The Design and Synthesis of a New Class of RTK/HDAC Dual-Targeted Inhibitors

Abstract
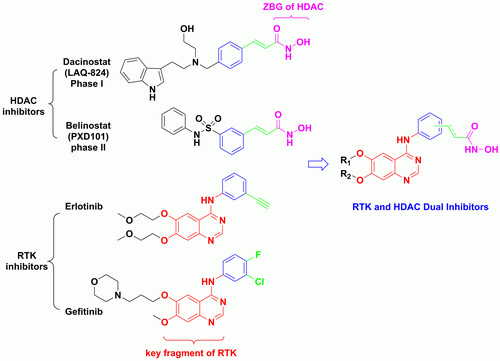
:
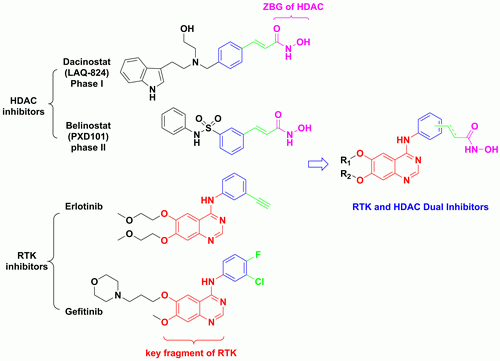
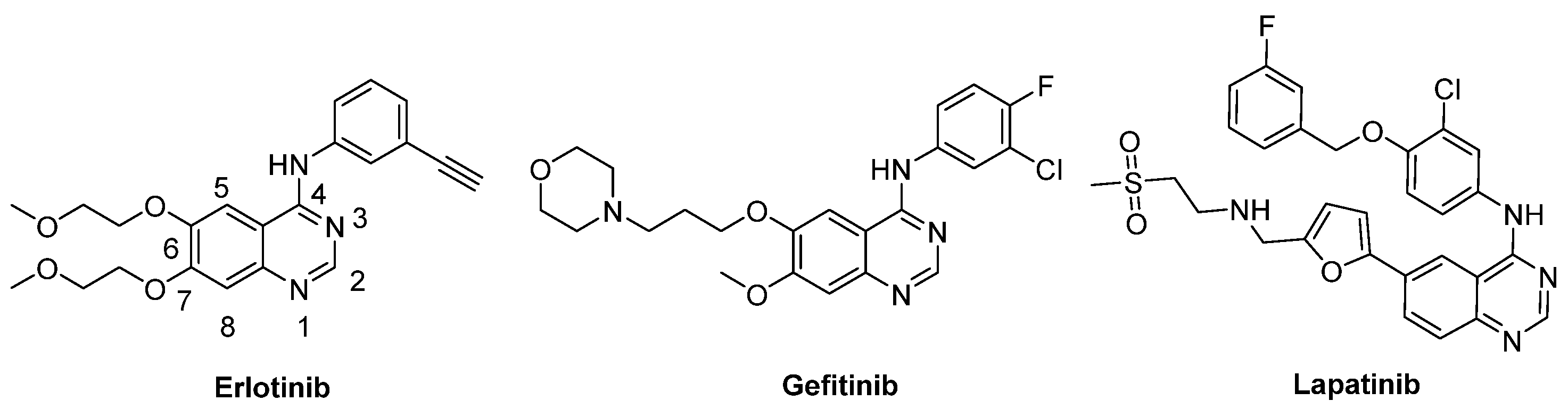
1. Introduction
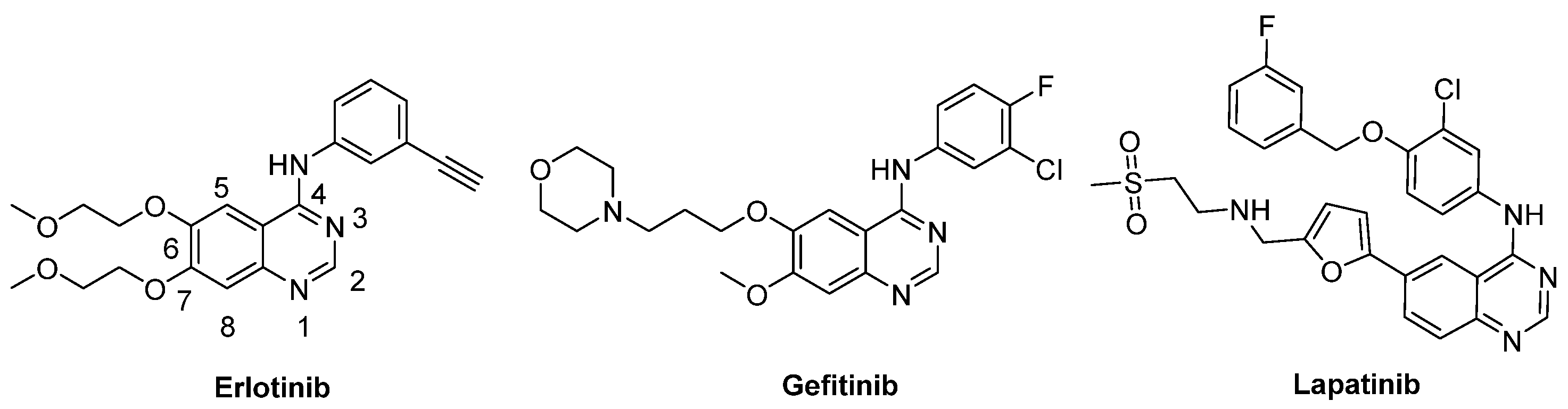
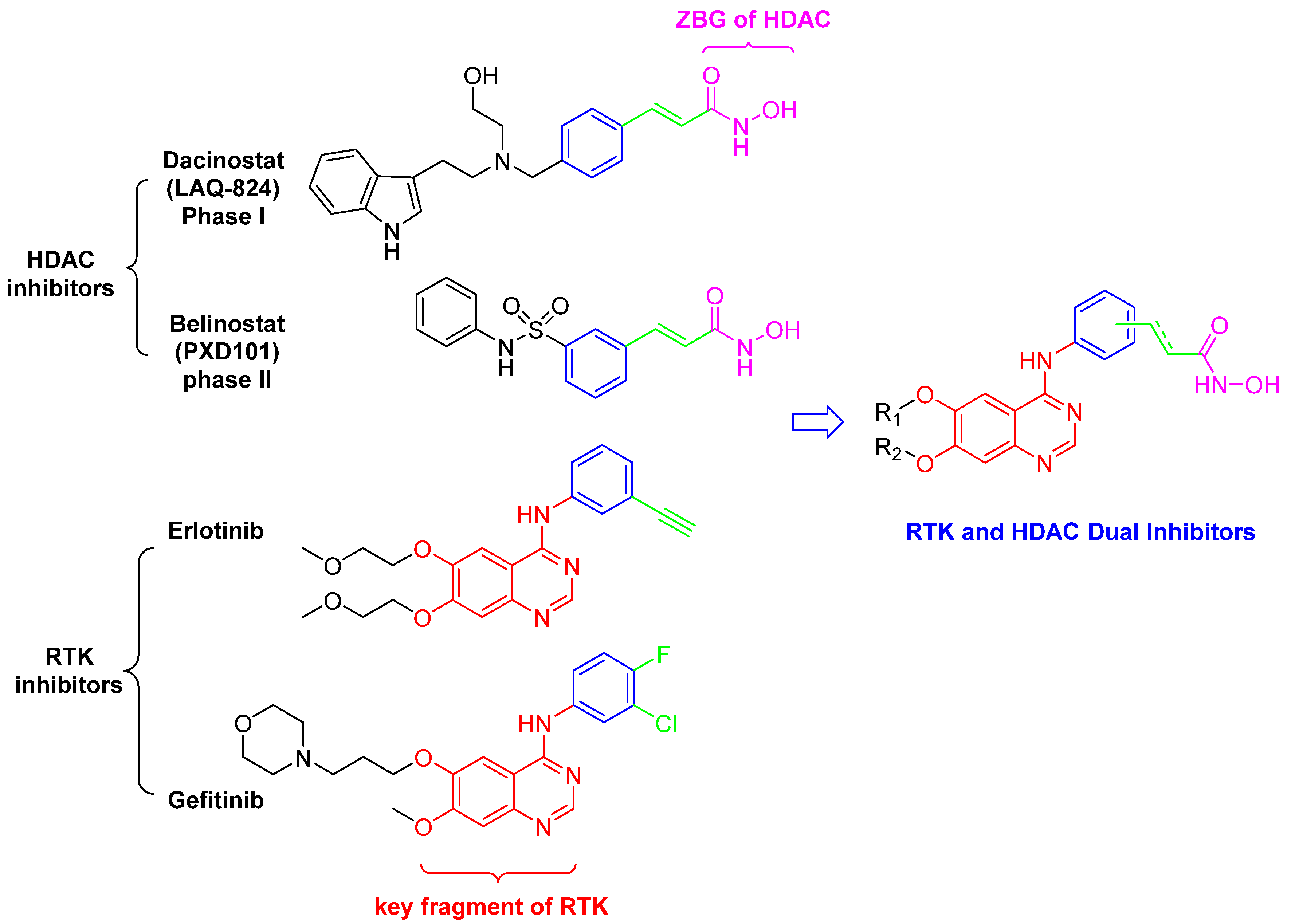
2. Results and Discussion
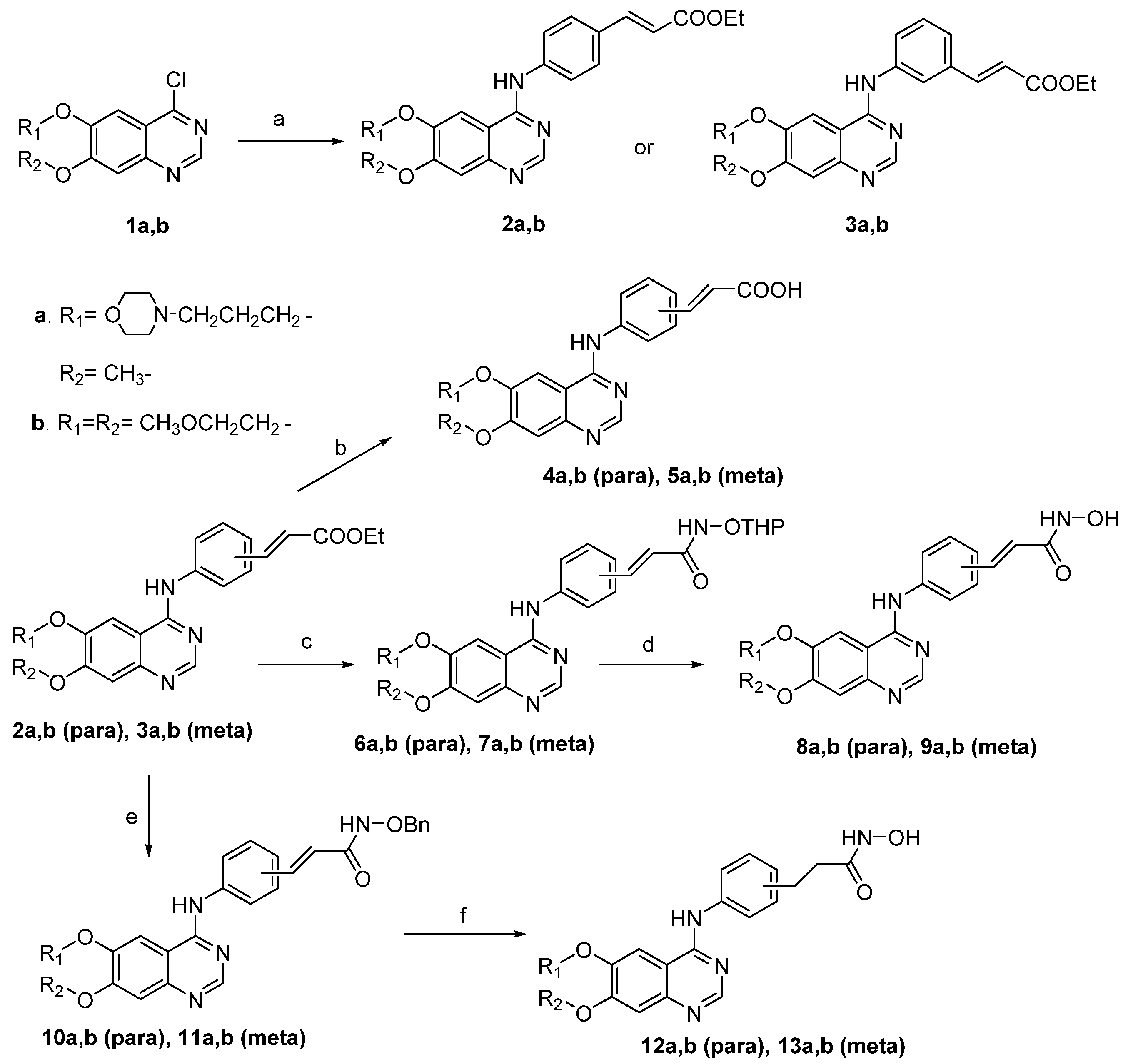
2.1. Chemisty
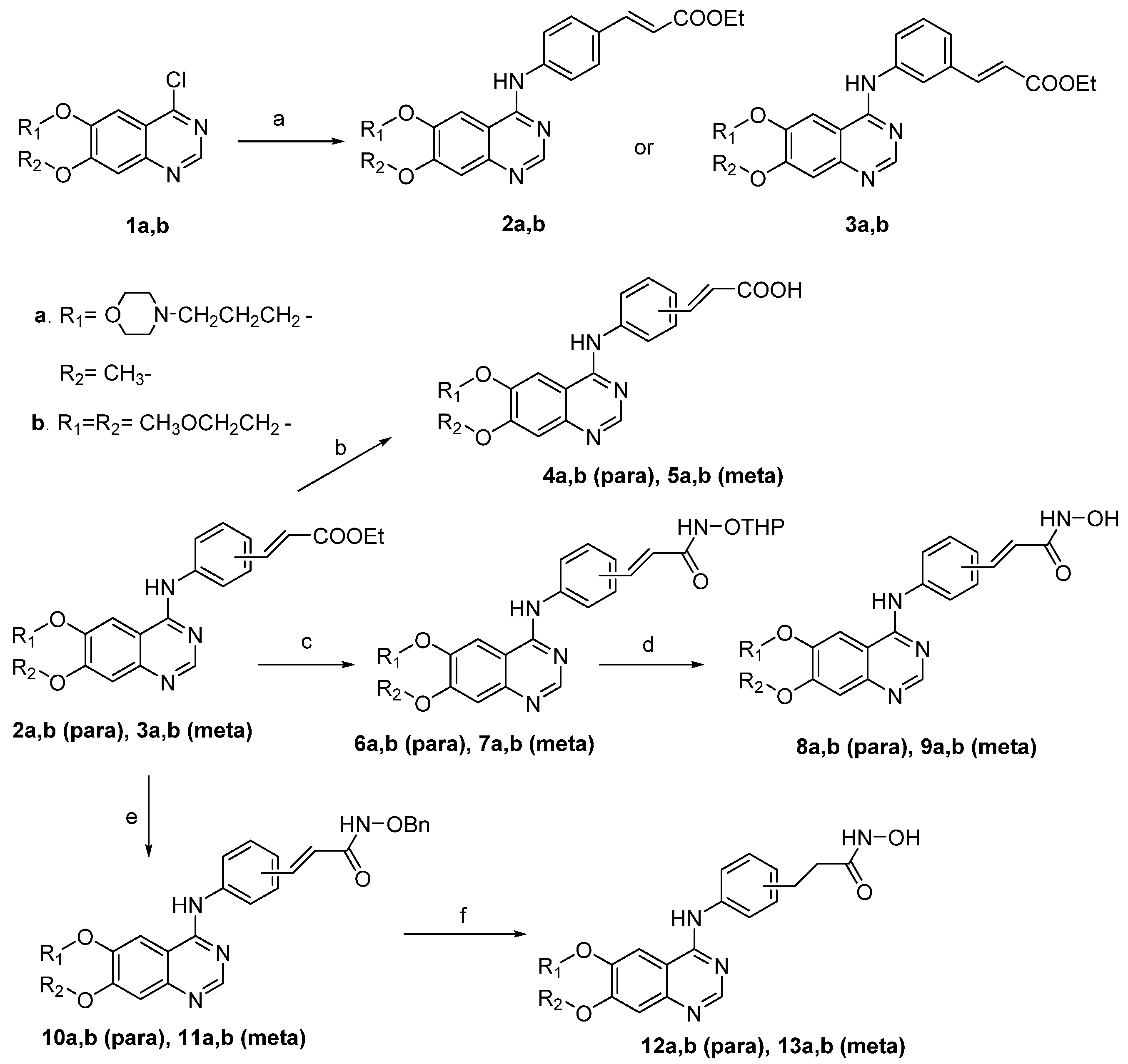
2.2. Results and Discussion
2.2.1. In Vitro HDAC Inhibition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | HDAC-1 | HDAC-3 | HDAC-6 |
|---|---|---|---|
| IC50 ± SD | IC50 ± SD | IC50 ± SD | |
| [μM] | [μM] | [μM] | |
| 4a | - a | - | - |
| 4b | - | - | - |
| 5a | - | - | - |
| 5b | - | - | - |
| 8a | 5.57 ± 1.87 | 2.38 ± 0.38 | 1.66 ± 0.19 |
| 8b | - | - | 2.77 ± 1.02 |
| 9a | 0.16 ± 0.02 | 0.18 ± 0.05 | 0.56 ± 0.06 |
| 9b | 0.29 ± 0.05 | 0.15 ± 0.02 | 0.56 ± 0.05 |
| 12a | 2.83 ± 0.65 | 3.29 ± 0.34 | 5.71 ± 0.99 |
| 12b | 5.82 ± 1.15 | 3.90 ± 0.78 | 7.21 ± 0.76 |
| 13a | 1.03 ± 0.27 | 2.11 ± 0.34 | 7.63 ± 0.99 |
| 13b | 2.43 ± 0.50 | 1.57 ± 0.17 | 4.82 ± 0.37 |
| SAHA | 0.25 ± 0.04 | 0.17 ± 0.02 | 0.23 ± 0.06 |
| Lapatinib | - | - | - |
2.2.2. In Vitro RTK Inhibition
| Compound | EGFR %Inhibition a | HER2 %Inhibition b |
|---|---|---|
| 4a | 6.1 | 18.2 |
| 4b | 20.1 | 0 |
| 5a | 20.9 | 0 |
| 5b | 19.0 | 2.6 |
| 8a | 63.6 | 74.0 |
| 8b | 70.1 | 84.6 |
| 9a | 9.7 | 47.2 |
| 9b | 8.5 | 64.2 |
| 12a | 0 | 0 |
| 12b | 44.6 | 0.1 |
| 13a | 42.6 | 0 |
| 13b | 20.4 | 0 |
| SAHA | 0 | 0 |
| Lapatinib | 92.7 | 92.0 |
3. Experimental
3.1. General
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of Compounds 2a, 2b, 3a, 3b
3.2.2. General Procedure for the Synthesis of Compounds 4a, 4b, 5a, 5b
3.2.3. General Procedure for the Synthesis of Compounds 6a, 6b, 7a, 7b
3.2.4. General Procedure for the Synthesis of Compounds 8a, 8b, 9a, 9b
3.2.5. General Procedure for the Synthesis of Compounds 10a, 10b, 11a, 11b
3.2.6. General Procedure for the Synthesis of Compounds 12a, 12b, 13a, 13b
3.3. Evaluation of Enzyme Activities
3.3.1. HDAC Enzymatic Assay in Vitro
3.3.2. EGFR and HER2 Inhibition Assay
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kim, M.S.; Blake, M.; Baek, J.H.; Kohlhagen, G.; Pommier, Y.; Carrier, F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003, 63, 7291–7300. [Google Scholar] [PubMed]
- Bruzzese, F.; Rocco, M.; Castelli, S.; Di Gennaro, E.; Desideri, A.; Budillon, A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol. Cancer Ther. 2009, 8, 3075–3087. [Google Scholar] [CrossRef] [PubMed]
- Bevins, R.L.; Zimmer, S.G. It’s about time: Scheduling alters effect of histone deacetylase inhibitors on camptothecin-treated cells. Cancer Res. 2005, 65, 6957–6966. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M.; Alajati, A.; Ganslmayer, M.; Zopf, S.; Luders, M.; Neureiter, D.; Hahn, E.G.; Schuppan, D.; Herold, C. The histone-deacetylase inhibitor SAHA potentiates proapoptotic effects of 5-fluorouracil and irinotecan in hepatoma cells. J. Cancer Res. Clin. Oncol. 2005, 131, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Kahali, S.; Chinnaiyan, P. Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J. Neurooncol. 2010, 99, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chen, C.S.; Lin, S.P.; Weng, J.R.; Chen, C.S. Targeting histone deacetylase in cancer therapy. Med. Res. Rev. 2006, 26, 397–413. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kim, Y.S.; Kummar, S.; Giaccone, G.; Trepel, J.B. Histone deacetylase inhibitors in cancer therapy. Curr. Opin. Oncol. 2008, 20, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.S.; Weng, S.C.; Tseng, P.H.; Lin, H.P.; Chen, C.S. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J. Biol. Chem. 2005, 280, 38879–38887. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, Epidermal growth factor receptor, And human epidermal growth factor receptor 2, Exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef] [PubMed]
- Mahboobi, S.; Dove, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Design of chimeric histone deacetylase- and tyrosine kinase-inhibitors: A series of imatinib hybrides as potent inhibitors of wild-type and mutant BCR-ABL, PDGF-Rbeta, and histone deacetylases. J. Med. Chem. 2009, 52, 2265–2279. [Google Scholar] [CrossRef] [PubMed]
- Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Ciossek, T.; Baer, T.; Maier, T.; Beckers, T. Novel chimeric histone deacetylase inhibitors: A series of lapatinib hybrides as potent inhibitors of epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and histone deacetylase activity. J. Med. Chem. 2010, 53, 8546–8555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Tong, L.; Luo, Y.; Su, M.; Zang, Y.; Li, J.; Lu, W.; Chen, Y. The discovery of colchicine-SAHA hybrids as a new class of antitumor agents. Bioorg. Med. Chem. 2013, 21, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.; Zhang, X. The ErbB kinase domain: structural perspectives into kinase activation and inhibition. Exp. Cell Res. 2009, 315, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4297. [Google Scholar] [CrossRef] [PubMed]
- Glick, R.D.; Swendeman, S.L.; Coffey, D.C.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; La Quaglia, M.P. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 1999, 59, 4392–4399. [Google Scholar] [PubMed]
- Butler, L.M.; Agus, D.B.; Scher, H.I.; Higgins, B.; Rose, A.; Cordon-Cardo, C.; Thaler, H.T.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 2000, 60, 5165–5170. [Google Scholar] [PubMed]
- Oyelere, A.K.; Chen, P.C.; Guerrant, W.; Mwakwari, S.C.; Hood, R.; Zhang, Y.; Fan, Y. Non-peptide macrocyclic histone deacetylase inhibitors. J. Med. Chem. 2009, 52, 456–468. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Mwakwari, S.C.; Patil, V.; Guerrant, W.; Oyelere, A.K. Macrocyclic Histone Deacetylase Inhibitors. Curr. Top. Med. Chem. 2010, 10, 1423–1440. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Kramer, O.H. Inhibitors of HDACs—Effective drugs against cancer? Curr. Cancer Drug Targets 2010, 10, 210–228. [Google Scholar] [CrossRef] [PubMed]
- Beckers, T.; Mahboobi, S.; Sellmer, A.; Winkler, M.; Eichhorn, E.; Pongratz, H.; Maier, T.; Ciossek, T.; Baer, T.; Kelter, G.; et al. Chimerically designed HDAC- and tyrosine kinase inhibitors. A series of erlotinib hybrids as dual-selective inhibitors of EGFR, HER2 and histone deacetylases. Med. Chem. Comm. 2012, 3, 829. [Google Scholar] [CrossRef]
- Chandregowda, V.; Rao, G.V.; Reddy, G.C. One-pot conversion of 2-nitrobenzonitriles to quinazolin-4(3H)-ones and synthesis of gefitinib and erlotinib hydrochloride. Heterocycles 2007, 71, 39–48. [Google Scholar] [CrossRef]
- Bradner, J.E.; Mak, R.; Tanguturi, S.K.; Mazitschek, R.; Haggarty, S.J.; Ross, K.; Chang, C.Y.; Bosco, J.; West, N.; Morse, E.; et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc. Natl. Acad. Sci. USA 2010, 107, 12617–12622. [Google Scholar] [CrossRef] [PubMed]
- Kramer, O.H. HDAC2: A critical factor in health and disease. Trends Pharmacol. Sci. 2009, 30, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.M.; Miao, Z.H.; Lin, L.P.; Gui, M.; Zhu, C.H.; Xie, H.; Duan, W.H.; Ding, J. BB, a new EGFR inhibitor, Exhibits prominent anti-angiogenesis and antitumor activities. Cancer Biol. Ther. 2009, 8, 1640–1647. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 4a,b, 5a,b, 8a,b, 9a,b, 12a,b and 13a,b are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, X.; Su, M.; Chen, Y.; Li, J.; Lu, W. The Design and Synthesis of a New Class of RTK/HDAC Dual-Targeted Inhibitors. Molecules 2013, 18, 6491-6503. https://doi.org/10.3390/molecules18066491
Zhang X, Su M, Chen Y, Li J, Lu W. The Design and Synthesis of a New Class of RTK/HDAC Dual-Targeted Inhibitors. Molecules. 2013; 18(6):6491-6503. https://doi.org/10.3390/molecules18066491
Chicago/Turabian StyleZhang, Xuan, Mingbo Su, Yi Chen, Jia Li, and Wei Lu. 2013. "The Design and Synthesis of a New Class of RTK/HDAC Dual-Targeted Inhibitors" Molecules 18, no. 6: 6491-6503. https://doi.org/10.3390/molecules18066491
APA StyleZhang, X., Su, M., Chen, Y., Li, J., & Lu, W. (2013). The Design and Synthesis of a New Class of RTK/HDAC Dual-Targeted Inhibitors. Molecules, 18(6), 6491-6503. https://doi.org/10.3390/molecules18066491