The Clip-Segment of the von Willebrand Domain 1 of the BMP Modulator Protein Crossveinless 2 Is Preformed

Abstract
:1. Introduction
2. Results and Discussion
2.1. Production of the N-Terminal von Willebrand Type C Domain of Crossveinless 2 for NMR Analysis
2.2. The Distribution of the NOE-Derived Distance Restraints Suggest Flexible N- and C-Termini in the CV2 VWC1 Domain

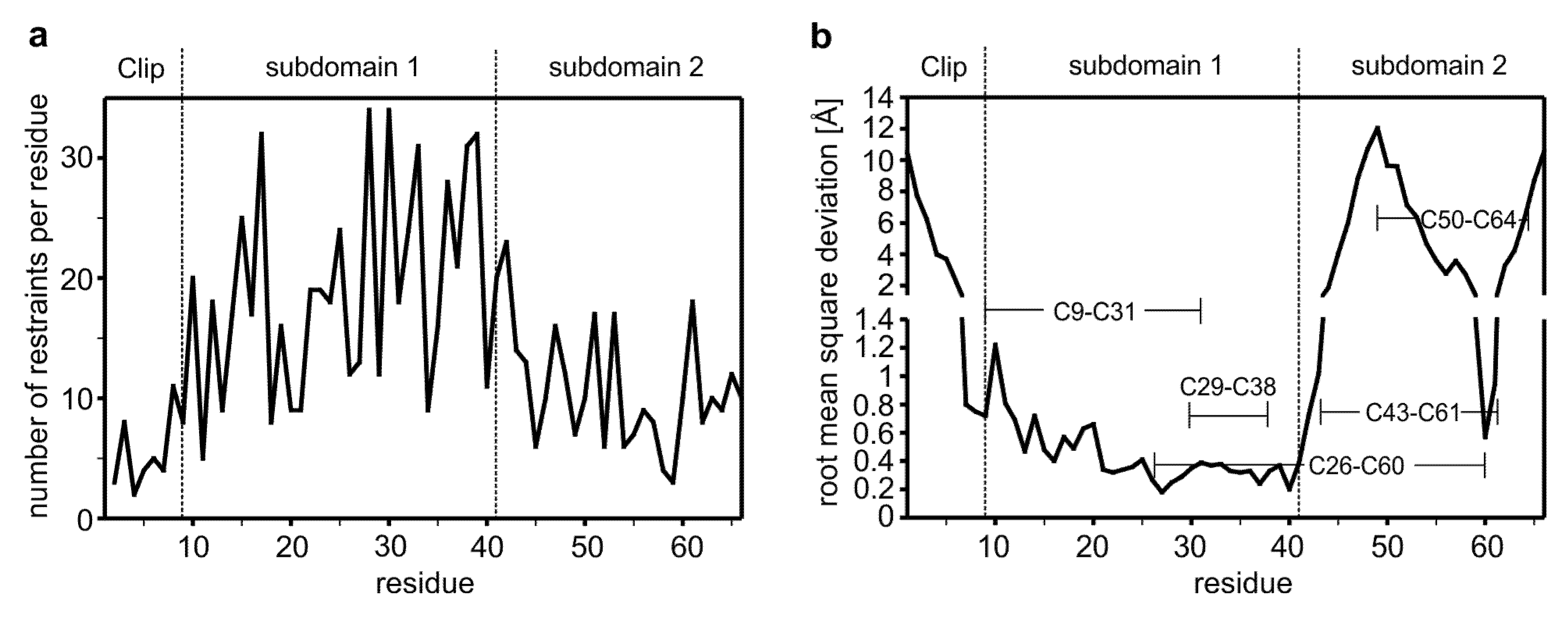{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance restraints | for residues Leu1 to Gly66 |
|---|---|
| NOE-derived | |
| Total | 958 |
| Sequential (|i-j| = 1) | 479 |
| Medium-range (|i-j| ≤ 4) | 168 |
| Long-range (|i-j| > 4) | 311 |
| Average NOE/residue | 14.3 |
| Hydrogen bonds | 3 |
| Structure statistics | |
| RMSD from experimental restraints (NOE) (Å) | 0.067 ± 0.005 |
| RMSD from idealized geometry | |
| Bonds (Å) | 0.006 ± 0.001 |
| Angles (deg.) | 0.664 ± 0.053 |
| Energies (kcal mol.1) | |
| Etotal | −82.1 ± 70.9 |
| Ebond | 32.9 ± 6.1 |
| Eangle | 121.1 ± 19.4 |
| EvdW | 47.2 ± 17.2 |
| Echemshift | 46.1 ± 11.1 |
| Eramachandran | −522.3 ± 23.1 |
| ENOE | 174.0 ± 18.5 |
| RMSD from average structure (Å) a | |
| Backbone atoms for residues 8–41 | 0.46 ± 0.19 |
| All heavy atoms for residues 8–41 | 0.92 ± 0.21 |
| Backbone atoms for residues 1–7 | 1.40 ± 0.40 |
| Backbone atoms for residues 42–66 | 1.99 ± 0.68 |
| PROCHECK analysis | |
| Residues in most favored regions (%) | 64.4 (380) b |
| Residues in additionally allowed regions (%) | 23.7 (140) |
| Residues in generously allowed regions (%) | 11.9 (70) |
| Residues in disallowed regions (%) | 0 (0) |

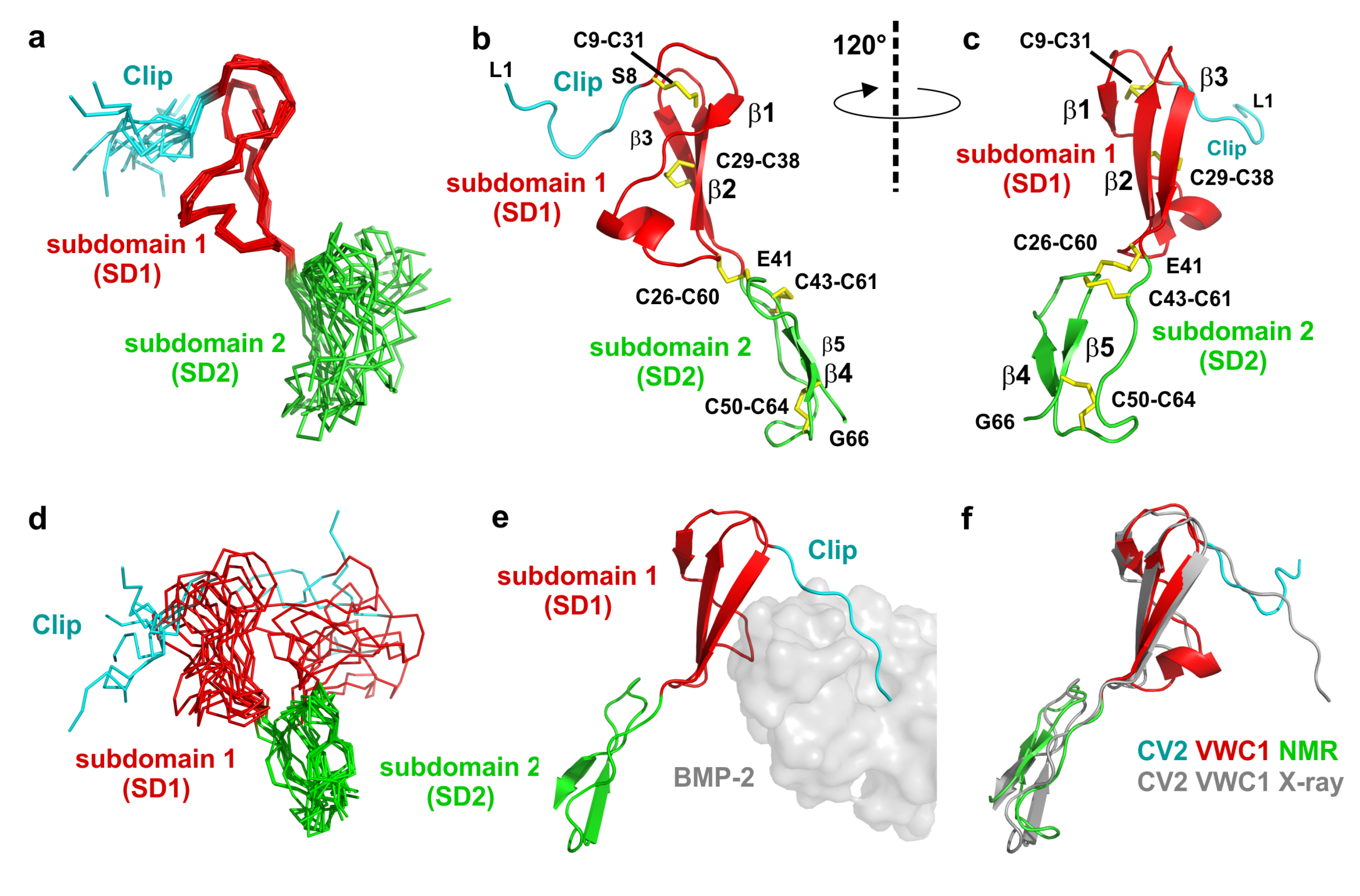
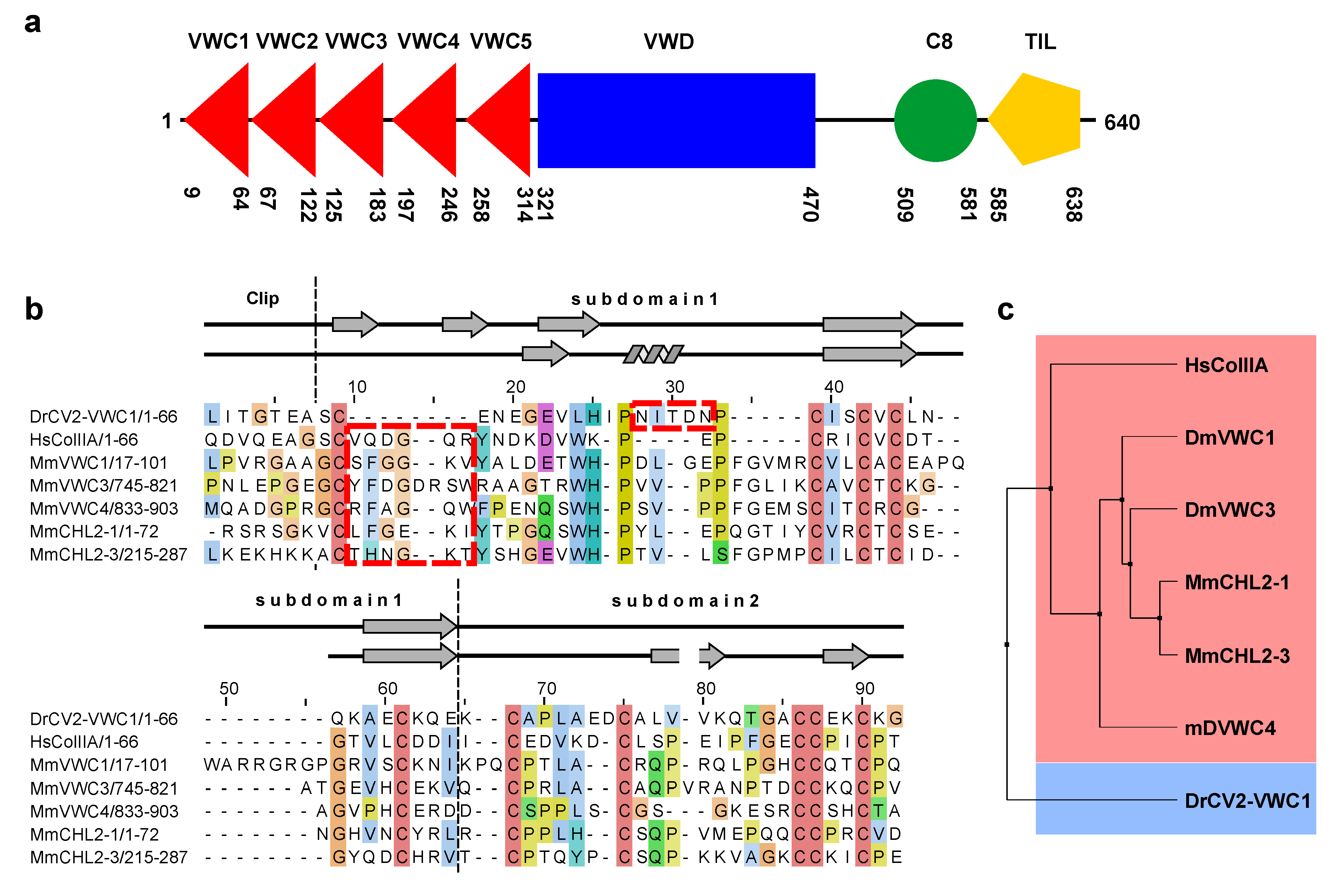
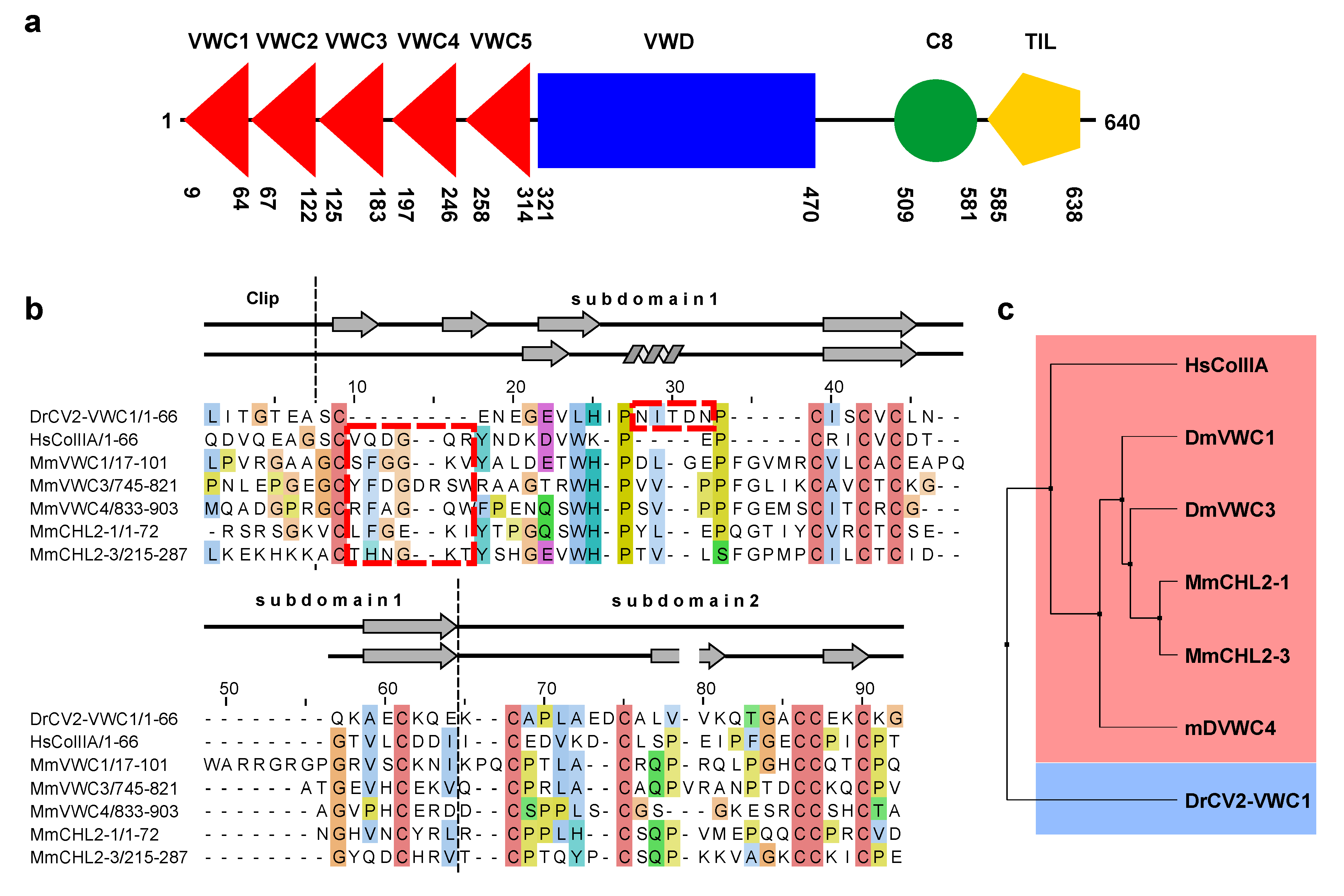
2.3. The Structure of CV2 VWC1 Reveals a Modular Architecture with Flexible and Rigid Segments

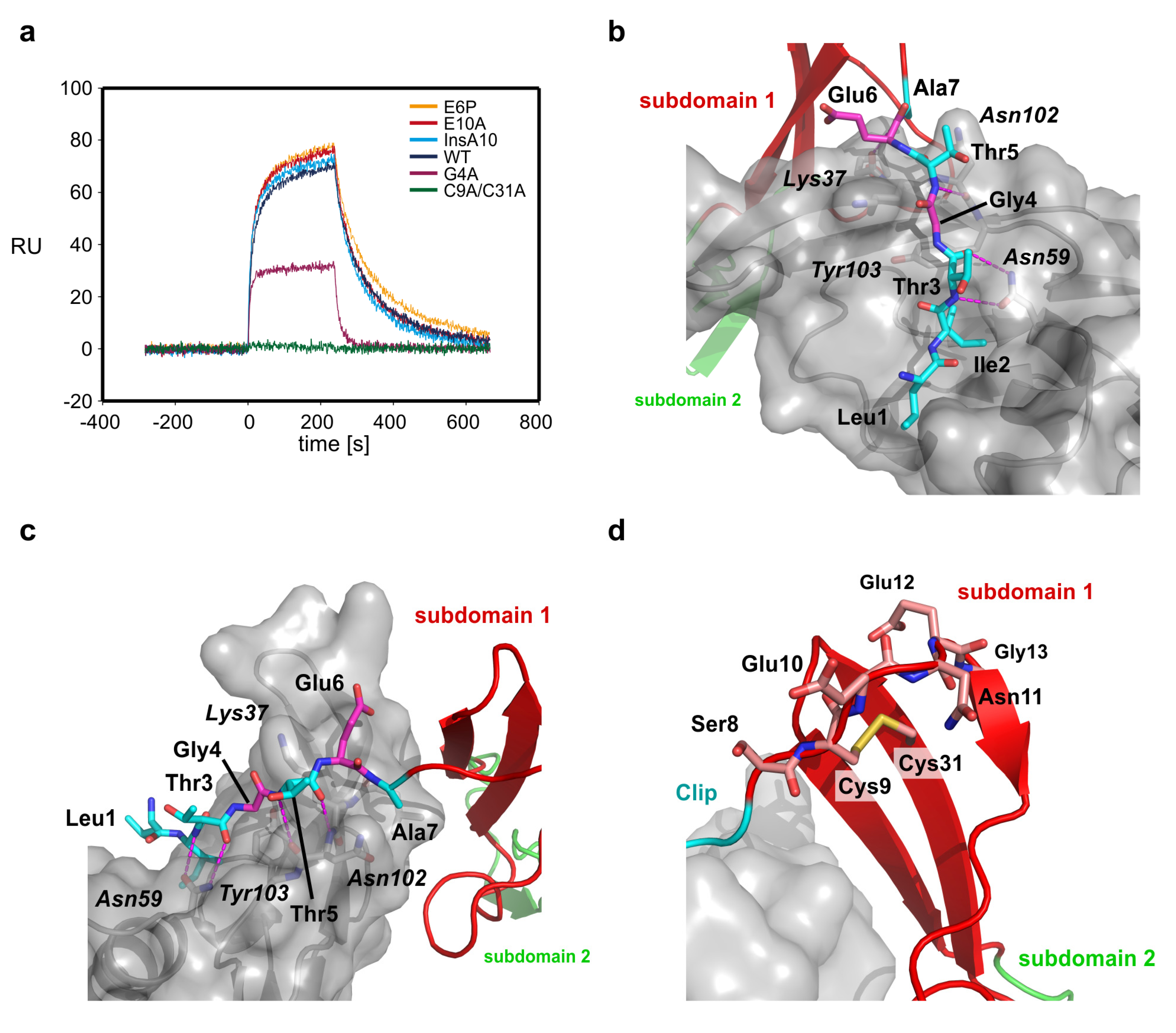
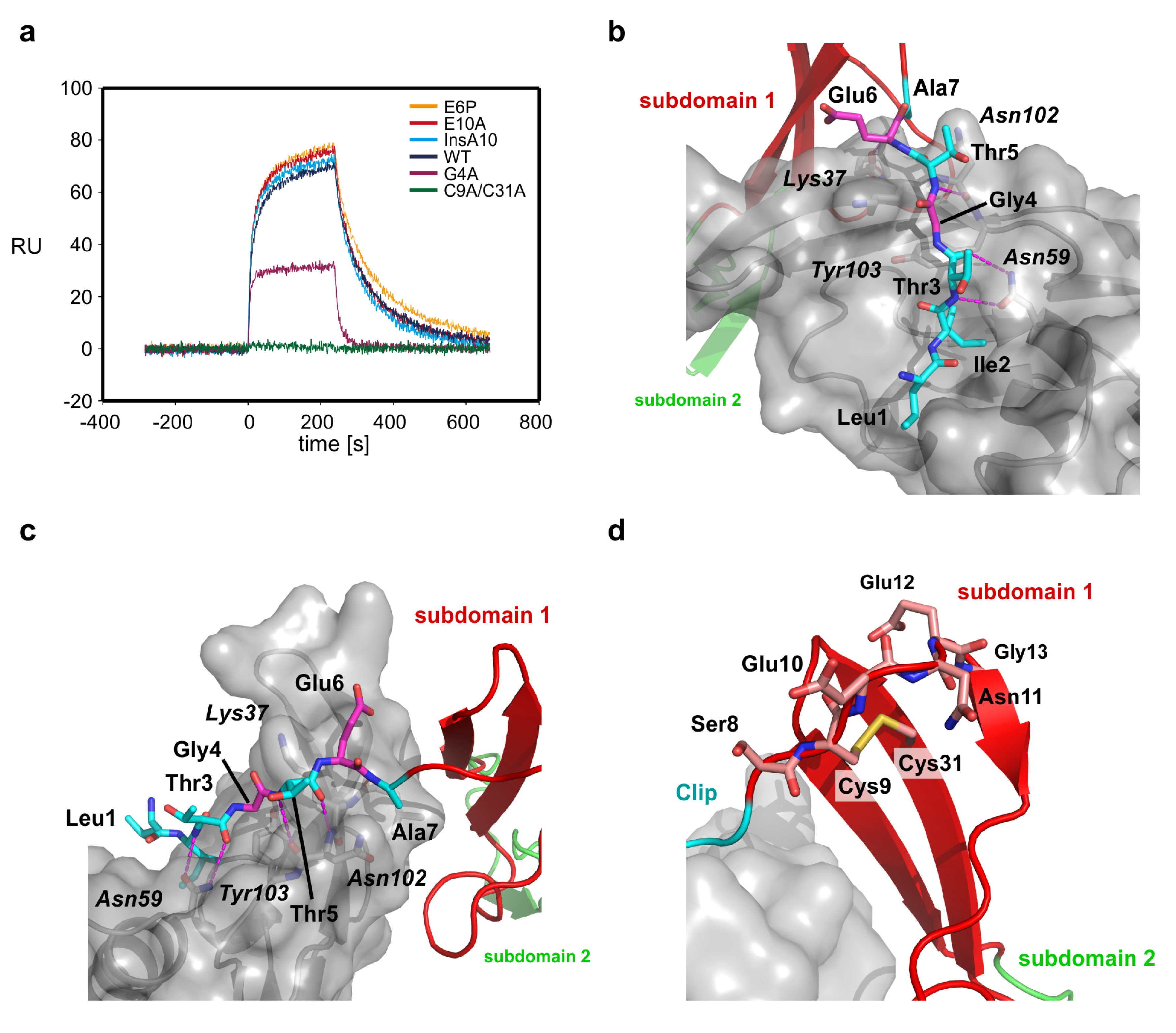
2.4. The Clip Segment Must be Pre-Oriented for High Affinity Binding to BMPs
| CV2 VWC protein | KD (eq) (nM) | kon × 105 (M−1s−1) | koff × 10−3 (s−1) | KD (
koff/kon) (nM) | KD (var)/KD (wildtype) |
|---|---|---|---|---|---|
| wildtype | - | 6.5 ± 0.2 | 8.7 ± 0.1 | 13 ± 4 | 1 |
| G4A | 239 ± 44 | - | ≥50 | - | 18.4 |
| E6P | - | 6.3 ± 0.2 | 7.3 ± 0.1 | 12 ± 3.6 | 0.9 |
| E10A | - | 7.5 ± 0.3 | 9.5 ± 0.1 | 13 ± 4 | 1 |
| InsA10 | - | 9.1 ± 0.2 | 1.2 ± 0.01 | 13 ± 4.4 | 1 |
| C9A/C31A | ≥3300 ± 1400 | - | ≥50 | - | 255 |
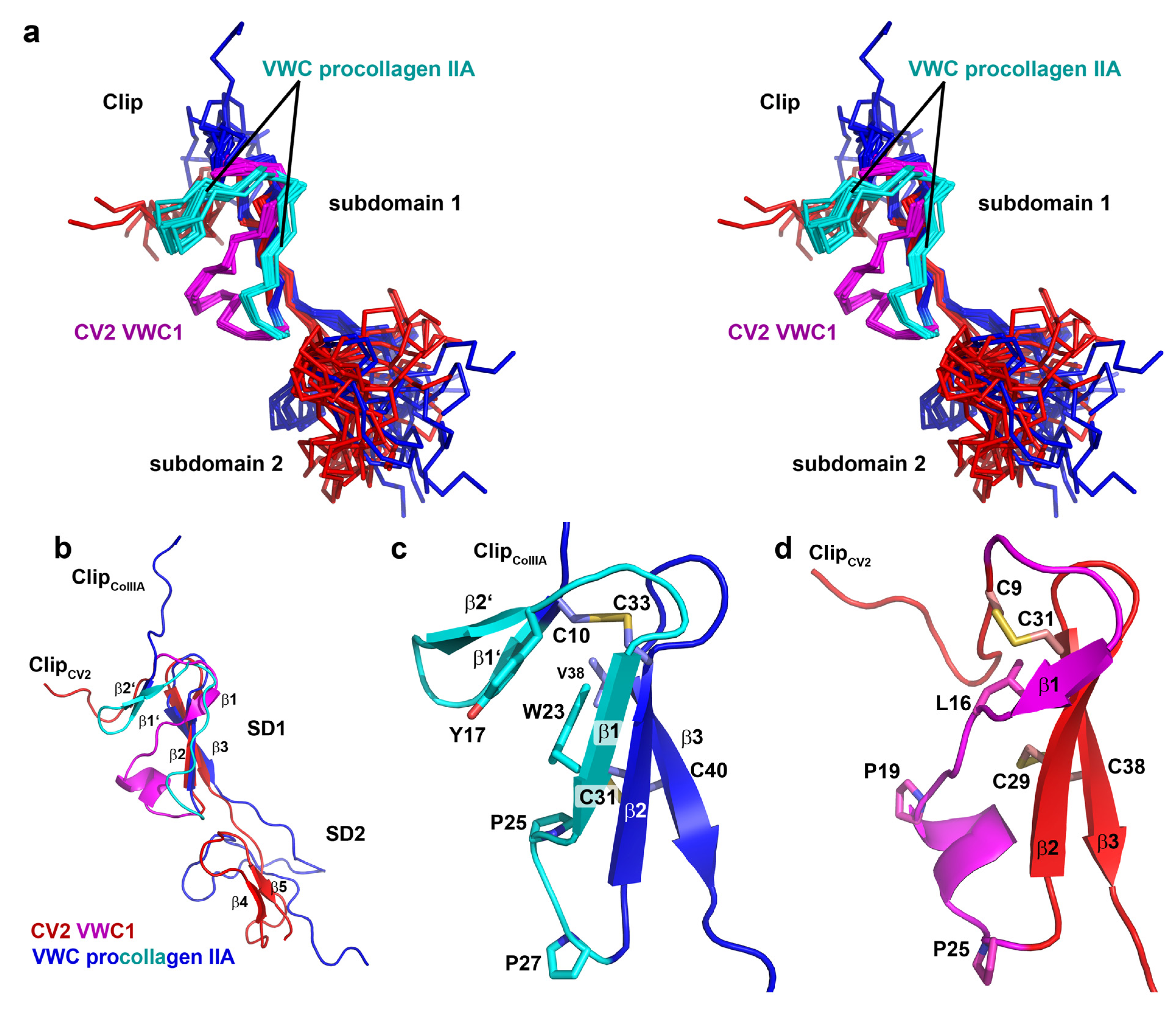
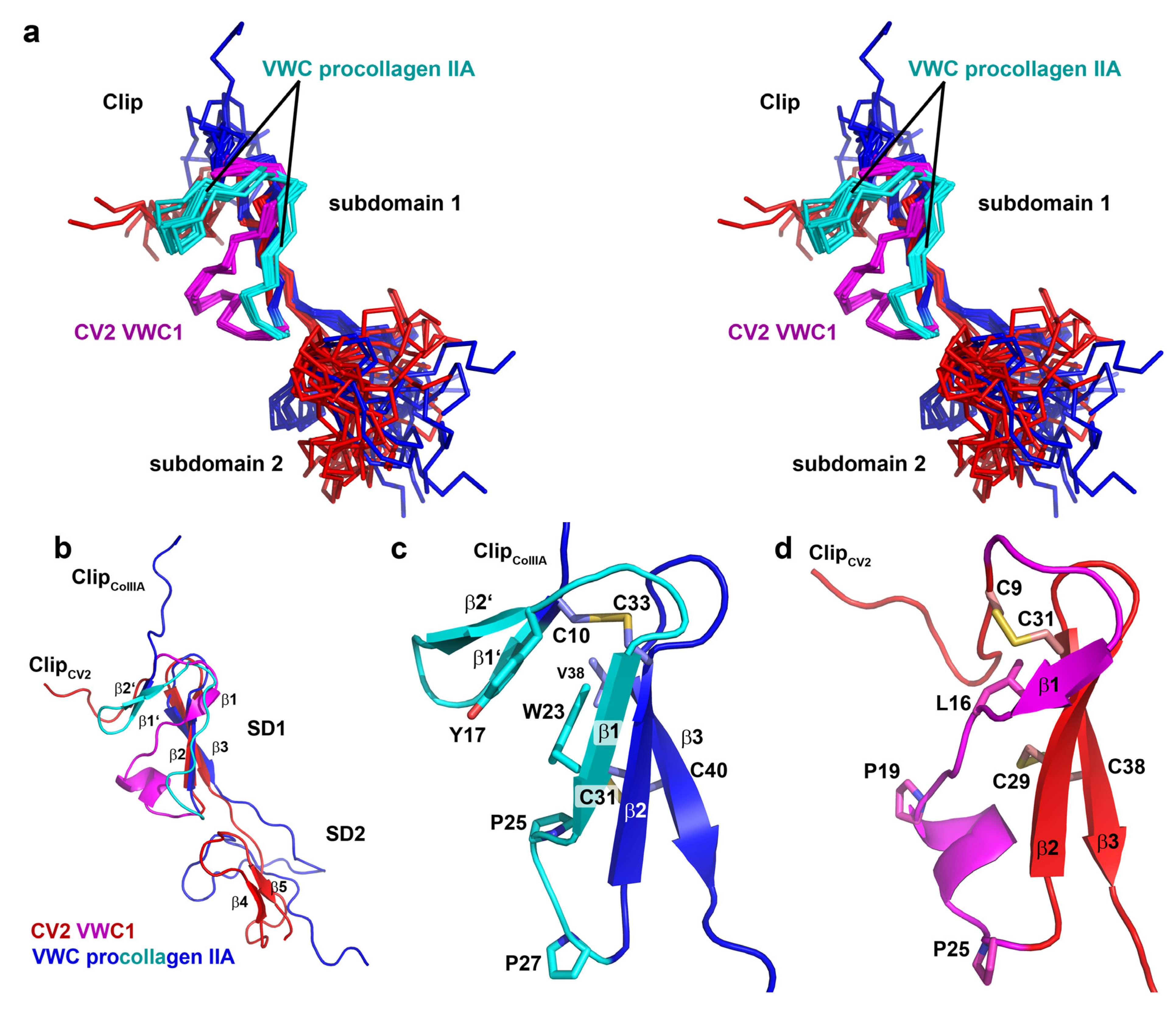
2.5. The First VWC Domain of Crossveinless 2 Possibly Unique Compared to Other VWC Domains
3. Experimental
3.1. Expression and Purification of the VWC1 Domain of Crossveinless 2
3.2. NMR Data Acquisition and Analysis
3.3. In Vitro Interaction Analysis Using Surface Plasmon Resonance
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Nickel, J.; Sebald, W.; Groppe, J.C.; Mueller, T.D. Intricacies of BMP receptor assembly. Cytokine Growth Factor Rev. 2009, 20, 367–377. [Google Scholar] [CrossRef]
- Mueller, T.D.; Nickel, J. Promiscuity and specificity in BMP receptor activation. FEBS Lett. 2012, 586, 1846–1859. [Google Scholar] [CrossRef]
- Graff, J.M. Embryonic patterning: To BMP or not to BMP, that is the question. Cell 1997, 89, 171–174. [Google Scholar] [CrossRef]
- Kishigami, S.; Mishina, Y. BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev. 2005, 16, 265–278. [Google Scholar] [CrossRef]
- Bond, A.M.; Bhalala, O.G.; Kessler, J.A. The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev. Neurobiol. 2012, 72, 1068–1084. [Google Scholar] [CrossRef]
- Botchkarev, V.A.; Sharov, A.A. BMP signaling in the control of skin development and hair follicle growth. Differentiation 2004, 72, 512–526. [Google Scholar] [CrossRef]
- Schneider, M.D.; Gaussin, V.; Lyons, K.M. Tempting fate: BMP signals for cardiac morphogenesis. Cytokine Growth Factor Rev. 2003, 14, 1–4. [Google Scholar] [CrossRef]
- Simic, P.; Vukicevic, S. Bone morphogenetic proteins in development and homeostasis of kidney. Cytokine Growth Factor Rev. 2005, 16, 299–308. [Google Scholar] [CrossRef]
- Zaret, K.S.; Watts, J.; Xu, J.; Wandzioch, E.; Smale, S.T.; Sekiya, T. Pioneer factors, genetic competence, and inductive signaling: Programming liver and pancreas progenitors from the endoderm. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 119–126. [Google Scholar] [CrossRef]
- Mundlos, S. The brachydactylies: a molecular disease family. Clin. Genet. 2009, 76, 123–136. [Google Scholar] [CrossRef]
- Reddi, A.H. Cartilage morphogenetic proteins: Role in joint development, homoeostasis, and regeneration. Ann. Rheum Dis. 2003, 62 (Suppl 2), ii73–ii78. [Google Scholar]
- Zhao, G.Q. Consequences of knocking out BMP signaling in the mouse. Genesis 2003, 35, 43–56. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Chakkalakal, S.A.; Shore, E.M. Fibrodysplasia ossificans progressiva: Mechanisms and models of skeletal metamorphosis. Dis. Model. Mech. 2012, 5, 756–762. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Yadav, P.S.; Prashar, P. BMP signaling in development and diseases: A pharmacological perspective. Biochem. Pharmacol. 2013, 85, 857–864. [Google Scholar] [CrossRef]
- Massague, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef]
- Miyazono, K.; Maeda, S.; Imamura, T. BMP receptor signaling: Transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005, 16, 251–263. [Google Scholar] [CrossRef]
- Hellmann, T.V.; Nickel, J.; Mueller, T.D. Missense Mutations in GDF5 Signaling: Molecular Mechanisms behind Skeletal Malformation. In Gene Mutation; Cooper, D., Chen, J.-M., Eds.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Gazzerro, E.; Canalis, E. Bone morphogenetic proteins and their antagonists. Rev. Endocr. Metab. Disord. 2006, 7, 51–65. [Google Scholar] [CrossRef]
- Cash, J.N.; Rejon, C.A.; McPherron, A.C.; Bernard, D.J.; Thompson, T.B. The structure of myostatin:follistatin 288: Insights into receptor utilization and heparin binding. EMBO J. 2009, 28, 2662–2676. [Google Scholar] [CrossRef]
- Groppe, J.; Greenwald, J.; Wiater, E.; Rodriguez-Leon, J.; Economides, A.N.; Kwiatkowski, W.; Affolter, M.; Vale, W.W.; Izpisua Belmonte, J.C.; Choe, S. Structural basis of BMP signalling inhibition by the cystine knot protein Noggin. Nature 2002, 420, 636–642. [Google Scholar] [CrossRef]
- Harrington, A.E.; Morris-Triggs, S.A.; Ruotolo, B.T.; Robinson, C.V.; Ohnuma, S.; Hyvonen, M. Structural basis for the inhibition of activin signalling by follistatin. EMBO J. 2006, 25, 1035–1045. [Google Scholar] [CrossRef]
- Thompson, T.B.; Lerch, T.F.; Cook, R.W.; Woodruff, T.K.; Jardetzky, T.S. The structure of the follistatin:activin complex reveals antagonism of both type I and type II receptor binding. Dev. Cell 2005, 9, 535–543. [Google Scholar] [CrossRef]
- Zhang, J.L.; Qiu, L.Y.; Kotzsch, A.; Weidauer, S.; Patterson, L.; Hammerschmidt, M.; Sebald, W.; Mueller, T.D. Crystal structure analysis reveals how the Chordin family member crossveinless 2 blocks BMP-2 receptor binding. Dev. Cell 2008, 14, 739–750. [Google Scholar] [CrossRef]
- Zhang, J.L.; Huang, Y.; Qiu, L.Y.; Nickel, J.; Sebald, W. Von Willebrand factor type C domain-containing proteins regulate bone morphogenetic protein signaling through different recognition mechanisms. J. Biol. Chem. 2007, 282, 20002–20014. [Google Scholar] [CrossRef]
- Mancuso, D.J.; Tuley, E.A.; Westfield, L.A.; Worrall, N.K.; Shelton-Inloes, B.B.; Sorace, J.M.; Alevy, Y.G.; Sadler, J.E. Structure of the gene for human von Willebrand factor. J. Biol. Chem. 1989, 264, 19514–19527. [Google Scholar]
- Bork, P. Shuffled domains in extracellular proteins. FEBS Lett. 1991, 286, 47–54. [Google Scholar] [CrossRef]
- Patthy, L. Detecting homology of distantly related proteins with consensus sequences. J. Mol. Biol. 1987, 198, 567–577. [Google Scholar] [CrossRef]
- Hunt, L.T.; Barker, W.C. Von Willebrand factor shares a distinctive cysteine-rich domain with thrombospondin and procollagen. Biochem. Biophys. Res. Commun. 1987, 144, 876–882. [Google Scholar] [CrossRef]
- Schultz, J.; Milpetz, F.; Bork, P.; Ponting, C.P. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc. Natl. Acad. Sci. USA 1998, 95, 5857–5864. [Google Scholar]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef]
- Garcia Abreu, J.; Coffinier, C.; Larrain, J.; Oelgeschlager, M.; de Robertis, E.M. Chordin-like CR domains and the regulation of evolutionarily conserved extracellular signaling systems. Gene 2002, 287, 39–47. [Google Scholar] [CrossRef]
- Gong, Y.; Krakow, D.; Marcelino, J.; Wilkin, D.; Chitayat, D.; Babul-Hirji, R.; Hudgins, L.; Cremers, C.W.; Cremers, F.P.; Brunner, H.G.; et al. Heterozygous mutations in the gene encoding noggin affect human joint morphogenesis. Nat. Genet. 1999, 21, 302–304. [Google Scholar] [CrossRef]
- Hirshoren, N.; Gross, M.; Banin, E.; Sosna, J.; Bargal, R.; Raas-Rothschild, A. P35S mutation in the NOG gene associated with Teunissen-Cremers syndrome and features of multiple NOG joint-fusion syndromes. Eur. J. Med. Genet. 2008, 51, 351–357. [Google Scholar] [CrossRef]
- Mangino, M.; Flex, E.; Digilio, M.C.; Giannotti, A.; Dallapiccola, B. Identification of a novel NOG gene mutation (P35S) in an Italian family with symphalangism. Hum. Mutat. 2002, 19, 308. [Google Scholar]
- Zhang, J.L.; Patterson, L.J.; Qiu, L.Y.; Graziussi, D.; Sebald, W.; Hammerschmidt, M. Binding between Crossveinless-2 and Chordin von Willebrand factor type C domains promotes BMP signaling by blocking Chordin activity. PLoS One 2010, 5, e12846. [Google Scholar]
- Qiu, L.Y.; Zhang, J.L.; Kotzsch, A.; Sebald, W.; Mueller, T.D. Crystallization and preliminary X-ray analysis of the complex of the first von Willebrand type C domain bound to bone morphogenetic protein 2. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2008, 64(Pt 4), 307–312. [Google Scholar]
- Ortenberg, R.; Gon, S.; Porat, A.; Beckwith, J. Interactions of glutaredoxins, ribonucleotide reductase, and components of the DNA replication system of Escherichia coli. Proc. Natl. Acad. Sci. USA 2004, 101, 7439–7444. [Google Scholar] [CrossRef]
- Ritz, D.; Lim, J.; Reynolds, C.M.; Poole, L.B.; Beckwith, J. Conversion of a peroxiredoxin into a disulfide reductase by a triplet repeat expansion. Science 2001, 294, 158–160. [Google Scholar] [CrossRef]
- Klages, J.; Kotzsch, A.; Coles, M.; Sebald, W.; Nickel, J.; Muller, T.; Kessler, H. The solution structure of BMPR-IA reveals a local disorder-to-order transition upon BMP-2 binding. Biochemistry 2008, 47, 11930–11939. [Google Scholar] [CrossRef]
- Watt, M.A.; Lo, R.Y.; Mellors, A. Refolding of recombinant Pasteurella haemolytica A1 glycoprotease expressed in an Escherichia coli thioredoxin gene fusion system. Cell Stress Chaperones 1997, 2, 180–190. [Google Scholar] [CrossRef]
- Cavanagh, J. Protein NMR Spectroscopy: Principles and Practice; Academic Press: San Diego, CA, USA, 1996; pp. xxiii, 587. [Google Scholar]
- Clore, G.M.; Gronenborn, A.M. New methods of structure refinement for macromolecular structure determination by NMR. Proc. Natl. Acad. Sci. USA 1998, 95, 5891–5898. [Google Scholar] [CrossRef]
- Kuszewski, J.; Gronenborn, A.M.; Clore, G.M. Improving the quality of NMR and crystallographic protein structures by means of a conformational database potential derived from structure databases. Protein Sci. 1996, 5, 1067–1080. [Google Scholar] [CrossRef]
- O'Leary, J.M.; Hamilton, J.M.; Deane, C.M.; Valeyev, N.V.; Sandell, L.J.; Downing, A.K. Solution structure and dynamics of a prototypical chordin-like cysteine-rich repeat (von Willebrand Factor type C module) from collagen IIA. J. Biol. Chem. 2004, 279, 53857–53866. [Google Scholar]
- Ambrosio, A.L.; Taelman, V.F.; Lee, H.X.; Metzinger, C.A.; Coffinier, C.; de Robertis, E.M. Crossveinless-2 Is a BMP feedback inhibitor that binds Chordin/BMP to regulate Xenopus embryonic patterning. Dev. Cell 2008, 15, 248–260. [Google Scholar] [CrossRef]
- Fujisawa, T.; Huang, Y.; Sebald, W.; Zhang, J.L. The binding of von Willebrand factor type C domains of Chordin family proteins to BMP-2 and Tsg is mediated by their SD1 subdomain. Biochem. Biophys. Res. Commun. 2009, 385, 215–219. [Google Scholar] [CrossRef]
- Ganderton, T.; Wong, J.W.; Schroeder, C.; Hogg, P.J. Lateral self-association of VWF involves the Cys2431-Cys2453 disulfide/dithiol in the C2 domain. Blood 2011, 118, 5312–5318. [Google Scholar] [CrossRef]
- Bork, P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993, 327, 125–130. [Google Scholar] [CrossRef]
- Larrain, J.; Bachiller, D.; Lu, B.; Agius, E.; Piccolo, S.; de Robertis, E.M. BMP-binding modules in chordin: A model for signalling regulation in the extracellular space. Development 2000, 127, 821–830. [Google Scholar]
- Zhou, Y.F.; Eng, E.T.; Zhu, J.; Lu, C.; Walz, T.; Springer, T.A. Sequence and structure relationships within von Willebrand factor. Blood 2012, 120, 449–458. [Google Scholar] [CrossRef]
- Marley, J.; Lu, M.; Bracken, C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 2001, 20, 71–75. [Google Scholar] [CrossRef]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor-NIH for NMR molecular structure determination. Progr. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. [Google Scholar]
- Brunger, A.T. XPLOR (Version 3.1) A System for X-ray Crystallography and NMR; Yale University, The Howard Hughes Medical Institute and Department of Molecular Biophysics and Biochemistry: New Haven, CT, USA, 1992. [Google Scholar]
- Kuszewski, J.; Qin, J.; Gronenborn, A.M.; Clore, G.M. The impact of direct refinement against 13C alpha and 13C beta chemical shifts on protein structure determination by NMR. J. Magn. Reson. B 1995, 106, 92–96. [Google Scholar] [CrossRef]
- Ruppert, R.; Hoffmann, E.; Sebald, W. Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur. J. Biochem. 1996, 237, 295–302. [Google Scholar]
- Sample Availability: Samples of the compound CV2 VWC1 are available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fiebig, J.E.; Weidauer, S.E.; Qiu, L.-Y.; Bauer, M.; Schmieder, P.; Beerbaum, M.; Zhang, J.-L.; Oschkinat, H.; Sebald, W.; Mueller, T.D. The Clip-Segment of the von Willebrand Domain 1 of the BMP Modulator Protein Crossveinless 2 Is Preformed. Molecules 2013, 18, 11658-11682. https://doi.org/10.3390/molecules181011658
Fiebig JE, Weidauer SE, Qiu L-Y, Bauer M, Schmieder P, Beerbaum M, Zhang J-L, Oschkinat H, Sebald W, Mueller TD. The Clip-Segment of the von Willebrand Domain 1 of the BMP Modulator Protein Crossveinless 2 Is Preformed. Molecules. 2013; 18(10):11658-11682. https://doi.org/10.3390/molecules181011658
Chicago/Turabian StyleFiebig, Juliane E., Stella E. Weidauer, Li-Yan Qiu, Markus Bauer, Peter Schmieder, Monika Beerbaum, Jin-Li Zhang, Hartmut Oschkinat, Walter Sebald, and Thomas D. Mueller. 2013. "The Clip-Segment of the von Willebrand Domain 1 of the BMP Modulator Protein Crossveinless 2 Is Preformed" Molecules 18, no. 10: 11658-11682. https://doi.org/10.3390/molecules181011658
APA StyleFiebig, J. E., Weidauer, S. E., Qiu, L.-Y., Bauer, M., Schmieder, P., Beerbaum, M., Zhang, J.-L., Oschkinat, H., Sebald, W., & Mueller, T. D. (2013). The Clip-Segment of the von Willebrand Domain 1 of the BMP Modulator Protein Crossveinless 2 Is Preformed. Molecules, 18(10), 11658-11682. https://doi.org/10.3390/molecules181011658