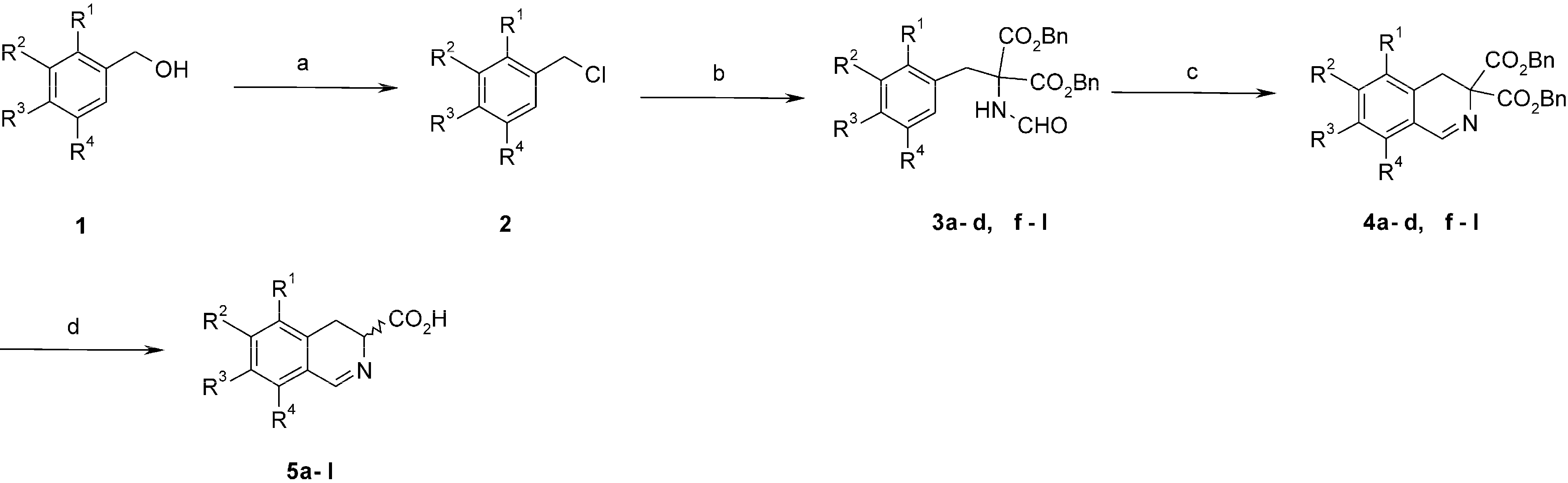
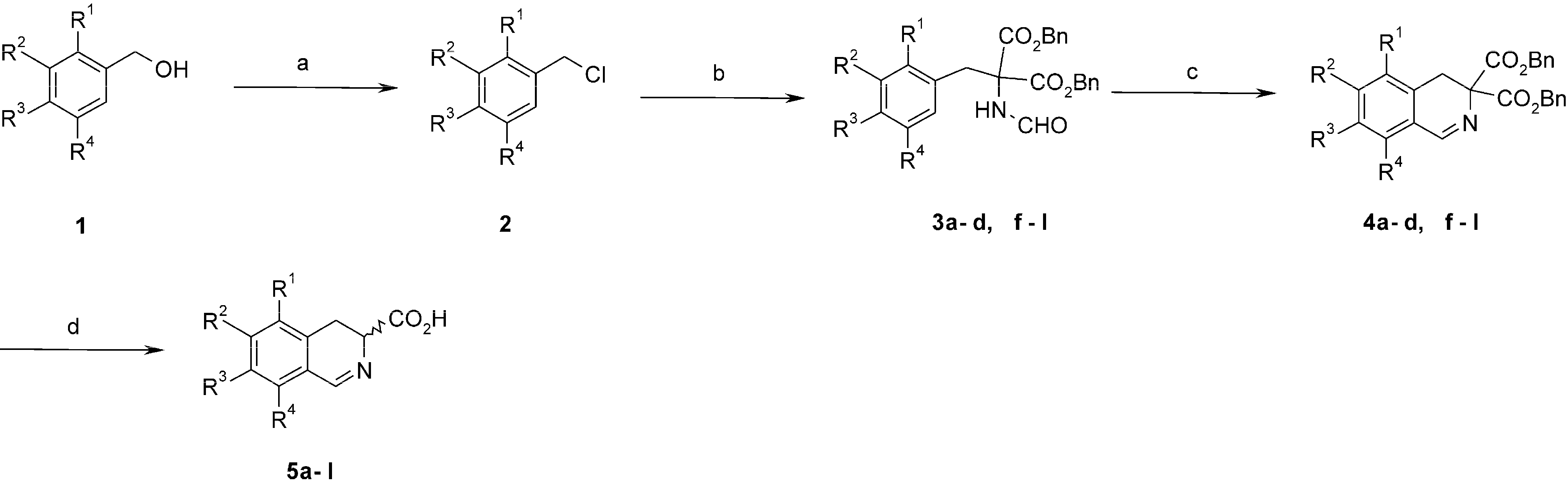
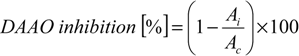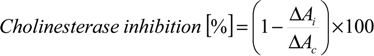3.2. Synthesis and Characterisation
3.2.1. General Procedure for the Synthesis of Benzyl Chlorides 2
Pyridine (0.36 mL) and thionyl chloride (0.37 mL) were added dropwise to a solution of protected benzyl alcohol 1 (4.78 mmol) in CH2Cl2 (15 mL). The resultant mixture was then stirred at rt for 2.5 h. The reaction was quenched with 10 mL of water and stirred for 0.5 h to destroy excess of thionyl chloride. The organic layer was removed, and the aqueous layer was extracted twice with CH2Cl2 (3 × 15 mL). The combined organic layers were dried over MgSO4 and evaporated. The products were used in the next step without purification.
3.2.2. General Procedure for Alkylation of Dibenzyl Formamidomalonate 3a–3l
Dibenzyl formamidomalonate (315 mg, 0.96 mmol) followed by K2CO3 (1.76 g, 12.75 mmol) and KI (478 mg, 2.88 mmol) were added to a solution of respective benzyl chlorides (0.96 mmol) in acetone (20 mL). The reaction mixture was stirred under reflux (oil bath, 60 °C) for 20 h. When complete, the reaction mixture was cooled and filtered through Celite. The solvent was evaporated to give a solid.
Dibenzyl 2-(2-bromo-5-benzyloxybenzyl)-2-formamidomalonate (3a). The compound was purified by chromatography on silica gel (EtOAc-hexane, 1:3). White crystals. Yield 0.363 g (63%). 1H-NMR (CDCl3) δ: 3.84 (s, 2H, CH2); 4.98 (s, 2H, OCH2Ph); 5.12 and 5.16 (AB, J = 14, 4H, CH2Ph); 6.58 (bs, 1H, NH); 6.69 (d, J = 3.3 Hz, 1H, C6-H); 6.76 (dd, J = 3.3, 8.8 Hz, 1H, C4-H); 7.40 (d, J = 8.8 Hz; 1H, C3-H); 7.16–7.38 (m, 15H, Ar); 7.94 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.6; 65.5; 68.5; 70.0; 116.0; 116.1; 118.7; 127.2; 128.1; 128.3; 128.5; 128.55; 128.6; 133.7; 134.5; 135.4; 136.5; 157.6; 160.1; 166.8. HR MS ESI calculated for C32H29BrNO6 (M+H) 602.1173. Found 602.1170.
Dibenzyl 2-(3-benzyloxy-4-bromobenzyl)-2-formamidomalonate (3b). This compound was used in the next step without purification. Beige solid. Yield 95%. 1H-NMR (CDCl3) δ: 3.84 (s, 2H, CH2); 4.98 (s, 2H, OCH2Ph); 5.12 and 5.16 (AB, J = 12, 4H, CH2Ph); 6.76 (dd, J = 2.9 Hz, J = 8.8, 1H, C6-H); 6.89 (d, J = 2.9, 1H, C2-H); 6.60 (bs, 1H, NH); 7.14–7.38 (m, 15H, Ar); 7.40 (d, J = 8.8, 1H, C5-H); 7.93 (d, J = 0.8 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.6; 65.5; 68.4; 69.9; 116.04; 116.05 118.6; 127.2; 128.0; 128.3; 128.5; 128,6; 133.7; 134.4; 135.4; 136.5; 157.6; 160.1, 166.8. HR MS ESI calculated for C32H29BrNO6 (M+H) 602.1172. Found 602.1170.
Dibenzyl (3,5-dibenzyloxybenzyl)-2-formamidomalonate (3c). The product was purified on silica gel (EtOAc-hexane 1:2.5). White solid. Yield 77%. 1H-NMR (CDCl3) δ: 3.59 (s, 2H, CH2); 4.93 (s, 4H, OCH2Ph); 5.08 and 5.12 (AB, J = 12 Hz, 4H, CH2Ph); 6.17 (d, J = 2.1 Hz, 2H, C2-H, C6-H); 6.52 (t, J = 2.1 Hz, 1H, C4-H); 6.55 (bs, 1H, NH); 7.16–7.44 (m, 20H, Ar); 7.91 (d, J = 0.8 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 38.2; 66.7; 68.3; 69.9; 101.2; 109.2; 127.3; 127.9; 128.3; 128.5; 128.61; 128.62; 134.5; 136.7; 136.8; 159.7; 159.8; 166.6. HR MS ESI calculated for C39H36NO7 (M+H) 630.2486. Found 630.2466.
Dibenzyl 2-(3-iodo-4-benzyloxy-5-methoxybenzyl)-2-formamidomalonate (3d). This compound was used in the next step without purification. Beige solid. Yield 65%. 1H-NMR (CDCl3) δ: 3.63 (s, 2H, CH2); 3.67 (s, 3H, OCH3); 4.97 (s, 2H, OCH2Ph); 5.13 and 5.20 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.51 (d, J = 1.8 Hz, 1H, C6-H); 6.80 (bs, 1H, NH); 6.99 (d, J = 1.8 Hz, 1H, C2-H); 7.24–7.40 (m, 13H, Ar); 7.53–7.57 (m, 2H, Ar); 8.20 (d, J = 1.1 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.2; 55.91; 55.94; 66.8; 68.5; 74.4; 92.8; 114.8; 128.0; 128.3; 128.37; 128.4; 128.69; 128.74; 131.6; 132.7; 134.4; 137.0; 147.2; 152.4; 159.9; 166.5. HR MS ESI calculated for C33H30INO7 (M+H) 680.1139. Found 680.1131.
Dibenzyl 2-(5-bromo-2,3-dimethoxybenzyl)-2-formamidomalonate (3f). The product was purified on silica gel (EtOAc-hexane 1:2). White solid. Yield 66%. 1H-NMR (CDCl3) δ: 3.66 (s, 2H, CH2); 3.66 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 5.13 and 5.17 (AB, J = 12 Hz, 4H, OCH2Ph); 6.67 (bs, 1H, NH); 6.75 (d, J = 2.5 Hz, 1H, C6-H); 6.93 (d, J = 2.5 Hz, 1H, C4-H); 7.21–7.38 (m, 10H, Ar); 8.12 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 33.0; 55.9; 60.6; 65.8; 68.4; 115.2; 116.1; 126.3; 128,2; 128.5; 128.6; 130.1; 134.6; 147.3; 153.3; 160.2; 166.9. HR MS ESI calculated for C27H27BrNO7 (M+H) 556.0965. Found 556.0960.
Dibenzyl 2-(3-bromo-4,5-dimethoxybenzyl)-2-formamidomalonate (3g). The product was purified on silica gel (EtOAc-hexane 1:2). White solid. Yield 65%. 1H-NMR (CDCl3) δ: 3.62 (s, 2H, CH2); 3.66 (s, 3H, OCH3); 3.81 (s, 3H, OCH3); 5.12 and 5.20 (AB, J = 12 Hz, 4H, OCH2Ph); 6.44 (d, J = 2.1 Hz, 1H, C2-H); 6.71 (d, J = 2.1 Hz, 1H, C6-H); 6.76 (bs, 1H, NH); 7.24–7.38 (m, 10H, Ar); 8.20 (d, J = 0.8 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.4; 55.9; 60.5; 66.8; 68.5; 113.6; 117.5; 125.8; 128.4; 128.7; 128,8; 131.7; 134.4; 145.8; 153.4; 159.9; 166.5. HR MS ESI calculated for C27H27BrNO7 (M+H) 558.0965. Found 558.0935.
Dibenzyl 2-(3-chloro-4,5-dimethoxybenzyl)-2-formamidomalonate (3h). This compound was purified on silica gel (EtOAc-hexane 1:2.5). White solid. Yield 57%. 1H-NMR (CDCl3) δ: 3.62 (s, 2H, CH2); 3.67 (s, 3H, OCH3); 3.82 (s, 3H, OCH3); 5.12 and 5.20 (AB, J = 12.1 Hz, 2H, OCH2Ph); 6.40 (d, J = 1.7 Hz, 1H, C6-H); 6.52 (d, J = 1.7 Hz, 1H, C2-H); 6.76 (s, 1H, NH); 7.25–7.37 (m, 10H, Ar); 8.20 (d, J = 1.3 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 37.5; 56.0; 60.6; 66.7; 68.5; 112.9; 123.0; 128.1; 128.4; 128.7; 128.8; 131.0; 134.4; 144.8; 153.5; 159.9; 166.6. HR MS ESI calculated for C27H27ClNO7 (M+H) 512.1470. Found 512.1467.
Dibenzyl 2-(3,4,5-trimethoxybenzyl)-2-formamidomalonate (3i). This compound was purified on silica gel (EtOAc-hexane 1:1.5). White solid. Yield 70%. 1H-NMR (CDCl3) δ: 3.63 (s, 6H, OCH3); 3.67 (s, 2H, CH2); 3.80 (s, 3H, OCH3); 5.10 and 5.20 (AB, J = 12.1 Hz, 4H, OCH2PH); 6.16 (s, 2H, C2-H, C6-H); 6.73 (bs, 1H, NH); 7.23–7.36 (m, 10H, Ar); 8.19 (d, J = 1.2 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 38.3; 55.9; 60.8; 66.9; 68.3; 106.9; 128.2; 128.67; 128.68; 130.1; 134.5; 137.3; 153.0; 159.8; 166.8. HR MS ESI calculated for C28H30NO8 (M+H) 508.1965. Found 508.1945.
Dibenzyl 2-(4-bromo-3,5-dibenzyloxybenzyl)-2-formamidomalonate (3j). This compound was purified on silica gel (EtOAc-hexane 1:4). Beige solid. Yield 75%. 1H-NMR (CDCl3) δ: 3.58 (s, 2H, CH2); 5.00 (s, 4H, OCH2Ph); 5.02 and 5.05 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.17 (s, 2H, C2-H, C6-H); 6.34 (bs, 1H, NH); 7.14–7.42 (m, 20H, Ar); 7.68 (d, J = 1.2 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 38.2; 66.6; 68.3; 70.6; 101.7; 108.5; 126.7; 127.8; 128.1; 128.5; 128.6; 128.7; 134.4; 134.9; 136.5; 155.9; 159.9; 166.4. HR MS ESI calculated for C39H35BrNO7 (M+H) 708.1591. Found 708.1593.
Dibenzyl (2-chloro-3,5-dibenzyloxybenzyl)-2-formamidomalonate (3k). This compound was used in the next step without purification. Beige solid. Yield 88%. 1H-NMR (CDCl3) δ: 3.87 (s, 2H, CH2); 4.94 (s, 2H, OCH2Ph); 5.07 (s, 2H, OCH2Ph); 5.11 and 5.16 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.30 (d, J = 2.9 Hz, 1H, C4-H); 6.56 (br, 1H, NH); 6.57 (d, J = 2.9 Hz, 1H, C6-H); 7.18–7.45 (m, 20H, Ar); 7.88 (d, J = 1.3, 1H, CHO). 13C-NMR (CDCl3) δ: 35.7; 65.5; 68.4; 70.1; 70.9; 101.7; 109.7; 116.3; 127.1; 127.2; 128.0; 128.1; 128.3; 128.5; 128.56; 128.59; 128.6; 134.47; 134.52; 136.2; 136.5; 155.0; 157.3; 160.1; 166.9. HR MS ESI calculated for C39H35ClNO7 (M+H) 664.2096. Found 664.2079.
Dibenzyl 2-(2-chloro-3,4-dimethoxybenzyl)-2-formamidomalonate (3l). This compound was purified on silica gel (EtOAc-hexane 1:2.5). White solid. Yield 56%. 1H-NMR (CDCl3) δ: 3.81 (s, 3H, OCH3); 3.82 (s, 2H, CH2); 3.83 (s, 3H, OCH3); 5.15 (s, 4H, CH2Ph); 6.63 and 6.70 (AB, J = 8.3 Hz, 2H, C6-H and C5-H); 6.66 (br, 1H, NH); 7.21–7.34 (m, 10H, Ar); 8.13 (s, 1H, CHO). 13C-NMR (CDCl3) δ: 35.1; 55.9; 60.5; 65.7; 68.4; 110.3; 125.4; 126.9; 128.4; 128.6; 129.6; 134.5; 145.4; 152.9; 154.4; 160.1; 166.9. HR MS ESI calculated for C27H27ClNO7 (M+H) 512.1470. Found 512.1448.
3.2.3. General Procedure for Bischler-Napieralski Cyclization. Preparation of Dibenzyl 3,4-Dihydro- isoquinolin-3,3-dicarboxylates 4a–4l
POCl3 (0.23 mL) was added to a stirred solution of the respective dibenzyl formamidomalonate 3a–3l (0.83 mmol) in acetonitrile (10 mL) and the mixture was heated under various conditions depending on the compound. The solvent was evaporated and residue was made alkaline with aqueous NaHCO3 (5 mL). The mixture was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were dried with MgSO4 and concentrated to give the product.
Dibenzyl 5-bromo-8-benzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4a). The reaction mixture was heated at 80 °C (temp. of oil bath) for 8 h. The compound was purified by chromatography on silica gel (EtOAc-hexane, 1:3). Brown oil. Yield 0.150 g (31%). 1H-NMR (CDCl3) δ: 3.44 (s, 2H C4-H); 5.12 (s, 2H, OCH2Ph); 5.10 and 5.20 (AB, J = 12.1 Hz, 4H, CH2PH); 6.76 (d, J = 8.7 Hz, 1H, C7-H); 7.48 (d, J = 8.7 Hz, 1H, C6-H); 7.14–7.43 (m, 15H, Ar); 8.95 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 30.8; 67.8; 70.3; 70.6; 113.2; 114.5; 118.8; 126.9; 127.1; 128.2; 128.3; 128.4; 128.5; 128.7; 134.4; 135.1; 135.8; 136.5; 155.7; 157.7; 168.4. MS ESI calculated for C32H27BrNO5 (M+H) 586.1. Found 586.1.
Dibenzyl 8-benzyloxy-7-bromo-3,4-dihydroisoquinolin-3,3-dicarboxylate (4b). The reaction mixture was heated at 73 °C for 15 h. The product was purified on silica gel (EtOAc-hexane 1:6). Brown oil. Yield 100 mg (26%). 1H-NMR (CDCl3) δ: 3.44 (s, 2H C4-H); 5.11 (s, 2H, OCH2Ph); 5.10 and 5.21 (AB, J = 12.5 Hz, 4H, CH2PH); 6.76 (d, J = 8.7 Hz, 1H, C5-H); 7.19–7.40 (m, 15H, Ar); 7.47 (d, J = 8.7 Hz, 1H, C6-H); 8.94 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 30.8; 67.8; 70.3; 70.6; 113.2; 114.5; 118.8; 127.1; 128.0; 128.2; 128.3; 128.4; 128.7; 134.4; 135.1; 135.8; 136.5; 155.7; 157.7; 168.4. MS ESI calculated for C32H27NO5Br (M+H) 584.1. Found 583.9.
Dibenzyl 6.8-dibenzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4c). The reaction mixture was heated at 70 °C for 8 h. Product was purified by extraction with hot cyclohexane. Orange solid. Yield 94%. 1H-NMR (CDCl3) δ: 3.31 (s, 2H C4-H); 5.00 (s, 2H, OCH2Ph); 5.07 (s, 2H, OCH2Ph); 5.10 and 5.19 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.33 (d, J = 2.1 Hz, 1H, C8-H); 6.42 (d, J = 2.1 Hz, 1H, C5-H); 7.20–7.44 (m, 20H, Ar); 8.87 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.7; 67.8; 68.3; 70.3; 99.1; 106.0; 109.1; 127.1; 127.6; 128.0; 128.2; 128.4; 128.7; 128.74; 134.4; 135.1; 135.8; 136.9; 158.0; 158.1. HR MS EI calculated for C39N33NO6 611.2308. Found 611.2292.
Dibenzyl 6-methoxy-7-benzyloxy-8-iodo-3,4-dihydroisoquinolin-3,3-dicarboxylate (4d). The reaction mixture was heated at 80 °C for 9 h. The product was purified on silica gel (EtOAc-hexane 1:3). Brown oil. Yield 94%. 1H-NMR (CDCl3) δ: 3.30 (s, 2H, C4-H); 3.86 (s, 3H, OCH3); 4.97 (s, 2H, OCH2Ph); 5.14 and 5.19 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.63 (s, 1H); 7.17–7.21 (m, 4H, Ar); 7.26–7.29 (m, 5H, Ar); 7.34–7.42 (m, 4H); 7.55–7.58 (m, 2H); 8.65 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.3; 56.1; 67.8; 70.3; 74.5; 98.3; 111.6; 122.4; 128.0; 128.18; 128.2; 128.3; 128.43; 128.44; 132.6; 135.1; 136.7; 147.0; 155.4; 165.2; 168.6. HR MS EI calculated for C33H29INO6 662.1034. Found 662.1037.
Dibenzyl 8-bromo-5,6-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4f). The reaction mixture was heated at 95 °C for 4 h. The product was purified on silica gel (EtOAc-hexane 1:3). Beige oil. Yield 58%. 1H-NMR (CDCl3) δ: 3.36 (s, 2H, C4-H); 3.67 (s, 3H, OCH3); 3.88 (s, 3H, OCH3); 5.14 and 5.17 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.95 (s, 1H, C7-H); 7.18–7.30 (m, 10H, Ar); 8.72 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 25.4; 56.0; 60.6; 67.7; 70.1; 114.9; 119.0; 119.7; 128.0; 128.2; 128.4; 129.3; 135.1; 145.2; 155.9; 160.8; 168.5. MS ESI calculated for C27H25BrNO6 (M+H) 538.1. Found 538.1.
Dibenzyl 8-bromo-6,7-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4g). The reaction mixture was heated at 80 °C for 9 h. Product was used in the next step without purification. Brown oil. Yield 94%. 1H-NMR (CDCl3) δ: 3.30 (s, 2H, C4-H); 3.83 (s, 3H, OCH3); 3.86 (s, 3H, OCH3); 5.14 and 5.19 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.59 (s, 1H, C5-H); 7.15–7.36 (m, 10H, Ar); 8.78 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.2; 56.1; 60.6; 67.8; 70.2; 110.5; 119.7; 119.8; 128.0; 128.2; 128.4; 132.1; 135.1; 145.7; 156.3; 160.8; 168.6. HR MS EI calculated for C27H24BrNO6 537.0787. Found 537.0774.
Dibenzyl 8-chloro-6,7-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4h). The reaction mixture was heated at 70 °C for 8 h. Product was used in the next step without purification. Brown oil. Yield 95%. 1H-NMR (CDCl3) δ: 3.31 (s, 2H, C4-H); 3.84 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 5.14 and 5.20 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.56 (s, 1H, C5-H); 7.19–7.38 (m, 10H, Ar); 8.85 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.1; 56.1; 60.7; 67.8; 70.2; 109.8; 118.4; 128.0; 128.2; 128.3; 128.4; 131.4; 135.1; 144.6; 156.5; 158.5; 168.6. HR MS ESI calculated for C27H25ClNO6 (M+H) 494.1365. Found 494.1360.
Dibenzyl 6,7,8-trimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4i). The reaction mixture was heated at 70 °C for 9 h. Product was used in the next step without purification. Brown oil. Yield 95%. 1H-NMR (CDCl3) δ: 3.66 (s, 2H, C4-H); 3.83 (s, 3H, OCH3), 3.99 (s, 3H, OCH3); 4.17 (s, 3H, OCH3); 5.20 and 5.27 (AB, J = 12.1 Hz, 4H, CH2Ph); 6.53 (s, 1H, C5-H); 7.21–7.39 (m, 10H, Ar); 9.02 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 32.4; 57.3; 61.5; 61.6; 66.3; 69.9; 107.4; 110.3; 128.6; 128.7; 128.9; 133.2; 133.8; 139.7; 157.0; 161.5; 163.8; 165.0. HR MS ESI calculated for C28H28NO7 (M+H) 490.1860. Found 490.1841.
Dibenzyl 7-bromo-6,8-dibenzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4j). The reaction mixture was heated at 80 °C for 7 h. Product was used in the next step without purification. Brown oil. Yield 85%. 1H-NMR (CDCl3) δ: 3.29 (s, 2H C4-H); 4.84 (s, 2H, OCH2Ph); 5.15 (s, 2H, OCH2Ph); 5.15 and 5.18 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.55 (s, 1H, C5-H); 7.10–7.55 (m, 20H, Ar); 8.69 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 31.4; 53.4; 67.8; 70.3; 71.1; 106.5; 108.5; 116.4; 127.0; 128.1; 128.26; 128.3; 128.4; 128.44; 128.57; 128.61; 128.7; 135.0; 135.1; 135.6; 135.8; 156.1; 157.8; 159.0; 168.6. HR MS ESI calculated for C39H33BrNO6 (M+H) 690.1485. Found 690.1464.
Dibenzyl 5-chloro-6,8-dibenzyloxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4k). The reaction mixture was heated at 75 °C for 10 h. Product was used in the next step without purification. Brown oil. Yield of crude product 94%. 1H-NMR (CDCl3) δ: 3.75 (s, 2H, C4-H); 5.18 and 5.26 (AB, J = 12.1 Hz, 4H, CH2Ph); 5.20 (s, 2H, OCH2Ph); 5.21 (s, 2H, OCH2Ph); 6.49 (s, 1H, C7-H); 7.22–7.46 (m, 20H, Ar); 9.07 (s, 1H, C1-H). HR MS EI calculated for C39H33ClNO6 646.1991. Found 646.1978.
Dibenzyl 5-chloro-6,7-dimethoxy-3,4-dihydroisoquinolin-3,3-dicarboxylate (4l). The reaction mixture was heated at 75 °C for 10 h. Product was used in the next step without purification. Brown oil. Yield of crude product 86%. 1H-NMR (CDCl3) δ: 3.44 (s, 2H, C4-H); 3.88 (s, 3H, OCH3); 3.89 (s, 3H, OCH3); 5.09 and 5.22 (AB, J = 12.1 Hz, 2H, CH2Ph); 6.84 (s, 1H, C8-H); 7.15–7.40 (m, 10H, Ar); 8.43 (s, 1H, C1-H). HR MS ESI calculated for C27H25ClNO6 (M+H) 494.1365. Found 494.1352.
3.2.4. General Procedure for Removal of Benzylic Groups. Preparation of 3,4-Dihydroisoquinolin-3-carboxylic Acids 5a–5l
BBr3 (0.2 mL, 2.13 mmol) was added dropwise under argon to a solution of respective dibenzyl 3,4-dihydroisoquinolin-3,3-dicarboxylate 4a–4l (0.17 mmol) in CH2Cl2 (10 mL) at −20 °C. The reaction mixture was stirred for 0.5 h at the same temperature and quenched with H2O (25 mL). The resultant mixture was then stirred at rt for 0.5 h. The water layer was separated and washed 3 times with CH2Cl2. The solution was concentrated under vacuum at rt and the residue was loaded on a column packed with ion exchange resin Dowex 50W X4 (ca. 30 mL, pretreated subsequently with water, 2 M HCl, and water to pH 6–7). The column was washed with water (200 mL), and the product was eluted with 2 M aqueous NH3. Brown to yellow colored fractions were collected. Ammonia was removed under water pump vacuum at rt and the solution was evaporated (water bath temp. 35 °C). The residue was acidified with 2 M aqueous HCl to pH 2, and purified on polyacrylamide (Biogel P-2) using 0.1% aqueous TFA.
5-Bromo-8-hydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5a). Beige powder. Yield 10 mg (22%). 1H-NMR (D2O/DCl) δ: 3.25 and 3.35 (ABX, J = 7.1 Hz, 8.8 Hz, 17.5 Hz, 2H, C4-H); 4.48 (t, J = 7.1 Hz, 1H, C3-H); 6.58 (d, J = 9.6 Hz, 1H, C7-H); 7.59 (d, J = 9.6 Hz, 1H, C6-H); 8.73 (s, 1H, C1-H). MS ESI calculated for C10H9NO3Br (M+H) 269.9. Found 269.9.
7-Bromo-8-hydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5b). Beige powder. Yield 87%. 1H-NMR (D2O/DCl) δ: 3.27 and 3.36 (ABX, J = 7.1 Hz, 8.8 Hz, 17.5 Hz, 2H, C4-H), 4.50 (t, J = 7.9 Hz, C3-H); 6.62 (d, J = 9.1, 1H, C5-H), 7.62 (d, J = 9.1, 1H, C6-H), 8.77 (s, 1H, C1-H). IR: 579; 617; 665; 698; 723; 750; 781; 804; 841; 897; 937; 993; 1040; 1084; 1136; 1184; 1234; 1277; 1292; 1310; 1344; 1364; 1400; 1452; 1601; 1634; 2250–3500 br; 2806; 3044; 3129. MS ESI calculated for C10H9NO3Br (M+H) 270.0. Found 269.9.
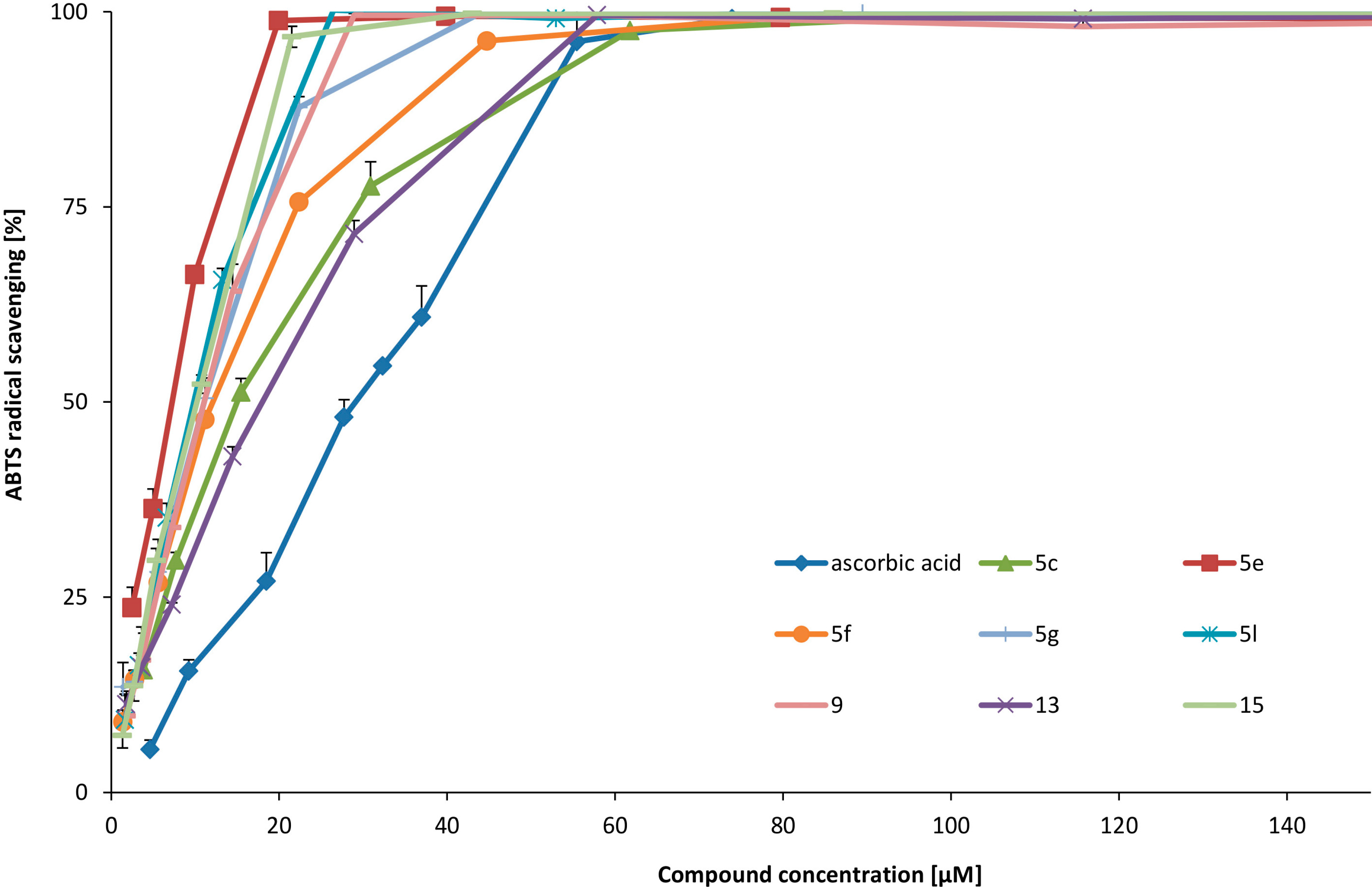
6,8-Dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5c). Beige powder. Yield 62%. 1H-NMR (D2O-H2O/DCl) δ: 3.19 and 3.31 (ABX, J = 7.0 Hz, 9.2 Hz, 16.9 Hz, 2H, C3-H); 4.44 (m, 1H, C3-H); 6.13 (d, J = 1.5 Hz, 1H, C7-H); 6.26 (s, 1H, C6-H); 8.64 (s, 1H, C1-H). IR: 586; 619; 633; 662; 704; 733; 770; 808; 843; 870; 901; 957; 986; 1015; 1069; 1113; 1169; 1188; 1202; 1250; 1314; 1348; 1385; 1404; 1504; 1541; 1582; 1609; 2250–3300 br; 2519; 2612; 2689; 2722; 2793; 3071; 3250. MS ESI calculated for C10H10NO4 (M+H) 208.0. Found 208.0.
6,7-Dihydroxy-8-iodo-3,4-dihydroisoquinolin-3-carboxylic acid (5d). The reaction mixture was kept at rt for 24 h. Beige powder. Yield 22%. 1H-NMR (DMSO) δ: 3.23 and 3.31 (ABX, J = 7.5; 8.3; 17.5 Hz, 2H, C4-H); 4.82 (t, J = 7.9 Hz, 1H, C3-H); 6.59 (s, 1H, C5-H); 8.48 (s, 1H, C1-H); 11.9 (bs, 1H, OH). IR: 598; 648; 667; 692; 741; 768; 795; 822; 870; 885; 908; 943; 966; 1013; 1032; 1057; 1128; 1177; 1192; 1219; 1256; 1281; 1298; 1371; 1485; 1508; 1557; 1587; 1634; 1721; 2250–3650 br; 2826; 2911; 2959; 3146. MS ESI calculated for C10H9INO4 (M+H) 333.9. Found: 333.9.
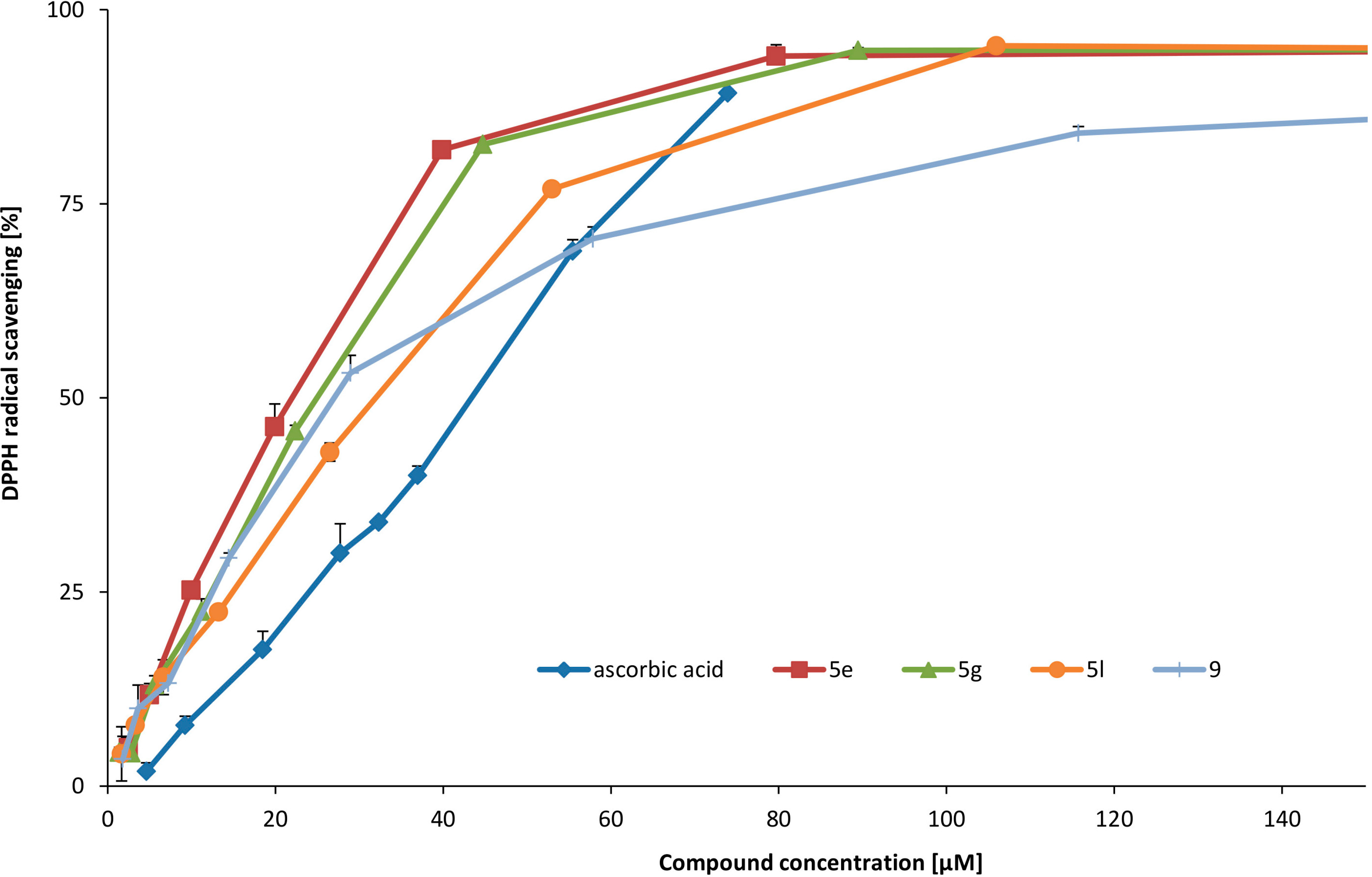
6,7-Dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (
5e). This compound was obtained from
l-DOPA according to published procedure [
9].
1H-NMR (D
2O-H
2O/DCl) δ: 3.28 and 3.38 (ABX,
J = 17.0, 8.6, 7.7, Hz, 1H, C4-H); 4.56 (dd,
J = 8.6 Hz, 7.7 Hz, 1H, C3-H); 6.91 (s, 1H, C5-H); 7.28 (s, 1H, C8-H); 8.64 (s, 1H, C1-H). IR: 586; 629; 648; 691; 725; 752; 773; 802; 843; 853; 883; 908; 937; 982; 1013; 1130; 1177; 1196; 1258; 1288; 1371; 1393; 1429; 1531; 1557; 2250–3500 br; 3038.
8-Bromo-5,6-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5f). The reaction mixture was kept at rt for 24 h. Beige powder. Yield 21%. 1H-NMR (D2O-H2O/DCl) δ: 3.13 and 3.21 (ABX, J = 16.9, 8.8, 7.0 Hz, 2H, C4-H); 4.30 (t, J = 8.1 Hz, 1H, C3-H) 6.73 (s, 1H, C7-H); 8.42 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 23.9; 54.9; 108.8; 120.1; 121.4; 126.2; 143.2; 158.4; 175.9. IR: 615; 669; 694; 735; 770; 797; 824; 853; 891; 962; 1009; 1096; 1128; 1202; 1219; 1234; 1298; 1348; 1377; 1404; 1420; 1474; 1499; 1560; 1595; 1719; 2250–3500 br; 2970; 3038; 3134. MS ESI calculated for C10H8BrNO4 (M+H) 286.0. Found 286.0.
8-Bromo-6,7-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5g). The reaction was kept at rt for 24 h. Beige powder. Yield 34%. 1H NMR (D2O-H2O/DCl) δ: 3.32 and 3.43 (ABX, J = 16.5, 8.8, 7.0 Hz, 2H, C4-H); 4.53 (t, J = 8.8 Hz, 1H, C3-H); 6.59 (s, 1H, C5-H); 8.64 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 32.7; 114.1; 114.9; 115.8; 133.9; 142.4; 155.7; 163.9; 170.3. IR: 600; 667; 696; 721; 762; 799; 839; 893; 1040; 1088; 1136; 1182; 1236; 1258; 1279; 1292; 1344; 1368; 1393; 1557; 1593; 2250–3500 br; 2806; 3044; 3125. MS ESI calculated for C10H8BrNO4 (M+H) 286.0. Found 286.0.
8-Chloro-6,7-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5h). The reaction mixture was kept at rt for 16 h. Beige powder. Yield 15%. 1H-NMR (D2O-H2O/DCl) δ: 3.15 and 3.25 (ABX, J = 7.1 Hz, J = 9.2 Hz, J = 17.1 Hz, 2H, C4-H); 4.37 (t, J = 7.9 Hz, 1H, C3-H); 6.41 (s, 1H, C5-H); 8.50 (s, 1H, C1-H). IR: 600; 640; 665; 696; 719; 735; 824; 851; 876; 908; 943; 968; 989; 1032; 1047; 1105; 1134; 1179; 1194; 1215; 1234; 1256; 1265; 1325; 1379; 1393; 1454; 1470; 1487; 1557; 1595; 1611; 1682; 1730; 2851; 2916; 2957; 3225; 3294. HR MS ESI calculated for C10H9ClNO4 (M+H) 242.0215. Found: 242.0213.
6,8-Dihydroxy-7-methoxy-3,4-dihydroisoquinolin-3-carboxylic acid (5i). The reaction mixture was kept at rt for 24 h. Product 5i could not be purified from 6- and 8-methoxy isomers. Beige powder. Yield 61 mg. Major isomer (60%). 1H-NMR (D2O-H2O/DCl) δ: 3.10 (m, 2H, C4-H), 3.82 (s, 3H, OCH3), 4.30 (m, 1H, C3-H), 6.33 (s, 1H, C5-H), 8.39 (s, 1H, C1-H). IR: 554; 600; 635; 671; 756; 777; 800; 901; 949; 991; 1080; 1098; 1138; 1202; 1265; 1304; 1354; 1373; 1454; 1522; 1589; 2250–3650 br; 2835; 3030; 3179. HR MS ESI calculated for C11H12NO5 (M+H) 238.0709. Found: 238.0702.
7-Bromo-6,8-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5j). Beige powder. Yield 53%. 1H-NMR (D2O-H2O/DCl) δ: 2.99 and 3.11 (ABX, J = 6.7, 9.6, 16.2 Hz, 2H, C4-H); 4.25 (dd, J = 9.6, 6.7 Hz; 1H, C3-H); 6.00 (s, 1H, C5-H); 8.33 (s, 1H, C1-H). 13C-NMR (D2O-H2O/DCl) δ: 27.7; 53.8; 97.5; 106.4; 109.0; 137.8; 159.6; 159.7; 164.5; 171.3. IR: 590; 660; 691; 746; 764; 797; 826; 854; 872; 945; 1011; 1030; 1061; 1096; 1123; 1173; 1229; 1246; 1283; 1329; 1369; 1514; 1589; 2250–3500 br; 2970; 3144. MS ESI calculated for C10H9NO4Br (M+H) 286.0. Found 285.9.
5-Chloro-6,8-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5k). The reaction mixture was kept at −20 °C for 1 h. Beige powder. Yield 17%. 1H-NMR (D2O-H2O/DCl) δ: 3.22 and 3.29 (ABX, J = 7.8, J = 8.6, 18.0 Hz, 2H, C4-H); 4.69 (ddd, J = 1.3, 7.8, 8.6 Hz, 1H, C3-H); 6.25 (s, 1H, C7-H); 8.68 (d, J = 1.3 Hz, 1H, C1-H). IR: 598; 633; 650; 671; 743; 781; 829; 858; 939; 1001; 1036; 1094; 1186; 1215; 1248; 1285; 1319; 1337; 1398; 1420; 1504; 1531; 1557; 2250–3650 br; 2602; 2745; 3065. MS ESI calculated for C10H9ClNO4 (M+H) 242.0. Found: 241.9.
5-Chloro-6,7-dihydroxy-3,4-dihydroisoquinolin-3-carboxylic acid (5l). The reaction mixture was kept at rt for 21 h. Beige powder. Yield 8%. 1H-NMR (D2O-H2O/DCl) δ: 2.77 and 2.92 (ABX. J = 7.9 Hz, J = 10.0, 17.5 Hz, 2H, C4-H); 4.42 (ddd, J = 7.9, 10.0, 1.7 Hz, 1H, C3-H); 6.64 (s, 1H, C8-H); 8.20 (d, J = 1.5 Hz, 1H, C1-H). 13C-NMR (D2O-H2O/DCl) δ: 53.4; 114.8; 119.9; 127.6; 143.8; 150.6; 150.7; 164.3; 169.7. IR: 606; 631; 654; 689; 718; 731; 758; 800; 826; 853; 868; 907; 930; 951; 976; 1020; 1142; 1171; 1254; 1265; 1308; 1342; 1396; 1429; 1454; 1472; 1514; 1557; 1593; 1639; 2250–3500 br; 2731; 2920; 3063; 3146; 3566. MS ESI calculated for C10H9ClNO4 (M+H) 242.0. Found 241.9.
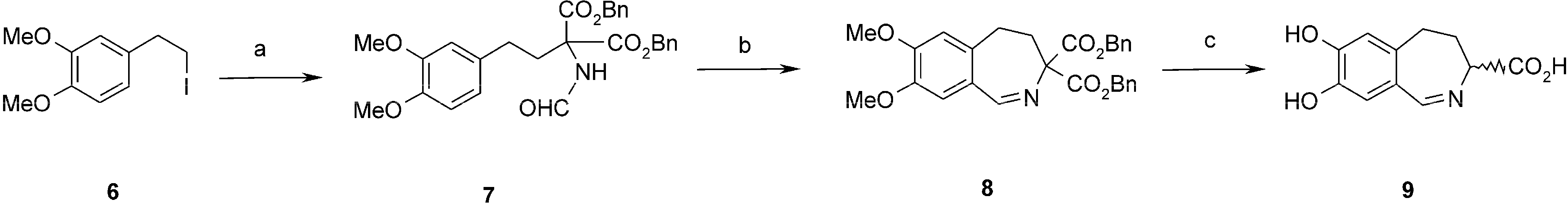
3.2.6. Preparation of Dibenzyl 7,8-Dimethoxy-4,5-dihydro-3H-2-benzazepine-3,3-dicarboxylate (8)
This compound was obtained according to the procedure 3.2.3. The reaction mixture was heated at 80 °C for 20 h. The product was purified by dissolving in boiling cyclohexane and cooling to 10 °C. Brown oil. Yield 52%. 1H-NMR (CDCl3) δ: 2.68 (m, 2H, C5-H); 2.85 (m, 2H, C4-H); 3.86 (s, 3H, OCH3); 3.87 (s, 3H, OCH3); 5.09 and 5.18 (AB, J = 12.5 Hz, 4H, CH2Ph); 6.58 (s, 1H, C7-H); 6.84 (s, 1H, C10-H); 7.20–7.30 (m, 10H, Ar); 8.41 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 30.6; 33.1; 55.9; 56.0; 67.5; 75.8; 112.2; 116.4; 125.4; 128.0; 128.1; 128.3; 134.8; 135.2; 147.2; 150.8; 163.7; 169.1. HR MS EI calculated for C28H27NO6 473.1838. Found: 473.1852.
3.2.7. Preparation of 7,8-Dihydroxy-4,5-dihydro-3H-2-benzazepine-3-carboxylic Acid (9)
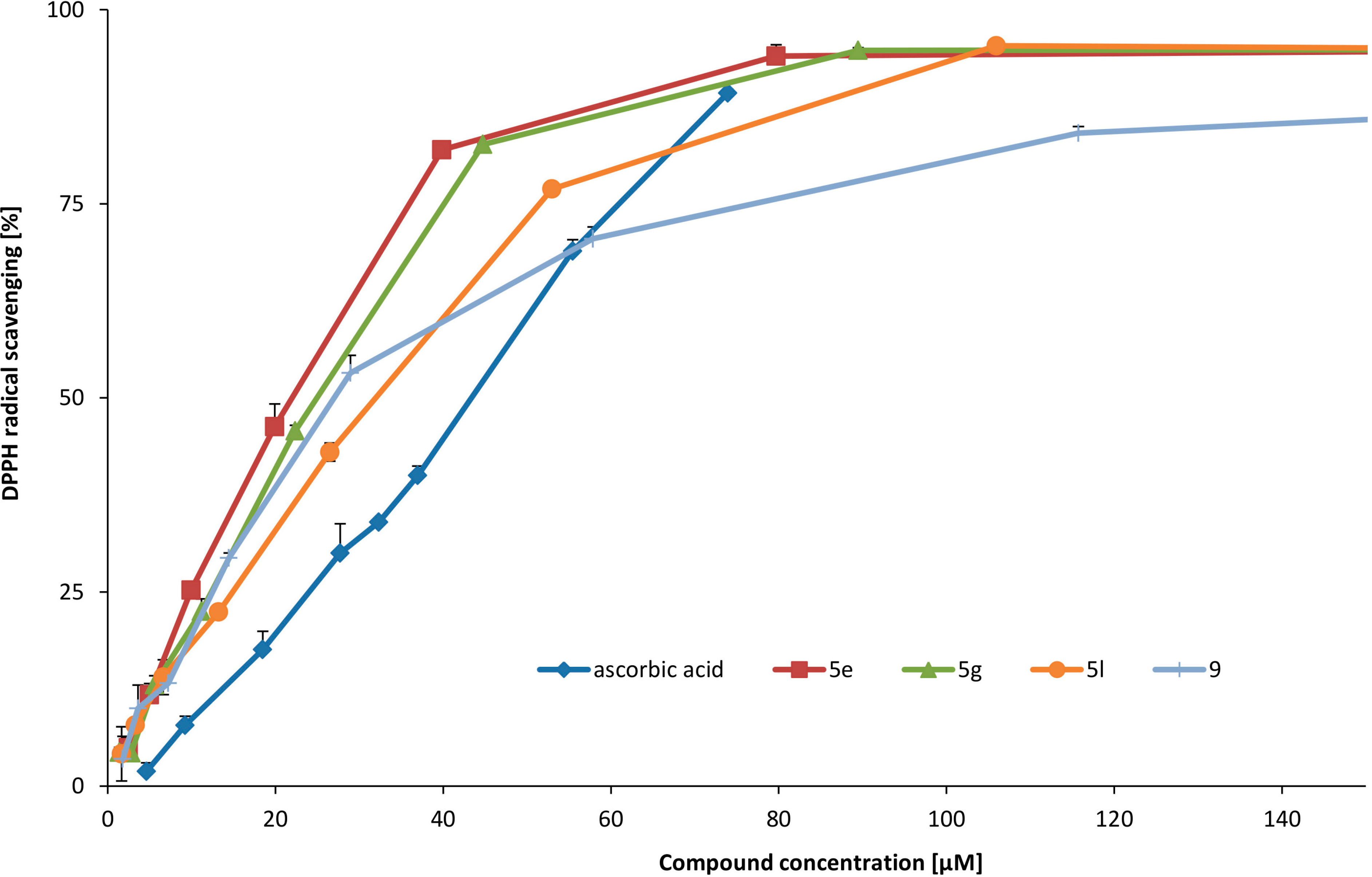
This compound was obtained according to the procedure 3.2.4. The reaction mixture was kept at rt for 24 h. Brown solid. Yield 58%. 1H-NMR (D2O-H2O/DCl) δ: 2.07–2.18 (m, 2H, C4-H); 2.70 and 2.86 (m, 2H, C5-H); 4.62 (t, J = 5.3 Hz, 1H, C3-H); 6.61 (s, 1H, C6-H); 6.99 (s, 1H, C9-H); 8.11 (s, 1H, C1-H). IR: 552; 565; 594; 667; 694; 752; 804; 843; 881; 941; 1038; 1103; 1138; 1171; 1238; 1252; 1281; 1373; 1456; 1506; 1558; 1576; 1601; 2250–3500 br; 2805; 2870; 3042; 3123. MS ESI calculated for C11H12NO4 (M+H) 222.0. Found 222.0.
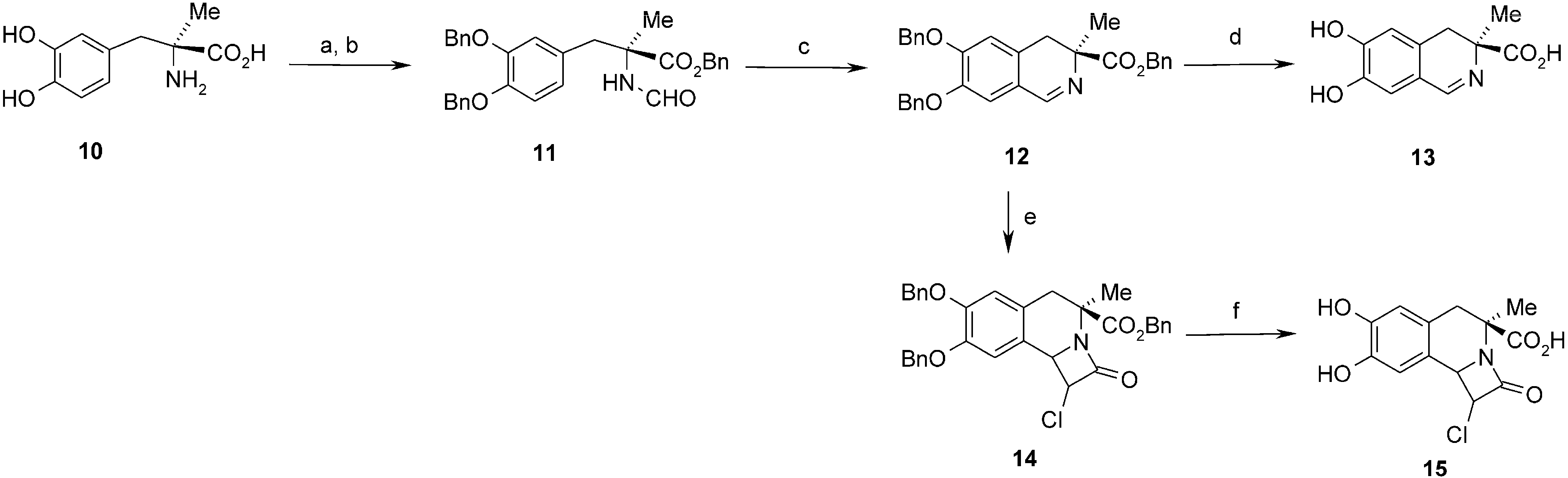
3.2.8. Preparation of (S)-Benzyl 2-(3,4-Dibenzyloxy)benzyl-2-formamidopropanoate (11)
Formylation of l-methyl-DOPA. Formyloxyacetonitrile (1.326 g, 15.6 mmol) was added to a solution of l-methyl-DOPA hydrate, (1.38 g, 5.8 mmol) in DMSO (10 mL) and the mixture was stirred at rt for 3 days. Solvent was removed under vacuum and the residue was dissolved in acetonitrile (5 mL). Dichloromethane (150 mL) and hexane (10 mL) were added to precipitate product. After filtration white solid was obtained (0.88 g). Yield 64%. 1H-NMR (DMSO) δ: 1.32 (s, 3H, CH3), 2.94 and 2.98 (AB, J = 13.3 Hz, 2H, CH2), 6.36 (dd, J1=1.9, 7.9 Hz, 1H, C6-H), 6.51 (d, J = 1.9 Hz, 1H, C2-H), 6.60 (d, J = 7.9 Hz, 1H, C5-H), 7.90 (br, 1H, NH), 7.93 (d, J = 1.7 Hz, 1H, CHO); 8.70 (br, 1H, OH). 13C-NMR (DMSO) δ: 22.7; 58.9; 115.1; 117.8; 117.8; 121.2; 127.2; 144.0; 144.7; 160.8; 174.6.
Benzylation of l-N-formylmethyl-DOPA. K2CO3, (12.4 g) and benzyl bromide (10.74 g, 62.8 mmol) were added to a solution of l-N-formylmethyl-DOPA (4.29 g, 17.9 mmol) in anh. acetonitrile (80 mL). The reaction mixture was stirred and heated at 60 °C for 16 h. The solvent was evaporated and the water was added to the residue. The mixture was extracted with CH2Cl2 (3 × 20 mL). Combined extracts were dried with Na2SO4 and evaporated. Product was purified on silica gel (EtOAc-CH2Cl2 1:1). White solid. Yield 244 mg (48%). 1H-NMR (CDCl3) δ: 1.68 (s, 3H, CH3), 3.10 (d, J = 13.8 Hz, 1H, CH2), 3.45 (d, J = 13.8 Hz, 1H, CH2), 5.05–5.14 (m, 6H, CH2Ph), 6.06 (bs, 1H, NH), 6.47 (dd, J = 8.3, 2.1 Hz, 1H, C6-H), 6.63 (d, J = 2.1 Hz, 1H, C2-H), 6.75 (d, J = 8.3 Hz, 1H, C5-H), 7.28–7.46 (m, 15H, Ar), 7.87 (d, J = 2.1 Hz, 1H, CHO). 13C-NMR (CDCl3) δ: 23.5; 40.9; 61.4; 67.6; 71.0; 71.2; 114.7; 117.0; 122.7; 127.1; 127.2; 127.3; 127.7; 127.8; 128.3; 128.4; 128.6; 128.7; 129.1; 134.9; 137.3; 148.1; 148.3; 160.4; 173.2. HR MS ESI calculated for C32H32NO5 (M+H) 510.2275. Found 510.2252.
3.2.9. Preparation of (S)-Benzyl 6,7-Dibenzyloxy-3-methyl-3,4-dihydroisoquinolin-3-carboxylate (12)
This compound was obtained according to the procedure 3.2.3. The reaction mixture was heated at 60 °C (oil bath) for 1.5 h. The product was purified on silica gel (first CH2Cl2 then CH2Cl2-EtOAc 1:1). Orange oil. Yield 1.479 g (88%). 1H-NMR (CDCl3) δ: 1.45 (s, 3H, CH3), 2.75 (d, 1H, J = 16.3 Hz, C4-H), 3.16 (d, 1H, J = 16.3 Hz, C4-H), 5.15 (s, 2H, CH2Ph); 5.17 (s, 2H, CH2Ph); 5.18 (s, 2H, CH2Ph); 6.72 (s, 1H, C5-H); 6.92 (s, 1H, C8-H); 7.25–7.45 (m, 15H, Ar); 8.22 (s, 1H, C1-H). 13C-NMR (CDCl3) δ: 23.6; 33.8; 62.7; 66.8; 71.0; 71.6; 113.8; 114.5; 120.7; 127.2; 127.3; 127.9; 127. 94; 128.0; 128.0; 128.2; 128.4; 128.5; 128.6; 135.9; 136.5; 136.9; 147.8; 152.0; 159.0; 174.5. HR MS ESI calculated for C32H30NO4 (M+H) 492.2169. Found 492.2169.
3.2.10. Preparation of (S)-6,7-Dihydroxy-3-methyl-3,4-dihydroisoquinolin-3-carboxylic Acid (13)
This compound was obtained according to the procedure 3.2.4. Green powder. Yield 88%. 1H-NMR (D2O-H2O/DCl) δ: 1.62 (s, 3H, CH3); 3.12 and 3.36 (AB, J = 17.1 Hz, 2H, C4-H); 6.79 (s, 1H, C5-H); 7.16 (s, 1H, C8-H); 8.57 (s, 1H, C1-H). 13C-NMR (D2O-H2O/DCl) δ: 21.8; 34.0; 60.8; 115.2; 116.0; 120.7; 131.7; 144.1; 155.8; 164.2; 173.8. IR: 554; 596; 627; 650; 719; 750; 797; 835; 883; 941; 966; 1126; 1180; 1233; 1277; 1331; 1381; 1447; 1516; 1557; 1568; 1614; 1634; 1643; 1667; 1732; 2250–3500 br; 2787; 2945; 3123; 3196. HR MS ESI calculated for C11H11NO4Na (M+Na) 244.0580. Found 244.0589.
3.2.11. Preparation of Benzyl 1-Chloro-7,8-dibenzyloxy-4-methyl-2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]isoquinoline-4-carboxylate (14)
Et3N (654 mg, 6.48 mmol) and solution of chloroacetyl chloride (549 mg, 4.86 mmol) in CH2Cl2 (2 mL) were added to a stirred solution of benzyl 6,7-dibenzyloxy-3,4-dihydroisoquinolin-3-methyl-3-carboxylate (400 mg, 0.81 mmol) in CH2Cl2 (10 mL). The reaction mixture was stirred at rt for 20 h and quenched with aqueous NaHCO3 (2 mL). Stirring was continued for next 1 h. Water layer was separated and extracted with CH2Cl2. Organic layers were dried over MgSO4, evaporated and purified by chromatography on silica gel (acetone/CH2Cl2 150:1). Brown oil. Yield 140 mg (47%). 1H-NMR (CDCl3) δ: 1.88 (s, 3H, CH3); 2.94 and 3.14 (AB, J = 15.8, 2H, C5-H); 4.41 (d, J = 1.7, 1H, C1-H); 4.69 (br, 1H, C9b-H); 5.09 (1/2AB, J = 12.1, 1H, CH2Ph); 5.10 (s, 2H, OCH2Ph); 5.12 (s, 2H, OCH2Ph); 5.13 (1/2AB, J = 12.1, 1H, CH2Ph); 6.64 (s, 1H, C6-H); 6.75 (s, 1H, C9-H); 7.13–7.46 (m, 15H, Ar). 13C-NMR (CDCl3) δ: 22.3; 39.3; 60.3; 60.4; 61.2; 67.3; 71.2; 71.4; 111.6; 115.4; 124. 2; 124.9; 127.2; 127.4; 127.8; 127.9; 128.0; 128.4; 128.5; 135.1; 136.6; 136.7; 148.5; 149.0; 162.9; 171.3. HR MS ESI calculated for C34H31ClNO5 (M+H) 568.1813. Found: 568.1887.
3.2.12. Preparation of 1-Chloro-7,8-dihydroxy-4-methyl-2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]-isoquinoline-4-carboxylic Acid (15)
Pd/C (10%, 15 mg) was added to a solution of benzyl 1-chloro-7,8-dibenzyloxy-4-methyl-2-oxo-1,4,5,9b-tetrahydro-2H-azeto[2,1-a]isoquinoline-4-carboxylate 14 (90 mg, 0.16 mmol) in EtOH (7 mL) and a balloon with hydrogen was connected. The suspension was stirred for 3 h. The catalyst was centrifuged and washed two times with EtOH (3 mL). Collected solution were evaporated and dried under vacuum. White solid. Yield 40 mg (85%). 1H-NMR (acetone-d6) δ: 1.80 (s, 3H, CH3); 2.94 and 3.13 (AB, J = 15.4 Hz, 2H, C5-H); 4.70 (br, 1H, C9b-H); 4.74 (d, J = 2.1 Hz, 1H, C1-H); 6.69 (s, 1H, C6-H); 6.77 (s, 1H, C9-H); 8.10 (br, 1H, OH); 8.15 (br, 1H, OH). 13C-NMR (acetone-d6) δ: 22.5; 39.5; 60.7; 61.1; 62.1; 112.8; 116.8; 124.1; 124.7; 145.5; 146.1; 163.3; 173.3. HR MS ESI calculated for C13H12ClNO5 (M+H) 298.0404. Found: 298.0478.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}