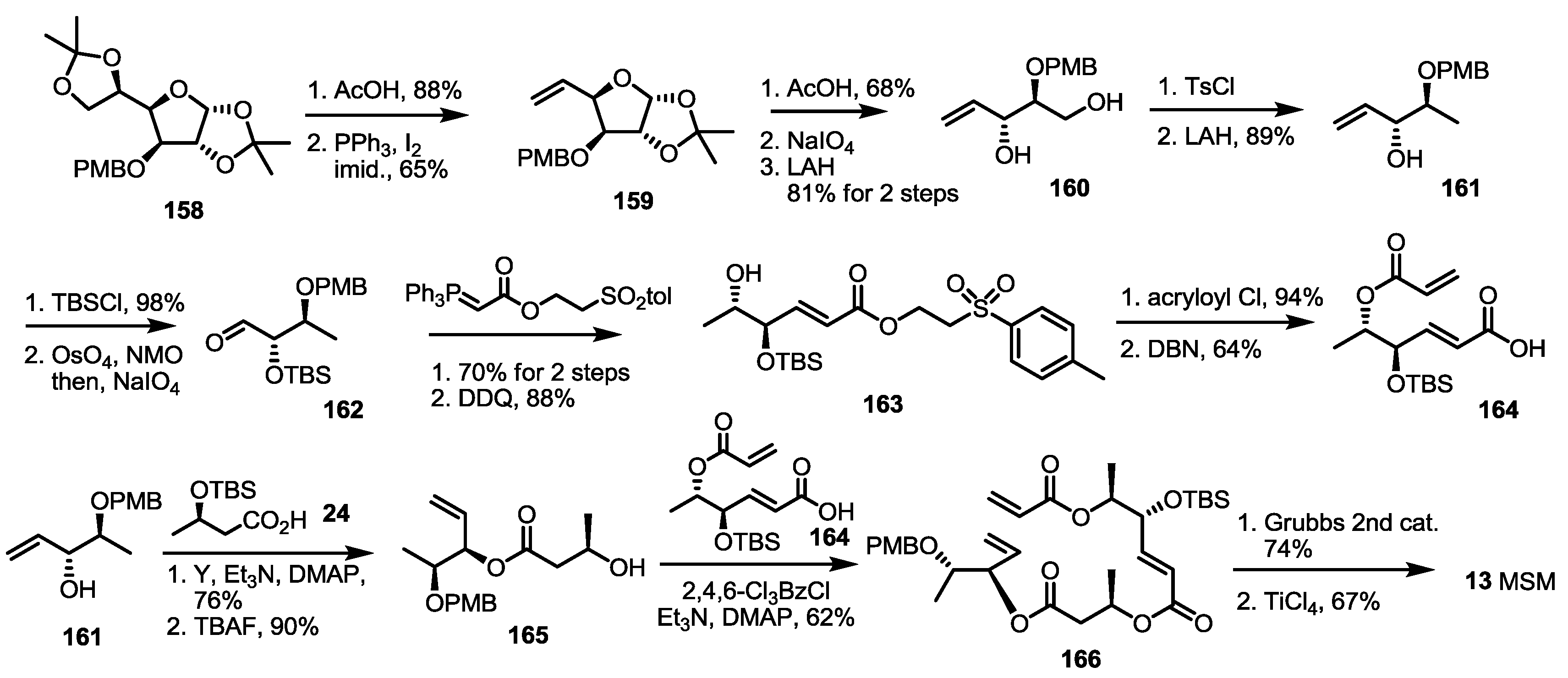
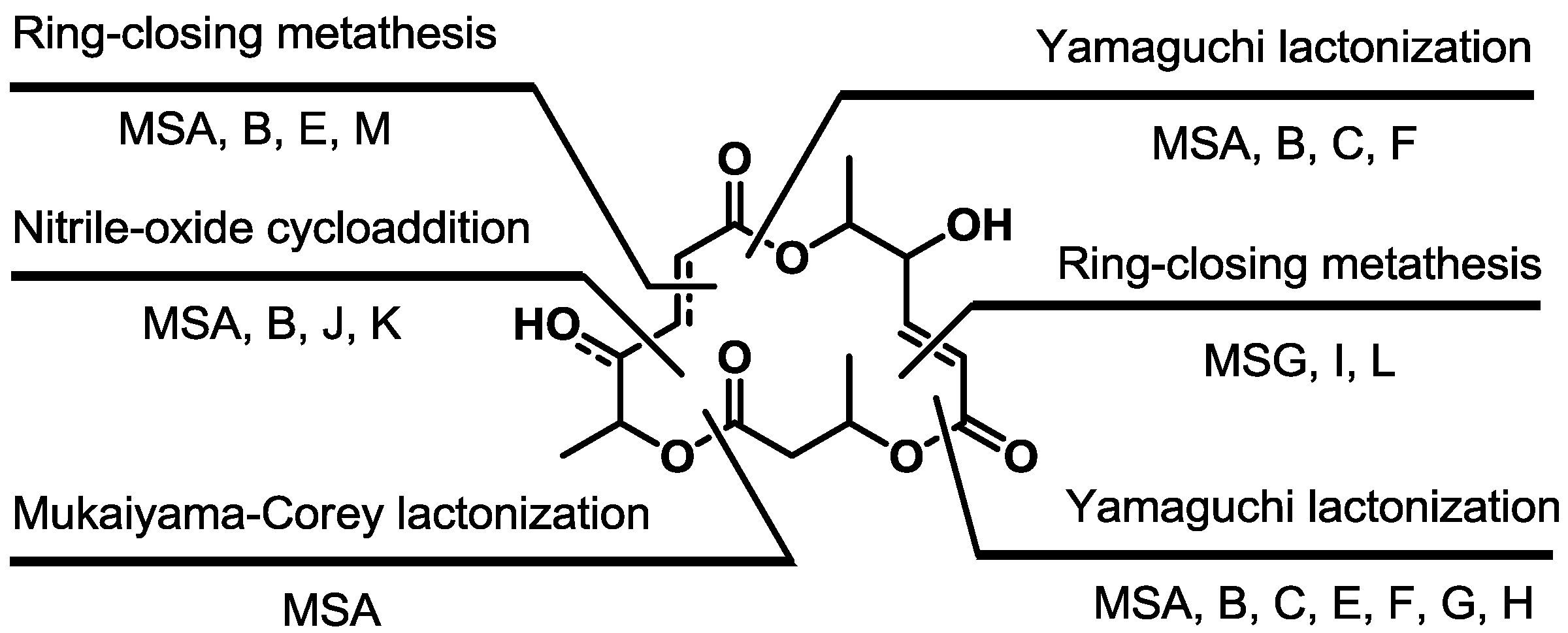
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hayashi, M.; Kim, Y.-P.; Hiraoka, H.; Natori, M.; Takamatsu, S.; Kawakubo, T.; Masuma, R.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 1995, 48, 1435–1439. [Google Scholar] [CrossRef]
- Takamatsu, S.; Kim, Y.-P.; Hayashi, M.; Hiraoka, H.; Natori, M.; Komiyama, K.; Omura, S. Macrosphelide, a novel inhibitor of cell-cell adhesion molecule II. Physicochemical properties and structural elucidation. J. Antibiot. 1996, 49, 95–98. [Google Scholar]
- Takamatsu, S.; Hiraoka, H.; Kim, Y.-P.; Hayashi, M.; Natori, M.; Komiyama, K.; Omura, S. Macrosphelides C and D, novel inhibitors of cell adhesion. J. Antibiot. 1997, 50, 878–880. [Google Scholar] [CrossRef] [PubMed]
- Numata, A.; Iritani, M.; Yamada, T.; Minoura, K.; Matsumura, E.; Yamori, T.; Tsuruo, T. Novel antitumour metabolites produced by a fungal strain from a sea hare. Tetrahedron Lett. 1997, 38, 8215–8218. [Google Scholar] [CrossRef]
- Fukami, A.; Taniguchi, Y.; Nakamura, T.; Rho, M.-C.; Kawaguchi, K.; Hayashi, M.; Komiyama, K.; Omura, S. New members of the macrosphelides from microsphaeropsis sp. FO-5050 IV. J. Antibiot. 1999, 52, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Iritani, M.; Doi, M.; Minoura, K.; Ito, T.; Numata, A. Absolute stereostructures of cell-adhesion inhibitors, macrosphelides C, E–G and I, produced by a Periconia species separated from an Aplysia sea hare. J. Chem. Soc. Perkin Trans. 1 2001, 3046–3053. [Google Scholar]
- Yamada, T.; Iritani, M.; Minoura, K.; Numata, A.; Kobayashi, Y.; Wang, Y.-G. Absolute stereostructures of cell adhesion inhibitors, macrosphelides H and L, from Periconia byssoides OUPS-N133. J. Antibiot. 2002, 55, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sunazuka, T.; Hirose., T.; Harigaya, Y.; Takamatsu, S.; Hayashi, M.; Komiyama, K.; Omura, S.; Sprengeler, P.A.; Smith, A.B. Relative and absolute stereochemistries and total synthesis of (+)-macrosphelides A and B, potent, orally bioavailable inhibitors of cell-cell adhesion. J. Am. Chem. Soc. 1997, 119, 10247–10248. [Google Scholar] [CrossRef]
- Omura, S.; Komiyama, K. Immunosuppressant Macrosphelide B. WO 0147516 A1, 5 July 2001. [Google Scholar]
- Fukami, A.; Iijima, K.; Hayashi, M.; Komiyama, K.; Omura, S. Macrosphelide B suppressed metastasis through inhibition of adhesion of sLex/E-Selectin molecules. Biochem. Biophy. Res. Commun. 2002, 291, 1065–1070. [Google Scholar]
- Tomprefa, N.; McQuilken, M.; Hill, R.; Whipps, J. Antimicrobial activity of Coniothyrium minitans and itsmacrolide antibiotic macrosphelide A. J. Appl. Microbiol. 2009, 106, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Akita, H.; Nakamura, H.; Ono, M. Total synthesis of (+)-macrosphelides A, C, E, F, and G based on enzymatic function. Chirality 2003, 15, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Matsuya, Y.; Nemoto, H. Recent advances in macrosphelide synthesis. Heterocycles 2005, 65, 1741–1749. [Google Scholar]
- Ivanova, V.; Kolarova, M.; Aleksieva, K.; Graefe, U.; Schlegel, B. Diphenylether and macrotriolides occurring in a fungal isolate from the antarctic lichen Neuropogon. Prep. Biochem. Biotechnol. 2007, 37, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar]
- Boden, E.P.; Keck, G.E. Proton-transfer steps in Steglich esterification: A very practical new method for macrolactonization. J. Org. Chem. 1985, 50, 2394–2395. [Google Scholar] [CrossRef]
- Sunazuka, T.; Hirose, T.; Chikaraishi, N.; Harigaya, Y.; Hayashi, M.; Komiyama, K.; Sprengeler, P.A.; Smith, A.B., III; Ōmura, S. Absolute stereochemistries and total synthesis of (+)/(−)-macrosphelides, potent, orally bioavailable inhibitors of cell-cell adhesion. Tetrahedron 2005, 61, 3789–3803. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kumar, B.G.; Kurachi, T. Total synthesis of macrosphelides B and A. Tetrahedron Lett. 2000, 41, 1559–1563. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kumar, G.B.; Kurachi, T.; Acharya, H.P.; Yamazaki, T.; Kitazume, T. Furan ring oxidation strategy for the synthesis of macrosphelides A and B. J. Org. Chem. 2001, 66, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Bal, B.S.; Childers, W.E., Jr.; Pinnick, H.W. Oxidation of α, β-unsaturated aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Acharya, H.P. First total synthesis of macrosphelides C and F. Tetrahedron Lett. 2001, 42, 2817–2820. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Matsuumi, M. First total synthesis of grahamimycin A. J. Org. Chem. 2000, 65, 7221–7224. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O.; Yamada, M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967, 40, 2380–2382. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Wang, Y.-G. Synthesis of macrosphelides H and G. Tetrahedron Lett. 2002, 43, 4381–4384. [Google Scholar] [CrossRef]
- Hareau, G.P.; Koiwa, M.; Hikichi, S.; Sato, F. Synthesis of optically active 5-(tert-butyldimethylsiloxy)-2-cyclohexenone and its 6-substituted derivatives as useful chiral building blocks for the synthesis of cyclohexane rings. Syntheses of carvone, penienone, and penihydrone. J. Am. Chem. Soc. 1999, 121, 3640–3650. [Google Scholar] [CrossRef]
- Ono, M.; Nakamura, H.; Konno, F.; Akita, H. Total syntheses of macrosphelides (+)-A, (−)-A and (+)-E. Tetrahedron: Asymmetry 2000, 11, 2753–2764. [Google Scholar] [CrossRef]
- Ono, M.; Nakamura, H.; Arakawa, S.; Honda, N.; Akita, H. Formal total synthesis of (+)-macrosphelide A based on regioselective hydrolysis using lipase. Chem. Pharm. Bull. 2002, 50, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Ono, M.; Shida, Y.; Akita, H. New total syntheses of (+)-macrosphelides C, F and G. Tetrahedron: Asymmetry 2002, 13, 705–713. [Google Scholar] [CrossRef]
- Nakamura, H.; Ono, M.; Makino, M.; Akita, H. Formal total synthesis of macrosphelide (+)-A, effect on macrolactonization depended upon the lactone formation position. Heterocycles 2002, 57, 327–336. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Mouli, C.C. The total synthesis of macrosphelides A and E from carbohydrate precursors. Tetrahedron Lett. 2002, 43, 9159–9161. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Mouli, C.C. A total synthesis of macrosphelides C and F from l-(+)-arabinose. Tetrahedron Lett. 2003, 44, 8161–8163. [Google Scholar] [CrossRef]
- Chakraborty, T.K.; Purkait, S.; Das, S. Synthesis of chiral 4-hydroxy-2,3-unsaturated carbonyl compounds from 3,4-epoxy alcohols by oxidation: application in the formal synthesis of macrosphelide A. Tetrahedron 2003, 59, 9127–9135. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kawaguchi, T.; Nemoto, H. New strategy for the total synthesis of macrosphelides A and B based on ring-closing metathesis. Org. Lett. 2003, 5, 2939–2941. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Funamori, N.; Matsuya, Y.; Nemoto, H. Total synthesis of macrosphelides A, B, and E: First application of ring-closing metathesis for macrosphelide synthesis. J. Org. Chem. 2004, 69, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Kusaka, S.-I.; Dohi, S.; Doi, T.; Takahashi, T. Total synthesis of macrosphelide A by way of palladium-catalyzed carbonylative esterification. Tetrahedron Lett. 2003, 44, 8857–8859. [Google Scholar] [CrossRef]
- Takahashi, T.; Kusaka, S.-I.; Doi, T.; Takayuki, T.; Sunazuka, T.; Omura, S. A combinatorial synthesis of a macrosphelide library utilizing a palladium-catalyzed carbonylation on a polymer support. Angew. Chem. Int. Ed. Engl. 2003, 42, 5230–5234. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Seo, S.-Y.; Kim, S.-H.; Jung, J.-W.; Lee, Y.-S.; Jung, J.-K.; Suh, Y.-G. Concise syntheses of (+)-macrosphelides A and B. Org. Lett. 2005, 7, 3159–3162. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Rajapakse, H.A.; Chiu, A.; Stenkamp, D. Asymmetric syntheses of pectenotoxins-4 and -8, Part II: Synthesis of the C20-C30 and C31-C40 subunits and fragment assembly. Angew. Chem. Int. Ed. Engl. 2002, 114, 4755–4758. [Google Scholar] [CrossRef]
- Suh, Y.-G.; Jung, J.-K.; Seo, S.-Y.; Min, K.-H.; Shin, D.-Y.; Lee, Y.-S.; Kim, S.-H.; Park, H.-J. Total synthesis of (+)-brefeldin A. J. Org. Chem. 2002, 67, 4127–4137. [Google Scholar] [CrossRef]
- Maezaki, N.; Hirose, Y.; Tanaka, T. Stereoselective synthesis of 1,4-bifunctional compounds by regioselective Pd-catalyzed allylic substitution reaction. Org. Lett. 2004, 6, 2177–2180. [Google Scholar] [CrossRef] [PubMed]
- Paek, S.-M.; Yun, H.; Kim, N.-J.; Jung, J.-W.; Chang, D.-J.; Lee, S.; Yoo, J.; Park, H.-J.; Suh, Y.-G. Concise syntheses of (+)-macrosphelides A and B: Studies on the macro-ring closure strategy. J. Org. Chem. 2009, 74, 554–561. [Google Scholar] [CrossRef]
- Paek, S.-M.; Suh, Y.-G. Synthetic studies on bioactive natural polyketides: Intramolecular nitrile oxide-olefin cycloaddition approach for construction of a macrolactone skeleton of macrosphelide B. Molecules 2011, 16, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.; Paek, S.-M.; Jung, J.-W.; Kim, N.-J.; Kim, S.-H.; Suh, Y.-G. First total syntheses of (−)-macrosphelides J and K and elucidation of their absolute configuration. Chem. Commun. 2009, 2463–2465. [Google Scholar]
- Rao, K.S.; Mukkanti, K.; Reddy, D.S.; Pal, M.; Iqbal, J. A simple procedure for the synthesis of γ-hydroxy-α, β-(E)-alkenoic esters: Formal synthesis of (+)-macrosphelides A and B. Tetrahedron Lett. 2005, 46, 2287–2290. [Google Scholar] [CrossRef]
- Sharma, G.V.M.; Babu, K.V. RCM mediated synthesis of macrosphelides I and G. Tetrahedron: Asymmetry 2007, 18, 2175–2184. [Google Scholar] [CrossRef]
- Hanessian, S.; Ugolini, A.; Dubé, D.; Glamyan, A. Facile access to (S)-1, 2, 4-butanetriol and its derivatives. Can. J. Chem. 1984, 62, 2146–2147. [Google Scholar] [CrossRef]
- Matsuya, Y.; Kobayashi, Y.; Kawaguchi, T.; Hori, A.; Watanabe, Y.; Ishihara, K.; Ahmed, K.; Wei, Z.-L.; Yu, D.-Y.; Zhao, Q.-L.; et al. Design, synthesis, and biological evaluation of artificial macrosphelides in the search for new apoptosis-inducing agents. Chem.-Eur. J. 2009, 15, 5799–5819. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.R.; Gutala, P. Enantioselective total synthesis of macrosphelides A and E. Tetrahedron 2011, 67, 4514–4520. [Google Scholar] [CrossRef]
- Colin-Messager, S.; Girard, J.-P.; Rossi, J.-C. Convenient method for the preparation of trityl ethers from secondary alcohols. Tetrahedron Lett. 1992, 33, 2689–2692. [Google Scholar] [CrossRef]
- Curran, D.P.; Sinha, M.K.; Zhang, K.; Sabatini, J.J.; Cho, D.-H. Binary fluorous tagging enables the synthesis and separation of a 16-stereoisomer library of macrosphelides. Nat. Chem. 2012, 4, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.V.M.; Reddy, P.S. Total synthesis of macrosphelide M from diacetone glucose. Eur. J. Org. Chem. 2012, 2012, 2414–2421. [Google Scholar] [CrossRef]
- Garegg, P.J.; Samuelsson, B. Conversion of vicinal diols into olefins using triphenylphosphine and triiodoimidazole. Synthesis 1979, 1979, 813–814. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paek, S.-M. Synthetic Advances in Macrosphelides: Natural Anticancer Agents. Molecules 2014, 19, 15982-16000. https://doi.org/10.3390/molecules191015982
Paek S-M. Synthetic Advances in Macrosphelides: Natural Anticancer Agents. Molecules. 2014; 19(10):15982-16000. https://doi.org/10.3390/molecules191015982
Chicago/Turabian StylePaek, Seung-Mann. 2014. "Synthetic Advances in Macrosphelides: Natural Anticancer Agents" Molecules 19, no. 10: 15982-16000. https://doi.org/10.3390/molecules191015982
APA StylePaek, S.-M. (2014). Synthetic Advances in Macrosphelides: Natural Anticancer Agents. Molecules, 19(10), 15982-16000. https://doi.org/10.3390/molecules191015982