One-Pot Synthesis of Novel Chiral β-Amino Acid Derivatives by Enantioselective Mannich Reactions Catalyzed by Squaramide Cinchona Alkaloids


Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
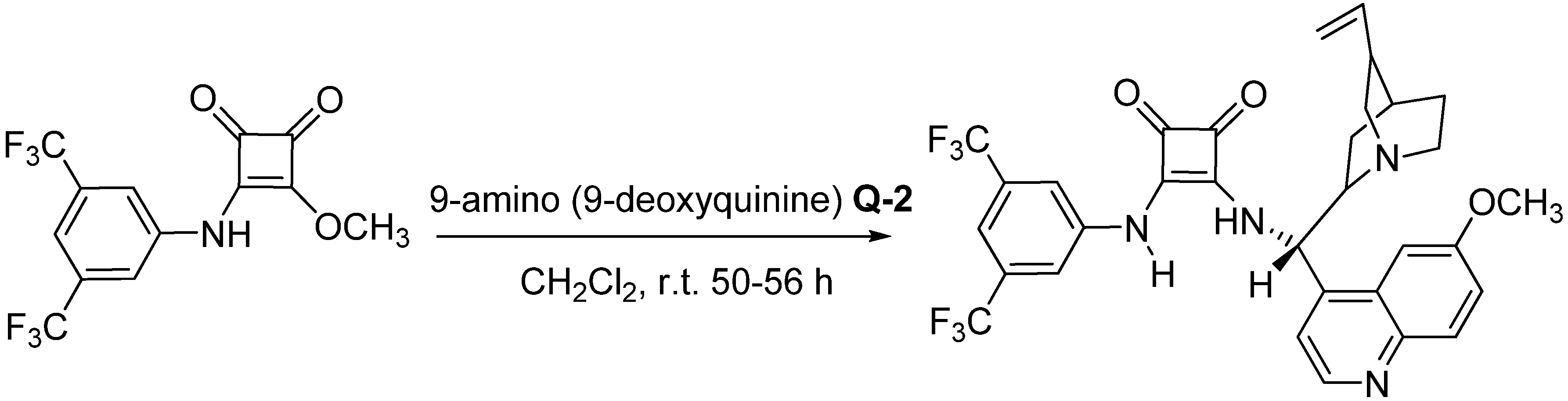
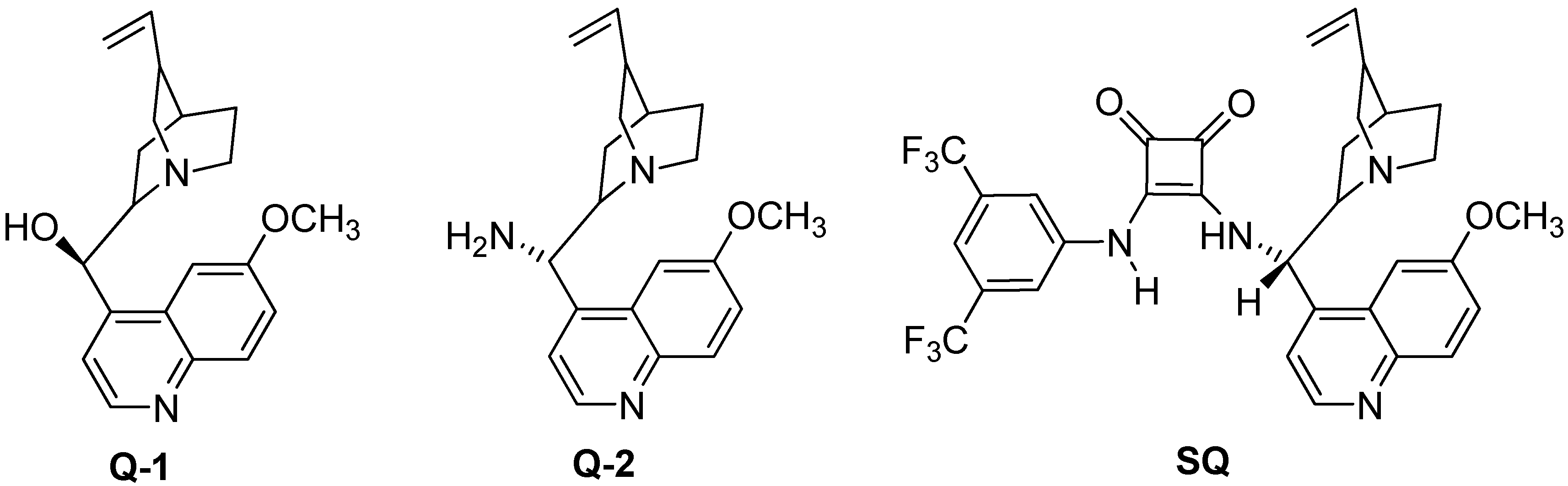


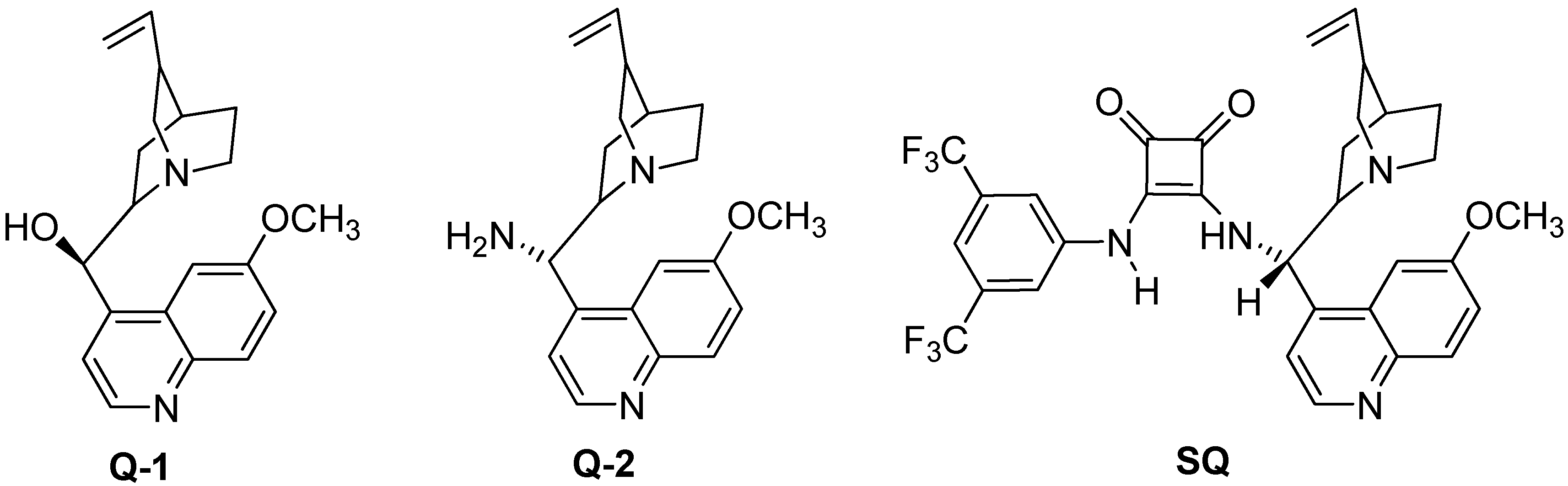{kind=link}
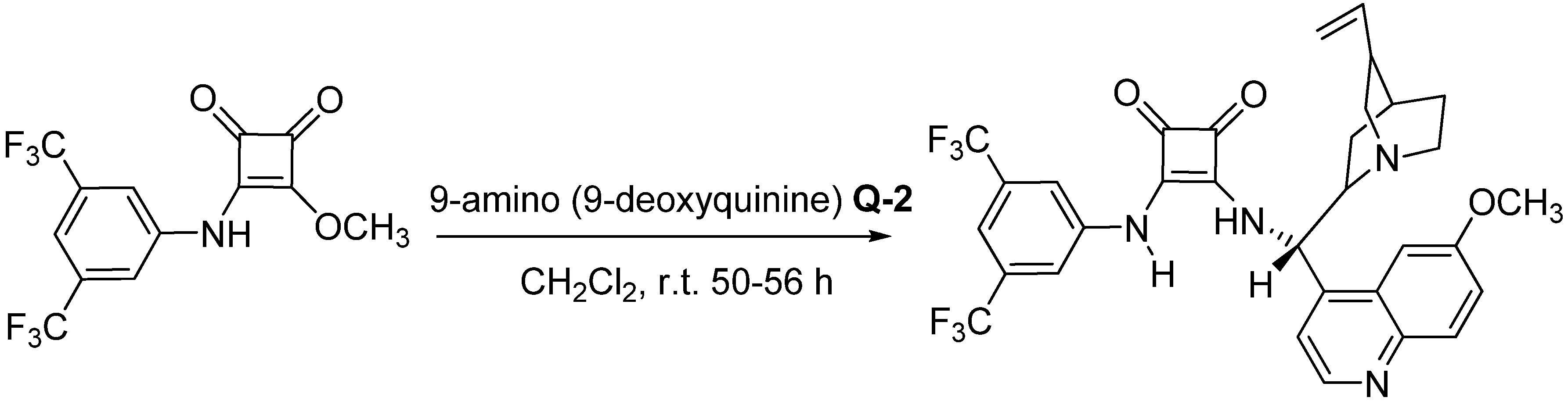{kind=link}
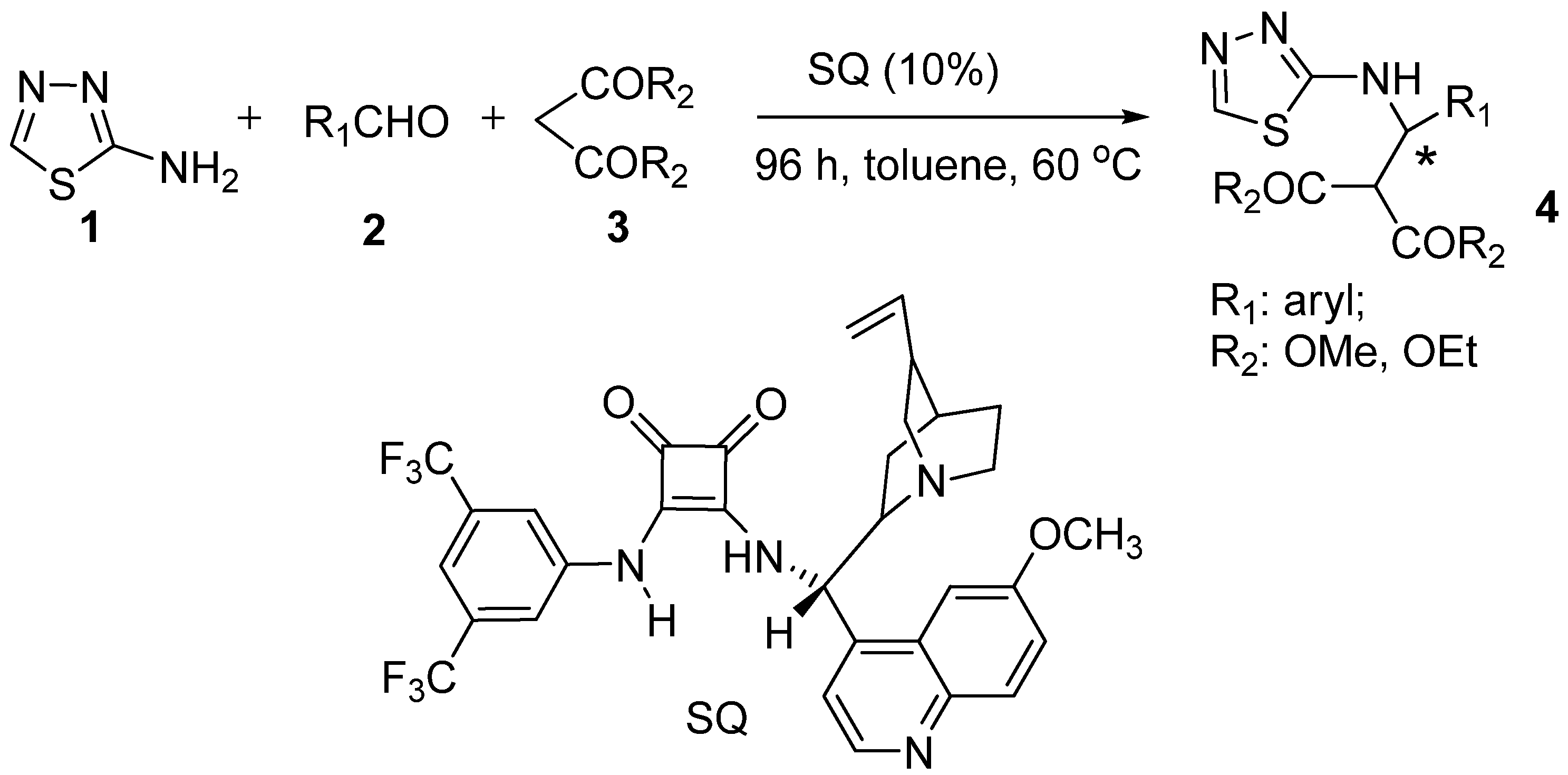{kind=link}
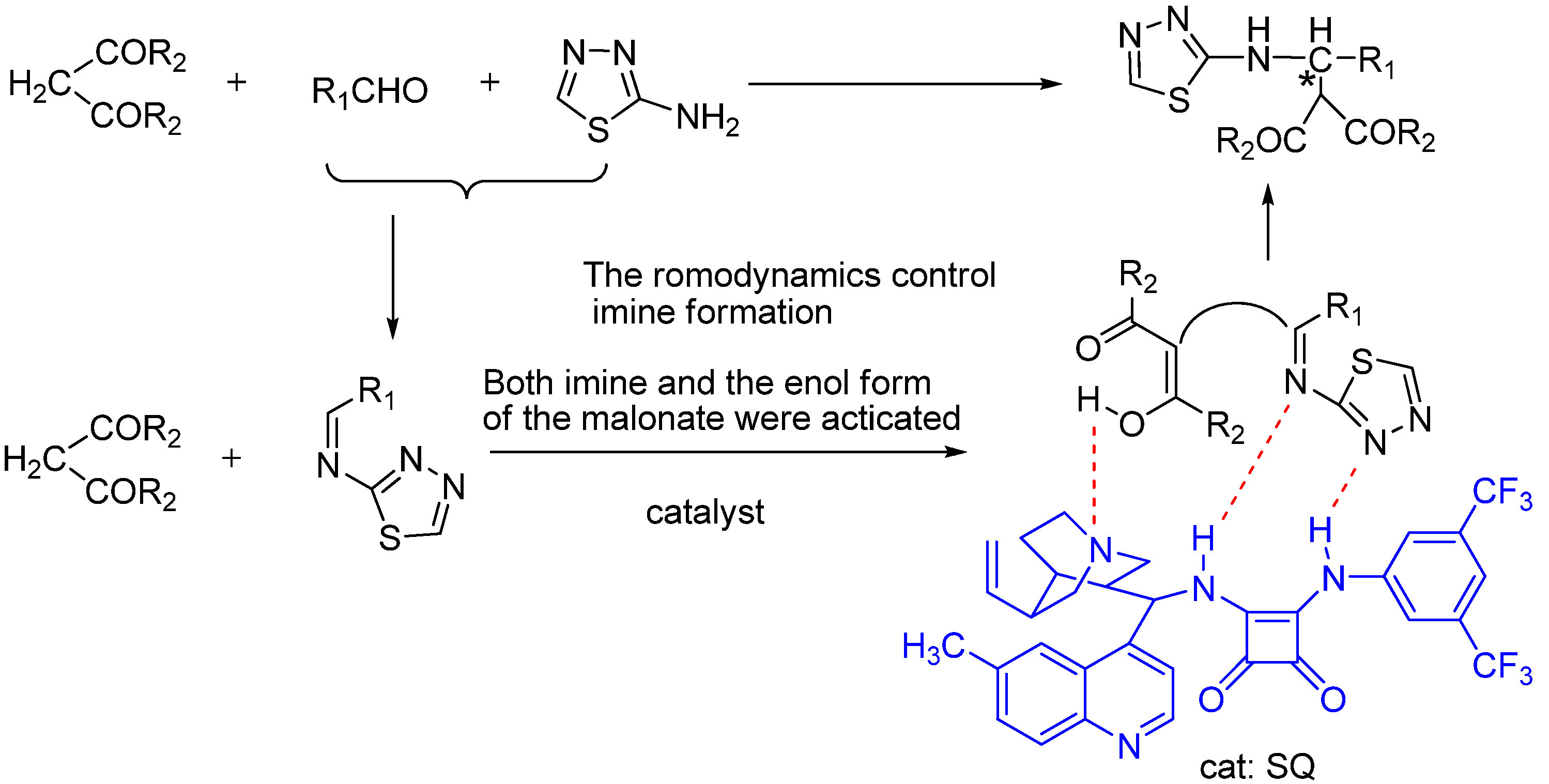{kind=link}
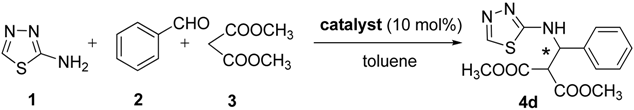 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Temp. (°C) | Solvent | Yield b (%) | ee c (%) |
| 1 | Q-1 | 60 | Toluene | 37 | 11 |
| 2 | Q-2 | 60 | Toluene | 39 | 37 |
| 3 | SQ | 60 | Toluene | 41 | 91 |
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst load (mol%) | Temp. (°C) | Solvent | Yield b (%) | ee c (%) |
| 1 | 10 | 60 | Methanol | 53 | 71 |
| 2 | 10 | 60 | Acetone | 41 | 75 |
| 3 | 10 | 60 | Chloroform | 38 | 83 |
| 4 | 10 | 60 | Toluene | 49 | 91 |
| 5 | 10 | r.t. | Toluene | 37 | 80 |
| 6 | 5 | 60 | Toluene | 45 | 70 |
| 7 | 2.5 | 60 | Toluene | 44 | 43 |
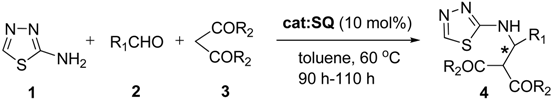 | ||||||
|---|---|---|---|---|---|---|
| Entry | 4 | R1 | R2 | Time (h) | Yield b (%) | ee c (%) |
| 1 | 4a | 2, 3-di-Cl-Ph- | OMe | 96 | 42 | 41 |
| 2 | 4b | 3-CF3- Ph- | OMe | 96 | 39 | 58 |
| 3 | 4c | 2, 3-di-OMe-Ph- | OMe | 97 | 44 | 48 |
| 4 | 4d | Ph- | OMe | 96 | 45 | 91 |
| 5 | 4e | 2, 4-di-Cl-Ph- | OMe | 96 | 61 | 42 |
| 6 | 4f | Ph- | OEt | 93 | 52 | 99 |

3. Experimental
3.1. General
3.2. Preparation of the SQ Catalyst (Scheme 1)
3.3. Preparation of Chiral Compounds 4a–4f
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Seebach, D.; Gardiner, J. Beta-peptidic peptidomimetics. Acc. Chem. Res. 2008, 41, 1366–1375. [Google Scholar] [CrossRef]
- Weiner, B.; Baeza, A.; Jerphagnon, T.; Feringa, B.L. Aldehyde selective wacker oxidations of phthalimide protected allylic amines: A new catalytic route to β-amino acids. J. Am. Chem. Soc. 2009, 131, 9473–9474. [Google Scholar] [CrossRef]
- Gellman, S.H. Foldamers: A manifesto. Acc. Chem. Res. 1998, 31, 173–180. [Google Scholar] [CrossRef]
- Gademann, K.; Hintermann, T.; Schreiber, J.V. Beta-peptides: Twisting and turning. Curr. Med. Chem. 1999, 6, 905–925. [Google Scholar]
- Hill, D.J.; Mio, M.J.; Prince, R.B.; Hughes, T.S.; Moore, J.S. A field guide to foldamers. Chem. Rev. 2001, 101, 3893–4011. [Google Scholar] [CrossRef]
- Cheng, R.P.; Gellman, S.H.; DeGrado, W.F. β-peptides: From structure to function. Chem. Rev. 2001, 101, 3219–3232. [Google Scholar] [CrossRef]
- Steer, D.L.; Lew, R.A.; Perlmutter, P.; Smith, A.I.; Aguilar, M.I. β-amino acids: Versatile peptidomimetics. Curr. Med. Chem. 2002, 9, 811–822. [Google Scholar] [CrossRef]
- Miyabe, H.; Fujii, K.; Naito, T. Highly diastereoselective radical addition to oxime ethers: Asymmetric synthesis of β-amino acids. Org. Lett. 1999, 1, 569–572. [Google Scholar] [CrossRef]
- Abele, S.; Seebach, D. Preparation of achiral and of enantiopure geminally disubstituted β-amino acids for β-peptide synthesis. Eur. J. Org. Chem. 2000, 1, 1–15. [Google Scholar] [CrossRef]
- Seki, M.; Shimizu, T.; Matsumoto, K. Stereoselective synthesis of β-benzyl-α-alkyl-β-amino acids from L-aspartic acid. J. Org. Chem. 2000, 65, 1298–1304. [Google Scholar] [CrossRef]
- Albertina, G.M.; Elena, M.; José, A.C.; Ángel, Á.L.; Graciela, Y.M.; Vicenc, B.; Rosa, M.O. Reaction between N-alkylhydroxilamines and chiral enoate esters: More experimental evidence for a cycloaddition-like process, a rationale based on DFT theoretical calculations and stereoselective synthesis of new enantiopure β-amino acids. J. Org. Chem. 2002, 67, 2402–2410. [Google Scholar]
- Luisi, R.; Capriati, V.; Degennaro, L.; Florio, S. Oxazolinyloxiranyllithium-mediated stereoselective synthesis of a-epoxy-b-amino acids. Org. Lett. 2003, 5, 2723–2726. [Google Scholar]
- Swiderska, M.A.; Stewart, J.D. Asymmetric bioreductions of β-nitro acrylates as a route to chiral β-amino acids. Org. Lett. 2006, 8, 6131–6133. [Google Scholar]
- Saavedra, C.; Hernández, R.; Boto, A.; Álvarez, E. Catalytic, One-pot synthesis of β-amino acids from α-amino acids. Preparation of α,β-peptide derivatives. J. Org. Chem. 2009, 74, 4655–4665. [Google Scholar]
- Juaristi, E.; Soloshonok, V.A. Enantioselective Synthesis of β-amino Acids, 2nd ed; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Juaristi, E.; Quintana, D.; Escalante, J. Enantioselective synthesis of β-amino acids. Aldrichim. Acta 1994, 27, 3–11. [Google Scholar]
- Cole, D.C. Recent stereoselective synthetic approaches to β-amino acids. Tetrahedron. 1994, 50, 9517–9582. [Google Scholar]
- Cardillo, G.; Tomasini, C. Asymmetric synthesis of ß-amino acids and α-substituted β-amino acids. Chem. Soc. Rev. 1996, 25, 117–128. [Google Scholar]
- Juaristi, E. Enantioselective Synthesis of β-amino Acids; Wiley-VCH: New York, NY, USA, 1997; chapters: 11–13. [Google Scholar]
- Juaristi, E.; López-Ruiz, H. Recent advances in the enantioselective synthesis of beta-amino acids. Curr. Med Chem. 1999, 6, 983–1004. [Google Scholar]
- Liu, M.; Sibi, M.P. Recent advances in the stereoselective synthesis of β-amino acids. Tetrahedron 2002, 58, 7991–8035. [Google Scholar] [CrossRef]
- Mohammad, A.; Harish, K.; Sadique, A.J. Synthesis and pharmacological evaluation of condensed heterocyclic 6-substituted-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazole derivatives of naproxen. Bioorg. Med. Chem. Lett. 2007, 17, 4504–4508. [Google Scholar]
- Mathew, V.; Keshavayya, J.; Vaidya, V.P. Heterocyclic system containing bridgehead nitrogen atomsynthesis and pharmacologicalactivities of some substituted 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles. Eur. J. Med. Chem. 2006, 41, 1048–1058. [Google Scholar] [CrossRef]
- Nasser, S.; Khalil, A.M. N- and S-a-L-arabinopyranosyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazoles. First synthesis and biological evaluation. Eur. J. Med. Chem. 2007, 42, 1193–1199. [Google Scholar] [CrossRef]
- Karegoudar, P.; Prasad, D.J.; Ashok, M.; Mahalinga, M.; Poojary, B.; Holla, B.S. Synthesis, antimicrobial and anti-inflammatory activities of some 1,2,4-triazolo[3,4-b][1,3,4]thiadiazoles and 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines bearing trichlorophenyl moiety. Eur. J. Med. Chem. 2008, 43, 808–815. [Google Scholar] [CrossRef]
- El-Barbary, A.A.; Abou-El-Ezz, A.Z.; Abdel-Kader, A.A.; El-Daly, M.; Nielsenc, C. Synthesis of some new 4-amino-1,2,4-triazole derivatives as potential anti-HIV and anti-HBV. Phosphorus, Sulfur Silicon. Relat. Elem. 2004, 179, 1497–1508. [Google Scholar] [CrossRef]
- Bai, S.; Liang, X.P.; Song, B.A.; Bhadury, P.S.; Hu, D.Y.; Yang, S. Asymmetric Mannich reactions catalyzed by cinchona alkaloid thiourea: Enantioselective one-pot synthesis of novel β-amino ester derivatives. Tetrahedron: Asymmetry. 2011, 22, 518–523. [Google Scholar] [CrossRef]
- Li, L.; Song, B.A.; Bhadury, P.S.; Zhang, Y.P.; Hu, D.Y.; Yang, S. Enantioselective synthesis of β-amino esters bearing a benzothiazole moiety via a Mannich-type reaction catalyzed by a cinchona alkaloid derivative. Eur. J. Org. Chem. 2011, 25, 4743–4376. [Google Scholar]
- Lin, P.; Song, B.A.; Bhadury, P.S.; Hu, D.Y.; Zhang, Y.P.; Jin, L.H.; Yang, S. Chiral cinchona alkaloid-thiourea catalyzed Mannich reaction for enantioselective synthesis of β-amino ketones bearing benzothiazol moiety. Chin. J. Chem. 2011, 29, 2433–2438. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem.Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef]
- Yang, W.; Du, D.M. Highly Enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Org. Lett. 2010, 12, 5450–5453. [Google Scholar] [CrossRef]
- Jang, H.B.; Rho, H.S.; Oh, J.S.; Nam, E.H.; Park, S.F.; Bae, H.Y.; Song, C.E. DOSY NMR for monitoring self aggregation of bifunctional organocatalysts: Increasing enantioselectivity with decreasing catalyst concentration. Org. Biomol. Chem. 2010, 8, 3918–3922. [Google Scholar] [CrossRef]
- Jiang, H.; Paixão, M.W.; Monge, D.; Jørgensen, K.A. Acyl phosphonates: Good hydrogen bond acceptors and ester/amide equivalents in asymmetric organocatalysis. J. Am. Chem. Soc. 2010, 132, 2775–2783. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, K.; Liang, X.; He, M.; Wu, J.; Zhang, Y.; Xue, W.; Jin, L.; Yang, S.; Hu, D. One-Pot Synthesis of Novel Chiral β-Amino Acid Derivatives by Enantioselective Mannich Reactions Catalyzed by Squaramide Cinchona Alkaloids. Molecules 2013, 18, 6142-6152. https://doi.org/10.3390/molecules18066142
Zhang K, Liang X, He M, Wu J, Zhang Y, Xue W, Jin L, Yang S, Hu D. One-Pot Synthesis of Novel Chiral β-Amino Acid Derivatives by Enantioselective Mannich Reactions Catalyzed by Squaramide Cinchona Alkaloids. Molecules. 2013; 18(6):6142-6152. https://doi.org/10.3390/molecules18066142
Chicago/Turabian StyleZhang, Kankan, Xueping Liang, Ming He, Jian Wu, Yuping Zhang, Wei Xue, Linhong Jin, Song Yang, and Deyu Hu. 2013. "One-Pot Synthesis of Novel Chiral β-Amino Acid Derivatives by Enantioselective Mannich Reactions Catalyzed by Squaramide Cinchona Alkaloids" Molecules 18, no. 6: 6142-6152. https://doi.org/10.3390/molecules18066142
APA StyleZhang, K., Liang, X., He, M., Wu, J., Zhang, Y., Xue, W., Jin, L., Yang, S., & Hu, D. (2013). One-Pot Synthesis of Novel Chiral β-Amino Acid Derivatives by Enantioselective Mannich Reactions Catalyzed by Squaramide Cinchona Alkaloids. Molecules, 18(6), 6142-6152. https://doi.org/10.3390/molecules18066142